

Abstract
Kinetoplastid-based infections are neglected diseases that represent a significant human health issue. Chemotherapeutic options are limited due to toxicity, parasite susceptibility, and poor patient compliance. In response, we studied a molecular target-directed approach involving intervention of hexokinase activity – a pivotal enzyme in parasite metabolism. A benzamidobenzoic acid hit with modest biochemical inhibition of T. brucei hexokinase 1 (TbHK1, IC50 = 9.1 μM), low mammalian cytotoxicity (IMR-90, EC50 > 25 μM), and no appreciable activity on whole BSF parasites was optimized to afford probe 4f with improved TbHK1 potency and, significantly, efficacy against whole BSF parasites (TbHK1, IC50 = 0.28 μM, BSF LD50 = 1.9 μM). Compound 4f and analogs also inhibited the hexokinase enzyme from Leishmania major (LmHK1), albeit with less potency compared to TbHK1, suggesting that inhibition of the glycolytic pathway may be a promising opportunity to target multiple, disease-causing trypanosomatid protozoa.
Keywords: trypanosomes, Leishmania, anti-parasitic, benzamidobenzamidines, sleeping sickness
TOC image
Power play: The discovery of novel, target-based, anti-parasitic agents is necessary to address the dearth of therapeutic options for neglected diseases such as sleeping sickness and Leishmaniasis. Inhibitors of hexokinase 1, a key metabolic enzyme in these parasites, have been identified and explored as a means to interrupting glucose metabolism on which these parasites rely. Low micromolar efficacy has been observed in whole blood-stream form trypanosomes, suggesting that this strategy useful in targeting glucose-dependent parasites.
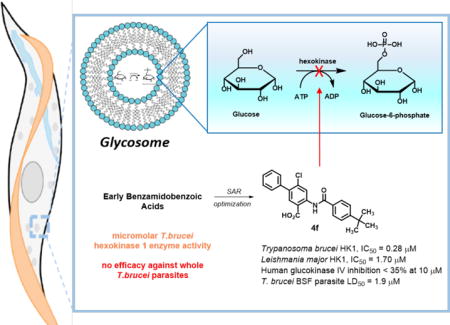
Introduction
Several parasitic species of kinetoplastid protozoa cause significant human disease. Notable examples include human African trypanosomiasis1 – otherwise known as HAT or sleeping sickness, Chagas disease,2 and leishmaniasis3 (caused respectively by Trypanosoma brucei spp., Trypanosoma cruzi, and over 20 species of Leishmania). These diseases are commonly transmitted by infected insects and progress to a debilitating phase in patients that, without intervention, is often fatal. Therapies have been slow to emerge as these are neglected diseases endemic to geographically poor regions of the world that have difficulty attracting therapeutic investment. Annual at-risk populations for HAT and leishmaniasis top 70 and 310 million people, respectively.4,5 While treatments have improved beyond agents that are limited by availability, cost, toxicity, resistance, and poor patient compliance, newer therapies still suffer from subspecies or disease-stage specificity, emerging resistance, or result in adverse reactions or refractory infection.6–7
In an effort to identify mechanistically novel antiparasitic agents, we focused on targeting enzymes within the trypanosomatid glycolytic pathway of these parasites. Unlike non-kinetoplastid organisms that execute glycolysis in the cytosol, a substantial segment of glucose metabolism in T. brucei and Leishmania spp. parasites is uniquely compartmentalized within a peroxisomal organelle called a glycosome. Hexokinase, the first enzyme in the sequence that converts glucose to glucose-6-phosphate, has been shown to be critical for the survival of T. brucei bloodsteam form (BSF) parasites8–10 and Leishmania promastigote parasite infection of macrophages.11,12 Both promastigote and amastigote stages of cultured Leishmania parasites preferentially use glucose in vitro, and Leishmania hexokinase has been shown to be important in the pathogenic intracellular amastigote stage even though leishmania amastigotes also partially utilize gluconeogenesis to derive nutrients.12,13 Consequently, we sought to exploit the metabolic dependence of these kinetoplastids on glycolysis to investigate new chemical starting points for potential chemotherapy development.
The genomes of T. brucei and most strains of Leishmania harbor two nearly-identical hexokinases.14–15 In T. brucei, these two hexokinases, T. brucei hexokinase 1 and 2 (TbHK1 and TbHK2, respectively) are 98% identical. L. major, a causative agent for human cutaneous leishmaniasis, also expresses two HKs, LmHK1 and LmHK2, which are >99% identical to each other, differing by a single residue. Cross-species variation can range more dramatically. For instance, TbHK1 shares 61% and sequence identity with LmHK1.15 There are four mammalian hexokinase isozymes, HK I-IV, which share limited (30–33%) sequence identity with TbHK1. Further, compared to mammalian hexokinase enzymes, the parasite enzymes have unusual biochemical properties (oligomerization into hexamers, for example16) suggesting that the development of selective trypanosomatid hexokinase inhibitors is feasible. While the study of T. brucei hexokinase 2 is complicated by the fact that recombinant TbHK2 (rTbHK2) lacks enzymatic activity,14 recombinant T. brucei hexokinase 1 (rTBHK1) is a functional enzyme that is well-suited for a high throughput platform. As such, a high throughput screen of 220,223 compounds from the Molecular Libraries Small Molecule Repository (MLSMR) was performed to find inhibitors of rTbHK1.17–18 This screening campaign revealed hit compound 4-methoxybenzamidochlorobenzoic acid 1 (Fig. 1) which was the subject of an optimization effort aimed at improving rTbHK1 potency to submicromolar levels, while assessing cytotoxicity in a mammalian IMR-90 cell line and liability against a related human hexokinase, glucokinase (hGlk, HK IV).17–18
Figure 1.
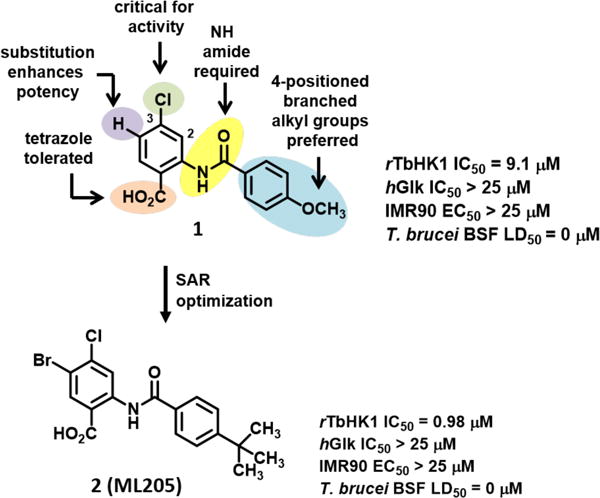
Overview of SAR study of hit 1 leading to probe 2 (ML205)
A rTbHK1 enzymatic assay18 was used to drive structure-activity relationship (SAR) optimization activities, along with a glucose-6-phosphate dehydrogenase counter-screen to triage false positives that interfered with the reporter enzyme in the primary assay coupled reaction. Our SAR evaluation of hit 1 surveyed several architectural regions (shaded areas, 1, Fig. 1) and delivered compound 2, designated ML205, which demonstrated a rTbHK1 IC50 = 0.98 μM, limited hGlk activity (IC50 = 48.3 μM), and no discernable mammalian toxicity in IMR90 cells (EC50 > 25 μM). Notably, ML205 was determined to be a mixed inhibitor of rTbHK1 with respect to ATP with a Ki of 0.70 μM. As typical kinase inhibitors are competitive ATP binding substrates, ML205 represented a novel, allosteric modulator of rTbHk1.
While ML205 was a useful proof-of-concept biochemical tool, nothing was known of its ADME characteristics or general promiscuity against other mammalian targets, and significantly, it lacked efficacy against T. brucei BSF parasites (6.9% growth inhibition at 10 μM). We hypothesized that the latter issue might be a result of inadequate permeability, thereby preventing the compounds from reaching the hexokinase within the glycosome. The carboxylic acid moiety was recognized as a limiting feature in this regard, though we knew from our primary SAR efforts that it was also critical for rTbHK1 potency. We found the only marginally acceptable surrogate for the carboxylic acid group was an N-H tetrazole. This particular analog of 2 showed rTbHK1 IC50 = 5.2 μM and a 40% inhibition of BSF parasites at 10 μM compound concentration. With these issues in mind, we embarked on an effort to derive a more suitable probe with efficacy against T. brucei parasites and then determine if the hexokinase 1 enzyme of Leishmania major was also inhibited by these same compounds. With respect to T. brucei, it was anticipated that a second generation prototype was within reach that (a) demonstrated a rTbHK1 IC50 < 500 nM with > 90% efficacy, (b) inhibited growth of T. brucei parasites with a BSF LD50 < 10 μM, and (c) showed limited cytotoxicity and liability against human glucokinase. Additionally, it was also desirable to assess lead compounds against Leishmania hexokinase 1 and better characterize the structural class in terms of its potential off-target effects and ADME profile.
Results and Discussion
In an effort to engineer improved TbHK1 potency and BSF growth inhibition within this chemical series, we explored compounds bearing different substituents in place of the C4 bromide of compound 2. Generally, C4-substituted analogs were prepared in 2–5 overall steps (Scheme 1). Commercially available starting materials 3a-c were N-acylated using 4-tert-butylbenzoyl chloride, followed by ester hydrolysis to afford 4a-c. From commercial bromide 3d, a modified Suzuki-Miyaura cross-coupling19–21 with alkyltrifluoroborate salts or arylboronic acids was employed to deliver C4-derivatized intermediates which were similarly N-acylated and hydrolyzed to deliver benzamidobenzoic acid derivatives 4d-k, 4n-z and 4bb. Synthesis of dimethylaniline analogs 4l and 4m required two additional steps involving a nitro group reduction, followed by reductive amination. Reordering the acylation and cross-coupling steps afforded an expedient preparation of nitrogen-linked heterocyclic derivatives22 4aa, 4cc and 4dd from iodide 3e. In these cases, ester hydrolysis occurred concomitantly with aryl coupling. The NH-tetrazole 4ee was generated from cyanation of 3e, followed by a click reaction. By N-methylating the tetrazole prior to ester hydrolysis, 2-N-methylated-tetrazole 4ff was obtained.
Scheme 1.
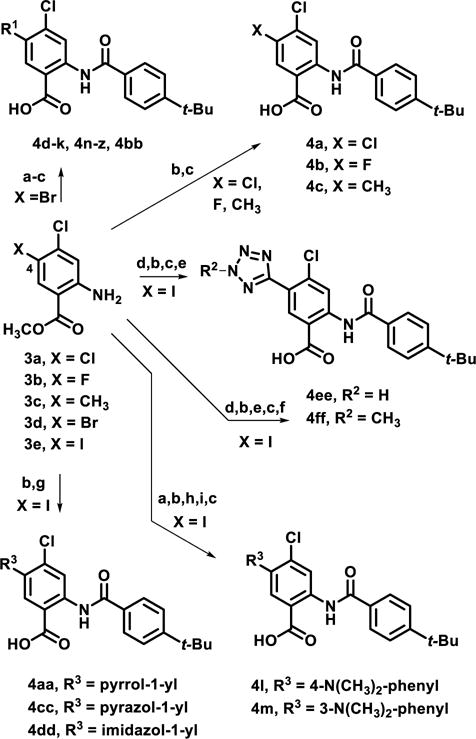
Synthesis of second generation, C4-derivatized analogs
Reagents and conditions: (a) coupling conditions, 21-92%; (b) 4-tert-butylbenzoyl chloride, CH3CN, 150°C, MW, 1 h, 25-95%; (c) LiOH, THF:H2O (1.5:1), rt, 18 h, 15-93%; (d) CuCN, DMF, 120 °C, 6 h, 78%; (e) TMSN3, CuI, DMF, MeOH, 18 h, 90 °C, 36%; (f) TMS-diazomethane, CH2Cl2:MeOH (1:1), rt, 2 h, 56-74%; (g) Cu powder, pyrrole or pyrazole, or imidazole, Cs2CO3, 150 °C, CH3CN, 27-58%; (h) Raney Ni, NaBH4, 0°C to rt, 17 h, 91%; (i) paraformaldehyde, NaCNBH3, rt, 18 h, 53%.
Compounds showing > 70% BSF growth inhibition at a compound concentration of 10 μM were evaluated in dose response format to help drive the optimization process. Analogs were tested for hGlk inhibition and IMR90 cell toxicity in parallel. Halide derivatives 4a–b17 did not appreciably alter the rTbHK1 potency compared to bromide 2 (Table 1). While bromide replacement with a methyl group was marginally tolerated, ethyl and cyclopropyl groups resulted in a nearly 7-fold loss in rTbHK1 potency compared to 2 (see 4c-4e). However, we were delighted to discover that the incorporation of a phenyl ring at the C4 position afforded analog 4f with a 3.5-fold improvement in rTbHK1 potency, high BSF growth inhibition efficacy (95%), and a BSF LD50 = 1.9 μM (entry 7, Table 1). Substituted phenyl derivatives were explored, generally revealing rTbHK1 potency ranging from 1.7 – 4.6 μM and BSF LD50 values of 1.9 – 3.2 μM for most analogs (entries 8-18). A notable exception included the 3-methoxyphenyl analog 4j which showed an rTbHK1 IC50 = 0.88 μM and BSF LD50 = 1.5 μM. Inhibition of human glucokinase for the set of compounds was below 50%, and mammalian cytotoxicity was not observed (> 25 μM).
Table 1.
SAR Summary of TbHK1, LmHK1 and whole parasite T. brucei BSF data for C4 analogs of 2
| entry | cmpd |
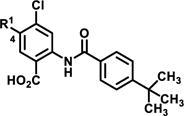
|
rTbHK1 potency (IC50 ± SEM, μM)[a] | rLmHK1 potency (IC50 ± SEM, μM)[a] | % T. brucei BSF growth inhibition (10 μM)[a] | T. brucei BSF potency (LD50 ± SEM, μM)[a] | % hGlk inhibition (10 μM)[a] | IMR90 toxicity (EC50, μM)[a] | cLogP[b] |
|---|---|---|---|---|---|---|---|---|---|
| R1 | |||||||||
| 1 | 2 | Br | 0.98 ± 0.07 | 1.10 ± 0.07 | 6.9 ± 3.0 | ND | 11.9 ± 7.2 | > 25.0 | 6.7 |
| 2 | 4a | Cl | 1.36 ± 0.06 | ND | 35.0 ± 11.0 | ND | BLD | > 25.0 | 6.6 |
| 3 | 4b | F | 2.79 ± 0.10 | ND | 32.0 ± 11.0 | ND | BLD | > 25.0 | 6.5 |
| 4 | 4c | CH3 | 2.70 ± 0.18 | ND | 97.5 ± 0.6 | 5.2 ± 1.8 | 12.5 ± 4.8 | > 25.0 | 6.5 |
| 5 | 4d | CH2CH3 | 7.40 ± 0.15 | ND | BLD | ND | 0.7 ± 1.4 | > 25.0 | 7.0 |
| 6 | 4e | cyclopropyl | 6.70 ± 0.38 | ND | 43.4 ± 16.8 | ND | BLD | > 25.0 | 6.9 |
| 7 | 4f | phenyl | 0.28 ± 0.002 | 1.70 ± 0.11 | 95.4 ± 0.8 | 1.9 ± 0.7 | 35.2 ± 8.3 | > 25.0 | 7.6 |
| 8 | 4g | 4-CH3-phenyl | 2.78 ± 0.03 | ND | 81.4 ± 5.6 | 3.2 ± 1.6 | 15.7 ± 3.0 | > 25.0 | 8.1 |
| 9 | 4h | 2-CH3-phenyl | ND | ND | 65.2 ± 0.2 | ND | 16.6 ± 2.3 | > 25.0 | 8.1 |
| 10 | 4i | 4-CH3O-phenyl | 1.72 ± 0.09 | ND | 82.5 ± 7.1 | 3.1 ± 0.2 | 22.2 ± 1.6 | > 25.0 | 7.5 |
| 11 | 4j | 3- CH3O-phenyl | 0.88 ± 0.01 | ND | 92.2 ± 2.9 | 1.5 ± 0.3 | 13.8 ± 1.9 | > 25.0 | 7.5 |
| 12 | 4k | 2- CH3O-phenyl | 4.60 ± 0.32 | ND | 90.7 ± 0.9 | 2.0 ± 0.2 | 18.8 ± 2.4 | > 25.0 | 7.5 |
| 13 | 4l | 4-N(CH3)2-phenyl | > 10.0 | ND | 58.0 ± 4.0 | 6.0 ± 0.2 | 27.8 ± 1.7 | > 25.0 | 7.8 |
| 14 | 4m | 3-N(CH3)2-phenyl | 4.00 ± 0.32 | 0.60 ± 0.09 | 82.8 ± 0.2 | 1.2 ± 0.1 | 43.7 ± 4.1 | > 25.0 | 7.8 |
| 15 | 4n | 4-F-phenyl | 2.44 ± 0.08 | ND | 76.7 ± 10.4 | 3.1 ± 0.7 | 15.1 ± 2.6 | > 25.0 | 7.8 |
| 16 | 4o | 3-F-phenyl | 4.60 ± 0.32 | ND | 93.7 ± 2.6 | 1.8 ± 0.3 | 16.1 ± 8.7 | > 25.0 | 7.8 |
| 17 | 4p | 2-F-phenyl | 2.08 ± 0.07 | ND | 92.8 ± 2.0 | 5.2 ± 0.9 | 24.5 ± 2.5 | > 25.0 | 7.8 |
| 18 | 4q | 3-Cl-phenyl | 2.70 ± 0.05 | 1.70 ± 0.22 | 90.7 ± 1.8 | 1.1 ± 0.2 | 30.9 ± 3.5 | > 25.0 | 8.3 |
| 19 | 4r | 4-CO2H-phenyl | 3.50 ± 0.33 | ND | 51.0 ± 14.3 | ND | 3.5 ± 7.1 | > 25.0 | 7.4 |
| 20 | 4s | pyridin-4-yl | > 10[c] | ND | 99.7 ± 1.4 | 3.5 ± 0.6 | 15.1 ± 3.2 | > 25.0 | 6.2 |
| 21 | 4t | pyridin-3-yl | ND[c] | ND | 57.7 ± 14.2 | ND | 16.7 ± 1.2 | > 25.0 | 6.2 |
| 22 | 4u | thiophene-2-yl | 0.47 ± 0.15 | 1.91 ± 0.04 | 69.1 ± 5.8 | ND | 12.5 ± 1.3 | > 25.0 | 7.5 |
| 23 | 4v | thiophen-3-yl | 0.33 ± 0.03 | 1.71 ± 0.03 | 68.5 ± 2.1 | ND | 24.8 ± 1.0 | > 25.0 | 7.5 |
| 24 | 4w | furan-2-yl | 0.42 ± 0.02 | 1.64 ± 0.22 | 59.0 ± 8.2 | > 10.0 | 10.4 ± 2.1 | > 25.0 | 7.0 |
| 25 | 4x | furan-3-yl | 0.51 ± 0.06 | ND | 73.7 ± 3.7 | 4.2 ± 0.4 | 3.1 ± 2.8 | > 25.0 | 7.0 |
| 26 | 4y | dihydrobenzo[1,4]dioxin-6-yl | 0.60 ± 0.09 | 0.61 ± 0.09 | 98.0 ± 1.2 | 3.3 ± 0.5 | 26.6 ± 0.4 | > 25.0 | 7.5 |
| 27 | 4z | benzo[1,3]dioxol-5-yl | 3.9 ± 0.1 | ND | 89.8 ± 1.1 | 3.0 ± 0.3 | 23.9 ± 2.5 | > 25.0 | 7.6 |
| 28 | 4aa | 1H-pyrrol-1-yl | 3.0 ± 0.2 | ND | 73.6 ± 1.0 | 6.9 ± 2.1 | 15.8 ± 3.0 | > 25.0 | 7.2 |
| 29 | 4bb | 1H-pyrrol-2-yl | 0.50 ± 0.05 | 2.06 ± 0.04 | 57.0 ± 7.1 | 6.6 ± 1.6 | 6.1 ± 0.1 | > 25.0 | 7.3 |
| 30 | 4cc | 1H-pyrazol-1-yl | 2.8 ± 0.2 | ND | 22.5 ± 12.3 | > 10.0 | 8.7 ± 4.4 | > 25.0 | 6.2 |
| 31 | 4dd | 1H-imidazol-1-yl | > 10.0 | ND | 16.4 ± 13.3 | ND | 1.7 ± 3.2 | > 25.0 | 5.9 |
| 32 | 4ee | 1H-tetrazol-5-yl | 0.14 ± 0.002 | 0.94 ± 0.16 | BLD | > 10.0 | 1.6 ± 0.5 | > 25.0 | 5.2 |
| 33 | 4ff | 2-CH3-2H-tetrazol-5-yl | > 10 | ND | BLD | ND | 14.0 ± 4.1 | > 25.0 | 5.5 |
Data were an average of n ≥ 3 experiments.
Data were calculated using CambridgeSoft ChemBioDraw Ultra 12.0.
Compound showed poor solubility which obscured analysis in this assay. ND = not determined. BLD = below level of detection.
Analogs were also examined that contained a heterocycle at the C4 position of 2 (Table 1, entries 20-33). Pyridine compounds 4st suffered from poor aqueous solubility that obscured their enzymatic potency. Thiophenyl and furyl derivatives 4u-x were more potent rTbHK1 inhibitors than bromide 2; however, BSF inhibition at 10 μM ranged from 59 – 74%. In the case ofdihydrobenzodioxine derivative 4y, high percent inhibition of BSF growth and submicromolar rTbHK1 potency was achieved; however, as with many of the analogs in this series, potency against the BSF parasite remained in the low micromolar range (entry 26, Table 1). To assess compounds with a different range of cLogP and hydrogen bonding capability, several analogs bearing a C4-positioned, 5-membered, nitrogen-containing, heterocyclic ring were also prepared (entries 28-33, Table 1). For this subset, the presence of an N-H functionality led to better rTbHK1 activity, culminating in the most potent rTbHk1 inhibitor, NH-tetrazole 4ee, with an IC50 = 140 nM. Despite the improvement in rTbHK1 potency, a corresponding robust inhibitory effect was not observed in BSF parasites, likely due to permeability limitations. The importance of the N-H tetrazole to rTbHK1 potency was underscored by the assessment of 2-N-methylated tetrazole 4ff which, along with the 1-N-methylated isomer (data not shown), lacked enzymatic potency and failed to inhibit BSF parasite growth. While we reasoned that a prodrug approach may facilitate compound transport, methyl esters of carboxylic acids 4f and 4ee and an acetoxymethylated tetrazole derivative of 4ee did not result in activity in either of our T. brucei focused assays (data not shown). Nonetheless, this effort revealed a number of compounds with submicromolar TbHK1 enzymatic activity that were worthy of assessment against Leishmania major hexokinase 1.
Study of Leishmania major hexokinase 1
To determine if hexokinases of other kinetoplastids would be inhibited by benzamidobenzamides engineered against T. brucei, we expressed, purified, and characterized recombinant Leishmania major hexokinase 1 (rLmHK1, see supporting information). Following characterization, we then assessed a subset of the compounds in hand against rLmHK1 (Table 1). Compounds showed inhibition against HK1 from both parasites, although generally, potency against rTbHK1 was better than that observed against rLmHK1. For the subset of compounds evaluated, a consistent trend was not observed between the two enzymes, although sub-to low micromolar potency was obtained for all of the tested agents on both enzymes, supporting our hypothesis that multiple members of the kinetoplastid family may be susceptible to hexokinase 1 inhibition if a suitable strategy can be employed to successfully deliver and/or retain these or other compounds to the glycosomal target.
In vitro ADME characterization of compound 4f
To benchmark ADME parameters against which future compounds might be compared, aqueous solubility, PAMPA permeability, plasma and microsomal stability, and plasma protein binding was determined for compound 4f as this was the first analog in the structural series to be distinguished by submicromolar rTbHK1 potency and efficacy against BSF parasites. If analogous parameters were available for the initial hit compound 1 and the prototype bromide 2, then they are provided (Table 2). Notably, the lipophilicity of the analogs derived from hit 1 increased, culminating in cLogP values in the 5-7 range. Accordingly, observing a high level of protein binding for 4f (cLogP = 7.6) was not surprising. Permeability, as determined by an in vitro PAMPA assay, reflected that permeability was poor due to passive transport at pH levels of 5.0 and 6.7 while moderate permeability was observed at pH 7.4. Consistent with this overall profile, the solubility of compound 4f in PBS buffer was determined to be modest at 9.6 μM – although significantly, this assessment shows that the compound was soluble at least 34- to 5-fold above the level of the observed IC50 and LD50 values, respectively. Some liability was noted in microsomes, as the percentage of parent remaining after 1 hour of exposure was nearly 50% in both mouse and human samples.
Table 2.
Physiochemical and in vitro ADME data for milestone compounds
| compound | ||||
|---|---|---|---|---|
| property or assessment | 1 | 2 | 4f | |
| cLogP[a] | 4.3 | 6.7 | 7.6 | |
| aqueous solubility (PBS buffer, pH 7.4, μM)[b] | > 274.3 | 62.3 | 9.6 | |
| PAMPA permeability (×10−6 cm/s)[d] | ND | < 1.9/7.6/120 | 0/1.6/49 | |
| microsomal stability, %[e] | mouse | ND | ND | 55.5 |
| human | ND | ND | 46.5 | |
| plasma stability, %[f] | mouse | ND | ND | 97.4 |
| human | ND | ND | 80.8 | |
| plasma protein binding, %[g] | 1 μM | ND | ND | 99.0 |
| 10 μM | ND | ND | 99.1 | |
Data were calculated using CambridgeSoft ChemBioDraw Ultra 12.0
Kinetic solubility method
Pe, membrane permeability, measured using an in vitro model for the passive transport from the GI into the blood system. Donor pH 5.0/6.2/7.4; acceptor pH 7.4. Controls: verapamil-HCl (highly permeable): 138; corticosterone (moderately permeable): 15; theophylline (poorly permeable): < 0.3;
Percent parent remaining after 1 h;
Percent parent remaining after 3 h;
mouse species; ND = not determined.
Probe compound 2 was evaluated against a 50-member kinase panel23 at a concentration of 5 μM to assess selectivity for the T. brucei hexokinase over mammalian kinases.17 Inhibition of any one mammalian kinase did not exceed 10%. Given this precedent, we decided to profile compound 4f against a broader range of biological targets to identify off-target liabilities associated with the chemical series (Table 3). Analog 4f was assessed in a 67-member, radioligand binding-based, PanLabs LeadProfilingScreen® that surveyed the inhibition profile over a diverse cross section of GPCRs, receptors, transporters, and ion channels.24 At a concentration of 10 μM, ≥ 50% inhibition was noted for several of the targets. Determination of IC50 values was not pursued; however, the outcome suggests that compound 4f may show undesirable, off-target effects that are in range of the observed potency for inhibition of the target hexokinases. Nonetheless, advancement of the benzamidobenzoic acid series would certainly require structural augmentation to improve the parasitic activity profile which would likely alter this off-target liability.
Table 3.
Percent inhibition of mammalian targets with compound 4f[a]
| entry | target | species | percent inhibition |
|---|---|---|---|
| 1 | Adrenergic α1B | rat | 53 |
| 2 | Dopamine D1 | human | 57 |
| 3 | Dopamine D3 | human | 86 |
| 4 | Histamine H2 | human | 98 |
| 5 | Leukotriene, cysteinyl CysLT1 | human | 87 |
| 6 | Serotonin (5-hydroxytryptamine) 5-HT1A | human | 90 |
| 7 | Thyroid hormone | rat | 65 |
| 8 | Dopamine transporter (DAT) | human | 88 |
| 9 | Norepinephrine transporter (NET) | human | 91 |
Experiments performed in duplicate at a concentration of 10 μM; the complete profile is included in the Supporting Information.
Conclusions
In an effort to identify target-specific inhibitors of a critical hexokinase in glycolysis-dependent trypanosomes, our team discovered a benzamidobenzamide scaffold with modest enzymatic activity against T. brucei hexokinase 1. First generation analogs showed improved enzymatic T. brucei HK1 inhibition, but failed to demonstrate efficacy against parasites. In this work, structural changes were explored to generate several structurally related compounds with improved, submicromolar biochemical activity against T. brucei HK1 and, notably, single-digit micromolar efficacy against whole T. brucei BSF parasites. We were further excited to discover that the hexokinase 1 of the related kinetoplastid, Leishmania major, demonstrated a similar susceptibility to these compounds. Nonetheless, some limitations were noted. Overall, our SAR survey revealed a relatively consistent T. brucei BSF LD50 of ~ 2.0 μM, despite a broad range of rTbHK1 IC50 values. This suggests that other factors may be relevant to the delivery and maintenance of inhibitor concentrations in the glycosome. Possibilities include the involvement of parasitic transport mechanisms that export the inhibitor or additional parasitic molecular targets. This, coupled with our evaluation of the SAR, in vitro ADME and pharmacology profile of the benzamidobenzamides and specifically, compound 4f, suggests that these particular compounds are not likely to reach the threshold of lead candidates for drug development. However, these results do represent a significant milestone in targeting trypanosomal hexokinases as a novel, potentially broad spectrum therapeutic approach to kinetoplastid diseases against which chemotypes with greater optimization potential may be launched.
Experimental Section
Chemistry
Purity of all final compounds was confirmed by HPLC/MS analysis and determined to be ≥95%. Analytical TLC experiments were performed on Hard Layer Silica Gel UNIPLATE™ (with organic binder) plates from Analtech, Inc. and analyzed with 254 nm UV light using diluted samples. 1H and 13C NMR spectra were recorded on a Bruker Avance 400 spectrometer (operating at 400 and 101 MHz respectively) or a Bruker Avance AVIII 500 spectrometer (operating at 500 and 126 MHz, respectively) and reported with either 0.05% TMS (1H = δ 0.00 ppm, 13C = δ 0.00 ppm) or residual solvent (CHCl3: 1H = δ 7.26 ppm, 13C = δ 77.16 ppm; CD3SOCD2H: 1H = δ 2.50 ppm, 13C = δ 39.52 ppm) as an internal standard. The chemical shifts (δ) reported are given in parts per million (ppm) and the coupling constants (J) are in Hertz (Hz). The spin multiplicities are reported as s = singlet, br s = broad singlet, d = doublet, t = triplet, q = quartet, p = pentuplet, dd = doublet of doublet, ddd = doublet of doublet of doublet, and m = multiplet. The LC-MS analysis was performed on an Agilent 1200 HPLC system with photodiode array UV detection and an Agilent 6224 TOF mass spectrometer. The chromatographic method utilized the following parameters: a Waters Acquity BEH C-18 2.1 × 50 mm, 1.7 μm column; UV detection wavelength = 214 nm; flow rate = 0.4 mL/min; gradient = 5-100% CH3CN over 3 minutes with a hold of 0.8 minutes at 100% CH3CN; the aqueous mobile phase contained 0.15% NH4OH. The mass spectrometer utilized the following parameters: an Agilent multimode source which simultaneously acquires ESI+/APCI+; a reference mass solution consisting of purine and hexakis(1H, 1H, 3H-tetrafluoropropoxy) phosphazine; and a make-up solvent of 90:10:0.1 MeOH/H2O/HCO2H which was introduced to the LC flow prior to the source to assist ionization. Melting points were determined on a Stanford Research Systems OptiMelt apparatus. Microwave irradiated (MWI) reactions were carried out using a Biotage Initiator Classic synthesizer. Flash chromatography separations were carried out using a Teledyne ISCO CombiFlash Rf 200 purification system with either silica gel columns (normal-phase) or RediSep Rf C-18 columns (reverse-phase). Common starting materials used in the synthesis of the benzamidobenzoic acid series included 2-amino-4-chlorobenzoic acid (CAS# 89-77-0), methyl 2-amino-5-bromo-4-chlorobenzoate 3d (CAS#765211-09-4), and methyl 2-amino-5-iodo-4-chlorobenzoate 3e (CAS#199850-56-1). All three reagents are available from Millipore-Sigma (formerly Sigma-Aldrich) and several other vendors. The synthesis of compound 2 (ML205) has been previously described.17
2-(4-tert-Butylbenzamido)-4-chloro-5-methylbenzoic acid (4c)
Step 1: synthesis of methyl 2-(4-(tert-butylbenzamido)-4-chloro-5-methylbenzoate
To a microwave vial was added methyl 2-amino-4-chloro-5-methylbenzoate (0.028 g, 0.13 mmol, CAS# 458533-69-2), acetonitrile (2 mL) and 4-(tert-butylbenzoyl chloride (0.029 g, 0.026 mL, 0.14 mmol). The reaction was heated to 150 °C in the microwave for 60 min. The reaction was cooled to rt, diluted with saturated NaHCO3 (10 mL), and extracted with EtOAc (2 × 10 mL). The organic layers were combined and dried (MgSO4), filtered, and adsorbed directly onto silica. Purification by reverse-phase MPLC (10 - 100% MeCN:water) produced methyl 2-(4-(tert-butylbenzamido)-4-chloro-5-methylbenzoate (0.156 g, 0.434 mmol, 99% yield). 1H NMR (400 MHz, CDCl3) δ 11.91 (s, 1H), 9.04 (s, 1H), 7.99 - 7.94 (m, 2H), 7.92 (s, 1H), 7.57 - 7.50 (m, 2H), 3.95 (s, 3H), 2.36 (s, 3H), 1.36 (s, 9H).
Step 2: Synthesis of 2-(4-(tert-butylbenzamido)-4-chloro-5-methylbenzoic acid (4c)
To a vial was added methyl 2-(4-(tert-butylbenzamido)-4-chloro-5-methylbenzoate (0.055 g, 0.14 mmol) and THF (2 mL). The LiOH (0.022 g, 0.92 mmol) was dissolved in water (2 mL) and the resulting solution was added to the reaction vial and stirred at rt for 18 h. The reaction was acidified to pH 2 - 3 with aqueous 1.0 M HCl and extracted with CH2Cl2 (3 × 10 mL). The organic layers were combined, dried with MgSO4, filtered and adsorbed onto silica. Purified by reverse-phase MPLC (10 – 100% MeCN:water) to produce 2-(4-tert-butylbenzamido)-4-chloro-5-methylbenzoic acid 4c (0.11 g, 0.33 mmol, 75% yield). 1H NMR (400 MHz, DMSO-d6) δ 12.12 (s, 1H), 8.83 (s, 1H), 8.02 (d, J = 0.9 Hz, 1H), 7.88 (d, J = 8.5 Hz, 2H), 7.62 (d, J = 8.5 Hz, 2H), 2.35 (s, 3H), 1.33 (s, 9H). 13C NMR (126 MHz, DMSO-d6) δ 169.39, 164.63, 155.38, 139.93, 138.83, 133.31, 131.36, 129.83, 126.92, 125.89, 119.61, 115.24, 34.81, 30.87, 18.94. LCMS Retention time: 1.44 min. LCMS purity 100%. HRMS (ESI) m/z calcd for C19H20ClNO3 [M+H]+ 346.1211, found 346.1166.
2-(4-tert-Butylbenzamido)-4-chloro-5-ethylbenzoic acid (4d)
Step 1: synthesis of methyl 2-amino-4-chloro-5-ethylbenzoate
To a vial was added methyl 2-amino-5-bromo-4-chlorobenzoate 3d (0.11 g, 0.433 mmol), potassium ethyltrifluoroborate (0.11 g, 0.774 mmol), palladium (II) acetate (4.8 mg, 0.021 mmol), RuPhos (0.020 g, 0.043 mmol) and cesium carbonate (0.42 g, 1.3 mmol). The vial was evacuated with argon 3 times and then degassed toluene (1.5 mL) and degassed water (0.5 mL) were added via syringe. The reaction contents was heated to 100 °C in the microwave for 80 min before it was cooled to rt, diluted with EtOAc (10 mL) and washed with saturated NaHCO3 (12 mL). The separated organic layer was dried (MgSO4), filtered and concentrated. The crude residue was purified by MPLC (0 – 25% EtOAc:hexanes) to provide methyl 2-amino-4-chloro-5-ethylbenzoate (0.033 g, 0.154 mmol, 36% yield). 1H NMR (400 MHz, CDCl3) δ 7.69 (s, 1H), 6.69 (s, 1H), 5.61 (br s, 2H), 3.86 (s, 3H), 2.62 (q, J = 7.5 Hz, 2H), 1.18 (t, J = 7.5 Hz, 3H).
Step 2: synthesis of methyl 2-(4-tert-butylbenzamido)-4-chloro-5-ethylbenzoate
To a microwave vial was added methyl 2-amino-4-chloro-5-ethylbenzoate (0.028 g, 0.13 mmol), acetonitrile (2 mL) and 4-tert-butylbenzoyl chloride (0.029 g, 0.026 mL, 0.14 mmol). The reaction was heated to 150 °C in the microwave for 60 min. The reaction was cooled to rt, diluted with saturated NaHCO3 (10 mL), and extracted with EtOAc (2 × 10 mL). The organic layers were combined and dried (MgSO4), filtered, and adsorbed directly onto silica. Purification by MPLC (0 - 15% EtOAc:hexanes) afforded the methyl 2-(4-tert-butylbenzamido)-4-chloro-5-ethylbenzoate which was carried into the next reaction.
Step 3: synthesis of 2-(4-tert-butylbenzamido)-4-chloro-5-ethylbenzoic acid (4d)
To a vial was added methyl 2-(4-(tert-butylbenzamido)-4-chloro-5-ethylbenzoate (0.049 g, 0.13 mmol) and THF (2 mL). The LiOH (0.022 g, 0.92 mmol) was dissolved in water (2 mL) and the resulting solution was added to the reaction vial and stirred at rt for 18 h. The reaction was acidified to pH 2 - 3 with aqueous 1.0 M HCl and extracted with CH2Cl2 (3 × 10 mL). The organic layers were combined, dried with MgSO4, filtered and adsorbed onto silica. Purification by reverse-phase MPLC (10 - 100% MeCN:water) produced 2-(4-(tert-butylbenzamido)-4-chloro-5-ethylbenzoic acid 4d (0.032 g, 0.089 mmol, 68% yield). 1H NMR (400 MHz, DMSO-d6) δ 12.16 (s, 1H), 8.83 (s, 1H), 8.01 (s, 1H), 7.89 (d, J = 8.5 Hz, 2H), 7.62 (d, J = 8.5 Hz, 2H), 2.73 (q, J = 7.5 Hz, 2H), 1.19 (t, J = 7.5 Hz, 3H). 13C NMR (126 MHz, DMSO-d6) δ 169.4, 164.7, 155.4, 139.9, 138.2, 135.3, 132.0, 131.4, 126.9, 125.9, 119.9, 115.6, 34.8, 30.9, 25.5, 14.0. LCMS Retention time: 2.564 min. LCMS purity 99.1%. HRMS (ESI) m/z calcd for C20H22ClNO3 [M+H]+ 360.1367, found 360.1354.
2-(4-(tert-Butylbenzamido)-4-chloro-5-cyclopropylbenzoic acid (4e)
Step 1: synthesis of methyl 2-amino-4-chloro-5-cyclopropylbenzoate
To a vial was added methyl 2-amino-5-bromo-4-chlorobenzoate 3a (0.055 g, 0.215 mmol), potassium cyclopropyltrifluoroborate (0.037 g, 0.252 mmol), Pd(PPh3)4 (4.8 mg, 4.2 μmol), and tribasic potassium phosphate (0.15 g, 0.671 mmol). After evacuating the vial with argon 3 times, degassed toluene (1.5 mL) and degassed water (0.5 mL) were added and the resulting mixture was heated at 100 °C for 17 h. The reaction was quenched with NaHCO3 (5 mL) and extracted with EtOAc (2 × 5 mL). The separated organic extracts were combined, dried (MgSO4), filtered, adsorbed to silica and purified by MPLC (20 min, 0 – 10% EtOAc:hexanes) to produce methyl 2-amino-4-chloro-5-cyclopropylbenzoate (0.020 g, 0.089 mmol, 42% yield). 1H NMR (400 MHz, CDCl3) δ 7.50 (s, 1H), 6.71 (s, 1H), 5.62 (br s, 2H), 3.86 (s, 3H), 2.00 – 1.92 (m, 1H), 0.93 – 0.87 (m, 2H), 0.62 – 0.54 (m, 2H). This intermediate was then advanced through analogous steps 2 and 3 as described for compound 4d. Isolated 4e in 11% yield (0.009 g, 0.023 mmol) after 3 steps. 1H NMR (400 MHz, DMSO-d6) δ 12.20 (s, 1H), 8.85 (s, 1H), 7.88 (d, J = 8.4 Hz, 2H), 7.65 (s, 1H), 7.62 (d, J = 8.4 Hz, 2H), 2.16 – 2.08 (m, 1H), 1.33 (s, 1H), 1.06 – 1.00 (m, 2H), 0.71 – 0.66 (m, 2H). 13C NMR (126 MHz, DMSO-d6) δ 169.3, 164.6, 155.4, 139.5, 139.4, 134.6, 131.4, 128.7, 126.9, 125.9, 119.7, 115.7, 34.8, 30.9, 12.7, 7.8. LCMS Retention time: 4.173 min. LCMS purity 100%. HRMS (ESI) m/z calcd for C21H22ClNO3 [M+H]+ 372.1367, found 372.1355.
4-(4-(tert-Butylbenzamido)-6-chloro-[1,1′-biphenyl]-3-carboxylic acid (4f)
Step 1: synthesis of methyl 4-amino-6-chloro-[1,1′-biphenyl]-3-carboxylate
To a microwave vial was added phenylboronic acid (0.051 g, 0.42 mmol), methyl 2-amino-5-bromo-4-chlorobenzoate 3d (0.093 g, 0.35 mmol), 1,1′-bis(di-t-butylphosphino)ferrocene palladium dichloride, (4.8 mg, 7.0 μmol) and K2CO3 (0.073 g, 0.53 mmol). The vial was evacuated with argon 3 times before degassed acetonitrile (1.5 mL) and water (1.5 mL) were added. The reaction stirred at 110 °C for 60 minutes in the microwave and then cooled to rt. After diluting with EtOAc (10 mL), the reaction mixture was washed with saturated NaHCO3 (12 mL), organic extracts were separated and dried with MgSO4, filtered and concentrated. The crude product was purified by reverse-phase MPLC (10 - 100% MeCN:water) to provide methyl 4-amino-6-chloro-[1,1′-biphenyl]-3-carboxylate (0.085 g, 0.32 mmol, 92% yield). 1H NMR (400 MHz, CDCl3) δ 7.85 (s, 1H), 7.65 (d, J = 7.9 Hz, 2H), 7.52 (d, J = 7.9 Hz, 2H), 6.82 (s, 1H), 5.88 (br s, 2H), 3.87 (s, 3H).
Step 2: synthesis of methyl 4-(4-(tert-butyl)benzamido)-6-chloro-[1,1′-biphenyl]-3-carboxylate
Synthesized as described for compound 4d (step 2). Isolated methyl 4-(4-(tert-butylbenzamido)-6-chloro-[1,1′-biphenyl]-3-carboxylate (0.35 g, 0.82 mmol, 91% yield). 1H NMR (400 MHz, CDCl3) δ 12.02 (s, 1H), 9.17 (s, 1H), 8.04 (s, 1H), 7.89 (d, J = 8.6 Hz, 2H), 7.54 (d, J = 8.6 Hz, 2H), 7.44 – 7.36 (m, 5H), 3.93 (s, 3H), 1.36 (s, 9H).
Step 3: synthesis of 4-(4-(tert-butylbenzamido)-6-chloro-[1,1′-biphenyl]-3-carboxylic acid (4f)
Synthesized as described for compound 4d (step 3). Isolated 4-(4-(tert-butylbenzamido)-6-chloro-[1,1′-biphenyl]-3-carboxylic acid 4f in (0.12 g, 0.28 mmol, 31% yield) as a white solid, mp 249 – 252 °C. 1H NMR (400 MHz, DMSO-d6) δ 12.25 (s, 1H), 8.98 (s, 1H), 8.02 (s, 1H), 7.92 (d, J = 8.4 Hz, 2H), 7.64 (d, J = 8.4 Hz, 2H), 7.51 – 7.44 (m, 5H), 1.34 (s, 9H). 13C NMR (126 MHz, DMSO-d6) δ 169.2, 164.8, 155.6, 141.0, 137.6, 136.8, 134.0, 133.6, 131.2, 129.2, 128.4, 128.0, 127.0, 126.0, 120.4, 115.7, 34.8, 30.9. LCMS Retention time: 2.87 min. LCMS purity 100%. HRMS (ESI) m/z calcd for C24H22ClNO3 [M+H]+ 408.1367, found 408.1371.
4-(4-(tert-Butylbenzamido)-6-chloro-4′-methyl-[1,1′-biphenyl]-3-carboxylic acid (4g)
Step 1: synthesis of methyl 4-amino-6-chloro-4′-methyl-[1,1′-biphenyl]-3-carboxylate
To a microwave vial was added methyl 2-amino-5-bromo-4-chlorobenzoate 3d (0.13 g, 0.473 mmol), p-tolylboronic acid (0.084 g, 0.61 mmol), bis(triphenylphosphine)palladium(II) dichloride (0.033 g, 0.047 mmol), K2CO3 (0.13 g, 0.945 mmol) and DMF:water (2.5 mL, 5:1). The vial was evacuated with argon 3 times and a degassed (bubbling argon × 30 min) mixture of DMF:water (1 mL, 5:1) was added. The reaction heated at 150 °C in the microwave for 10 min. After cooling to rt, EtOAc (2 mL) was added, and the reaction was washed sequentially with water (2 mL) and brine (2 mL). The organic layer was separated, dried (MgSO4), filtered and concentrated. The crude product was purified by reverse-phase MPLC (10 - 100% MeCN:water) provide methyl 4-amino-6-chloro-4′-methyl-[1,1′-biphenyl]-3-carboxylate (0.044 g, 0.16 mmol, 33% yield). 1H NMR (400 MHz, CDCl3) δ 7.83 (s, 1H), 7.33 (d, J = 8.8 Hz, 2H), 6.94 (d, J = 8.8 Hz, 2H), 6.79 (s, 1H), 5.77 (s, 2H), 3.85 (s, 3H), 3.84 (s, 3H). This intermediate was then advanced through analogous steps 2 and 3 as described for compound 4d. Isolated 4-(4-(tert-butylbenzamido)-6-chloro-4′-methyl-[1,1′-biphenyl]-3-carboxylic acid 4g in (0.047 g, 0.11 mmol, 88% yield) as a white solid, mp 260 - 262 °C. 1H NMR (400 MHz, DMSO-d6) δ 12.26 (s, 1H), 8.97 (s, 1H), 8.00 (s, 1H), 7.92 (d, J = 8.5 Hz, 2H), 7.65 (d, J = 8.5 Hz, 2H), 7.38 (d, J = 8.1 Hz, 2H), 7.31 (d, 8.1 Hz, 2H), 2.38 (s, 3H), 1.34 (s, 9H). 13C NMR (126 MHz, DMSO-d6) δ 169.2, 164.8, 155.5, 140.8, 137.4, 136.8, 134.7, 134.0, 133.5, 131.3, 129.1, 129.0, 127.0, 125.9, 120.4, 34.8, 30.9, 20.8. LCMS Retention time: 4.435 min. LCMS purity 100%. HRMS (ESI) m/z calcd for C25H24ClNO3 [M+H]+ 422.1524, found 422.1491.
4-(4-(tert-Butylbenzamido)-6-chloro-2′-methyl-[1,1′-biphenyl]-3-carboxylic acid (4h)
Synthesized as described for compound 4f. Isolated 4-(4-(tert-butylbenzamido)-6-chloro-2′-methyl-[1,1′-biphenyl]-3-carboxylic acid 6h (0.014 g, 0.034 mmol, 15% yield). 1H NMR (400 MHz, DMSO-d6) δ 13.99 (br s, 1H), 8.93 (s, 1H), 7.96 (d, J = 8.5 Hz, 2H), 7.91 (br s, 1H), 7.61 (d, J = 8.5 Hz, 2H), 7.35 – 7.25 (m, 3H), 7.17 – 7.13 (m, 1H), 2.10 (s, 3H), 1.34 (s, 9H). 13C NMR (126 MHz, DMSO-d6) δ 168.8, 164.7, 155.0, 141.0, 138.4, 135.8, 133.5, 133.3, 131.8, 129.8, 129.5, 128.0, 127.1, 125.8, 125.7, 119.0, 34.8, 30.9. LCMS Retention time: 4.323 min. LCMS purity 100%. HRMS (ESI) m/z calcd for C25H24ClNO3 [M+H]+ 422.1524, found 422.1511.
4-(4-(tert-Butylbenzamido)-6-chloro-4′-methoxy-[1,1′-biphenyl]-3-carboxylic acid (4i)
Synthesized as described for compound 4g. Isolated 4-(4-(tert-butylbenzamido)-6-chloro-4′-methoxy-[1,1′-biphenyl]-3-carboxylic acid 4i (0.056 g, 0.13 mmol, 71% yield) as a white solid, mp 237 – 239 °C. 1H NMR (400 MHz, DMSO-d6) δ 12.25 (s, 1H), 8.96 (s, 1H), 8.00 (s, 1H), 7.91 (d, J = 8.4 Hz, 2H), 7.65 (d, J = 8.2 Hz, 2H), 7.42 (d, J = 8.6 Hz, 2H), 7.05 (d, J = 8.6 Hz, 2H), 3.82 (s, 3H), 1.34 (s, 9H). 13C NMR (126 MHz, DMSO-d6) δ 169.2, 164.38, 159.0, 155.5, 140.6, 136.8, 133.7, 133.5, 131.3, 130.5, 129.8, 127.0, 125.9, 120.4, 115.7, 113.8, 55.2, 34.8, 30.9. LCMS Retention time: 4.361 min. LCMS purity 97.4%. HRMS (ESI) m/z calcd for C25H24ClNO4 [M+H]+ 438.1473, found 438.1469.
4-(4-(tert-Butylbenzamido)-6-chloro-3′-methoxy-[1,1′-biphenyl]-3-carboxylic acid (4j)
Synthesized as described for compound 4f. Isolated 4-(4-(tert-butylbenzamido)-6-chloro-3′-methoxy-[1,1′-biphenyl]-3-carboxylic acid 4j (0.050 g, 0.11 mmol, 79% yield). 1H NMR (400 MHz, DMSO-d6) δ 12.26 (s, 1H), 8.97 (s, 1H), 8.02 (s, 1H), 7.91 (d, J = 8.5 Hz, 2H), 7.65 (d, J = 8.5 Hz, 2H), 7.44 – 7.39 (m, 1H), 7.05 – 7.00 (m, 3H), 3.82 (s, 3H), 1.34)s, 9H). 13C NMR (125 MHz, DMSO-d6) δ 169.2, 164.8, 159.6, 155.6, 141.0, 138.9, 136.8, 133.9, 133.5, 131.2, 129.5, 127.0, 126.0, 121.5, 120.4, 115.7, 114.9, 113.5, 55.2, 34.8, 30.9. LCMS Retention time: 4.201 min. LCMS purity 100%. HRMS (ESI) m/z calcd for C25H24ClNO4 [M+H]+ 438.1473, found 438.1455.
4-(4-(tert-Butylbenzamido)-6-chloro-2′-methoxy-[1,1′-biphenyl]-3-carboxylic acid (4k)
Synthesized as described for compound 4f. Isolated 4-(4-(tert-butylbenzamido)-6-chloro-2′-methoxy-[1,1′-biphenyl]-3-carboxylic acid 4k (0.075 g, 0.17 mmol, 93% yield). 1H NMR (400 MHz, DMSO-d6) δ 12.23 (s, 1H), 8.93 (s, 1H), 7.93 (d, J = 8.5 Hz, 2H), 7.92 (s, 1H), 7.65 (d, J = 8.5 Hz, 2H), 7.47 – 7.41 (m, 1H), 7.22 (dd, J2 = 7.5 Hz, J2 = 1.8 Hz, 1H), 7.16 – 7.13 (m, 1H), 7.08 – 7.03 (m, 1H), 3.76 (s, 3H), 1.34 (s, 9H). 13C NMR (125 MHz, DMSO-d6) δ 169.2, 164.8, 156.4, 155.5, 140.9, 138.6, 133.9, 131.7, 131.3, 130.6, 130.0, 127.0, 126.5, 125.9, 120.4, 119.8, 115.2, 111.4, 55.5, 34.8, 30.9. LCMS Retention time: 4.096 min. LCMS purity 99.2%. HRMS (ESI) m/z calcd for C25H24ClNO4 [M+H]+ 438.1473, found 438.1458.
4-(4-(tert-Butylbenzamido)-6-chloro-4′-(dimethylamino)-[1,1′-biphenyl]-3-carboxylic acid (4l)
Step 1: synthesis of methyl 4-amino-6-chloro-4′-nitro-[1,1′-biphenyl]-3-carboxylate
Synthesized from methyl 2-amino-5-bromo-4-chlorobenzoate 3d as described for compound 4f, step 1, to afford methyl 4-amino-6-chloro-4′-nitro-[1,1′-biphenyl]-3-carboxylate (0.15 g, 0.50 mmol, 68% yield). 1H NMR (400 MHz, CDCl3) δ 8.26 (d, J = 8.9 Hz, 2H), 7.87 (s, 1H), 7.59 (d, J = 8.9 Hz, 2H), 6.84 (s, 1H), 3.88 (s, 3H).
Step 2: synthesis of methyl 4-(4-(tert-butylbenzamido)-6-chloro-4′-nitro-[1,1′-biphenyl]-3-carboxylate
Synthesized as described for compound 4d (step 2) to afford methyl 4-(4-(tert-butylbenzamido)-6-chloro-4′-nitro-[1,1′-biphenyl]-3-carboxylate (0.20 g, 0.43 mmol, 87% yield). 1H NMR (400 MHz, CDCl3) δ 12.07 (br s, 1H), 9.25 (s, 1H), 8.32 (d, J = 8.8 Hz, 2H), 8.08 (s, 1H), 8.01 (d, J = 8.6 Hz, 2H), 7.64 (d, J = 8.8 Hz, 2H), 7.58 (d, J = 8.6 Hz, 2H), 3.99 (s, 3H), 1.38 (s, 9H).
Step 3: synthesis of methyl 4′-amino-4-(4-(tert-butylbenzamido)-6-chloro-[1,1′-biphenyl]-3-carboxylate
To a vial containing MeOH (4 mL) and CH2Cl2 (4 mL) was added methyl 4-(4-(tert-butylbenzamido)-6-chloro-4′-nitro-[1,1′-biphenyl]-3-carboxylate (0.162 g, 0.347 mmol). The reaction was cooled to 0 °C, and Raney Nickel (2.0 mg, 0.035 mmol) was added. The NaBH4 (0.033 g, 0.87 mmol) was then added portion-wise at 0 °C. After continued stirring at rt for 17 h, the reaction contents was filtered through Celite, diluted with CH2Cl2 (10 mL), and then washed with water (15 mL). The separated organic layer was dried (MgSO4), filtered, adsorbed to silica and purified by MPLC (0 - 10% MeOH:CH2Cl2) to provide methyl 4′-amino-4-(4-(tert-butylbenzamido)-6-chloro-[1,1′-biphenyl]-3-carboxylate (0.14 g, 0.316 mmol, 91% yield). 1H NMR (400 MHz, CDCl3) δ 12.00 (s, 1H), 9.15 (s, 1H), 8.01 (s, 1H), 7.98 (d, J = 8.4 Hz, 2H), 7.55 (d, J = 8.6 Hz, 2H), 7.34 (d, J = 8.5 Hz, 2H), 7.04 (d, J = 8.5 Hz, 2H), 3.96 (s, 3H), 1.36 (s, 9H).
Step 4: synthesis of methyl 4-(4-(tert-butylbenzamido)-6-chloro-4′-(dimethylamino)-[1,1′-biphenyl]-3-carboxylate
To a vial was added the methyl 4′-amino-4-(4-(tert-butylbenzamido)-6-chloro-[1,1′-biphenyl]-3-carboxylate (0.037 g, 0.085 mmol) and acetic acid (1.0 mL). Addition of 37% w/v paraformaldehyde (0.063 mL, 0.85 mmol) solution in water was followed by the addition of sodium cyanoborohydride (0.016 g, 0.25 mmol), and the resulting reaction was stirred at rt for 18 h. After concentrating the reaction mixture under reduced pressure, the residue was diluted EtOAc (5 mL), washed with saturated NaHCO3 (5 mL) and extracted with EtOAc (3 × 5 mL). The separated organic layers were combined, dried (MgSO4), filtered, and concentrated. The crude product was purified by reverse-phase MPLC (10 - 100% MeCN:water) to produce methyl 4-(4-(tert-butylbenzamido)-6-chloro-4′-(dimethylamino)-[1,1′-biphenyl]-3-carboxylate (0.021 g, 0.045 mmol, 53% yield). 1H NMR (400 MHz, CDCl3) δ 11.98 (s, 1H), 9.14 (s, 1H), 8.06 (s, 1H), 7.99 (d, J = 8.5 Hz, 2H), 7.56 (d, J = 8.4 Hz, 2H), 7.36 (d, J = 8.6 Hz, 2H), 6.79 (br d, J = 8.2 Hz, 2H), 3.95 (s, 3H), 3.02 (s, 6H), 1.37 (s, 9H).
Step 5: Synthesis of 4-(4-(tert-butylbenzamido)-6-chloro-4′-(dimethylamino)-[1,1′-biphenyl]-3-carboxylic acid (4l)
Methyl 4-(4-(tert-butylbenzamido)-6-chloro-4′-(dimethylamino)-[1,1′-biphenyl]-3-carboxylate was hydrolyzed as described for compound 6d (step 3) to afford, after purification, 4-(4-(tert-butylbenzamido)-6-chloro-4′-(dimethylamino)-[1,1′-biphenyl]-3-carboxylic acid 4l (0.010 g, 0.022 mmol, 49% yield). 1H NMR (400 MHz, DMSO-d6) δ 11.92 (s, 1H), 9.16 (s, 1H), 8.14 (s, 1H), 7.95 (d, J = 8.0 Hz, 2H), 7.52 (d, J = 8.0 Hz, 2H), 7.37 (t, J = 8.0 Hz, 1H), 6.82 (d, J = 8.4 3H), 3.01 (s, 6H), 1.34 (s, 9H). 13C NMR (126 MHz, DMSO-d6) δ 170.93, 165.73, 155.84, 149.78, 141.02, 140.07, 135.06, 133.89, 131.51, 130.27, 127.25, 126.54, 125.84, 121.60, 113.13, 112.33, 53.42, 40.72, 31.13. LCMS Retention time: 4.281 min. LCMS purity 95.8%. HRMS (ESI) m/z calcd for C26H27ClN2O3 [M+H]+ 451.1789, found 451.1779.
4-(4-(tert-Butylbenzamido)-6-chloro-3′-(dimethylamino)-[1,1′-biphenyl]-3-carboxylic acid (4m)
Synthesized as described for compound 4l. Isolated 4-(4-(tert-butylbenzamido)-6-chloro-3′-(dimethylamino)-[1,1′-biphenyl]-3-carboxylic acid 4m (0.011 g, 0.024 mmol, 54.0% yield). 1H NMR (400 MHz, DMSO-d6) δ 12.40 (s, 1H), 9.17 (s, 1H), 8.19 (s, 1H), 7.99 (d, J = 8.5 Hz, 2H), 7.52 (d, J = 8.4 Hz, 2H), 7.34 (t, J = 7.9 Hz, 1H), 7.05 – 7.01 (m, 2H), 6.96 – 6.92 (m, 1H), 3.01 (s, 6H), 1.35 (s, 9H). 13C NMR (126 MHz, DMSO-d6) δ 170.9, 165.8, 155.7, 149.6, 141.6, 139.5, 138.8, 134.7, 134.0, 131.6, 128.8, 127.3, 125.8, 121.7, 121.3, 117.3, 114.6, 114.0, 53.4, 50.7, 35.0, 31.1. LCMS Retention time: 4.256 min. LCMS purity 96.1%. HRMS (ESI) m/z calcd for C26H27ClN2O3 [M+H]+ 451.1789, found 451.1782.
4-(4-(tert-Butylbenzamido)-6-chloro-4′-fluoro-[1,1′-biphenyl]-3-carboxylic acid (4n)
Synthesized as described for compound 4g. Isolated 4-(4-(tert-butylbenzamido)-6-chloro-4′-fluoro-[1,1′-biphenyl]-3-carboxylic acid 4n (0.053 g, 0.124 mmol, 70% yield) as an off-white solid, mp 282 - 284 °C. 1H NMR (400 MHz, DMSO-d6) δ 14.26 (br s, 1H), 8.93 (s, 1H), 8.03 (s, 1H), 7.96 (d, J = 8.5 Hz, 2H), 7.61 (d, J = 8.5 Hz, 2H), 7.51 (d, J = 8.8 Hz, 1H), 7.50 (d, J = 8.8 Hz, 1H), 7.32 (d, J = 8.8 Hz, 1H), 7.30 (d, J = 8.8 Hz, 1H), 1.33 (s, 9H). 13C NMR (126 MHz, DMSO-d6) δ 168.5, 164.7, 162.6, 160.7, 155.0, 141.1, 134.7 (d, J = 2.5 Hz), 133.8, 132.1, 131.8, 131.3 (d, J = 6.5 Hz), 127.1, 125.7, 119.5, 115.1 (d, J = 17.1 Hz), 67.0, 34.8, 30.9. LCMS Retention time: 2.696 min. LCMS purity 99.5%. HRMS (ESI) m/z calcd for C24H21ClFNO3 [M+H]+ 426.1273, found 426.1159.
4-(4-(tert-Butylbenzamido)-6-chloro-3′-fluoro-[1,1′-biphenyl]-3-carboxylic acid (4o)
Synthesized as described for compound 4g. Isolated 4-(4-(tert-butylbenzamido)-6-chloro-3′-fluoro-[1,1′-biphenyl]-3-carboxylic acid 4o (0.051 g, 0.12 mmol, 69% yield) as a white solid, mp 259 - 260 °C. 1H NMR (400 MHz, DMSO-d6) δ 12.25 (br s, 1H), 8.98 (s, 1H), 8.05 (s, 1H), 7.91 (d, J = 8.6 Hz, 2H), 7.64 (d, J = 8.5 Hz, 2H), 7.58 – 7.51 (m, 1H), 7.37 – 7.26 (m, 3H), 1.34 (s, 9H). 13C NMR (126 MHz, DMSO-d6) δ 169.1, 164.9, 161.8 (d, J = 244.2 Hz), 155.6, 141.3, 139.7, (d, J = 8.2 Hz), 136.7, 132.6, 132.6 (d, J = 2.1 Hz), 131.2, 130.4 (d, J = 8.5 Hz), 127.0, 125.9, 125.5 (d, J = 2.7 Hz), 120.3, 116.2 (d, J = 22.1 Hz), 115.7, 114.9 (d, J = 20.9 Hz), 34.8, 30.9. LCMS Retention time: 4.308 min. LCMS purity 100%. HRMS (ESI) m/z calcd for C24H21ClFNO3 [M+H]+ 426.1273, found 426.1260.
4-(4-(tert-Butylbenzamido)-6-chloro-2′-fluoro-[1,1′-biphenyl]-3-carboxylic acid (4p)
Synthesized as described for compound 4g. Isolated 4-(4-(tert-butylbenzamido)-6-chloro-2′-fluoro-[1,1′-biphenyl]-3-carboxylic acid 4p (0.037 g, 0.087 mmol, 59% yield). 1H NMR (400 MHz, DMSO-d6) δ 12.31 (s, 1H), 9.00 (s, 1H), 8.02 (s, 1H), 7.93 (d, J = 8.5 Hz, 2H), 7.65 (d, J = 8.5 Hz, 2H), 7.57 – 7.50 (m, 1H), 7.47 – 7.42 (m, 1H), 7.39 – 7.32 (m, 1H), 1.34 (s, 9H). 13C NMR (125 MHz, DMSO-d6) δ 169.0, 164.9, 159.0 (d, J = 245.5 Hz), 155.6, 141.7, 138.0, 134.0, 131.6 (d, J = 2.6 Hz), 131.2, 130.8 (d, J = 8.1 Hz), 128.4, 127.0, 126.0, 125.2 (d, J = 15.7 Hz), 124.6 (d, J = 3.4 Hz), 120.0, 115.8, 115.6 (d, J = 4.0), 34.8, 30.9. LCMS Retention time: 4.140 min. LCMS purity 97.3%. HRMS (ESI) m/z calcd for C24H21ClFNO3 [M+H]+ 426.1273, found 426.1257.
4-(4-(tert-Butylbenzamido)-3′,6-dichloro-[1,1′-biphenyl]-3-carboxylic acid (4q)
Synthesized as described for compound 4f. Isolated 4-(4-(tert-butylbenzamido)-3′,6-dichloro-[1,1′-biphenyl]-3-carboxylic acid 4q (0.0276 g, 0.062 mmol, 54% yield) as a white solid. 1H NMR (400 MHz, DMSO-d6) δ 12.26 (s, 1H), 8.99 (s, 1H), 8.02 (s, 1H), 7.92 (d, J = 8.5 Hz, 2H), 7.65 (d, J = 8.5 Hz, 2H), 7.57 – 7.55 (m, 1H), 7.54 – 7.52 (m, 2H), 7.47 – 7.44 (m, 1H), 1.34 (s, 9H). 13C NMR (126 MHz, DMSO-d6) δ 169.1, 164.9, 155.6, 141.4, 139.5, 136.7, 133.6, 133.0, 132.5, 131.2, 130.3, 129.0, 128.1, 128.0, 127.0, 126.0, 120.4, 115.6, 34.8, 30.9. LCMS Retention time: 2.693 min. LCMS purity 95.5%. HRMS (ESI) m/z calcd for C24H21Cl2NO3 [M+H]+ 442.0977, found 442.0961.
4-(4-(tert-Butylbenzamido)-6-chloro-[1,1′-biphenyl]-3,4′-dicarboxylic acid (4r)
Synthesized as described for compound 4f. Isolated 4-(4-(tert-butylbenzamido)-6-chloro-[1,1′-biphenyl]-3,4′-dicarboxylic acid 4r (0.014 g, 0.031 mmol, 32% yield). 1H NMR (400 MHz, DMSO-d6) δ 13.01 (br s, 1H), 12.38 (br s, 1H), 9.01 (s, 1H), 8.06 (s, 1H), 8.05 (d, J = 8.4 Hz, 2H), 7.93 (d, J = 8.6 Hz, 2H), 7.65 (d, J = 8.5 Hz, 2H), 7.63 (d, J = 8.4 Hz, 2H), 1.34 (s, 9H). 13C NMR (126 MHz, DMSO-d6) δ 169.0, 167.0, 164.9, 155.6, 141.8, 141.4, 136.5, 133.6, 132.9, 131.2, 130.2, 129.6, 129.3, 127.0, 125.9, 120.4, 34.8, 30.9. LCMS Retention time: 3.821 min. LCMS purity 100%. HRMS (ESI) m/z calcd for C25H22ClNO5 [M+H]+ 452.1266, found 452.1280.
2-(4-(tert-Butylbenzamido)-4-chloro-5-(pyridin-4-yl)benzoic acid (4s)
Prepared as described for compound 4g. Isolated 2-(4-(tert-butylbenzamido)-4-chloro-5-(pyridin-4-yl)benzoic acid 4s (0.017 g, 0.042 mmol, 39% yield) as a yellow solid, mp 221 – 223 °C. 1H NMR (400 MHz, DMSO-d6) δ 12.58 (s, 1H), 9.01 (s, 1H), 8.70 (d, J = 6.1 Hz, 2H), 8.07 (s, 1H), 7.93 (d, J = 8.5 Hz, 2H), 7.65 (d, J = 8.5 Hz, 2H), 7.55 (d, J = 6.1 Hz, 2H), 1.34 (s, 9H). 13C NMR (126 MHz, DMSO-d6) δ 168.9, 164.9, 155.6, 149.7 (2), 145.2, 141.9, 136.1, 133.5, 131.2, 131.1, 127.1, 125.9, 124.2, 120.4, 38.8, 30.9. LCMS Retention time: 3.701 min. LCMS purity 97.3%. HRMS (ESI) m/z calcd for C23H21ClN2O3 [M+H]+ 409.1320, found 409.1315.
2-(4-(tert-Butylbenzamido)-4-chloro-5-(pyridin-3-yl)benzoic acid (4t)
Prepared as described for compound 4f. Isolated 2-(4-(tert-butyl)benzamido)-4-chloro-5-(pyridin-3-yl)benzoic acid 4t (0.031 g, 0.076 mmol, 29% yield) as a white solid, mp 262 – 264 °C. 1H NMR (400 MHz, DMSO-d6) δ 12.45 (br s, 1H), 9.01 (s, 1H), 8.70 – 8.68 (m, 1H), 8.66 – 8.64 (m, 1H), 8.07 (s, 1H), 7.96 – 7.91 (m, 3H), 7.65 (d, J = 8.5 Hz, 2H), 7.56 – 7.52 (m, 1H), 1.34 (s, 9H). 13C NMR (126 MHz, DMSO-d6) δ 169.0, 164.9, 155.6, 149.5, 149.0, 141.6, 136.9, 136.8, 133.8, 133.5, 131.2, 130.6, 127.0, 126.0, 123.4, 120.3, 116.3, 34.8, 30.9. LCMS Retention time: 3.753 min. LCMS purity 100%. HRMS (ESI) m/z calcd for C23H21ClN2O3 [M+H]+ 409.1320, found 409.1308.
2-(4-(tert-Butylbenzamido)-4-chloro-5-(thiophen-2-yl)benzoic acid (4u)
Prepared as described for previous analogs although the coupling method was modified as follows: To a microwave vial was added methyl 2-amino-4-chloro-5-iodobenzoate 3e (0.056 g, 0.180 mmol), 2-dicyclohexylphosphino-2′,6′-di-i-propoxy-1,1′-biphenyl (10 mg, 0.022 mmol), palladium (II) acetate (2.4 mg, 10.8 μmol), potassium trifluoro(thiophen-2-yl)borate (0.036 g, 0.189 mmol) and sodium carbonate (0.038 g, 0.360 mmol). The vial was evacuated with argon 3 times and then degassed ethanol (1 mL) was added. The reaction heated at 100 °C for 30 min in the microwave. After cooling to rt, the reaction was diluted with EtOAc (10 mL) and washed with saturated NaHCO3 (10 mL). The EtOAc layer was separated, dried (MgSO4), filtered and concentrated. Purification by reverse-phase MPLC (10 - 100% MeCN:water) produced methyl 2-amino-4-chloro-5-(thiophen-2-yl)benzoate (0.026 g, 0.097 mmol, 54% yield). 1H NMR (400 MHz, CDCl3) δ 8.01 (s, 1H), 7.32 (dd, J = 5.2, 1.2 Hz, 1H), 7.20 (dd, J = 3.6, 1.2 Hz, 1H), 7.07 (dd, J = 5.2, 3.6 Hz, 1H), 6.80 (s, 1H), 5.81 (s, 2H), 3.87 (s, 3H). This intermediate was acylated as described for compound 4d (step 2) to afford methyl 2-(4-(tert-butylbenzamido)-4-chloro-5-(thiophen-2-yl)benzoate. 1H NMR (400 MHz, CDCl3) δ 12.02 (s, 1H), 9.20 (s, 1H), 8.24 (s, 1H), 7.99 (d, J = 8.5 Hz, 2H), 7.56 (d, J = 8.5 Hz, 2H), 7.41 (dd, J = 5.2, 1.2 Hz, 1H), 7.36 (dd, J = 3.6, 1.2 Hz, 1H), 7.12 (dd, J = 5.1, 3.6 Hz, 1H), 3.98 (s, 3H), 1.37 (s, 9H). Hydrolysis of methyl 2-(4-(tert-butyl)benzamido)-4-chloro-5-(thiophen-2-yl)benzoate 5u as described for compound 4d (step 3) afforded, after reverse-phase MPLC (10 – 100% MeCN:water) purification, 2-(4-(tert-butylbenzamido)-4-chloro-5-(thiophen-2-yl)benzoic acid 4u (0.012 g, 0.029 mmol, 83% yield). 1H NMR (400 MHz, DMSO-d6) δ 12.67 (s, 1H), 8.98 (s, 1H), 8.22 (s, 1H), 7.92 (d, J = 8.5 Hz, 2H), 7.71 (dd, J = 5.1, 1.2 Hz, 1H), 7.63 (d, J = 8.5 Hz, 2H), 7.46 (dd, J = 3.6, 1.2 Hz, 1H), 7.20 (dd, J = 5.1, 3.6 Hz, 1H), 1.34 (s, 9H). 13C NMR (126 MHz, DMSO-d6) δ 168.84, 164.81, 155.50, 140.88, 138.22, 135.65, 133.45, 131.30, 128.03, 127.67, 127.41, 127.04, 126.50, 125.92, 120.67, 34.84, 30.88. LCMS Retention time: 1.79 min. LCMS purity 97.0%. HRMS (ESI) m/z calcd for C22H20ClNO3S [M+H]+ 414.0931, found 414.0918.
2-(4-(tert-Butylbenzamido)-4-chloro-5-(thiophen-3-yl)benzoic acid (4v)
Prepared as described for compound 4f. Isolated 2-(4-(tert-butylbenzamido)-4-chloro-5-(thiophen-3-yl)benzoic acid 4v (0.012 g, 0.029 mmol, 20% yield). 1H NMR (400 MHz, DMSO-d6) δ 14.69 (s, 1H), 8.90 (s, 1H), 8.13 (s, 1H), 7.96 (d, J = 8.5 Hz, 2H), 7.71 (dd, J = 3.0, 1.3 Hz, 1H), 7.65 (dd, J = 5.0, 3.0 Hz, 1H), 7.60 (d, J = 8.5 Hz, 2H), 7.36 (dd, J = 5.0, 1.4 Hz, 1H), 1.34 (s, 9H). 13C NMR (126 MHz, DMSO-d6) δ 168.48, 164.58, 154.87, 140.78, 138.50, 133.59, 132.00, 128.64, 127.91, 127.09, 125.90, 125.66, 124.26, 119.50, 34.75, 30.92. LCMS Retention time: 1.67 min. LCMS purity 100%. HRMS (ESI) m/z calcd for C22H20ClNO3S [M+H]+ 414.0931, found 414.0922.
2-(4-(tert-Butylbenzamido)-4-chloro-5-(furan-2-yl)benzoic acid (4w)
Prepared as described for compound 4v. Isolated 2-(4-(tert-butylbenzamido)-4-chloro-5-(furan-2-yl)benzoic acid 4w (0.007 g, 0.018 mmol, 40% yield). 1H NMR (400 MHz, DMSO-d6) δ 13.41 (s, 1H), 8.95 (s, 1H), 8.52 (s, 1H), 7.93 (d, J = 8.4 Hz, 2H), 7.86 (dd, J = 1.8, 0.7 Hz, 1H), 7.61 (d, J = 8.5 Hz, 2H), 7.14 - 7.09 (m, 1H), 6.68 (dd, J = 3.5, 1.8 Hz, 1H), 1.33 (s, 9H). 13C NMR (126 MHz, DMSO-d6) δ 168.83, 164.72, 155.26, 148.73, 143.15, 140.42, 132.56, 131.53, 130.23, 127.03, 125.82, 122.45, 120.53, 112.11, 110.36, 34.79, 30.88. LCMS Retention time: 2.82 min. LCMS purity 98.9%. HRMS (ESI) m/z calcd for C22H20ClNO4 [M+H]+ 398.1160, found 398.1149.
2-(4-(tert-Butylbenzamido)-4-chloro-5-(furan-3-yl)benzoic acid (4x)
Prepared as described for compound 4f. Isolated 2-(4-(tert-butylbenzamido)-4-chloro-5-(furan-3-yl)benzoic acid 4x (0.008 g, 0.020 mmol, 75% yield). 1H NMR (400 MHz, DMSO-d6) δ 13.82 (s, 1H), 8.92 (s, 1H), 8.18 (s, 1H), 8.16 - 8.09 (m, 1H), 7.93 (d, J = 8.4 Hz, 1H), 7.80 (t, J = 1.7 Hz, 1H), 7.60 (d, J = 8.5 Hz, 1H), 6.88 (dd, J = 1.9, 0.9 Hz, 1H), 1.33 (s, 8H). 13C NMR (126 MHz, DMSO-d6) δ 168.82, 164.63, 155.08, 143.42, 141.19, 140.46, 134.36, 132.66, 131.73, 127.04, 125.74, 124.40, 122.44, 120.02, 110.89, 34.77, 30.89. LCMS Retention time: 2.70 min. LCMS purity 95.7%. HRMS (ESI) m/z calcd for C22H20ClNO4 [M+H]+ 398.1160, found 398.1145.
2-(4-(tert-Butylbenzamido)-4-chloro-5-(2,3-dihydrobenzo[b][1,4]dioxin-6-yl)benzoic acid (4y)
Prepared as described for compound 4f. Isolated 2-(4-(tert-butylbenzamido)-4-chloro-5-(2,3-dihydrobenzo[b][1,4]dioxin-6-yl)benzoic acid 4y (0.042 g, 0.090 mmol, 92% yield). 1H NMR (400 MHz, DMSO-d6) δ 12.21 (s, 1H), 8.93 (s, 1H), 7.97 (s, 1H), 7.90 (d, J = 8.5 Hz, 2H), 7.63 (d, J = 8.5 Hz, 2H), 6.98 - 6.88 (m, 3H), 4.29 (s, 4H), 1.33 (s, 9H). 13C NMR (126 MHz, DMSO-d6) δ 169.21, 164.78, 155.54, 143.34, 143.02, 140.68, 136.81, 133.53, 133.48, 131.25, 130.58, 126.99, 125.94, 122.24, 120.38, 117.89, 117.00, 115.59, 64.18, 64.12, 34.84, 30.87. LCMS Retention time: 1.59 min. LCMS purity 96.5%. HRMS (ESI) m/z calcd for C26H24ClNO5 [M+H]+ 466.1422, found 466.1401.
2-(4-(tert-Butylbenzamido)-4-chloro-5-(benzo[d][1,3]dioxol-5-yl)benzoic acid (4z)
Prepared as described for compound 4f. Isolated 2-(4-(tert-butylbenzamido)-4-chloro-5-(benzo[d][1,3]dioxol-5-yl)benzoic acid 4z (0.025 g, 0.055 mmol, 92% yield). 1H NMR (400 MHz, DMSO-d6) δ 12.21 (s, 1H), 8.94 (s, 1H), 7.98 (s, 1H), 7.90 (d, J = 8.5 Hz, 2H), 7.63 (d, J = 8.5 Hz, 2H), 7.04 (d, J = 1.7 Hz, 1H), 7.02 (d, J = 8.0 Hz, 1H), 6.91 (dd, J = 8.0, 1.8 Hz, 1H), 6.09 (s, 2H), 1.33 (s, 9H). 13C NMR (126 MHz, DMSO-d6) δ 169.20, 164.79, 155.55, 147.15, 147.08, 140.76, 136.95, 133.73, 133.60, 131.31, 131.23, 126.99, 125.94, 122.99, 120.36, 115.52, 109.71, 108.27, 101.32, 34.84, 30.87. LCMS Retention time: 1.59 min. LCMS purity 96.8%. HRMS (ESI) m/z calcd for C25H22ClNO5 [M+H]+ 452.1265, found 452.1269.
2-(4-(tert-Butylbenzamido)-4-chloro-5-(1H-pyrrol-1-yl)benzoic acid (4aa)
To a vial was added methyl 2-(4-(tert-butylbenzamido)-4-chloro-5-iodobenzoate (0.027 g, 0.056 mmol), copper powder (1.0 mg, 0.016 mmol), pyrrole (6.0 μL, 0.084 mmol), cesium carbonate (0.064 g, 0.197 mmol) and acetonitrile (0.5 mL). The vial was then purged with argon for 15 minutes then heated at 80 °C for 21 h. The reaction was cooled to rt, diluted with EtOAc (5 mL), filtered through Celite, concentrated and purified with reverse-phase MPLC (10 - 100% MeCN:water) to directly produce 2-(4-(tert-butylbenzamido)-4-chloro-5-(1H-pyrrol-1-yl)benzoic acid 4aa without need for hydrolysis (0.013 g, 0.033 mmol, 58% yield). 1H NMR (400 MHz, DMSO-d6) δ 14.00 (s, 1H), 8.97 (s, 1H), 7.99 (s, 1H), 7.95 (d, J = 8.4 Hz, 2H), 7.61 (d, J = 8.5 Hz, 2H), 7.01 (t, J = 2.1 Hz, 2H), 6.26 (t, J = 2.1 Hz, 2H), 1.33 (s, 9H). 13C NMR (126 MHz, DMSO-d6) δ 164.71, 155.17, 140.55, 131.96, 131.62, 130.10, 127.09, 125.79, 125.76, 122.28, 120.00, 109.21, 34.78, 30.89. LCMS Retention time: 2.678 min. LCMS purity 95.8%. HRMS (ESI) m/z calcd for C22H21ClN2O3 [M+H]+ 397.1320, found 397.1316.
2-(4-(tert-Butylbenzamido)-4-chloro-5-(1H-pyrrol-2-yl)benzoic acid (4bb)
Step 1: synthesis of tert-butyl 2-(4-amino-2-chloro-5-(methoxycarbonyl)phenyl)-1H-pyrrole-1-carboxylate
To a microwave vial was added the methyl 2-amino-4-chloro-5-iodobenzoate 3e (0.056 g, 0.180 mmol), 2-dicyclohexylphosphino-2′,6′-di-i-propoxy-1,1′-biphenyl (10 mg, 0.022 mmol), palladium (II) acetate (2.4 mg, 11 μmol), potassium (1-(tert-butoxycarbonyl)-1H-pyrrol-2-yl)trifluoroborate (0.052 g, 0.189 mmol) and sodium carbonate (0.038 g, 0.360 mmol). The vial was evacuated with argon 3 times and then degassed ethanol (3 mL) was added. The reaction was heated at 100 °C for 30 min in the microwave, followed by cooling to rt. The reaction mixture was diluted with EtOAc (12 mL) and washed with saturated NaHCO3 (12 mL). The EtOAc layer was separated, dried with MgSO4, filtered and concentrated. The reaction was purified by reverse-phase MPLC (10 - 100% MeCN:water) to produce tert-butyl 2-(4-amino-2-chloro-5-(methoxycarbonyl)phenyl)-1H-pyrrole-1-carboxylate (0.043 g, 0.123 mmol, 68% yield). 1H NMR (400 MHz, CDCl3) δ 7.83 (s, 1H), 7.37 (dd, J = 3.4, 1.8 Hz, 1H), 6.75 (s, 1H), 6.23 (t, J = 3.3 Hz, 1H), 6.13 (dd, J = 3.3, 1.8 Hz, 1H), 3.85 (s, 3H), 1.37 (s, 9H).
Step 2: Synthesis of methyl 2-(4-(tert-butylbenzamido)-4-chloro-5-(1H-pyrrol-2-yl)benzoate
Tert-butyl 2-(4-amino-2-chloro-5-(methoxycarbonyl)phenyl)-1H-pyrrole-1-carboxylate was treated with 4-(tert-butylbenzoyl chloride as described for compound 4d (step 2) which directly afforded the un-BOC-protected product, methyl 2-(4-(tert-butylbenzamido)-4-chloro-5-(1H-pyrrol-2-yl)benzoate in (0.021 g, 0.051 mmol, 42% yield). 1H NMR (400 MHz, CDCl3) δ 11.94 (s, 1H), 9.10 (s, 2H), 8.24 (s, 1H), 8.01 - 7.91 (m, 2H), 7.59 - 7.49 (m, 2H), 6.99 - 6.86 (m, 1H), 6.66 - 6.53 (m, 1H), 6.38 - 6.24 (m, 1H), 3.96 (s, 3H), 1.37 (s, 9H).
Step 3: Synthesis of 2-(4-(tert-butylbenzamido)-4-chloro-5-(1H-pyrrol-2-yl)benzoic acid (4bb)
Methyl 2-(4-(tert-butylbenzamido)-4-chloro-5-(1H-pyrrol-2-yl)benzoate was hydrolyzed as described for compound 4f (step 3). Isolated 2-(4-(tert-butylbenzamido)-4-chloro-5-(1H-pyrrol-2-yl)benzoic acid 4bb (0.020 g, 0.050 mmol, 99% yield). 1H NMR (400 MHz, DMSO-d6) δ 12.16 (s, 1H), 11.37 (s, 1H), 8.92 (s, 1H), 8.24 (s, 1H), 7.90 (d, J = 8.5 Hz, 2H), 7.63 (d, J = 8.5 Hz, 2H), 6.96 - 6.89 (m, 1H), 6.63 - 6.56 (m, 1H), 6.22 - 6.15 (m, 1H), 1.34 (s, 9H). 13C NMR (126 MHz, DMSO-d6) δ 169.29, 164.64, 155.42, 139.08, 134.87, 131.33, 126.92, 126.44, 126.28, 125.88, 119.74, 115.81, 109.97, 108.71, 34.79, 30.85. LCMS Retention time: 2.60 min. LCMS purity 96.5%. HRMS (ESI) m/z calcd for C22H21ClN2O3 [M+H]+ 397.1320, found 397.1309.
2-(4-(tert-Butylbenzamido)-4-chloro-5-(1H-pyrazol-1-yl)benzoic acid (4cc)
Synthesized as described for compound 4aa. Purification by reverse-phase MPLC (10 - 100% MeCN:water) produced 2-(4-(tert-butylbenzamido)-4-chloro-5-(1H-pyrazol-1-yl)benzoic acid 6cc (0.010 g, 0.024 mmol, 38% yield). 1H NMR (400 MHz, DMSO-d6) δ 12.42 (s, 1H), 9.04 (s, 1H), 8.20 (dd, J = 2.5, 0.6 Hz, 1H), 8.15 (s, 1H), 7.93 (d, J = 8.5 Hz, 2H), 7.79 (dd, J = 1.9, 0.6 Hz, 1H), 7.65 (d, J = 8.5 Hz, 2H), 6.60 - 6.51 (m, 1H), 1.34 (s, 9H). 13C NMR (126 MHz, DMSO-d6) δ 168.50, 164.93, 155.67, 141.08, 140.94, 132.88, 132.17, 132.15, 131.10, 130.00, 127.07, 125.97, 120.68, 107.03, 34.86, 30.87. LCMS Retention time: 4.037 min. LCMS purity 95.8%. HRMS (ESI) m/z calcd for C21H20ClN3O3 [M+H]+ 398.1272, found 398.1266.
2-(4-(tert-Butylbenzamido)-4-chloro-5-(1H-imidazol-1-yl)benzoic acid (4dd)
Prepared as described for compound 4aa. Isolated 2-(4-(tert-butylbenzamido)-4-chloro-5-(1H-imidazol-1-yl)benzoic acid 4dd (0.007 g, 0.018 mmol, 27% yield). 1H NMR (400 MHz, DMSO-d6) δ 13.10 (s, 1H), 9.04 (s, 1H), 8.22 (s, 1H), 8.11 (s, 1H), 7.98 - 7.89 (m, 2H), 7.67 - 7.62 (m, 2H), 7.60 (s, 1H), 7.27 (s, 1H), 1.34 (s, 9H). 13C NMR (126 MHz, DMSO-d6) δ 168.14, 164.93, 155.58, 141.81, 137.83, 133.31, 131.19, 130.46, 128.41, 127.36, 127.12, 125.91, 121.65, 120.33, 34.84, 30.87. LCMS Retention time: 3.015 min. LCMS purity 100%. HRMS (ESI) m/z calcd for C21H20ClN3O3 [M+H]+ 398.1272, found 398.1259.
2-(4-(tert-Butylbenzamido)-4-chloro-5-(1H-tetrazol-5-yl)benzoic acid (4ee)
Step 1: synthesis of methyl 2-amino-4-chloro-5-cyanobenzoate
To a vial was added the methyl 2-amino-4-chloro-5-iodobenzoate 3e (0.29 g, 0.931 mmol) and copper (I) cyanide (0.17 g, 1.86 mmol) in DMF (15 mL). After heating at 120 °C for 6 h, the reaction was cooled to rt, diluted with saturated NH3Cl in water (30 mL) and extracted with EtOAc (3 × 30 mL). The separated EtOAc layers were combined and washed with sat. NH3Cl (1 × 100mL), water (3 × 100 mL) and dried (MgSO4), filtered, adsorbed to silica then purified by MPLC (0 - 30% EtOAc:hexanes) to produce methyl 2-amino-4-chloro-5-cyanobenzoate (0.15 g, 0.722 mmol, 78% yield). 1H NMR (400 MHz, CDCl3) δ 8.19 (s, 1H), 6.75 (s, 1H), 6.35 (s, 2H), 3.90 (s, 3H).
Step 2: synthesis of methyl 2-(4-(tert-butylbenzamido)-4-chloro-5-cyanobenzoate
Methyl 2-amino-4-chloro-5-cyanobenzoate was treated with 4-(tert-butylbenzoyl chloride as described for compound 4d (step 2) which afforded, after purification, methyl 2-(4-tert-butyl)benzamido)-4-chloro-5-cyanobenzoate (0.038 g, 0.102 mmol, 53% yield). 1H NMR (400 MHz, CDCl3) δ 12.23 (s, 1H), 9.26 (s, 1H), 8.39 (s, 1H), 7.96 (d, J = 8.5 Hz, 2H), 7.57 (d, J = 8.5 Hz, 2H), 4.02 (s, 3H), 1.37 (s, 9H).
Step 3: synthesis of 2-(4-tert-butylbenzamido)-4-chloro-5-cyanobenzoic acid
Hydrolyzed as described for compound 4d (step 3) that afforded, after purification, 2-(4-tert-butylbenzamido)-4-chloro-5-cyanobenzoic acid (0.005 g, 0.014 mmol, 87% yield). 1H NMR (400 MHz, DMSO-d6) δ 12.59 (s, 1H), 9.04 (s, 1H), 8.52 (s, 1H), 7.91 (d, J = 8.5 Hz, 2H), 7.65 (d, J = 8.5 Hz, 2H), 1.34 (s, 9H). 13C NMR (126 MHz, DMSO-d6) δ 168.03, 165.24, 156.09, 145.36, 140.32, 137.45, 130.70, 127.20, 126.07, 119.80, 116.22, 115.56, 105.39, 34.90, 30.84. LCMS Retention time: 2.536 min. LCMS purity 100%. HRMS (ESI) m/z calcd for C19H17ClN2O3 [M+H]+ 357.1007, found 357.0995.
Step 4: synthesis of 2-(4-(tert-butylbenzamido)-4-chloro-5-(1H-tetrazol-5-yl)benzoic acid (4ee)
To a vial was added 2-(4-(tert-butylbenzamido)-4-chloro-5-cyanobenzoic acid (0.030 g, 0.083 mmol), copper(I) iodide (0.8 mg, 4 μmol) and trimethylsilyl azide (0.016 mL, 0.124 mmol). The DMF (0.25 mL) and MeOH (0.028 mL) were added and the reaction was purged with argon then heated to 90 °C for 18 h. After cooling to rt, the reaction was filtered through Celite, and the filtrate was collected, concentrated and purified by RP MPLC (10 - 100% MeCN:water). Isolated 2-(4-tert-butylbenzamido)-4-chloro-5-(1H-tetrazol-5-yl)benzoic acid 4ee (0.012 g, 0.030 mmol, 36% yield). 1H NMR (400 MHz, DMSO-d6) δ 15.21 (s, 1H), 8.87 (s, 1H), 8.49 (s, 1H), 7.97 (d, J = 8.4 Hz, 2H), 7.59 (d, J = 8.5 Hz, 2H), 1.34 (s, 9H). 13C NMR (126 MHz, DMSO-d6) δ 169.32, 164.52, 154.69, 140.91, 133.99, 132.86, 132.27, 127.11, 125.61, 119.28, 34.30, 30.96. LCMS Retention time: 2.07 min. LCMS purity 96.8%. HRMS (ESI) m/z calcd for C19H18ClN5O3 [M+H]+ 400.1177, found 400.1173.
2-(4-(tert-Butylbenzamido)-4-chloro-5-(2-methyl-2H-tetrazol-5-yl)benzoic acid (4ff)
Prepared from methyl 2-(4-tert-butylbenzamido)-4-chloro-5-(1H-tetrazol-5-yl)benzoate, obtained through the reaction sequence for compound 4ee that was not hydrolyzed.
Step 1: synthesis of both methyl 2-(4-(tert-butylbenzamido)-4-chloro-5-(1-methyl-1H-tetrazol-5-yl)benzoate and methyl 2-(4-tert-butylbenzamido)-4-chloro-5-(2-methyl-2H-tetrazol-5-yl)benzoate
To a vial containing methyl 2-(4-(tert-butylbenzamido)-4-chloro-5-(1H-tetrazol-5-yl)benzoate (0.017 g, 0.040 mmol) was added CH2Cl2 (0.30 mL) with MeOH (0.20 mL). TMS-diazomethane (0.036 mL, 0.072 mmol) was added at rt, and the resulting mixture was stirred at rt for 18 h. After quenching with saturated NaHCO3 (2 mL), the aqueous phase was extracted with CH2Cl2 (3 × 2 mL), dried (MgSO4), filtered, adsorbed to Celite and purified by reverse-phase MPLC (10 - 100% MeCN:water). Two peaks were isolated, corresponding to the two methylated isomers. First fraction isolated was identified as methyl 2-(4-tert-butylbenzamido)-4-chloro-5-(1-methyl-1H-tetrazol-5-yl)benzoate (0.006 g, 0.015 mmol, 37% yield). 1H NMR (400 MHz, CDCl3) δ 12.07 (s, 1H), 9.20 (s, 1H), 8.14 (s, 1H), 7.88 (d, J = 8.5 Hz, 2H), 7.46 (d, J = 8.5 Hz, 2H), 3.90 (s, 3H), 3.86 (s, 3H), 1.25 (s, 9H). The second peak isolated corresponded to methyl 2-(4-tert-butylbenzamido)-4-chloro-5-(2-methyl-2H-tetrazol-5-yl)benzoate (0.0094 g, 0.022 mmol, 55% yield). 1H NMR (400 MHz, CDCl3) δ 12.01 (s, 1H), 9.15 (s, 1H), 8.63 (s, 1H), 7.87 (d, J = 8.5 Hz, 2H), 7.44 (d, J = 8.5 Hz, 2H), 4.33 (s, 3H), 3.87 (s, 3H), 1.25 (s, 9H).
Step 2: synthesis of 2-(4-tert-butylbenzamido)-4-chloro-5-(2-methyl-2H-tetrazol-5-yl)benzoic acid
Prepared as described for compound 4d, (step 3). Purified by reverse-phase MPLC (10 – 100% MeCN:water) to afford 2-(4-(tert-butylbenzamido)-4-chloro-5-(2-methyl-2H-tetrazol-5-yl)benzoic acid 4ff (0.008 g, 0.018 mmol, 84% yield). 1H NMR (400 MHz, DMSO-d6) δ 12.68 (s, 1H), 9.06 (s, 1H), 8.63 (s, 1H), 7.93 (d, J = 8.5 Hz, 2H), 7.65 (d, J = 8.5 Hz, 2H), 4.48 (s, 3H), 1.34 (s, 9H). 13C NMR (126 MHz, DMSO-d6) δ 168.67, 165.00, 155.65, 142.88, 133.69, 131.18, 127.26, 127.10, 125.95, 120.82, 119.71, 34.85, 30.87, 29.01. LCMS Retention time: 2.56 min. LCMS purity 95.6%. HRMS (ESI) m/z calcd for C20H20ClN5O3 [M+H]+ 414.1334, found 414.1299.
Biochemical Assays
Recombinant TbHK1 was expressed and purified as described.18 Recombinant LmHK was expressed and purified to 99% homogeneity (as determined by Coomassie Blue staining of an SDS-PAGE gel) using a similar approach, with the only modification being the elimination of detergent in the lysis buffer, a step replaced sonication to lyse the cells. HK assays were performed in in triplicate as described using a coupled reaction to measure enzyme activity.25 In short, the coupled assay employed glucose-6-phosphate dehydrogenase (G6PDH) to convert glucose-6-phosphate (G6-P) generated by HK to 6-phosphogluconate with coincident reduction of NADP to NADPH, which was monitored spectrophotometrically at 340 nm. Kinetic analyses were performed using KaleidaGraph 4.1 (Synergy Software, Reading, PA). To test inhibitors, enzyme in 195 μL of assay buffer (5.25 mM ATP, 3.3 mM MgCl2, 0.75 mM NADP, 2 units/μL G6PDH, and 50 mM triethanolamine (pH 7.4)) was mixed with test and control compounds. Negative (vehicle) controls contained 1% DMSO, positive controls contained either ebselen or SID 17387000, a structural analog of ebselen.18 Following incubation (15′, RT), glucose (20 mM) was added to initiate the reaction. Enzyme activity as reflected in change in absorbance at OD340 was then measured on a Synergy H1 Hybrid Reader (Biotek, Winooski, VT). Kinetic analyses were performed using KaleidaGraph 4.1 (Synergy Software, Reading, PA). Specificity assays were performed as described previously using human HK 4 (human glucokinase, hGlk, GenBank accession no. BC001890).18 To identify compounds that interfered with the reporter enzyme, counterassays were assembled without HK but with G6-P and G6PDH in reaction buffer and the impact of inhibitor scored on the reaction.
Whole Parasite Viability Assays
To determine the impact of TbHK1 inhibitors on cell growth, we seeded 5 × 103 bloodstream form (BSF) parasites (cell line 90-13, a 427 strain) into 96-well clear-bottomed polystyrene plates in 200 μL HMI-9 supplemented with 10% fetal bovine serum and 10% Serum Plus (Sigma-Aldrich, St. Louis, MO) in the presence of compound (2 μL) or equivalently diluted carrier for 3 days in 5% CO2 at 37 °C. CellTiter Blue (Promega, Madison WI) was added and the plates incubated an additional 3 hr under standard culture conditions. Fluorescence emission at A585 was then measured after excitation at A546. DMSO solvent was maintained at or below 1%, with 1% causing a 16% reduction in cell number at the end of the three-day assay. Averages of the triplicates were calculated and fit to dose-response curves for the determination of EC50 values.
Human Cell Line Toxicity Testing
Toxicity of compounds against IMR-90 human cells was scored as previously described.18 Briefly, viability was determined in 25 μL final volume assays using a 384-well plate-based assay with 1,000 cells/22 μL seeded into each well. Cells were cultured in complete growth medium (according to ATCC specifications, ATCC, Manassas VA). Test and control compounds (3 μL) were then added, with vehicle and positive controls (1% and 10% DMSO, respectively). Plates were incubated 44-46 h (37 °C, 5% CO2) and 5 μL CellTiter Blue (Promega) reagent added. Following an additional 2-4 h incubation period, data were captured on a SpectraMax M5 (Molecular Devices, LLC., Sunnyvale, CA) with EC50 values calculated using GraphPad Prism 6.0.
Supplementary Material
Acknowledgments
J.C. Morris thanks Dr. Cuixiang Wan and Mark Griffith for their expert technical support in this project. The authors gratefully acknowledge funding from the following sources. Medicinal chemistry efforts were carried out by J.E. Golden at the University of Kansas Specialized Chemistry Center with NIH support U54HG005031 to J. Aubé. Support for the University of Kansas NMR instrumentation was provided by NIH Shared Instrumentation Grant S10RR024664 and NSF Major Research Instrumentation Grant 0320648. Parasitology assays supporting post-ML205 development were supported by NIH grants 1 R03 MH082340-01A1 and 1R15AI075326 to J. C. Morris. Support for the Leishmania parasite-based and mammalian cell line-based growth inhibition assays was provided by the Fiske Drug Discovery Laboratory at the University of Virginia.
Abbreviations
- ADME
BSF, bloodstream form
- cLogP
calculated partition coefficient of a compound in octanol and water
- HAT
human African trypanosomiasis
- hGlk
human glucokinase
- LmHK
Leishmania major hexokinase
- MLSCN
Molecular Libraries Screening Centers Network
- MLSMR
Molecular Libraries Small Molecule Repository
- MW
microwave irradiation
- PBS
phosphate buffered saline
- rTbHK1
recombinant Trypanosoma brucei hexokinase 1
- TbHK
Trypanosoma brucei hexokinase
- SAR
structure activity relationship
Footnotes
Supporting information for this article is given via a link at the end of the document.
References
- 1.Kioy D, Jannin J, Mattock N. Nat Rev Micro. 2004;2(3):186–187. doi: 10.1038/nrmicro848. [DOI] [PubMed] [Google Scholar]
- 2.Lockman JW, Hamilton AD. Curr Med Chem. 2005;12(8):945–59. doi: 10.2174/0929867053507289. [DOI] [PubMed] [Google Scholar]
- 3.WHO. Leishmaniasis Fact sheet N°375. 2014 Jan; [Google Scholar]
- 4.WHO. Leishmaniasis. 2014 Jan; [Google Scholar]
- 5.WHO. Trypanosomiasis, human African (sleeping sickness) Fact sheet N°259. 2014 Mar; [Google Scholar]
- 6.Barrett MP, Croft SL. Brit Med Bull. 2012;104(1):175–196. doi: 10.1093/bmb/lds031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ferrins L, Gazdik M, Rahmani R, Varghese S, Sykes ML, Jones AJ, Avery VM, White KL, Ryan E, Charman SA, Kaiser M, Bergstrom CA, Baell JB. J Med Chem. 2014;57(15):6393–402. doi: 10.1021/jm500191u. [DOI] [PubMed] [Google Scholar]
- 8.Chambers JW, Fowler ML, Morris MT, Morris JC. Mol Biochem Parasitol. 2008;158(2):202–7. doi: 10.1016/j.molbiopara.2007.12.013. [DOI] [PubMed] [Google Scholar]
- 9.Trinquier M, Perie J, Callens M, Opperdoes F, Willson M. Bioorg Med Chem. 1995;3(11):1423–7. doi: 10.1016/0968-0896(95)00129-5. [DOI] [PubMed] [Google Scholar]
- 10.Willson M, Sanejouand YH, Perie J, Hannaert V, Opperdoes F. Chem Biol. 2002;9(7):839–47. doi: 10.1016/s1074-5521(02)00169-2. [DOI] [PubMed] [Google Scholar]
- 11.Burchmore RJ, Rodriguez-Contreras D, McBride K, Merkel P, Barrett MP, Modi G, Sacks D, Landfear SM. Proc Natl Acad Sci U S A. 2003;100(7):3901–6. doi: 10.1073/pnas.0630165100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.McConville MJ, Naderer T. Ann Rev Microbiol. 2011;65(1):543–561. doi: 10.1146/annurev-micro-090110-102913. [DOI] [PubMed] [Google Scholar]
- 13.Naderer T, Heng J, Saunders EC, Kloehn J, Rupasinghe TW, Brown TJ, McConville MJ. PLOS Pathogens. 2015;11(9):e1005136. doi: 10.1371/journal.ppat.1005136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Joice AC, Lyda TL, Sayce AC, Verplaetse E, Morris MT, Michels PAM, Robinson DR, Morris JC. Int J Parasitol. 2012;42(4):401–409. doi: 10.1016/j.ijpara.2012.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Umasankar PK, Jayakumar PC, Shouche YS, Patole MS. J Parasitol. 2005;91(6):1504–9. doi: 10.1645/GE-502R1.1. [DOI] [PubMed] [Google Scholar]
- 16.Chambers JW, Kearns MT, Morris MT, Morris JC. J Biol Chem. 2008;283(22):14963–14970. doi: 10.1074/jbc.M802124200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sharlow E, Golden JE, Dodson H, Morris M, Hesser M, Lyda T, Leimgruber S, Schroeder CE, Flaherty DP, Weiner WS, Simpson D, Lazo JS, Aubé J, Morris JC. In: Probe Reports from the NIH Molecular Libraries Program [Internet] (US), N. C. f. B. I., editor. National Center for Biotechnology Information (US); [PubMed] [Google Scholar]
- 18.Sharlow ER, Lyda TA, Dodson HC, Mustata G, Morris MT, Leimgruber SS, Lee K-H, Kashiwada Y, Close D, Lazo JS, Morris JC. PLoS Neg Trop Dis. 2010;4(4):e659. doi: 10.1371/journal.pntd.0000659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.van den Hoogenband A, Lange JHM, Terpstra JW, Koch M, Visser GM, Visser M, Korstanje TJ, Jastrzebski JTBH. Tetrahedron Lett. 2008;49(26):4122–4124. [Google Scholar]
- 20.Moseley JD, Murray PM, Turp ER, Tyler SNG, Burn RT. Tetrahedron. 2012;68(30):6010–6017. [Google Scholar]
- 21.Molander GA, Canturk B. Angew Chem Int Ed. 2009;48(49):9240–9261. doi: 10.1002/anie.200904306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhu R, Xing L, Wang X, Cheng C, Su D, Hu Y. Adv Synth Cat. 2008;350(9):1253–1257. [Google Scholar]
- 23.Luceome Luceome KinaseSeeker, mixed kinase panel. 2016 http://www.luceome.com/index.php/technology/kinaseseeker.
- 24.Eurofins; Panlabs, LeadProfiling Screen. 2015 [Google Scholar]
- 25.Morris MT, DeBruin C, Yang Z, Chambers JW, Smith KS, Morris JC. Euk Cell. 2006;5(12):2014–23. doi: 10.1128/EC.00146-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.