

Abstract
Alzheimer’s disease (AD) is the leading cause of dementia worldwide, and currently no disease-modifying therapy is available to slow or prevent AD, underscoring the urgent need for neuroprotective therapies. Selective M1 muscarinic acetylcholine receptor (mAChR) activation is an attractive mechanism for AD therapy since M1 mediates key effects on memory, cognition, and behavior and has potential for disease-modifying effects on Aβ formation and tau phosphorylation. To validate M1 as a neuroprotective treatment target for AD, the M1-selective agonist, VU0364572, was chronically dosed to 5XFAD mice from a young age preceding Aβ pathology (2 months) to an age where these mice are known to display memory impairments (6 months). Chronic M1 activation prevented mice from becoming memory-impaired, as measured by Morris water maze (MWM) testing at 6 months of age. Additionally, M1 activation significantly reduced levels of soluble and insoluble Aβ40,42 in the cortex and hippocampus of these animals, as measured by ELISA and immunohistochemistry. Moreover, soluble hippocampal Aβ42 levels were strongly correlated with MWM memory impairments and M1 activation with VU0364572 abolished this correlation. Finally, VU0364572 significantly decreased oligomeric (oAβ) levels in the cortex, suggesting one mechanism whereby VU0364572 may be exerting its neuroprotective effects is by reducing the available oAβ pool in the brain. These findings suggest that chronic M1 activation has neuroprotective potential for preventing memory impairments and reducing neuropathology in AD. M1 activation therefore represents a promising avenue for preventative treatment, as well as a promising opportunity to combine symptomatic and disease-modifying effects for early AD treatment.
Keywords: Muscarinic, acetylcholine, Alzheimer’s, amyloid, memory, hippocampus
Graphical Abstract
INTRODUCTION
Alzheimer’s disease is a progressive, neurodegenerative disease, which is the most common cause of dementia worldwide and accounts for 60–70% of all cases of dementia.1,2 At present, there is not a single disease-modifying available therapy for AD patients that slows or prevents the course of the disease.2–4 AD is characterized by the presence of insoluble Aβ plaques and neurofibrillary tangles composed of hyperphosphorylated tau.5–15 While many anti-amyloid therapies have shown promising effects in mouse models of AD, so far none have translated into an effective therapy in human AD patients.2–4 Data from recent human biomarker studies now clearly suggest that asymptomatic AD cascades begin up to two decades prior to symptom onset; this has refocused therapeutic efforts from symptomatic treatment to disease prevention and underscores the urgent need for developing neuroprotective therapies that slow or prevent AD onset.2,16,17 Moreover, there is an unmet need for therapies with both symptomatic and disease-modifying effects for individuals with mild cognitive deficits or early stage dementia.
The only currently United States Food and Drug Administration (FDA)-approved therapies for AD are AChEIs (e.g., donepezil) and the NMDAR antagonist memantine, both of which only provide modest symptomatic effects.2–4,18,19 Cholinergic neurotransmission is complex, whereby ACh acts on multiple subtypes of both nicotinic and muscarinic ACh receptors to mediate its effects, many of which functionally oppose one another.3,4,20–22 Thus, it is not surprising that nonselective activators such as AChEIs suffer from modest efficacy and dose-limiting side effects.23,24 Selective M1 activation is a strategy considered to be highly attractive for AD therapy given the importance of M1 in mediating crucial effects on memory, cognition, and behavior.3,4 The M1 receptor is a Gq-coupled G-protein-coupled receptor (GPCR) expressed heavily throughout the hippocampus and neocortex, and M1 knockout animals show memory deficits.25–33 Previous studies have shown that selective M1 activators can both improve memory as measured behaviorally as well as modulate hippocampal synaptic plasticity.34–38 The mechanism underlying the disease-modifying effects of M1 activation for AD has been well-established through both pharmacological and genetic studies, as M1 activation shifts the balance of amyloid precursor protein (APP) processing toward nonamyloidogenic sAPP formation and decreases Aβ production by modulating cellular secretase machinery.21,22,33,39 The ability of M1 to directly modulate APP processing has important implications for AD therapy, as the accumulation of Aβ into insoluble neuritic plaques and soluble pools of oligomeric Aβ (oAβ) is widely believed to play a key role in AD pathogenesis.41–56 Additionally, activating M1 receptors decreases tau pathology by signaling through GSK3β.40 Taken together, the available data indicate that selective M1 activators have the potential to simultaneously bolster the function of memory circuitry while protecting against the development of AD neuropathology.
The current study sought to evaluate M1 as a neuroprotective target for AD therapy. The failure of all disease-modifying clinical trials to date, along with the recognition that disease pathology begins 20 or more years prior to the onset of clinical symptoms, has produced a paradigm-shift in AD treatment efforts. Specifically, the field is increasingly focused on earlier intervention, including in asymptomatic individuals with pathology already present (i.e., secondary prevention), and individuals with mild cognitive impairment due to AD. Therefore, we took a similar view in the current study by designing a preventative treatment trial that models exactly what is now being pursued in the leading NIH and industry-sponsored human trials. 5XFAD mice were chronically dosed with the orally bioavailable M1-selective agonist, VU0364572, from an age before mice display Aβ pathology (2 months) to an age where mice are known to display memory impairments (6 months).37,38,57 Subsequent behavioral and neuropathological analyses confirmed that chronic M1 activation by VU0364572 prevented the development of memory impairments and significantly decreased levels of both soluble and insoluble Aβ40,42 in the neocortex and hippocampus of 5XFAD mice. These results validate M1 activators as a promising neuroprotective strategy to stem AD progression.
RESULTS
Chronic M1 Activation by VU0364572 Preserves Hippocampal Memory in 5XFAD Transgenic Alzheimer’s Mice
Given the potentially disease-modifying role of M1 activation for AD, we designed a preclinical proof-of-mechanism trial to assess the neuroprotective efficacy of chronic M1 activation and simulate the design of a prevention trial in humans (Figure 1A). We dosed the M1 agonist, VU0364572, to selectively drive M1 activation in the aggressive 5XFAD mouse model of AD, taking the view that such a model would provide a rigorous test of predictive validity for possible translation to clinical efficacy in humans.57 5XFAD animals were dosed with placebo (regular drinking water) or M1 agonist (approximately 10 mg/kg/day VU0364572 in drinking water) chronically from 2 months of age to 6 months (Figure 1A). Because M1 activators like VU0364572 have symptomatic effects on memory, we incorporated a drug-washout period of at least 24 h at the end of the treatment phase to avoid confounding the detection of disease-modifying effects. The 24 h washout period was deemed sufficient based upon previously published pharmacokinetic data demonstrating that VU0364572 has a half-life of 45 min.37
Figure 1.

M1 Activation by VU0364572 preserves hippocampal memory when dosed chronically in 5XFAD transgenic Alzheimer’s mice. VU0364572 preserves memory when dosed chronically prior to the initiation of Morris water maze training, but does not benefit nonmemory measures of Morris water maze performance, such as swim speed. (A) Schematic depicting design of chronic treatment trial of 5XFAD mice with M1 agonist, VU0364572. (B) Following dosing VU0364572 at 10 mg/kg to 5XFAD mice for 4 months, drug-treated mice were significantly improved in their performance (36.7% time in probe quadrant) on the probe trial of the Morris water maze relative to vehicle-treated (26.7% time in probe quadrant) 5XFAD mice (*p < 0.05, t = 2.11, df = 24.49). 5XFAD mice were found to be significantly impaired relative to vehicle-treated WT littermate controls (**p < 0.01, t = 2.93, df = 26.17). (C) VU0364572 does not influence swim speed of animals during training. No statistically significant difference was observed during training in swim speed for either WT mice (N = 13), 5XFAD vehicle-treated mice (N = 17), or 5XFAD mice receiving 10 mg/kg VU0364572 (N = 12). Error bars show ± SEM across all mice within a treatment group. (D) Representative swim paths depicting probe trial performance of WT littermate controls (left), 5XFAD vehicle-treated mice (center), and drug-treated 5XFAD mice (right) with maze quadrants depicted as red, purple, blue, and orange quadrants, respectively, and the submerged platform location (platform not present for probe trial) depicted as a red dot.
Following placebo treatment and behavioral testing at 6 months of age, 5XFAD mice displayed a significant deficit (mean probe trial performances of 26.7%; **p < 0.01, t = 2.93, df = 26.17) in Morris water maze probe trial performance relative to WT littermate controls (42.6% for WT; Figure 1B). These results are in good agreement with previous results of the magnitude and rate of cognitive decline in 5XFAD mice.63 Following chronic drug treatment, the performance of VU0364572-treated animals on the memory task was significantly improved (mean probe trial performance of 36.8%; *p < 0.05, t = 2.11, df = 24.49) relative to 5XFAD vehicle-treated animals, and not significantly different from WT animals (Figure 1B). Swim speed was not impacted relative to WT littermate controls by either genotype or treatment (Figure 1C), indicating no effect of drug treatment on motor behavior. All mice were also tested on cued and contextual fear conditioning tasks, which are amygdalo-hippocampal memory tests. While 5XFAD mice were found to be impaired in contextual fear conditioning, no impairment was observed in tests of cued fear conditioning and no drug effect was noted for either task (Figure S1). Measures of freezing between vehicle-treated and drug-treated 5XFAD animals were not found to be different between groups on either task.
VU0364572 Decreases Soluble and Insoluble Aβ40 and Aβ42 Levels in 5XFAD Mice
A truly effective disease-modifying treatment for AD would delay cognitive decline by reducing the neuropathological burden, including Aβ accumulation. In chronically treated 5XFAD mice, Aβ40 and Aβ42 levels were analyzed separately in both the SDS-soluble and formic-acid-insoluble fractions for hippocampus and total cortex (neocortex + entorhinal cortex) by ELISA following chronic treatment with VU0364572 or placebo. There was a significant VU0364572-mediated decrease in soluble hippocampal Aβ40 levels (40.4%; *p < 0.05, t = 2.15, df = 24.55), but no significant effect noted on cortical Aβ40 levels versus vehicle-treated 5XFAD mice (n.s., p > 0.05, t = 1.30, df = 23.84) (Figure 2A). Insoluble Aβ40 levels were found to be significantly decreased in both the hippocampus (43.3%; *p < 0.05, t = 2.69, df = 22.35) and cortex (38.9%; *p < 0.05, t = 2.47, df = 19.60) (Figure 2B). Furthermore, there was a trend toward a decrease in soluble Aβ42 levels in the hippocampus following VU0364572 treatment (23.4%; p < 0.1, t = 1.87, df = 25.66) and a significant decrease observed in the cortex (34.2%; *p < 0.01, t = 2.54, df = 20.68) (Figure 2C). Insoluble Aβ42 levels were also significantly decreased in the cortex (34.2%; p = 0.05, t = 1.98, df = 25.62) with no significant effect observed in the hippocampus (n.s., p > 0.05, t = 1.30, df = 25.21) following VU0364572 treatment (Figure 2D). Aβ40 and Aβ42 levels were also found to be decreased by VU0364572 treatment in ex vivo studies in primary hippocampal and cortical neurons from WT mice transduced with hAPP695WT (Figure S2), suggesting the amyloid-modifying effects are directly mediated by neurons. Thus, chronic M1 activation by VU0364572 robustly decreased Aβ40 and Aβ42 levels in the soluble and insoluble fractions cortex and hippocampus.
Figure 2.

M1 activation by VU0364572 decreases soluble and insoluble Aβ40 and Aβ42 levels in cortex and hippocampus of 5XFAD mice. Total cortex and hippocampus were microdissected from 5XFAD animals and WT littermate controls and biochemically fractionated. Fractions were then subjected to Aβ40 and Aβ42 ELISA analysis. (A) VU0364572 significantly decreases soluble Aβ40 levels in the hippocampus of 5XFAD mice by 40.4% relative to vehicle-treated 5XFAD animals (*p < 0.05, t = 2.15, df = 24.55). No significant decrease of soluble Aβ40 was observed in the cortex. (B) VU0364572 significantly decreases insoluble Aβ40 levels in both the neocortex (38.9%; *p < 0.05, t = 2.47, df = 19.60) and hippocampus (43.3%; *p < 0.05, t = 2.69, df = 22.35) of 5XFAD mice relative to vehicle-treated 5XFAD animals. (C) VU0364572 significantly decreases soluble Aβ42 levels in both the cortex (34.2%; *p < 0.01, t = 2.54, df = 20.68) with a trend toward a decrease in the hippocampus (23.4%; p < 0.1, t = 1.87, df = 25.66) of 5XFAD mice relative to vehicle-treated 5XFAD animals. (D) VU0364572 significantly decreases insoluble Aβ42 levels in the cortex of 5XFAD mice relative to vehicle-treated 5XFAD animals (34.2%; p = 0.05, t = 1.98, df = 25.62), with no significant decrease in hippocampus (23.4%; n.s., p > 0.05, t = 1.30, df = 25.21). Error bars show ± SEM across all mice within a treatment group.
M1 Activation by VU0364572 Reduces Aβ40 and Aβ42 Neuropathology in 5XFAD Mice
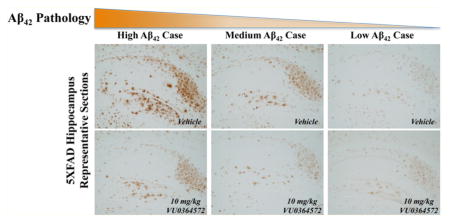
To determine if the strong effects of VU0364572 on insoluble Aβ40 and Aβ42 levels translated into reduced amyloid plaque burden, brain sections from VU0364572- and vehicle-treated 5XFAD mice (Figures 3 and S3), and WT littermate controls (Figure S4) were stained with antibodies against human Aβ42 (Figure 3) and Aβ40 (Figure S3). Qualitatively, there was a striking decrease in Aβ42 plaque immunoreactivity throughout the hippocampus and cortex of VU0364572-treated 5XFAD mice (Figure 3D–F, J–L) relative to vehicle-treated 5XFAD mice (Figure 3A–C, G–I). In the hippocampus, substantial decreases were noted across all hippocampal subfields: subiculum, dentate gyrus, CA3, and CA1 (Figure 3J–L). In order to quantify VU0364572-induced changes, Aβ42 immunoreactivity was measured in a blinded manner in equivalent regions of cortex and hippocampus. 5XFAD vehicle-treated animals showed a heavy burden of Aβ42 pathology that was significantly mitigated by VU0364572 treatment in both the hippocampus (14.9% decrease; ****p < 0.0001, t = 4.82, df = 24.88) and cortex (36.1% decrease; ***p < 0.001, t = 4.58, df = 24.18) (Figure 3M). Similarly, VU0364572 globally decreased Aβ40 immunoreactivity in plaques across hippocampus and cortex in 5XFAD mice (Figure S3D–F, J–L) relative to vehicle-treated 5XFAD mice (Figure S3A–C, G–I). Quantification of Aβ40 immunoreactivity in a similar fashion yielded a substantial decrease in Aβ40 pathology following VU0364572 treatment in both the neocortex (32.8% decrease; ***p < 0.001, t = 3.81, df = 24.49) and hippocampus (28.1% decrease; **p < 0.01, t = 3.87, df = 17.26) (Figure S3M). Representative sections stained from WT littermate controls with the Aβ42 and Aβ40 antibodies used to assess neuropathology in this study showed no evidence of positive immunoreactivity, indicating that the observed Aβ42 (Figure 3) and Aβ40 (Figure S3) staining is highly specific (Figure S4).
Figure 3.

M1 Activation by VU0364572 reduces Aβ42 neuropathology in the brains of 5XFAD mice. Serial sections at 50 μm were taken from 5XFAD mice and WT littermate controls, and six sections per animal were then subjected to Aβ42 immunohistochemical analysis with anti-human Aβ42 antibody (1:1000; Biosource) in order to examine Aβ42 pathology in the cortex and hippocampus. Vertical columns show representative sections for cases displaying high (A, D, G, J), medium (B, E, H, K), and low (C, F, I, L) Aβ42 pathology. Horizontal rows denote cortical (A–F) and hippocampal (G–L) sections for both vehicle-treated (A–C, G–I) animals and VU0364572-treated animals (D–F, J–L). 5XFAD vehicle-treated animals show marked cortical (A–C) and hippocampal (G–I) Aβ42 pathology that is substantially mitigated by VU0364572 treatment across the neocortex (D–F) and all hippocampal subfields (J–L). (M) Total Aβ42 immunoreactive surface area was measured for 6 consecutive slices taken at the same anterior-posterior coordinates per animal for cortical and hippocampal regions, with the mean immunoreactive surface area plotted for individual animals. 5XFAD vehicle-treated animals showed robust Aβ42 pathology that is significantly mitigated by VU0364572 treatment in both the neocortex (36.1% decrease; ***p < 0.001, t = 4.58, df = 24.18) and hippocampus (14.9% decrease; ****p < 0.0001, t = 4.82, df = 24.88). Error bars show ± SEM across all mice within a treatment group.
Soluble Aβ42 Levels are Strongly Correlated with Memory Impairment in 5XFAD Animals
Since higher soluble Aβ42 levels are known to correlate with worse memory performance in human AD patients, we assessed the correlation between Aβ40 or Aβ42 pathology in 5XFAD mice and memory performance in the Morris water maze.41–53 To this end, pairwise correlations were performed between Aβ40 and Aβ42 fractions (soluble and insoluble, cortical and hippocampal) and Morris water maze probe trial performance for all mice (Figures 4 and S5). The strongest correlation was obtained between soluble Aβ42 levels in the hippocampus and memory performance, with increasing levels of soluble hippocampal Aβ42 highly correlated (R2 = 0.44; ***p < 0.001) to worsening behavioral performance (Figure 4C). The correlation of soluble Aβ42 levels with memory impairment is consistent with a similar correlation in APP/PS1 mice and with the notion that Aβ42 is the predominant species giving rise to soluble Aβ oligomers which profoundly disrupt synaptic plasticity and memory.63
Figure 4.

Aβ42 Levels in the hippocampus and neocortex are correlated with memory performance. Behavioral performance of 5XFAD mice on the probe trial of the Morris water maze was correlated with soluble (A, C) and insoluble (B, D) Aβ42 levels from the cortex (A, B) and hippocampus (C, D) of these mice. (A) The strongest correlation was observed between soluble hippocampal Aβ42 levels and probe trial performance in the Morris water maze (C), which was very highly significant and the strongest correlation observed in the study for any Aβ40 or Aβ42 species with memory performance (R2 = 0.44; ***p < 0.001). (B) There was also a strong correlation (B) noted between insoluble cortical Aβ42 levels and probe trial performance (R2 = 0.37; ***p < 0.001). While significant, correlations between soluble cortical Aβ42 levels and insoluble hippocampal Aβ42 levels were substantially less robust (A, D). All correlations reflect the inclusion of WT animals that received vehicle alone.
Apart from soluble Aβ species, levels of insoluble Aβ40 (R2 = 0.36; **p < 0.01) and Aβ42 (R2 = 0.37 ***p < 0.001) in the cortex were found to correlate significantly with Morris water probe trial performance (Figures 4B, D and S5B, D). Treatment with VU0364572 abolished both insoluble Aβ40 (R2 = 0.089; n.s.) and Aβ42 (R2 = 0.069; n.s.) correlations with probe trial performance. There were no significant perturbations in soluble or insoluble Aβ40/42 ratios in VU0364572-treated samples relative to controls (Figure S6).
Since soluble oAβ levels have been linked most directly to the memory-deficits and impairments in synaptic plasticity, we also evaluated the effects of chronic M1 treatments on soluble oAβ in the brain. VU0364572 significantly lowered oAβ levels in the cortex (20.1% decrease; *p < 0.05, t = 3.04, df = 11.77), with a similar but nonsignificant trend observed in the hippocampus (Figure 5). These observations suggest that VU0364572 may improve memory, in part, by decreasing the size of the available oAβ pool in the brain.
Figure 5.
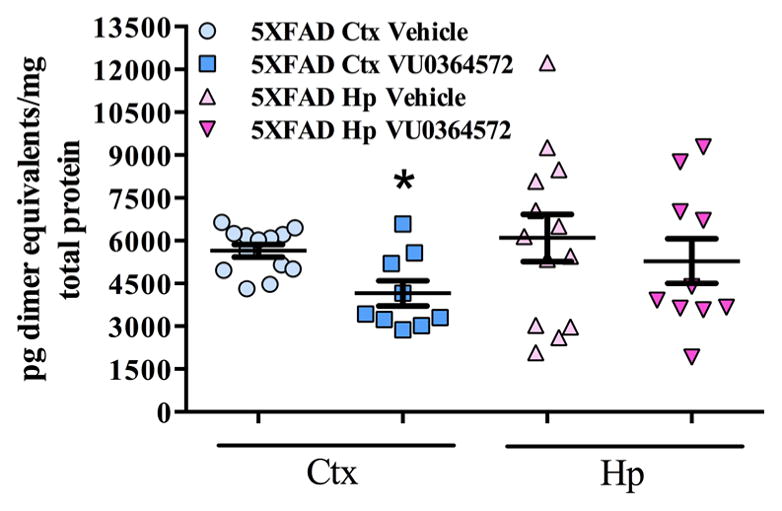
M1 Activation by VU0364572 reduces oligomeric aβ neuropathology in the cortex of 5XFAD mice. Total cortex (neocortex + entorhinal cortex) and hippocampus were microdissected from 5XFAD animals and WT littermate controls and biochemically fractionated. Soluble Aβ fractions were then subjected to ELISA analysis for oAβ detection as described previously.59 VU0364572 was found to significantly decrease oAβ levels in the cortex (20.1% decrease; *p < 0.05, t = 3.04, df = 11.77), however, hippocampal samples did not reach significance, as much more variability was present. Error bars show ± SEM across all mice within a treatment group.
DISCUSSION
The M1 receptor has long been regarded as a promising target for disease modification in AD. Many studies have described the ability of M1 stimulation to exert nonamyloidogenic effects on APP processing in vitro, and M1 knockout mice have demonstrated the importance of the M1 receptor for regulating amyloid levels in vivo. However, a lack of truly selective M1 activators that display druggable characteristics acceptable for in vivo dosing has hampered the investigation into whether chronic M1 activation can prevent AD pathology and preserve memory by modifying underlying Aβ pathology. While key proof of concept studies with M1 activators to-date have demonstrated the disease-modifying effects of M1 activation on Aβ and tau pathologies, these studies were conducted with nonselective M1 receptor ligands and were performed in mouse models of disease with a very mild phenotype (e.g., in 3xTg mice).39,64,65 Many treatments have advanced to the clinical trials after showing improvements in such animal models with mild disease phenotypes and so far none have translated into an effective treatment that slows or blocks the clinical progression of AD. Since the overproduction and accumulation of Aβ species is widely viewed as a proximal pathological event in the course of AD, preventative efficacy in an aggressive rodent AD model may offer higher translational potential.
The present work shows for the first time that chronic activation of M1 with the selective M1 agonist VU0364572 protects against neuropathology and prevents memory impairments from forming in an aggressive 5XFAD mouse model of AD. Specifically, chronic M1 activation by VU0364572 from 2 to 6 months of age prevented development of memory impairments in 5XFAD transgenic AD mice, as measured by Morris water maze testing. Importantly, at 2 months of age, 5XFAD mice display little detectable amyloid, whereas at 6 months these mice are known to display significant amyloid accumulation along with impairments in the Morris water maze. The Morris water maze is a sensitive test of hippocampal-dependent memory function and deficits in this task are thought to model the disruption of memory encoding and recognition memory processes that occurs in humans with AD.
Chronic M1 activation with VU0364572 was found to exert sustained and lasting benefits on lowering soluble and insoluble Aβ40 and Aβ42 levels and pathology in the cortex and hippocampus of 5XFAD mice, which translated to the prevention of hippocampal memory impairments in animals tested on the Morris water maze. The significant correlation of higher soluble Aβ42 levels with worse memory performance is consistent with recent findings on the role of soluble oAβ species in AD. A number of studies have attributed the memory impairments that accompany early AD to the accumulation of oAβ that has been shown to profoundly disrupt synaptic plasticity. The significant decrease in both cortical oAβ levels as well as hippocampal Aβ42 pathology observed in our studies and the finding that VU0364572 abolishes the correlation between soluble Aβ42 levels and memory performance highlights the potential of M1 activation by VU0364572 to lower the circulating levels of oAβ that disrupt synaptic plasticity.
While improved performance was observed in the MWM, we observed no evidence of a VU0364572-mediated benefit in reversing contextual fear conditioning deficits at the dose tested. Most parsimoniously MWM, cued fear conditioning, and contextual fear conditioning are all distinct behavioral paradigms that depend upon partially overlapping but distinct brain circuitry, including the hippocampus. While the MWM is a sensitive test of hippocampal memory and cued fear conditioning is a sensitive test of amygdala function (with involvement of hippocampus also), it has become increasingly clear that interaction between hippocampus, amygdala and prefrontal cortex are crucial for other varieties of fear learning such as contextual (tested in the current study) and trace conditioning.66 Although we do not have mechanistic data that directly speaks to engagement of these individual regions, it is possible that 5XFAD mice may exhibit greater prefrontal-hippocampal pathology that impairs contextual fear learning that the current degree of M1 activation is unable to rescue at the dose and age tested. While M1 receptors are expressed across all of the above brain regions, the levels are substantially higher in the amygdala and hippocampus than in the prefrontal cortex. In this vein, previous studies have demonstrated hippocampal efficacy of VU0364572 at doses below those required to elicit prefrontal efficacy.38 Therefore, since the doses in the present study were based upon previously observed hippocampal MWM effects it may be that the dose was simply too low to result in beneficial effects on PFC-dependent processes. Future work in an independent cohort may help to confirm the present results and clarify these findings.
The observed efficacy of VU0364572 together with its novel pharmacological properties offer several advantages for clinical translation. Unlike conventional M1 agonists that were extensively developed by the pharmaceutical industry to target the orthosteric acetylcholine binding site, M1 allosteric and bitopic agonists, including VU0364572, do not appear to couple as efficiently to the intracellular β-arrestin signaling cascades involved in desensitization of M1 receptor signaling.22,38,67,68 This property is beneficial, as receptor densensitization limits the ability to dose treatments chronically, as is necessary for AD. Also, extensive molecular pharmacological characterization of VU0364572 reveals this compound has excellent selectivity for M1, but rather low intrinsic activity. Thus, the present findings indicate that modest but selective M1 activation with allosteric or bitopic ligands may be therapeutically beneficial. The lower intrinsic activity of a partial M1 agonist would also reduce seizure liability and off-target side effects elicited by peripheral mAChR activation.
Our findings support further development of M1 activators for early intervention for presymptomatic and prodromal AD. There is increasing recognition that the decades-long preclinical period of AD offers a large window for disease-modifying treatment. Unlike anti-amyloid treatments, which have substantial risk of amyloid-related vasogenic edema (ARIA-E), chronic M1 treatment would not be expected to have this risk—an important consideration given that preventive treatments will be given to large numbers of asymptomatic individuals. Individuals with subjective memory loss, i.e., memory symptoms but no objective deficits on neuropsychological testing, and mild cognitive impairment, are also excellent groups to target for clinical translation of M1 activators. Given there are currently no FDA-approved drugs with symptomatic benefits on memory for age-associated memory loss or for mild cognitive impairment, M1 activators would provide urgently needed symptom relief and disease modification.
METHODS
Subjects for Transgenic AD Mouse Studies
5XFAD transgenic AD mice from Jackson Laboratories were utilized for this study.57 The 5XFAD strain is a double transgenic APP/PS1 strain that expresses five AD mutations. These mice bear Florida, London, and Swedish familial AD mutations in the gene coding for APP. Additionally, these mice bear M146L and L286 V familial AD mutations in the PS1 gene. These mutations result in higher overall levels of Aβ, as well as increased production of Aβ42, the major plaque forming species in AD. The 5XFAD mice show intraneuronal Aβ42 and amyloid deposition beginning around 2 months of age, and memory deficits at 6 months.58 All procedures involving mice were approved by the Emory University Institutional Animal Care and Use Committee. Mice were chronically dosed with VU0364572 ad libitum in drinking water for 4 months (from age 2 to 6 months). At 6 months, a 24 h drug washout was performed followed by behavioral testing. The 24 h washout period was deemed sufficient based upon previously published pharmacokinetic data demonstrating that VU0364572 has a half-life of 45 min.37 Thus, no drug was onboard for any behavioral testing performed. Immediately after testing, the mice were perfused and brains were harvested, as described below.
A previously published study treated wild type animals with VU0364572 and showed a beneficial effect on memory in the Morris water maze,38 indicating that indeed VU0364572 has procognitive symptomatic effects. Since this experiment has already been performed and published, our primary goal was to model a treatment regimen as would be performed in humans. The prevention trials that seek to modify the course of disease begin treatment in individuals with existing amyloid pathology and/or at high risk for the disease (e.g., carrying genetic mutations). Thus, we only pursued drug treatment in transgenic animals carrying autosomal dominant mutations.
Drugs for Transgenic AD Mouse Studies
VU0364572 was synthesized as a mono-HCl salt and provided as a jet-milled powder to aid in drug solubilization and systemic absorption. VU0364572 was kindly provided by P. Jeffrey Conn and Craig Lindsley in the Vanderbilt Center for Neuroscience Drug Discovery (Vanderbilt University, Nashville, TN), and central penetrance verified as described in the Supporting Information and detailed in Table S1. Due to the documented oral bioavailability of VU0364572 in rat, drug was delivered to transgenic mice ad libitum in their drinking water (0.075 mg/mL concentration) from 2 months of age to 6 months of age. Mice had continuous access to drug-treated water at all times during this 4 month dosing window. Mouse cages were coded so that experimenters testing transgenic 5XFAD mice were blinded to treatment.
Determination of CNS VU0364572 Concentrations for Chronic Dosing
VU0364572 was delivered ad libitum to a group of six wild type B6SJL (same genetic background of 5XFAD mice) mice for 5 days. Calculation of the administered dose (approximately 10 mg/kg/day) was based upon the average weight of mice (30 g) and the average volume of drinking water a mouse was found to consume during a given 24 h period (4 mL; compound concentration = 0.075 mg/mL). With regard to the behavioral readouts in the current study, previously published data demonstrates that doses up to 56.6 mg/kg VU0364572 have no effect on baseline locomotor activity.38 Following dosing to steady-state, mice were decapitated, brains removed and immediately washed with ice-cold phosphate-buffered saline, and brains then immediately frozen on dry ice until analysis. Trunk blood was immediately collected in EDTA Vacutainer tubes and plasma was separated by centrifugation and stored at −80 °C until analysis. Bioanalysis of plasma and tissue samples to quantitate VU0364572 concentration was as described previously.36,37
Biochemical Tissue Fractionation
Fresh-frozen sagittal hemibrains were removed from −80 °C storage after which neocortex and total hippocampus were microdissected from each hemisphere. Wet tissue weight for each cortex and hippocampus sample was recorded and tissue homogenized using a Konte’s Dounce tissue grinder in phosphate-buffered saline with 1× protease inhibitor cocktail (Roche, Indianapolis, IN). Cortical samples were homogenized to a concentration of 150 mg/mL, whereas hippocampal samples were homogenized to a concentration of 100 mg/mL. Total homogenate was then sonicated for ~30 s using a microtip sonicator at 20% total amplitude. 2% SDS was then added to the homogenate in order to enable soluble amyloid extraction. Homogenates were spun for 1 h at 53 000g at −4 °C (Optima TLX Ultracentrifuge, Beckman-Coulter, Fullerton, CA) to separate soluble from insoluble amyloid species. The supernatant containing soluble amyloid was then collected and the pellet containing insoluble amyloid resuspended in 70% formic acid. Once resuspended, the insoluble fraction was resonicated for ~30s at 20% total amplitude. Individual tissue fractions were analyzed fresh and never subjected to more than one freeze–thaw cycle. All brains were randomized prior to microdissection so that experimenters were blinded during downstream biochemical analyses (e.g., Aβ40 and Aβ42 ELISAs).
Immunohistochemistry
Sagittal hemibrains were removed from storage in 30% sucrose and serially sectioned at a thickness of 50 μm on a freezing stage sliding microtome. Sections were immediately submerged in cryoprotectant and placed at −20 °C until analysis. For immunohistochemical staining, six consecutive tissue sections were taken from equivalent depths across all mice enrolled in the 5XFAD chronic dosing trial.
For immunohistochemical analysis, free-floating brain sections were then rinsed in 0.1 M phosphate buffer pH 7.2 (PB) 5 times × 5 min. Next, PB was used to dilute 30% H2O2 (Sigma) to 3%. Tissue was washed with 3% H2O2 for 15 min in order to remove any endogenous peroxidase activity, after which the sections were again rinsed with PB 5 × 5 min. Sections were then blocked with a solution of 10 μg/mL avidin (1:1), 8% normal serum, and 0.1% Triton-X in TBS. Sections were blocked for 1 h at 4 °C, then rinsed with TBS 3 × 5 min. Primary antibody incubation then took place in a solution of 50 μg/mL biotin, 2% normal serum, and α-hAβ1–40 (rabbit polyclonal, BioSource Invitrogen, Carlsbad, CA, 1:5000) or α-hAβ1–42 (rabbit polyclonal, BioSource Invitrogen, Carlsbad, CA, 1:1000) in TBS. Incubation in the primary antibody solution took place for 48 h at 4 °C with shaking.
Following primary incubation, tissue was rinsed with TBS 4 × 5 min. Sections were then incubated with a biotinylated secondary antibody (bGαRb) for 3 h at 4 °C with shaking and washed again with TBS 4 × 5 min. Secondary signal was then visualized using the avidin–biotin-peroxidase complex (ABC) method (ABC kit; Vector Laboratories, Burlingame, CA). ABC reagent was prepared according to the manufacturer’s instructions in TBS and allowed to stand on ice for 30 min prior to use. Sections were then incubated in the ABC solution for 1 h with shaking at 4 °C. Sections were then rinsed 4 × 5 min with TBS and stained with diaminobenzidine (DAB). Following DAB staining, tissue was removed and rinsed with TBS 4 × 5 min. Brain sections were then mounted on precleaned Superfrost Plus slides (Fisher) in 0.1 M NaNO3. Slides were then allowed to air-dry after which they were then immersed in dH2O 3 min, 70% ethanol 3 min, 95% ethanol 2 × 3 min, 100% ethanol 2 × 3 min, and Histoclear 3 × 3 min. Slides were then coverslipped with DPX and allowed to dry overnight.
To quantify hAβ40 and hAβ42 immunoreactivity, cortex and hippocampus were photographed at low power (4×) from each of six consecutive sections stained for either hAβ40 or hAβ42. Total Aβ40 and Aβ42 immunoreactive surface area was then measured in a blinded manner using MetaMorph 5.0 software (Molecular Devices).
Aβ40, Aβ42, and oAβ ELISAs
hAβ40 and hAβ42 levels in conditioned media from primary neuronal cultures as well as soluble and insoluble Aβ40 and Aβ42 tissue homogenates from biochemically fractionated mouse brains were measured using human Aβ40 and Aβ42 ELISA kits according to the manufacturer’s protocols (Biosource, Invitrogen). Insoluble amyloid fractions from mouse tissue homogenates containing 70% formic acid were first neutralized by performing a 1:100 dilution in a solution of 1.0 M Tris (pH 11) prior to performing dilution series in ELISA diluent buffer supplied with ELISA kits. Soluble amyloid fractions were diluted as normal directly in ELISA diluent buffer. Plates were read at 450 nm using a Spectra Max Plus plate reader (Molecular Devices, Sunnyvale, CA). ELISA-based detection of oAβ was performed as described previously.59 For detection of oAβ, total tissue homogenates in PBS were utilized for oAβ ELISAs, as the addition of detergent is known to induce artifactual Aβ oligomerization. Moreover, albuminized tubes were used for the handling of total tissue homogenates to block nonspecific oAβ binding sites on the plastic tubes.
Statistical Analyses
For all behavioral, primary culture data and ELISA data, graphs were generated using GraphPad Prism 5.0. Due to the small and unequal sample sizes of the study, as well as to account for the likelihood of different population variances, Welch’s unequal variance t-test was used.60,61 All p-values reported for these tests are two-tailed, Welch’s-corrected, p-values. Due to the small number of subjects in the drug treatment trial of 5XFAD mice, outlier detection/exclusion was performed using a test of Median Absolute Deviation (MAD), which is less biased by any outliers themselves than tests which rely upon standard deviations from the mean.62 Samples with a MAD ≥ 2.0 were excluded as outliers. For MWM data, fear-conditioning data, and immunohistochemistry data, one animal was excluded from the vehicle-treated WT group and one animal from the vehicle-treated 5XFAD group. For the soluble and insoluble Aβ40 and Aβ42 ELISA data and subsequent correlations with MWM data, two animals from the Ctx-Veh group were excluded, four animals from the Ctx-M1 group, two animals from the Hp-Veh group, and four animals from the Hp-M1 group. For oAβ data, three animals were excluded from the Ctx-Veh group, three animals from the Ctx-M1 group, three animals from the Hp-Veh group, and two animals from the Hp-M1 group.
Supplementary Material
Acknowledgments
Funding
The authors wish to acknowledge the Vanderbilt Center for Neuroscience Drug Discovery for graciously providing VU0364572 utilized for this study. The present work was funded by Grants U54 MH084659 and R01 MH082867 to C.W.L., Grants R01 MH073676 and U01 MH087965 to P.J.C., the Cure Alzheimer’s Fund and Grant R01 NS065069 to D.L.B., Grant R01 NS030454 to A.I.L., and by Grant T32NS007480-12 to E.P.L.
All behavioral tasks were performed by the Emory Rodent Behavioral Core, which is subsidized by the Emory University School of Medicine and is one of the Emory University Integrated Core Facilities. The authors wish to acknowledge Jay Li for his technical assistance in conducting the present studies.
ABBREVIATIONS
- Aβ
amyloid beta
- AChEI
acetylcholinesterase inhibitor
- ACh
acetylcholine
- AD
Alzheimer’s disease
- APP
amyloid precursor protein
- Ctx
cortex
- ELISA
enzyme-linked immunosorbent assay
- GPCR
G protein-coupled receptor
- Hp
hippocampus
- mAChR
muscarinic acetylcholine receptor
- MWM
Morris water maze
- oAβ
oligomeric amyloid beta
- PS1
presenilin 1
- sAPP
soluble amyloid precursor protein
- WT
wild type
Footnotes
Author Contributions
E.P.L., J.P.S., T.J.E., T.M.B., C.W.L., P.J.C., D.L.B., J.S.D., and A.I.L. designed research; E.P.L., J.P.S., T.J.E., and T.M.B. conducted research; E.P.L., J.P.S., T.H.E., and T.M.B. analyzed data; E.P.L., T.M.B., C.W.L., P.J.C., D.L.B., and A.I.L. wrote the manuscript.
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acschemneur-o.6b00278.
Supplementary methods; contextual fear conditioning in 5XFAD mice with VU0364572; effect of VU0364572 on Aβ40/42 levels in primary cortical and hippocampal neurons; effect of VU0364572 on Aβ40 neuropathology in neocortex and hippocampus of 5XFAD mice; Aβ40 and Aβ42 immunohistochemistry in 5XFAD WT littermate control sections; correlation of Aβ40 levels with memory impairment in 5XFAD mice; effects of VU0364572 on Aβ40/42 ratios in 5XFAD mice; satellite pharmacokinetic study with VU0364572 (PDF)
Notes
E.P.L. performed experiments comprising this study while a student at Emory University, prior to his current employment by Pfizer, Inc.
The authors declare the following competing financial interest(s): C.W.L. receives, or has received, funding from Janssen, BMS, and AZ for the development of allosteric modulators for CNS Disorders and consults for AbbVie, Sanofi, and multiple foundations. P.J.C. has been funded by NIH, Johnson and Johnson, AstraZeneca, Bristol-Myers Squibb, Michael J. Fox Foundation, and Seaside Therapeutics. Over the past 3 years he has served on the Scientific Advisory Boards of Seaside Therapeutics, Michael J. Fox Foundation, Stanley Center for Psychiatric Research Broad Institute (MIT/Harvard), Karuna Pharmaceuticals, Lieber Institute for Brain Development (Johns Hopkins University), Clinical Mechanism and Proof of Concept Consortium, and Neurobiology Foundation for Schizophrenia and Bipolar Disorder. C.W.L., P.J.C., and E.P.L. are inventors on patents that protect different classes of muscarinic receptor allosteric modulators. D.L.B. has served as a consultant for Pfizer, Inc., Intellectual Ventures, Signum, Nutralogix, Kypha, Inc., Sage Therapeutics, iPerian, Inc., Avid Radiopharmaceuticals (Eli Lilly & Co.), the St. Louis County Public Defender, the United States Attorneys Office, the St. Louis County Medical Examiner, and has given testimony in over 50 medicolegal cases. D.L.B. receives research funding from the Department of Defense, NIH, and Bright Focus Foundation, and has received research funding from the Cure Alzheimers Fund, Health South, the National Football League Charities, Pfizer, Inc., Burroughs Wellcome Fund, Hope Center at Washington University, and Thrasher Research Fund.
References
- 1.Barker WW, Luis CA, Kashuba A, Luis M, Harwood DG, Loewenstein D, Waters C, Jimison P, Shepherd E, Sevush S, Graff-Radford N, Newland D, Todd M, Miller B, Gold M, Heilman K, Doty L, Goodman I, Robinson B, Pearl G, Dickson D, Duara R. Relative frequencies of Alzheimer disease, Lewy body, vascular and frontotemporal dementia, and hippocampal sclerosis in the State of Florida Brain Bank. Alzheimer Dis Assoc Disord. 2002;16(4):203–212. doi: 10.1097/00002093-200210000-00001. [DOI] [PubMed] [Google Scholar]
- 2.Holtzman DM, Morris JC, Goate AM. Alzheimer’s disease: the challenge of the second century. Sci Transl Med. 2011;3(77):77sr1–77sr1. doi: 10.1126/scitranslmed.3002369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Conn PJ, Jones CK, Lindsley CW. Subtype-selective allosteric modulators of muscarinic receptors for the treatment of CNS disorders. Trends Pharmacol Sci. 2009;30(3):148–155. doi: 10.1016/j.tips.2008.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Conn PJ, Christopoulos A, Lindsley CW. Allosteric modulators of GPCRs: a novel approach for the treatment of CNS disorders. Nat Rev Drug Discovery. 2009;8(1):41–54. doi: 10.1038/nrd2760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Alzheimer A. Uber eine eijenartige Erkrankung der Hirnride. Allg Z Psychiatr. 1907;64:146–148. [Google Scholar]
- 6.Gandy SE, Caporaso GL, Buxbaum JD, Da Cruz e Silva O, Iverfeldt K, Nordstedt C, Suzuki T, Czernik AJ, Nairn AC, Greengard P. Protein phosphorylation regulates relative utilization of processing pathways for Alzheimer beta/A4 amyloid precursor protein. Ann N Y Acad Sci. 1993;695:117–21. doi: 10.1111/j.1749-6632.1993.tb23038.x. [DOI] [PubMed] [Google Scholar]
- 7.Selkoe DJ. Normal and abnormal biology of the beta-amyloid precursor protein. Annu Rev Neurosci. 1994;17(1):489–517. doi: 10.1146/annurev.ne.17.030194.002421. [DOI] [PubMed] [Google Scholar]
- 8.Selkoe DJ, Yamazaki T, Citron M, Podlisny MB, Koo EH, Teplow DB, Haass C. The role of APP processing and trafficking pathways in the formation of amyloid beta-protein. Ann N Y Acad Sci. 1996;777:57–64. doi: 10.1111/j.1749-6632.1996.tb34401.x. [DOI] [PubMed] [Google Scholar]
- 9.Selkoe DJ. Alzheimer’s disease: genes, proteins, and therapy. Phys Rev. 2001;81(2):741–766. doi: 10.1152/physrev.2001.81.2.741. [DOI] [PubMed] [Google Scholar]
- 10.Buckner RL, Snyder AZ, Shannon BJ, LaRossa G, Sachs R, Fotenos AF, Mintun MA. Molecular, structural, and functional characterization of Alzheimer’s disease: evidence for a relationship between default activity, amyloid, and memory. J Neurosci. 2005;25(34):7709–7717. doi: 10.1523/JNEUROSCI.2177-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.deToledo-Morrell L, Stoub TR, Wang C. Hippocampal atrophy and disconnection in incipient and mild Alzheimer’s disease. Prog Brain Res. 2007;163:741–823. doi: 10.1016/S0079-6123(07)63040-4. [DOI] [PubMed] [Google Scholar]
- 12.Suzuki T, Nakaya T. Regulation of amyloid beta-protein precursor by phosphorylation and protein interactions. J Biol Chem. 2008;283(44):29633–7. doi: 10.1074/jbc.R800003200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Thinakaran G, Koo EH. Amyloid precursor protein trafficking, processing, and function. J Biol Chem. 2008;283(44):29615–9. doi: 10.1074/jbc.R800019200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Palop JJ, Mucke L. Amyloid-[beta]-induced neuronal dysfunction in Alzheimer’s disease: from synapses toward neural networks. Nat Neurosci. 2010;13(7):812–818. doi: 10.1038/nn.2583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Small SA, Schobel SA, Buxton RB, Witter MP, Barnes CA. A pathophysiological framework of hippocampal dysfunction in ageing and disease. Nat Rev Neurosci. 2011;12(10):585–601. doi: 10.1038/nrn3085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jack CR, Jr, Knopman DS, Jagust WJ, Shaw LM, Aisen PS, Weiner MW, Petersen RC, Trojanowski JQ. Hypothetical model of dynamic biomarkers of the Alzheimer’s pathological cascade. Lancet Neurol. 2010;9(1):119–128. doi: 10.1016/S1474-4422(09)70299-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sperling RA, Rentz DM, Johnson KA, Karlawish J, Donohue M, Salmon DP, Aisen P. The A4 Study: Stopping AD Before Symptoms Begin? Sci Transl Med. 2014;6(228):228fs13. doi: 10.1126/scitranslmed.3007941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Reisberg B, Doody R, Stöffler A, Schmitt F, Ferris S, Möbius HJ. Memantine in moderate-to-severe Alzheimer’s disease. N Engl J Med. 2003;348(14):1333–1341. doi: 10.1056/NEJMoa013128. [DOI] [PubMed] [Google Scholar]
- 19.Thomas SJ, Grossberg GT. Memantine: a review of studies into its safety and efficacy in treating Alzheimer’s disease and other dementias. Clin Interventions Aging. 2009;4:367. doi: 10.2147/cia.s6666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Farber SA, Nitsch RM, Schulz JG, Wurtman RJ. Regulated secretion of beta-amyloid precursor protein in rat brain. J Neurosci. 1995;15(11):7442–7451. doi: 10.1523/JNEUROSCI.15-11-07442.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Davis AA, Fritz JJ, Wess J, Lah JJ, Levey AI. Deletion of M1 muscarinic acetylcholine receptors increases amyloid pathology in vitro and in vivo. J Neurosci. 2010;30(12):4190–4196. doi: 10.1523/JNEUROSCI.6393-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Davis AA, Heilman CJ, Brady AE, Miller NR, Fuerstenau-Sharp M, Hanson BJ, Lindsley CW, Conn PJ, Lah JJ, Levey AI. Differential effects of allosteric M1 muscarinic acetylcholine receptor agonists on receptor activation, arrestin 3 recruitment, and receptor downregulation. ACS Chem Neurosci. 2010;1(8):542–551. doi: 10.1021/cn100011e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Thompson S, Lanctôt KL, Herrmann N. The benefits and risks associated with cholinesterase inhibitor therapy in Alzheimer’s disease. Expert Opin Drug Saf. 2004;3(5):425–440. doi: 10.1517/14740338.3.5.425. [DOI] [PubMed] [Google Scholar]
- 24.Mangialasche F, Solomon A, Winblad B, Mecocci P, Kivipelto M. Alzheimer’s disease: clinical trials and drug development. Lancet Neurol. 2010;9(7):702–716. doi: 10.1016/S1474-4422(10)70119-8. [DOI] [PubMed] [Google Scholar]
- 25.Levey AI, Kitt CA, Simonds WF, Price DL, Brann MR. Identification and localization of muscarinic acetylcholine receptor proteins in brain with subtypespecific antibodies. J Neurosci. 1991;11:3218–3226. doi: 10.1523/JNEUROSCI.11-10-03218.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Levey AI, Edmunds SM, Heilman CJ, Desmond TJ, Frey KA. Localization of muscarinic M3 receptor protein and M3 receptor binding in rat brain. Neuroscience. 1994;63:207–221. doi: 10.1016/0306-4522(94)90017-5. [DOI] [PubMed] [Google Scholar]
- 27.Levey AI, Edmunds SM, Koliatsos V, Wiley RG, Heilman CJ. Expression of m1-m4 muscarinic acetylcholine receptor proteins in rat hippocampus and regulation by cholinergic innervation. J Neurosci. 1995;15:4077–4092. doi: 10.1523/JNEUROSCI.15-05-04077.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Levey AI. Muscarinic acetylcholine receptor expression in memory circuits: implications for treatment of Alzheimer disease. Proc Natl Acad Sci U S A. 1996;93(24):13541–13546. doi: 10.1073/pnas.93.24.13541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rouse ST, Levey AI. Expression of m1-m4 muscarinic acetylcholine receptor immunoreactivity in septohippocampal neurons and other identified hippocampal afferents. J Comp Neurol. 1996;375(3):406–416. doi: 10.1002/(SICI)1096-9861(19961118)375:3<406::AID-CNE5>3.0.CO;2-6. [DOI] [PubMed] [Google Scholar]
- 30.Wess J. Molecular biology of muscarinic acetylcholine receptors. Crit Rev Neurobiol. 1996;10(1):69. doi: 10.1615/critrevneurobiol.v10.i1.40. [DOI] [PubMed] [Google Scholar]
- 31.Wess J. Muscarinic Acetylcholine Receptor Knockout Mice: Novel Phenotypes and Clinical Implications. Annu Rev Pharmacol Toxicol. 2004;44:423–450. doi: 10.1146/annurev.pharmtox.44.101802.121622. [DOI] [PubMed] [Google Scholar]
- 32.Wess J, Eglen RM, Gautam D. Muscarinic acetylcholine receptors: mutant mice provide new insights for drug development. Nat Rev Drug Discovery. 2007;6(9):721–733. doi: 10.1038/nrd2379. [DOI] [PubMed] [Google Scholar]
- 33.Anagnostaras SG, Murphy GG, Hamilton SE, Mitchell SL, Rahnama NP, Nathanson NM, Silva AJ. Selective cognitive dysfunction in acetylcholine M1 muscarinic receptor mutant mice. Nat Neurosci. 2002;6(1):51–58. doi: 10.1038/nn992. [DOI] [PubMed] [Google Scholar]
- 34.Ma L, Seager MA, Wittmann M, Jacobson M, Bickel D, Burno M, Jones K, Kuzmick Graufelds V, Xu G, Pearson M, McCampbell A, Gaspar R, Shughrue P, Danziger A, Regan C, Flick R, Pascarella D, Garson S, Doran S, Kreatsoulas C, Veng L, Lindsley CW, Shipe W, Kuduk S, Sur C, Kinney G, Seabrook GR, Ray WJ. Selective activation of the M1 muscarinic acetylcholine receptor achieved by allosteric potentiation. Proc Natl Acad Sci U S A. 2009;106(37):15950–15955. doi: 10.1073/pnas.0900903106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Jones CK, Byun N, Bubser M. Muscarinic and nicotinic acetylcholine receptor agonists and allosteric modulators for the treatment of schizophrenia. Neuropsychopharmacology. 2012;37(1):16–42. doi: 10.1038/npp.2011.199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lebois EP, Bridges TM, Lewis LM, Dawson ES, Kane AS, Xiang Z, Jadhav SB, Yin H, Kennedy JP, Meiler J, Niswender CM, Jones CK, Conn PJ, Weaver CD, Lindsley CW. Discovery and characterization of novel subtype-selective allosteric agonists for the investigation of M1 receptor function in the central nervous system. ACS Chem Neurosci. 2010;1(2):104–121. doi: 10.1021/cn900003h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lebois EP, Digby GJ, Sheffler DJ, Melancon BJ, Tarr JC, Cho HP, Miller NR, Morrison R, Bridges TM, Xiang Z, Daniels JS, Wood MR, Conn PJ, Lindsley CW. Development of a highly selective, orally bioavailable and CNS penetrant M1 agonist derived from the MLPCN probe ML071. Bioorg Med Chem Lett. 2011;21(21):6451–6455. doi: 10.1016/j.bmcl.2011.08.084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Digby GJ, Noetzel M, Bubser MJ, Utley TJ, Walker AG, Byun NE, Lebois EP, Xiang Z, Sheffler DJ, Cho HP, Davis AA, Nemirovsky NE, Mennenga SE, Camp BW, Bimonte-Nelson HA, Bode J, Italiano K, Morrison R, Daniels JS, Niswender CM, Olive MF, Lindsley CW, Jones CK, Conn PJ. Novel allosteric agonists of M1 muscarinic acetylcholine receptors induce brain region-specific responses that correspond with behavioral effects in animal models. J Neurosci. 2012;32(25):8532–8544. doi: 10.1523/JNEUROSCI.0337-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Caccamo A, Oddo S, Billings LM, Green KN, Martinez-Coria H, Fisher A, LaFerla FM. M1 receptors play a central role in modulating AD-like pathology in transgenic mice. Neuron. 2006;49(5):671–682. doi: 10.1016/j.neuron.2006.01.020. [DOI] [PubMed] [Google Scholar]
- 40.Fisher A. M1Muscarinic Receptor Agonists Target Major Hallmarks of Alzheimer’s Disease - an Update. Curr Alzheimer Res. 2007;4(5):577–580. doi: 10.2174/156720507783018163. [DOI] [PubMed] [Google Scholar]
- 41.Kuo YM, Emmerling MR, Vigo-Pelfrey C, Kasunic TC, Kirkpatrick JB, Murdoch GH, Ball MJ, Roher AE. Water-soluble Abeta (N-40, N-42) oligomers in normal and Alzheimer disease brains. J Biol Chem. 1996;271(8):4077–81. doi: 10.1074/jbc.271.8.4077. [DOI] [PubMed] [Google Scholar]
- 42.Kostylev MA, Kaufman AC, Nygaard HB, Patel P, Haas LT, Gunther EC, Vortmeyer A, Strittmatter SM. Prion-Protein-Interacting Amyloid-β Oligomers of High Molecular Weight Are Tightly Correlated with Memory Impairment in Multiple Alzheimer Mouse Models. J Biol Chem. 2015;290:17415–38. doi: 10.1074/jbc.M115.643577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lue LF, Kuo YM, Roher AE, Brachova L, Shen Y, Sue L, Beach T, Kurth JH, Rydel RE, Rogers J. Soluble amyloid beta peptide concentration as a predictor of synaptic change in Alzheimer’s disease. Am J Pathol. 1999;155(3):853–62. doi: 10.1016/s0002-9440(10)65184-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.McLean CA, Cherny RA, Fraser FW, Fuller SJ, Smith MJ, Beyreuther K, Bush AI, Masters CL. Soluble pool of Abeta amyloid as a determinant of severity of neurodegeneration in Alzheimer’s disease. Ann Neurol. 1999;46(6):860–6. doi: 10.1002/1531-8249(199912)46:6<860::aid-ana8>3.0.co;2-m. [DOI] [PubMed] [Google Scholar]
- 45.Naslund J, Haroutunian V, Mohs R, Davis KL, Davies P, Greengard P, Buxbaum JD. Correlation between elevated levels of amyloid beta-peptide in the brain and cognitive decline. JAMA. 2000;283(12):1571–7. doi: 10.1001/jama.283.12.1571. [DOI] [PubMed] [Google Scholar]
- 46.Walsh DM, Klyubin I, Fadeeva JV, Cullen WK, Anwyl R, Wolfe MS, Rowan MJ, Selkoe DJ. Naturally secreted oligomers of amyloid beta protein potently inhibit hippocampal long-term potentiation in vivo. Nature. 2002;416(6880):535–9. doi: 10.1038/416535a. [DOI] [PubMed] [Google Scholar]
- 47.Bitan G, Kirkitadze MD, Lomakin A, Vollers SS, Benedek GB, Teplow DB. Amyloid β-protein (Aβ) assembly: Aβ40 and Aβ42 oligomerize through distinct pathways. Proc Natl Acad Sci U S A. 2003;100(1):330–335. doi: 10.1073/pnas.222681699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Oddo S, Caccamo A, Kitazawa M, Tseng BP, LaFerla FM. Amyloid deposition precedes tangle formation in a triple transgenic model of Alzheimer’s disease. Neurobiol Aging. 2003;24(8):1063–1070. doi: 10.1016/j.neurobiolaging.2003.08.012. [DOI] [PubMed] [Google Scholar]
- 49.Takahashi RH, Almeida CG, Kearney PF, Yu F, Lin MT, Milner TA, Gouras GK. Oligomerization of Alzheimer’s beta-amyloid within processes and synapses of cultured neurons and brain. J Neurosci. 2004;24(14):3592–9. doi: 10.1523/JNEUROSCI.5167-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Billings LM, Oddo S, Green KN, McGaugh JL, LaFerla FM. Intraneuronal Aβ causes the onset of early Alzheimer’s disease-related cognitive deficits in transgenic mice. Neuron. 2005;45(5):675–688. doi: 10.1016/j.neuron.2005.01.040. [DOI] [PubMed] [Google Scholar]
- 51.Glabe CC. Amyloid accumulation and pathogensis of Alzheimer’s disease: significance of monomeric, oligomeric and fibrillar Abeta. Subcell Biochem. 2005;38:167–77. doi: 10.1007/0-387-23226-5_8. [DOI] [PubMed] [Google Scholar]
- 52.Walsh DM, Klyubin I, Shankar GM, Townsend M, Fadeeva JV, Betts V, Podlisny MB, Cleary JP, Ashe KH, Rowan MJ, Selkoe DJ. The role of cell-derived oligomers of Abeta in Alzheimer’s disease and avenues for therapeutic intervention. Biochem Soc Trans. 2005;33(5):1087–90. doi: 10.1042/BST20051087. [DOI] [PubMed] [Google Scholar]
- 53.Walsh DM, Townsend M, Podlisny MB, Shankar GM, Fadeeva JV, El Agnaf O, Hartley DM, Selkoe DJ. Certain inhibitors of synthetic amyloid betapeptide (Abeta) fibrillogenesis block oligomerization of natural Abeta and thereby rescue long-term potentiation. J Neurosci. 2005;25(10):2455–62. doi: 10.1523/JNEUROSCI.4391-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Haass C, Selkoe DJ. Soluble protein oligomers in neurodegeneration: lessons from the Alzheimer’s amyloid β-peptide. Nat Rev Mol Cell Biol. 2007;8(2):101–112. doi: 10.1038/nrm2101. [DOI] [PubMed] [Google Scholar]
- 55.Li S, Hong S, Shepardson NE, Walsh DM, Shankar GM, Selkoe D. Soluble oligomers of amyloid β protein facilitate hippocampal long-term depression by disrupting neuronal glutamate uptake. Neuron. 2009;62(6):788–801. doi: 10.1016/j.neuron.2009.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Sperling RA, LaViolette PS, O’Keefe K, O’Brien J, Rentz DM, Pihlajamaki M, Marshall G, Hyman BT, Selkoe DJ, Hedden T, Buckner RL, Becker JA, Johnson KA. Amyloid deposition is associated with impaired default network function in older persons without dementia. Neuron. 2009;63(2):178–188. doi: 10.1016/j.neuron.2009.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Oakley H, Cole SL, Logan S, Maus E, Shao P, Craft J, Guillozet-Bongaarts A, Ohno M, Disterhoft J, Van Eldik L, Berry R, Vassar R. Intraneuronal β-amyloid aggregates, neurodegeneration, and neuron loss in transgenic mice with five familial Alzheimer’s disease mutations: potential factors in amyloid plaque formation. J Neurosci. 2006;26(40):10129–10140. doi: 10.1523/JNEUROSCI.1202-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Eimer WA, Vassar R. Neuron loss in the 5XFAD mouse model of Alzheimer’s disease correlates with intraneuronal Aβ42 accumulation and Caspase-3 activation. Mol Neurodegener. 2013;8(2):2–12. doi: 10.1186/1750-1326-8-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Esparza TJ, Zhao H, Cirrito JR, Cairns NJ, Bateman RJ, Holtzman DM, Brody DL. Amyloid-beta oligomerization in Alzheimer dementia versus high-pathology controls. Ann Neurol. 2013;73(1):104–119. doi: 10.1002/ana.23748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Moser BK, Stevens GR. Homogeneity of Variance in the Two-Sample Means Test. Am Stat. 1992;46(1):19–21. [Google Scholar]
- 61.Ruxton GD. The unequal variance t-test is an underused alternative to Student’s t-test and the Mann-Whitney U test. Behav Ecology. 2006;17(4):688–690. [Google Scholar]
- 62.Leys C, Ley C, Klein O, Bernard P, Licata L. Detecting outliers: Do not use standard deviation around the mean, use absolute deviation around the median. J Exp Soc Psych. 2013;49(4):764–766. [Google Scholar]
- 63.Zhang Z, Liu X, Schroeder JP, Chan CB, Song M, Yu SP, Weinshenker D, Ye K. 7,8-Dihydroxyflavone Prevents Synaptic Loss and Memory Deficits in a Mouse Model of Alzheimer’s Disease. Neuropsychopharmacology. 2014;39:638–50. doi: 10.1038/npp.2013.243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Langmead CJ, Austin NE, Branch CL, Brown JT, Buchanan KA, Davies CH, Forbes IT, Fry VA, Hagan JJ, Herdon HJ, Jones GA, Jeggo R, Kew JNC, Mazzali A, Melarange R, Patel N, Pardoe J, Randall AD, Roberts C, Roopun A, Starr KR, Teriakidis A, Wood MD, Whittington M, Wu Z, Watson J. Characterization of a CNS penetrant, selective M1 muscarinic receptor agonist, 77-LH-28-1. Br J Pharmacol. 2008;154(5):1104–1115. doi: 10.1038/bjp.2008.152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Jones CK, Brady AE, Davis AA, Xiang Z, Bubser M, Tantawy MN, Kane AS, Bridges TM, Kennedy JP, Bradley SR, Peterson TE, Sib Ansari M, Baldwin RM, Kessler RM, Deutch AY, Lah JJ, Levey AI, Lindsley CW, Conn PJ. Novel selective allosteric activator of the M1 muscarinic acetylcholine receptor regulates amyloid processing and produces antipsychotic-like activity in rats. J Neurosci. 2008;28(41):10422–10433. doi: 10.1523/JNEUROSCI.1850-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Gilmartin MR, Balderston NL, Helmstetter FJ. Prefrontal cortical regulation of fear learning. Trends Neurosci. 2014;37(8):455–464. doi: 10.1016/j.tins.2014.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Thomas RL, Langmead CJ, Wood MD, Challiss RJ. Contrasting effects of allosteric and orthosteric agonists on m1 muscarinic acetylcholine receptor internalization and down-regulation. J Pharmacol Exp Ther. 2009;331(3):1086–1095. doi: 10.1124/jpet.109.160242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Avlani VA, Langmead CJ, Guida E, Wood MD, Tehan BG, Herdon HJ, Watson JM, Sexton PM, Christopoulos A. Orthosteric and allosteric modes of interaction of novel selective agonists of the M1 muscarinic acetylcholine receptor. Mol Pharm. 2010;78(1):94–104. doi: 10.1124/mol.110.064345. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.