

Abstract
Tumor necrosis factor related apoptosis-inducing ligand (TRAIL) was initially described to induce apoptosis of tumor cells and/or virally infected cells while sparing normal cells and has been implicated in the pathogenesis of HIV disease. We previously identified TRAILshort, a TRAIL splice variant, in HIV infected patients and characterized it as being a dominant negative ligand to subvert TRAIL-mediated killing. Herein using single cell genomics we demonstrate that TRAILshort is produced by HIV infected cells, as well as by uninfected bystander cells, and that the dominant stimulus which induces TRAILshort production are type I interferons and Toll Like Receptor 7, 8, and 9 agonists. TRAILshort has a short half-life by virtue of containing a PEST domain, which targets the protein towards the ubiquitin proteasome pathway for degradation. Further we show that TRAILshort binds preferentially to TRAIL receptors 1 and 2 with significantly reduced interaction with the decoy TRAIL receptors 3 and 4. Recombinant TRAILshort is sufficient to protect cells against TRAIL-induced killing, while immunodepletion of TRAILshort with a specific antibody restores TRAIL sensitivity. Importantly we show that TRAILshort is shed in microvesicles into the cellular microenvironment and therefore confers TRAIL resistance not only on the cell which produces it, but also upon neighboring bystander cells. These results establish a novel paradigm for understanding and overcoming TRAIL resistance, in particular how HIV infected cells escape immune elimination by the TRAIL:TRAILshort receptor axis.
Introduction
TNF-related apoptosis-inducing ligand (TRAIL) is an immuno-regulatory protein, which can kill virally infected or malignant cells through binding to TRAIL receptor R1 or TRAIL-R2 on target cells. TRAIL has been implicated in immune surveillance and contributes to the control of adaptive T cell responses. The role of TRAIL in immune surveillance is poignantly demonstrated in mouse studies wherein TRAIL or TRAIL receptor deficient mice spontaneously develop stromal and lymphoid tumors (1, 2). TRAIL can bind to one of five cognate receptors, TRAIL-R1, R2, R3, R4, or osteoprotegerin, yet only binding to TRAIL-R1 or R2 induces death through apoptosis of the receptor bearing cell (3). Homo-oligomerization of TRAIL and its cognate death receptors leads to recruitment of multiple proteins, including the initiator procaspase-8, into the death inducing signaling complex (DISC). During apoptosis, clustering of procaspase-8 near the death receptors leads to their autocatalytic cleavage and activation, which in turn directly or indirectly cleaves and activates caspase-3, the central executioner caspase, leading to the phenotypic and biochemical events of apoptosis.
Concerning the role of TRAIL in HIV immunopathogenesis, two seemingly disparate observations were made: TRAIL mediates the depletion of B cells (4) and uninfected CD4T cells (5), while other cell subsets become HIV resistant when infected with HIV (6). Furthermore, while there is abundant TRAIL expressed by cells from HIV infected patients (7), cells which contain HIV are not eliminated during natural infection; conversely, treatment of HIV infected CD4 T cells or HIV infected macrophages with supra-physiologic levels of TRAIL agonists caused the preferential killing of HIV infected cells resulting in decreased HIV reservoir size ex vivo (8), arguing that these cells might resist the physiologic levels of TRAIL seen in an infected host. This seeming paradox is what led us to discover the presence of TRAILshort (9).
TRAILshort is a novel splice variant of TRAIL, a 101-amino acid polypeptide that shares the first 90 amino acids with full length TRAIL (TRAILFL). TRAIL is encoded by 5 exons. The splicing event that generates TRAILshort consists of excision of exons 3 and 4 and the introduction of a frameshift in exon 5 resulting in a unique 11 amino acid carboxyl terminus and a premature stop codon. TRAILshort lacks apoptosis-inducing activity and acts as an antagonist of TRAILFL possibly explaining why HIV infected cells are not eradicated by endogenously produced TRAIL, which is increased during HIV infection (9).
Herein we evaluate whether HIV infected or uninfected cells produce TRAILshort and show that both infected and uninfected cells produce the splice variant, largely in response to type I interferons (IFN) and Toll Like Receptor 7, 8, and 9 agonists. We further demonstrate that the C terminus of TRAILshort is extracellular to the plasma membrane, where it interacts preferentially with death inducing TRAIL receptors TRAIL-R1 and R2, and significantly less with the decoy TRAIL receptors TRAIL-R3 and R4. TRAILshort alone is sufficient to confer TRAIL resistance, and depleting TRAILshort with a specific monoclonal antibody converts a TRAILshort expressing TRAIL resistant cell into a TRAIL sensitive cells. Importantly, TRAILshort is incorporated into extracellular vesicles, allowing TRAILshort-mediated resistance to be conferred onto neighboring cells within the microenvironment. These findings underscore the significance of TRAILshort in regulating TRAIL sensitivity, suggest that TRAILshort is of pathophysiologic relevance in disease states characterized by elevated type I IFN expression, and identify a novel approach to restoring TRAIL sensitivity through monoclonal antibody inhibition of TRAILshort.
Materials and Methods
Isolation of CD4 cells from Donor Patients
Blood samples from HIV positive donors were obtained through a Mayo Clinic Institutional Review Board approved protocol, following informed consent. Peripheral blood mononuclear cells were isolated by density gradient centrifugation using Ficoll Paque. Primary CD4 T cells were isolated using negative selection by RosetteSep with Human CD4+ T Cell Enrichment Cocktail (Stemcell Technologies) per manufacture’s protocol.
DNA Plasmid construction
We have previously described our plasmids for TRAIL short expression (9). TRAILshort with an N-terminal Ruby tag was PCR subcloned into mRuby2-C1 (Addgene, #54768) using the Bgl II and SalI restriction sites and TRAILshort with a single HA tag was generated in pcDNA3. A moiety with a deleted transmembrane domain was additionally generated with Pfu DNA polymerase (MBL International). TRAILshort-ECD:Fc fusion was constructed through PCR cloning and subcloned into pFUSE-hIgG4-Fc1 (InvivoGen) at EcoRI and XhoI sites. TRAILshort ECD:Fc fusion protein was purified from transiently transfected HEK293T cells on a HiTrap Protein G HP column (1 mL, GE Healthcare) following manufacturer’s instruction. TRAILshort with the internal c-myc epitope was cloned into pcDNA3 using the Bam HI and NotI restriction sites via overlap extension PCR. All final sequences were verified by direct sequencing. The following primers were used: Ruby-TRAILshort, 5’-CAGCAGATCTATGGCTATGATGGAGGTCC-3’ and 5’-GTTTGTCGACGCTACTATTTTGCGGCCCAGAGCC-3’; Ruby-TRAILshortΔTM, 5’-CTGGGACAGACCTGCGTCACCAACGAGCTGAAGCAGATG-3’ and 5’-CATCTGCTTCAGCTCGTTGGTGACGCAGGTCTGTCCCAG-3’; TRAILshort-ECD:Fc, 5’-GTGAATTCATGTTTTGCGGCCCAGAGCCTTTTC-3’ and 5’-TGCTCGAGTGACCAACGAGCTGAAGCAGATGCAG-3’; c-myc-TRAILshort, 5’-GTGTTGGGATCCGTCATGGCTATGATGGAGGTCC-3’, 5’-CCTCTTCTGAGATGAGTTTTTGTTCTTGCCACTTGACTTG-3’, 5’-CAAAAACTCATCTCAGAAGAGGATCTCCGTCAGCTCGTTAG-3’, and 5’-CATTGCGGCCGCTACTATTTTGCGGCCGAGA-3’.
Cell Transfections
HEK293T cells, Jurkat T cells, or HeLa cells were transfected with standard methods using calcium phosphate or lipofectamine.
Western analysis and immunoprecipitation
For Western analysis cell lysates were resolved on 12.5% a SDS-PAGE (25 mM Tris, 192 mM Glycine, and 0.05% SDS) then transferred onto PVDF, blocked with 2% BSA in TBST (50mM Tris, 150mM NaCL, 0.2% Tween), incubated at room temperature overnight with primary antibodies in TBST, washed then incubated with HRP-conjugated and detected by enhanced chemiluminescence (SuperSignal West Pico Chemiluminescent Substrate) For reprobing of blots, antibody was stripped for 15 min in 50 mM Tris pH 6.8 buffer containing 1% SDS and 115 μM β-mercaptoethanol.
For immunoprecipitation, pre cleared lysates were incubated with anti-HA or anti-FLAG pre-bound to Protein A/G PLUS agarose for 1 hour at 4°C while rotating. Bound proteins were eluted from the resin with 1x concentrated loading buffer.
Cellular immunofluorescence microscopy
Cover slips with attached cells were washed with PBS, fixed for 30 minutes with 2.5% paraformaldehyde and counterstained with 1 μg/mL of DAPI. Coverslips were washed a final time with deionized distilled water (ddH2O) and mounted on glass slides with Prolong Gold mounting solution. Cells were then visualized using a Zeiss Axio Observer D.1 inverted fluorescent microscope with a 63x objective with 1.4NA oil lens. Images were captured with a Hamamatsu ORCA II ERG digital camera driven by iVision software (BioVision Technologies). Microparticles from eGFP TRAILFL and Ruby-TRAILshort expressing cells HEK293T cells were analyzed in a similar manner.
Single cell transcriptional analysis
Sorted live cells were counted and measured for size and viability using the Vi-Cell XR Cell Viability Analyzer (Beckman-Coulter PN 383556). A C1 Single-Cell Array Integrated Fluidics Circuit (IFC; cell size 5-10 μm; Fluidigm PN 100-5757) was primed and cells were pipetted into the IFC and cell lysis, reverse transcription and pre-amplification reagents were added along with primers specific for the desired transcripts. An average of 50 individual pre-amplified cDNA samples per IFC were harvested and diluted at a ratio of 1:5. All samples were interrogated in qPCR experiments using the Fluidigm BioMark system and custom primers for the desired gene transcripts. Raw CT values were exported into a spreadsheet or analyzed by the Fluidigm Real-Time PCR Analysis software (version 4.1.3).
Isolation of extracellular vesicles and exosomes from conditioned media of primary CD4 and transfected cells
Primary CD4+ cells were isolated as described and maintained for 24 hours in RPMI with 10% FBS which was pre-cleared of particulate matter and endogenous vesicles by centrifugation at 112,000 × g for 16 to 18 hours. Following incubation, the cultures were centrifuged to remove cells and large debris, then supernatant centrifuged 24,000 × g for 16 hours, and the pellet containing extracellular vesicles retained. Indicated exosomes were isolated using ExoQuick-TC (System Biosciences) isolation reagent according to manufacturer’s protocol.
Transmission electron microscopy of extracellular vesicles
For transmission electron microscopy (TEM), extracellular vesicle pellets were suspended in fixative (4% paraformaldehyde + 1% glutaraldehyde in 0.1 M phosphate buffer, pH 7.0) and 4 μL of fixed vesicles were loaded onto 200 mesh, formvar/carbon coated, copper grids (EMS, Hatfield, PA). Grids were rinsed twice with a drop of ddH2O and stained with 1% phosphotungstic acid in ddH2O, pH 7.0 (EMS). Images were collected on a JEOL, J1400+ operating at 80kV. Images were analyzed using ImageJ (http://imagej.nih.gov) to determine the diameters of the vesicles.
Flow cytometric analyses
Flow cytometric analyses were performed on a FACSCalibur (BD Bioscience) flow cytometer with the appropriate filter sets for the different fluorescent stains being detected with BD FACS Diva Software (Core Facility in Mayo Clinic) and analyzed with FlowJo v10.2 (FlowJo, LLC). Gating against each detection channel are as indicated in the figures. Microparticles with fluorescent TRAIL fusion proteins were analyzed on a BD Canto X flow cytometer (BD Biosciences). Where indicated cells were permeabilized in PBS containing 0.1% NP-40 and 2% BSA. The following antibodies were used: monoclonal – rabbit anti-HA tag (AB_1587101, Millipore), rat anti-HA-HRP (AB_390917, Roche), mouse anti-FLAG-M2 HRP (AB_439702, Sigma), mouse anti-human TRAILshort (Mayo Clinic Hybridoma core), mouse anti-human TRAIL-R1 (AB_468187, eBioscience), mouse anti-human TRAIL-R2 (AB_10668836, eBioscience), mouse anti-human TRAIL-R3 (AB_468067, eBioscience); polyclonal – rabbit anti-c-myc (AB_631274, Santa Cruz), DyLight 649 donkey anti-rabbit IgG (AB_1057590, BioLegend), goat anti-actin (AB_630836, Santa Cruz), rabbit anti-human TRAIL (AB_227032, Proteintech); control – rabbit IgG (AB_73719, Santa Cruz).
For experiments involving assessment of effect of TRAILshort containing extracellular vesicles, HeLa cells were grown in FBS that was pre-cleared of particulate matter and endogenous vesicles as above. Extracellular vesicles that were collected as above were was added to the HeLa cells for 1 h, then 1 ng/mL SuperKiller TRAIL (sk-TRAIL; Enzo Life sciences) was added. To measure cell death, the IncuCyte Casapse 3/7 apoptosis assay reagent (Essen Bioscience Cat# 4440) was added to the cells at a dilution of 1:1000. For live time analysis of cell death, the Incucyte (Essen bioscience) was used to capture real time images of the cells every 2 hours. Analysis of the data was done using the Incucyte Zoom software (2016A). Each treatment was done in triplicate.
For experiments assessing effects of anti-TRAILshort antibody and TRAILshort-ECD:Fc fusion proteins, Jurkat cells were preincubated with the either anti-TRAILshort antibody or the TRAILshort ECD-Fc fusion protein at the indicated concentrations (5, 2.5, and 1 μg/mL) for one hour then sk-TRAIL was added. The IncuCyte Casapse 3/7 apoptosis assay reagent (Essen Bioscience Cat# 4440) was added to the cells at a dilution of 1:1000 and cell death kinetics was measured as above.
ELISA analyses
96 well Flat Bottom plates (Immulon 4HBX, Thermo Fisher Scientific) were coated with 100 μl of TRAIL R1-Fc, R2-Fc, R3-Fc, or R4-Fc fusion proteins (R&D systems, 1 μg/ml) in triplicate overnight at 4°C, then washed three times with 200 μl PBS containing 0.05% Tween20. The ability of TRAIL receptors binding to TRAIL short was determined with by adding 100 μl of a peptide (Phoenix Pharmaceuticals) corresponding to the extracellular amino acids 30-101 of TRAILshort (10 ng/ml in PBS containing 0.05% Tween20) at room temperature for 2 hours. Bound TRAILshort peptides were detected with biotin-labeled anti-TRAILshort antibody (9) (0.01 μg/ml in PBS containing 0.05% Tween20) and HRP conjugated streptavidin (1:120000 in PBS containing 0.05% Tween20). After washing with 200 μl PBS containing 0.05% Tween20 and twice with PBS, 100 μl of the substrate OPD solution (1mg/ml in 0.05M citric acid, 0.05M sodium phosphate; pH 5 and 0.03% hydrogen peroxide). The absorbance was read at 490 nm with a plate reader (Synergy HT, BioTek) with software Gen5 1.11 after adding 100 μl of stop solution (2N H2SO4).
Confocal microscopy
Putative colocalization of TRAIL receptors with TRAILshort was assessed by co-transfecting either with pEGFPC1, pEGFPC1-TRAILshort and with sequentially pEZ-M14-Flag-tagged TRAIL receptor R1, R2, R3 and R4 (GenCopoeia) using Polyjet (SignaGen Laboratories). After incubation for 12 hours, cells were stained with Alexa Fluor® 594-conjugated antibodies against either human TRAIL R1, R2, R3 or R4 (R&D systems) at room temperature for 1 hour then counterstained with DAPI (4′,6-diamidino-2-phenylindole) and fixed. Cells were imaged at 555 nm, 488 nm, and 405 nm by confocal microscopy (Magnification 40X, ZEISS, Mayo Imaging Core Facility) using ZEN 2012 image software (ZEISS).
Statistical Analysis
Statistical analysis was performed using GraphPad Prism 7.00 (GraphPad Software Inc). Where appropriate, figure legends define the statistical test and associated parameters used to analyze data displayed in the corresponding figure. Data are judged to be statistically significant when p<0.05 in applied statistical analyses (Student’s t-test).
Results
TRAILshort is produced by both uninfected and HIV infected cells
We previously reported that HIV infection of primary CD4 T cells results in the production of TRAILshort message and protein, and that TRAILshort is present in the soluble (supernatant) fraction of HIV infected cultures as well as in the plasma of HIV infected patients (9). Yet it remained unknown whether production of TRAILshort in an individual cell requires productive HIV infection. Given that TRAILshort is present in plasma, it may be bound both to cells that produce it as well as to cells that do not. Therefore, tracking the protein is unlikely to discriminate TRAILshort producing cells from TRAILshort binding cells. To circumvent that possibility, we assessed TRAILshort production in HIV infected versus uninfected cells using single cell mRNA analysis. In these experiments single CD4 T cells from HIV infected or uninfected donors were separated by an integrated fluidic circuit and individual cell analyses were performed using PCR for HIV specific targets, TRAILshort and TRAIL receptor/ ligand, and markers of cell lineage and activation status (Fig. 1A).
Figure 1. TRAILshort is produced in both uninfected and HIV infected cells.
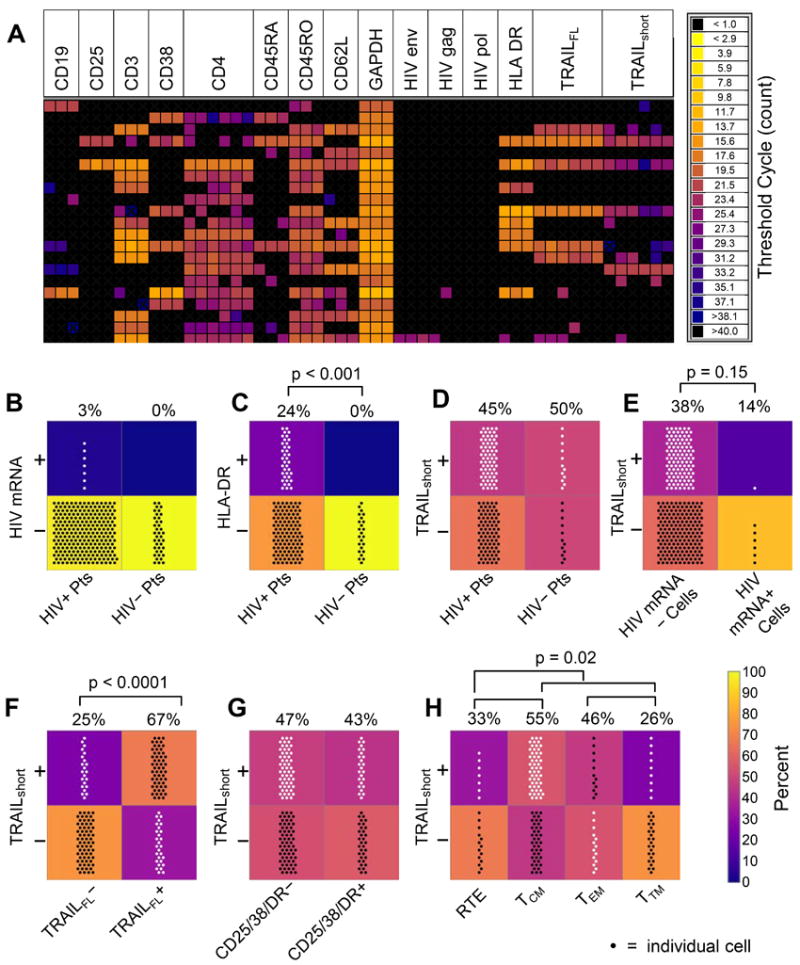
Single cell gene expression in primary CD4 T cells from HIV positive patients (HIV+; N=6) and HIV negative controls (HIV−; N=3) was assessed by Fluidigm technology. (A) Representative heat map of expression of mRNA transcripts for TRAIL, TRAILshort, mRNA levels, and cell surface markers in single cells from a single HIV positive patient. (B-H) Number of cells in each group shown by number of points; percentages of cells in top panels vs. bottom panels indicated above panel pairs; background color of panels also represents percentage. (B) HIV mRNA gene expression in cells of HIV+ and HIV− patients (Pts). (C) HLA-DR gene expression in cells of HIV+ and HIV− Pts. (D) TRAILshort message expression in cells of HIV+ and HIV− Pts. (E) TRAILshort expression in HIV+ and HIV− cells, as determined by cell expression of HIV genes. (F) TRAILshort expression in single cells expressing TRAIL or not expressing TRAIL. (G) TRAILshort expression in resting (CD25−, CD38− and HLA-DR−) or activated (any CD25, CD38, and/or HLA-DR expression). (H) TRAILshort expression in T cell subsets – RTE: recent thymic emigrants; TCM: central memory T cells; TEM: effector memory T cells; TTM: transitional memory T cells. P<0.05 considered significant (Fisher’s Exact test).
The specificity of the assay was confirmed by observing that HIV uninfected donors had no cells which contained HIV transcripts, and that very few of the CD4 T cells from HIV infected donors contained HIV transcripts, consistent with known biology (Fig. 1B) (10). The known inappropriate and excessive immune activation observed in patients with HIV infection was also observed (11), that is, more cells from HIV infected donors expressed the activation marker HLA-DR than cells from HIV uninfected donors (Fig. 1C). We found that both cells from HIV infected as well as uninfected donors contained transcripts for TRAILshort (Fig. 1D), and that both HIV infected (HIV mRNA+) as well as HIV uninfected (HIV mRNA−) cells from HIV infected donors expressed TRAILshort (Fig. 1E), indicating that HIV infection per se is not required for production of TRAILshort. We also observed that the majority of individual cells expressed either TRAILFL and TRAILshort, or neither TRAILFL nor TRAILshort, consistent with TRAILshort being a splice variant of TRAILFL (Fig. 1F). In order to gain insight into what stimuli induce TRAILshort expression, we assessed whether such expression was correlated with cellular activation and found that TRAILshort message was not associated with immune activation as determined by the presence of transcripts for CD25, CD38, or HLA-DR (Fig. 1G). Within the CD4 T cell subsets represented in this analysis, more of the central memory CD4 T cells (TCM) contained TRAILshort transcripts than recent thymic emigrants (RTE), effector memory CD4 T cells (TEM), or transitional memory CD4 T cells (TTM) (Fig. 1H), consistent with the known apoptosis resistant phenotype of TCM (12). Thus while we have previously characterized TRAILshort to be expressed in HIV infected cultures and in samples from HIV infected patients, our new data indicate that TRAILshort can be expressed in individual cells not infected by HIV.
Type I interferons drive production of TRAILshort
Since HIV uninfected cells are capable of making TRAILshort, it becomes relevant to understand which stimuli are responsible for driving the production of TRAILshort; HIV proteins, HIV induced cytokines, and/or IFN might be responsible for inducing TRAILshort expression. HIV infection produces a number of bioactive HIV encoded proteins that have pleotropic effects on host cells. HIV infection is also associated with diffuse perturbation of the cytokine milieu (13). Finally, HIV infection of individual cells can be recognized by a range of Pattern Recognition Receptors (PRR), including TLR and RIG, which culminate in production of type I IFN and consequent induction of interferon-stimulated genes (ISG) (14). To determine which of these stimuli drives TRAILshort production, we screened a panel of cytokines, IFN, and HIV proteins that are present in the plasma of infected patients to assess which if any of these stimuli can induce a resting (CD25−, CD69−, HLA-DR−) CD4 T cell to produce TRAILshort. IFNα14 and IFNβ significantly induced TRAILshort message by ~50 fold (Fig. 2A & 2E) while a panel of cytokines, including inflammatory cytokines, modestly but non-significantly increased the expression of TRAILshort message only by several fold (Fig. 2B). Similarly TNFα, LPS, and gp120 minimally impacted TRAILshort expression (Fig. 2C). We found similar effects of type I interferons compared to other stimuli when we analyzed TRAILshort protein levels in treated samples (Fig. 2E, and supplementary figure 1). In the same samples we also measured TRAILFL message and found a tight linear correlation between TRAILFL expression and TRAILshort expression (Fig. 2D), consistent with figure 1F. Together these data support the conclusion that TRAILshort is a de facto ISG, which is therefore likely expressed in diverse conditions characterized by type I interferon signaling.
Figure 2. Type I interferons and TLR agonists drive production of TRAILshort.
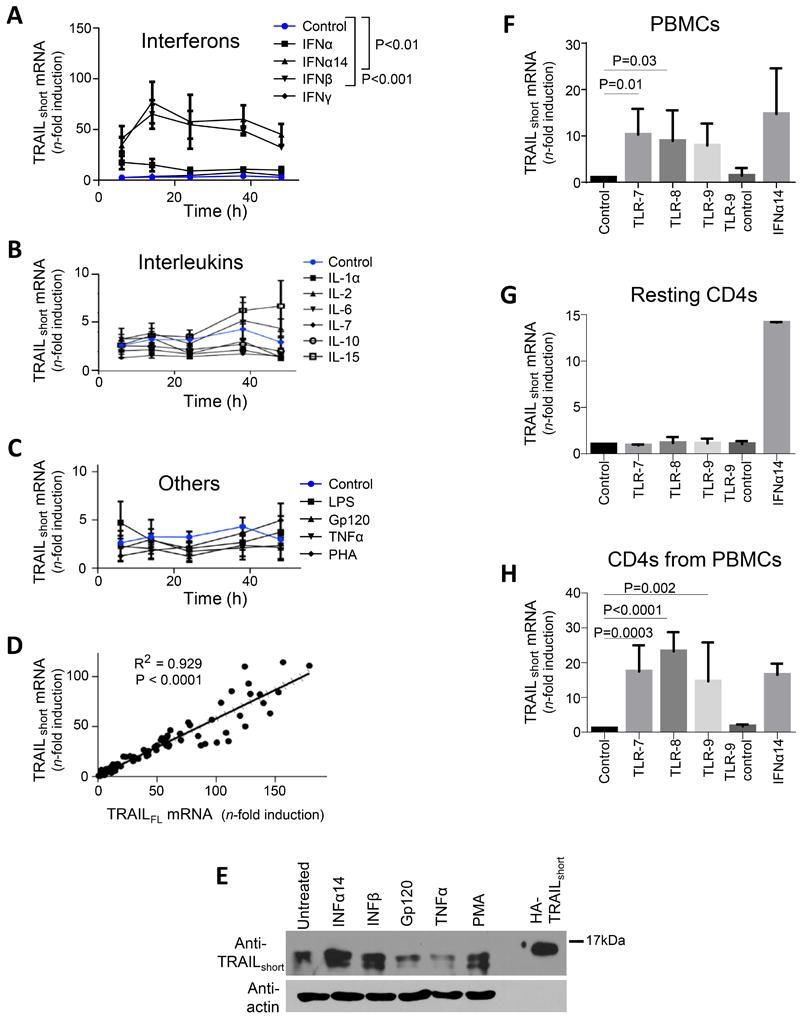
(A, B, and C) Primary uninfected resting (CD25−, CD69−, HLA-DR−) CD4 T cells were either unstimulated, or stimulated with interferons (A), interleukins (B), or other biologically active proteins (C) and TRAILshort mRNA measured by qRT-PCR. (D) Concomitant TRAIL and TRAILshort mRNA expression was compared across samples. (E) Resting CD4 T cells were stimulated as depicted and TRAILshort protein expression assessed by western blot. Representative blot is shown from three independent experiments. (F) PBMCs were treated with vehicle control, TLR 7, 8, or 9 agonists or TL9 inactive control for 24 hours and TRAILshort mRNA expression measured. (G) Resting isolated CD4 T were treated similarly as the PBMCs in (F) and TRAILshort mRNA measured. (H) PBMCs were treated as in (F), and after 24 hours, CD4 T cells were separated and TRAILshort mRNA measured in the CD4 subset. Data represent means (SEM) of 5 independent experiments per treatment. P<0.05 considered statistically significant.
Innate immune defense against invading pathogens can be mediated, in part, by detection of Pathogen Associated Molecular Patterns (PAMPs) through Pattern Recognition Receptors (PRR) expressed by immune cells. One family of PRR are Toll Like Receptors (TLR), which in humans comprise 10 member receptors. TLR expressed predominantly by phagocytic cells recognize a range of pathogen encoded ligands, including lipid based lipopolysaccharides, protein products including flagellin, and nucleic acids including ss or ds RNA. Activation of the intracellular TLR’s 7, 8, or 9 in particular induces a robust type I interferon response (15). Having shown that TRAILshort expression can be driven by type I IFN we next assessed if TRAILshort expression was induced by TLR 7, 8, or 9 agonists.
Uninfected bulk PBMCs were treated with vehicle control, imiquimod (TLR 7 agonist), TL8-506 (TLR 8 agonist), CPG ODN2395 (TLR9 agonist), inactive ODN-2395 control, or IFNα14 for 24 hours and TRAILshort mRNA expression was measured. TRAILshort expression in PBMCs was significantly increased by imiquimod (P = 0.013) and TL8-506 (P = 0.032) compared to vehicle control (Fig. 2F). However, TRAILshort mRNA expression was not induced by TLR 7, 8, or 9 agonists in isolated resting CD4 T cells from the same donors (Fig. 2G).
We next questioned whether TLR 7, 8, or 9 agonist treatment of PBMCs would induce TRAILshort expression in the CD4 T cells contained therein. Bulk PBMCs were treated as above for 24 hours and then CD4 T cells were separated by negative selection and TRAILshort mRNA expression was measured in those CD4 T cells. TRAILshort expression was significantly increased in CD4 T cells isolated from PBMCs treated with TLR 7 agonist (P=0.0003), TLR 8 agonist (P<0.0001), and TLR 9 agonist (P=0.002) compared to control or to an inactive TLR agonist (Fig. 2H). Together, these data indicate that stimulation of PBMC’s with TLR 7, 8, or 9 agonists, induces TRAILshort expression in CD4 T cell subset.
The carboxyl terminus of TRAILshort is externalized on the plasma membrane and interacts with TRAIL receptors R1 & R2 but less so with TRAIL receptors R3 & R4
TRAILFL is expressed as a single pass type II transmembrane protein with an extracellular C-terminal domain. The C-terminal region of TRAILFL containing amino acids ~95-281 is responsible for pro-apoptotic function and it forms homotrimers coordinated around a central zinc ion (16). TRAILshort occurs as a consequence of a splicing event in which exons 3 and 4 are deleted and a frameshift is introduced in exon 5 resulting in a premature stop codon and a novel 11 amino acid C terminus (Fig. 3A).
Figure 3. C-terminus of TRAILshort is extracellular and interacts with TRAIL “death” receptors, but not with TRAIL “decoy” receptors.
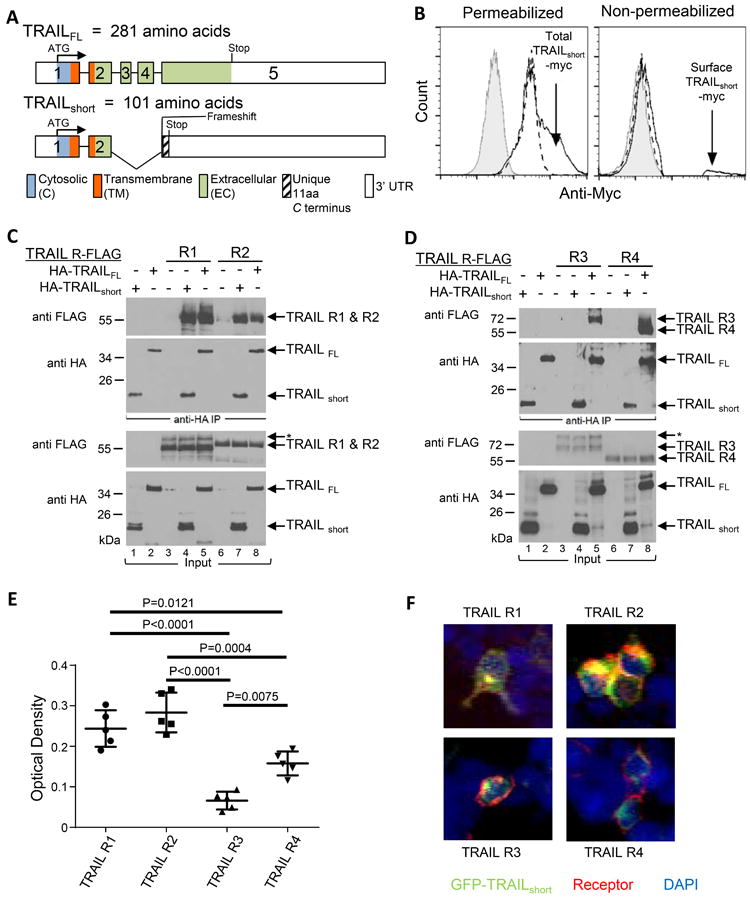
(A) Schematic alignment of the exons, cytosolic, transmembrane, extracellular, and 3’ untranslated regions (UTR) of TRAILFL and TRAILshort. (B) 293T cells transiently expressing TRAILshort with c-myc cloned into the EC domain were stained with an anti-c-myc antibody or an isotype control. Key: Grey filled (eGFP + TRAILshort myc with Isotype control); Dashed line (eGFP, anti-myc); Solid line (eGFP + TRAILshort, anti-myc). (C) Lysates from 293T cell transiently expressing C-terminal FLAG-tagged TRAIL-R1 or R2 were combined with lysates from cells expressing N-terminal HA-tagged TRAILFL or N-terminal HA-tagged TRAILshort (as indicated at top of panels), then immunoprecipitated with anti-HA antibody. The complexed proteins were resolved and immunoblotted for FLAG or for HA. Total input lysates are shown in the lower two panels. (D) As in (C), except lysates from 293T cells expressing decoy TRAIL receptors R3 and R4 were used. Data shown is representative of four independent experiments. (E) Binding of TRAILshort to TRAIL receptors R1, R2, R3 and R4 were determined by ELISA and (F) visualized by confocal microscopy.
We evaluated whether TRAILshort traffics to the plasma membrane and is present on the exterior surface of the cell. To do so, a myc tag was inserted within the novel C terminus of TRAILshort and epitope accessibility was analyzed in non-permeabilized or permeabilized transfected cells using flow cytometry. Whereas permeabilized control transfected cells contained detectable myc (likely reflecting endogenous myc), TRAILshort-myc transfected cells showed additional myc staining consistent with transfected protein expression (Fig. 3B left panel). Non permeabilized samples of the same cells showed no surface myc staining of control transfected cells, yet a distinct population of surface myc positive cells was detectable following TRAILshort-myc expression (Fig. 3B right panel) supporting the interpretation that the carboxyl terminus of TRAILshort is accessible in the extracellular environment.
TRAILFL exerts its pro-apoptotic effect through binding to members of the TRAIL receptor family, of which there are four distinct member proteins. TRAIL-R1 and R2 contain Death Domains, which following ligand binding, recruit Fas-Associated protein with Death Domain (FADD), and lead to caspase-8 activation. TRAIL-R3 and R4 do not contain functional Death Domains, do not lead to caspase-8 activation, and are often considered “decoy” receptors. TRAIL receptor family members are also distinguished one from another by their varying abilities to bind TRAILFL. One isothermal titration calorimetry study showed TRAILFL binding affinity to TRAIL-R2 was at least 35-fold higher than to TRAIL-R1 and 100-fold higher than to TRAIL-R3 (TRAIL-R4 was not assessed) (17), while another showed TRAIL R4 > R2 > R3 > R1, with dissociation constants ranging from 0.869 to 4.08 nM (18). These studies highlight TRAIL-R2 as the apoptosis inducing receptor with the highest affinity for TRAILFL.
We previously described that TRAILshort binds to TRAIL-R2 (9), yet we did not assess whether TRAILshort also interacts with any other TRAIL receptor family members. To address that possibility, cell lysates from 293T cells expressing C-terminal FLAG-tagged TRAIL-R1, R2, R3, or R4 were combined with cell lysates from cells expressing N-terminal HA-tagged TRAILFL or N-terminal HA-tagged TRAILshort. The mixtures were immunoprecipitated with anti-HA and analyzed for FLAG containing proteins. Both FLAG-TRAIL-R1 and R2 were immunoprecipitated via HA-TRAILFL (Fig. 3C). We found that FLAG-TRAIL-R1 and R2 also co-immunoprecipitated HA-TRAILshort to a similar extent (Fig. 3C). However, while TRAILFL associated with both TRAIL-R3 and R4, TRAILshort notably did not (Fig. 3D). TRAILshort therefore binds preferentially to the death inducing TRAIL-R1 and R2, while sparing the decoy receptors R3 and R4, suggesting that selective receptor binding may be central to the biologic effects of TRAILshort.
Differential binding of TRAILshort to TRAIL receptors was further explored using two different orthogonal assays, as follows. First we performed ELISA using fusion proteins of Immunoglobulin Fc fused to the extracellular domains of TRAIL receptors 1, 2, 3, or 4 (as capture reagents), then a synthetic peptide corresponding to the extracellular domain of TRAILshort was added, and biotin-labeled anti-TRAIL short antibody was used as a detection reagent. These experiments revealed greater binding of TRAILshort to TRAIL receptors 1 and 2 compared to TRAIL receptors 3 and 4 (Fig. 3E, binding of TRAILshort to TRAIL R1 vs R3 p = 0.0001, R1 vs R4 p = 0.0121, R2 vs R3 p < 0.0001, R2 vs R4 p = 0.0004). Finally we assessed TRAILshort co-localization with TRAIL receptors by confocal microscopy using 293T cells transfected with GFP-TRAILshort and either TRAIL-R1, R2, R3 or R4. TRAIL receptor expression was detected with Alexa Fluor conjugated anti-TRAIL-R1, R2, R3 or R4 antibodies. In these experiments, co-localization of GFP-TRAILshort and Alexafluor TRAIL receptors was readily apparent for TRAIL R1 and R2, but less apparent for R3 and R4 (Fig. 3F and Supplementary figure 2). Thus by three independent measures TRAILshort binds preferentially to TRAIL receptors 1 and 2 compared to TRAIL receptors 3 and 4.
TRAILshort Contains a PEST Domain and is Ubiquitinated and Degraded by the Proteasome
Many proteins which impact cell survival, proliferation, and apoptosis are known to have a short half-life by virtue of targeted proteasome mediated proteolytic removal (19). During our studies examining the subcellular localization of TRAILshort (Fig. 1A) we noted a seemingly short half-life for TRAILshort. To confirm and flesh out this observation we transfected 293T cells with HA-TRAILshort and evaluated the protein half-life in the absence and presence of the protein synthesis inhibitor cycloheximide (Fig. 4A). We found that ~50% of the protein was lost within the first ~60 minutes, indicating a rapid protein turnover. We identified a putative PEST domain in TRAILshort between amino acids 59 and 81 (Fig. 4B). The substitution of Proline 76 to Alanine markedly prolonged the half-life of TRAILshort (Fig. 4C), confirming a functional PEST domain and suggesting subsequent ubiquitinaton and proteasome mediated degradation.
Figure 4. TRAILshort contains a PEST domain and is ubiquitinated and degraded by the proteasome.
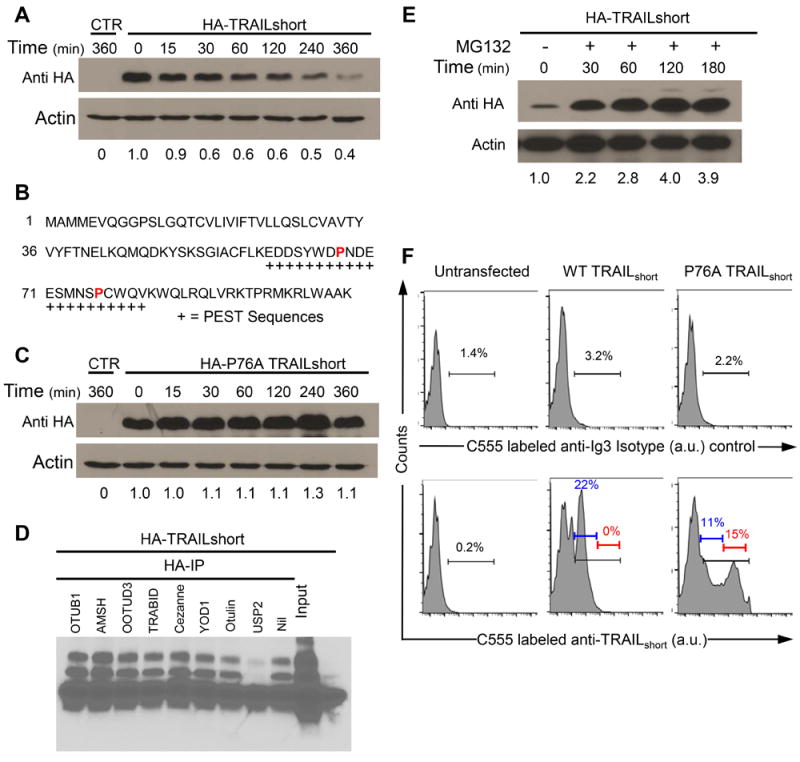
(A) 293T cells were transfected with N-terminal HA-tagged TRAILshort, treated with cycloheximide (100 μg/mL) and analyzed at the times indicated by western analysis with an anti-HA antibody. (B) Putative PEST domains in TRAILshort are indicated by “+” below the peptide sequence of TRAILshort. (C) A proline 76 to alanine (P76A) mutation was engineered into the PEST domain of TRAILshort and cells were transfected and analyzed in the presences of cycloheximide as in (A). (D) HA-tagged TRAILshort was transfected into 293T cells and immunoprecipated with anti-HA antibody, then immunoprecipitates were treated with various de-ubiquitinating enzymes (DUBs) that distinguish between Lys48 and Lys63 ubiquitin linkages (OTUB1, AMSH, OOTUD3, TRABID, OTUD7B, YOD1, Otulin, or USP2). The treated samples were separated by SDS-PAGE then immunoblotted for ubiquitin. (E) 293T cells transfected with TRAILshort were incubated in the presence of MG132 for the times indicated, then harvested and analyzed by western with anti-TRAILshort. (F) TRAILshort expression is increased in the P76A mutant. TRAILshort and P76A TRAILshort were transfected into 293T cells and analyzed by flow cytometry for TRAILshort (bottom panels) or with an isotype control antibody (top panels). All data are representative of at least three independent experiments.
To verify that TRAILshort is ubiquitinated, HA-tagged TRAILshort was expressed in 293T cells and anti-HA pulldowns and immunoblotting for ubiquitin were performed. These analyses revealed an abundance of ubiquitinated proteins migrating at sizes corresponding to polyubiquitinated HA-TRAILshort. Modification of proteins with ubiquitin at their Lys48 linkages can drive proteasome degradation, whereas ubiquitination at Lys63 linkages results in modified protein protein interactions and alteration of signal transduction pathways (20). To assess whether ubiquitinated TRAILshort is targeted for degradation via Lys48, we used specific de-ubiquitinating enzymes (DUBs) that distinguish between Lys48 and Lys63 ubiquitin linkages (21). Ubiquitination of HA-TRAILshort was reversed by the Lys48-specific DUB USP2 (Fig. 4D), indicating that TRAILshort is ubiquitinated in a manner that is consistent with it being targeted to the proteasome for degradation. This was confirmed by measuring TRAILshort levels in cells treated with or without the proteasome inhibitor MG132, revealing that proteasome inhibition greatly increases TRAILshort expression levels (Fig. 4E). Consistent with these observations and with the hypothesis that the ubiquitin proteasome pathway is governing expression levels of TRAILshort, expression of the PEST mutant P76A TRAILshort resulted in increased levels of TRAILshort on individual cells; while the total number of cells containing TRAILshort did not increase appreciably (Fig. 4F).
TRAILshort is contained within extracellular vesicles
We previously observed TRAILshort protein in the supernatant fraction of cell cultures as well as in the plasma of HIV patients (9), yet it remained unknown how TRAILshort is released into that compartment. To explore this process, Jurkat T cells transfected with GFP or GFP-TRAILshort were analyzed by confocal microscopy. TRAILshort was observed to localize to the plasma membrane (Fig. 5A), which was confirmed by immunoblotting the heavy-membrane fraction of HIV-infected or control cells (Fig. 5B) (purity of fractions was confirmed by cytosol specific HSP70). Consistent with our prior observations, we found TRAILshort in the supernatant fraction of infected cultures. Moreover, supernatant associated TRAILshort migrated similarly to cell associated TRAILshort, suggesting that supernatant associated TRAILshort is unlikely to have been generated by a proteolytic cleavage event of TRAILFL (Fig. 5C).
Figure 5. TRAILshort is contained in microvesicles.
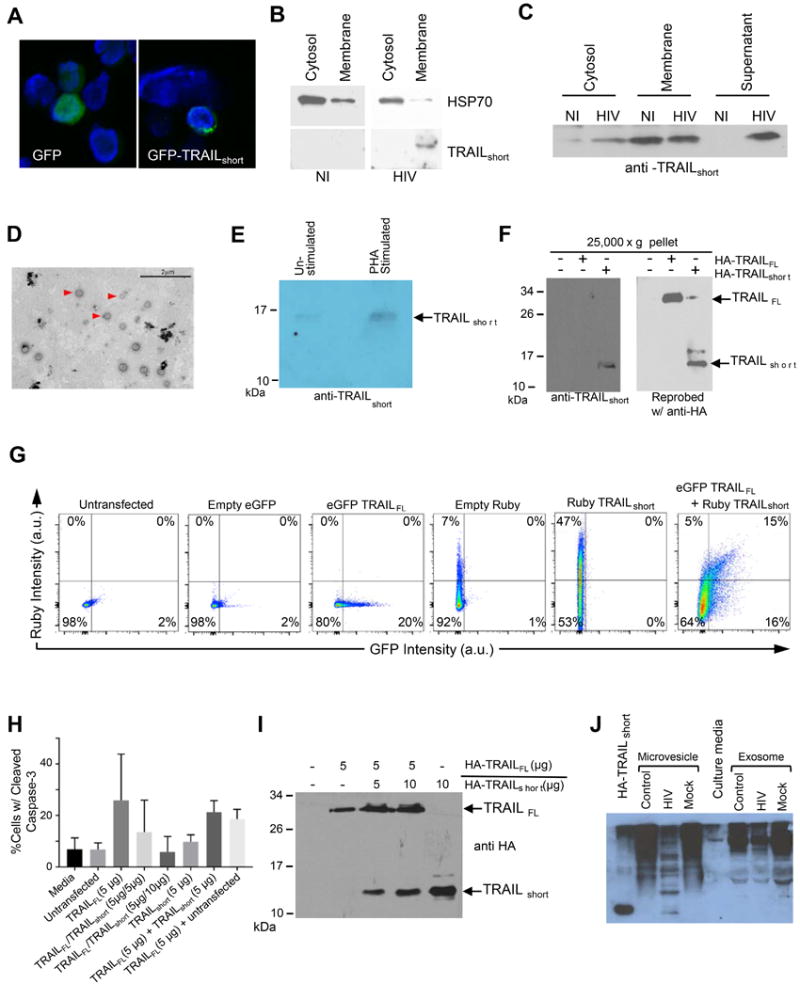
(A) Jurkat T cells transiently expressing GFP-TRAILshort were analyzed by confocal microscopy and cells were counter stained with DAPI. (B) The membrane or cytosolic fractions of HIV infected and non-infected (NI) Jurkat T cells were analyzed for HSP 70 (cytosol selective protein) or for TRAILshort. (C) Cytosolic, membrane, and supernatant fractions were prepared from HIV infected and non-infected (NI) Jurkat T cells and analyzed by western blotting with anti-TRAILshort antibody. (D) Extracellular vesicles from PHA-stimulated CD4+ cells were fixed and stained with phosphotungstic acid, then analyzed by electron microscopy. Scale bar, 2 μm. (E) Extracellular vesicles from PHA-stimulated and unstimulated CD4+ cells were analyzed by western blotting for TRAILshort. (F) Western blot analysis of purified extracellular vesicle preparations from 293T cells transiently expressing HA-TRAILFL and HA-TRAILshort. (G) Flow cytometric analysis of extracellular vesicle preparations from supernatants of 293T cells transfected with eGFP-TRAILshort alone, Ruby-TRAILFL alone. (H) Extracellular vesicles from supernatant of 293T cells transfected with HA-TRAILFL alone, HA-TRAILshort alone, or co-transfected with 1:1 or 1:2 ratios of HA-TRAILFL/HA-TRAILshort were used to treat target Jurkat T-cells. Apoptotic activity was determined by staining Jurkat cells for cleaved caspase 3. Represented is mean (SD) of three independent experiments. (I) Western blot confirmation of HA-TRAILFL and HA-TRAILshort expression in extracellular vesicles in 293T cells from (H). (J) Supernatants from control, HIV infected, and mock infected primary CD4 T cells in culture were separated into microvesicle or exosome fractions as indicated and analyzed by Western blotting with TRAILshort specific monoclonal antibody.
Finding TRAILshort, which contains a transmembrane domain, in the supernatant fraction of cell cultures and in plasma led us to postulate that TRAILshort might be incorporated into extracellular vesicles similar to what has been previously reported for TRAILFL (22). Extracellular vesicles from PHA-stimulated and unstimulated CD4+ cells were harvested by ultracentrifugation, fixed, and analyzed for size by electron microscopy revealing a range of 50 nm to ~400 nm with a median diameter of 136 nm (Fig. 5D). These results suggest the presence of both microvesicles (>100 nm) and exosomes (<100 nm). Western blot analysis of the same extracellular vesicles was performed using anti-TRAILshort antibody (see supplemental Fig. S1 for TRAILshort antibody validation data). This revealed a band at ~15 kDa (Fig. 5E) consistent with the extracellular vesicles containing TRAILshort. Whole cell lysates and purified extracellular vesicle preparations from 293T cells transfected with HA-TRAILFL and HA-TRAILshort produced a similar pattern by western blot (Fig. 5F). We further verified the presence of extracellular vesicle associated TRAILshort by transfecting 293T cells with eGFP-TRAILFL and/or Ruby-TRAILshort and analyzing the supernatants with flow cytometry. Untransfected cell supernatants contained virtually no GFP or Ruby signal, whereas cells transfected with eGFP-TRAILFL or Ruby-TRAILshort alone showed detectable expression of each protein individually. When cells were co-transfected with eGFP-TRAILFL and Ruby-TRAILshort, vesicles were detectable with eGFP-TRAILFL alone, Ruby-TRAILshort alone, and both eGFP-TRAILFL and Ruby-TRAILshort. (Fig. 5G).
Microvesicle localized TRAILFL has been shown to be bioactive (23), and therefore we questioned whether TRAILshort in extracellular vesicles is bioactive as well. 293T cells were transfected with HA-TRAILFL alone, HA-TRAILshort alone, both in a 1:1 ratio, or both in a 1:2 HA-TRAILFL:HA-TRAILshort ratio. Concentrated supernatants containing TRAILshort microvesicles were collected. Jurkat cells were incubated in either fresh media, media conditioned from untransfected 293T cells, or media conditioned from one of the four varieties of transfected 293T cells and were analyzed by flow cytometry for cleaved caspase-3 as a marker of cell death by apoptosis. Jurkat cells incubated with fresh media or media from untransfected 293T cells had low levels of basal apoptosis (~5%). By contrast, Jurkat cells incubated with media from 293T cells transfected with TRAILFL had >20% apoptosis consistent with TRAILFL being active in microvesicle fractions. Significantly, when 293T cells were co-transfected with TRAILFL and TRAILshort, the degree of Jurkat T cell killing by the microvesicle preparations was reduced, and was lowest when greater amounts of TRAILshort was present (TRAILFL 5ug versus TRAILFL 5ug/TRAILshort 10ug P=0.03; Fig. 5H & 5I).
It is well described that HIV infected cells produce microvesicles that contain host cell proteins (24). We tested whether TRAILshort is similarly associated with microvesicles and not exosomes as TRAILshort is a membrane associated protein with a transmembrane domain. Supernatants from HIV infected primary CD4 T cell cultures were fractionated into microvesicles by ultracentrifugation or into exosomes by resin based extraction, previously shown to concentrate exosomes containing CD63, CD9, Cd81 and Hsp70 (25). Microvesicle preparations and exosomes were analyzed by immunoblot using our TRAILshort specific antibody, and identified TRAILshort only in the microvesicle preparations (Fig. 5J).
TRAILshort is necessary and sufficient to cause TRAIL resistance
Having previously shown that TRAILshort siRNA knockdown abrogates TRAIL resistance, we next assessed whether the more therapeutically relevant immunodepletion of TRAILshort is sufficient to mitigate TRAIL resistance. To test this Jurkat T cells which constitutively express TRAILshort were induced to die by sk-TRAIL. Consistent with our prior observations, sk-TRAIL treatment resulted in robust killing of the Jurkat T cells (Fig. 6A). Congruent with TRAILshort being an antagonist of TRAIL, sk-TRAIL treatment in the presence of increasing doses of TRAILshort antibody induced a dose dependent increase in Jurkat killing, which at the highest dose of antibody tested of 5 μg/ml, effectively doubled the number of dead cells despite the TRAILshort antibody alone having no intrinsic cytotoxicity. Altogether therefore in this model system the presence of TRAILshort is both necessary and sufficient for TRAIL resistance, and our data indicate that the anti-TRAIL effects of TRAILshort can be effectively inhibited by TRAILshort specific antibody.
Figure 6. TRAILshort is both necessary and sufficient to cause TRAIL resistance.
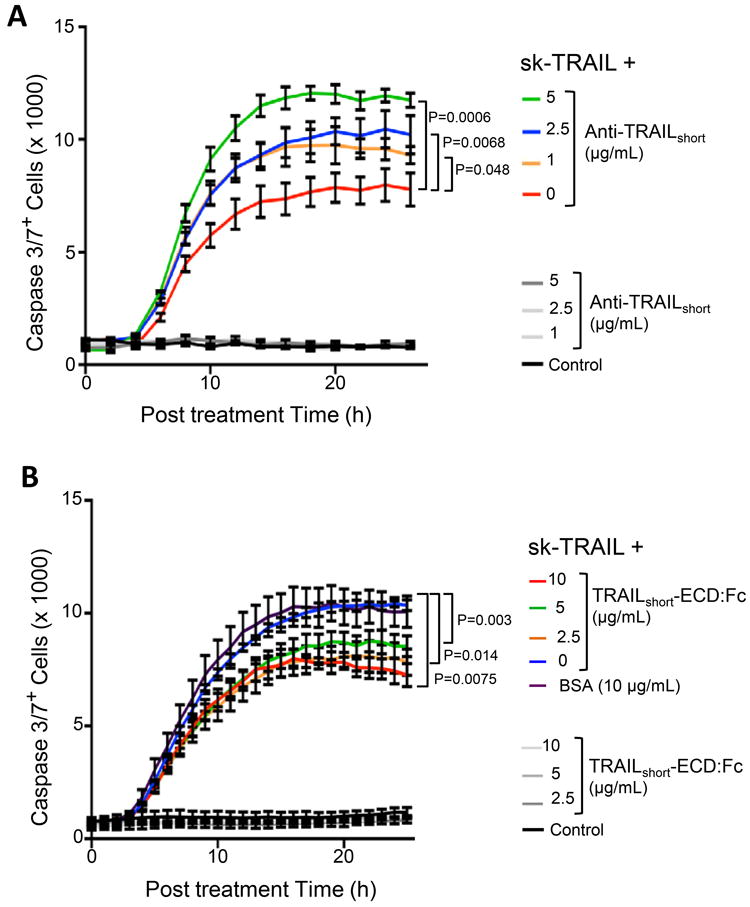
(A) Jurkat T cells, which constitutively express TRAILshort, were induced to die by the addition of sk-TRAIL, in the presence or absence of increasing amounts of anti-TRAILshort antibody. Cell death was measured by active caspase 3 staining. Control cells were treated with increasing anti-TRAILshort antibody alone. (B) Jurkat T cells, which do not express TRAILshort, were stimulated to die by the addition of sk-TRAIL in the absence or presence of increasing amounts of a fusion protein consisting of the extracellular domain of TRAILshort fused to Fc (TRAILshort-ECD:Fc) or with bovine serum albumin (BSA) control and analyzed as in (A). Additional control cells were treated with increasing TRAILshort-ECD:Fc alone. Data are representative of six independent experiments. P<0.05 considered statistically significant (linear regression).
Our cumulative data indicate that TRAILshort fundamentally impacts cell death/survival, and now we assessed whether TRAILshort alone is responsible for these effects. To accomplish this, we generated a recombinant construct of the extracellular domain of TRAILshort fused to the Fc domain of immunoglobulin G (TRAILshortECD:Fc). Following purification, TRAILshortECD:Fc was added to Jurkat T cells alone or to Jurkat T cells treated with sk-TRAIL. While TRAILshortECD:Fc was not toxic, it did prevent sk-TRAIL-mediated killing of Jurkat T cells in a dose dependent manner (Fig. 6B).
TRAILshort protection from TRAIL can be transferred to neighboring cells
To assess the biological impact of TRAILshort expression on the cellular microenvironment, TRAILFL, TRAILshort, or both were expressed in 293T cells. Target Jurkat T cells, which express TRAIL-R2, were labelled with Cell Tracker Orange (CTO), and mixed with 293T cells. Following co-incubation cell death was determined in the CTO+ Jurkat population gating on the CTO positive populations (Fig. 7A). Jurkats died following coincubation with TRAILFL expressing 293T cells, and this was reduced by the 293 T cells coexpressing TRAILshort (Fig 7A). Jurkat cell killing increased with increasing amounts of TRAILFL plasmid expressed by 293 T cells ranging from 7.1% to 32.8% with transfection of 1 μg to 20 μg HA-TRAILFL (Fig. 7B & 7D). When effector 293T cells were transfected with 10 μg of HA-TRAILFL and increasing amounts of HA-TRAILshort, increasing expression of TRAILshort was observed (Fig. 7C) resulting in decreased amounts of Jurkat killing (Fig. 7E). Therefore, TRAILshort co-expression with TRAILFL antagonizes the apoptosis inducing activity of TRAILFL in a dose dependent manner.
Figure 7. TRAILshort protection from TRAIL can be transferred to neighboring cells.
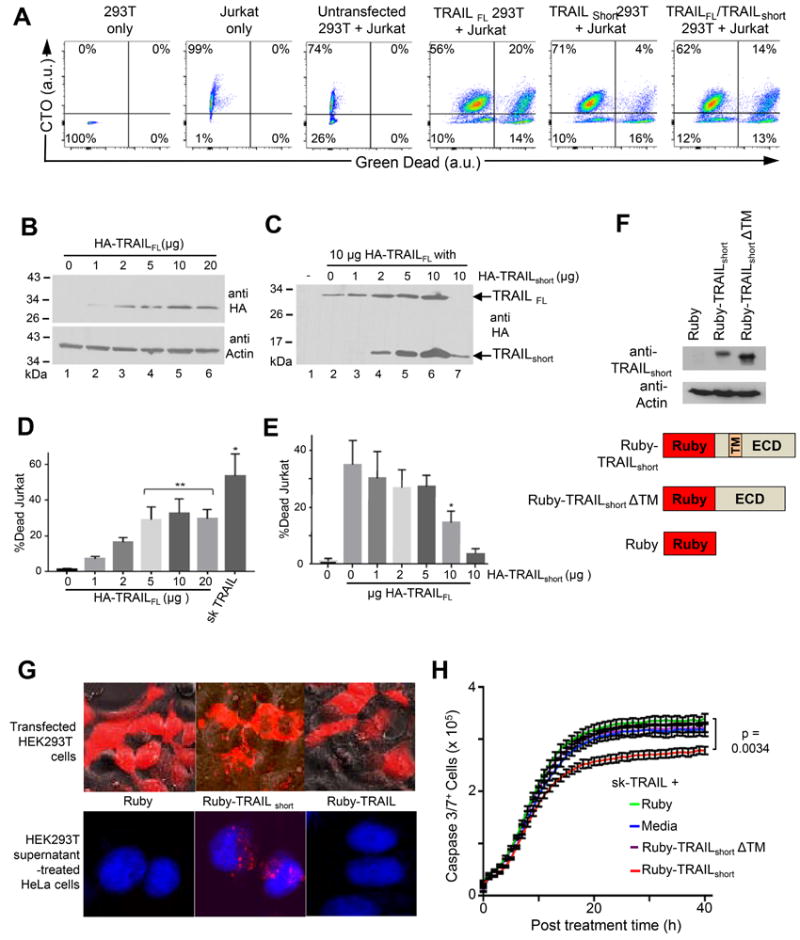
(A) Effector 293T cells expressing either TRAILFL, TRAILshort, or both were mixed with CTO-labeled target Jurkat T cells. Following incubation cells were analyzed by flow cytometry, gating on CTO positive (Jurkat) populations and staining for Live/Dead cells (Green). (B) 293T cells were transfected with 1, 2, 5, 10, or 20 μg of N-terminal HA-tagged TRAILFL alone and analyzed by Western with an anti-HA antibody. (C) 293T cells were co transfected with 1, 2, 5, or 10 μg of TRAILshort and a constant 10 μg TRAILFL and analyzed by Western blot. (D) 293T effector cells transfected with increasing amounts of TRAILFL from (B) were co-cultured with CTO+ target Jurkat cells and the percentage CTO+ Jurkat cell killing was determined. (E) 293T effector cells from (D), co-transfected with increasing amounts of TRAILshort at constant TRAILFL, were co-cultured with target cell and analyzed as in (A). D, E represent mean (SD) of four independent experiments. (F) Schematic of plasmids encoding N-terminal Ruby-tagged-TRAILshort (Ruby-TRAILshort), a construct missing the transmembrane domain (Ruby-TRAILshortΔTM), and the Ruby tag alone (bottom panel). Western analysis with anti-TRAILshort antibody and anti-actin controls of lysates from 293T cells transfected with these constructs (top panel). (G) 293T cells were transfected with the constructs in (F) and representative micrographs are shown (top panel). Supernatants from the 293T transfected cells were harvested and used to treat target HeLa cells which were then analyzed by confocal microscopy. Representative micrographs superimposing the blue (DAPI stained HeLa nuclei) and red (ruby, ruby-TRAILshort or Ruby-TRAILshortΔTM) filter channels are shown (bottom panel). (H) HeLa cells from (G) that were pre-treated with supernatants from 293T transfected with Ruby-TRAILshort or Ruby-TRAILshortΔTM were treated with sk-TRAIL. Cell killing was quantified by active caspase 3 staining (Green) over time. Data representative of four independent experiments. P<0.05 considered statistically significant by linear regression.
We hypothesized that, as TRAILshort is present in microvesicles, TRAILshort-mediated resistance to TRAIL killing would be transferrable from TRAILshort producing cells to bystander cells present in the microenvironment that do not produce TRAILshort. To test the hypothesis we generated expression constructs of ruby tagged TRAILshort (ruby-TRAILshort) and ruby tagged TRAILshort missing the transmembrane domain (ruby-TRAILshortΔTM – which should not be incorporated into membranes, nor into microvesicles) and verified robust expression of each construct (Fig. 7F). 293T cells transfected with ruby-TRAILshort demonstrated perimembrane expression of ruby, whereas, the ruby-TRAILshortΔTM expressing cells showed diffuse cytoplasmic expression (Fig. 7G top panel). Supernatants from transfected cells were harvested and used to treat HeLa cells. Treated HeLa cells showed uptake of ruby-TRAILshort, but not ruby-TRAILshortΔTM (Fig. 7G bottom panel), which is consistent with the understanding that the transmembrane domain is necessary for membrane localization of TRAILshort, and also indicates that microvesicles containing TRAILshort are taken up by neighboring cells that do not produce TRAILshort. We next tested whether these microvesicle treated HeLa cells acquired resistance to TRAIL-mediated killing. Untreated HeLa cells died in a time dependent manner following treatment with sk-TRAIL. Addition of supernatants from ruby transfected or ruby-TRAILshortΔTM transfected 293T cells did not appreciably alter sk-TRAIL killing, whereas, addition of supernatants from ruby-TRAILshort transfected cells significantly reduced sk-TRAIL induced killing (Fig. 7H). Thus, TRAILshort expression by one cell can confer TRAIL resistance on another cell mediated by microvesicle transfer of TRAILshort.
Discussion
When a pathogenic virus infects a cell, it is in the interest of the host to eradicate the infected cell before the virus can replicate and propagate. Consequently, humans have evolved mechanisms to sense invading pathogens including TLRs and RIG-I. For example, in the case of HIV infection, the host cell can sense HIV DNA through IFI16, and DNA nicking mediated by HIV integrase can be sensed by DNA-PK (26, 27). Some viruses have concomitantly evolved mechanisms to resist cell death. For example, HBV, HCV, EBV, HSV, and HPV all encode viral proteins that directly or indirectly inhibit apoptosis (28). These virally encoded apoptosis inhibitors may be necessary for viral persistence. For example, EBV deleted for antiapoptotic proteins BALF1 and/or BHRF1 replicate efficiently, yet compared to wild type virus these isolates do not establish viral persistence (29). Similarly, CRISPR/Cas9 disruption of HPV antiapoptotic proteins E6 or E7 generates viruses which replicate efficiently, yet all infected cells die thereby preventing HPV from establishing persistence (30).
HIV is currently incurable because a subset of infected cells enter a state of virologic latency. These cells are unaffected by antiretroviral agents and host immune effector mechanisms and persist for years. Such persistence occurs despite HIV proteins not having intrinsic antiapoptotic properties. However, it is increasingly recognized that expression of HIV proteins Vpr, Nef, and Tat alter the transcriptional profile of infected cells to upregulate endogenous apoptosis inhibitory proteins achieving an analogous effect. For example, Tat increases cFLIP in T cells (31); Vpr increases Bcl2 and decreases Bax (32); and chronically HIV infected ACH2 cells, U937 cells, and Jurkat T cells have increased XIAP expression (33). It is also increasingly recognized that HIV infected cells are phenotypically resistant to apoptosis induction by diverse stimuli including TRAIL. Specifically, TRAIL-R1 and TRAIL-R2 expressing cells from HIV infected donors are paradoxically resistant to TRAIL (6) including CD4 T cells (34). Our studies illuminate a mechanistic basis for that observation.
Innate sensing of invading pathogens through TLR and other pattern recognition receptors is followed by activation of a variety of host defense mechanisms, principally the interferons. In its original description, interferon was described as a factor produced by one virally infected cell that inhibited viral replication in a neighboring cell; the identified factor was named interferon because of this interference (35). In the intervening decades three distinct families of IFN have been described (types I, II, and III). Type I IFNs, which include IFN-α and IFN-β, regulate and activate immune responses, including upregulating TRAIL expression in effector cells of the immune system including CD8 cells and NK cells (36). Type I IFN upregulation of TRAIL occurs via JAK-STAT dependent transcriptional upregulation, and this pathway also upregulates hundreds of ISG that together enhance antitumor and antiviral immunity by polarizing CD4 and CD8 T cells towards aTh1 phenotype (37), by augmenting dendritic cell and monocyte antigen presentation, and by enhancing NK and B cell responses (38).
In addition to inducing transcriptional upregulation of ISG, IFN signaling can also promote alternative splicing that results in increased biologic diversity of encoded proteins and may impart additional biologic behavior to the novel protein variants. For example, tryptophanyl-tRNA synthetase normally catalyzes the aminoacylation of tRNA by tryptophan, yet when its expression is induced by type I IFN, an alternative, shorter splice variant is produced that has additional anti-proliferative and anti-angiogenic effects (39). Our data indicate that type I IFN similarly induce alternative splicing of TRAIL resulting in both TRAILFL and TRAILshort being produced. Moreover, since the same stimulus produces both a prodeath (TRAIL) and antideath (TRAILshort) protein variant from the same gene, one may suspect that other variables concomitant with production stimuli dictate the resultant biologic effect. That is, timing, duration, and/or magnitude of IFN stimulation may result in differing biologic effect in the case of TRAIL/TRAILshort production.
That variations in timing and duration of type I IFN exposure can influence immunologic outcome, as we postulate for TRAIL and TRAILshort, has been observed in other systems. For example, IFN exposure coincident with antigen exposure is T cell stimulatory, whereas exposure during a chronic infection is immunosuppressive (40). In support of this SIV infected primates who have chronically increased IFN levels have more rapid disease progression than those with lower IFN responses (41), and HIV infected patients with high IFN levels have more rapid disease progression than patients with low IFN levels (42). Moreover, inhibiting IFNα/β signaling during chronic HIV infection reverses immune dysfunction, improves HIV specific T cell responses, and reduces HIV-1 reservoir size in humanized mouse models (43, 44). Our finding that TRAILshort is induced by type I interferons (Fig. 2) and is present in cells from HIV infected patients with chronic disease (Fig. 1) aligns with these models, and argues that TRAILshort may contribute to the immunodeficiency associated with chronic HIV.
The binding of TRAIL to TRAIL receptors is best understood through the prism of the interaction of the extracellular domain of TRAIL with the extracellular domain of TRAIL receptor 2 as this complex has been crystallized (45). The crystal structure reveals that TRAIL adopts two antiparallel β sheets and that three TRAIL molecules trimerize around a central zinc ion bound by cysteine residues at position 230. C230S mutations of TRAIL exhibit significantly reduced apoptosis inducing activity, attesting to the importance of this trimerization event (46). The external surface of the TRAIL trimer is hydrophobic, and is bound by three TRAIL receptor 2 molecules, with the region of greatest interaction occurring on TRAIL receptor 2 residues Glu 147, Glu 151, Arg 154, and Asp 175. These residues are conserved in other TRAIL receptors, suggesting their involvement in TRAIL binding to the other TRAIL receptors as well. Less is known about the binding domains which govern the differential affinity of TRAIL for the various TRAIL receptors. What limited data exists shows that while wild type TRAIL can bind to both TRAIL receptors 1 and 2, TRAIL containing D269H/T214R mutations maintains TRAIL R2 binding yet abrogates TRAIL R1 binding (47). That TRAILshort shares only the first 88 amino acids with full length TRAIL (Fig. 3A) yet shows differential binding affinities for the various TRAIL receptors (Fig. 3E) also suggests the presence and importance of additional residues in both TRAILshort and TRAIL that dictate this differential binding. It will be of interest to model the binding of TRAILshort to TRAIL receptors 1 and 2 to further define these determinants as well as to gain additional insights into full length TRAIL stoichiometry.
The observations presented herein are sufficient to draw meaningful conclusions regarding TRAIL resistance. Since TRAILshort (I) is present as a transmembrane protein, (II) is contained principally in microvesicles, (III) binds to TRAIL-R1 and R2 but less so toTRAIL-R3 and R4, (IV) confers resistance to TRAIL-mediated killing in cells within the microenvironment which bind TRAILshort, and (V) is alone sufficient to confer TRAIL resistance, in can be concluded that TRAILshort is an effective antagonist of TRAIL and that TRAILshort-mediated TRAIL resistance can be conferred upon cells that do not produce TRAILshort. Results showing that anti-TRAILshort antibody can reverse TRAIL resistance, may have implications for the treatment of HIV as well as other chronic viral infections and diseases.
Supplementary Material
Acknowledgments
We thank Drs. Jin Jen and Vernadette Simon, from Genome Analysis Core, Medical Genome Facility for Fluidigm C1 analyses. Finally, we are indebted to the patients who graciously donated blood samples for our analyses.
This work was supported by National Institute of Health grants R01AI110173 and R01AI120698 to A.B. and through Clinician Investigator support from Mayo Clinic and foundation.
Footnotes
Author Contributions
Conceptualization, A.D.B.; Methodology, Z.N, F.A., S.N., S.P.B., G.D.B., D.J.K., N.W.C., and A.D.B.; Formal Analysis, Z.N., F.A., S.N., S.P.B., A.L.K., G.D.B., N.C., G.R., and A.D.B.; Investigation, Z.N., F.A., S.N., S.P.B., A.L.K., G.D.B., M.K.S., G.R., and N.W.C.; Resources, D.J.K., G.R., and A.D.B.; Data Curation, T.D.Y.C.; Writing – Original Draft, A.D.B.; Writing – Review & Editing, T.D.Y.C., J.R.A., and A.D.B.; Visualization, J.R.A. and A.D.B.; Project Administration, T.D.Y.C. and A.D.B.; Funding Acquisition, A.D.B.
Conflict of Interest Statement
AB is an inventor on a granted patent for the therapeutic use of TRAILshort and on a pending, unpublished patent application related to TRAILshort antibodies.
References
- 1.Finnberg N, Klein-Szanto AJ, El-Deiry WS. TRAIL-R deficiency in mice promotes susceptibility to chronic inflammation and tumorigenesis. J Clin Invest. 2008;118:111–123. doi: 10.1172/JCI29900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Zerafa N, Westwood JA, Cretney E, Mitchell S, Waring P, Iezzi M, Smyth MJ. Cutting edge: TRAIL deficiency accelerates hematological malignancies. J Immunol. 2005;175:5586–5590. doi: 10.4049/jimmunol.175.9.5586. [DOI] [PubMed] [Google Scholar]
- 3.Wang S, El-Deiry WS. TRAIL and apoptosis induction by TNF-family death receptors. Oncogene. 2003;22:8628–8633. doi: 10.1038/sj.onc.1207232. [DOI] [PubMed] [Google Scholar]
- 4.van Grevenynghe J, Cubas RA, Noto A, DaFonseca S, He Z, Peretz Y, Filali-Mouhim A, Dupuy FP, Procopio FA, Chomont N, Balderas RS, Said EA, Boulassel MR, Tremblay CL, Routy JP, Sekaly RP, Haddad EK. Loss of memory B cells during chronic HIV infection is driven by Foxo3a- and TRAIL-mediated apoptosis. J Clin Invest. 2011;121:3877–3888. doi: 10.1172/JCI59211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Miura Y, Misawa N, Maeda N, Inagaki Y, Tanaka Y, Ito M, Kayagaki N, Yamamoto N, Yagita H, Mizusawa H, Koyanagi Y. Critical contribution of tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) to apoptosis of human CD4+ T cells in HIV-1-infected hu-PBL-NOD-SCID mice. J Exp Med. 2001;193:651–660. doi: 10.1084/jem.193.5.651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Melki MT, Saidi H, Dufour A, Olivo-Marin JC, Gougeon ML. Escape of HIV-1-infected dendritic cells from TRAIL-mediated NK cell cytotoxicity during NK-DC cross-talk--a pivotal role of HMGB1. PLoS Pathog. 2010;6:e1000862. doi: 10.1371/journal.ppat.1000862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Katsikis PD, Garcia-Ojeda ME, Torres-Roca JF, Tijoe IM, Smith CA, Herzenberg LA, Herzenberg LA. Interleukin-1 beta converting enzyme-like protease involvement in Fas-induced and activation-induced peripheral blood T cell apoptosis in HIV infection. TNF-related apoptosis-inducing ligand can mediate activation-induced T cell death in HIV infection. J Exp Med. 1997;186:1365–1372. doi: 10.1084/jem.186.8.1365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lum JJ, Pilon AA, Sanchez-Dardon J, Phenix BN, Kim JE, Mihowich J, Jamison K, Hawley-Foss N, Lynch DH, Badley AD. Induction of cell death in human immunodeficiency virus-infected macrophages and resting memory CD4 T cells by TRAIL/Apo2l. J Virol. 2001;75:11128–11136. doi: 10.1128/JVI.75.22.11128-11136.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Schnepple DJ, Shepard B, Bren GD, Cummins NW, Natesampillai S, Trushin S, Algeciras-Schimnich A, Meng XW, Sainski AM, Rizza SA, Kaufmann SH, Badley AD. Isolation of a TRAIL antagonist from the serum of HIV-infected patients. J Biol Chem. 2011;286:35742–35754. doi: 10.1074/jbc.M111.274639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Schnepple T-W, Carruth L, Finzi D, Shen X, DiGiuseppe JA, Taylor H, Hermankova M, Chadwick K, Margolick J, Quinn TC. Quantification of latent tissue reservoirs and total body viral load in HIV-1 infection. Nature. 1997;387:183–188. doi: 10.1038/387183a0. [DOI] [PubMed] [Google Scholar]
- 11.Fauci AS. Multifactorial nature of human immunodeficiency virus disease: implications for therapy. Science. 1993;262:1011–1018. doi: 10.1126/science.8235617. [DOI] [PubMed] [Google Scholar]
- 12.Cummins NW, Sainski AM, Dai H, Natesampillai S, Pang YP, Bren GD, de Araujo Correia MC, Sampath R, Rizza SA, O’Brien D, Yao JD, Kaufmann SH, Badley AD. Prime, Shock, and Kill: Priming CD4 T Cells from HIV Patients with a BCL-2 Antagonist before HIV Reactivation Reduces HIV Reservoir Size. J Virol. 2016;90:4032–4048. doi: 10.1128/JVI.03179-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kinter A, Arthos J, Cicala C, Fauci AS. Chemokines, cytokines and HIV: a complex network of interactions that influence HIV pathogenesis. Immunol Rev. 2000;177:88–98. doi: 10.1034/j.1600-065x.2000.17708.x. [DOI] [PubMed] [Google Scholar]
- 14.Zitvogel L, Galluzzi L, Kepp O, Smyth MJ, Kroemer G. Type I interferons in anticancer immunity. Nat Rev Immunol. 2015;15:405–414. doi: 10.1038/nri3845. [DOI] [PubMed] [Google Scholar]
- 15.Kawai T, Akira S. Toll-like receptors and their crosstalk with other innate receptors in infection and immunity. Immunity. 2011;34:637–650. doi: 10.1016/j.immuni.2011.05.006. [DOI] [PubMed] [Google Scholar]
- 16.Cha SS, Kim MS, Choi YH, Sung BJ, Shin NK, Shin HC, Sung YC, Oh BH. 2.8 A resolution crystal structure of human TRAIL, a cytokine with selective antitumor activity. Immunity. 1999;11:253–261. doi: 10.1016/s1074-7613(00)80100-4. [DOI] [PubMed] [Google Scholar]
- 17.Truneh A, Sharma S, Silverman C, Khandekar S, Reddy MP, Deen KC, McLaughlin MM, Srinivasula SM, Livi GP, Marshall LA, Alnemri ES, Williams WV, Doyle ML. Temperature-sensitive differential affinity of TRAIL for its receptors. DR5 is the highest affinity receptor. J Biol Chem. 2000;275:23319–23325. doi: 10.1074/jbc.M910438199. [DOI] [PubMed] [Google Scholar]
- 18.Lang I, Fullsack S, Wyzgol A, Fick A, Trebing J, Arana JA, Schafer V, Weisenberger D, Wajant H. Binding Studies of TNF Receptor Superfamily (TNFRSF) Receptors on Intact Cells. J Biol Chem. 2016;291:5022–5037. doi: 10.1074/jbc.M115.683946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Eden E, Geva-Zatorsky N, Issaeva I, Cohen A, Dekel E, Danon T, Cohen L, Mayo A, Alon U. Proteome half-life dynamics in living human cells. Science. 2011;331:764–768. doi: 10.1126/science.1199784. [DOI] [PubMed] [Google Scholar]
- 20.Deng L, Wang C, Spencer E, Yang L, Braun A, You J, Slaughter C, Pickart C, Chen ZJ. Activation of the IkappaB kinase complex by TRAF6 requires a dimeric ubiquitin-conjugating enzyme complex and a unique polyubiquitin chain. Cell. 2000;103:351–361. doi: 10.1016/s0092-8674(00)00126-4. [DOI] [PubMed] [Google Scholar]
- 21.Komander D, Clague MJ, Urbe S. Breaking the chains: structure and function of the deubiquitinases. Nat Rev Mol Cell Biol. 2009;10:550–563. doi: 10.1038/nrm2731. [DOI] [PubMed] [Google Scholar]
- 22.Martinez-Lorenzo MJ, Anel A, Gamen S, Monle n I, Lasierra P, Larrad L, Pineiro A, Alava MA, Naval J. Activated human T cells release bioactive Fas ligand and APO2 ligand in microvesicles. J Immunol. 1999;163:1274–1281. [PubMed] [Google Scholar]
- 23.Huber V, Fais S, Iero M, Lugini L, Canese P, Squarcina P, Zaccheddu A, Colone M, Arancia G, Gentile M, Seregni E, Valenti R, Ballabio G, Belli F, Leo E, Parmiani G, Rivoltini L. Human colorectal cancer cells induce T-cell death through release of proapoptotic microvesicles: role in immune escape. Gastroenterology. 2005;128:1796–1804. doi: 10.1053/j.gastro.2005.03.045. [DOI] [PubMed] [Google Scholar]
- 24.Kadiu I, Narayanasamy P, Dash PK, Zhang W, Gendelman HE. Biochemical and biologic characterization of exosomes and microvesicles as facilitators of HIV-1 infection in macrophages. J Immunol. 2012;189:744–754. doi: 10.4049/jimmunol.1102244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Taylor DD, Zacharias W, Gercel-Taylor C. Exosome isolation for proteomic analyses and RNA profiling. Methods Mol Biol. 2011;728:235–246. doi: 10.1007/978-1-61779-068-3_15. [DOI] [PubMed] [Google Scholar]
- 26.Cooper A, Garcia M, Petrovas C, Yamamoto T, Koup RA, Nabel GJ. HIV-1 causes CD4 cell death through DNA-dependent protein kinase during viral integration. Nature. 2013;498:376–379. doi: 10.1038/nature12274. [DOI] [PubMed] [Google Scholar]
- 27.Doitsh G, Galloway NL, Geng X, Yang Z, Monroe KM, Zepeda O, Hunt PW, Hatano H, Sowinski S, Munoz-Arias I, Greene WC. Cell death by pyroptosis drives CD4 T-cell depletion in HIV-1 infection. Nature. 2014;505:509–514. doi: 10.1038/nature12940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Galluzzi L, Brenner C, Morselli E, Touat Z, Kroemer G. Viral control of mitochondrial apoptosis. PLoS Pathog. 2008;4:e1000018. doi: 10.1371/journal.ppat.1000018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Oudejans JJ, van den Brule AJ, Jiwa NM, de Bruin PC, Ossenkoppele GJ, van der Valk P, Walboomers JM, Meijer CJ. BHRF1, the Epstein-Barr virus (EBV) homologue of the BCL-2 protooncogene, is transcribed in EBV-associated B-cell lymphomas and in reactive lymphocytes. Blood. 1995;86:1893–1902. [PubMed] [Google Scholar]
- 30.Kennedy EM, Kornepati AV, Goldstein M, Bogerd HP, Poling BC, Whisnant AW, Kastan MB, Cullen BR. Inactivation of the human papillomavirus E6 or E7 gene in cervical carcinoma cells by using a bacterial CRISPR/Cas RNA-guided endonuclease. J Virol. 2014;88:11965–11972. doi: 10.1128/JVI.01879-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Badley AD, Parato K, Cameron DW, Kravcik S, Phenix BN, Ashby D, Kumar A, Lynch DH, Tschopp J, Angel JB. Dynamic correlation of apoptosis and immune activation during treatment of HIV infection. Cell Death Differ. 1999;6:420–432. doi: 10.1038/sj.cdd.4400509. [DOI] [PubMed] [Google Scholar]
- 32.Conti L, Rainaldi G, Matarrese P, Varano B, Rivabene R, Columba S, Sato A, Belardelli F, Malorni W, Gessani S. The HIV-1 vpr protein acts as a negative regulator of apoptosis in a human lymphoblastoid T cell line: possible implications for the pathogenesis of AIDS. J Exp Med. 1998;187:403–413. doi: 10.1084/jem.187.3.403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Berro R, de la Fuente C, Klase Z, Kehn K, Parvin L, Pumfery A, Agbottah E, Vertes A, Nekhai S, Kashanchi F. Identifying the membrane proteome of HIV-1 latently infected cells. J Biol Chem. 2007;282:8207–8218. doi: 10.1074/jbc.M606324200. [DOI] [PubMed] [Google Scholar]
- 34.Chehimi J, Papasavvas E, Tomescu C, Gekonge B, Abdulhaqq S, Raymond A, Hancock A, Vinekar K, Carty C, Reynolds G, Pistilli M, Mounzer K, Kostman J, Montaner LJ. Inability of plasmacytoid dendritic cells to directly lyse HIV-infected autologous CD4+ T cells despite induction of tumor necrosis factor-related apoptosis-inducing ligand. J Virol. 2010;84:2762–2773. doi: 10.1128/JVI.01350-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Isaacs A, Lindenmann J. Virus interference. I. The interferon. Proc R Soc Lond B Biol Sci. 1957;147:258–267. [PubMed] [Google Scholar]
- 36.Kayagaki N, Yamaguchi N, Nakayama M, Eto H, Okumura K, Yagita H. Type I interferons (IFNs) regulate tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) expression on human T cells: A novel mechanism for the antitumor effects of type I IFNs. J Exp Med. 1999;189:1451–1460. doi: 10.1084/jem.189.9.1451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gonzalez-Navajas JM, Lee J, David M, Raz E. Immunomodulatory functions of type I interferons. Nat Rev Immunol. 2012;12:125–135. doi: 10.1038/nri3133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.McNab F, Mayer-Barber K, Sher A, Wack A, O’Garra A. Type I interferons in infectious disease. Nat Rev Immunol. 2015;15:87–103. doi: 10.1038/nri3787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Liu J, Shue E, Ewalt KL, Schimmel P. A new gamma-interferon-inducible promoter and splice variants of an anti-angiogenic human tRNA synthetase. Nucleic Acids Res. 2004;32:719–727. doi: 10.1093/nar/gkh240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Marshall HD, Urban SL, Welsh RM. Virus-induced transient immune suppression and the inhibition of T cell proliferation by type I interferon. J Virol. 2011;85:5929–5939. doi: 10.1128/JVI.02516-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Manches O, Bhardwaj N. Resolution of immune activation defines nonpathogenic SIV infection. J Clin Invest. 2009;119:3512–3515. doi: 10.1172/JCI41509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Rotger M, Dalmau J, Rauch A, McLaren P, Bosinger SE, Martinez R, Sandler NG, Roque A, Liebner J, Battegay M, Bernasconi E, Descombes P, Erkizia I, Fellay J, Hirschel B, Miro JM, Palou E, Hoffmann M, Massanella M, Blanco J, Woods M, Gunthard HF, de Bakker P, Douek DC, Silvestri G, Martinez-Picado J, Telenti A. Comparative transcriptomics of extreme phenotypes of human HIV-1 infection and SIV infection in sooty mangabey and rhesus macaque. J Clin Invest. 2011;121:2391–2400. doi: 10.1172/JCI45235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zhen A, Rezek V, Youn C, Lam B, Chang N, Rick J, Carrillo M, Martin H, Kasparian S, Syed P. Targeting type I interferon mediated activation restores immune function in chronic HIV infection. The Journal of Clinical Investigation. 2016;127 doi: 10.1172/JCI89488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Cheng L, Ma J, Li J, Li D, Li G, Li F, Zhang Q, Yu H, Yasui F, Ye C, Tsao LC, Hu Z, Su L, Zhang L. Blocking type I interferon signaling enhances T cell recovery and reduces HIV-1 reservoirs. J Clin Invest. 2017;127:269–279. doi: 10.1172/JCI90745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Mongkolsapaya J, Grimes JM, Chen N, Xu XN, Stuart DI, Jones EY, Screaton GR. Structure of the TRAIL-DR5 complex reveals mechanisms conferring specificity in apoptotic initiation. Nat Struct Biol. 1999;6:1048–1053. doi: 10.1038/14935. [DOI] [PubMed] [Google Scholar]
- 46.Bodmer JL, Meier P, Tschopp J, Schneider P. Cysteine 230 is essential for the structure and activity of the cytotoxic ligand TRAIL. J Biol Chem. 2000;275:20632–20637. doi: 10.1074/jbc.M909721199. [DOI] [PubMed] [Google Scholar]
- 47.van der Sloot AM, Tur V, Szegezdi E, Mullally MM, Cool RH, Samali A, Serrano L, Quax WJ. Designed tumor necrosis factor-related apoptosis-inducing ligand variants initiating apoptosis exclusively via the DR5 receptor. Proc Natl Acad Sci U S A. 2006;103:8634–8639. doi: 10.1073/pnas.0510187103. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.