

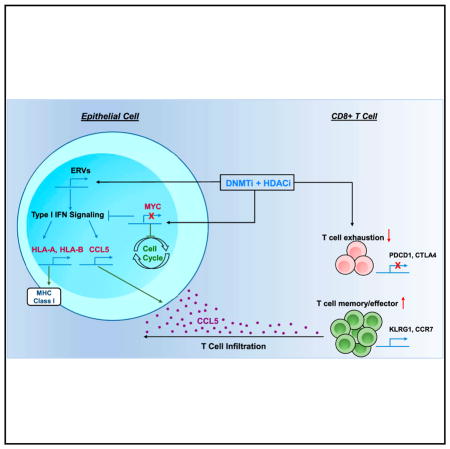
SUMMARY
Combining DNA-demethylating agents (DNA methyltransferase inhibitors [DNMTis]) with histone deacetylase inhibitors (HDACis) holds promise for enhancing cancer immune therapy. Herein, pharmacologic and isoform specificity of HDACis are investigated to guide their addition to a DNMTi, thus devising a new, low-dose, sequential regimen that imparts a robust anti-tumor effect for non-small-cell lung cancer (NSCLC). Using in-vitro-treated NSCLC cell lines, we elucidate an interferon α/β-based transcriptional program with accompanying upregulation of antigen presentation machinery, mediated in part through double-stranded RNA (dsRNA) induction. This is accompanied by suppression of MYC signaling and an increase in the T cell chemoattractant CCL5. Use of this combination treatment schema in mouse models of NSCLC reverses tumor immune evasion and modulates T cell exhaustion state towards memory and effector T cell phenotypes. Key correlative science metrics emerge for an upcoming clinical trial, testing enhancement of immune checkpoint therapy for NSCLC.
In Brief
Myc depletion through combined epigenetic therapy reverses immune evasion and enables effective treatment of lung cancer.
INTRODUCTION
The development of effective therapeutic approaches for patients with lung cancer, the world’s leading cause of cancer-related death, is a critical medical need. The advent of immune checkpoint therapy has been a tremendous step forward (Borghaei et al., 2015; Garon et al., 2015; Reck et al., 2016), but only a minority of unselected patients benefit. Thus, identifying rational combinations to augment anti-tumor immunity and increase the response to immune checkpoint therapy represents a major challenge for the field. Durable responses were observed in patients with non-small-cell lung cancer (NSCLC) who were initially treated with a DNA methyltransferase inhibitor (DNMTi) combined with intermittent histone deacetylase inhibitor (HDACi), followed by immune checkpoint therapy (Juergens et al., 2011). Combination epigenetic therapy regimens most commonly utilize DNMTis in combination with HDACis on the premise that the latter can enhance re-expression of abnormally silenced genes mediated by abnormal gene promoter DNA methylation (Cameron et al., 1999; Jones et al., 2016). However, little attention has been paid to examining the specific pharmacologic features of HDACi, optimal dosing strategy, and underlying mechanisms mediating maximal synergy with DNMTis. The zinc-chelating HDACis comprise 3 major classes: benzamide, hydroxamic acid, and cyclic tetrapeptides (Bradner et al., 2010; Falkenberg and Johnstone, 2014; Rasheed et al., 2007; Zahnow et al., 2016). Benzamide class HDACis target class I HDAC isoforms, while hydroxamic acid class HDACis target both class I (HDAC1, 2, and 3) and class IIb (HDAC6), although differences in isoform targeting can be noted (Bradner et al., 2010).
We have previously shown that low doses of DNMTi impart durable changes in both genome-wide DNA methylation and transcriptome while avoiding acute cytotoxicity (Tsai et al., 2012). Additionally, we and others have discovered that DNMTis can induce an epithelial immune signature for NSCLC and other cancers (Li et al., 2014; Moreno-Bost et al., 2011; Oi et al., 2009; Weiser et al., 2001; Wrangle et al., 2013). A key mediator of this signaling is a cytoplasmic, double-stranded RNA (dsRNA)-mediated interferon response induced through the activation of endogenous retrovirus (ERV) transcription (Chiappinelli et al., 2015; Roulois et al., 2015). How the distinct classes of HDACis contribute to enhancing immune therapy specifically is unknown.
Herein, testing individual HDACis based on both their isoform-specific inhibitory constant (Ki) and pharmacokinetic parameters, we demonstrate the best pairing of these agents with Aza for achieving anti-tumor effects, which include immune responses. The combination of HDACi + Aza achieves robust anti-tumor responses mediated by reducing MYC-driven cell proliferation with enhancement of immune signaling. We hypothesize dual epigenetic therapy will synergize with immune checkpoint blockade in NSCLC patients to drive a potent anti-tumor response.
RESULTS
Combination Aza + ITF-2357 Induces Profound Drug Synergy when Applied to Human NSCLC
We tested HDACis by pairing these agents with low-dose (500 nM) Aza. Clinically relevant HDACis (ITF-2357, MGCD-0103, and MS-275) were selected and paired with Aza based on their known pharmacokinetic parameters (serum maximum concentration [Cmax] and half-life) and enzymatic Ki against key HDAC isoforms (Figure S1A) (Bradner et al., 2010; Falkenberg and Johnstone, 2014; Furlan et al., 2011; Gore et al., 2008; Rasheed et al., 2007; Ryan et al., 2005; Zahnow et al., 2016). We initially examined the effect of sequential Aza + HDACi on cell proliferation across a panel of 16 cell lines representing the diverse oncogenotypes of NSCLC (Figures 1A, S1B, and S1C). These data revealed that the antiproliferative effect of this treatment regimen correlated with the subtype and mutational status of the NSCLC cell line queried. The hydroxamic acid class HDACi (ITF-2357) in combination with Aza demonstrated the most potent antiproliferative phenotype. Among our four most responsive cell lines, three were RAS mutant subtype. Therefore, we selected these cell lines for further studies of therapeutic scheduling and HDAC isoform targeting delineation. Using a comparative analysis, wherein Aza and ITF-2357 were both sequentially and concurrently employed (Figure S1C), only the former schedule induced both synergy and robust potentiation of HDACi by Aza (Figures 1B, 1C, S1D, and S1E). We thus used this schema of sequential treatment in our further studies. To delineate the enhanced antiproliferative effect of ITF-2357 over the others, we next tested the function of the individual HDAC isoforms in the Aza priming response to HDACi. We found, through the application of HDAC isoform-specific inhibitors, an Aza induced priming to both class I and IIb targeting HDACis (Figures 1D and 1E). These data suggest the observed Aza + ITF-2357 antiproliferative phenotype to be the result of broader targeting of HDACs, including both nuclear (HDAC1, 2, and 3) and non-nuclear isoforms (HDAC6).
Figure 1. Azacitidine Synergizes with Sequential HDACi for Reducing Cell Proliferation.

(A) Composite representation of ITF-2357 IC50 as determined by four parameter dose-response analysis in the presence of mock or 500 nM Aza pre-treatment. Individual dose-response curves are in Figures 1B, S1B, and S1E. H1650 not depicted due to lack of sensitivity to epigenetic agents deployed in study.
(B) Sequential treatment with (mock or 500 nM Aza) + ITF-2357 log dose-response curves for growth inhibition of A549 and H460 cells (day 11, n = 5, data represented as mean ± SEM).
(C) Combination index (CI) plots for sequential application of Aza + ITF-2357 in A549 and H460 cells (n = 5).
(D) Sequential treatment with (mock or 500 nM Aza) + MS-275, RGFP996 (HDAC3i), or Tubastatin A (HDAC6i) log dose-response curves for growth inhibition of A549 and H460 cells (day 11, n = 3, data represented as mean ± SEM).
(E) Sequential treatment (mock or 500 nM Aza) with MS-275 plus either RGFP996 or Tubastatin A drug dose-response matrix for growth inhibition of A549 and H460 cells (day 11, n = 3, color gradation indicates percentage viability at the indicated dose combination).
(F and G) Mean volumes of tumor xenografts obtained from NOD-SCID mice subcutaneously injected with H460 cells (F) or H1299 cells (G) and treated with the agents as indicated in the figure. Data are presented as mean ± SEM (n = 5). *p < 0.05 calculated using two tailed t test.
(H) Tumor weights for patient-derived xenografts treated with the agents as outlined in the figure (28 days of treatment duration; data are presented as mean ± SEM; n = 6 mock and n = 7 Aza + ITF-2357). *p value < 0.05 calculated using two-tailed t test.
See also Figure S1.
As a further characterization of the antiproliferative effects of Aza in combination with HDACi, we deployed equal molar concentrations of MS-275 and ITF-2357 and assessed cell proliferation specifically. In agreement with the previous data, we found 100 nM ITF-2357, a concentration roughly one-third Cmax (Furlan et al., 2011), demonstrated a more robust inhibition of proliferation than 100 nM MS-275 (Figure S1F). We next sought to define the contribution of individual HDAC isoforms to the antiproliferative effect noted from 100 nM ITF-2357. To accomplish this end, we first determined equivalent doses based on isoform pharmacodynamic targets, these being histone acetylation for HDAC1 and 2 and tubulin acetylation for HDAC6 (Asthana et al., 2013; Fournel et al., 2008; Khan et al., 2008). The resulting equivalent doses were 200 nM MGCD0103 and 1,000 nM Tubastatin A to mimic 100 nM ITF-2357 (Figure S1G). Selection of 2,000 nM RGFP996 for HDAC3 inhibitor was based on extrapolation of Ki, due to a lack of known pharmacodynamic markers for these isoforms (Malvaez et al., 2013). These normalized doses of HDAC isoform-specific inhibitors identified HDAC1, 2, and 3 as key arbiters of the Aza + ITF-2357 antiproliferative actions (Figure S1H). Additionally, to elucidate the specificity of Aza priming to HDACi, we deployed small hairpin RNA (shRNA)-mediated DNMT1 depletion, the major pharmacodynamic target of Aza (Cai et al., 2014). We observed potent augmentation of the HDACi antiproliferative effect when utilized in combination with DNMT1 depletion (Figures S1I and S1J).
Finally, we tested the efficacy of sequential Aza + HDACi in vivo using immune-incompetent mice. We observed a significant reduction of tumor burden across 3 xenograft models, two for established NSCLC cell lines and one using a KRAS, KDR, TP53 mutant primary patient-derived xenograft (PDX) model when utilizing Aza + ITF-2357 (Figures 1F–1H). The combination of Aza + MS-275 did not have significant efficacy in the H460 xenograft model, the most sensitive cell line tested, at the doses used in our study (Figure 1F). Aza + MGCD0103 achieved significant efficacy in H460 xenografts (Figure S1K) and modest efficacy in the patient-derived xenograft (Figure S1L).
Effects of the Drug Combination Paradigm on the Transcriptome in NSCLC Lines
Assessment of drug combination-induced effects on gene expression yields important insight into signaling pathway alterations that may relate to eventual clinical efficacy. The combination epigenetic treatment induced an increase in differential gene expression relative to HDACi mono-treatment conditions, with a clear advantage noted from the application of Aza + ITF-2357 (Figures 2A, S2A, and S2B). Clustering of the top 500 differentially expressed genes showed a divergence between HDACi mono-treatment and Aza-containing regimens (Figure 2B). Database for Annotation, Visualization and Integrated Discovery (DAVID) pathway analysis revealed the most differentially expressed pathways to be immune, MYC, and metabolism related (Figure 2C). Gene set enrichment analysis (GSEA) identified three significantly induced pathways by combination epigenetic treatment, two of which are related to immune signaling (Figures 2D and 2E). The interferon alpha/beta (IFNα/β)-signaling pathway was the most prominent of these altered pathways (Figure S2C). Comparative analysis of combination epigenetic treatment-downregulated pathways by GSEA revealed a conserved cell cycle repression signature across the drug combinations tested, with a noted advantage for Aza + ITF-2357 (Figures 2F, 2G, and S2D). The above transcriptional downregulation of cell cycle-related pathways elicited by combinatorial epigenetic treatment matched with the observed proliferative arrest induced by combination epigenetic treatment (Figures S1F).
Figure 2. Epigenetic Treatment of NSCLC Cell Lines Induces Robust Alteration of Cell Transcriptome.

(A) Quantitation of differentially expressed genes (cutoff Log2 fold change over mock >0.5) for each treatment condition.
(B) Unsupervised hierarchical clustering of relative RNA expression by median absolute deviation (MAD). RNA expression Log2 fold change over mock; blue to red color gradation is based on the ranking of each condition from minimum (blue) to maximum (red). The top 500 genes are depicted.
(C) DAVID analysis of the top 500 MAD genes using Kyoto Encyclopedia of Genes and Genomes (KEGG) gene ontology.
(D) Venn diagrams depicting GSEA-derived overlapping and unique pathways induced by combination treatment in at least 3 cell lines with the respective HDACi (normalized enrichment score [NES] > 2.0, false discovery rate [FDR] < 0.25).
(E) Venn diagram of pathways commonly upregulated by combination treatment with Aza and the respective HDACi.
(F) Venn diagrams depicting GSEA-derived overlapping and unique pathways downregulated by combination treatment in at least 3 cell lines with the respective HDACi (NES < 2.0, FDR < 0.25).
(G) Venn diagram of pathways commonly downregulated by combination treatment with Aza and the respective HDACi.
The above data are derived from microarray analysis of RNA from cells treated with 500 nM Aza, 100 nM ITF-2357, and 100 nM MS-275.
See also Figure S2.
The Potential of Combinatorial Epigenetic Treatment to Stimulate Specific Immune-Related Genes
A primary goal for combined epigenetic therapy is to increase the efficacy of immune checkpoint and other immunotherapies. We found the combination of Aza + ITF-2357 to be the most effective relative to other conditions tested in terms of induction of IFNα/β pathway-related genes, including those associated with antigen presentation (Figures 3A, 3B, and S3A). To demonstrate whether this efficacy of immune gene induction by Aza + ITF-2357 was potency or isoform targeting based, we used isoform-specific HDACi at concentrations mimicking 100 nM ITF-2357. We observed the inhibition of HDAC1, 2 (MGCD0103), and HDAC6 (Tubastatin A) in combination with Aza, induced IFNα/β pathway-related genes, including antigen presentation, while HDAC3- (RGFP996) specific inhibitors demonstrated no significant induction of these genes (Figures 3C–3F, S3B, and S3C).
Figure 3. Combination Epigenetic Treatment Augments IFNα/β Pathway-Associated Immune Genes and ERV Transcription.

(A) Heatmap of relative RNA expression for IFNα/β-signaling pathway core-enriched genes for the indicated cell lines (microarray, day 8; 500 nM Aza, 100 nM MS-275, and 100 nM ITF-2357).
(B) Quantification of IFNα/β pathway core-enriched genes differentially expressed by the indicated conditions (microarray, day 8; 500 nM Aza, 100 nM MS-275, and 100 nM ITF-2357; differential gene expression cutoff Log2 fold change over mock >0.5).
(C and D) Expression of viral defense gene subset of IFNα/β pathway (PCR genecard, day 8; 500 nM Aza, 100 nM ITF-2357, 200 nM MGCD0103, and 1,000 nM Tubastatin A) in H23 (C) and A549 (D) cells.
(E and F) Quantitation of selected major histocompatibility complex (MHC) class I genes of the IFNα/β pathway in response to Aza and/or HDACi in H23 (E) and A549 (F) cells (qRT-PCR, day 8; 500 nM Aza, 100 nM ITF-2357, 200 nM MGCD0103, 2,000 nM RGFP996, and 1,000 nM Tubastatin A; n = 3).
(G and H) Quantitation of ERV transcripts in response to Aza and/or HDACi in H23 (G) and A549 (H) cells (qRT-PCR, day 8; 500 nM Aza, 100 nM ITF-2357, and 100 nM MS-275; n = 4).
(I and J) Quantitation of ERV9-1 in response to Aza and/or HDACi in H23 (I) and A549 (J) cells (500 nM Aza, 100 nM ITF-2357, 200 nM MGCD0103, 2,000 nM RGFP996, and 1,000 nM Tubastatin A; n = 3).
Data are presented as mean ± SEM (*p < 0.05 relative to mock and #p value < 0.05 relative to Aza; p value determined by two-tailed t test).
See also Figures S3 and S4.
We also compared the efficacy of ITF-2357 against another hydroxamic acid derivative HDACi (SAHA). Using doses at equal ratio of Cmax for the respective inhibitors, we observed a superior augmentation of interferon genes by ITF-2357 when deployed in combination with Aza (Figures S3D and S3E). The above data demonstrates an induction of interferon-stimulated genes by Aza, which are further augmented by HDACi’s targeting isoforms 1, 2, and 6.
Cancer/testis antigens are epigenetic-treatment responsive and have an established role in facilitating the recognition of tumors through immune surveillance (Moreno-Bost et al., 2011; Oi et al., 2009; Weiser et al., 2001). We found, in agreement with previous studies, the expression of cancer/testis antigens are Aza inducible, but a significant additional benefit is noted with the combination of Aza + ITF-2357 (Figures S4A–S4D).
The augmentation of the IFNα/β pathway by Aza partially relies on increasing dsRNA species, including ERV transcripts (Chiappinelli et al., 2015; Roulois et al., 2015; Stengel et al., 2010; Strissel et al., 2012). Indeed, combination HDACi with Aza transcriptionally induced multiple ERVs across all cell lines tested (Figures 3G, 3H, S4E, and S4F). We found ERV9-1 to be the most differentially expressed ERV induced by Aza + ITF-2357. Using isoform-specific inhibitors in combination with Aza, we determined its transcription is potentiated most robustly by inhibition of HDAC6 (a cytoplasmic HDAC) (Figures 3I and 3J). Further, we demonstrated using DNMT1-targeting shRNA that ERV9-1 induction specifically resulted from DNMT1 depletion in our combination treatment (Figure S4G). The inducibility of interferon signaling from ERV transcription was linked in our NSCLC cells, as single ERV overexpression induced transcription of OASL, an interferon-responsive gene (Figure S4H).
In summary, HDACis enhance Aza-induced interferon-responsive gene transcription, particularly with the inhibition of HDAC1, 2, and 6. Class I antigen presentation is augmented through the inhibition of HDAC1, 2, 3, and 6 in combination with Aza. The induction of cancer/testis antigen transcription requires potent HDACis, such as ITF-2357, to mediate significant upregulation over Aza alone.
Perturbation of MYC by Aza Drives Sensitization to HDACi
As described earlier, DAVID analysis of the top 500 differentially expressed genes revealed that three of the top eight pathways are MYC related (Figure 2C), suggesting the perturbation of MYC as a key target of our epigenetic therapy. Critically, in the tested NSCLC cell lines, Aza produced a significant transcriptional downregulation of MYC, with an at least 1.4-fold reduction observed across all cell lines assayed (Figure 4A). These transcription data correlated with downregulation of MYC protein in A549 and H460 cells (Figure 4A). Additional assessment of MYC protein downregulation by Aza and priming to sequential HDACi across a panel of NSCLC cell lines of varying oncogenotypes revealed a significant correlation between the degree of downregulation and HDACi sensitization by Aza (Figures 4A and S5A). Combinatorial Aza + HDACi acted to further deplete MYC over Aza treatment alone (Figure S5B). The application of Aza and Aza + HDACi treatment induced potent repression of the top 200 MYC targets (Figure 4B). Importantly, knockdown of MYC protein phenocopied the Aza sensitization of NSCLC to HDACi-induced cytostasis (Figures 4C and S5C). The stable over-expression of MYC partially rescued the Aza-induced sensitization to HDACi-induced cell depletion (Figures 4D and S5D). These data indicated an Aza-induced MYC depletion as a potent sensitizer to HDACi-induced proliferative arrest, and, accordingly, they implicate depletion of MYC signaling as a driver of these events.
Figure 4. MYC Perturbation Drives Aza Sensitization to HDACi and IFNα/β Pathway Gene Augmentation.

(A) Top panel: quantitation of relative MYC RNA expression in NSCLC cell lines following 500 nM Aza treatment (microarray, day 8). Bottom panel: immunoblot shows expression of MYC protein on day 9 of treatment. β-actin was used as a loading control (500 nM Aza, n = 3).
(B) Unsupervised hierarchical clustering of relative RNA expression for GSEA HALLMARK MYC TARGETS. Color gradation is based on Z score ranking of log2 fold change over mock (microarray, day 8; 500 nM Aza, 100 nM MS-275, and 100 nM ITF-2357).
(C and D) A549 and H460 cell line quantitation of colorimetric absorbance as an indicator of proliferating cell number, normalized to untreated control. (C) Percentage proliferation for GFP and shMYC vector-infected cells treated with the indicated HDACi for 5 days (n = 3). (D) Percentage proliferation for empty vector (EV) or MYC overexpression vector containing cells treated with the indicated HDACi for 5 days (n = 3 overexpression clones).
(E and F) Relative RNA expression of IFNα/β pathway-responsive genes in A549 cells infected with EV or MYC overexpression construct and treated with mock or 500 nM Aza + 100 nM ITF-2357. (E) Quantitation of relative RNA expression for IFNα/β pathway-viral defense gene subset (genecard, day 8). (F) Quantitation of relative RNA expression for IFNα/β pathway-MHC class I genes (qRT-PCR, day 8, n = 3).
Data are presented as mean ± SEM (*p < 0.05 relative to mock and #p < 0.05 relative to EV + Epigenetic treatment; p value determined by two-tailed t test).
See also Figure S5.
High MYC tumors have been reported to display resistance to IFNγ signaling and the action of cytotoxic T lymphocytes (Casey et al., 2016; Schlee et al., 2007a, 2007b). In this regard, exogenous MYC overexpression resulted in antagonism of interferon-stimulated gene induction and antigen presentation induced by epigenetic treatment (Figures 4E and 4F). Thus, the MYC depletion signature imparted by Aza sensitizes to the actions of HDACi-induced cytostasis and augments NSCLC interferon signaling.
Combination Epigenetic Treatment Exerts a Robust Anti-tumor Effect and Decreases myc and myc Pathway Signaling in Mouse Models of NSCLC
The translational potential for utilizing our chronic schema of alternating, low-dose 1-week Aza followed by 1-week ITF-2357 was apparent from its robust efficacy when tested in two mouse models of NSCLC. The first, the LSL-KrasG12D mouse model of NSCLC, represented an interception model, as we began treatment when lung adenomas emerged 16 weeks following the lung-specific activation of the mutation (Jackson et al., 2001). Mice were treated with the combination drug treatment for 3 months using the schema as shown in Figure S6A. The therapy was well tolerated, and, while mock-treated mice developed large adenocarcinoma lesions in the lungs, combination epigenetic treatment prevented the occurrence of these macroscopic lesions and caused over a 60% reduction of tumor area in the treated mice (Figures 5A, 5B, and S6B). Consistent with all the studies in the human NSCLC cells, proliferation as assessed by Ki67 positivity was markedly reduced in any remaining histologic, adenomatous lesions (Figure 5C). To evaluate the implications of this treatment regimen in combination with immune checkpoint blockade, we added treatment with anti-PD1 antibody to the described regimen (Figure S6C). However, the combination epigenetic therapy alone proved as above to be so efficacious that it fully dominated the therapeutic effect (Figure S6D).
Figure 5. Combination Epigenetic Treatment Reduces Lung Tumor Burden and Progression in Mouse Models of NSCLC.

(A) Representative H&E-stained images of lung sections from mice treated with mock or Aza + ITF-2357. Scale bar, 100 μm.
(B) Quantitation of total tumor area occupied by lesions in lungs of LSL-KrasG12D mice treated with mock or Aza + ITF-2357. Data are presented as mean ± SEM (p value determined by two-tailed t test; n = 6 mock and n = 7 Aza + ITF-2357 mice per group/2 sections analyzed per mouse).
(C) Representative Ki67-stained IHC images of lung sections from mice treated with mock or Aza + ITF-2357 (n = 5 per group). Scale bar, 100 μm.
(D) Lewis Lung Carcinoma (LLC) tumor weights of subcutaneous explants from 1-month mock- and Aza + ITF-2357-treated mice (n = 19 mice per group; error bars, SEM).
(E) Representative H&E-stained images of lung metastasis from LLC mice obtained from 1-month mock- and Aza + ITF-2357-treated mice (n = 12 mice per group), with the indicated percentage frequency of metastasis.
(F) Volcano plot of relative RNA expression from LSL-KrasG12D mouse lung tumors treated for 3 months with Aza + ITF-2357 as compared to mock mice. Genes in upper left and right quadrants are significantly differentially expressed (microarray, n = 2 per group).
(G) GSEA (KEGG, REACTOME, and HALLMARK) pathway distribution for Aza + ITF-2357 versus mock tumors from LSL-KrasG12D mice. Horizontal line denotes FDR significance cutoff of 0.25. Immune- and cell cycle-related gene sets are demarcated by green and red dots, respectively (microarray, n = 2 per group).
(H) Gene sets upregulated in LSL-KrasG12D mice (FDR < 0.25 and NES > 1.5) by Aza + ITF-2357. Color gradation is based on GSEA NES.
(I) Representative upregulated GSEA plots with corresponding core-enriched genes. Color gradation is representative of Log2 fold change over mock.
(J) Gene sets downregulated in LSL-KrasG12D mice (FDR < 0.25 and NES < 1.5) by Aza + ITF-2357. Color gradation is based on GSEA NES.
(K) Representative downregulated GSEA plot with core-enriched genes. Color gradation is representative of Log2 fold change over mock-treated RNA expression.
*p < 0.05 calculated using two tailed t test. See also Figure S6.
The translational potential for our combination epigenetic drug efficacy for established, aggressive NSCLC was apparent utilizing a second mouse model of NSCLC, Lewis Lung Carcinoma (LLC). This murine model has demonstrated high correlation between response in the animal and response in the clinic for 2 therapeutic regimens (Kellar et al., 2015). Treatment with our alternating schedule of Aza + ITF-2357 and of Aza + MGCD0103 for 1 month not only reduced growth of established and rapidly growing primary tumor explants of the LLC cells but also dramatically reduced metastatic occurrence and burden (Figures 5D, 5E, S6E, and S6F).
Importantly, as outlined below, the above efficacy of our combination therapy was accompanied by profound immune effects. First, in the LSL-KrasG12D mice, the combination epigenetic treatment profoundly altered the tumor transcriptome with 5,167 significantly upregulated and 4,540 downregulated genes (Figure 5F). This corresponded to 18 gene sets upregulated and 52 gene sets downregulated by GSEA, with the former predominantly associated with immune parameters and the latter with cell cycle pathways (Figure 5G). Similar to the results for human NSCLC lines, 12 of the 18 gene sets upregulated involved strong induction of cytokine- and interferon-related gene sets (Figure 5H) as well as for 24 genes involved with inflammatory responses (Figure 5I). Concomitantly, as for the human NSCLC lines, most of the downregulated cell cycle pathways involved myc pathway signaling (Figure 5J), with decreases in 30 myc pathway-related genes (Figure 5K). Likewise, we confirmed the downregulation of MYC protein in the LLC cells (Figure S6G). These data in total demonstrate a potent reduction in tumor proliferation, growth, and burden is induced by the combination epigenetic treatment, which is mediated by repression of myc and myc-related signaling.
Combination Epigenetic Treatment Alters the Tumor Immune Microenvironment
In the above mouse models, deployment of epigenetic treatment reversed tumor immune evasion status, and a significant perturbation of tumor-associated immune cell subsets appeared to underlie the efficacy of the drug combinations. First, there was a robust alteration in the distribution of F4/80+ macrophages, which have been shown to have an established role in the progression of the LSL-KrasG12D mouse model (Okayama et al., 2013). These cells have been shown to basally infiltrate tumors induced in this model, and our combination treatment resulted in a significant decrease in their numbers (Figure 6A). Transcriptional profiling of tumor-associated macrophages (TAMs) isolated by fluorescence-activated cell sorting (FACS) of F4/80hi, CD11b+ macrophages from the lungs, following the 3 months of combination epigenetic treatment, revealed differential expression of 1,104 genes (Figure 6B), including significant downregulation of angiogenesis and hypoxia-responsive genes and gene sets (Figure 6C). These observed perturbations appeared to be a direct effect of the treatment and not a function of tumor stage, as treatment of normal bone marrow-derived macrophages (BMDMs) in vitro induced angiogenic signature downregulation as part of the 2,295 differentially expressed genes (Figures 6D and 6E).
Figure 6. Effect of Combination Epigenetic Treatment on Tumor-Associated Immune Populations and Their Functional Status.

(A) Representative IHC staining of F4/80+ macrophages in LSL-KrasG12D lung tumor sections treated with mock or Aza + ITF-2357 for 3 months. Upper panel: representative F4/80+ IHC. Lower panel: positive pixel transformation of IHC images in upper panel using Aperio Imagescope software. Scale bar, 100 μm.
(B) Volcano plot of relative RNA expression of CD45+CD11b+F4/80hi macrophages sorted via FACS and isolated from tumor-bearing lungs from 3-month mock- or Aza + ITF-2357-treated LSL-KrasG12D mice. Genes in the upper left and right quadrants are significantly differentially expressed (microarray, n = 2 per group). Hypoxia- and angiogenic pathway-associated genes are highlighted.
(C) Key affected pathways obtained from GSEA of CD45+CD11b+F4/80hi macrophage RNA isolated from tumor-bearing lungs from 3-month Aza + ITF-2357-treated LSL-KrasG12D mice as compared to mock-treated mice.
(D) Log2 fold relative RNA probe distribution showing differential gene expression from bone marrow-derived macrophages (BMDMs) treated in vitro with mock or Aza + ITF-2357. Angiogenic pathway-associated genes are highlighted (microarray, BMDM data representative of n = 3 mice).
(E) GSEA enrichment plot for angiogenesis pathway from BMDM RNA expression.
(F) CD8+ IHC in lung tumors of mock- and Aza + ITF-2357-treated LSL-KrasG12D mice following 3 months of treatment (scale bar, 100 μm). The graph on the right indicates the average number of CD8+ T cells counted per field of view (FOV) intra-tumor for mock and treated mice (n = 6 mock and n = 7 Aza + ITF-2357 mice).
(G) Relative RNA expression-based enrichment plot for hallmark interferon gamma response gene set in tumors from LSL-KrasG12D mice treated with mock and Aza + ITF-2357. Color gradation is representative of Log2 fold change over mock RNA expression (microarray, n = 2 per group).
(H) Percentage IFNγ+ CD8+/CD3+ TILs by FACS in LLC subcutaneous tumors from 1-month mock- and Aza + ITF-2357-treated mice.
(I) Volcano plot of relative RNA expression for CD45+CD3+CD8+ FACS-obtained lymphocytes isolated from tumor-bearing lungs of 3-month Aza + ITF-2357-treated LSL-KrasG12D mice as compared to mock mice. Genes in the upper left and right quadrants are significantly differentially expressed (microarray, n = 2 per group). Highlighted genes are involved in T cell fate determination.
(J and K) Fold change in expression of selected differentially expressed genes in FACS-obtained T cells from 3-month Aza + ITF-2357-treated mice. Genes shown are those that overlapped with exhaustion versus memory signatures (J) or exhaustion versus effector signatures (K) queried from Wherry et al. (2007) as defined in the Results. Genes on the left of each panel were differentially expressed by Aza + ITF-2357 and directionality in the gene set queried is on the right (red, upregulated; and blue, downregulated. The associated p value for the probability of overlap as derived by hypergeometric probability calculation is depicted above each panel. See also Figure S6.
The above altered pathways in TAMs may have implications for mediating CD8+ T cell intra-tumor infiltration. The inhibition of angiogenesis has proved efficacious in the MMTV mouse model through facilitating an increase in CD8+ tumor-infiltrating lymphocytes (Huang et al., 2012). This anti-angiogenic-based strategy for the induced infiltration of T lymphocytes depends on the concept of vascular normalization (Huang et al., 2013). Therefore, the repression of angiogenic programs in macrophages may act locally in the tumor to create a more immune infiltrate-permissive environment in the LSL-KrasG12D.
Critically, there were increased CD8+ tumor-infiltrating lymphocytes (TILs) in the tumor microenvironment of treated mice (Figure 6F). This enhanced TIL accumulation was accompanied by an IFNγ-responsive signature in the transcriptional profile of LSL-KrasG12D whole-tumor lysates (Figure 6G). As a validation of this signature, we profiled TILs by FACS in the LLC model, and, although we did not observe changes in total CD8 numbers in this short-term treatment model, there was a 2.25-fold increase in CD8+ IFNγ positivity, as the result of exposure to epigenetic treatment (Figures 6H, S6H, and S6I).
Transcriptional profiling of TILs from control and treated tumors in the LSL-KrasG12D mice suggested a key role of epigenetic therapy on these cells. TILs are known to acquire an exhaustion state in response to continuous stimuli and interaction with inhibitory ligands (Wherry and Kurachi, 2015; Zehn and Wherry, 2015). Therefore, the reversion or prevention of this exhausted state remains a major hurdle for durable T cell-mediated anti-tumor responses. A recent study by Ghoneim et al. (2017) established the utility of demethylating agents to reverse CD8+ T cell exhaustion, thereby allowing this population to be actionable by PD-1 blockade. Our epigenetic treatment regimen induced alteration of the TIL transcriptome. Comparative analysis of our 3,698 differentially expressed TIL genes revealed downregulation of exhaustion-associated genes and induction of activation- and memory-associated genes, as defined by Wherry et al. (2007). Our gene signature reflected an admixture of increased memory and/or effector T cells (Figures 6I–6K). The expansion of effector/memory T cell population is associated with durable responses to immune checkpoint blockade (Ribas et al., 2016). Thus, through the deployment of epigenetic treatment in two animal models, we confirmed epigenetic treatment can both increase activated CD8s in an inflamed tumor and induce the attraction of CD8s to an immune desert tumor.
Combination Epigenetic Treatment Inhibits Tumor Growth through CD8+ T Cells and MYC-Dependent Mechanisms
The above results demonstrated that epigenetic therapy can potentially reduce a key barrier to immune therapy, namely, the failure of tumors to attract functional CD8+ T cells. Depleting CD8+ T cells in the LLC mouse model attenuated the efficacy of epigenetic treatment on the reduction of primary tumor burden (Figures 7A and S6J). Relative to the Aza + ITF-2357 efficacy established in Figure 5, we also observed an increase in both metastatic frequency and burden as the result of CD8 depletion. In the CD8-depleted environment, we noted a metastatic frequency of 33% in mock- and 20% in Aza + ITF-2357-treated mice (Figure 7B).
Figure 7. The Functional Role of Immune Parameters for Anti-tumor Effects of Combination Epigenetic Treatment.

(A) Weights of subcutaneous LLC tumors from 1-month mock and Aza + ITF-2357 treated mice in the presence of CD8a-depleting antibody (n = 7 mock and n = 5 treated mice). NS, non-significant p value calculated by two tail t test.
(B) Representative H&E-stained images of lung metastases from the above 1-month mock and Aza + ITF-2357 treated mice in the presence of CD8a-depleting antibody (n = 7 mock and n = 5 treated mice).
(C) Expression of ccl5 RNA in lung tumors from LSL-KrasG12D mice in response to 3 months of treatment with mock or Aza + ITF-2357 (microarray, n = 2, p value < 0.05 calculated by two-tailed t test).
(D) Quantitation of ccl5 protein levels in bronchoalveolar lavage from LSL-KrasG12D mice treated with mock or Aza + ITF-2357 (n = 3; error bars, SD; *p value < 0.05 calculated by two-tailed t test).
(E) Relative fold change for CCL5 RNA expression in empty vector (EV) or MYC-overexpressing A549 human NSCLC cells treated with 500 nM Aza and/or 100 nM ITF-2357 (qRT-PCR, day 8, n = 3). Data are presented as mean ± SEM (*p value < 0.05 relative to EV and #p value < 0.05 relative to EV + Aza + ITF-2357, p values calculated by two-tailed t test).
(F) Profile interaction plots of TCGA RNA sequencing data for CCL5 and MYC expression across primary LUAD samples.
(G) Somatic copy number status of the MYC locus in NSCLC tumors. A 1.13-Mb segment overlapping MYC harbored a 12-fold amplification in CGLU117T1, the tumor that did not derive durable clinical benefit from immune checkpoint blockade. Orange lines represent segments of constant copy number. Circle and triangle markers indicate genomic bins in coding (target) and non-coding (off-target) regions, respectively.
(H) Copy number status of chromosome 8. The heatmap depicts segmental copy ratios after tumor purity correction, highlighting the amplification of the MYC locus for CGLU117T1 (arrow), but not for the other tumors analyzed.
See also Figure S6.
Additionally, in response to our combined epigenetic treatment, there was transcriptional augmentation of CCL5, a secreted chemokine involved in lymphocyte attraction (Figures 7C and S6K). Importantly, protein levels of CCL5 also accumulated in bronchoalveolar lavage of mice treated with combination epigenetic therapy (Figure 7D). This protein and its ligand have been identified as the primary factors influencing T cell infiltration of melanoma post-chemotherapy (Hong et al., 2011), thus suggesting that the enhanced levels of CCL5 induced by the epigenetic treatment may contribute to the increased number of CD8+ T cells observed in tumors of treated mice.
Several key findings suggest the importance of the above CCL5 results and link this parameter with the combination treatment-mediated downregulation of MYC signaling. First, expression of exogenous MYC in human A549 NSCLC cells acts as a potent repressor of CCL5 transcriptional induction in response to epigenetic treatment (Figure 7E). This links perturbation of MYC by epigenetic treatment to the alteration of immune correlates. Second, the potentially repressive role of MYC on CCL5 is also apparent in The Cancer Genome Atlas (TCGA) project primary NSCLC, lung adenocarcinoma (LUAD) samples, wherein there is an inverse relationship between CCL5 and MYC RNA transcription (Figure 7F).
Finally, our new findings in a small, previously reported cohort of patients with NSCLC treated with immune checkpoint therapy (Anagnostou et al., 2017) provide a suggestion for the importance of the present pre-clinical findings for the role of MYC signaling in response to immune therapy. In these four patients, all but one achieved durable clinical benefit, with partial responses for greater than 6 months as defined by RECIST 1.1. Genome-wide copy-number variation analysis on the tumors of these patients revealed a significant amplification of the MYC locus only in the one patient who did not achieve durable benefit (Figures 7G and 7H). Of note, the tumors from all four patients had characteristics predicting favorable response to checkpoint blockade therapy (Anagnostou et al., 2017), including high PD-L1 protein expression and high mutation density (Rizvi et al., 2015; Tumeh et al., 2014). These data, which must be validated in larger patient cohorts, suggest a predictive value when considering both the genetic and expression status of MYC for monitoring outcomes and practicing precision medicine for patients with NSCLC and other cancers receiving immune checkpoint therapy.
DISCUSSION
We have derived pre-clinical data supporting a role for epigenetic therapy in decreasing MYC signaling and driving a substantial anti-tumor response associated with a reversion of an immune evasion phenotype. We describe a novel regimen using careful selection of specific HDACis in combination with Aza. We find the inhibition of HDAC1, 2, and 3 isoforms is key for inducing a profound proliferative arrest (Figure S1H). The inhibition of HDAC6 appears to play a more important role in interferon signaling (Figures 3C–3F), possibly involving its known function for regulating Stat-1-dependent nuclear factor κB (NF-κB) signaling (Krämer et al., 2006).
We find these HDACis can complement the therapeutic effects of Aza across a diverse panel of NSCLC cell lines, thus suggesting its applicability for the treatment of NSCLC (Figures 1A and S1B). Our observation of a strong correlation between both MYC basal levels and degree of downregulation by Aza suggests the importance of MYC assessment for the delineation of patients most likely to respond to epigenetic treatment (Figures 4A and S5A).
The best indication of the translational potential for our combination epigenetic therapy are the results achieved by treating two mouse models of NSCLC (Figure 5). These models allow for our observations that the epigenetic treatment alters the intra-tumor localization and functional status of key immune cell populations (Figure 6). This occurs concurrently with a decrease in MYC pathway signaling (Figures 5K and S6G). The importance of MYC in driving tumor formation in the LSL-KrasG12D mouse model has been established, wherein the inhibition of MYC and its targets has been shown to eradicate lung tumor lesions (Soucek et al., 2013).
Although the above anti-tumor responses are partly driven by the direct growth inhibitory effects of the therapy as observed in immune-incompetent mice (Figures 1F–1H, S1K, and S1L), we also observed a significant alteration of immune populations mediated by our therapeutic paradigm when treating immune-competent mouse models (Figures 6A–6K, 7A, 7B, S6I, and S6J). The crucial role of the CD8+ T cells in driving the anti-tumor response is apparent from the significant attenuation of the anti-tumor response in the immune-competent mice following CD8+ T cell depletion (Figures 7A, 7B, and S6J). Our observation of decreased angiogenic potential of TAMs post-treatment has implications in tumor vascularization, which is essential for the growth of large adenocarcinoma lesions (Figures 6B and 6C). Key among these findings is the effect of epigenetic treatment duration on the modulation of T cell fate. A critical observation is that application of epigenetic treatment for limited duration induces the accumulation of activated T effector cells (Figure 6H and S6I), while chronic, long-term treatment induces modulation away from an exhausted T cell phenotype, inducing a profile more indicative of the formation of effector-memory T cells (Figures 6J and 6K). This acquisition of a memory phenotype coupled with intra-tumor accumulation of activated T cells imparts an immunological landscape primed for durable response to immune checkpoint therapy (Ribas et al., 2016).
A final point for high translational implications is our linking epigenetic therapy-induced attraction of CD8+ T cells to the tumor microenvironment and the relationship of this event with decreased MYC signaling. An important parameter altered, which may have implications for the induced infiltration of CD8+ T cells into the tumor microenvironment, is the simultaneous upregulation of CCL5 protein secretion into the bronchoalveolar compartment (Figure 7D). The forced overexpression of this secreted protein has been found to induce recruitment of T cells (Lavergne et al., 2004). Notably, others have found higher levels of CCL5 to be an independent prognostic indicator for longer overall survival, with an induction of an active tumor-associated lymphocyte compartment in NSCLC patients (Moran et al., 2002).
In patient tumors, CCL5 augmentation by epigenetic therapy may prove crucial both functionally and as a biomarker in pre- and post-epigenetic treatment. As a biomarker, these data indicate that such findings for basal levels of CCL5 may be especially valuable when combined with the other markers studied, such as the genetic and expression status of MYC. Considering these relationships may enable the prediction of those patients most likely to respond to a combination of epigenetic and immune therapy. Specifically, based on our corollary hypothesis, patients with MYC-high, CCL5-low tumors could be those who most benefit from this combination.
In summary, we believe that combination epigenetic therapy-induced depletion of MYC can remove a barrier to interferon responsiveness, potentiate T cell attraction, and cause direct antiproliferative actions. This coupled with the reversion of T cell exhaustion and acquisition of a T cell memory fate suggests the potential for use of epigenetic therapy in combination with immune checkpoint blockade. Our early findings for MYC amplification in the one of four patients who had a much less robust initial response to immune checkpoint response than the others who achieved durable clinical benefit are in keeping with the need to further pursue these findings in the clinic (Figures 7G and 7H). In this regard, the work in our present study and the hypotheses derived are being pursued in the aforementioned clinical trial for patients with NSCLC, testing our combined epigenetic therapy with the DNA-demethylating agent Guadecitabine plus the HDACi Mocetinostat plus anti-PD1 (NCT03220477).
STAR★METHODS
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Mouse monoclonal anti-β-Actin | Sigma-Aldrich | Cat#A5441; RRID: AB_476744 |
| Rat monoclonal anti-CD8a | BioXCell | Cat#BE0061; RRID: AB_1125541 |
| Rat monoclonal anti-CD16/CD32 | BD Biosciences | Cat#553141; RRID: AB_394656 |
| BV510 Rat monoclonal anti-CD45 | BD Biosciences | Cat#563891 |
| Rat monoclonal anti-CD3 | BD Biosciences | Cat#560527; RRID: AB_1727463 |
| FITC Rat monoclonal anti-CD8a | Biolegend | Cat#100706; RRID: AB_312745 |
| Brilliant Violet Rat monoclonal anti-F4/80 | Biolegend | Cat#123137; RRID: AB_2563102 |
| APC Rat monoclonal anti-CD11b | Biolegend | Cat#101212; RRID: AB_312795 |
| APC Rat IgG2b, kappa Isotype Ctrl antibody | Biolegend | Cat#400611; RRID: AB_326555 |
| Rat monoclonal anti-IFNγ | BD Biosciences | Cat#562018; RRID: AB_10896992 |
| Rat anti-PD-1 IgG2a (RMP1-14) | BioXCell | Cat#BE0146; RRID: AB_10949053 |
| Rat anti-IgG2a Isotype Control Clone 2A3 | BioXCell | Cat#BE0089; RRID: AB_1107769 |
| Rat monoclonal anti-CD8a | eBioscience | Cat#14-0808; RRID: AB_2572860 |
| Rat monoclonal anti-F4/80 | Serotec | Cat#MCAP497; RRID: AB_2335598 |
| Rabbit monoclonal anti-Ki67 | Cell signaling | Cat#12202; RRID: AB_2620142 |
| Rabbit monoclonal anti-c-MYC | Cell signaling | Cat#13987; RRID: AB_2631168 |
| Rabbit monoclonal anti-Acetyl α Tubulin | Cell signaling | Cat# 5335; RRID: AB_10544694 |
| Rabbit monoclonal anti-Acetylated Histone 3 | Cell signaling | Cat#4499; RRID: AB_10544537 |
| Rabbit polyclonal anti-Histone H3 | Cell signaling | Cat#9715; RRID: AB_331563 |
| Rabbit polyclonal anti-DNMT1 | Sigma Aldrich | Cat#D4692; RRID: AB_262096 |
| Anti-rabbit secondary (western blot) | GE Healthcare | Cat#NA934V |
| Anti-mouse secondary (western blot) | GE Healthcare | Cat#NA931V |
| Mouse monoclona IgG1 Isotype Control | Cell Signaling | Cat#5415; RRID: AB_10829607 |
| Rabbit anti-rat Biotinylated IgG | Vector | Cat#BA-4001; RRID: AB_10015300 |
| Bacterial and Virus Strains | ||
| Ad-CMV-Cre | Viral Vector Core, University of Iowa | Cat#VVC-U-IOWA-5 |
| Biological Samples | ||
| Patient-derived xenografts (PDX) | The Jackson Laboratory | Cat#TM00302 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Azacidine | Sigma-Aldrich | Cat#A2385 |
| ITF-2357 | Apexbio | Cat#A4093 |
| MS-275 | Syndax | MTA |
| MCGD0103 | Apexbio | Cat#A4089 |
| RGFP996 | Apexbio | Cat#A8803 |
| Tubastatin | Apexbio | Cat#A4101 |
| Vorinostat | Cell signaling | Cat#12520 |
| Puromycin | Sigma-Aldrich | Cat#P7255 |
| DMSO | Sigma | Cat#D2660 |
| RPMI1640 | Corning | Cat#10-040 |
| Fetal Bovine Serum | Hyclone | Cat#SH30910.03 |
| 0.25% Trypsin | Corning | Cat#25-053 |
| DMEM | Corning | Cat#10-013 |
| Pen/Strep | GIBCO | Cat#15140-122 |
| m-CSF murine | Peprotech | Cat#315-02-100UG |
| ECM gel | Sigma Aldrich | Cat#E2170-10ML |
| Lipofectamine 2000 | Invitrogen | Cat#11668-022 |
| Fixable Viability Dye | eBioscience | Cat# 65-0865-14 |
| eBiosciences Stim cocktail | eBioscience | Cat# 00-4970-03 |
| Propidium Iodide | eBioscience | Cat#00-6990-50 |
| Critical Commercial Assays | ||
| RNeasy Mini kit | QIAGEN | Cat#74104 |
| RNeasy Microkit | QIAGEN | Cat#74004 |
| RNase free DNase Set | QIAGEN | Cat#79254 |
| qScript cDNA SuperMix | Quanta | Cat#95048-025 |
| iTaq universal sybr green supermix | BioRad | Cat#1725124 |
| Homogenizer mini column | Omega | Cat#HCR003 |
| Purelink midiprep | Invitrogen | Cat#K210014 |
| Cell titer AQueous one | Promega | Cat#G3582 |
| BrdU Cell Proliferation ELISA | Calbiochem | Cat#QIA58 |
| Mouse Cytokine Array Q1 | Raybiotech | Cat#QAM-CYT-1-1 |
| Sigma Whole Transcriptome Amplification kit | Sigma Aldrich | Cat#WTA2-10RXN |
| Deposited Data | ||
| Raw and analyzed data for gene expression | This paper | GEO: GSE104244 |
| Experimental Models: Cell Lines | ||
| NCI-H1755 ( | ATCC | RRID: CVCL_1492 |
| NCI-H520 | ATCC | RRID: CVCL_1566 |
| NCI-H1650 | ATCC | RRID: CVCL_1483 |
| NCI-H1975 | ATCC | RRID: CVCL_1511 |
| NCI-H441 | ATCC | RRID: CVCL_1561 |
| NCI-H661 | ATCC | RRID: CVCL_1577 |
| NCI-H596 | ATCC | RRID: CVCL_1571 |
| HCC4006 | ATCC | RRID: CVCL_1269 |
| NCI-H1703 | ATCC | RRID: CVCL_1490 |
| NCI-H838 | ATCC | RRID: CVCL_1594 |
| NCI-H2170 | ATCC | RRID: CVCL_1535 |
| NCI-H1792 | ATCC | RRID: CVCL_1495 |
| NCI-H460 | ATCC | RRID: CVCL_0459 |
| NCI-H23 | ATCC | RRID: CVCL_1547 |
| A-549 | ATCC | RRID: CVCL_0023 |
| NCI-H1299 | ATCC | RRID: CVCL_0060 |
| HCC827 | ATCC | RRID: CVCL_DH92 |
| LLC1 | ATCC | RRID: CVCL_4358 |
| Experimental Models: Organisms/Strains | ||
| Mouse: B6.129S4-Krastm4Tyj/J | The Jackson Laboratory | RRID: IMSR_JAX:008179 |
| Oligonucleotides | ||
| shMYC (TRCN0000010391) | RNAi Consortium | Cat# TRCN0000010391 |
| Primers for qRT-PCR, see Table S1 | This paper | N/A |
| shDNMT1 | ||
| MYC vector | (Cheng et al., 2013) | Addgene plasmid # 46970 |
| pLV-AcGFP-N1 | Clontech | Cat#632154 |
| Recombinant DNA | ||
| Syncytin-1 (5′ LTR-Syn-1-pGL3) | (Chiappinelli et al., 2015) | N/A |
| ERV-3 (p3Xflag-CMV-14) | (Chiappinelli et al., 2015) | N/A |
| pE-GFP-N1 | Clontech Laboratories | Cat#632469 |
| pMD2.G | Addgene # 12259 | |
| psPAX2 | Addgene | Addgene plasmid #12260 |
| Software and Algorithms | ||
| GraphPad Prism | GraphPad Software | https://www.graphpad.com. RRID: SCR_002798 |
| Gene set enrichment Analysis | (Subramanian et al., 2005) | http://www.broadinstitute.org/gsea. RRID: SCR_003199 |
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Stephen B. Baylin (sbaylin@jhmi.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Cell lines and cell culture
Non small cell lung cancer cell lines used in the study were obtained from ATCC. The cells were cultured in RPMI1640 media (Corning) containing FBS (Hyclone) at 10% v/v. Cells were maintained at 37°C in the presence of 5% CO2. Cells were passaged every 3 days. Human Non Small Cell Lung Cancer cell lines: NCI-H1755 (female), NCI-H520 (male), NCI-H1650 (male), NCI-H1975 (female), NCI-H441 (male), NCI-H661 (male), NCI-H596 (male), HCC4006 (male), NCI-H1703 (male), NCI-H838 (male), NCI-H2170 (male), NCI-H1792 (male), NCI-H460 (male), NCI-H23 (male), A-549 (male), NCI-H1299 (male), HCC827 (female)
In Vivo Mouse Studies
Xenograft studies in NOD/SCID mice
We used male NOD-SCID mice (5–6 weeks old) for these studies. Mice were bred and housed at the Johns Hopkins Animal care facility. All animal experiments were approved by the Johns Hopkins Animal Care and Use Committee. All animal care and protocols followed were in accordance with guidelines of the institutional Animal Care and Use Committee (IACUC). Mice were injected subcutaneously in the flank with 2.0 × 105 viable cells for H1299 and 5.0 × 104 viable cells H460 cells in 0.2 mL of RPMI with Matrigel (1:1). Drug treatments were started 10 days post injection for H460 and 14 days post injection for H1299, when palpable tumors could be discerned and continued as indicated in the figure. Mice were treated with the dosing schedules shown in (Figure S6A). Treatment was continued for the entire duration of the study and mice were sacrificed before tumors volume exceeded 2000 mm3. Tumor volume determined by measurements obtained from digital caliper and calculated as V = 0.5(LxWxH).
PDX mouse model
A PDX mouse model (TM00302) was obtained from Jackson Laboratories. This model was derived from metastatic lung adenocarcinoma in a female patient with the following mutations KRAS G12D, KDR Q472H, TP53 R158L. The tumor was harvested from a JAX provided donor mouse and sectioned into 2 mm3 fragments. These fragments were then implanted subcutaneously in the flank of male NSG mice (5–6 weeks old). Once implanted tumors were palpable, mice were randomized into three treatment groups and dosed as depicted in Figure S6A. Mice were treated for the duration of the study and at culmination of treatment mice were sacrificed. Tumors were excised from the animals and weighed as a measure of disease burden.
LSL-KrasG12D mouse model
LSL-KrasG12D mice (Jackson et al., 2001) were obtained from Jackson laboratories and bred with wild-type C57BL/6 mice to generate heterozygous LSL-KrasG12D mice. LSL-KrasG12D mice (8–10 weeks, both genders) were infected intratracheally with Adenoviral vectors encoding Cre (obtained from University of Iowa) to activate Kras. LSL-KrasG12D mice harbor a latent point mutant allele of Kras (KrasG12D) and infection with an adenoviral vector encoding Cre leads to Cre-mediated deletion of a transcriptional termination sequence (Lox-Stop-Lox, LSL) and the oncogenic Kras production, thereby resulting in very high frequency of lung tumors (Jackson et al., 2001). All experimental animal protocols were performed in accordance with guidelines approved by the animal care and use committee at the Johns Hopkins University (Baltimore, MD). Mice were examined for presence of disease at 16 weeks post infection with AdenoCre. Mice were sacrificed and lungs were examined histologically. At this stage most of the mice showed presence of significant regions of hyperplasia as well as adenomas. Drug treatments were started as per the schedule shown in Figure S6A. Mice were divided in two groups. One group was injected with vehicle control while the other was injected with combination epigenetic therapeutics. Mice were uniformly distributed into the two treatment groups so that each group had comparable numbers of age and weight matched male and female mice. Drug treatments were continued for 12 weeks, following which mice were sacrificed and the lungs processed for histological studies, RNA extraction and analysis of cytokine levels in lung bronchoalveolar lavage (BAL) fluid as described as described in main methods.
Lewis lung Carcinoma (LLC) model
LLC cells (LLC1, ATCC) 2.0 × 104 were injected subcutaneously in flanks of male C57BL6 mice (6–8 weeks old) obtained from Harlan. LLC1 cells were suspended in 100uL RPMI + Matrigel (1:1). Drug treatments were started 7 days post injection when palpable tumors could be discerned. Mice were injected with drugs as per the dosing schema in Figure S6A. Treatment was continued for a month and at the culmination of study mice were sacrificed, tumors were harvested and weighed. Lungs of these mice were inflated and fixed in formaldehyde for 24 hours prior to sectioning and staining with H&E stains.
LLC model CD8 depletion model
CD8a antibody (BioXcell, 2.43 clone) mediated CD8 depletion of male C57BL6 mice was initiated 3 days prior to onset of treatment (Day 4 post implantation). Implantation and therapeutic treatment of CD8 depleted LLC model is described in Lewis Lung Carcinoma model section of methods. For CD8 depletion mice were injected IP with 150ug of CD8a antibody (BioXcell, 2.43 clone), three times per week throughout the duration of study. CD8 depletion was verified by assessment of CD8% of CD45 population in the spleen of depleted sample versus control samples. Treatment was continued for one month and at the culmination of study mice were sacrificed, tumors were harvested and weighed. Lungs of these mice were inflated and fixed in formaldehyde for 24 hours prior to sectioning and staining with H&E stains.
METHODS DETAILS
Drug Reagents
Azacitidine (Sigma) was dissolved in PBS at 500uM (in vitro) and 7.1 mg/mL (in vivo), aliquoted, and stored at −80C for single use. ITF-2357 (Apexbio) was dissolved in DMSO to concentrations of: 1mM, 500uM, 250uM, 100uM, 50uM and 25uM (in vitro) and 50mg/mL (in vivo), aliquoted and stored at −20C. MS-275 (Syndax) was dissolved in DMSO to concentrations of: 2.5mM, 1mM, 500uM, 250uM, 125uM (in vitro) and 50mg/mL (in vivo), aliquoted and stored at −20C. MGCD0103 (Apexbio) was dissolved in DMSO to concentrations of: 1mM, 500uM, 250uM, 100uM, 50uM and 25uM (in vitro) and 10mg/mL (in vivo), aliquoted and stored at −20C. RGFP996 (Apexbio) was dissolved in DMSO to concentrations of: 10mM, 5mM, 2.5uM, 1uM, 500uM and 100uM (in vitro), aliquoted and stored at −20C. Tubastatin A (Apexbio) was dissolved in DMSO to concentrations of: 10mM, 5mM, 2.5mM, 1mM, 500uM and 250uM (in vitro), aliquoted and stored at −20C. Vorinostat (Cell signaling) was dissolved in DMSO to concentrations of 300uM (in vitro), aliquoted, and stored at −20C. Puromycin (Sigma), dissolved in PBS at 1mg/mL, aliquoted and stored −20C.
Azacitidine drug treatments
Cell lines were plated at the following densities for Azacitidine treatments per T75 flask: NCI-H1755 (2.0*105), NCI-H520 (2.0*105), NCI-H1650 (2.0*105), NCI-H1975 (1.0*105), NCI-H441 (2.0*105), NCI-H661 (2.0*105), NCI-H596 (2.0*105), HCC4006 (2.0*105), NCI-H1703 (2.0*105), NCI-H838 (1.0*105), NCI-H2170 (2.0*105), NCI-H1792 (1.0*105), NCI-H460 (1.0*105), NCI-H23 (2.0*105), A-549 (1.0*105), NCI-H1299 (1.0*105), HCC827 (2.0*105). Cells were allowed to adhere for 24 hours before the onset of treatment. Cells were treated with 500uM Azacitidine diluted 1:1000 or PBS vehicle in complete RPMI1640 media every 24 hours for the indicated treatment duration. Drug treatments with HDACi are assay specific and are detailed in the relevant sections.
Cell Viability Assays (MTS colorimetric)
Cells treated as described in Azacitidine treatment section, were trypsinized, enumerated, and assessed for viability by trypan blue exclusion assay. Equal numbers of viable cells were plated in 96 well plates at the following densities per well, in technical triplicate: NCI-H1755 (2.0*103), NCI-H520 (2.0*103), NCI-H1650 (2.0*103), NCI-H1975 (1.0*103), NCI-H441 (2.0*103), NCI-H661 (2.0*103), NCI-H596 (2.0*103), HCC4006 (2.0*103), NCI-H1703 (2.0*103), NCI-H838 (1.0*103), NCI-H2170 (2.0*103), NCI-H1792 (1.0*103), NCI-H460 (1.0*103), NCI-H23 (2.0*103), A-549 (1.0*103), NCI-H1299 (1.0*103), HCC827 (2.0*103). Cells were allowed to adhere for 24 hours in the presence of complete media prior to the onset of treatment. Adhered cells were incubated with 100uL drug supplemented media changed every 3 days, treated with DMSO(vehicle) at 0.1% or the following drugs/concentrations standardized to 0.1% DMSO final concentration. ITF-2357: 25nM, 50nM, 100nM, 250nM, 500nM, 1uM; MGCD0103: 25nM, 50nM, 100nM, 250nM, 500nM, 1uM; RGFP996: 250nM, 500nM, 1uM, 2.5uM, 5.0uM and 10uM; Tubastatin A: 250nM, 500nM, 1uM, 2.5uM, 5.0uM and 10uM; MS-275: 125nM, 250nM, 500nM, 1uM, 2.5uM. Treatments were applied for a period of 5 days, afterwhich, MTS colorimetric assay (promega) was conducted. MTS reagent was diluted 20uL per mL in complete media and vortexed to ensure the resultant solution was homogeneous. Drug treated media was gently removed from treated cells by multichannel pipette and replaced with MTS supplemented media. Cells were incubated in 37C incubator to allow the colorimetric reaction to occur. After incubation 490nm absorbance was read using the BioRad iMark microplate reader for media+MTS, vehicle treated+MTS and drug treated+MTS cells. Absorbance values were imported to Graphpad prism. Data were normalized and drug doses were log transformed. These normalized, log transformed data were analyzed by 4 parameter nonlinear regression to generate log dose response curves for each cell line and drug condition and determination of drug effect at dose. The resultant dose response curves are a representation of these data, with the indicated biological replicates being a representation of the mean for each condition. These dose response data were utilized for drug synergy analysis (Compusyn). The fractional effect data for each drug condition were imported for the inhibitors deployed both alone and in combination with Aza. These data were analyzed and used to generate combination index output as an indicator of drug synergy.
BrdU Cell Proliferation Assay, colorimetric ELISA
Cells were plated in technical triplicate in 96 well format at the densities described in the Cell viability section. Vehicle or Aza treated cells were allowed to adhere for 24 hr prior to the onset of treatment. Cell were subsequently treated with the following inhibitors for 3 days. MGCD0103: 200nM, ITF-2357: 100nM, Tubastatin A: 1uM, RGFP996: 2uM. On day 3 of treatment, Stock Brdu (Calbiochem BrdU Cell Proliferation Assay) was diluted 1:1000 in complete media and added to 100uL of drug treated media. Cells were incubated with BrdU at 37C for 5 hours to allow for DNA synthesis based incorporation. The remainder of protocol was carried out as described in Calbiochem protocol, with the lot specific 1:2400 dilution of secondary antibody. 450nm absorbance values derived from ELISA were measured using the Biorad iMark plate reader. The triplicate measurements per condition were imported to graphpad prism and the data background subtracted, normalized to vehicle treated controls to generate the normalized proliferation. Error bars depicted as SEM between the means of experimental replicates. Statistical significance is the result of two way t test conducted between the experimental conditions depicted.
Gene expression analysis by microarray
RNA expression analyses were conducted by microarray for the conditions described in the relevant figures. The R/Bioconductor package limma was used to process expression data. Within- and between-array normalizations were performed using the loess and aquantile methods, respectively. The normexp option was used for background correction. Raw files read in using the read.maimages function. Log2 fold change in transcription for drug treated conditions over mock treated was obtained for each sample at each time point studied. Ranked lists of log2 fold change were analyzed using Gene Set Enrichment Analysis (GSEA) by the Broad Institute and data packages (Subramanian et al., 2005). Pathways enriched with a false discovery rate less than 0.25 were selected with cutoff of 2.0 normalized enrichment score in human and 1.5 normalized enrichment score cutoff in mouse studies. Median absolute deviation analysis of Log2 fold change transcription data were obtained using Morpheus program (Broad). Top 500 genes were selected and depicted as heatmap representative of minimum to maximum value ranking for each gene across cell lines and conditions.
DAVID analysis of median absolute deviation derived genes (KEGG Pathways)
Top 500 genes derived by median absolute deviation analysis were obtained as described in Gene expression by microarray methods section and were analyzed for KEGG Gene Ontology (GO) enrichment using the DAVID Bioinformatics resources database (Huang da et al., 2009a, 2009b). Only categories that were below the DAVID p value of 0.05, and containing at least 5 genes per pathway are reported.
MYC targets heatmap
MYC targets heatmaps are based on hierarchical clustering of Log2 fold change over mock using Euclidean distance and complete linkage of MYC hallmarks gene set v2.
ERV absolute quantitative real time PCR (qPCR)
Eleven codogenic and partially codogenic ERV envelope (env) gene families (see Figure 3 and Figure S4 for ERV env genes used in this investigation), one ERV gag (ERV-W5) and two ERV pols (ERVFXA34, erv9-1) were quantified using qPCR and have been previously described (Chiappinelli et al., 2015). Additionally, some of these ERV primers could potentially hybridize with other ERV transcripts among the ERV family members, e.g., the ERV-K env primers could hybridize with 8 transcripts derived from ERV-K102 (1q22), −107 (5q33.3), −108 (7p22.1), −109 (6q14.1), −113 (19p13.11), −115 (8p23.1), −17833 (19q12), −74261 (12q14.1) according to sequence alignments (Chiappinelli et al., 2015).
The qPCR methodology detecting the ERV genes has been previously described (Chiappinelli et al., 2015). Briefly, ERV genes were amplified by qPCR from 40 ng of cell line cDNA with SYBR-green technology and then analyzed with an ABI7300 (ABI, Darmstadt, Germany). Expression values were calculated as molecules per ng total RNA using a standard curve of each cloned gene determined by real time PCR and calculated as mean ± SEM. TBP and beta-actin were used as housekeeping genes where a mean from both genes was used for normalization of NSCLC cell lines.
IFNα/β qPCR
qPCR methodology for detecting IFNα/β related genes (ISG15, OASL, HLA-A, HLA-B) differential expression was conducted on 20ng of cell line cDNA with SYBR green technology. Expression values were calculated as fold change over mock by delta-delta ct. Beta-actin was used as housekeeping gene for normalization of NSCLC cell line expression.
Taqman microfluidic genecard
1ug of total RNA was used for cDNA reaction and diluted to 50uL total volume in molecular grade water. Diluted cDNA was combined with 50uL of Taqman universal PCR mastermix and loaded onto a Taqman microfluidic genecard with the following primer/probe sets preloaded: IRF7, IFI27, STAT1, IFNB1, MX1, OASL, IFI6, beta actin, TBP. Expression values were calculated as fold change over mock by delta-delta ct. Beta actin and TBP were used as housekeeping genes for normalization of NSCLC cell line expression.
ERV Overexpression and OASL qPCR
Syncytin-1, ERV-3, and empty vectors were transfected using standard lipofectamine 2000 methods. Vectors for ERV overexpression assay are described in Chiappinelli et al., 2015. Four days post transfection of either empty or indicated ERV vector assessment of OASL and ERV3 or Syn1 RNA transcription by qPCR was conducted.
Immunoblotting
Protein was extracted by 4% SDS mediated lysis, followed by processing of lysates through homogenizer column (Omega). Protein concentrations were estimated by BCA (Pierce Biotechnology). Lysates were processed using PAGE with 4%–12% Bis-Tris BOLT gel (Life Technologies) and transferred to PVDF membrane (Millipore). Membranes were blocked in 10% milk/TBST and immunoblotted with the following antibodies.: rabbit monoclonal c-MYC (Cell Signaling, 1:1000), Rabbit monoclonal anti-Acetyl α Tubulin (Cell Signaling, 1:1000), Rabbit monoclonal anti-Acetylated Histone 3 (Cell Signaling, 1:1000), Rabbit polyclonal anti-Histone H3 (Cell Signaling, 1:1000), Rabbit polyclonal anti-DNMT1 (Sigma Aldrich, 1:2000), Mouse monoclonal anti-β-Actin (Sigma Aldrich, 1:10000). The loading control antibodies (anti-β-Actin, anti-Histone H3) in all cases were applied after membrane stripping.
DNMT1 Knockdown Cell Viability Assay
DNMT1 knockdown was initiated see reference for sequence (Cai et al., 2017) and control vector shGFP 5′-GCAAGCTGACCCT GAAGTTCAT3-3′. Clonal selection was initiated with puromycin selection. Selected clones were plated at cell densities, treated and analyzed as described in Cell viability Assay section of methods. Error bars are representative of mean ± SEM between experimental replicates.
MYC Knockdown Cell Viability Assay
MYC knockdown was initiated using Broad TRCN0000010391 clone and control vector shGFP 5′-GCAAGCTGACCCTGAAGTT CAT3-3′. Clonal selection was initiated with puromycin selection. Selected clones were plated at the cell densities and treated as described in Cell viability assay section of methods. MTS was used to quantify numbers of proliferating cells. Bar graphs depicted are representative of the percent of MYC knockdown cells present as a percentage of the empty vector treated controls for same dose of drug. Error bars are representative of mean ± SEM between experimental replicates. Statistical significance was determined by two tailed t test between empty vector and MYC shRNA treated cells at each dose.
MYC Overexpression Cell Viability Assay
MYC vector (Addgene plasmid # 46970) (Cheng et al., 2013) and empty vector (Clontech cat# 632154). Clonal selection was initiated with puromycin, 2 days post infection. Selected clones were plated at cell densities and treated as described in Cell viability assay section of methods. MTS was used to quantify numbers of proliferating cells. Bar graphs depicted are representative of the percent of MYC overexpression cells present as a percentage of the empty vector treated controls for same dose of drug. Error bars are representative of mean ± SEM between experimental replicates. Statistical significance was determined by two tailed t test between empty vector and MYC overexpression vector treated cells at each dose.
In-vivo Drug Studies
Mice were treated as follows: Azacitidine 0.5 mg/kg (PBS vehicle) IP injected daily for the first 5 days of every 14 day treatment cycle. In vivo HDACi was applied on day 7 of 14 day treatment cycle and injected daily for 5 days of 14 day cycle. ITF-2357 2.0 mg/kg (1% DMSO in PBS vehicle) IP injected daily for 5 days sequentially, every 14 days. MS-275 2.0 mg/kg (1% DMSO in PBS vehicle) IP injected daily for 5 days sequentially, every 14 days. MGCD0103 4.0 mg/kg (5% DMSO in PBS vehicle) IP injected daily for 5 days sequentially, every 14 days. InVivoMAb mouse Anti-PD1 (BioXcell RMPI 14 clone) injected 10ug/kg on the 12th day of a 14 day cycle. InVivoMAb Rat IgG2a Isotype control (BioXcell 2A3 clone) injected 10ug/kg on the 12th day of a 14 day cycle. CD8a depletion antibody (BioXcell, 2.43 clone), 3 times per week at 150ug per animal, see LLC CD8 depletion model for details about the treatment schema. For xenograft studies, treatment was initiated when palpable tumors could be discerned and continued for the duration of study. For Kras G12D mouse model studies, drug application initiated 16 weeks post Ad-Cre instillation, and applied for 12 weeks as described in the Kras G12D animal model section. For LLC model treatment was initiated 7 days after subcutaneous implant of flank tumor and mice were treated as described in (Figure S6A). Mice were injected IP with 150ug of CD8a antibody (BioXcell), three times per week throughout the duration of study. CD8 depletion was verified by assessment of CD8% of CD45 population in the spleen of depleted sample versus control samples.
Assessment of lung tumor formation
Mice were sacrificed and the left lung was fixed in formalin for histologic examination. The right lung in some mice were lavaged by instilling with 1.0ml of sterile PBS to collect Bronchoalveolar lavage (BAL) fluid. In some mice right lung was used to macro dissect tumors for RNA extraction.
Bronchoalveolar lavage (BAL) fluid cytokine analysis
1.0 mL of BAL fluid was instilled into the right lung of tumor bearing KrasG12D mice. 50uL of this BAL was utilized for cytokine analysis as described by the manufacturer (Raybiotech, QAM-CYT-1). Cytokine array was scanned by the manufacturer. Cytokine quantification was conducted using a standard curve of each cytokine present at known concentrations supplied by the manufacturer. p values for cytokine data were derived using Graphpad Prism software and were defined as < 0.05 by unpaired two tailed t test when comparing treated condition versus control, biological replicates were used for the derivation of significance. Data are mean ± SD between biological replicates.
Histological analysis of mouse tissues
Lungs tissues were fixed in 10% formalin overnight, and subsequently transferred into 70% ethanol, embedded in paraffin and sectioned (5 μM) at 3 levels at regular intervals. Sections were stained with Hematoxylin and eosin (H&E) and scanned using the Aperio whole slide scanning system (Scanscope CS) The entire left lung lobe was scanned for presence of adenomas/adenocarcinomas using the Aperio ImageScope analysis software. For each sample, sections from 3 levels were analyzed. Unstained sections of the left lung were also used for Immunohistochemistry (IHC) staining. All Images were scanned at 20x magnification using the Aperio Scanscope system.
Immunohistochemistry (IHC)
Immunostaining of lung sections was performed with the PowerVision kit according to the manufacturer’s protocol (Leica Bio-systems). Briefly, slides were heated at 60°C for 10 min,deparaffinized and hydrated through xylene, graded ethyl alcohols, dH2O, dH2O with 20% Tween 20 (P-7949, Sigma-Aldrich). After antigen retrieval (45 minutes of steaming in Target Retrieval Solution (Dako S170084-2) using Black and Decker Handy Steamer Plus), sections were treated 5 minutes with Dual Endogenous Enzyme Block (S2003, Dako). Sections with primary antibodies Ki67 (Cell signaling, 9101, 1:500) were incubated at room temperature for 45 minutes. Additional blocking steps were used for CD8 and F4/80 slides using DakoCytomation Biotin Blocking System (X0590). Antibody incubations for CD8 (eBiosciences, 14-0808, 1:800) and F4/80 (Serotec, MCAP497, 1:1000) were carried out at room temperature for 45 minutes, soaked an additional 45 min in PBS-Tween, and followed by mouse adsorbed biotinylated anti Rat IgG (Vector, BA-4001, 1:500) for 15 minutes. For all, the secondary used was anti-rabbit IgG-reagent provided in the Powervision kit (PV6119, Leica Biosystems) for 30 minutes. Immunostaining was visualized with DAB chromogen (D4293, Sigma-Aldrich) and sections were counterstained with Mayer’s hematoxylin. Control slides: No primary for each, CD8 (spleen), F480 (tumor)
F4/80 Intratumor Macrophage Analysis
F4/80 IHC stained sections were scanned used the Aperio system as described earlier, these images were analyzed using image-scope software. To generate the false color images depicted used to demarcate pixel saturation by macrophage infiltration, the images were analyzed by the positive pixel v4 algorythm.
Isolation of murine bone marrow derive macrophages (BMDMs)
Mice (6–8) week old were euthanized and hind legs were harvested under aseptic conditions. Femurs and tibia were obtained and flushed with RPMI medium. Cells were then centrifuged and treated with ACK buffer to lyse RBC’s. Cell pellets were then washed with and resuspended in RPMI medium and plated in six well plates in the presence of M-CSF. Medium was changed every 2 days. Treatments with Aza and HDACi were started after 4 days and the in vitro drug treatment schema was 5 days of 500nM Aza followed by either vehicle or 100 nM ITF-2357 for 5 days. Cells were then collected and RNA extracted.
TCGA Analysis
Broad Morpheus tool was used to analyze TCGA LUAD data for the relationship between CCL5 and MYC. First the 2 genes were selected from available RNA seq data of 576 patients available. These data were used to generate a new heatmap. This new heatmap was sorted based on CCL5 and MYC expression and the samples were separated into low, intermediate and high expression groups. These subsets were than compared using the profile interaction tool to generate the depicted plots. 576 LUAD samples were ranked by expression of CCL5 and MYC into: low, intermediate and high expression groups. These 3 expression categories for MYC and CCL5 were then compared as follows on the y axis, where gene(y) is sorted based on low, intermediate or high expression of other gene: MYC high:CCL5(y), MYCintermediate:CCL5(y), MYClow:CCL5(y), MYC(y):CCL5high, MYC(y):CCL5intermediate, MYC(y):CCL5low.
Patient and sample characteristics used for MYC amplification studies
The studies were conducted in accordance with the Declaration of Helsinki and were approved by the Institutional Review Board (IRB), and patients provided written informed consent for sample acquisition for research purposes. We analyzed pre-treatment exome data derived from a recently published set of NSCLC patients treated with immune checkpoint blockade (Anagnostou et al., 2017). Patient and sample characteristics as well as treatment and response assessments are described in detail elsewhere (Anagnostou et al., 2017). In brief, our study group consisted of two NSCLC patients treated with PD-1 blockade (CGLU117T1 and CGLU127T1) and two NSCLC patients treated with combined PD-1 and CTLA4 blockade (CGLU116T1 and CGLU161T1). All patients achieved a partial response of more than six months in duration (durable clinical benefit-DCB) apart from CGLU117T1, who achieved stable disease (22% tumor regression by RECIST 1.1) of 4 months duration before he developed disease progression (non-durable clinical benefit-NCB). An additional NSCLC tumor sample from a patient with sustained response to PD-1 blockade was obtained for comparative analysis of CD8+ T-cell density.
Somatic Copy Number Analysis
Whole exome sequence data from a previously published cohort of four patients with NSCLC (Anagnostou et al., 2017) was evaluated to determine the somatic copy number of MYC locus in pre-treatment tumor samples. The sequencing depth of coverage of the tumor sample in bins spanning coding and non-coding regions of the genome were compared to the matched normal sample from each patient. The log2 read ratio values were corrected for biases resulting from the exome capture process and sequence composition using CNVKit (0.7.6) (Talevich et al., 2016), and were adjusted to account for variable normal cell contamination as previously described (Anagnostou et al., 2017).
FACS sorting and profiling of macrophages and T cells LSL-KrasG12D
For FACS sorting of macrophages and CD8 T cells from LSL-KRasG12D mice, lungs were harvested from mock or treated mice following three months of treatment. Tissue was then digested for 30 minutes at 37°C in digestion buffer (RPMI, FBS (10%), Collagenase Type 1 (0.2%), Collagenase Type 2 (0.2%) and DNase I (50u/mL). Tissue was then minced and strained through a 40um cell strainer to obtain a single cell suspension. RBC’s were then lysed with ACK buffer and the cell pellet was washed twice in RPMI prior to suspension in RPMI medium. Cells were then counted and blocked with Rat monoclonal anti-CD16/CD32 (Fc block Antibody) in PBS for 30 min at 4°C. Cells were then stained with antibodies against CD45, Cd11b and F4/80 for macrophages or CD45, CD3, CD8a for CD8+ T cells. Propidium Iodide was used as a viability marker (5uL/100uL total volume). Cells were sorted and pellets were lysed and RNA extracted using RNeasy kit. RNA was then quantitated and amplified using the Sigma Whole Transcriptome Amplification kit. Amplified RNA was then processed and analyzed on Mouse Agilent 4×44 single color microarray. Post sort purity checks were utilized and the sorted populations were determined to be more than 95% pure population.
FACS profiling T cells LLC
For FACS profiling of CD8+ T cells from LLC mice, primary tumors were harvested from mock or treated mice following one month of treatment. Tissue was then digested using a mixture of collagenase, hyaluronidase and DNase. The resulting single cell suspension was counted and plated in complete media (RPMI+10% FBS) with or without eBiosciences stimulation cocktail (1:1000) for 4 hours. Cells were then collected and blocked with Rat monoclonal anti-CD16/CD32 (Fc block Antibody) in PBS for 30 min at 4°C. Cells were then stained with antibodies against Live/dead, CD45, CD3, CD8a, and IFNγ for CD8+ T cells. Cells were then profiled by FACS.
QUANTIFICATION AND STATISTICAL ANALYSIS
Dose response data
Four parameter, non-linear regression of normalized and log transformed colorimetric absorbance response to increasing concentrations of therapeutics as described in each figure. Data analyzed using Graphpad prism, error bars are mean ± SEM between biological replicates unless indicated otherwise.
BrdU proliferation data
Colorimetric absorbance was used as an indicator of proliferating cell numbers. Bar graphs depicted are representative of the percent of treated cells normalized to untreated controls. Error bars are representative of mean ± SEM between experimental replicates. Statistical significance was determined using Graphpad Prism software and are two tailed t test between control and Aza treated cells or Aza + MS275 and Aza + ITF-2357 at dose indicated in the figure.
Microarray data
Differential gene expression was defined as 0.5 log2 fold change in treated condition versus control. Significantly enriched gene sets were defined using an FDR cutoff < 0.25. p values for microarray data were derived using R programming language, limma package, and were defined as < 0.05 when comparing treated condition versus control.
qRT-PCR data
Differential gene expression was defined as 0.5 log2 fold change in treated condition versus control. p values for qRT-PCR data were derived using Graphpad Prism software and were defined as < 0.05 by unpaired two tailed t test when comparing treated condition versus control, biological replicates were used for the derivation of significance.
MYC knockdown/overexpression data
Colorimetric absorbance was used as an indicator of proliferating cell numbers. Bar graphs depicted are representative of the percent of MYC knockdown cells present as a percentage of the empty vector treated controls for same dose of drug. Error bars are representative of mean ± SEM between experimental replicates. Statistical significance was determined using Graph-pad Prism software and are two tailed t test between empty vector and MYC knockdown or overexpression treated cells at each dose.
Tumor area quantification data
Tumor area was quantitated as total area of left lung presenting with lesions in LSL-KrasG12D mice using Aperio Imagescope software. p values for tumor area quantification data were derived using Graphpad Prism software and defined as < 0.05 by unpaired two tailed t test when comparing treated condition versus control, independent animals were used for the derivation of significance. Error bars are representative of mean ± SEM between experimental replicates.
Immune cell infiltration data
IHC quantification- tumor infiltrating CD8 T cells were enumerated in field of view for hotspots of infiltration for the conditions indicated in the figure. The n values depicted indicate individual animals scored for infiltration. p values for IHC quantification data were derived using Graphpad Prism software and were defined as < 0.05 by unpaired two tailed t test when comparing treated condition versus control, independent animals were used for the derivation of significance. FACS infiltration data- tumor infiltrating CD8 T cells were profiled from whole digested tumors. The n values depicted indicate individual animals profiled by FACS. p values for FACS data were derived using Graphpad Prism software and were defined as < 0.05 by unpaired two tailed t test when comparing treated condition versus control, independent animals were used for the derivation of significance.
Hypergeometric test of cumulative distribution function
The number of overlapping differentially expressed genes present in the Wherry et al., 2007 and our RNA expression dataset (> 0.5 log2 fold change over mock treated, p value < 0.05) were used to calculate over representation of these genes. p values were calculated using hypergeometric test of cumulative distribution function using p value/representation calculator (http://systems.crump.ucla.edu/hypergeometric/).
Cytokine data
Data are array based fluorescence intensity. Cytokine quantification was conducted using a standard curve of each cytokine present at known concentrations supplied by the manufacturer p values for cytokine data were derived using Graphpad Prism software and were defined as < 0.05 by unpaired two tailed t test when comparing treated condition versus control, n value represents biological replicates and data are mean ± SD between biological replicates.
TCGA data
Broad Morpheus tool was used to define low, medium and high groups for MYC and CCL5 expression. These groups were then compared as defined in the figure using the built-in profile interaction tool (Morpheus).
MYC amplification data
Whole exome sequencing depth of coverage of the tumor sample in bins spanning coding and non-coding regions of the genome were compared to the matched normal sample from each patient. The log2 read ratio values were corrected for biases resulting from the exome capture process and sequence composition using CNVKit (0.7.6) (Talevich et al., 2016), and were adjusted to account for variable normal cell contamination as previously described (Anagnostou et al., 2017).
DATA AND SOFTWARE AVAILABILITY
The accession number for microarray data reported in this paper is GEO: GSE104244.
ADDITIONAL RESOURCES
Clinical trial identifier: NCT03220477: https://clinicaltrials.gov/ct2/show/NCT03220477
Supplementary Material
Figure S1. Sequential but Not Concurrent Aza + HDACi Potently Enhances HDACi Effect on Cell Proliferation, Related to Figure 1
(A) Table of pharmacokinetic and pharmacodynamic parameters for clinically relevant HDAC inhibitors used in study.
(B) Log dose response curves of MTS data from screen of NSCLC cell lines treated sequentially with either vehicle or 500nM Aza followed by the HDAC inhibitors indicated in the figure. (day 11, data are mean ± SD between n = 3 technical replicates).
(C) In vitro treatment schema for sequential and concurrent application of Aza and/or HDACi to NSCLC cell lines.
(D) Log dose response curve of MTS data for H460 cells treated concurrently with Aza and variable concentrations of ITF-2357 (day 5, data are mean ± SEM, n = 3).
(E) Log dose response curves of MTS data for sequential treatment with Aza or vehicle and various concentrations of ITF-2357 in H23 and H1299 cells (day 11, data are mean ± SEM, n = 3).
(F) Normalized BrdU percent positivity for Aza ± 100nM MS-275 or 100nM ITF-2357 (day 9, n = 4 (MS-275), n = 6 (ITF-2357)). Data are presented as mean ± SEM. *p value < 0.05 relative to mock, # p value < 0.05 relative to Aza + MS-275.
(G) Evaluation of pharmacologic targets of HDAC1/2: histone acetylation and for HDAC6: tubulin acetylation by immunoblotting in A549 cell line. Histone 3 and β-Actin used as loading controls. Drug and drug concentrations are indicated in the figure. (24-hour treatment, ITF = ITF-2357, MGCD = MGCD0103, TubA = Tubastatin A, n = 2).
(H) Normalized BrdU percent positivity for mock or Aza treated cells in combination with the indicated HDACi’s (day 9, 200nM MGCD0103, 2000nM RGFP996, 1000nM Tubastatin A; data are mean ± SEM, n = 6).
(I) Immunoblotting for DNMT1 in A549 and H460 cells infected with empty vector or shDNMT1 vector. β-Actin used as loading control (day 5, n=2).
(J) Log dose response for A549 and H460 cells infected with empty vector or shDNMT1 vector and treated with HDACi as indicated in figure (5-day HDACi exposure, n=2, data are mean ± SEM).
(K) Average volumes of tumor xenografts obtained from NOD-SCID mice subcutaneously injected with H460 cells (Day 0 equals onset of treatment, data are mean ± SEM, n = 4 mock, n = 5 Aza + MGCD0103).
(L) Tumor weights for patient derived xenografts obtained from NSG mice and treated with the agents as outlined in the figure, mock data are also presented in Figure 1H (28 days of treatment duration, n = 6 mock n = 7 Aza + MGCD0103, data are mean ± SEM)
*p value < 0.05 relative to mock obtained by two tail t test.
Figure S2. Combination Epigenetic Treatment Induces Significant Differential Gene Expression, Related to Figure 2
(A) Quantitation of the differentially expressed genes for each treatment condition (differential gene expression cutoff Log2 fold change over mock > 0.5, day 8, microarray, 500nM Aza, 100nM MS275, 100nM ITF-2357).
(B) Relative RNA expression correlation matrix for inhibitor conditions and cell lines indicated in panel A (microarray, day 8, 500nM Aza, 100nM MS275, 100nM ITF-2357).
(C) GSEA enrichment plots for top upregulated pathway (Interferon αβ) following combination epigenetic treatment with the indicated HDACi.
(D) GSEA enrichment plots for top downregulated pathway (DNA replication) following combination epigenetic treatment with the indicated HDACi.
Figure S3. HDACi Isoform-Specific Induction of Interferon Signaling, Related to Figure 3
(A) Expression of Interferon stimulated genes induced by Aza and/or the indicated HDACi in various NSCLC cell lines (qRT-PCR, day 8, 500nM Aza, 100nM ITF-2357, and 200nM MGCD0103).
(B and C) Expression of Interferon stimulated genes induced by Aza and/or the indicated HDACi in A549 and H23 cells (PCR genecard, day 8, 500nM Aza, 1000nM Tubastatin A, 2000nM RGFP996).
(D and E) Expression of Interferon stimulated genes induced by Aza and/or the indicated HDACi in A549 and H23 cells (PCR genecard, day 8, 500nM Aza, 100nM ITF-2357, 200nM MGCD0103, 300nM SAHA).
Figure S4. Sequential Aza + HDACi Imparts Significant Amplification of Aza Response, Related to Figure 3
(A) Heatmap of relative RNA expression for cancer/testis antigen genes across NSCLC cell lines (microarray, day 8, 500nM Aza, 100nM MS275, 100nM ITF-2357).
(B) Quantification of cancer/testis antigen transcriptional induction by combination Aza + HDACi treatment over Aza alone across NSCLC cell lines (microarray, day 8, 500nM Aza, 100nM MS275, 100nM ITF-2357; differential gene expression cutoff Log2 fold change over Aza > 0.5).
(C and D) Relative RNA expression of selected cancer/testis antigen transcripts in response to Aza and/or HDACi (qRT-PCR, day 8, 500nM Aza, 100nM ITF-2357, 200nM MGCD0103, 2000nM RGFP996, 1000nM Tubastatin A, n = 3, data are mean ± SEM).
(E and F) ERV expression in H1299 and H460 cells treated sequentially with Aza and/or HDACi (day 8, qRT-PCR, 500nM Aza, 100nM MS275, 100nM ITF-2357, n = 4, data are mean ± SEM).
(G) Relative ERV9-1 expression in A549 cells treated with HDACi in the presence of empty vector or shDNMT1 vector (2-day HDACi exposure, 100nM ITF-2357, 1000nM Tubastatin A, 2000nM RGFP996, 200nM MGCD0103, n=2). Data represented as mean ± SEM.
(H) Effect of ERV overexpression on transcription of the interferon stimulated gene, OASL, in A549 and H23 cells (96-hour post transfection, qRT-PCR, ERV3 overexpressed in A549, SYN1 overexpressed in H23, n = 4, data are mean ± SEM).
*p < 0.05 relative to mock and #p < 0.05 relative to Aza. p values calculated by two tail t test.
Figure S5. Validation of MYC Perturbations by Epigenetic Treatment, Related to Figure 4
(A) Immunoblot showing MYC expression in the indicated cell lines treated with Aza. β-Actin used as loading control. NR = non-responding cell line with no significant change in ITF-2357 IC50 by Aza pretreatment dose response data, PR = partial responder, less than a 25% reduction in ITF-2357 IC50 by 500nM Aza, R = responding cell line, greater than or equal to 50% ITF-2357 IC50 reduction by Aza pretreatment (Day 9, 500nM Aza).
(B) Immunoblot showing MYC expression in H460 cells treated with Aza with or without HDACi. β-Actin used as loading control (Day 9, 500nM Aza, 100nM ITF-2357, 200nM MGCD0103, 2000nM RGFP996, 1000nM Tubastatin A, n = 2).
(C) Immunoblot showing MYC expression in H460 and A549 cells infected with empty or MYC shRNA vector. β-Actin used as loading control. (day 4 of knockdown, n = 2).
(D) Immunoblotting for MYC in H460 and A549 cells infected with empty vector or MYC overexpression vector. β-Actin used as loading control (day 5 of overexpression, n = 2)
Figure S6. Combination Epigenetic Treatment Reduces Disease Burden In Vivo, Related to Figures 5, 6, and 7
(A) In vivo treatment schema for Aza + ITF2357 applied to murine models of NSCLC.
(B) Representative macroscopic images of LSL-KrasG12D lungs treated with vehicle or Aza and ITF-2357, for 3 months.
(C) In vivo treatment schema for addition of anti-PD1 to LSL-KrasG12D.
(D) Representative H&E sections of lungs from anti-PD1 and Aza + ITF-2357 + anti-PD1 treated mice (n = 2 anti-PD1, n = 5 anti-PD1 + Aza + ITF-2357).
(E) Representative macroscopic images of lungs assessed for lung metastases from mice with subcutaneous, flank Lewis Lung Carcinoma tumors, 5 weeks after subcutaneous implantation and 1 month after treatment initiation with the indicated treatment.
(F) Left panel, Lewis lung carcinoma (LLC) tumor weights for subcutaneous explants from one-month mock and Aza + MCCD0103 treated mice (n = 19 mock, same mice represented in Figure 5D, and n = 12 Aza + MGCD0103). Right panel, Representative H & E stained images of lung metastasis from these mice. Percentage frequency of this event shown on the right. Mock mice similar to those shown in Figure 5E.
(G) Immunoblotting for myc in LLC cells treated with Aza and/or HDACi. β-Actin used as loading control (Day 9, 500nM Aza, 100nM ITF-2357, 200nM MGCD0103).
(H) FACS analysis of CD8+/CD3+ TILs from LLC subcutaneous tumors from 1-month mock and Aza + ITF-2357 or MGCD0103 treated mice (n = 4 mice per group ± SEM, *p value < 0.05 calculated by two tail t test).
(I) FACS analysis of percentage ifnγ+ CD8+/CD3+ TILs from LLC subcutaneous tumors from 1-month mock and Aza + MGCD0103 treated mice (n = 4 mice per group, mock mice represented in Figure 6H, mean ± SEM, *p value < 0.05 calculated by two tail t test).
(J) Weights of subcutaneous LLC tumors from one-month mock and Aza + MGCD0103 mice treated in the presence of CD8a depleting antibody (n = 7 mock and n = 5 treated mice, mock mice represented in Figure 7A, mean ± SEM, NS = non-significant p value calculated by two tail t test).
(K) Relative CCL5 RNA expression in human NSCLC cell lines treated in vitro with Aza and/or HDACi (day 8, microarray, 500nM Aza, 100nM MS275, 100nM ITF-2357).
Highlights.
Azacitidine depletes MYC and sensitizes NSCLC to HDACi
Combination epigenetic treatment induces a potent anti-tumor response in vivo
Epigenetic treatment potentiates anti-tumor responses by modulating T cell phenotypes
MYC status determines tumor immunophenotype
Acknowledgments
This work was supported by grants from The Hodson Trust, The Dr. Miriam and Sheldon G. Adelson Medical Research Foundation, The Commonwealth Foundation, Stand Up To Cancer Jim Toth Sr. Breakthrough Prize in Lung Cancer, and the SWCRF Collaboration for a Cure. This work is also supported by The Defense Health Program (DOD0), through the Department of Defense Ovarian Cancer Research Program, Teal Innovator Award No. OC130454/W81XWH-14-1-0385 (opinions, interpretations, conclusions, and recommendations are those of the author and not necessarily endorsed by the Department of Defense). Research reported in this publication was supported by the National Cancer Institute under award numbers CA121113, CA006973, and CA180950. The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH. Research funding was also provided by the Van Andel Research Institute through the Van Andel Research Institute – Stand Up To Cancer Epigenetics Dream Team. Stand Up To Cancer is a program of the Entertainment Industry Foundation, administered by AACR. We acknowledge Mrs. Elizabeth Stiegler and Lauren Murphy for expert technical help. We thank Kathy Bender for help with manuscript preparation. V.E.V. is a founder of Personal Genome Diagnostics, a member of its Scientific Advisory Board and Board of Directors, and owns Personal Genome Diagnostics stock, which is subject to certain restrictions under university policy. V.E.V. is also on the Scientific Advisory Board for Ignyta.
Footnotes
Supplemental Information includes six figures and one table and can be found with this article online at https://doi.org/10.1016/j.cell.2017.10.022.
AUTHOR CONTRIBUTIONS
M.J.T., M.V., and S.B.B. designed experiments, performed data analyses, and wrote the manuscript. M.J.T., M.V., and K.B.C. performed experiments. P.L.S. and R.S. analyzed ERV data. C.D.S., A.W., and J.H. performed immunohistochemistry (IHC). V.A., N.N., and V.E.V. analyzed patient data. M.B. and P.T.T. performed and analyzed computed tomography (CT) scans. C.A.Z. assisted with xenograft studies. M.D.H. and M.S. helped in manuscript review. R.-W.C.Y. provided cell lines used in study.
References
- Anagnostou V, Smith KN, Forde PM, Niknafs N, Bhattacharya R, White J, Zhang T, Adleff V, Phallen J, Wali N, et al. Evolution of Neoantigen Landscape during Immune Checkpoint Blockade in Non-Small Cell Lung Cancer. Cancer Discov. 2017;7:264–276. doi: 10.1158/2159-8290.CD-16-0828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asthana J, Kapoor S, Mohan R, Panda D. Inhibition of HDAC6 deacetylase activity increases its binding with microtubules and suppresses microtubule dynamic instability in MCF-7 cells. J Biol Chem. 2013;288:22516–22526. doi: 10.1074/jbc.M113.489328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borghaei H, Paz-Ares L, Horn L, Spigel DR, Steins M, Ready NE, Chow LQ, Vokes EE, Felip E, Holgado E, et al. Nivolumab versus Docetaxel in Advanced Nonsquamous Non-Small-Cell Lung Cancer. N Engl J Med. 2015;373:1627–1639. doi: 10.1056/NEJMoa1507643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bradner JE, West N, Grachan ML, Greenberg EF, Haggarty SJ, Warnow T, Mazitschek R. Chemical phylogenetics of histone deace-tylases. Nat Chem Biol. 2010;6:238–243. doi: 10.1038/nchembio.313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai Y, Geutjes EJ, de Lint K, Roepman P, Bruurs L, Yu LR, Wang W, van Blijswijk J, Mohammad H, de Rink I, et al. The NuRD complex cooperates with DNMTs to maintain silencing of key colorectal tumor suppressor genes. Oncogene. 2014;33:2157–2168. doi: 10.1038/onc.2013.178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai Y, Tsai HC, Yen RC, Zhang YW, Kong X, Wang W, Xia L, Baylin SB. Critical threshold levels of DNA methyltransferase 1 are required to maintain DNA methylation across the genome in human cancer cells. Genome Res. 2017;27:533–544. doi: 10.1101/gr.208108.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cameron EE, Bachman KE, Myöhänen S, Herman JG, Baylin SB. Synergy of demethylation and histone deacetylase inhibition in the re-expression of genes silenced in cancer. Nat Genet. 1999;21:103–107. doi: 10.1038/5047. [DOI] [PubMed] [Google Scholar]
- Casey SC, Tong L, Li Y, Do R, Walz S, Fitzgerald KN, Gouw AM, Baylot V, Gütgemann I, Eilers M, Felsher DW. MYC regulates the antitumor immune response through CD47 and PD-L1. Science. 2016;352:227–231. doi: 10.1126/science.aac9935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng Z, Gong Y, Ma Y, Lu K, Lu X, Pierce LA, Thompson RC, Muller S, Knapp S, Wang J. Inhibition of BET bromodomain targets genetically diverse glioblastoma. Clin Cancer Res. 2013;19:1748–1759. doi: 10.1158/1078-0432.CCR-12-3066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiappinelli KB, Strissel PL, Desrichard A, Li H, Henke C, Akman B, Hein A, Rote NS, Cope LM, Snyder A, et al. Inhibiting DNA Methylation Causes an Interferon Response in Cancer via dsRNA Including Endogenous Retroviruses. Cell. 2015;162:974–986. doi: 10.1016/j.cell.2015.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falkenberg KJ, Johnstone RW. Histone deacetylases and their inhibitors in cancer, neurological diseases and immune disorders. Nat Rev Drug Discov. 2014;13:673–691. doi: 10.1038/nrd4360. [DOI] [PubMed] [Google Scholar]
- Fournel M, Bonfils C, Hou Y, Yan PT, Trachy-Bourget MC, Kalita A, Liu J, Lu AH, Zhou NZ, Robert MF, et al. MGCD0103, a novel isotype-selective histone deacetylase inhibitor, has broad spectrum antitumor activity in vitro and in vivo. Mol Cancer Ther. 2008;7:759–768. doi: 10.1158/1535-7163.MCT-07-2026. [DOI] [PubMed] [Google Scholar]
- Furlan A, Monzani V, Reznikov LL, Leoni F, Fossati G, Modena D, Mascagni P, Dinarello CA. Pharmacokinetics, safety and inducible cytokine responses during a phase 1 trial of the oral histone deacetylase inhibitor ITF2357 (givinostat) Mol Med. 2011;17:353–362. doi: 10.2119/molmed.2011.00020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garon EB, Rizvi NA, Hui R, Leighl N, Balmanoukian AS, Eder JP, Patnaik A, Aggarwal C, Gubens M, Horn L, et al. KEYNOTE-001 Investigators. Pembrolizumab for the treatment of non-small-cell lung cancer. N Engl J Med. 2015;372:2018–2028. doi: 10.1056/NEJMoa1501824. [DOI] [PubMed] [Google Scholar]
- Ghoneim HE, Fan Y, Moustaki A, Abdelsamed HA, Dash P, Dogra P, Carter R, Awad W, Neale G, Thomas PG, Youngblood B. De Novo Epigenetic Programs Inhibit PD-1 Blockade-Mediated T Cell Rejuvenation. Cell. 2017;170:142–157. e19. doi: 10.1016/j.cell.2017.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gore L, Rothenberg ML, O’Bryant CL, Schultz MK, Sandler AB, Coffin D, McCoy C, Schott A, Scholz C, Eckhardt SG. A phase I and pharmacokinetic study of the oral histone deacetylase inhibitor, MS-275, in patients with refractory solid tumors and lymphomas. Clin Cancer Res. 2008;14:4517–4525. doi: 10.1158/1078-0432.CCR-07-1461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong M, Puaux AL, Huang C, Loumagne L, Tow C, Mackay C, Kato M, Prévost-Blondel A, Avril MF, Nardin A, Abastado JP. Chemotherapy induces intratumoral expression of chemokines in cutaneous melanoma, favoring T-cell infiltration and tumor control. Cancer Res. 2011;71:6997–7009. doi: 10.1158/0008-5472.CAN-11-1466. [DOI] [PubMed] [Google Scholar]
- Huang da W, Sherman BT, Lempicki RA. Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res. 2009a;37:1–13. doi: 10.1093/nar/gkn923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang da W, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc. 2009b;4:44–57. doi: 10.1038/nprot.2008.211. [DOI] [PubMed] [Google Scholar]
- Huang Y, Yuan J, Righi E, Kamoun WS, Ancukiewicz M, Nezivar J, Santosuosso M, Martin JD, Martin MR, Vianello F, et al. Vascular normalizing doses of antiangiogenic treatment reprogram the immunosuppressive tumor microenvironment and enhance immunotherapy. Proc Natl Acad Sci USA. 2012;109:17561–17566. doi: 10.1073/pnas.1215397109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Y, Goel S, Duda DG, Fukumura D, Jain RK. Vascular normalization as an emerging strategy to enhance cancer immunotherapy. Cancer Res. 2013;73:2943–2948. doi: 10.1158/0008-5472.CAN-12-4354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson EL, Willis N, Mercer K, Bronson RT, Crowley D, Montoya R, Jacks T, Tuveson DA. Analysis of lung tumor initiation and progression using conditional expression of oncogenic K-ras. Genes Dev. 2001;15:3243–3248. doi: 10.1101/gad.943001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones PA, Issa JP, Baylin S. Targeting the cancer epigenome for therapy. Nat Rev Genet. 2016;17:630–641. doi: 10.1038/nrg.2016.93. [DOI] [PubMed] [Google Scholar]
- Juergens RA, Wrangle J, Vendetti FP, Murphy SC, Zhao M, Coleman B, Sebree R, Rodgers K, Hooker CM, Franco N, et al. Combination epigenetic therapy has efficacy in patients with refractory advanced non-small cell lung cancer. Cancer Discov. 2011;1:598–607. doi: 10.1158/2159-8290.CD-11-0214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kellar A, Egan C, Morris D. Preclinical Murine Models for Lung Cancer: Clinical Trial Applications. BioMed Res Int. 2015;2015:621324. doi: 10.1155/2015/621324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan N, Jeffers M, Kumar S, Hackett C, Boldog F, Khramtsov N, Qian X, Mills E, Berghs SC, Carey N, et al. Determination of the class and isoform selectivity of small-molecule histone deacetylase inhibitors. Biochem J. 2008;409:581–589. doi: 10.1042/BJ20070779. [DOI] [PubMed] [Google Scholar]
- Krämer OH, Baus D, Knauer SK, Stein S, Jäger E, Stauber RH, Grez M, Pfitzner E, Heinzel T. Acetylation of Stat1 modulates NF-kappaB activity. Genes Dev. 2006;20:473–485. doi: 10.1101/gad.364306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lavergne E, Combadière C, Iga M, Boissonnas A, Bonduelle O, Maho M, Debré P, Combadiere B. Intratumoral CC chemokine ligand 5 overexpression delays tumor growth and increases tumor cell infiltration. J Immunol. 2004;173:3755–3762. doi: 10.4049/jimmunol.173.6.3755. [DOI] [PubMed] [Google Scholar]
- Li H, Chiappinelli KB, Guzzetta AA, Easwaran H, Yen RW, Vatapalli R, Topper MJ, Luo J, Connolly RM, Azad NS, et al. Immune regulation by low doses of the DNA methyltransferase inhibitor 5-azacitidine in common human epithelial cancers. Oncotarget. 2014;5:587–598. doi: 10.18632/oncotarget.1782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malvaez M, McQuown SC, Rogge GA, Astarabadi M, Jacques V, Carreiro S, Rusche JR, Wood MA. HDAC3-selective inhibitor enhances extinction of cocaine-seeking behavior in a persistent manner. Proc Natl Acad Sci. 2013;110:2647–2652. doi: 10.1073/pnas.1213364110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moran CJ, Arenberg DA, Huang CC, Giordano TJ, Thomas DG, Misek DE, Chen G, Iannettoni MD, Orringer MB, Hanash S, Beer DG. RANTES expression is a predictor of survival in stage I lung adenocarcinoma. Clin Cancer Res. 2002;8:3803–3812. [PubMed] [Google Scholar]
- Moreno-Bost A, Szmania S, Stone K, Garg T, Hoerring A, Szymonifka J, Shaughnessy J, Jr, Barlogie B, Prentice HG, van Rhee F. Epigenetic modulation of MAGE-A3 antigen expression in multiple myeloma following treatment with the demethylation agent 5-azacitidine and the histone deacetlyase inhibitor MGCD0103. Cytotherapy. 2011;13:618–628. doi: 10.3109/14653249.2010.529893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oi S, Natsume A, Ito M, Kondo Y, Shimato S, Maeda Y, Saito K, Wakabayashi T. Synergistic induction of NY-ESO-1 antigen expression by a novel histone deacetylase inhibitor, valproic acid, with 5-aza-2′-deoxycytidine in glioma cells. J Neurooncol. 2009;92:15–22. doi: 10.1007/s11060-008-9732-0. [DOI] [PubMed] [Google Scholar]
- Okayama H, Saito M, Oue N, Weiss JM, Stauffer J, Takenoshita S, Wiltrout RH, Hussain SP, Harris CC. NOS2 enhances KRAS-induced lung carcinogenesis, inflammation and microRNA-21 expression. Int J Cancer. 2013;132:9–18. doi: 10.1002/ijc.27644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rasheed WK, Johnstone RW, Prince HM. Histone deacetylase inhibitors in cancer therapy. Expert Opin Investig Drugs. 2007;16:659–678. doi: 10.1517/13543784.16.5.659. [DOI] [PubMed] [Google Scholar]
- Reck M, Rodrìguez-Abreu D, Robinson AG, Hui R, Csöszi T, Fülöp A, Gottfried M, Peled N, Tafreshi A, Cuffe S, et al. KEYNOTE-024 Investigators. Pembrolizumab versus Chemotherapy for PD-L1-Positive Non-Small-Cell Lung Cancer. N Engl J Med. 2016;375:1823–1833. doi: 10.1056/NEJMoa1606774. [DOI] [PubMed] [Google Scholar]
- Ribas A, Shin DS, Zaretsky J, Frederiksen J, Cornish A, Avramis E, Seja E, Kivork C, Siebert J, Kaplan-Lefko P, et al. PD-1 Blockade Expands Intratumoral Memory T Cells. Cancer Immunol Res. 2016;4:194–203. doi: 10.1158/2326-6066.CIR-15-0210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rizvi NA, Hellmann MD, Snyder A, Kvistborg P, Makarov V, Havel JJ, Lee W, Yuan J, Wong P, Ho TS, et al. Cancer immunology. Mutational landscape determines sensitivity to PD-1 blockade in non-small cell lung cancer. Science. 2015;348:124–128. doi: 10.1126/science.aaa1348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roulois D, Loo Yau H, Singhania R, Wang Y, Danesh A, Shen SY, Han H, Liang G, Jones PA, Pugh TJ, et al. DNA-Demethylating Agents Target Colorectal Cancer Cells by Inducing Viral Mimicry by Endogenous Transcripts. Cell. 2015;162:961–973. doi: 10.1016/j.cell.2015.07.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryan QC, Headlee D, Acharya M, Sparreboom A, Trepel JB, Ye J, Figg WD, Hwang K, Chung EJ, Murgo A, et al. Phase I and pharmacokinetic study of MS-275, a histone deacetylase inhibitor, in patients with advanced and refractory solid tumors or lymphoma. J Clin Oncol. 2005;23:3912–3922. doi: 10.1200/JCO.2005.02.188. [DOI] [PubMed] [Google Scholar]
- Schlee M, Hölzel M, Bernard S, Mailhammer R, Schuhmacher M, Reschke J, Eick D, Marinkovic D, Wirth T, Rosenwald A, et al. C-myc activation impairs the NF-kappaB and the interferon response: implications for the pathogenesis of Burkitt’s lymphoma. Int J Cancer. 2007a;120:1387–1395. doi: 10.1002/ijc.22372. [DOI] [PubMed] [Google Scholar]
- Schlee M, Schuhmacher M, Hölzel M, Laux G, Bornkamm GW. c-MYC impairs immunogenicity of human B cells. Adv Cancer Res. 2007b;97:167–188. doi: 10.1016/S0065-230X(06)97007-9. [DOI] [PubMed] [Google Scholar]
- Soucek L, Whitfield JR, Sodir NM, Massó-Vallés D, Serrano E, Karnezis AN, Swigart LB, Evan GI. Inhibition of Myc family proteins eradicates KRas-driven lung cancer in mice. Genes Dev. 2013;27:504–513. doi: 10.1101/gad.205542.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stengel S, Fiebig U, Kurth R, Denner J. Regulation of human endogenous retrovirus-K expression in melanomas by CpG methylation. Genes Chromosomes Cancer. 2010;49:401–411. doi: 10.1002/gcc.20751. [DOI] [PubMed] [Google Scholar]
- Strissel PL, Ruebner M, Thiel F, Wachter D, Ekici AB, Wolf F, Thieme F, Ruprecht K, Beckmann MW, Strick R. Reactivation of codogenic endogenous retroviral (ERV) envelope genes in human endometrial carcinoma and prestages: Emergence of new molecular targets. Oncotarget. 2012;3:1204–1219. doi: 10.18632/oncotarget.679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, Mesirov JP. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci USA. 2005;102:15545–15550. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Talevich E, Shain AH, Botton T, Bastian BC. CNVkit: Genome-Wide Copy Number Detection and Visualization from Targeted DNA Sequencing. PLoS Comput Biol. 2016;12:e1004873. doi: 10.1371/journal.pcbi.1004873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai HC, Li H, Van Neste L, Cai Y, Robert C, Rassool FV, Shin JJ, Harbom KM, Beaty R, Pappou E, et al. Transient low doses of DNA-demethylating agents exert durable antitumor effects on hematological and epithelial tumor cells. Cancer Cell. 2012;21:430–446. doi: 10.1016/j.ccr.2011.12.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tumeh PC, Harview CL, Yearley JH, Shintaku IP, Taylor EJ, Robert L, Chmielowski B, Spasic M, Henry G, Ciobanu V, et al. PD-1 blockade induces responses by inhibiting adaptive immune resistance. Nature. 2014;515:568–571. doi: 10.1038/nature13954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiser TS, Guo ZS, Ohnmacht GA, Parkhurst ML, Tong-On P, Marincola FM, Fischette MR, Yu X, Chen GA, Hong JA, et al. Sequential 5-Aza-2 deoxycytidine-depsipeptide FR901228 treatment induces apoptosis preferentially in cancer cells and facilitates their recognition by cytolytic T lymphocytes specific for NY-ESO-1. J Immunother. 2001;24:151–161. doi: 10.1097/00002371-200103000-00010. [DOI] [PubMed] [Google Scholar]
- Wherry EJ, Kurachi M. Molecular and cellular insights into T cell exhaustion. Nat Rev Immunol. 2015;15:486–499. doi: 10.1038/nri3862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wherry EJ, Ha SJ, Kaech SM, Haining WN, Sarkar S, Kalia V, Subramaniam S, Blattman JN, Barber DL, Ahmed R. Molecular signature of CD8+ T cell exhaustion during chronic viral infection. Immunity. 2007;27:670–684. doi: 10.1016/j.immuni.2007.09.006. [DOI] [PubMed] [Google Scholar]
- Wrangle J, Wang W, Koch A, Easwaran H, Mohammad HP, Vendetti F, Vancriekinge W, Demeyer T, Du Z, Parsana P, et al. Alterations of immune response of Non-Small Cell Lung Cancer with Azacytidine. Oncotarget. 2013;4:2067–2079. doi: 10.18632/oncotarget.1542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zahnow CA, Topper M, Stone M, Murray-Stewart T, Li H, Baylin SB, Casero RA., Jr Inhibitors of DNA Methylation, Histone Deacetylation, and Histone Demethylation: A Perfect Combination for Cancer Therapy. Adv Cancer Res. 2016;130:55–111. doi: 10.1016/bs.acr.2016.01.007. [DOI] [PubMed] [Google Scholar]
- Zehn D, Wherry EJ. Immune Memory and Exhaustion: Clinically Relevant Lessons from the LCMV Model. Adv Exp Med Biol. 2015;850:137–152. doi: 10.1007/978-3-319-15774-0_10. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Sequential but Not Concurrent Aza + HDACi Potently Enhances HDACi Effect on Cell Proliferation, Related to Figure 1
(A) Table of pharmacokinetic and pharmacodynamic parameters for clinically relevant HDAC inhibitors used in study.
(B) Log dose response curves of MTS data from screen of NSCLC cell lines treated sequentially with either vehicle or 500nM Aza followed by the HDAC inhibitors indicated in the figure. (day 11, data are mean ± SD between n = 3 technical replicates).
(C) In vitro treatment schema for sequential and concurrent application of Aza and/or HDACi to NSCLC cell lines.
(D) Log dose response curve of MTS data for H460 cells treated concurrently with Aza and variable concentrations of ITF-2357 (day 5, data are mean ± SEM, n = 3).
(E) Log dose response curves of MTS data for sequential treatment with Aza or vehicle and various concentrations of ITF-2357 in H23 and H1299 cells (day 11, data are mean ± SEM, n = 3).
(F) Normalized BrdU percent positivity for Aza ± 100nM MS-275 or 100nM ITF-2357 (day 9, n = 4 (MS-275), n = 6 (ITF-2357)). Data are presented as mean ± SEM. *p value < 0.05 relative to mock, # p value < 0.05 relative to Aza + MS-275.
(G) Evaluation of pharmacologic targets of HDAC1/2: histone acetylation and for HDAC6: tubulin acetylation by immunoblotting in A549 cell line. Histone 3 and β-Actin used as loading controls. Drug and drug concentrations are indicated in the figure. (24-hour treatment, ITF = ITF-2357, MGCD = MGCD0103, TubA = Tubastatin A, n = 2).
(H) Normalized BrdU percent positivity for mock or Aza treated cells in combination with the indicated HDACi’s (day 9, 200nM MGCD0103, 2000nM RGFP996, 1000nM Tubastatin A; data are mean ± SEM, n = 6).
(I) Immunoblotting for DNMT1 in A549 and H460 cells infected with empty vector or shDNMT1 vector. β-Actin used as loading control (day 5, n=2).
(J) Log dose response for A549 and H460 cells infected with empty vector or shDNMT1 vector and treated with HDACi as indicated in figure (5-day HDACi exposure, n=2, data are mean ± SEM).
(K) Average volumes of tumor xenografts obtained from NOD-SCID mice subcutaneously injected with H460 cells (Day 0 equals onset of treatment, data are mean ± SEM, n = 4 mock, n = 5 Aza + MGCD0103).
(L) Tumor weights for patient derived xenografts obtained from NSG mice and treated with the agents as outlined in the figure, mock data are also presented in Figure 1H (28 days of treatment duration, n = 6 mock n = 7 Aza + MGCD0103, data are mean ± SEM)
*p value < 0.05 relative to mock obtained by two tail t test.
Figure S2. Combination Epigenetic Treatment Induces Significant Differential Gene Expression, Related to Figure 2
(A) Quantitation of the differentially expressed genes for each treatment condition (differential gene expression cutoff Log2 fold change over mock > 0.5, day 8, microarray, 500nM Aza, 100nM MS275, 100nM ITF-2357).
(B) Relative RNA expression correlation matrix for inhibitor conditions and cell lines indicated in panel A (microarray, day 8, 500nM Aza, 100nM MS275, 100nM ITF-2357).
(C) GSEA enrichment plots for top upregulated pathway (Interferon αβ) following combination epigenetic treatment with the indicated HDACi.
(D) GSEA enrichment plots for top downregulated pathway (DNA replication) following combination epigenetic treatment with the indicated HDACi.
Figure S3. HDACi Isoform-Specific Induction of Interferon Signaling, Related to Figure 3
(A) Expression of Interferon stimulated genes induced by Aza and/or the indicated HDACi in various NSCLC cell lines (qRT-PCR, day 8, 500nM Aza, 100nM ITF-2357, and 200nM MGCD0103).
(B and C) Expression of Interferon stimulated genes induced by Aza and/or the indicated HDACi in A549 and H23 cells (PCR genecard, day 8, 500nM Aza, 1000nM Tubastatin A, 2000nM RGFP996).
(D and E) Expression of Interferon stimulated genes induced by Aza and/or the indicated HDACi in A549 and H23 cells (PCR genecard, day 8, 500nM Aza, 100nM ITF-2357, 200nM MGCD0103, 300nM SAHA).
Figure S4. Sequential Aza + HDACi Imparts Significant Amplification of Aza Response, Related to Figure 3
(A) Heatmap of relative RNA expression for cancer/testis antigen genes across NSCLC cell lines (microarray, day 8, 500nM Aza, 100nM MS275, 100nM ITF-2357).
(B) Quantification of cancer/testis antigen transcriptional induction by combination Aza + HDACi treatment over Aza alone across NSCLC cell lines (microarray, day 8, 500nM Aza, 100nM MS275, 100nM ITF-2357; differential gene expression cutoff Log2 fold change over Aza > 0.5).
(C and D) Relative RNA expression of selected cancer/testis antigen transcripts in response to Aza and/or HDACi (qRT-PCR, day 8, 500nM Aza, 100nM ITF-2357, 200nM MGCD0103, 2000nM RGFP996, 1000nM Tubastatin A, n = 3, data are mean ± SEM).
(E and F) ERV expression in H1299 and H460 cells treated sequentially with Aza and/or HDACi (day 8, qRT-PCR, 500nM Aza, 100nM MS275, 100nM ITF-2357, n = 4, data are mean ± SEM).
(G) Relative ERV9-1 expression in A549 cells treated with HDACi in the presence of empty vector or shDNMT1 vector (2-day HDACi exposure, 100nM ITF-2357, 1000nM Tubastatin A, 2000nM RGFP996, 200nM MGCD0103, n=2). Data represented as mean ± SEM.
(H) Effect of ERV overexpression on transcription of the interferon stimulated gene, OASL, in A549 and H23 cells (96-hour post transfection, qRT-PCR, ERV3 overexpressed in A549, SYN1 overexpressed in H23, n = 4, data are mean ± SEM).
*p < 0.05 relative to mock and #p < 0.05 relative to Aza. p values calculated by two tail t test.
Figure S5. Validation of MYC Perturbations by Epigenetic Treatment, Related to Figure 4
(A) Immunoblot showing MYC expression in the indicated cell lines treated with Aza. β-Actin used as loading control. NR = non-responding cell line with no significant change in ITF-2357 IC50 by Aza pretreatment dose response data, PR = partial responder, less than a 25% reduction in ITF-2357 IC50 by 500nM Aza, R = responding cell line, greater than or equal to 50% ITF-2357 IC50 reduction by Aza pretreatment (Day 9, 500nM Aza).
(B) Immunoblot showing MYC expression in H460 cells treated with Aza with or without HDACi. β-Actin used as loading control (Day 9, 500nM Aza, 100nM ITF-2357, 200nM MGCD0103, 2000nM RGFP996, 1000nM Tubastatin A, n = 2).
(C) Immunoblot showing MYC expression in H460 and A549 cells infected with empty or MYC shRNA vector. β-Actin used as loading control. (day 4 of knockdown, n = 2).
(D) Immunoblotting for MYC in H460 and A549 cells infected with empty vector or MYC overexpression vector. β-Actin used as loading control (day 5 of overexpression, n = 2)
Figure S6. Combination Epigenetic Treatment Reduces Disease Burden In Vivo, Related to Figures 5, 6, and 7
(A) In vivo treatment schema for Aza + ITF2357 applied to murine models of NSCLC.
(B) Representative macroscopic images of LSL-KrasG12D lungs treated with vehicle or Aza and ITF-2357, for 3 months.
(C) In vivo treatment schema for addition of anti-PD1 to LSL-KrasG12D.
(D) Representative H&E sections of lungs from anti-PD1 and Aza + ITF-2357 + anti-PD1 treated mice (n = 2 anti-PD1, n = 5 anti-PD1 + Aza + ITF-2357).
(E) Representative macroscopic images of lungs assessed for lung metastases from mice with subcutaneous, flank Lewis Lung Carcinoma tumors, 5 weeks after subcutaneous implantation and 1 month after treatment initiation with the indicated treatment.
(F) Left panel, Lewis lung carcinoma (LLC) tumor weights for subcutaneous explants from one-month mock and Aza + MCCD0103 treated mice (n = 19 mock, same mice represented in Figure 5D, and n = 12 Aza + MGCD0103). Right panel, Representative H & E stained images of lung metastasis from these mice. Percentage frequency of this event shown on the right. Mock mice similar to those shown in Figure 5E.
(G) Immunoblotting for myc in LLC cells treated with Aza and/or HDACi. β-Actin used as loading control (Day 9, 500nM Aza, 100nM ITF-2357, 200nM MGCD0103).
(H) FACS analysis of CD8+/CD3+ TILs from LLC subcutaneous tumors from 1-month mock and Aza + ITF-2357 or MGCD0103 treated mice (n = 4 mice per group ± SEM, *p value < 0.05 calculated by two tail t test).
(I) FACS analysis of percentage ifnγ+ CD8+/CD3+ TILs from LLC subcutaneous tumors from 1-month mock and Aza + MGCD0103 treated mice (n = 4 mice per group, mock mice represented in Figure 6H, mean ± SEM, *p value < 0.05 calculated by two tail t test).
(J) Weights of subcutaneous LLC tumors from one-month mock and Aza + MGCD0103 mice treated in the presence of CD8a depleting antibody (n = 7 mock and n = 5 treated mice, mock mice represented in Figure 7A, mean ± SEM, NS = non-significant p value calculated by two tail t test).
(K) Relative CCL5 RNA expression in human NSCLC cell lines treated in vitro with Aza and/or HDACi (day 8, microarray, 500nM Aza, 100nM MS275, 100nM ITF-2357).