

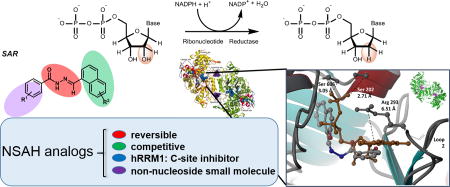
Abstract
Ribonucleotide reductase (RR), an established cancer target is usually inhibited by antimetabolites which display multiple cross-reactive effects. Recently, we discovered a naphthyl salicyl acyl hydrazone-based inhibitor (NSAH or E-3a) of human RR (hRR) binding at the catalytic site (C-site) and inhibiting hRR reversibly. We herein report the synthesis and biochemical characterization of 25 distinct analogs. We designed each analog through docking to the C-site of hRR based on our 2.7Å X-ray crystal structure (PDB ID: 5TUS). Broad tolerance to minor structural variations preserving inhibitory potency is observed. E-3f (82% yield) displayed an in vitro IC50 of 5.3 ± 1.8 µM against hRR, making it the most potent in this series. Kinetic assays reveal that E-3a, E-3c, E-3t and E-3w bind and inhibit hRR through a reversible and competitive mode. Target selectivity towards the R1 subunit of hRR is established, providing a novel way of inhibition of this crucial enzyme.
Graphical Abstract
Introduction
Ribonucleotide reductase (RR) catalyzes the conversion of ribonucleoside substrates into deoxyribonucleosides (Figure 1A), the rate-limiting step in the formation of all deoxyribonucleoside 5'-triphosphates (dNTPs) as building blocks for DNA synthesis and replication.1 Therefore, RR crucially maintains a balanced nucleotide pool in the cell. Consequently, RR has been an established target for many proliferative diseases, including cancer.2 Additionally, RR is a relatively complex cellular target for anti-cancer therapeutic development due to the fact that inhibition of this critical enzyme is likely to cause cell death even in normal cells. This presents an additional layer of challenge to develop RR inhibitors that can target cancer cells selectively.
Figure 1.

A. Conversion of ribonucleoside diphosphates into deoxyribonucleoside diphosphates by RR and its redox coupled activity with thioredoxin; B. Comparison of lead compound Naphthyl Salicyl Acyl Hydrazone (NSAH or E-3a) and natural substrate GDP bound to the C-site of hRR (PDB ID: 5TUS)10; C. Overall approach leading to identification of novel hRR inhibitor whose analogs (E-3a-z) are synthesized and biochemically characterized in this report.
Foundational knowledge on allosteric regulation of this multi-subunit enzyme has been extensively probed through a series of studies conducted on prokaryotic and some eukaryotic RRs.1 Mechanistic details of substrate regulation through allostery also remains a focus of ongoing investigations.3 Recent studies have provided structural insights underlying allosterically driven dNTP regulation by RR.1d–e, 4, 5c Though previous studies establish the molecular details of multimerization as a basis for development of novel anticancer agents,5 there has been little focus on identifying non-nucleosidic, reversible and competitive inhibitors of RR.
With the exceptions of hydroxyurea, triapine, and related radical scavengers that target the R2 subunit, most small-molecule anticancer agents targeting RR belong to the antimetabolite class of antineoplastic agents (see Table S1 for a list of FDA-approved drugs). For example gemcitabine, FDA approved for treatment of breast, ovarian, non-small cell lung and pancreatic cancer, is a nucleoside analog that structurally mimics ribonucleoside diphosphate substrates.6 Gemcitabine inactivates the RR enzymatic machinery by covalently and irreversibly modifying an active site nucleophilic Cys sulfhydryl moiety, causing significant abrogation of enzyme function.6 There is little distinction if any for the action of gemcitabine between RRs in normal cells versus RRs in cancer cells, resulting in toxic side effects in chemotherapy.6 Clofarabine and others inhibit hRR through a non-covalent mode; however, in general the antimetabolite class of inhibitors significantly cross react with essential nucleotide metabolic enzymes causing indiscriminate toxicity. For example, clofarabine triphosphate causes chain termination of DNA polymerase activity.7
Given this background, we hypothesized that reversible, non-covalent inhibitors of hRR that are non-nucleosidic may show reduced cross reactivity, and thereby reduced side effects and toxicity. In 2015, we reported an integrated approach consisting of in silico rapid throughput screening of small molecules, in vitro biochemical assays against hRR, cellular growth inhibition studies against select cancer cell lines, and X-ray crystallography to guide us with future rational design.8 This integrated approach afforded identification of several new classes of non-nucleosidic modulators of hRR with in vitro hRR inhibitory potencies in the micromolar range. Detailed X- ray crystallography revealed the binding of a phthalimide-containing class of modulators maintaining non-covalent stabilizing interactions at the hexamerization interface of hRR.8,9
Subsequent to the 2015 report, we discovered a lead compound, Naphthyl Salicyl Acylhydrazone (NSAH or E-3a) that binds to the catalytic site (C-site) of hRRM1 (Figure 1B).10 E-3a demonstrated significant levels of selective cytotoxicity against multiple cancer cell lines while displaying little cytotoxicity against normal mobilized peripheral blood progenitor cells.10 This result presented us with a lead compound possessing favorable properties worthy of further exploration. In the present study we use the previously determined 2.7 Å crystal structure of this lead inhibitor (E-3a) in complex with hRRM1 (PDB ID: 5TUS)10 as a template for structure-based ligand optimization. We designed suitable analogs of E-3a, docked them to the C-site of hRR using the Schrödinger software suite, and evaluated their docking poses to predict which analogs would show increased inhibitory efficacy relative to the lead compound, E-3a. We then synthesized and biochemically characterized a series of twenty five analogs (E-3a-z) as hRR inhibitors and established preliminary structure-activity evaluations (Figure 1C). A five step, modular synthetic scheme was developed that generally afforded analogs in yields ranging between 63% and 85%, with few exceptions. E-3f was obtained in 82% yield and displayed an in vitro IC50 of 5.3 ± 1.8 µM against hRR, making it the most potent in this series of analogs. Ranking of the analogs by their enzymatic IC50 value reveals that the inclusion of a polar substituent in the ortho position of the salicylic moiety is critical for biological activity, while the substitution of an indole ring for a naphthalene ring showed no loss in activity. Detailed kinetics assays reveal that these inhibitors bind and inactivate hRR through a reversible and competitive mode, consistent with our recently reported finding for the lead inhibitor E-3a.10 Remarkably, this class of compounds are the first non-nucleosidic inhibitors of hRR displaying a competitive, reversible mode of inhibition, consistent with C-site binding.
Results
In silico modeling based upon the X-ray crystal structure of E-3a in complex with hRRM1 (PDB ID: 5TUS) was used to design a focused library of hydrazone-based hRR inhibitors.10 Approximately one hundred distinct analogs with unique substitution patterns surrounding the central hydrazone core were docked to the C-site, where the grid was defined as a 5 Å cube centered around the lead hydrazone ligand. Evaluation of the docking poses for interactions with residues previously established to interact with nucleoside substrates, such as Ser 202, Ser 606, Gly 246, Cys 218, Cys 429 and Ser 217, helped define a subset of twenty five analogs most likely to bind at the C-site and inhibit the enzyme. We noted the key residues involved in NDP binding in the C-site of hRR and catalysis, such as: Glu 431, Cys 429, Cys 444, Cys 218 and Asn 427. Upon comparing the close hydrogen bonding contacts between parent inhibitor (E-3a) at the C-site of hRR in our recent X-ray structure (PDB ID: 5TUS)10, Ser 217 and Cys 218 made relevant contacts through hydrogen bonds with E-3a. Cys 429 offered weaker Van der Waals contact with inhibitor E-3a, however it did not contribute strongly to stabilize binding to the inhibitor. The docking poses along with predicted affinities of representative candidates to the C-site of hRR, are presented in Figure 2A–F.
Figure 2.

Predicted binding interactions for top-ranked NSAH candidates at the C-site of hRR based on the X-ray recently published structure PDB ID: 5TUS.10 A. E-3a; B. E-3c; C. E-3f; D. E-3s; E. E-3t; F. E-3u.
The library of compounds (E-3a-z) were docked against the S-site and A-site of hRRM1 to determine if any analogs were likely to bind to allosteric sites other than the C-site. The Schrödinger docking scores for all sites are reported in Table S3. In general, a lower score is considered favorable, with scores between 0 and -5 considered poor and scores of -10 or lower considered an indication of strong binding interactions. The docking scores for the C-site ranged from −6.26 to −7.76, indicating favorable interactions between the ligands and C-site residues. The scores for the A-site ranged from −2.99 to −5.23, with only two compounds reporting a score below -5 (E-3m and E-3j). Scores for the S-site ranged from −1.95 to −5.41 with all but one compound (E-3r) reporting poor docking scores. While E-3m, E-3j, and E-3r showed some favorable interactions with either the A-site or S-site, all analogs reported much higher docking scores for the C-site, suggesting that these analogs are more likely to inhibit hRR by binding to the C-site.
By using the now well-defined pan-assay interference compounds11 (PAINS) filter as a gateway parameter, we monitored the presence or absence of potentially problematic functional groups in our analog design. Despite being a potential PAINS candidate, the validity of parent E-3a as a lead compound was greatly strengthened by the crystal structure data that showed binding of E-3a to the C-site of RR.10 A subset of analogs, E-3a, E-3c, E-3f, E-3s, E-3t and E-3u, displayed favorable docking poses as shown in Figure 2 upon docking to the C-site of hRR. They were primary synthetic targets in our design leading to twenty additional analogs.
Synthesis of Naphthyl Salicylacylhydrazone analogs
We constructed a library of salicyl-derived acyl hydrazones (Scheme 1A) based upon in silico screening results. The synthetic method yielded a library of analogs through a five-step sequence from commercially available acids in high overall yields. Specifically, salicylic acid derivatives (1) were converted to their corresponding methyl esters and subsequently transformed into corresponding acylhydrazides (2) under the treatment with hydrazine hydrate in methanol. Naphthyl ring containing acids (4), were reduced to their primary alcohol using lithium aluminum hydride in diethyl ether and the resulting alcohols were re-oxidized with pyridinium chlorochromate (PCC) in dichloromethane at room temperature to yield several aldehydes as represented by 5. The resulting aldehydes were condensed with acyl hydrazides (2) with catalytic amount of glacial acetic acid under reflux over 3–4 h. This step resulted in the generation of E-3a as the major product whose identity was verified using 1H, 13C-NMR and high resolution mass spectrometry (See SI). Detailed step-wise description of the synthesis is provided in the SI. Our synthetic pathway, by virtue of employing higher temperatures during the final condensation step involving 2 and 5, uniformly afforded the thermally more stable E-isomer in all cases. Though Scheme 1 outlines the theoretical possibility of a photoisomerization equilibrium that may exist between the two configurational isomers of 3A (E and Z isomers), in practice we observe only E-isomers to be formed. We observe a possible influence of severe allylic (A1,3) strain in the Z-isomer (which forces 6 atoms including the hydrazone nitrogens and the naphthyl carbons and the hydroxyl group to be in one plane) thermodynamically favoring the E-isomer to be the sole product in our synthesis.
Scheme 1.

A. Modular synthesis of hRR inhibitor library.; Yields and other structural characterization per each analog is provided in the experimental section. Thermodynamic control of the condensation step leading to E-isomer is observed. B. UV profile for E-3a. λmax at 320 nm is attributed to acyl hydrazone in trans configuration. The varying concentrations for E-3a are 2.0, 4.0, 8.0, 16.0 and 32.0 µM respectively as measured in MeOH as a solvent. Data corrected for solvent and background.
Recently, acylhydrazones with unique λmax per geometrical isomers were characterized as new class of highly tunable photoswitching compounds.12 Through UV emission spectroscopy, we measured a λmax of 320 nm attributable to the acyl hydrazone functionality in the trans configuration (Scheme 1B). It is the E-isomer that crystallized with hRRM1 in the recent report we published and used as a template herein.10 The E and Z isomers are expected to show distinct biological efficacy, and therefore throughout this work we have characterized the major E-isomer as a single component, in each analog case, and depict them with the E-notation.
Synthesis of most of the analogs followed the sequence as shown in Scheme 1, with the exception of sulfonyl hydrazones (E-3s, E-3o, E-3r and E-3q) that were obtained by a slightly modified procedure (See SI). A few diimino hydrazones were obtained through a bi-directional imine condensation with aldehydes that deviated slightly from general procedures described for reactions in Scheme 1 (See SI). Nevertheless, all analogs were neatly obtained as purified products in moderate to high yields (> 60–90 %) and the reactions were easy to perform on multi-gram quantities. Overall, twenty-five analogs were prepared for biological evaluations and their inhibitory properties are listed in Table 1.
Table 1.
Structures, % yields, in vitro IC50 values, and predicted solubility and permeability properties for E-3a and its analogs. Cellular IC50 values are averaged from HCT116 and MDA-MB-231 cell lines. ClogP and membrane permeability parameters were predicted using Qikprop. Permeability is reported as a diffusion rate in nanometers/second. Other related properties are provided in Table S2 (SI).

|
Enzymatic inhibition assays reveal favourable substitutions for increasing potency
The parent inhibitor and its analogs (E-3a-z) were subjected to enzymatic inhibition assays and ranked according to their IC50s. All of the analogs that were evaluated reported IC50s in the micromolar range, with subtle variations in structure leading to observable differences in potencies. Of the top ranked hydrazones based on their IC50 values, five compounds (E-3f, E-3c, E-3t, E-3s and E-3z as listed in Table 1, group A (above) showed ~ 2–4-fold improvement from the initial IC50 of 19 µM for our lead compound E-3a. Pyridyl ring-containing E-3f (IC50 ~ 5.3 µM), E-3c consisting of a 6-hydroxy naphthyl ring (IC50 ~ 7.3 µM), p-methyl substituted aryl sulfonamide containing E-3s (IC50 ~ 6.8 µM), and p-amino-m-chloro substituted E-3t (IC50 ~ 6.1 µM) analogs showed the greatest improvement from E-3a with IC50s below 10 µM, as listed in Table 1, group A (above). Interestingly, E-3z (IC50 ~ 10.2 µM) consisting of an C3-derived indole ring system in place of the naphthyl ring, retained its potency despite the change in the heterocyclic ring system. The single point structural change occurring on either of the two flanking aromatic rings on either side of the hydrazone core impacted IC50s of analogs. The importance of these substitutions were predicted earlier through the in silico screening, where these five analogs were identified as top choices. For example, the C-site Ser 606 and Thr 607 display shorter hydrogen bonding interactions with the acyl group of E-3a and at least one more stabilizing interaction comes from an additional hydrogen bond due to the presence of polar hydroxyl or amino groups on the aromatic ring system on these inhibitors as depicted in Figure 2B–F (above). For E-3s, a sulfonyl hydrazone, both O atoms connected to S engage in hydrogen bonding interactions favoring the binding of these analogs at the C-site of hRR. For naphthalene substitutions containing a C6-hydroxy group (E-3c), the main chain of Pro 294 engages in a stronger hydrogen bonding interaction as depicted in Figure 2B. E-3w, E-3u, E-3v, E-3y and E-3n showed modest improvements from the lead compound as ranked in Table 1, group B (above). Analogs E-3y, E- 3n and E-3z (from group A) contain an indole ring system replacing the naphthalene core of lead compounds. This change resulted in preserving, or even improving (E-3z), the efficacy afforded by the naphthalene ring system. These analogs are a favorable choice for further refinement because it is easier to derivatize indole ring-containing compounds than the naphthalene ring core of these inhibitors. Besides the six most potent inhibitors shown in Figure 2, docking poses revealing contacts between the remaining nine inhibitors and the hRR C-site are presented in Figures S1–S9 (see SI). Together, these data provide a roadmap for designing second generation non-nucleosidic RR inhibitors.
Seven of the 12 analogs reporting IC50s below 24 µM have a polar group positioned ortho to the hydrazone chain on the benzene ring, while another three feature a polar group in the meta position, as shown in Table 1, group C (above). The majority of para-substituted analogs, however, showed a marked decrease in activity. This suggests that the ortho-polar substitution is critical to enhancing hRR inhibition. While E-3s showed improvement in potency towards hRR, all other sulfonyl hydrazones became less potent than the lead compound, suggesting that this functional change is sensitive to the pendant hydrophobic groups present on either side of the hydrazone core. Moving the hydroxyl group of the naphthalene ring from carbon 2 to carbon 6 improved the potency toward RR by approximately 3-fold, supporting the prediction made by structure guided design that this substitution would hydrogen bond more favorably within the catalytic C-site of hRR. Additionally, substituting the naphthalene ring with an indole derivative showed no loss in potency, suggesting that the two ring systems are equivalent toward hRR inhibition. Of the five halogenated analogs tested, four showed an increased potency toward RR.
By these observations, a broad pharmacophore for these analogs can be developed incorporating ortho substitution of polar groups on both the benzene and naphthalene ring systems. Substitutions which interact with Ser 606, Thr 607, and Pro 294 are considered favorable for improving inhibitory potency. Additionally, substitution of the naphthalene ring for an indole ring can be used to access additional substitutions not readily available from a naphthalene ring system while fragment growth can be directed from the para position of the benzene ring system. Together, these observations provide a guide for future lead optimization of this class of hRR inhibitors.
NSAHs do not inhibit hRR through sequestering catalytically essential Fe
There are multiple reports of small molecule agents that inhibit RR through the scavenging of free radicals that are required for the reduction of nucleoside diphosphates. Nitric oxide13 and hydroxyurea14 (that quench the tyrosyl free radicals), and iron chelating small molecules15 such as desferrioxamine (that irreversibly chelate to Fe (II) that is essential for housing the free radical) are a few examples. Because the basic scaffold of these inhibitors possessed a possible chelating functional group that involves the 2-OH substituent on the naphthyl ring along with the hydrazonyl-imino N, we wondered if a metal chelating mechanism may contribute to its inhibitory mode of action against hRR. Therefore, to test this hypothesis, the propensity of E-3a to bind to Fe3+ and Fe2+ was studied by UV spectroscopy. In 10 mM ammonium acetate buffer (pH 7.0) under anaerobic conditions, a 100 µM solution of E-3a was treated with FeCl2·6H2O or FeCl3·6H2O at varying concentrations of metal ranging from 100 µM to 5 mM. The stoichiometry of E-3a: MX+ tested were 1:1, 1:2, 1:3, 1:4, 1:5, 1:6, 1:7, 1:8, 1:9, 1:10, 1:20, and 1:50. A scanning UV emission spectrum was recorded between 200–800 nm at each concentration. Noticeable changes occurred when E-3a was subjected to a titration with FeCl2·6H2O at concentrations of metal ranging from 400 µM to 1 mM. At concentrations above 1 mM, the UV detector was saturated and the UV profile of the ligand-metal complex could not be readily obtained. Once the E-3a:Fe2+ ion stoichiometry reached, or exceeded a 1:4 ratio, the UV emission spectrum revealed mild changes reflecting that the ligand was in a bound state with Fe2+. Similarly, a titration experiment was carried against Fe3+ salt at concentrations of metal ranging from 100 µM to 5 mM at stoichiometry of 1:1, 1:2, 1:3, 1:4, 1:5, 1:6, 1:7, 1:8, 1:9, 1:10, 1:20, and 1:50. There were no noticeable changes in UV emission spectra at any of these stoichiometry for Fe3+ ions, indicating that E-3a does not chelate Fe3+ ions. Taken together, these results support the conclusion that while E-3a does not inhibit hRR through metal chelation or sequestration of Fe3+, E-3a may participate in a chelation mechanism in the presence of excess Fe2+. Results supporting these observations are presented in Figures S10–S11 (See SI). We reviewed the potential of these inhibitors to chelate Mg2+ ions as well, because these ions are present in the in vitro enzyme assay buffer for hRR inhibition. Per spectra shown in Figure S12 (See SI) we conclusively show that these compounds do not bind and sequester Mg2+ ions.
NSAH analogs inhibit hRR competitively and reversibly
In a recently published study10, using multiple lines of investigations involving steady-state inhibition kinetics, a jump-dilution assay and an in vitro hRRM1 fluorescence-quenching assay, we have gathered evidence that the lead inhibitor, E-3a inhibited hRR in a reversible, competitive manner with an IC50 of 32 ± 10.2 µM and a dissociation constant (KD) of 37 ± 4.7 µM.10 We initially suspected that due to the presence of an unsubstituted electrophilic azomethine carbon (C=N) on the parent pharmacophore, this inhibitor could irreversibly inactivate the enzyme, by forming covalent bonds with nucleophilic residues at the C-site. Though the 2.7 Ǻ X-ray structure did not support this model, further detailed kinetic analyses were undertaken for further confirmation. In order to verify the potential of E-3a to reversibly inhibit hRRM1 at its C-site, a preincubation-dilution experiment was performed as outlined in the parallel study.10 Results from that analysis point to reversible inhibition of the enzyme whose activity could be resuscitated upon diluting away the inhibitor. Further analyses using steady state-kinetics revealed a model consistent with competitive inhibition, and correspondingly the Ki value of E-3a was evaluated to be 5.0 µM.
To verify that new analogs of the parent inhibitor (E-3a) follow the same mode of inhibition of hRRM1in this study, we subjected the most potent analogs, E-3a (as positive control), E-3c, E-3t, and E-3w analysis by steady-state inhibition kinetics. Double-reciprocal plots were prepared for wild type hRRM1 at varying substrate concentrations and hRRM1 in the presence of these each of the four inhibitors, independently at concentrations of 100 µM, 50 µM, 25 µM, 10 µM, and 5 µM respectively. As shown in Figure 3A (below), the double-reciprocal plot of E-3a inhibition against hRRM1 indicates that all data sets converge upon a common y-intercept near the origin, indicative of the fact that Km of the inhibitor is dependent upon inhibitor concentration [I] while Vmax is independent of [I], consistent with a competitive mechanism of inhibition. As shown in Figure 3B, C and D (below), a similar trend was observed for E-3c, E-3t, and E-3w respectively, indicating that like E-3a, the Km of these analogs is dependent upon [I] while Vmax is independent of [I]. Overall, these observations suggest that E-3c, E-3t, and E-3w are competitive inhibitors in the same manner as the parent inhibitor E-3a.
Figure 3.

A. Double-reciprocal plot for E-3a. All data sets converge upon a common y- intercept, supporting a competitive mechanism of inhibition. B. Double-reciprocal plot for E-3c also follows a competitive model. C. Double-reciprocal plot for E-3t, depicting competitive inhibition. D. Double-reciprocal plot for E-3w supports a competitive model of inhibition.
For the kinetic analysis outlined above, the sigmoidal dose response curves for E-3a (as positive control), E-3c, E-3t, and E-3w are provided in Figure S13 (see SI). It is noteworthy that our recent discovery of the crystal structure data of E-3a in complex with hRRM1 shows the inhibitor bound at the C-site of the enzyme, corroborating our kinetic model of inhibition.10 Because ADP-reductase activity is one of the lowest among all NDPs (<50 U/mg) and thereby usually has a low dynamic range, we further tested these inhibitors with a 3H-CDP assay method. Specific activity of wild-type R1 (ranging between 450–481 nmol min-1 mg-1) as well as IC50 values calculated for inhibitors E-3a, E-3c, E-3t, and E-3w correlate with those values obtained through the 14C-ADP substrate method, negating the need to repeat the remaining 14C-ADP IC50 measurements. Data supporting this correlation is provided in the supporting information (Table S4).
As the acylhydrazone core of this library of inhibitors is structurally similar to the thiosemicarbazone functionality present in the known R2 inhibitor and metal chelator triapine, it was necessary to determine if the overall mode of inhibition for these inhibitors in this series involve targeting of hRRM2. In order to probe this aspect, enzymatic inhibition assays were used to determine the enzymatic IC50 values for triapine (as a control) and compare its efficacy to that of E-3a against hRRM2. The specific activity of wild type hRRM2 in the presence of triapine (at varying concentrations of 100, 50, 5, 0.2, 0.50, and 0.10 µM) and E-3a (at varying concentrations of 5,000, 400, 100, 50, 5, 0.2, 0.50, and 0.10 µM) were normalized, then plotted against the logarithm [I]. The data were fitted to a sigmoidal dose-response curve to determine the IC50 values of triapine and E-3a, as 0.185 µM and 123 µM respectively (Figure 4, below). As the IC50 for E-3a is approximately 1,000-fold that observed for triapine, and approximately 6 times greater than the IC50 value for E-3a against hRRM1, it is unlikely that hRRM2 is a significant cellular target of E-3a.
Figure 4.

Sigmoidal dose-response curves for triapine and E-3a against hRRM2. The IC50 values of triapine and E-3a are 0.185 and 123 µM, respectively.
siRNA knockdown approach further points to RR targeting mode of action in vivo
Previous work from our group demonstrated changes in dNTP pools and cell cycle progression after treatment with (NSAH) E-3a.10 Although generally measurements of perturbations to cellular dNTP pool, in tandem with cell cycle arrest, has been used to identify cellular RR activity16 (Bianchi 1986; Lagergren 1987) additional confirmation can be derived using siRNA by Reid et al.17 or using inducible shRNA by Wisitpitthaya et al.4d to repress RR. The siRNA approach measuring the level of sensitization that a drug renders to RR under knocked-down conditions has been previously utilized to verify the cellular inhibition of RRM1 by gemcitabine and RRM2 by hydroxyurea17 and to delineate the role of RR oligomerization in the activity of clinically relevant inhibitors4d. To identify optimal levels of RRM1 repression for NSAH sensitivity testing, we transfected a range of validated siRNAs (specific for RRM1, 3 RefSeqs, ThermoFisher Silencer® Select, s12358; and control scrambled siRNAs), and assessed cell growth over a 48-hour period (SI Figure S14). As expected, significant inhibition of RRM1 resulted in growth inhibition, with an IC50 of approximately 3 pmoles/well, while the control scrambled siRNAs showed less than 50% growth inhibition even at 20 pmole/well, the highest amount tested. This guided the selection of the range of siRNA amounts (1–5 pmoles siRNA/well) used in the inhibitor treatment studies. For drug sensitivity study, cells were transfected with RRM1 or control scrambled siRNAs, followed 48 hours later by administration of E-3a. Relative growth inhibition was assessed after incubation for an additional 5 days. As shown in Figure 5, the relative NSAH-inhibitor induced growth inhibition in cells with siRNA-mediated knockdown of RRM1 was greater than that in cells with endogenous levels of RRM1.
Figure 5.

A and B. Sensitization of MDA-MB-231 cell line toward NSAH inhibitor under siRNA transfection and RRM1 knockdown conditions. A: Using RRM1 siRNA and B: Using Control-scrambled siRNA. Line displayed in A at the 4.0 pmol level represents the maximal sensitization occurring in the presence of E-3a, operating within an optimized siRNA concentration range.
The relative differences in DNA content in respective treatments were measured and are shown in SI Figure S15. The relative growth inhibition in cells treated with 4 pmol siRNA (IC50 = 0.347 µM) was greater than that observed with lesser amounts of RRM1 siRNA (2 pmol IC50 = 1.43 µM) or any quantity of Control siRNA (4 pmol IC50 = 1.94 µM), or no siRNA (IC50 = 1.49 µM), indicating that suppression of RRM1 caused sensitization to NSAH, further supporting its RRM1 specific mode of action.
Cytotoxicity studies of NSAH inhibitor analogs
Similar to a recently published study from our group10, the entire suite of inhibitors (E-3a-z) were concurrently evaluated for their cellular effects against both human colon cancer (HCT116) and human breast cancer (MDA-MB-231) cell lines. Continuous 72-hour exposure of E-3a and gemcitabine (as a control) demonstrated growth inhibition in both human cancer cell lines. IC50s for gemcitabine ranged between 30 and100 nM. E-3a displays cytotoxicity against HCT116 cells with an IC50 of 225 nM while displaying cytotoxicity against MDA-MB-231 cells with an IC50 of 300 nM.10 Analogs E-3o and E-3v display cytotoxicity against HCT116 cells with IC50’s of 10 and 1 µM respectively. The remaining analogs did not exhibit significant cytotoxicity. Their net cytotoxicity effect is summarized in Table S2 (see SI). As illustrated in Table S2, not all inhibitors in this series exhibited significant levels of cytotoxicity. This observation correlated partly with the fact that not all inhibitors in this series were sufficiently soluble in cell culture media, with compound precipitation resulting in insignificant cytotoxicity for many analogs. Therefore, detailed cellular toxicity results could be obtained only for E-3a, E-3o and E-3v. In many cases, a cancer cell cytoxicity with cellular IC50 of >10 µM may primarily be an effect due to high insolubility of the inhibitor, underscoring the importance of solubility (in biologically relevant environments) for future improvements in potency.
Discussion and Conclusion
Although acylhydrazones display a wide range of biological activities, including inhibition of metazoan proteases18, anti-Leishmania properties19, herbicidal effects20, and inhibition of proliferating cellular nuclear antigen (PCNA)21, we were the first to discover that naphthyl Salicyl acylhydrazones (especially the lead inhibitor E-3a) inhibit hRR through binding at the C-site of RRM1.10 In this study, we establish a hydrazone pharmacophore inactivating hRR in a reversible, competitive mode through binding at the C-site of the enzyme. As such, we rationalized that structure-based design could be used in tandem with structure-activity relationship studies to further develop our understanding of how acylhydrazones could be used to target hRR for cancer treatment.
Most of the clinically used RR inhibitors are nucleoside analogs such as gemcitabine, which is currently used to treat pancreatic cancer. Gemcitabine has been shown to inhibit RR in a synergistic manner, with irreversible hRR inhibition and depleted nucleotide pools allowing for gemcitabine triphosphate to become incorporated into growing DNA strands by DNA polymerase, causing delayed chain termination and triggering apoptosis. This mechanism does not distinguish between normal and cancerous cells, and therefore gemcitabine is associated with serious side effects due to toxicity towards normal cells resulting from DNA chain termination, irreversible inhibition of hRR, and inhibition of other enzymes such as topoisomerase-1, thymidylate synthase, deoxycytidine deaminase, CTP-synthase, which recognize phosphate moieties.6,7 This combination of indiscriminate cross-reactivity, irreversible inhibition, and DNA chain termination produces undesirable cytotoxicity in normal cells and thereby results in a relatively low therapeutic index when use in the treatment of patients. We therefore hypothesize that identification and characterization of specific, reversible, non-nucleoside RR inhibitors could prove advantageous by reducing the cytotoxicity towards normal cells caused by nucleoside analogs.
While previous RR inhibitors display a diverse range of mechanisms of inhibition, there have been few efforts to identify reversible, non-nucleosidic small molecules that inhibit the hRRM1 subunit. In Ahmad et.al. 2015, we reported a rapid throughput screening method integrating in silico docking, cell-based assays, and biochemical experiments which identified ten novel classes of non-nucleosidic RR inhibitors.8 This study marked the first compounds identified to inhibit RR by binding to a protein-protein interface (M-site) and inducing formation of catalytically inactive hexamers. Recently, we reported the identification of the first non-nucleoside identified to bind to the C-site of hRR (PDB ID: 5TUS).10 This lead inhibitor E-3a, an acylhydrazone, was determined to inhibit RR in a reversible, competitive manner and exhibit potent cytotoxicity towards multiple cancer cell lines.
Using the crystal structure of E-3a in complex with hRRM1 (PDB ID: 5TUS)10 as a template, a focused library of analogs was designed using in silico modeling. In silico docking with the Schrödinger software suite was used to evaluate 100 analogs and the top 25 were selected based upon the predicted docking poses. A modular, E-isomer selective synthetic pathway was implemented which afforded hydrazones on a multi-gram scale in five steps with isolated yields ranging between 65 and 85% typically, with a few exceptions. In vitro RR inhibition assays identified five analogs with 2–4 fold improvement in IC50 from the lead compound, E-3a. All five analogs were predicted to be more potent toward RR during the in silico modeling, as all of these analogs showed shorter hydrogen bonding interactions with residues Ser 606, Thr 607, and Ser 217, which are known to hydrogen bond with natural nucleoside substrates. It was also determined that the ortho substitution of a polar group on either ring system was critical for RR inhibition, while substitution of the naphthalene ring system for an indole ring resulted in no loss in activity, providing potential access to a broader range of chemical derivatives. Upon evaluating in vitro inhibitory potencies against hRR, broadly a subset of 15 inhibitors were identified within a narrow range of IC50s.
Due to the presence of a hydrazone functionality on the parent inhibitor structure, we were concerned that this group had the propensity to be attacked by an active site nucleophile (e.g. Cys 429) present in hRR, thereby irreversibly inactivating the enzyme. The data presented in Figure 3 provided us with a mechanistic model of inhibition for these inhibitors. The participation of the hydrazone azomethine nitrogen and the C=N bond through a covalent mode can be ruled out, and reversible inhibitory modes can be invoked. Metal chelation assays determined that E-3a does not chelate Fe3+ ions, however Fe2+ complexation was observed in the presence of excess Fe2+(4-fold excess and higher). While the X-ray crystal data obtained in a previous study strongly suggests that the biological activity of E-3a-like compounds can be attributed to inhibition of RR, a separate metal chelation mechanism cannot be completely ruled out, especially when relatively high Fe2+ ion concentrations may be present in cells (typically > 1 mM). Importantly, analogs E-3c, E-3t, and E-3w were all determined to inhibit hRR through similar competitive, reversible mechanisms as the lead compound (E-3a), suggesting that novel analogs possessing this pharmacophore follow the same mechanism of inhibition as the lead compound.
The relatively large difference between the mid-micromolar enzymatic IC50 (in vitro) and nanomolar cellular IC50 (against cancer cells in vivo) suggested that E-3a may interact with multiple targets. This raised a concern regarding whether hRR is indeed a cellular target. In a recently published study, we evaluated the perturbation of cellular dNTP levels specifically in the presence of E-3a, and compared them to levels in cells treated with hydroxyurea, at their respective IC50s.10 The addition of E-3a was shown to induce a pattern of dNTP depletion with greatest depletion of dATP and dGTP, with little or no effect on dCTP or dTTP levels. This was followed by an arrest of cells in early S-phase. These observations are similar to patterns observed with gemcitabine and hydroxyurea and comprise a signature for a cellular hRR inhibitor.22 Using mobilized peripheral blood progenitor cells (a common target for dose limiting toxicity during gemcitabine therapy), E-3a was shown to have a superior therapeutic index compared to gemcitabine.10 These observations point to promising selective cytotoxicity attributable to hRR inhibition by E-3a. Analogs E-3o and E-3v, showing similar cytotoxicity profile as E-3a, may also be expected to target hRR in vivo. However, we are cognizant of the possibility that inhibitors like E-3a and its analogs may possibly engage multiple cellular targets that are yet to be identified.
It is generally agreed that RR is a challenging therapeutic target for cancer, as inhibitors are unlikely to distinguish between malignant and normal cells. We recently demonstrated that in blood progenitor cells, inhibitor E-3a showed a superior therapeutic index in comparison with gemcitabine, suggesting a possible discrimination between transformed and untransformed cells.10 We wish to further exploit this new class of inhibitors for the possible treatment of cancer, especially against gemcitabine-resistant forms. In the long term, identification of non-nucleoside, reversible RR inhibitors may provide an avenue towards developing safer alternatives to irreversible nucleoside analogs as chemotherapeutic agents. Furthermore, considering the fact that RR broadly impacts multiple pathologies, these reversible agents may impact drug discovery for treatment of a wider range of proliferative diseases.
Experimental Section
In Silico docking with Schrödinger suite for predicting C-site binding affinity
In silico docking of each hydrazone analog was performed using the Glide docking module of the Schrödinger 9.3 modeling software suite as previously described in Ahmed and Huff et al.8 The docking site was defined as a box of 5 Å centered on the original hydrazone ligand in the hR1 complex (PDB ID: 5TUS). Ligands were then docked to the catalytic site using Glide XP. The generated docking poses were evaluated for interactions with residues that commonly bind to natural substrates, such as Ser 202, Ser 606, Gly 246, and Arg 293. Ligands that showed favorable interactions with these residues were selected for synthesis. Generally, most inhibitors screened here showed very minimal perturbation or binding interaction with C-site residues involved in catalysis, such as Glu 431, Cys 429, Cys 444, Cys 218 and Asn 427. Therefore, these sites were omitted from depiction in the docked poses with inhibitors. Specific docking poses for inhibitors that showed lower than 5–10 µM potency (IC50) are depicted in Figures S1-S9 (see SI).
In Silico docking with Schrödinger suite for predicting site selectivity
Proteins and ligands were prepared as described below. Docking of the S-site and A-site was performed using the previously determined ATP-TTP-hRRM1 crystal structure (PDB ID: 3HNE). The S-site docking grid was defined as a 5 Å cube (125 Å3) centered on the TTP ligand. The A-site docking grid was defined as a 5 Å cube (125 Å3) centered on the ATP ligand. Docking scores for each inhibitor docked to each of the three sites are reported in Table S3.
Human ribonucleotide reductase protein expression and purification
The hRRM1 protein was expressed in E. coli BL21-codon plus (DE3)-RIL cells and purified using peptide affinity chromatography as previously described in Fairman et. al.5c The hRRM2 protein was also expressed in E. coli BL21-codon plus (DE3) cells and purified to homogeneity using Ni-NTA affinity chromatography as described in Fairman et. al.5c Iron was loaded into the hRRM2 subunit as described in Ahmed and Huff et. al.8 Concentration of the homogeneous protein was quantified using UV spectroscopy.
Ribonucleotide reductase in vitro inhibition assay
[14C]-ADP reduction assay
The in vitro specific activity of human ribonucleotide reductase was determined by 14C-ADP reduction assays using a reaction mixture of 0.3 µM hRRM1 and 2.1 µM hRRM2 in an activity assay buffer containing 50 mM HEPES (pH 7.6), 15 mM MgCl2, 1 mM EDTA, 100 mM KCl, 5 mM DTT, 3 mM ATP, 100 µM dGTP and 1 mM 14C-ADP (~3000 cpm/nmol). The reaction mixture was pre-incubated for 3 min at 37 °C, and 30 µL aliquots were sampled at fixed time intervals after initiation of the reaction. Reactions were quenched by immersion in a boiling water bath. The aliquots were then cooled and treated with alkaline phosphatase for 2 hours. The product 14C-dADP that formed during the reaction was separated from substrate 14C-ADP using boronate affinity chromatography. Amount of [14C]-dADP formed from the reaction was quantified by liquid scintillation counting using a Beckman LS6500 liquid scintillation counter. The assays were repeated in triplicate and the averaged specific activities were used for IC50 calculations.
[3H]-CDP reduction assay
Assays to determine the IC50 values for compounds 3a, 3c, 3t, and 3w in the presence of 3H-CDP substrate were performed with a reaction mixture consisting of 0.3 µM hRRM1 and 2.1 µM hRRM2 in an activity assay buffer of 50 mM HEPES (pH 7.6), 15 mM MgCl2, 1 mM EDTA, 100 mM KCl, 5 mM DTT, 3 mM ATP, 100 µM dGTP and 1 mM 3H-CDP (~2000 cpm/nmol). After processing of the reaction similar to the procedure described above, the amount of [3H]-dCDP formed from the reaction was quantified by liquid scintillation counting using a Beckman LS6500 liquid scintillation counter. The assays were repeated in triplicate and the raw specific activities are reported below in Table S2 (see SI).
[14C]-ADP assays to determine inhibition of the hRRM2 subunit
These assays were performed as with reaction mixture of 2.1 µM hRRM1 and 0.3 µM hRRM2 in an activity assay buffer of 50 mM HEPES pH 7.6, 15 mM MgCl2, 1 mM EDTA, 100 mM KCl, 5 mM DTT, 3 mM ATP, 100 µM dGTP and 1 mM 14C-ADP (~3000 cpm/nmol). The specific activity was determined for wild type hRRM2, and hRRM2 in the presence of triapine (at 100 µM, 50 µM, 5 µM, 0.2 µM, 0.05 µM, and 0.01 µM concentrations) or 3A (at 5,000 µM, 400 µM, 100 µM, 50 µM, 0.2 µM, 0.05 µM, and 0.01 µM concentrations) respectively. After processing of the reaction similar to the procedure described above, the amount of [14C]-dADP formed from the reaction was quantified by liquid scintillation counting using a Beckman LS6500 liquid scintillation counter. All assays were repeated in triplicate and reported values are averaged.
Assays in the presence of E-3a inhibitor analogs
All compounds were dissolved in 100% DMSO and diluted to the appropriate concentration so that the total DMSO volume in the reaction mixture did not exceed 0.2%. At this concentration of DMSO, the loss of RR activity is less than 1%. The IC50 was defined as the concentration of any compound that reduced the specific activity of hRRM1 to 50% of the control activity. A two-point assay for IC50 determination was adopted from the procedure described in Ahmed et. al.8 for most inhibitors tested. Based on this method, concentrations of 5 and 100 µM of the inhibitor were used for estimating the IC50. All assays were repeated in triplicate and reported values are averaged.
Assays to Determine Mode of RR Inhibition
The IC50s of 3a, 3c, 3t, and 3w were determined as previously described using inhibitor concentrations of 5 µM, 10 µM, 25 µM, 50 µM, and 100 µM respectively. The IC50s for these three analogs were determined for substrate concentrations of 0.5 mM, 1.0 mM, and 1.5 mM of 14C-ADP to determine if changes in substrate concentration influenced the observed IC50. All assays were repeated in triplicate and averages are reported.
Compound Characterization
Supporting information provides details on solvent purification and general methods followed for synthesis and purification of each analog reported herein. Each analog reported as a novel compound in this study was purified using column chromatography or crystallization methods before characterization of their identity and purity. All reported yields for final products were for compounds that are >95% pure. The purity was evaluated to be > 95% using NMR and HPLC methods whose copies are provided in the supporting information.
2-hydroxy-N'-((2-hydroxynaphthalen-1-yl)methylene)benzohydrazide (3a)
According to general procedure D (see SI), a 1:1 molar ratio of salicylic hydrazide (760 mg, 5 mmol) and 2-hydroxy-1-naphthaldehyde (861 mg, 5 mmol) were dissolved in 30 mL of MeOH and heated to reflux. Several drops of glacial acetic acid were added to catalyze the reaction and the flask was heated at reflux for 3–4 h. The reaction mixture was filtered to collect the crude product as a yellow precipitate, and the compound 3a was purified by silica gel column chromatography (1.38 g, 4.51 mmol; 90% yield). IR: 3500-3200 (broad, OH), 3046 (aromatic C-H), 2114 (C=N), 1639 (C=O), 1550 (C=C), 1236 (C-O), 744 (o substitution). 1H-NMR: (CD3CN, 500 MHz) 12.90 (s, 1H), 11.85 (s, 1H), 10.94 (s, 1H), 9.52 (s, 1H), 8.21 (d, J = 8.6 Hz, 1H), 8.02 (d, J = 8.7, 1H), 7.97 (d, J = 8.7 Hz, 1H), 7.92 (m, 1H), 7.72 (t, J = 7.8 Hz, 1H), 7.61 (t, J = 7.7 Hz, 1H), 7.53 (t, J = 7.6 Hz, 1H), 7.33 (m, 1H), 7.12 (m, J = 8.2 Hz, 2H). 13C-NMR: (CD3CN, 125 MHz) 164.9, 159.6, 159.0, 148.5, 135.4, 133.8, 132.6, 129.8, 129.6, 128.4, 127.5, 124.3, 120.6, 119.7, 119.6, 118.5, 116.6, 109.5. HRMS (ESI, [M+Na]+) m/z calcd. for C18H14N2O3Na 329.0902, found: 329.0895.
2-hydroxy-N'-((6-hydroxynaphthalen-2-yl)methylene)benzohydrazide (3c)
Prepared according to general procedures described in the SI. Yield 1.07 g, 3.50 mmol; 72% yield. Yellow solid, mp 230 °C. IR: 3415 (secondary NH), 3300-3200 (broad, OH), 2250 (C=N), 1628 (C=O), 1543 (C=C), 1204 (C-O). 1H-NMR: ((CD3)2SO, 500 MHz) δ 12.50 (s, 1H), 10.21 (s, 1H), 10.04 (s, 1H), 8.49 (s, 1H), 7.98 (d, J = 8.7 Hz, 1H), 7.86 (d, J = 8.4 Hz, 1H), 7.84 (d, J = 8.4 Hz, 1H), 7.78 (t, J = 8.4 Hz, 1H), 7.37 (ddd, J = 8.5, 7.2, 1.7 Hz, 1H), 7.21-7.12 (m, 3H), 6.92-6.80 (m, 2H). 13C-NMR: (CD3CN, 125 MHz) 168.4, 167.0, 160.1, 158.2, 137.6, 134.9, 132.2, 131.5, 127.4, 127.0, 126.4, 125.6, 124.1, 120.1, 119.7, 118.6, 114.8, 109.9. HRMS (ESI, [M+Na]+) m/z calcd. for C18H14N2O3Na 329.0902, found: 329.0915.
2-amino-N'-((2-hydroxynaphthalen-1-yl)methylene)benzohydrazide (3e)
Prepared according to general procedures described in the SI. Yield 1.16 g, 3.82 mmol; 76%. Yellow solid, mp 229 °C. IR: 3451 (primary NH), 3300-3250 (broad, OH), 2117 (C=N), 1618 (C=O), 1577 (C=C), 1240 (C-N), 1182 (C-O). 1H-NMR: ((CD3)2SO, 500 MHz) δ 12.89 (s, 1H), 10.00 (s, 1H), 9.02 (s, 1H), 8.65 (d, J = 8.7 Hz, 1H), 8.04 (d, J = 9.0 Hz, 1H), 7.92 (d, J = 8.0 Hz, 1H), 7.62 (t, J = 7.8 Hz, 1H), 7.52 (t, J = 7.4 Hz, 1H), 7.44 (t, J = 7.4 Hz, 1H), 7.29 (d, J = 9.0 Hz, 1H), 7.12 (d, J = 8.2 Hz, 1H), 6.92-6.89 (m, 2H), 5.34-5.27 (m, 2H). 13C-NMR: ((CD3)2SO,125 MHz) 168.1, 165.2, 148.4, 143.3, 134.9, 133.5, 132.2, 129.3, 128.2, 128.1, 127.6, 123.9, 120.1, 119.4, 119.2, 118.7, 113.7, 109.0. HRMS (ESI, [M+Na]+) m/z calcd. for C18H15N3O2Na 328.1056, found: 328.1061.
N'-((2-hydroxynaphthalen-1-yl)methylene)nicotinohydrazide (3f)
Prepared according to general procedures described in the SI. Yield 1.19 g, 4.08 mmol; 82%. Yellow solid; mp 231 °C. IR: 3415 (secondary NH), 3300-3200 (broad, OH), 2107 (C=N), 1619 (C=O), 1577 (C=C), 1140 (C-O). 1H-NMR: ((CD3)2SO, 500 MHz) δ 12.60 (s, 1H), 12.37 (s, 1H), 9.47 (s, 1H), 9.15 (s, 1H), 8.81 (d, J = 4.7 Hz, 1H), 8.33 (d, J = 8.0 Hz, 1H), 8.30 (d, J = 8.0 Hz, 1H), 7.96 (d, J = 8.5 Hz, 1H), 7.91 (d, J = 8.5 Hz, 1H), 7.63 (dd, J = 8.1, 5.1 Hz, 2H), 7.43 (t, J = 7.5 Hz, 1H), 7.25 (d, J = 8.5, 1H). 13C-NMR: ((CD3)2SO, 100 MHz) 171.2, 166.3, 150.2, 148.8, 148.2, 144.7, 138.0, 135.0, 129.3, 128.1 (2C), 126.8, 123.9, 122.3, 120.1, 119.2 108.9. HRMS (ESI, M+) m/z calcd. for C17H13N3O2 291.1002, found: 291.1006.
1-((E)-(((E)-4-hydroxy-3-methoxybenzylidene)hydrazono)methyl)naphthalen-2-ol (3g)
Commercially obtained vanillin was reacted with anhydrous hydrazine as shown in General Method B, followed by condensation with 2-hydroxy-1-naphthaldehyde to obtain 3g. Yield 0.70 g, 2.19 mmol; 44%. Yellow solid; mp 230 °C. IR: 3415 (secondary NH), 3300-3200 (broad, OH), 2150 (C=N), 1619 (C=O), 1577 (C=C), 1465 (C-H methyl), 1182 (C-O). 1H-NMR: (CDCl3, 500 MHz) δ 13.01 (s, 1H), 9.99 (s, 1H), 8.50 (s, 1H), 8.40 (s, 1H), 8.16 (d, J = 8.5 Hz, 1H), 8.05 (d, J= 9.0 Hz, 1H), 7.92 (d, J = 8.0 Hz, 1H), 7.85 (t, J = 7.9 Hz, 1H), 7.44 (t, J = 7.4 Hz, 1H), 7.34 (d, J = 8.2 Hz, 1H), 7.28 (t, J = 9.0 Hz, 1H), 7.19 (d, J = 8.2 Hz, 1H), 6.95 (d, J = 8.6 Hz, 1H), 3.97 (s, 3H). 13C-NMR: (CDCl3, 125 MHz) 170.8, 150.9, 149.4, 149.4, 135.0, 134.8, 129.3, 128.7, 128.1, 126.9, 124.6, 123.9, 122.9, 120.1, 119.2, 117.1, 114.3, 108.3, 56.1. HRMS (ESI, M+) m/z calcd. for C19H16N2O3 320.1155, found: 320.1157.
4-(dimethylamino)-N'-((2-hydroxynaphthalen-1-yl)methylene)benzohydrazide (3h)
Commercially obtained p-(dimethylamino)benzaldehyde was reacted with anhydrous hydrazine as shown in General Method B, followed by condensation with 2-hydroxy-1-naphthaldehyde to obtain 3h. Yield 1.28 g, 3.85 mmol; 77%. Orange solid; mp 231 °C. IR: 3500-3200 (broad, OH), 2901 (sp3 C-H), 2115 (C=N), 1629 (C=O), 1578 (C=C), 1319 (N, N-dimethyl), 1281 (C-N), 1241 (C-O). 1H-NMR: (CDCl3, 500 MHz) δ 13.56 (s, 1H), 9.69 (s, 1H), 8.61 (s, 1H), 8.21 (d, J = 8.5 Hz, 1H), 7.85 (d, J = 8.9 Hz, 1H), 7.8 (d, J = 8.9 Hz, 1H), 7.77 (d, J = 8.5 Hz, 1H), 7.57 (ddd, J = 8.4, 6.9, 1.4 Hz, 1H), 7.39 (ddd, J = 8.0, 6.9, 1.0 Hz, 1H), 7.26 (d, J = 9.0 Hz, 2H), 6.78 (d, J = 9.0 Hz, 2H), 3.10 (s, 6H). 13C-NMR: (CDCl3, 125 MHz) 169.9, 159.0, 153.2, 150.8, 133.7, 132.1, 131.4 (2C), 129.1, 128.9, 127.6, 123.5, 121.4, 120.2, 119.2, 111.7 (2C), 108.2, 40.2 (2C). HRMS (ESI, M+) m/z calcd. for C20H19N3O 318.1606, found: 318.1608.
N'-((2-hydroxynaphthalen-1-yl)methylene)-4-methoxybenzohydrazide (3i)
Commercially obtained p-anisaldehyde was reacted with anhydrous hydrazine as shown in General Method B, followed by condensation with 2-hydroxy-1-naphthaldehyde to obtain 3i. Yield 1.43 g, 4.46 mmol; 89%. Yellow solid; mp 230 °C. IR: 3420 (secondary NH), 3300-3200 (broad, OH), 2113 (C=N), 1618 (C=O), 1577 (C=C), 1465 (C-H methyl), 1182 (C-O). 1H-NMR: (CDCl3, 500 MHz) δ 13.56 (s, 1H), 13.01 (s, 1H), 9.66 (s, 1H), 8.55 (s,1H), 8.16 (d, J = 8.6 Hz, 1H), 7.79 (t, J= 9.3 Hz, 1H), 7.69 (d, J = 7.4 Hz, 2H), 7.59 (t, J = 7.6 Hz, 1H), 7.56 (t, 7.6 Hz, 1H), 7.38 (t, J = 7.4 Hz, 1H), 6.71 (d, J = 8.5 Hz, 2H), 3.06 (s, 3H). 13C-NMR: (CDCl3, 125 MHz) 171.0, 161.1, 159.0, 135.0, 133.7, 130.4, 129.9, 129.3, 129.1, 128.1, 127.6, 123.9, 123.4, 120.2, 120.1, 119.2 (2C), 111.7, 50.2. HRMS (ESI, M+) m/z calcd. for C19H16N2O2 304.1207, found: 304.1206.
N'-((2-hydroxynaphthalen-1-yl)methylene)-4-methylbenzohydrazide (3j)
Commercially obtained p-tolualdehyde was reacted with anhydrous hydrazine as shown in General Method B, followed by condensation with 2-hydroxy-1-naphthaldehyde to obtain 3j. Yield 1.00 g, 3.29 mmol; 66% yield. Yellow solid; mp 228 °C. IR: 3300-3200 (broad, OH), 2113 (C=N), 1619 (C=O), 1577 (C=C), 1416 (C-C), 1281 (C-N), 1240 (C-O). 1H-NMR: ((CD3)2SO, 400 MHz) δ 12.89 (br s, 1H), 9.98 (s, 1H), 8.64, (d, J = 8.3 Hz, 1H), 8.01 (t, J = 8.3 Hz, 1H), 7.90 (d, J = 8.0 Hz, 1H), 7.75 (d, J = 8.0 Hz, 2H), 7.60 (t, J = 7.6 Hz, 1H), 7.43 (t, J = 7.3 Hz, 1H), 7.36 (t, J = 7.3 Hz, 1H), 7.32 (d, J = 7.6 Hz, 2H), 7.25 (d, J = 7.3, 1H), 2.35 (s, 3H). 13C-NMR: (CDCl3, 100 MHz) 169.7, 161.2, 149.9, 135.0, 132.6, 132.1, 129.7, 129.5, 129.3, 129.2, 129.1 (2C), 128.1, 123.9, 120.1, 119.2 (2C), 108.4, 21.4. HRMS (ESI, M+) m/z calcd. for C19H16N2O 289.1341 found: 289.1353.
1-((2-benzylhydrazono)methyl)naphthalen-2-ol (3k)
Prepared according to general procedures described in the SI. Yield 1.24 g, 4.47 mmol; 89%. Yellow solid; mp 230 °C. IR: 3313 (secondary N-H), 3010 (sp2 C-H), 2360 (C=N), 1618 (C=O), 1592 (C=C), 1467 (C-C), 1277 (C-N), 1253 (C-O). 1H-NMR: ((CD3)2SO, 400 MHz) δ 11.84 (br s, 1H), 10.50 (s, 1H), 8.90 (s, 1H), 8.33 (d, J = 8.6 Hz, 1H), 7.85-7.82 (m, 2H), 7.53 (t, J = 7.9 Hz, 1H), 7.34 (t, J = 7.9 Hz, 1H), 7.30-7.21(m, 5H), 7.17 (d, J = 8.9 Hz, 1H), 3.91 (s, 2H). 13C-NMR (CDCl3, 100 MHz) 157.1, 143.6, 138.1, 131.6, 131.4, 129.8, 129.3, 128.6, 127.3, 123.5, 121.1, 120.1, 119.2, 112.9, 109.1, 58.7, 18.7. HRMS (ESI, M+) m/z calcd. for C17H16N2O 262.1101 found: 262.1094.
1-((2-(4-bromophenyl)hydrazono)methyl)naphthalen-2-ol (3l)
Prepared according to general procedures described in the SI. Yield 1.23 g, 3.62 mmol; 72%. Yellow solid; mp 230 °C. IR: 3322 (secondary NH), 2118 (C=N), 1619 (C=O), 1589 (C=C), 1286 (C-N), 1251 (C-O), 650 (C-Br). 1H-NMR: ((CD3)2SO, 400 MHz) δ 11.57 (br s, 1H), 10.64 (s, 1H), 8.89 (s, 1H), 8.45 (d, J = 8.5 Hz, 1H), 7.83 (d, J = 8.4 Hz, 1H), 7.78 (t, J = 7.3 Hz, 1H), 7.55 (t, J= 7.5 Hz 1H), 7.40 (d, 8.1 Hz, 2H), 7.35 (t, J = 7.0 Hz, 1H), 7.16 (d, J = 8.9 Hz, 1H), 6.93 (d, J = 8.8 Hz, 2H). 13C-NMR (CDCl3, 100 MHz) 160.1, 141.9, 138.7, 133.0, 132.4, 132.2, 132.1, 131.6, 129.2, 127.2, 126.6, 123.4, 119.8, 118.9, 118.8, 114.2 108.1. HRMS (ESI, M+) m/z calcd. for C17H13BrN2O 340.0206, found 340.0207.
1-((2-(4-nitrophenyl)hydrazono)methyl)naphthalen-2-ol (3m)
Prepared according to general procedures described in the SI. Yield 1.23 g, 4.01 mmol; 80% yield. Red solid; mp 230 °C. IR: 3290 (secondary N-H), 2117 (C=N), 1589 (C=C), 1310 (C-O), 1267, 1296 (N-O). 1H-NMR: ((CD3)2SO, 400 MHz) δ 11.41 (s, 1H), 11.14 (s, 1H), 8.98 (s, 1H), 8.75 (d, J = 8.6 Hz, 1H), 8.19 (d, J = 9.1 Hz, 2H), 7.86 (m, 2H), 7.60 (ddd, J = 8.4, 6.8, 1.3 Hz, 1H), 7.35 (m, 1H), 7.22 (d, J = 8.9 Hz, 1H), 7.11 (d, J = 8.9 Hz, 2H). 13C NMR: (CD3CN, 100 MHz) 152.0, 148.6, 142.6, 134.1, 133.7, 132.8, 132.4, 129.0, 128.0, 126.6, 124.2 (2C), 120.8, 119.9, 111.6, 111.6, 108.4. HRMS (ESI, [M+Na]+) m/z calcd. for C17H13N3O3Na 330.0854, found: 330.0870.
2-hydroxy-N'-((5-hydroxy-1H-indol-3-yl)methylene)benzohydrazide (3n)
Prepared according to general procedures described in the SI. Yield 0.95 g, 3.21 mmol; 64% yield. Red solid. mp 230 °C. IR: 3290 (secondary N-H), 3300-3200 (broad, OH), 2117 (C=N), 1692 (C=O), 1589 (C=C), 1310 (C-O). 1H NMR: (CD3CN, 500 MHz): δ 11.21 (s, 1H), 11.04 (s, 1H), 10.50 (s, 1H), 9.71 (s, 1H), 8.60 (s, 1H), 7.89 (d, J = 8.6 Hz, 1H), 7.85 (d, J = 5.3 Hz, 1H), 7.64 (d, J = Hz, 1H), 7.46 (t, J = 6.8, 1H), 7.40 (d, J = 8.9 Hz, 1H), 6.99 (d, J = 8.9 Hz, 1H), 7.95 (t, J = 6.8 Hz, 1H), 6.92 (d, J = 5.4 Hz, 1H). 13C NMR: (CD3CN, 125 MHz) 163.2, 157.1, 152.4, 146.2, 134.6, 130.4, 127.1, 125.1, 124.7, 124.2, 121.8, 118.3 (2C),114.3, 114.2, 104.0. HRMS (ESI, M+) m/z calcd. for C16H13N3O3 279.1002, found: 279.1006.
N'-((2-hydroxynaphthalen-1-yl)methylene)benzenesulfonohydrazide (3o)
Commercially obtained benzene sulfonyl hydrazine was condensed with 2-hydroxy-1-naphthaldehyde as described in General Method D to obtain 3o. Yield 1.47 g, 4.49 mmol; 89.8 %. Yellow solid; mp 230 °C. IR: 3156 (secondary N-H), 2117 (C=N), 1601 (C=C), 1309 (asymmetrical, S-O),1074-972 (symmetrical, S-O), 683 (S-N), 638 (C-S). 1H-NMR: : ((CD3)2SO, 400 MHz) δ 11.61 (s, 1H), 11.06 (s, 1H), 8.77 (s, 1H), 8.36 (d, J = 8.6 Hz, 1H), 7.92 (d, J = 7.0 Hz, 2H), 7.80 (d, J = 7.5 Hz, 1H), 7.76 (d, J = 7.0 Hz, 2H), 7.73-55 (m, 2H), 7.42 (ddd, J = 8.5, 6.8, 1.0 Hz,1H), 7.33 (ddd, J = 8.0, 6.9, 1.0 Hz, 1H), 7.14 (d, J = 8.9 Hz, 1H). 13C-NMR: (CDCl3, 100 MHz) 151.5, 143.2, 139.2, 134.2, 133.9, 133.9, 132.1, 129.5, 129.2, 129.2, 128.3, 128.0, 127.9, 123.8, 119.7, 119.0 108.9. HRMS (ESI, [M+Na]+) m/z calcd. for C17H14N2O3SNa 349.0623, found 349.0634.
N'-((2-hydroxynaphthalen-1-yl)methylene)-1-phenylmethanesulfonohydrazide (3p)
Commercially obtained phenylmethyl-sulfonyl fluoride was reacted with anhydrous hydrazine as shown in General Method B, followed by condensation with 2-hydroxy-1-naphthaldehyde to obtain 3p. Yield 0.31 g, 0.90 mmol; 18%. Yellow solid; mp 230° C. IR: 3156 (secondary N-H), 2117 (C=N), 1601 (C=C), 1309 (asymmetrical, S-O). 1H-NMR: ((CD3)2SO, 400 MHz) δ 12.84 (br s, 1H), 9.96 (s, 1H), 8.77 (s, 1H), 8.61 (d, J = 8.6 Hz, 1H), 7.99 (d, J = 8.4 Hz, 1H), 7.88 (d, J = 9.0 Hz, 1H), 7.58 (t, J = 8.5 Hz, 1H), 7.40 (t, J = 8.5 Hz, 1H), 7.36 (t, J = 8.5 Hz, 1H), 7.24 (m, 5H), 4.32 (s, 2H). 13C-NMR: (CDCl3, 100 MHz) 150.5, 143.2, 139.2, 135.0, 133.8, 133.9, 132.1, 129.5, 129.3, 129.3, 128.3, 128.1, 127.9, 123.9, 120.1, 119.2 108.9, 63.9. HRMS (ESI, [M+Na]+) m/z calcd. for C18H16N2O3SNa 363.0774, found: 363.0781.
N'-((2-hydroxynaphthalen-1-yl)methylene)-4-nitrobenzenesulfonohydrazide (3q)
Commercially obtained 4-nitrobenznesulfonylhydrazine was reacted with anhydrous hydrazine as shown in General Method B, followed by condensation with 2-hydroxy-1-naphthaldehyde to obtain 3q. Yield 1.59 g, 4.29 mmol; 86%. Yellow solid; mp 230 °C. IR: 3311 (secondary N-H), 2116 (C=N), 1577 (C=C), 1280 (C-N), 1240 (C-O), 953-869 (S-O), 679 (S-N), 657 (C-S). 1H-NMR: ((CD3)2SO, 500 MHz) δ 11.02 (s, 1H), 9.98 (s, 1H), 8.77 (s, 1H), 8.64 (d, J = 8.6 Hz, 2H), 8.19 (d, J = 8.4 Hz, 1H), 8.02 (d, J = 9.0 Hz, 2H), 7.91 (d, J = 8.1 Hz, 1H), 7.85 (t, J = 8.1 Hz, 1H), 7.61 (t, J = 8.0 Hz, 1H), 7.43 (t, J = 7.5 Hz, 1H), 7.27 (d, J = 8.9 Hz, 1H). 13C-NMR: (CDCl3, 125 MHz) 157.2, 151.7, 150.9, 147.4, 136.0, 133.7, 129.3, 128.5 (2C), 128.1, 126.1, 124.2, 123.9, 123.9, 120.1, 119.7, 118.5. HRMS (ESI, [M+Na]+) m/z calcd. for C17H13N3O5SNa 393.0468, found: 393.0474.
N'-((2-hydroxynaphthalen-1-yl)methylene)-4-methylbenzenesulfonohydrazide (3r)
Commercially obtained d-10 camphoresulfonic acid was converted to its ethyl ester as shown in General Method A (see SI). General methods B and D (see SI) were then followed as previously described to obtain 3r. Yield 0.55 g, 1.63 mmol; 33%. Yellow solid; mp 230 °C. IR: 2117 (C=N), 1680 (C=O), 1577 (C=C), 1465 (C-C), 1318-1281 (N-O), 1240 (C-O), 679 (S-N), 656 (C-S). 1H-NMR: ((CD3)2SO, 400 MHz) δ 12.67 (s, 1H), 9.99 (s, 1H), 8.70 (s, 1H), 8.03 (d, J = 9.0 Hz, 1H), 7.91 (d, J = 8.1 Hz, 1H), 7.61 (t, J = 7.6 Hz, 1H), 7.49-7.39 (m, 1H), 7.29 (dd, J = 14.1, 8.4 Hz, 1H), 7.12-7.05 (m, 1H), 3.38 (d, J = 15.1 Hz, 1H), 3.04 (d, J = 15.1 Hz, 1H), 2.52 (dd, J = 4.9, 2.7 Hz, 1H), 2.16 (t, J = 4.6 Hz, 1H), 2.15-1.99 (m, 3H), 1.44 (d, J = 8.7, 3.8 Hz, 1H), 0.93 (d, J = 9.3 Hz, 6H). 13C-NMR: ((CD3)2SO, 100 MHz) 144.9, 135.0, 129.3, 128.5, 124.2, 122.4, 119.2, 119.0, 53.6, 53.2, 49.7, 49.4, 48.4, 35.3, 28.3, 27.4, 20.6, 19.8, 18.1, 18.0, 15.4. HRMS (ESI, [M+Na]+) m/z calcd. for C21H24N2O4SNa 423.1349, found: 423.1358.
4-amino-3-chloro-N'-((2-hydroxynaphthalen-1-yl)methylene)benzohydrazide (3s)
Commercially obtained p-toluenesulfonylhydrazine was condensed with 2-hydroxy-1-naphthaldehyde as described in General Method D (see SI) to obtain 3s. Yield 0.85 g, 2.51 mmol; 50%. Yellow solid; mp 230 °C. IR: 3163 (secondary N-H), 3036 (aromatic C-H), 2104 (C=N), 1620 (primary NH2), 1578 (C=C), 1281 (C-N), 1241 (C-O), 666 (C-Cl). 1H-NMR: (CDCl3, 500 MHz) δ 12.18 (s, 1H), 10.81 (s, 1H), 9.67 (s, 1H), 8.16 (d, J = 8.5 Hz, 1H), 7.97 (t, J = 8.5 Hz,1H), 7.88 (d, J = 9.0 Hz, 2H), 7.80 (t, J = 9.7 Hz, 1H), 7.59 (t, J = 7.5 Hz, 1H), 7.40 (t, J = 7.5 Hz, 1H), 7.20 (d, J = 9.0 Hz, 2H), 7.13 (d, J = 9.7 Hz, 1H), 2.39 (s, 3H). 13C-NMR: (CDCl3, 125 MHz) 150.9, 143.3, 143.2, 135.0, 133.8, 132.8, 129.7, 129.5, 129.3, 128.3, 128.1, 126.6, 123.9, 120.1, 119.2, 118.5, 108.9, 30.7. HRMS (ESI, [M+Na]+) m/z calcd. for C18H16N2O3SNa 363.0774, found: 363.0771.
3-chloro-N'-((2-hydroxynaphthalen-1-yl)methylene)-4-nitrobenzohydrazide (3t)
Prepared according to general procedures described in the SI. Yield 1.36 g, 3.69 mmol; 74%. Orange solid; mp 230 °C. IR: 3415 (secondary NH), 3300-3200 (broad, OH), 2117 (C=N), 1618 (C=O), 1577 (C=C), 1318 1280 (symmetrical N-O), 1182 (C-O), 721 (C-Cl). 1H NMR: (CDCl3, 500 MHz) δ 13.01 (s, 1H), 9.67 (s, 1H), 8.16 (d, J = 8.5 Hz, 1H), 7.88 (d, J = 9.0 Hz, 1H), 7.80 (dd, J = 8.1, 1.4 Hz, 2H), 7.66-7.55 (m, 2H), 7.40 (ddd, J = 8.0, 6.8, 1.0 Hz, 2H), 7.25 (d, J = 9.0 Hz, 1H) 7.17 (d, J = 9.0 Hz, 1H), 2.5 (s, 2H). 13C NMR: (CDCl3, 125 MHz) 169.5, 163.4, 147.8, 143.0, 135.0, 133.3, 129.3, 128.1 (2C), 127.8, 125.5, 125.4, 123.9, 120.1, 119.7, 119.1, 118.9, 118.6. HRMS (ESI, M+) m/z calcd. For C18H14ClN3O2 339.0055, found 339.0059.
5-bromo-N'-((2-hydroxynaphthalen-1-yl)methylene)-2-methoxybenzohydrazide (3u)
Prepared according to general procedures described in the SI. Yield 1.25 g, 3.14 mmol; 63%. Yellow solid; mp 229 °C. IR: 3272 (secondary N-H), 3171 (sp2 C-H), 2996 (sp3 C-H), 2094 (C=N), 1639 (C=O), 1550 (C=C), 1235 (C-O), 635 (C-Br). 1H-NMR: (C2D6SO, 400 MHz) δ 12.76 (s, 1H), 11.76 (s, 1H), 9.43 (s, 1H), 8.25 (d, J = 8.6 Hz, 1H), 7.92 (d, J = 8.5 Hz, 1H), 7.88 (d, J = 9.0 Hz, 1H), 7.68 (d, J = 8.1 Hz, 1H), 7.58 (ddd, J = 8.4, 6.9, 1.3 Hz, 1H), 7.44-7.27 (m, 3H), 7.22 (d, J = 9.0 Hz, 1H). 13C-NMR: (CDCl3, 100 MHz) 168.4, 164.1, 151.0, 143.1, 143.0, 135.0, 133.3, 129.7, 129.5, 124.1, 129.3, 128.1, 127.8, 123.9, 120.1, 119.7, 119.2, 118.9. HRMS (ESI, [M+Na]+) m/z calcd. for C18H12ClN3O4Na 369.0511, found: 363.0516.
5-bromo-N'-((2-hydroxynaphthalen-1-yl)methylene)-2-methoxybenzohydrazide (3v)
Prepared according to general procedures described in the SI. Yield 1.70 g, 4.26 mmol; 85%. Yellow solid; mp 230 °C. IR: 3273 (secondary N-H), 2116 (C=N), 1642 (C=O), 1583 (C=C), 1462 (C-C), 1233 (C-O), 648 (C-Br). 1H-NMR: (CD3CN, 500 MHz) δ 13.07 (br s, 1H), 11.11 (s, 1H), 9.47 (s, 1H), 8.29 (d, J = 8.5 Hz, 1H), 8.13 (d, J = 8.4 Hz, 1H), 8.00 (m, 1H), 7.96 (m, 1H), 7.70 (ddd, J = 8.5, 6.9, 1.4 Hz, 1H), 7.55-7.48 (m, 2H), 7.42 (dd, J = 8.4, 1.8 Hz, 1H), 7.32 (d, J = 9.0 Hz, 1H) 4.21 (s, 3H). 13C NMR (CD3CN, 125 MHz) 161.3, 159.2, 157.3, 148.3, 134.1, 133.8, 129.8, 128.5, 128.5, 125.3, 124.5, 121.2, 120.0, 119.5, 116.5, 114.9, 112.7, 109.4, 30.8. HRMS (ESI, M+) m/z calcd. for C19H15BrN2O3 421.0164, found 421.0164.
4-bromo-N'-((2-hydroxynaphthalen-1-yl)methylene)-2-methoxybenzohydrazide (3w)
Prepared according to general procedures described in the SI. Yield 1.70 g, 4.26 mmol; 85%. Yellow solid; mp 230 °C. IR: 3273 (secondary N-H), 2116 (C=N), 1642 (C=O), 1583 (C=C), 1462 (C-C), 1233 (C-O), 648 (C-Br). 1H-NMR: (CD3CN, 500 MHz) δ 13.07 (br s, 1H), 11.11 (s, 1H), 9.47 (s, 1H), 8.29 (d, J = 8.5 Hz, 1H), 8.13 (d, J = 8.4 Hz, 1H), 8.00 (m, 1H), 7.96 (m, 1H), 7.70 (ddd, J = 8.5, 6.9, 1.4 Hz, 1H), 7.55-7.48 (m, 2H), 7.42 (dd, J = 8.4, 1.8 Hz, 1H), 7.32 (d, J = 9.0 Hz, 1H) 4.21 (s, 3H). 13C NMR (CD3CN, 125 MHz) 161.5, 160.1, 157.4, 148.3, 134.1, 133.8, 129.8, 128.5, 125.3, 124.5, 121.2, 120.0, 119.4, 118.7, 116.5, 114.3, 112.4, 109.1, 30.8. HRMS (ESI, M+) m/z calcd. For C19H15BrN2O3 421.0164, found 421.0174.
Benzyl2-((2-hydroxynaphthalen-1-yl)methylene)hydrazine-1-carboxylate (3x)
Prepared according to general procedures described in the SI. Yield 0.25 g, 0.78 mmol; 16%. Yellow solid; mp 230 °C. IR: 2987 (sp3 C-H), 2117 (C=N), 1694 (ester C=O), 1578 (C=C), 1467 (C-C), 1281 (C-N), 1239 (C-O). 1H-NMR: (CD3CN, 500 MHz) δ 12.46 (s, 1H), 9.52 (br. s, 1H), 9.11 (s, 1H), 8.12 (d, J = 8.6 Hz, 1H), 7.98-7.91 (m, 2H), 7.66 (ddd, J = 8.5, 6.9, 1.4 Hz, 1H), 7.60 – 7.43 (m, 5H), 7.27 (d, J = 9.0 Hz, 1H), 7.17 (d, J = 8.9 Hz, 1H), 5.35 (s, 2 H). 13C-NMR (CD3CN, 125 MHz) 165.2, 153.5, 143.6, 133.3, 132.9, 129.8, 129.5 (2C), 129.1, 128.9, 128.5 (2C), 128.0, 127.9, 124.5, 120.9, 119.7, 108.9, 67.8. HRMS (ESI, [M+Na]+) m/z calcd. for C19H16N2O3Na 343.1059, found 343.1049.
2-hydroxy-N'-((7-methyl-1H-indol-3-yl)methylene)benzo-hydrazide (3z)
Prepared according to general procedures described in the SI. Yield 0.21 g, 0.73 mmol; 15%. Yellow solid. mp 230 °C. IR: 3300-3200 (broad, OH), 2987 (sp3 C-H), 2117 (C=N), 1694 (C=O), 1578 (C=C), 1467 (C-C), 1281 (C-N), 1239 (C-O). 1H-NMR: (CD3CN, 500 MHz) δ 12.36 (s, 1H), 11.04 (s, 1H), 9.80 (br. s, 1H), 8.57 (s, 1H), 8.18 (d, J = 8.6 Hz, 1H), 7.79 (d, J = 5.5 Hz, 1H), 7.71 (d, J = 6.9 Hz, 1H), 7.49 (t, J = 9.1 Hz, 1H), 7.18 (t, J = 9.0 Hz, 1H), 7.12 (d, J = 8.9 Hz, 1H), 7.00 (d, J = 6.8 Hz, 1H), 6.96 (d, J = 5.4 Hz, 1H), 2.55 (s, 3H). 13C-NMR (CD3CN, 125 MHz) 163.0, 161.2, 146.2, 134.6, 132.3, 130.4, 127.1, 126.0, 124.4, 124.2, 121.8, 120.0, 119.1, 118.3, 118.2, 109.5, 16.2. HRMS (ESI, M+) m/z calcd. for C17H15N3O2 293.1159, found: 293.1162.
Growth Inhibitory Assays against Cancer Cell lines
Growth inhibition assays to assess cellular toxicity were performed in the Case Comprehensive Cancer Center, CWRU, in a manner similar to recently reported data.10 Human colon cancer (HCT116) and human breast cancer (MDA-MB-231) cell lines were maintained in standard growth media consisting of (RPMI1640 + 10% FBS + 2 mM glutamine + 100 U/mL penicillin, 100 µg/mL streptomycin). Cell growth was monitored and shown to be negative for mycoplasma contamination using the Mycoplasma Detection kit (MycoAlert™, Lonza, Basel, Switzerland). For growth inhibition assays, cells were harvested by trypsinization and seeded into 96-well tissue culture plates at 2500 cells/mL. The following day an equal volume of 2x-inhibitor containing medium was added to each well. The cells were cultured for 3 additional days at 37 °C in a 5% CO2 humidified incubator. HCT-116 and MDA-MB-231 cell lines were treated with inhibitor concentrations of 10.0, 1.0, and 0.1 µM respectively. Cell growth (and its inhibition) was assessed by measuring total DNA content per well using the method of Labarca and Paigen23 and reported as cellular IC50 values. Results of this evaluation are presented in Table S2 (see SI).
Sensitization of MDA-MB-231 cell line toward NSAH inhibitor under siRNA transfection and RRM1 knockdown conditions
All reagents necessary to perform siRNA transfection and knockdown studies were purchased from ThermoFisher, Grand Island, NY, unless otherwise noted. siRNA transfection was performed using the Lipofectamine® 2000 reagent per manufacturer’s instructions. siRNA pools (RRM1 Silencer® Select, 3 RefSeqs; or control scrambled siRNA) at 0–50 pmole/well concentrations, were diluted in serum-free Opti-MEM media, then mixed with Lipofectamine® 2000 to allow formation of respective complexes. This solution was then serially diluted in Opti-MEM media and pipetted into wells of a standard 96-well tissue culture plate. An equal number of MDA-MB-231 breast cancer cells (1000/well) diluted in RPMI1640 + 10% FBS (without antibiotics) and were then added to each well and cells were incubated for 48 hours at 37 °C in a 5% CO2 humidified tissue culture incubator to allow for repression of RRM1. Specific inhibitor 3a was then added as 5X concentrated stock, diluted in cell culture media, and cells were allowed to grow for an additional 5 days. Each series of siRNAs (0, 0.25, 0.5, 1.0, 2.0 & 4.0 pmole/well) were treated with a dose range of 3a in duplicate (20 µM to 0.075 µM, with an additional inhibitor-free control; in 2-fold dilution increment). DNA concentration was then measured using an adaptation of the method of Labarca and Paigen.23 Media was removed, cells washed briefly in 0.25X PBS, followed by addition of 100 µL of ddH2O water to each well. The plates were subjected to a cycle of freezing and thawing to induce hypotonic lysis. DNA dye (Hoechst 33258, bisbenzamide, Sigma Aldrich, St. Louis, MO) diluted in 2M NaCl, 10 mM Tris-HCl, pH 7.4 was then added to each well and allowed to incubate in the dark at room temperature for 2 hours. Intensity in the wells in each Plate was then read using a SpectraMax i3 fluorescence plate reader (Molecular Devices, Sunnyvale, CA) using excitation/emission wavelengths of 370/460 nm, respectively. A standard curve using purified salmon DNA was included to allow for determination of DNA amounts per well. Cell growth inhibition is calculated relative to untreated controls; individual curves were generated for each siRNA amount.
Supplementary Material
Acknowledgments
Authors thank Dr. William E. Harte (School of Medicine, CWRU) for insightful comments and suggestions during the development of this project. Authors thank Tyler Jenkins for synthesis of aldehyde precursor involved in the synthesis of 3c. The authors thank Dr. Johnathon Karty and Angela Hansen from Indiana University for providing high resolution mass spectrometry analysis on all analogs. The authors thank the Univ. of Cincinnati library (former P&G library), and specifically Dr. William Seibel, for providing access to the high throughput screening library.
Funding Sources
This study is partly funded by NIH funding to PI: Dr. Chris G. Dealwis (R01GM100887). RV thanks the Department of Chemistry for funding portions of this project. The authors thank the National Science Foundation for major research instrumentation funding through the grant to the Department of Chemistry (NSF MRI-1334048). This research was supported in part by the Translational Research Shared Resource of the Case Comprehensive Cancer Center (P30 CA043703). The authors acknowledge these funding sources for their support.
Abbreviations
- ADP
adenosine diphosphate
- Anhyd.
anhydrous
- ATP
adenosine triphosphate
- CDCl3
chloroform-d
- CD3CN
acetonitrile-d
- (CD3)OD
dimethylsulfoxide-d
- DEPT
distortionless enhancement by polarization transfer
- DMF
dimethylformamide
- DMSO
dimethylsulfoxide
- DTT
dithiothreitol
- EDTA
ethylenediaminetetraacetic acid
- GTP
guanosine triphosphate
- HEPES
4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid
- HPLC
high performance liquid chromatography
- HRMS
high resolution mass spectrometry
- hRRM1
human ribonucleotide reductase mammalian 1
- hRRM2
human ribonucleotide reductase mammalian 2
- H2SO4
sulfuric acid
- IC50
half maximal inhibitory concentration
- IR
infrared
- LiAlH4
lithium aluminum hydride
- MeOH
methanol
- MHz
megahertz
- MP
melting point
- NaHCO3
sodium bicarbonate
- NaOH
sodium hydroxide
- NMR
nuclear magnetic resonance
- PCC
pyridinium chlorochromate
- RR
ribonucleotide reductase
- siRNA
small interfering RNA
- TLC
thin layer chromatography
- UV
ultraviolet
Footnotes
Associated Content
Molecular formula strings as SMILES entries is provided as Supporting Information file. General synthetic methods, docking poses for some inhibitors at the C-site, experimental methods for metal chelation studies, copies of spectra, enzyme assay tabulated data and dose-response curves for 3a, 3c, 3t and 3w are provided in the Supporting Information file. The Supporting Information file is available free of charge on the ACS Publications website at DOI:
Competing Financial Interests
The authors declare no competing financial interests
Author Contributions
CGD, MEH and RV designed the project. SEH, FAM, JP and MY performed experiments. JP performed all cell-based experiments. RV, SEH, MEH, JP and CGD wrote the manuscript. Prashansa Agarwal contributed to collection and helped in analysis of NMR spectral data for compounds.
References
- 1.(a) Brown NC, Reichard P. Role of effector binding in allosteric control of ribonucleoside diphosphate reductase. J. Mol. Biol. 1969;46:39–55. doi: 10.1016/0022-2836(69)90056-4. [DOI] [PubMed] [Google Scholar]; (b) Thelander L, Reichard P. Reduction of ribonucleotides. Annu. Rev. Biochem. 1979;48:133–158. doi: 10.1146/annurev.bi.48.070179.001025. [DOI] [PubMed] [Google Scholar]; (c) Eriksson M, Uhlin U, Ramaswamy S, Ekberg M, Regnström K, Sjöberg BM, Eklund H. Binding of allosteric effectors to ribonucleotide reductase protein R1: reduction of active-site cysteines promotes substrate binding. Structure. 1997;5:1077–1092. doi: 10.1016/s0969-2126(97)00259-1. [DOI] [PubMed] [Google Scholar]; (d) Larsson KM, Jordan A, Eliasson R, Reichard P, Logan DT, Nordlund P. Structural mechanism of allosteric substrate specificity regulation in a ribonucleotide reductase. Nat. Struct. Mol. Biol. 2004;11:1142–1149. doi: 10.1038/nsmb838. [DOI] [PubMed] [Google Scholar]; (e) Xu H, Faber C, Uchiki T, Fairman JW, Racca J, Dealwis CG. Structures of eukaryotic ribonucleotide reductase I provide insights into dNTP regulation. Proc. Natl. Acad. Sci. 2006;103:4022–4027. doi: 10.1073/pnas.0600443103. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Wang J, Lohman GJ, Stubbe J. Enhanced subunit interactions with gemcitabine-5′-diphosphate inhibit ribonucleotide reductases. Proc. Natl. Acad. Sci. 2007;104:14324–14329. doi: 10.1073/pnas.0706803104. [DOI] [PMC free article] [PubMed] [Google Scholar]; (g) Kashlan OB, Cooperman BS. Comprehensive model for allosteric regulation of mammalian ribonucleotide reductase: refinements and consequences. Biochemistry. 2003;42:1696–1706. doi: 10.1021/bi020634d. [DOI] [PubMed] [Google Scholar]
- 2.(a) Rosell R, Crino L, Danenberg K, Scagliotti G, Bepler G, Taron M, Alberola V, Provencio M, Camps C, De MF, Sanchez JJ, Penas R. Targeted therapy in combination with gemcitabine in non-small cell lung cancer. Semin. Oncol. 2003;30:19–25. doi: 10.1016/s0093-7754(03)00281-1. [DOI] [PubMed] [Google Scholar]; (b) Rosell R, Fossella F, Milas L. Molecular markers and targeted therapy with novel agents: prospects in the treatment of non-small cell lung cancer. Lung Cancer. 2002;38:43–49. doi: 10.1016/s0169-5002(02)00171-x. [DOI] [PubMed] [Google Scholar]; (c) Rosell R, Taron M, Sanchez JM, Moran T, Reguart N, Besse B, Isla D, Massuti B, Alberola V, Sanchez JJ. The promise of pharmacogenomics: gemcitabine and pemetrexed. Oncology. 2004;18:70–76. [PubMed] [Google Scholar]
- 3.(a) Knappenberger AJ, Ahmad MF, Viswathanan R, Dealwis CG, Harris ME. Nucleotide analog triphosphates allosterically regulate human ribonucleotide reductase and identify chemical determinants that drive substrate specificity. Biochemistry. 2016;55:5884–5896. doi: 10.1021/acs.biochem.6b00594. [DOI] [PubMed] [Google Scholar]; (b) Fu Y, Lin H, Wisitpitthaya S, Blessing WA, Aye Y. A fluorometric readout reporting the kinetics of nucleotide-induced human ribonucleotide reductase oligomerization. ChemBioChem. 2014;15:2598–2604. doi: 10.1002/cbic.201402368. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Rofougaran R, Vodnala M, Hofer A. Enzymatically active mammalian ribonucleotide reductase exists primarily as an a6b2 octamer. J. Biol. Chem. 2006;281:27705–27711. doi: 10.1074/jbc.M605573200. [DOI] [PubMed] [Google Scholar]
- 4.(a) Ando N, Brignole EJ, Zimanyi CM, Funk MA, Yokoyama K, Asturias FJ, Stubbe J, Drennan CL. Structural interconversions modulate activity of Escherichia coli ribonucleotide reductase. Proc. Natl. Acad. Sci. U.S.A. 2011;108:21046–21051. doi: 10.1073/pnas.1112715108. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Ando N, Li H, Brignole EJ, Thompson S, McLaughlin MI, Page JE, Asturias FJ, Stubbe J, Drennan CL. Allosteric inhibition of human ribonucleotide reductase by dATP entails the stabilization of a hexamer. Biochemistry. 2016;55:373–381. doi: 10.1021/acs.biochem.5b01207. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Zimanyi CM, Chen PYT, Kang G, Funk MA, Drennan CL. Molecular basis for allosteric specificity regulation in class IA ribonucleotide reductase from Escherichia coli. eLife. 2016;5:e07141. doi: 10.7554/eLife.07141. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Wisitpitthaya S, Zhao Y, Long MJ, Li M, Fletcher EA, Blessing WA, Weiss RS, Aye Y. Cladribine and fludarabine nucleotides induce distinct hexamers defining a common mode of reversible RNR inhibition. ACS Chem. Biol. 2016;11:2021–2032. doi: 10.1021/acschembio.6b00303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.(a) Ahmad MF, Dealwis CG. The structural basis for the allosteric regulation of ribonucleotide reductase. In: Giraldo J, Ciruela F, editors. Oligomerization in Health and Disease. Vol. 117. Academic Press; Cambridge, MA: 2013. pp. 389–410. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Cooperman BS, Dealwis CG. Allosteric regulation and inhibition of class 1a ribonucleotide reductase activity. In: Anderson K, editor. Riboncleotide Reductase. Nova Science Publishers, Inc; Hauppauge, NY: 2008. pp. 99–124. Ch. 4. [Google Scholar]; (c) Fairman JW, Wijerathna SR, Ahmad MF, Xu H, Nakano R, Jha S, Prendergast J, Welin RM, Flodin S, Roos A, Nordlund P, Li Z, Walz T, Dealwis CG. Structural basis for allosteric regulation of human ribonucleotide reductase by nucleotide-induced oligomerization. Nat. Struct. Mol. Biol. 2011;18:316–322. doi: 10.1038/nsmb.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Xu H, Fairman JW, Wijerathna SR, Kreischer NR, LaMacchia J, Helmbrecht E, Cooperman BS, Dealwis CG. The structural basis for peptidomimetic inhibition of eukaryotic ribonucleotide reductase: A conformationally flexible pharmacophore. J. Med. Chem. 2008;51:4653–4659. doi: 10.1021/jm800350u. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Aye Y, Li M, Long MJ, Weiss RS. Ribonucleotide reductase and cancer: biological mechanisms and targeted therapies. Oncogene. 2015;34:2011–2021. doi: 10.1038/onc.2014.155. [DOI] [PubMed] [Google Scholar]
- 6.(a) Heinemann V, Schulz L, Issels RD, Plunkett W. Gemcitabine: a modulator of intracellular nucleotide and deoxynucleotide metabolism. Semin. Oncol. 1995;22:11–18. [PubMed] [Google Scholar]; (b) Baker CH, Banzon J, Bollinger JM, Stubbe J, Samano V, Robin MJ, Lippert B, Jarvi E, Resvick R. 2'-Deoxy-2'-methylenecytidine and 2'-deoxy-2',2'-difluorocytidine 5'-diphosphates: potent mechanism-based inhibitors of ribonucleotide reductase. J. Med. Chem. 1991;34:1879–1884. doi: 10.1021/jm00110a019. [DOI] [PubMed] [Google Scholar]; (c) Wang J, Lohman GJ, Stubbe J. Mechanism of inactivation of human ribonucleotide reductase with p53R2 by gemcitabine 5'-diphosphate. Biochemistry. 2009;48:11612–11621. doi: 10.1021/bi901588z. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Plunkett W, Huang P, Gandhi V. Preclinical characteristics of gemcitabine. Anticancer Drugs. 1995;6:7–13. doi: 10.1097/00001813-199512006-00002. [DOI] [PubMed] [Google Scholar]
- 7.(a) Heinemann V, Xu YZ, Chubb S, Sen A, Hertel LW, Grindey GB, Plunkett W. Cellular elimination of 2',2'-difluorodeoxycytidine 5'-triphosphate: a mechanism of self-potentiation. Cancer Res. 1992;52:533–539. [PubMed] [Google Scholar]; (b) Huang P, Plunkett W. Phosphorolytic cleavage of 2-fluoroadenine from 9-β-D-arabinofuranosyl-2-fluoroadenine by Escherichia coli: A pathway for 2-fluoro-ATP production. Biochem. Pharmacol. 1987;36:2945–2950. doi: 10.1016/0006-2952(87)90207-3. [DOI] [PubMed] [Google Scholar]; (c) Gandhi V, Plunkett W. Modulatory activity of 2',2'-difluorodeoxycytidine on the phosphorylation and cytotoxicity of arabinosyl nucleosides. Cancer Res. 1990;50:3675–3680. [PubMed] [Google Scholar]; (d) Gandhi V, Huang P, Xu YZ, Heinemann V, Plunkett W. Metabolism and action of 2',2'-difluorodeoxycytidine: self-potentiation of cytotoxicity. Adv. Exp. Med. Biol. 1991;309:125–130. doi: 10.1007/978-1-4899-2638-8_28. [DOI] [PubMed] [Google Scholar]
- 8.Ahmad MF, Huff SE, Pink J, Alam I, Zhang A, Perry K, Harris ME, Misko T, Porwal SK, Oleinick NL, Miyagi M, Viswanathan R, Dealwis CG. Identification of non-nucleoside human ribonucleotide reductase modulators. J. Med. Chem. 2015;58:9498–9509. doi: 10.1021/acs.jmedchem.5b00929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dealwis CG. Method of modulating ribonucleotide reductase. 009,4314A1. U.S. Patent 2015. 2015 Apr 2;
- 10.Ahmad MF, Alam I, Huff SE, Pink J, Flanagan S, Shewach DS, Misko TA, Oleinick NL, Harte W, Viswanathan R, Harris ME, Dealwis CG. Potent competitive inhibition of human ribonucleotide reductase by a non-nucleoside small molecule. Proc. Natl. Acad. Sci. U.S.A. 2017;114:8241–8246. doi: 10.1073/pnas.1620220114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.(a) Baell JB, Holloway GA. New substructure filters for removal of pan assay interference compounds (PAINS) from screening libraries and for their exclusion in bioassays. J. Med. Chem. 2010;53:2719–2740. doi: 10.1021/jm901137j. [DOI] [PubMed] [Google Scholar]; (b) Baell JB, Ferrins L, Falk H, Nikolakopoulos G. PAINS: Relevance to tool compound discovery and fragment-based screening. Australian J. Chem. 2013;66:1483–1494. [Google Scholar]; (c) Whitty A. Growing PAINS in academic drug discovery. Future Med. Chem. 2011;3:797–801. doi: 10.4155/fmc.11.44. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Aldrich C, Bertozzi C, Georg GI, Kiessling L, Lindsley C, Liotta D, Merz KM, Schepartz A, Wang S. The ecstasy and agony of assay interference compounds. ACS Central Sci. 2017;3:143–147. doi: 10.1021/acscentsci.7b00069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.van Dijken DJ, Kovaricek P, Ihrig SP, Hecht S. Acylhydrazones as widely tunable photoswitches. J. Am. Chem. Soc. 2015;137:14982–14991. doi: 10.1021/jacs.5b09519. [DOI] [PubMed] [Google Scholar]
- 13.(a) Lepoivre M, Flaman JM, Bobe P, Lemaire G, Henry Y. Quenching of the tyrosyl free radical of ribonucleotide reductase by nitric oxide: relationship to cytostasis induced in tumor cells by cytotoxic macrophages. J. Biol. Chem. 1994;269:21891–21897. [PubMed] [Google Scholar]; (b) Szalai VA, Brudvig GW. Reversible binding of nitric oxide to tyrosyl radicals in photosystem II: Nitric oxide quenches formation of the S3 EPR signal species in acetate-inhibited photosystem II. Biochemistry. 1996;35:15080–15087. doi: 10.1021/bi961117w. [DOI] [PubMed] [Google Scholar]
- 14.Heinemann V, Plunkett W. Modulation of deoxynucleotide metabolism by the deoxycytidylate deaminase inhibitor 3,4,5,6-tetrahydrodeoxyuridine. Biochem. Pharmacol. 1989;38:4115–21. doi: 10.1016/0006-2952(89)90693-x. [DOI] [PubMed] [Google Scholar]
- 15.(a) Nyholm S, Mann GJ, Johansson AG, Bergeron RJ, Graeslund A, Thelander L. Role of ribonucleotide reductase in inhibition of mammalian cell growth by potent iron chelators. J. Biol. Chem. 1993;268:26200–26205. [PubMed] [Google Scholar]; (b) Hershko C. Control of disease by selective iron depletion: a novel therapeutic strategy utilizing iron chelators. Baillieres Clin. Haematol. 1994;7:965–1000. doi: 10.1016/s0950-3536(05)80133-7. [DOI] [PubMed] [Google Scholar]; (c) Lundberg JH, Chitambar CR. Interaction of gallium nitrate with fludarabine and iron chelators: effects on the proliferation of human leukemic HL60 cells. Cancer Res. 1990;50:6466–6470. [PubMed] [Google Scholar]; (d) Mescher AL, Munaim SI. Transferrin and the growth-promoting effect of nerves. Int. Rev. Cytol. 1988;110:1–26. doi: 10.1016/s0074-7696(08)61846-x. [DOI] [PubMed] [Google Scholar]; (e) Nocentini G, Federici F, Franchetti P, Barzi A. 2,2'-bipyridyl-6-carbothioamide and its ferrous complex: Their in vitro antitumoral activity related to the inhibition of ribonucleotide reductase R2 subunit. Cancer Res. 1993;53:19–26. [PubMed] [Google Scholar]
- 16.(a) Bianchi V, Pontis E, Reichard P. Changes of deoxyribonucleoside triphosphate pools induced by hydroxyurea and their relation to DNA synthesis. J. Biol. Chem. 1986;261:16037–16042. [PubMed] [Google Scholar]; (b) Lagergren J, Reichard P. Purine deoxyribonucleosides counteract effects of hydroxyurea on deoxyribonucleoside triphosphate pools and DNA synthesis. Biochem. Pharmacol. 1987;36:2985–2991. doi: 10.1016/0006-2952(87)90213-9. [DOI] [PubMed] [Google Scholar]
- 17.Reid G, Wallant NC, Patel R, Antonic A, Saxon-Aliifaalogo F, Cao H, Webster G, Watson JD. Potent subunit-specific effects on cell growth and drug sensitivity from optimized siRNA-mediated silencing of ribonucleotide reductase. J. RNAi Gene Silencing. 2009;5:321–330. [PMC free article] [PubMed] [Google Scholar]
- 18.(a) Cohen FE, McKerrow JH, Ring CS, Rosenthal PJ, Kenyon GL, Li Z. Preparation of Hydrazides as Inhibitors of Metazoan Parasite Proteases. 5610192A. U.S. Patent. 1997 Mar 11;; (b) Cohen FE, McKerrow JH, Ring CSY, Rosenthal PJ, Kenyon GL, Li Z. Inhibitors of Metazoan Parasite Proteases. WO 9406280A1. 1994 Mar 31;
- 19.Munoz DL, Cardona DP, Cardona A, Carrillo LM, Quinones W, Echeverri F, Velez ID, Robledo SM. Effect of hydrazones against intracellular amastigotes of Leishmania panamensis and a parasitic cysteine protease. Vitae. 2006;13:5–12. [Google Scholar]
- 20.Drewes MW, Haug M, Luerssen K, Santel HJ, Schmidt RR. Herbicidal naphthalene Derivatives: Preparation of 2-(4,6-dimethoxypyrimidin-2-yloxy)-1-naphthaldehyde Hydrazones as Herbicides. 07/680,495. U.S. Patent. 1993 Feb 9;
- 21.(a) Tan Z, Wortman M, Dillehay KL, Seibel WL, Evelyn CR, Smith SJ, Malkas LH, Zheng Y, Lu S, Dong Z. Small-molecule targeting of proliferating cell nuclear antigen chromatin association inhibits tumor cell growth. Mol. Pharmacol. 2012;81:811–819. doi: 10.1124/mol.112.077735. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Dillehay KL, Seibel WL, Zhao D, Lu S, Dong Z. Target validation and structure-activity analysis of a series of novel PCNA inhibitors. Pharmacol. Res. Perspect. 2015;3:1–14. doi: 10.1002/prp2.115. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Dong Z, Wortman M, Tan Z, Dillehay K. Methods for Identification of Proliferating Cell Nuclear Antigen (PCNA) Targeting Compounds for Cancer Therapy and PCNA Function Regulation. WO2012033938A2. 2012 Mar 15;
- 22.Flanagan SA, Robinson BW, Krokosky CM, Shewach DS. Mismatched nucleotides as the lesions responsible for radiosensitization with gemcitabine: a new paradigm for antimetabolite radiosensitizers. Mol. Cancer Ther. 2007;6:1858–1868. doi: 10.1158/1535-7163.MCT-07-0068.(b) In a personal communication, Dr. Shewach further reaffirmed their view of NSAH inhibiting hRR to cause perturbations in cellular dNTP levels leading to results published earlier (reference 10).
- 23.Labarca C, Paigen KA. Simple, rapid, and sensitive DNA assay procedure. Anal. Biochem. 1980;102:344–352. doi: 10.1016/0003-2697(80)90165-7. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.