

Summary
Somatic mutations of ERBB2 (HER2) and ERBB3 (HER3) are found in a wide range of cancers. Preclinical modelling suggests that a subset lead to constitutive HER2 activation, but most remain biologically uncharacterized. We sought to prospectively define the biologic and therapeutic significance of known oncogenic HER2 and HER3 mutations and variants of unknown biological significance by conducting a multi-histology, genomically selected, ‘basket’ study utilizing the pan-HER kinase inhibitor neratinib (SUMMIT; Clinicaltrials.gov NCT01953926). Efficacy in HER2-mutant cancers varied as a function of both tumour type and mutant allele to a degree not predicted by preclinical models, with the greatest activity seen in breast, cervical and biliary cancers and with tumours harbouring kinase domain missense mutations. This study demonstrates how a molecularly driven clinical trial can be used to further refine our biological understanding of both characterized and novel genomic alterations with potential broad applicability for advancing the paradigm of genome-driven oncology.
Introduction
Genomic profiling of human cancers has identified recurrent somatic mutations of HER2 (ERBB2) and HER3 (ERBB3), typically occurring in the absence of gene amplification1–3. Mutations in HER2 are clustered in the extracellular, transmembrane, and kinase domains. Unlike other mutant oncogenes, eg BRAF or KRAS, no single mutant allele predominates and the precise distribution of mutations varies by tumour type4. In contrast, HER3 mutations cluster primarily in the extracellular domain and to a lesser extent in the kinase domain. Although HER2 and HER3 mutations are found in a wide variety of cancers, their overall prevalence does not exceed 10% in any individual tumour type and more typically the rate is <5% for HER2 and <1% for HER3.
Biological modelling has yielded conflicting findings addressing the functional consequences of HER2 and HER3 mutations. Substantial data suggest that a subset of these mutations lead to ligand-independent constitutive HER2 receptor signalling and promotes oncogenesis5–7. The mechanism of these oncogenic effects appears to differ by variant, with some causing enhanced HER2 kinase activity and others receptor dimerization5,8. Mutations in HER3, which in its wild-type configuration has impaired kinase function, appear to rely on wild-type HER2 to exert its oncogenic effects7. Most preclinical data exploring the functional consequences of HER2 and HER3 mutations have been generated using engineered models that overexpress the mutation and thus the results may be confounded by the known oncogenic effects of HER2 overexpression. Further enforcing the potential importance of this confounding variable, models of HER2 mutation generated by gene-editing techniques have failed to demonstrate a malignant phenotype in the absence of mutations in other oncogenes such as PIK3CA9.
Given the significant diversity of HER2 and HER3 mutations, as well as the challenge of generating preclinical models that recreate their true biology in human cancers, we sought to define the therapeutic significance of HER2 and HER3 mutations by conducting SUMMIT – a global, multicentre, multi-histology ‘basket’ study in patients with tumours harbouring these mutations (Extended Data Fig. 1). Patients were treated with neratinib, an irreversible pan-HER tyrosine kinase inhibitor, which potently inhibits the growth of HER2-mutant tumours in preclinical models5. Tumour tissue and plasma were collected to facilitate the detailed genomic characterization of patients. Here, we present the results of this study with a focus on the insights it provides into the biological and therapeutic significance of HER2 and HER3 mutations in cancer patients.
Results
Patient and mutation characteristics
Baseline patient demographics are shown in Table 1 and Extended Data Table 1. In total, 141 patients (125 with HER2-mutant tumours, 16 with HER3-mutant tumours) received neratinib. These patients were diagnosed with one of 21 unique cancer types, the most common being breast, lung, bladder and colorectal cancer (61% of patients treated). As has been seen in other basket studies10,11, we identified and enrolled a number of orphan tumour types including cancers of the biliary tract, salivary gland, small bowel and vagina, as well as extramammary Paget’s disease (13% of all patients). Patients tended to be heavily pretreated with approximately half having received ≥3 prior lines of systemic therapy.
Table 1.
Patient demographics
| Patient characteristic | HER2 mutant (n=125) | HER3 mutant (n=16) | Total (n=141) |
|---|---|---|---|
| Age | |||
| Median (range), years | 61 (30–83) | 66 (39–82) | 61 (30–83) |
| <65 years, n (%) | 81 (64.8) | 7 (43.8) | 88 (62.4) |
| ≥65 years, n (%) | 44 (35.2) | 9 (56.3) | 53 (37.6) |
| Sex, n (%) | |||
| Female | 80 (64.0) | 12 (75.0) | 92 (65.2) |
| Male | 45 (36.0) | 4 (25.0) | 49 (34.8) |
| ECOG performance status, n (%) | |||
| 0 | 37 (29.6) | 1 (6.3) | 38 (27.0) |
| 1 | 83 (66.4) | 12 (75.0) | 95 (67.4) |
| 2 | 5 (4.0) | 3 (18.8) | 8 (5.7) |
| Prior systemic treatment lines, n (%) | |||
| Any | 121 (96.8) | 16 (100) | 137 (97.2) |
| 1 | 33 (26.4) | 1 (6.3) | 34 (24.1) |
| 2 | 30 (24.0) | 11 (68.8) | 41 (29.1) |
| ≥3 | 58 (46.4) | 4 (25.0) | 62 (44.0) |
| Median time from metastasis to enrolment, years (range) | 1.02 (0.0–15.0) | 1.13 (0.3–4.5) | 1.03 (0.0–15.0) |
| Tumour type, n (%) | |||
| Lung | 26 (20.8) | 0 (0) | 26 (18.4) |
| Breast | 25 (20.0) | 0 (0) | 25 (17.7) |
| Bladder | 16 (12.8) | 2 (12.5) | 18 (12.8) |
| Colorectal | 12 (9.6) | 5 (31.3) | 17 (12.1) |
| Biliary tract | 9 (7.2) | 2 (12.5) | 11 (7.8) |
| Endometrial | 7 (5.6) | 1 (6.3) | 8 (5.7) |
| Cervical | 5 (4.0) | 0 (0) | 5 (3.5) |
| Gastroesophageal | 5 (4.0) | 2 (12.5) | 7 (5.0) |
| Ovarian | 4 (3.2) | 1 (6.3) | 5 (3.5) |
| Other | 16 (12.8) | 3 (18.8) | 19 (13.5) |
ECOG, Eastern Cooperative Oncology Group.
Enrolled patients had 31 unique HER2 and 11 unique HER3 mutations (Extended Data Fig. 2). The most frequent HER2 mutations were S310, L755, Y772_A775dup and V777 alleles. The HER2 kinase domain was most commonly mutated (66%), followed by the extracellular (26%) and transmembrane/juxtamembrane (8%) domains. The anticipated relationships between the mutated HER2 domain and tumour type were observed, with extracellular domain mutations predominant in bladder cancer, kinase domain missense mutations in breast and colon cancer, and kinase domain insertions in lung cancer4. Missense mutations were the most common class of genomic alteration (74%) followed by in-frame insertions (22%), the latter exclusively affecting the kinase domain. Two patients harboured insertions/deletions and one an in-frame kinase domain-retaining fusion (GRB7-ERBB2)12,13. HER3 mutations were all missense variants and clustered in the extracellular furin-like and receptor domains. In total, 87% (109/125) of HER2 and 75% (12/16) of HER3 mutations were at positions now known to be mutational hotspots4. This pattern of HER2 and HER3 mutations was comparable to the spectrum of non-truncating HER2 and HER3 mutations observed in previously published genomic landscape studies, including TCGA and ICGC4, although HER2 V777L and Y772_A755dup were more common in our study cohort (13.6% vs 5.3% and 12.0% vs 2.7%, respectively, Extended Data Fig. 3).
Treatment outcomes
When stratified by tumour type, we observed responses to neratinib in patients with HER2-mutant breast, non-small-cell lung, cervical, biliary and salivary cancers, which led to expanded enrollment in several of these tumour types (Fig. 1a, Extended Data Table 1). Neratinib exhibited the greatest degree of activity in patients with breast cancer (n=25 total, objective response rate at week 8 [ORR8] 32%, 95% confidence interval [CI] 15–54%) with responses observed in patients with missense mutations involving the extracellular and kinase domains, as well as insertions in the kinase domain. All breast cancer patients were classified as HER2 negative (non-amplified) at the time of enrolment per established guidelines14. Responses were observed in both estrogen receptor (ER)+ (30%, 6/20) and ER- (40%, 2/5) tumours. Overall, these breast cancer data are generally consistent with a prior report15. In patients with lung cancer (n=26), where exon 20 insertions predominate, we observed only one objective response. Of note, HER2 exon 20 insertions are paralogous of EGFR exon 20 insertions, which are resistant to first- and second-generation EGFR tyrosine kinase inhibitors16. Interestingly, the only patient with lung cancer to achieve a RECIST response had a kinase domain missense mutation (L755S). Despite the low response rate, the median progression-free survival in recurrent lung cancer was 5.5 months with 6 patients remaining on therapy for greater than 1 year, which compares favourably to second-line chemotherapy and immune checkpoint inhibitors176, suggesting that neratinib may still be having a positive impact on the natural history of this disease. Responses were also observed in biliary and cervical cancers, and enrolment is ongoing in these cohorts to better define this activity. No responses were observed in bladder cancer (n=16) or colorectal cancer (n=12), suggesting lineage-dependent resistance to single-agent pan-HER kinase inhibition in these tumour types. In summary, among the HER2-mutants cohorts, breast cancer met the primary endpoint for efficacy, while lung, colorectal and bladder cancers did not. For the remaining tumour-specific cohorts, enrolment is ongoing and they have therefore not undergone final efficacy analysis. Despite preclinical data suggesting that HER3 mutations can be oncogenic drivers, no responses to neratinib were observed in patients with HER3-mutant tumours.
Figure 1. Individual treatment outcome and response for 141 patients grouped by a) tumour cohort and b) mutant allele/domain.
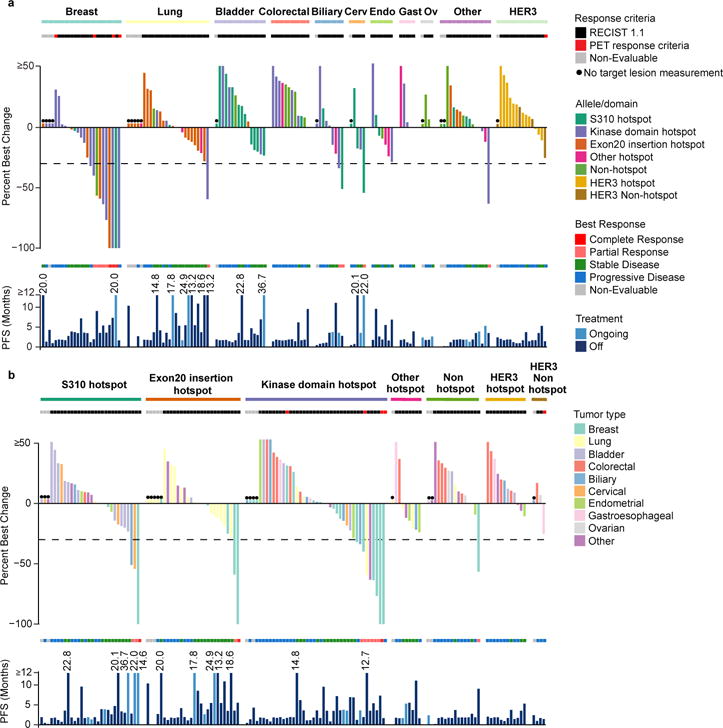
For each panel: The top graph shows percent best change from baseline in the target lesion assessed by the appropriate response criteria (RECIST version 1.1 or PET). Each bar is colour coded according to its a) mutation allele/domain or b) tumour type. The middle section shows best overall response. The bottom graph shows PFS colour coded by treatment status.
*Non-evaluable.
Cerv, cervical; endo, endometrial; gastro, gastroesophageal; Ov, ovarian; PET, positron-emission tomography; PFS, progression-free survival; RECIST, Response Evaluation Criteria in Solid Tumors.
When stratified by mutant allele, responses were observed in patients with tumours harbouring HER2 S310, L755, V777, G778_P780dup and Y772_A775dup mutations (Fig. 1b). Among patients with HER2 kinase domain hotspot missense mutations (n=42), responses were noted in four unique tumour types (breast, biliary, lung and salivary gland). By allele, we observed responses in several kinase domain mutants L755S (n=4), V777L (n=4) and L869R (n=1). In patients with HER2 hotspot extracellular domain mutations (S310, n=30), responses were observed in breast, cervical and biliary cancers (n=1 for each), but not in bladder cancer where these mutations predominate. Similarly, in patients with HER2 exon 20 insertions (n=28), responses were observed in two patients with breast cancer but none were seen in patients with lung cancer where this class of alterations is most common. In exon 20 insertions, preservation of glycine at the 770 position, which appears to facilitate binding of covalent HER kinase inhibitors such as neratinib, did not predict for response as previously suggested by preclinical modelling (Extended Data Fig. 4)18. Similarly, the number of amino acids involved in the insertion did not appear to predict outcome, with responses observed in patients with both 3 (G788_P780dup) and 4 (Y722_A755dup) amino acid insertions. Finally, among the 15 patients with HER2 mutations not known to be hotspots, only one responded to neratinib. Interestingly, this response occurred in a patient with breast cancer and a complex insertion/substitution (L755_E757delinsS) which, to our knowledge, has not been observed previously. While this case illustrates that some patients may be addicted to truly private oncogenic drivers (those arising in only a single patient), it is also noteworthy that this insertion occurs in a domain that is the target of recurrent insertions. The absence of clinical activity in the remaining 14 patients with cancers with non-hotspot mutations suggests that while the recurrence of a mutation in HER2 is insufficient to define it as sensitizing to a HER2 kinase inhibitor, the absence of recurrence (ie mutations that do not occur at hotspot positions) provides circumstantial evidence that the alteration is unlikely to be a driver.
While the overall numbers of patients in each subgroup preclude formal statistical comparison, integrating efficacy, mutational and lineage data, we observed that clinical benefit from neratinib therapy appeared to vary as a function of both mutational and disease context (Fig. 2). In tumour types sensitive to neratinib therapy, such as breast, biliary and cervical cancers, responses were collectively observed across all types and classes of HER2 mutations. In contrast, in lung cancer, a tumour type that exhibits modest sensitivity to neratinib, response was limited to a patient with a HER2 kinase domain missense mutation – a class of mutations with greater in vitro sensitivity to neratinib5. Finally, in tumour types with intrinsic lineage-based resistance to neratinib, such as bladder and colorectal cancers, RECIST responses were not observed regardless of the HER2 mutation, type or class.
Figure 2. Integrated efficacy by tumour type and HER2 allele/domain.
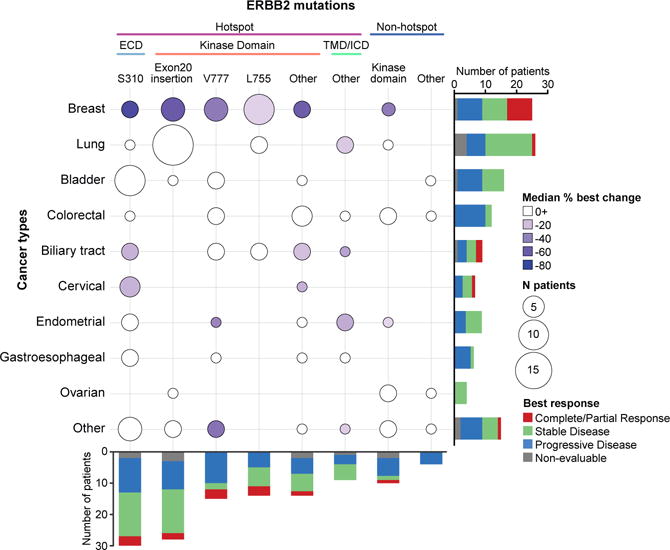
The y-axis represents the ten tumour types and the x-axis represents the mutated allele/domain and hotspot status. The hotspot mutations are further broken down into the various domains. The size of the circle is proportional to the frequency of the tumour type and allele/domain; the colour of the circle reflects the median percent best change in the target lesion (any positive median change is indicated in white). The stacked bars represent the best overall change for the tumour type or domain/allele, as indicated in the legend.
ECD, extracellular domain; ICD, intracellular domain; TMB, transmembrane domain.
Safety
All patients received neratinib with mandatory anti-diarrhoeal prophylaxis. With this regimen, the rate of grade 3 diarrhoea was 22% (Extended Data Table 2), consistent with previous experience19. Among patients who developed grade 3 diarrhoea, the median time to onset was 10 days and the median duration of the diarrhoea episode 2 days. Patients were typically managed with dose interruption and reduction, with only 2.8% permanently discontinuing therapy due to diarrhoea. The remainder of adverse events were low grade.
Central confirmation of HER2 and HER3 mutations
There is active debate within the cancer research community as to whether central confirmation of mutational status before study entry is optimal for determining trial eligibility for precision medicine studies. To define the reproducibility of local mutational testing, DNA from archival formalin-fixed paraffin-embedded tumour and plasma samples were re-sequenced (see Methods). Thirty-three patients (26 with HER2-mutant, 7 with HER3-mutant) were excluded from this concordance analysis because the local test used was the same as the central tumour assay being evaluated. Of the remaining 99 patients with HER2 mutations, adequate material for tumour genomic testing was unobtainable for 26 patients. Overall, concordance in the remaining patients based on central tumour and/or plasma sequencing was 95% (69/73), with 38 patients assessed by tissue and plasma, 14 by tissue alone, and 21 by plasma alone. Central testing identified one locally reported mutation (V773M) as a germline polymorphism and this patient, with renal cell carcinoma, had progressive disease at first scan. Central testing in the four cases where the HER2 mutation could not be confirmed passed all quality-control metrics but in two patients was performed on material collected ≥3 years after the tissue used for local testing, raising the possibility that tumour heterogeneity played a role in the discordance. None of the patients with discordant HER2 results responded to neratinib, and their median progression-free survival was only 43 days (range: 5–58 days). Among the 9 patients eligible for concordance testing with HER3 mutations, tumour tissue was available for central sequencing in eight patients, and overall concordance was 75% (6/8).
Genomic modifiers of response
Given the variability of treatment response, even among patients with the same tumour lineage and HER2-mutant allele, we sought to identify additional genomic modifiers of response through broader genomic characterization of tumour-derived DNA (see Methods). First, we explored the relationship between HER2 amplification and outcome, as this is a well-established predictor of response to HER2-targeted therapies in patients lacking HER2 mutations. In total, 17% of patients (15/86) had concurrent ERBB2 mutations and gene amplification. Amplifications preferentially targeted the mutant allele locus (86%, 12/14 evaluable). Using a dichotomous definition of clinical benefit (stable disease or partial response lasting ≥24 weeks), ERBB2 amplification did not correlate with outcome (p=0.50; Fig. 3), suggesting that in the presence of ERBB2 mutations, amplification may not confer additional sensitivity to irreversible HER kinase inhibitors. We also explored the relationship of ERBB2 mutation clonality on outcomes. In the 74 patients with adequate material to allow definitive assessment of ERBB2 mutant clonality, the HER2 mutation was clonal in 95% (70/74, Extended Data Fig. 5a). None of four patients with a subclonal HER2 mutation achieved clinical benefit.
Figure 3. Genomic modifiers of response and outcome by treatment duration.
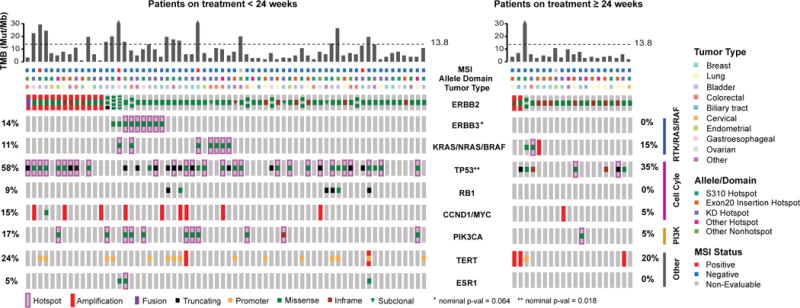
a) Comprehensive OncoPrint of the dichotomous clinical benefit groups for 86 patients with broad profiling data (left: no benefit (n=66, biologically independent samples), right: clinical benefit (n=20, biologically independent samples)). From top to bottom: TMB with the dotted line indicating the threshold for high TMB at 13.8 mutations per megabase, MSI status, allele/domain, tumour type, HER2 (ERBB2) status showing amplification, clonality and multiple mutations, and co-alterations in genes associated with key pathways. *Nominal Fisher’s p-values unadjusted for multiple hypothesis testing shown. Statistical significance is lost when corrected for multiple hypothesis testing.
MSI, microsatellite instability; TMB, tumour mutational burden.
Hypothesizing that tumours with an increased tumour mutational burden (TMB) might be more likely to acquire HER2 mutations without developing oncogenic dependence (ie passenger mutations), we evaluated whether overall TMB status affected outcome. Using a previously validated cut-off (≥13.8 non-synonymous mutations per megabase of DNA2), 20% of patients (17/86) met criteria for high TMB. In total, 24% of patients (16/66) without clinical benefit versus 5% of patients (1/20) with benefit met criteria for high TMB, a trend that did not reach statistical significance (p=0.10).
Next, we evaluated whether the pattern of co-mutations affected clinical benefit in the subset of patients where broader profiling was available (n=86). In patients with HER2-mutant disease, coincident mutations in TP53 and HER3 were enriched in patients with no clinical benefit (nominal p=0.018 and p=0.064, respectively; Fig. 3). While not significant after correcting for multiple hypothesis testing potentially due to the relatively small sample size, it is noteworthy that no patients with clinical benefit possessed co-mutation of HER2 and HER3. Concurrent mutation of these genes was observed in multiple cancer types (breast n=3, bladder n=2, gastroesophageal n=2, colorectal n=1 and pancreatic n=1) and involved a variety of unique HER2 and HER3 mutations (n=8 and n=9, respectively). Expanding our analysis to genomic activation at the pathway level, we identified somatic mutations of known oncogenic potential and grouped them by those involving the RTKs/RAS/RAF and PIK3CA/AKT/MTOR pathways, and cell cycle checkpoints (Extended Data Fig. 5b). In this analysis, aberrations in cell cycle checkpoints were associated with lack of clinical benefit (p=0.043), while activation of RTK/RAS/RAF also trended towards a worse outcome (p=0.060). The association between the cell cycle pathway and lack of clinical benefit appears to be primarily driven by TP53 mutations, losing significance upon removal of TP53 mutations (p=0.769). Interestingly, activation of the PI3K/AKT/mTOR pathway, an established negative predictor of response to HER2-targeted therapy in HER2-amplified breast cancer20–22, did not adversely affect the likelihood of clinical benefit (p=0.753). It is possible that the clinical impact of concurrent gene/pathway activation may vary by tumour type, and future disease-specific studies are needed to better define these associations. Although these were exploratory analyses that will require confirmation, our results suggest that concurrent activation of specific genes as well as pathways may act as an additional modifier of response beyond cancer type and specific HER2 mutant allele.
Discussion
The ability to comprehensively profile cancer at the point of care has made possible the opportunity to personalize therapy for each patient based on the compendium of genomic alterations identified23. Despite the promise of this approach, implementing this paradigm in clinical practice has been hampered by significant gaps in knowledge regarding the biological and clinical significance of the majority of genomic variants identified24. This challenge is exemplified by the marked diversity and wide distribution of HER2 and HER3 mutations in human cancers, as well as by the difficulty of generating preclinical models of these mutations that faithfully recreate their biology in patients. SUMMIT provides the first comprehensive dataset on the clinical actionability of HER2 and HER3 mutations. We found that HER2 mutations are associated with HER2-dependence in a subset of patients with HER2 mutant tumors, but that response to HER kinase inhibition varies a function of the individual mutant variant, the tumour types as well as the pattern of co-mutations present.
Although we identified promising preliminary activity for neratinib in breast, biliary and cervical cancers, the response rate in these tumours was still lower than with approved therapies targeting oncogenic alterations in EGFR, ALK, ROS1, and BRAF. The low response rate in lung cancer, where HER2 mutations exhibit mutually exclusivity of other known drivers25, is also striking and may in part reflect a lower potency of neratinib inhibition in Y772_A775dup compared to others insertions or missense mutants.18 It is noteworthy that successfully targeting HER2 activation in other contexts has historically necessitated drug combinations. For example, single-agent trastuzumab has a response rate of only ~20% in ERBB2-amplified breast cancer26,27. In contrast, overall survival in ERBB2-amplified breast and gastroesophageal cancers is markedly improved by adding trastuzumab to chemotherapy28,29. More recently, intensification of HER2 inhibition through combination of two HER-targeted agents has been shown to result in synergistic efficacy in patients with ERBB2-amplified breast30–32 or colorectal33,34 cancers, as well as in HER2-mutant colorectal cancer xenografts6. Cumulatively, these data suggest that combining neratinib with another HER-targeted therapy is a rational next step, and SUMMIT has been amended to evaluate this approach in multiple HER2-mutant tumour types.
SUMMIT represents a continued evolution in the design of basket studies, which enrol patients on the basis of qualifying mutations rather than tumour type. The initial generation of these studies focused on evaluating individual somatic mutations that were already clinically validated in one cancer (eg BRAF V600 in melanoma) in other tumour types10,35. More recently, basket studies have been used to generate initial or even practice-changing clinical data of truly novel genomic biomarkers, especially when these genomic alterations occur at low frequency across a wide distribution of cancer types11,36,37. SUMMIT extends this concept one step further by demonstrating for the first time how a single study can be used to simultaneously evaluate a range of individual variants in HER2 and HER3, each with varying degrees of prior biologic characterization. This permissive enrolment strategy allowed us to treat patients harbouring mutations that at the time of enrolment had not been characterized preclinically as gain of function but were either recurrent or paralogous to known activating mutations in homologous genes. For example, patients with previously uncharacterized HER2 variants, such as V697, D769N, and L869R, were included in this manner and responded to treatment, thus providing initial clinical proof-of-concept that these mutations confer a gain-of-function phenotype even before formal biologic characterization. The approach of pairing a permissive enrolment strategy with allele prioritization based on recurrence, paralogy and other readily computable features has potentially broad applicability to implementing genomic-driven oncology24. This strategy will take on even greater importance as clinical testing moves from targeted sequencing to whole exome or even whole genome sequencing, techniques that will allow for evaluation of an even greater number of therapeutic hypothesis but will also exponentially grow the number of uncharacterized alleles we routinely identify.
SUMMIT provides additional insights into the conduct of molecularly driven oncology studies. Our ability to understand the complex interactions between tumour lineage, individual HER2 variant, and response to neratinib was only possible because of the relatively large size of this study (n=141). By comparison, many of the ‘master/umbrella’ protocols currently underway are designed to enroll a maximum of 30–40 patients into each genomically defined treatment arm. Our experience suggests that many of studies of this size may be inadequately powered to identify the subgroups with true efficacy, assuming that most genomic alterations will not predict for tumour-type agnostic efficacy. SUMMIT also demonstrates the feasibility of enrolling patients based on local testing with patients enrolled on the basis of 30 unique sequencing assays performed in 25 different laboratories. Despite this, concordance on retrospective central review was extremely high (96%).
An important impediment to progress in oncology has been the limited availability of preclinical model systems that accurately recreate the complex biology of human cancer. While significant strides have been made, the wide-scale profiling of cancer in the clinic provides the potentially transformative opportunity to rapidly interrogate cancer biology at the bedside in a manner previously only possible at the bench. Here, we demonstrate how this opportunity can be leveraged to probe the biology of a diverse set of HER2 and HER3 mutations across a variety of solid tumours through pharmacological HER kinase inhibition in patients. In doing so, we found that response to pharmacological inhibition was based on the characteristics of both tumour type and genomic variant to a degree that was not predicted by established preclinical models. In summary, SUMMIT demonstrates how the clinical trial can become an important tool in refining our understanding of the biological dependencies in human cancers.
Methods
Patients
Eligible patients had histologically confirmed advanced solid tumours harbouring HER2 or HER3 mutations, an Eastern Cooperative Oncology Group (ECOG) performance score of 0-2 and an unlimited number of prior therapies. Patients with prior exposure to HER kinase inhibitors and unstable brain metastases were excluded. HER2 and HER3 mutations were determined by local tumour testing as routinely performed or ordered by each participating site. In total, 85% (120/141) of enrolled patients were identified by next-generation sequencing assays. In 81% of cases (97/120), the next-generation sequencing assay included full exon coverage for ERBB2 or ERBB3, while in 19% (23/120) of cases only select exons or hotspots were included in the assay design. The remaining 15% (21/141) of patients were enrolled via RT-PCR, Sanger, pyrosequencing, or mass spectrometry-based sequencing methods. The study was approved by the institutional review board or independent ethics committee at each site and complied with the International Ethical Guidelines for Biomedical Research Involving Human Subjects, Good Clinical Practice guidelines, the Declaration of Helsinki, and local laws. Written informed consent was obtained from all participants.
Study design, treatment and endpoints
This was a multi-cohort basket study of patients with solid tumours harbouring HER2 and HER3 mutations. Patients with HER2-mutant tumours were enrolled into one of several disease-specific cohorts or an “Other” cohort for tumour types not otherwise specified; all patients with HER3-mutant tumours were enrolled to one cohort. Patients known to harbour both HER2 and HER3 mutations at the time of enrolment were assigned to the HER2-mutant cohort. Patients were treated with neratinib 240 mg daily on a continuous basis with mandatory loperamide prophylaxis during cycle 1. The primary endpoint was ORR8, as assessed by investigators according to Response Evaluation Criteria in Solid Tumors (RECIST; version 1.1). Secondary endpoints included best overall response, progression-free survival, overall survival and safety. Patients who were not evaluable by RECIST were permitted to enrol and were evaluated for response by 18F-fluorodeoxyglucose positron-emission tomography (PET) according to a modified version of the original PET Response Criteria in Solid Tumors (PERCIST; version1.0)38, referred to here as PET Response Criteria (PRC, Extended Data Table 3).
Assessments
Disease assessments with computed tomography (CT), magnetic resonance imaging or PET/CT (for those evaluated by PRC) were performed at baseline and then every 8 weeks until disease progression, death or withdrawal. Adverse events were graded by the investigator according to the Common Terminology Criteria for Adverse Events (version 4.0) until day 28 after discontinuation of study treatment.
Genomic biomarker studies
All samples were assigned anonymized identifiers by the study sponsor based on the order of study enrolment. Both tumour DNA and tumour-derived cell-free (cf)DNA in plasma were collected with the goals of confirming locally reported HER2/3 mutations as well as evaluating how ERBB2/3 copy number and clonality as well as co-mutational pattern affected outcome. Collection of archival tumour and plasma samples was mandatory for all patients. Next-generation sequencing was performed utilizing targeted sequencing of pretreatment DNA from formalin-fixed paraffin-embedded tumour and matched blood specimens (preferentially) and cfDNA (if tumour was not available or was inadequate). A custom single-gene ERBB2 capture next-generation sequencing test was also performed on pretreatment cfDNA in a subset of patients with HER2-mutant disease.
Central sequencing confirmation
For patients with adequate material, DNA from formalin-fixed paraffin-embedded (n=91) or tumour-derived cell-free DNA from plasma (n=15) and matched germline DNA (n=102) underwent targeted next-generation sequencing assay using Memorial Sloan Kettering-Integrated Mutation Profiling of Actionable Cancer Targets (MSK-IMPACT)39, producing an average of 738-fold coverage per tumour (range: 253–1383). Briefly, this assay utilizes a hybridization-based exon capture designed to capture all protein-coding exons and select introns of oncogenes, tumour suppressor genes and key members of pathways that may be actionable by targeted therapies. In this study, either 341 (n=18) or 410 (n=88) key cancer-associated genes were used (Supplementary Information). Sequencing data were analysed as previously described to identify somatic single-nucleotide variants, small insertions and deletions, copy number alterations and structural arrangements40. Additionally, hotspot alterations were identified using an adaptation of a previously described method41 applied to a cohort of 24,592 sequenced human cancers42. For gene level analysis, select genes within our targeted 341/410 MSK-IMPACT panel involved in the RTK/RAS/RAF, PIK3CA/AKT/MTOR, and cell cycle checkpoint pathways were selected using the KEGG pathway database43. For pathway level analysis, only potentially oncogenic alterations in the selected genes were included and determined to be oncogenic by OncoKB (version September 2017), a curated knowledge base of the oncogenic effects and treatment implications of mutations and cancer genes (oncokb.org44).
HER2 amplification and clonality analysis
For patients in the HER2-mutant arm with MSK-IMPACT sequencing data (with matched germline DNA, n=74), the Fraction and Allele-Specific Copy Number Estimates from Tumour Sequencing (FACETS) algorithm (version 0.3.9) was used to estimate tumour purity and ploidy, total and allele-specific copy number45. Tumour samples with purity <20% were excluded from the analysis. Focal HER2 amplifications for tumours with MSK-IMPACT and FACETS data were inferred using the following criteria: fold change ≥1.5 (MSK-IMPACT tumour:normal sequencing coverage ratio) and total HER2 copy number ≥4 copies (FACETS-derived total copy number). To infer clonality of each HER2 mutation, cancer cell fractions (CCFs) were estimated with 95% confidence intervals by integrating FACETS-derived joint segmentation and MSK-IMPACT mutation data as input into the ABSOLUTE algorithm46 (version 1.0.6). Mutations were classified as either clonal or subclonal based on the following criteria: clonal if the estimated CCF >0.85, otherwise subclonal. For patients with HER2 amplification, the mutation copy number (mutation multiplicity) was calculated as previously described47 to infer amplification of the mutant allele when the mutation multiplicity was greater than half of the total HER2 copy number.
Tumour mutational burden and microsatellite instability
Tumour mutational burden (TMB), defined as the number of non-synonymous mutations per megabase, was calculated for patients with MSK-IMPACT sequencing data (n=106)6. Microsatellite instability (MSI) was assessed for patients with HER2-mutant tumours with matched germline DNA sequencing data (n=89) using an orthogonal bioinformatics tool, MSIsensor48. Additionally, mutations were decomposed into the thirty constituent mutational signatures as described previously49. Briefly, MSIsensor scores <10 were classified as microsatellite stable and >10 were considered MSI-High using a previously validated cut-off score50. Those with a MSIsensor score of <10 but having evidence of a dominant mismatch repair mutational signature were also considered MSI45,49.
Statistical analysis
For each HER2-mutant tumour type and the HER3-mutant cohort, a Simon optimal two-stage design with a true ORR8 ≤10% was considered unacceptable (null hypothesis) whereas a true ORR8 ≥30% (alternative hypothesis) merited further study. Efficacy in each cohort was analysed independently and the study was not designed to formally compare efficacy across cohorts. All patients who received at least one dose of neratinib were included in the safety and efficacy cohorts. All data reflect an interim data-cut taken on 10 Mar 2017 from patients enrolled up to 16 Dec 2016 (Extended Data Fig. 6). Most patients were off therapy at the time of data analysis (Extended Data Table 4). Progression-free survival was estimated using the Kaplan-Meier method. The study is registered at Clinicaltrials.gov, NCT01953926. Individual associations among genomic changes and response were assessed by either Fisher’s exact or chi-squared tests (where appropriate) and corrected for multiple hypothesis testing using Benjamini-Hochberg correction.
Chi-squared or Fisher’s exact tests were performed to compare gene-level and pathway-level associations between the dichotomous clinical benefit groups. P-values were corrected for multiple hypothesis testing using Benjamini-Hochberg correction. HER2 and HER3 lollipop distribution plots were generated using ProteinPaint51. All other figures were generated using R software (http://www.R-project.org/).
Data availability
All datasets generated during and/or analysed during the current study, including patient-level clinical data as well as all sequencing data have been deposited and are publically available in the cBioPortal for Cancer Genomics under the accession code “SUMMIT, Nature, 2018” (http://www.cbioportal.org/study?id=summit_2018). All figure source data are also provided at www.nature.com/nature.
Extended Data
Extended Data Figure 1. Design of SUMMIT study.
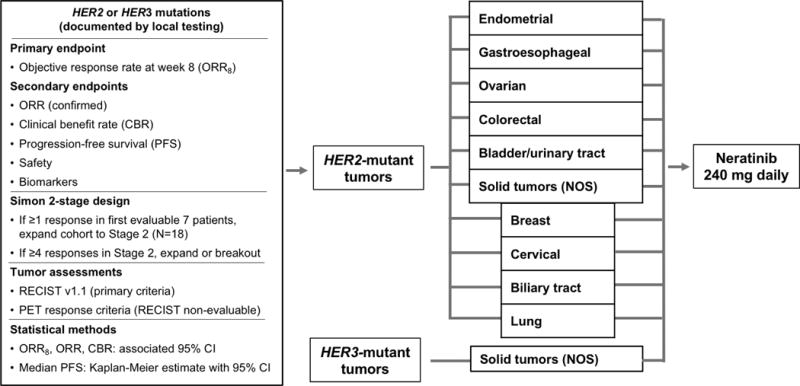
Five tumour-specific HER2 (ERBB2)-mutant cohorts were pre-specified (endometrial, gastroesophageal, ovarian, colorectal and bladder/urinary tract). In addition, a sixth “Solid tumour (NOS)” HER2-mutant cohort allowed for enrollment of patients with any other cancer types. A sufficient number of patients with breast, cervical, biliary and lung cancer were enrolled in the “Solid tumours (NOS)” cohort to permit independent efficacy analysis using the same design as the pre-specified cohorts. Patients with HER3 (ERBB3)-mutant tumours were enrolled in a HER3-specific cohort regardless of tumour type.
CBR, clinical benefit rate; cfDNA, cell-free [tumour] DNA; CI, confidence interval; FFPE, formalin-fixed paraffin-embedded; MSKCC, Memorial Sloan Kettering Cancer Center; MSK-IMPACT, Memorial Sloan Kettering-Integrated Mutation Profiling of Actionable Cancer Targets; NGS, next-generation sequencing; NOS, not otherwise specified; ORR, objective response rate; ORR8, objective response rate at week 8; PET, positron-emission tomography; PFS, progression-free survival; RECIST, Response Evaluation Criteria in Solid Tumors.
Extended Data Figure 2. Distribution of a) 125 HER2 and b) 16 HER3 mutations positioned by their amino acid co-ordinates across the respective protein domains.
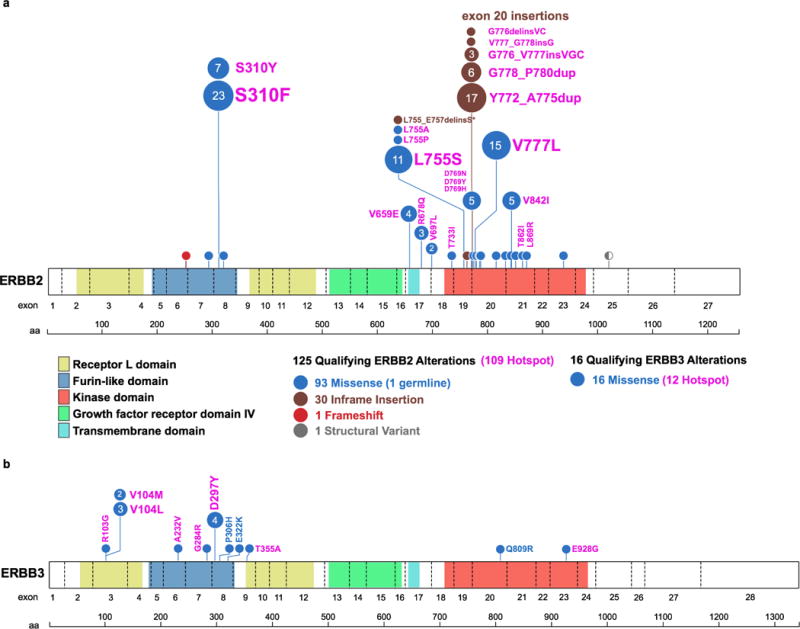
Each unique mutation is represented by a circle, with the circle size and number representing the frequency, and coloured to show the mutation class as indicated in the legend. The corresponding amino acid change and common hotspot mutations (shown in pink) are labelled next to the circles.
Extended Data Figure 3. Spectrum of HER2 and HER3 Mutations Observed in Neratinib Study versus TCGA, ICGC, and other Public Datasets.
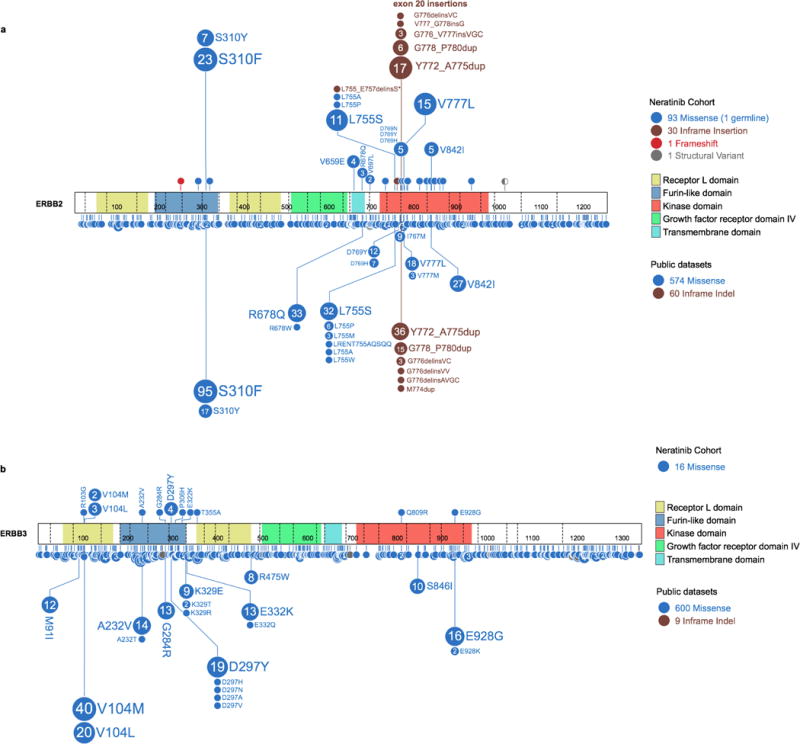
Distribution of a) HER2 and b) HER3 mutations observed across our cohort in comparison to the spectrum of HER2 and HER3 mutations (reflected lollipop) from publically available datasets (TCGA, ICGC, other published studies).
Extended Data Figure 4. Distribution and outcome of 28 HER2 exon 20 insertions.
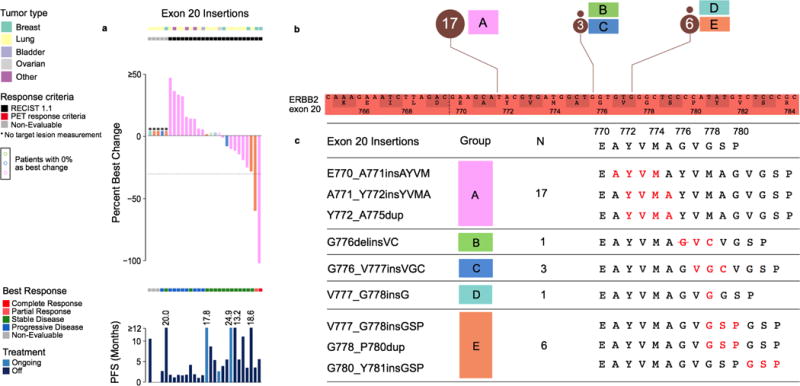
a) Percent best change and PFS plots corresponding to each type of exon 20 insertion (colour coded by synonymous amino acid change). Three cases with no change are indicated in colour-coded circles above the x-axis. b) Zoomed-in schematic of all exon 20 insertions positioned by their amino acid co-ordinates and frequencies. c) Five unique types of exon 20 insertions observed in the study with the resulting full amino acid sequences (insertion indicated in red).
PET, positron-emission tomography; PFS, progression-free survival; RECIST, Response Evaluation Criteria in Solid Tumors.
Extended Data Figure 5. Genomic modifiers of response and outcome by treatment duration.
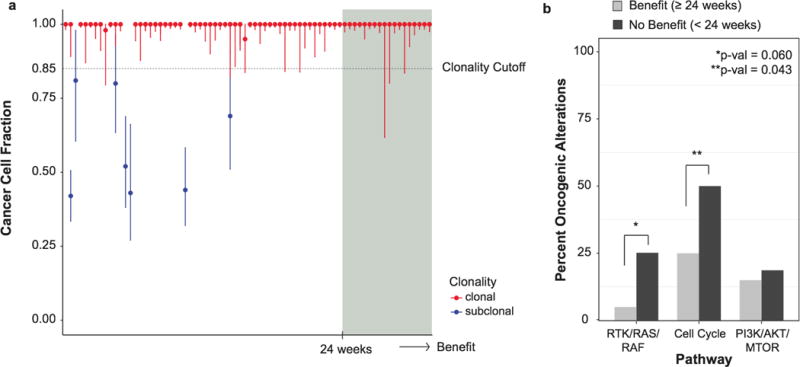
a) Cancer cell fractions with 95% confidence intervals and clonality status of all HER2 mutations in 74 patients with sufficient sequencing data ordered by increasing clinical benefit (weeks on therapy). b) Comparison of the percent activation of known oncogenic alterations in the three pathways between the patients of clinical benefit (n=20, biologically independent samples) and no benefit (n=66, biologically independent samples). Nominal Fisher’s p-values shown.
Extended Data Figure 6.
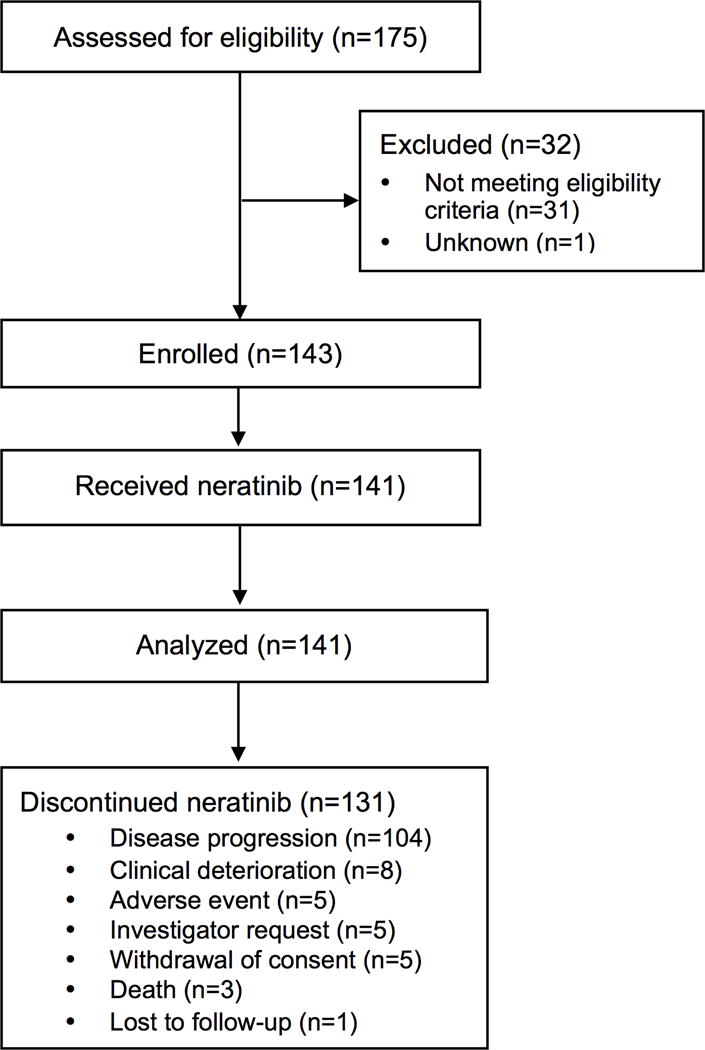
SUMMIT Consort Diagram.
Extended Data Table 1.
Patient demographics and efficacy by cohort CI, confidence interval; ORR, objective response rate; PFS, progression-free survival.
| Characteristic | HER2 | HER3 | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
|
| |||||||||||
| Breast (n=25) |
Lung (n=26) |
Bladder (n=16) |
Colorectal (n=12) |
Biliary
tract (n=9) |
Cervical (n=5) |
Endometrial (n=7) |
Gastro
esophageal (n=5) |
Ovarian (n=4) |
NOS (n=16) |
NOS (n=16) |
|
| Median (range), years | 57.0 (37–80) | 62.0 (46–74) | 65.0 (48–83) | 65.0 (30–81) | 66.0 (57–78) | 49.0 (42–56) | 57.0 (54–74) 5 | 67.0 (36–70) | 56.5 (38–58) | 59.0 (32–80) | 66.0 (39–82) |
| <65 years, n (%) | 19 (76.0) | 18 (69.2) | 8 (50.0) | 6 (50.0) | 2 (22.2) | 5 (100) | (71.4) | 1 (20.0) | 4 (100) | 13 (81.3) | 7 (43.8) |
| ≥65 years, n (%) | 6 (24.0) | 8 (30.8) | 8 (50.0) | 6 (50.0) | 7 (77.8) | 0 (0) | 2 (28.6) | 4 (80.0) | 0 (0) | 3 (18.8) | 9 (56.3) |
| Sex, n (%) | |||||||||||
| Female | 24 (96.0) | 17 (65.4) | 3 (18.8) | 6 (50.0) | 5 (55.6) | 5 (100) | 7 (100) | 2 (40.0) | 4 (100) | 7 (43.8) | 12 (75.0) |
| Male | 1 (4.0) | 9 (34.6) | 13 (81.3) | 6 (50.0) | 4 (44.4) | 0 (0) | 0 (0) | 3 (60.0) | 0 (0) | 9 (56.3) | 4 (25.0) |
| ECOG PS, n (%) | |||||||||||
| 0 | 7 (28.0) | 11 (42.3) | 6 (37.5) | 5 (41.7) | 2 (22.2) | 1 (20.0) | 2 (28.6) | 0 (0) | 0 (0) | 3 (18.8) | 1 (6.3) |
| 1 | 17 (68.0) | 14 (53.8) | 10 (62.5) | 7 (58.3) | 6 (66.7) | 4 (80.0) | 5 (71.4) | 5 (100) | 4 (100) | 11 (68.8) | 12 (75.0) |
| 2 | 1 (4.0) | 1 (3.8) | 0 (0) | 0 (0) | 1 (11.1) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 2 (12.5) | 3 (18.8)’ |
| Prior systemic lines, n (%) | 0 (0) | 1 (3.8) | 1 (6.3) | 0 (0) | 1 (11.1) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 1 (6.3) | 0 (0) |
| None | 3 (12.0) | 12 (46.2) | 2 (12.5) | 4 (33.3) | 3 (33.3) | 0 (0) | 1 (14.3) | 2 (40.0) | 0 (0) | 6 (37.5) | 1 (6.3) |
| 1 | 2 (8.0) | 6 (23.1) | 9 (56.3) | 3 (25.0) | 2 (22.2) | 3 (60.0) | 2 (28.6) | 1 (20.0) | 0 (0) | 2 (12.5) | 11 (68.8) |
| 2 | 20 (80.0) | 7 (26.9) | 4 (25.0) | 5 (41.7) | 3 (33.3) | 2 (40.0) | 4 (57.1) | 2 (40.0) | 4 (100) | 7 (43.8) | 4 (25.0) |
| ≥3 | |||||||||||
| Median time from metastasis to enrolment, years (range) | 2.64 (0.1–15.0) |
0.83 (0.1–3.1) |
0.69 (0.2–2.3) |
1.14 (0.0–2.7) |
1.00 (0.0–2.8) |
1.40 (0.3–4.5) |
0.43 (0.2–4.4) |
0.80 (0.4–4.3) |
7.54 (1.1–7.7) |
1.35 (0.0–5.4) |
1.13 (0.3–4.5) |
|
| |||||||||||
| Outcome | HER2 | HER3 | |||||||||
|
|
|||||||||||
| Breast (n=25) |
Lung (n=26) |
Bladder (n=16) |
Colorectal (n=12) |
Biliary tract (n=9) |
Cervical (n=5) |
Endometrial (n=7) |
Gastroesophageal (n=5) |
Ovarian (n=4) |
NOS (n=16) |
NOS (n=16) |
|
|
| |||||||||||
| ORR at week 8, n
(%) [95% CI] |
8
(32.0) [14.9–53.5] |
1
(3.8) [0.1–19.6] |
0
(0.0) [0.0–20.6] |
0
(0.0) [0.0–26.5] |
2
(22.2) [2.8–60.0] |
1
(20.0) [0.5–71.6] |
0
(0.0) [0.0–41.0] |
0
(0.0) [0.0–52.2] |
0
(0.0) [0.0–60.2] |
1
(6.3) [0.2–30.2] |
0
(0.0) [0.0–20.6] |
| ORR, n
(%) [95% CI] |
6
(24.0) [9.4–45.1] |
1
(3.8) [0.1–19.6] |
0
(0.0) [0.0–20.6] |
0
(0.0) [0.0–26.5] |
0
(0.0) [0.0–33.6] |
1
(20.0) [0.5–71.6] |
0
(0.0) [0.0–41.0] |
0
(0.0) [0.0–52.2] |
0
(0.0) [0.0–60.2] |
0
(0.0) [0.0–20.6] |
0
(0.0) [0.0–20.6] |
| Clinical benefit rate, n
(%) [95% CI] |
10
(40.0) [21.1–61.3] |
11
(42.3) [23.4–63.1] |
3
(18.8) [4.0–45.6] |
1
(8.3) [0.2–38.5] |
3
(33.3) [7.5–70.1] |
3
(60.0) [14.7–94.7] |
2
(28.6) [3.7–71.0] |
1
(20.0) [0.5–71.6] |
0
(0.0) [0.0–60.2] |
3
(18.8) [4.0–45.6] |
2
(12.5) [1.6–38.3] |
| Median PFS, months | 3.5 | 5.5 | 1.8 | 1.8 | 2.8 | 20.1 | 2.6 | 1.7 | 2.1 | 1.9 | 1.7 |
Extended Data Table 2.
Treatment-emergent adverse events (occurring in ≥10% of patients)
| Adverse event, n (%) | Neratinib monotherapy
(N=141) |
|
|---|---|---|
| Any grade | Grade ≥3 | |
| Diarrhoea | 104 (73.8) | 31 (22.0)* |
| Nausea | 61 (43.3) | 3 (2.1) |
| Vomiting | 58 (41.1) | 3 (2.1) |
| Constipation | 49 (34.8) | 2 (1.4) |
| Fatigue | 45 (31.9) | 5 (3.5) |
| Decreased appetite | 40 (28.4) | 1 (0.7) |
| Abdominal pain | 33 (23.4) | 7 (5.0) |
| Anaemia | 22 (15.6) | 10 (7.1) |
| Dyspnoea | 18 (12.8) | 5 (3.5) |
| Dehydration | 17 (12.1) | 8 (5.7) |
| Aspartate aminotransferase increased | 15 (10.6) | 5 (3.5) |
| Asthenia | 15 (10.6) | 1 (0.7) |
| Weight decreased | 15 (10.6) | 0 |
|
| ||
| Characteristics of diarrhoea | ||
|
| ||
| Action taken with neratinib, n (%) | ||
| Permanent discontinuation | 4 (2.8) | |
| Serious† diarrhoea, n (%) | 15 (10.6) | |
| Median (range) number of grade 3 diarrhoea episodes per patient | 1 (1–12) | |
| Median (range) duration of grade 3 diarrhoea episode, days | 2 (1–8) | |
| Median (range) time to first grade 3 diarrhoea episode, days | 10 (4–87) | |
Extended Data Table 3.
PET response criteria CT, computed tomography; FDG-PET, 18F-fluorodeoxyglucose positron-emission tomography; SUVmax, maximum standardised uptake value.
| Response category | Based on sum of SUVmax from 1 to 5 target lesions. Each target lesion with initial SUVmax of >1.5 × normal liver background SUVmax |
|---|---|
| Complete metabolic response (CMR) | • Reduction of SUVmax of all
target lesions to less than normal liver background SUVmax (for
non-brain lesions) or less than normal brain background SUVmax (for
brain lesions) AND • The reduction of all other FDG-avid lesions consistent with disease to less than normal liver background SUVmax |
| Partial metabolic response (PMR) | • Sum of SUVmax of all
target lesions is decreased by ≥30% compared to
baseline sum of SUVmax of all target
lesions AND • No new lesions |
| Stable metabolic disease (SMD) | Not satisfying the criteria for CMR. PMR. PMD, or NE |
| Progressive metabolic disease (PMD) | • Sum of SUVmax, of all
target lesions is Increased by
≥30% OR • Appearance of one or more unequivocal new FDG avid lesions |
| Not evaluable (NE) | • Missing FDG-PET series or
incomplete anatomy at follow-up timepoint • A PET/CT scanner change from baseline • Variation In FDG uptake time ≥15 minutes compared to baseline • Change in reconstruction algorithm |
Extended Data Table 4.
Patient disposition by cohort NOS, not otherwise specified.
| Characteristic | HER2 | HER3 | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
|
| |||||||||||
| Breast (n=25) |
Bladder (n=16) |
Lung (n=26) |
Colorectal (n=12) |
Biliary
tract (n=9) |
Cervical (n=5) |
Endometrial (n=7) |
Gastro
esophageal (n=5) |
Ovarian (n=4) |
NOS (n=16) |
NOS (n=16) |
|
| Patients continuing on treatment, n (%) | 1 (4) | 1 (6.2) | 1 (3.8) | 0 (0) | 1 (11.1) | 2 (40) | 1 (14.3) | 0 (0) | 1 (25) | 2 (12.5) | 0 (0) |
| Treatment discontinuation, n (%) | 24 (96.0) | 15 (93.8) | 25 (96.2) | 12 (100) | 8 (88.9) | 3 (60.0) | 6 (85.7) | 5 (100) | 3 (75.0) | 14 (87.5) | 16 (100) |
| Death | 0 (0) | 1 (6.3) | 0 (0) | 0 (0) | 1 (11.1) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 1 (6.3) |
| Disease progression | 22 (88.0) | 12 (75.0) | 18 (69.2) | 11 (91.7) | 5 (55.6) | 2 (40.0) | 4 (57.1) | 4 (80.0) | 2 (50.0) | 9 (56.3) | 15 (93.8) |
| Clinical deterioration | 0 (0) | 0 (0) | 3 (11.5) | 0 (0) | 1 (11.1) | 0 (0) | 0 (0) | 1 (20.0) | 1 (25.0) | 2 (12.5) | 0 (0) |
| Adverse Event | 0 (0) | 0 (0) | 2 (7.7) | 0 (0) | 1 (11.1) | 0 (0) | 1 (14.3) | 0 (0) | 0 (0) | 1 (6.3) | 0 (0) |
| Investigator Request | 0 (0) | 1 (6.3) | 0 (0) | 1 (8.3) | 0 (0) | 0 (0) | 1 (14.3) | 0 (0) | 0 (0) | 2 (12.5) | 0 (0) |
| Withdrawal of consent | 2 (8.0) | 0 (0) | 2 (7.7) | 0 (0) | 0 (0) | 1 (20.0) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 0 (0) |
| Lost to follow-up | 0 (0) | 1 (6.3) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 0 (0) |
| Subjects ended study, n (%) | 15 (60.0) | 14 (87.5) | 16 (61.5) | 9 (75.0) | 6 (66.7) | 1 (20.0) | 6 (85.7) | 5 (100) | 3 (75.0) | 7 (43.8) | 14 (87.5) |
| Death | 12 (48.0) | 13 (81.3) | 13 (50.0) | 8 (66.7) | 6 (66.7) | 1 (20.0) | 5 (71.4) | 3 (60.0) | 3 (75.0) | 7 (43.8) | 11 (68.8) |
| Withdrawal of consent | 2 (8.0) | 0 (0) | 2 (7.7) | 0 (0) | 0 (0) | 0 (0) | 1 (14.3) | 0 (0) | 0 (0) | 0 (0) | 2 (12.5) |
| Lost to follow-up | 1 (4.0) | 1 (6.3) | 1 (3.8) | 1 (8.3) | 0 (0) | 0 (0) | 0 (0) | 1 (20.0) | 0 (0) | 0 (0) | 1 (6.3) |
| Other | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 1 (20.0) | 0 (0) | 0 (0) | 0 (0) |
Supplementary Material
Acknowledgments
We thank patients and their families for participating in this study. Editorial support, not including writing, was provided by Lee Miller of Miller Medical Communications Ltd, UK. This work was funded by Puma Biotechnology Inc, Los Angeles, CA, USA.
Funding support
Supported by grants from the National Institutes of Health (Grants No. P30 CA008748, P30 CA016672, P30 CA014089, R01 CA204749, R01 CA80195, T32 CA009207, 1U01 CA180964, UL1 TR000371), the National Institutes of Health/National Cancer Institute (Breast SPORE Grant P50 CA098131), Cycle for Survival, Marie-Josée and Henry R. Kravis Center for Molecular Oncology, The Cancer Prevention and Research Institute of Texas (RP1100584), the Sheikh Khalifa Bin Zayed Al Nahyan Institute for Personalized Cancer Therapy, Nellie B. Connally Breast Cancer Research Endowment, the Breast Cancer Research Foundation, and Puma Biotechnology Inc, Los Angeles, CA, USA.
Footnotes
Supplementary Information
Supplementary Information is linked to the online version of the paper at www.nature.com/nature.
Author contributions
D.M.H., H.W., M.F.B., R.E.C, F.X., A.B., L.D.E., G.M., C.F., A.S.L., R.P.B., J.B., and D.B.S designed the study and supervised the analyses. R.E.C., F.X., L.D.E., G.M., C.F., A.S.L., and R.P.B., helped to collect and monitor the clinical outcome data. D.M.H., S.A.P., J.R., C.S., G.I.S., D.J., D.I.Q., V.M., B.D., I.A.M., V.B., E.C., S.L., A.C.L., J.P.E., B.T.L., A.J.H., R.M., A.M.S., A.D., L.M.S., K.J., G.I., J.J.H., C.L.A., F.M.B., J.B., and D.B.D. enrolled patients and provided patient samples. G.U., developed the PET response criteria and performed radiographic response assessments. B.S.T., J.P., J.T., S.D.S., N.B., M.M., M.F.B., J.B., and D.B.S. performed the tumour and plasma sequencing, provided computational infrastructure, and made final variant calls. D.M.H., H.W., M.S., B.S.T., J.P., J.T., H.B., M.F.B., and D.B.S. analysed clinical and genomic data and performed the integrated efficacy analyses. F.X. performed biostatistical analyses of the clinical efficacy data. D.M.H., H.W., B.S.T., C.L.A., F.M.B., and D.B.S. wrote the manuscript with input from all authors.
Competing financial interests include the following: R.E.C., F.X., L.D.E., G.M., C.F., A.S.L., and R.P.B are employees of Puma Biotechnology. D.M.H., M.S., and J.B. receive research support from Puma Biotechnology, B.T.L. and M.S. receive research funding from Diachi, A.D. receives personal fees from Roche, and D.S. received personal fees from Loxo Oncology and Pfizer.
References
- 1.Chmielecki J, et al. Oncogenic alterations in ERBB2/HER2 represent potential therapeutic targets across tumors from diverse anatomic sites of origin. Oncologist. 2015;20:7–12. doi: 10.1634/theoncologist.2014-0234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Zehir A, et al. Mutational landscape of metastatic cancer revealed from prospective clinical sequencing of 10,000 patients. Nat Med. 2017;23:703–713. doi: 10.1038/nm.4333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Schram A, et al. Landscape of somatic ERBB2 mutations: Findings from AACR GENIE and comparison to ongoing ERBB2 mutant basket study. Cancer Res. 2017;77(13 Supplement):LB-103. [Google Scholar]
- 4.Chang MT, et al. Identifying recurrent mutations in cancer reveals widespread lineage diversity and mutational specificity. Nat Biotechnol. 2016;34:155–163. doi: 10.1038/nbt.3391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bose R, et al. Activating HER2 mutations in HER2 gene amplification negative breast cancer. Cancer Discov. 2013;3:224–237. doi: 10.1158/2159-8290.CD-12-0349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kavuri SM, et al. HER2 activating mutations are targets for colorectal cancer treatment. Cancer Discov. 2015;5:832–841. doi: 10.1158/2159-8290.CD-14-1211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jaiswal BS, et al. Oncogenic ERBB3 mutations in human cancers. Cancer Cell. 2013;23:603–617. doi: 10.1016/j.ccr.2013.04.012. [DOI] [PubMed] [Google Scholar]
- 8.Chumsri S, et al. Prolonged response to trastuzumab in a patient with HER2-nonamplified breast cancer with elevated HER2 dimerization harboring an ERBB2 S310F Mutation. J Natl Compr Canc Netw. 2015;13:1066–1070. doi: 10.6004/jnccn.2015.0132. [DOI] [PubMed] [Google Scholar]
- 9.Zabransky DJ, et al. HER2 missense mutations have distinct effects on oncogenic signaling and migration. Proc Natl Acad Sci USA. 2015;112:E6205–E6214. doi: 10.1073/pnas.1516853112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hyman DM, et al. Vemurafenib in multiple nonmelanoma cancers with BRAF V600 Mutations. N Engl J Med. 2015;373:726–736. doi: 10.1056/NEJMoa1502309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hyman DM, et al. AKT inhibition in solid tumors with AKT1 mutations. J Clin Oncol. 2017;35:2251–2259. doi: 10.1200/JCO.2017.73.0143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ross JS, et al. A high frequency of activating extracellular domain ERBB2 (HER2) mutation in micropapillary urothelial carcinoma. Clin Cancer Res. 2014;20:68–75. doi: 10.1158/1078-0432.CCR-13-1992. [DOI] [PubMed] [Google Scholar]
- 13.Ross JS, et al. Relapsed classic E-cadherin (CDH1)-mutated invasive lobular breast cancer shows a high frequency of HER2 (ERBB2) gene mutations. Clin Cancer Res. 2013;19:2668–2676. doi: 10.1158/1078-0432.CCR-13-0295. [DOI] [PubMed] [Google Scholar]
- 14.Wolff AC, et al. Recommendations for human epidermal growth factor receptor 2 testing in breast cancer: American Society of Clinical Oncology/College of American Pathologists clinical practice guideline update. J Clin Oncol. 2013;31:3997–4013. doi: 10.1200/JCO.2013.50.9984. [DOI] [PubMed] [Google Scholar]
- 15.Ma CX, et al. Neratinib efficacy and circulating tumor DNA detection of HER2 mutations in HER2 non-amplified metastatic breast cancer. Clin Cancer Res. 2017 doi: 10.1158/1078-0432.CCR-17-0900. pii: clincanres.0900.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yasuda H, et al. Structural, biochemical, and clinical characterization of epidermal growth factor receptor (EGFR) exon 20 insertion mutations in lung cancer. Sci Transl Med. 2013;5:216ra177. doi: 10.1126/scitranslmed.3007205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Borghaei H, et al. Nivolumab versus docetaxel in advanced nonsquamous non-small-cell lung cancer. N Engl J Med. 2015;373:1627–1639. doi: 10.1056/NEJMoa1507643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kosaka T, et al. Response heterogeneity of EGFR and HER2 exon 20 insertions to covalent EGFR and HER2 inhibitors. Cancer Res. 2017;77:2712–2721. doi: 10.1158/0008-5472.CAN-16-3404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Freedman RA, et al. Translational Breast Cancer Research Consortium (TBCRC) 022: A Phase II trial of neratinib for patients with human epidermal growth factor receptor 2-positive breast cancer and brain metastases. J Clin Oncol. 2016;34:945–952. doi: 10.1200/JCO.2015.63.0343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Shi W, et al. Pathway level alterations rather than mutations in single genes predict response to HER2-targeted therapies in the neo-ALTTO trial. Ann Oncol. 2017;28:128–135. doi: 10.1093/annonc/mdw434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Loibl S, et al. PIK3CA mutations are associated with reduced pathological complete response rates in primary HER2-positive breast cancer: pooled analysis of 967 patients from five prospective trials investigating lapatinib and trastuzumab. Ann Oncol. 2016;27:1519–1525. doi: 10.1093/annonc/mdw197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Baselga J, et al. Biomarker analyses in CLEOPATRA: a phase III, placebo-controlled study of pertuzumab in human epidermal growth factor receptor 2-positive, first-line metastatic breast cancer. J Clin Oncol. 2014;32:3753–3761. doi: 10.1200/JCO.2013.54.5384. [DOI] [PubMed] [Google Scholar]
- 23.Schram AM, Berger MF, Hyman DM. Precision oncology: Charting a path forward to broader deployment of genomic profiling. PLoS Med. 2017;14:e1002242. doi: 10.1371/journal.pmed.1002242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hyman DM, Taylor BS, Baselga J. Implementing genome-driven oncology. Cell. 2017;168:584–599. doi: 10.1016/j.cell.2016.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jordan EJ, et al. Prospective Comprehensive Molecular Characterization of Lung Adenocarcinomas for Efficient Patient Matching to Approved and Emerging Therapies. Cancer Discov. 2017;7:596–609. doi: 10.1158/2159-8290.CD-16-1337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Baselga J, et al. Phase II study of weekly intravenous recombinant humanized anti-p185HER2 monoclonal antibody in patients with HER2/neu-overexpressing metastatic breast cancer. J Clin Oncol. 1996;14:737–744. doi: 10.1200/JCO.1996.14.3.737. [DOI] [PubMed] [Google Scholar]
- 27.Vogel CL, et al. Efficacy and safety of trastuzumab as a single agent in first-line treatment of HER2-overexpressing metastatic breast cancer. J Clin Oncol. 2002;20:719–726. doi: 10.1200/JCO.2002.20.3.719. [DOI] [PubMed] [Google Scholar]
- 28.Slamon DJ, et al. Use of chemotherapy plus a monoclonal antibody against HER2 for metastatic breast cancer that overexpresses HER2. N Engl J Med. 2001;344:783–792. doi: 10.1056/NEJM200103153441101. [DOI] [PubMed] [Google Scholar]
- 29.Bang YJ, et al. Trastuzumab in combination with chemotherapy versus chemotherapy alone for treatment of HER2-positive advanced gastric or gastro-oesophageal junction cancer (ToGA): a phase 3, open-label, randomised controlled trial. Lancet. 2010;376:687–697. doi: 10.1016/S0140-6736(10)61121-X. [DOI] [PubMed] [Google Scholar]
- 30.Swain SM, et al. Pertuzumab, trastuzumab, and docetaxel in HER2-positive metastatic breast cancer. N Engl J Med. 2015;372:724–734. doi: 10.1056/NEJMoa1413513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Blackwell KL, et al. Randomized study of lapatinib alone or in combination with trastuzumab in women with ErbB2-positive, trastuzumab-refractory metastatic breast cancer. J Clin Oncol. 2010;28:1124–1130. doi: 10.1200/JCO.2008.21.4437. [DOI] [PubMed] [Google Scholar]
- 32.Baselga J, et al. Lapatinib with trastuzumab for HER2-positive early breast cancer (NeoALTTO): a randomised, open-label, multicentre, phase 3 trial. Lancet. 2012;379:633–640. doi: 10.1016/S0140-6736(11)61847-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bertotti A, et al. A molecularly annotated platform of patient-derived xenografts (“xenopatients”) identifies HER2 as an effective therapeutic target in cetuximab-resistant colorectal cancer. Cancer Discov. 2011;1:508–523. doi: 10.1158/2159-8290.CD-11-0109. [DOI] [PubMed] [Google Scholar]
- 34.Sartore-Bianchi A, et al. Dual-targeted therapy with trastuzumab and lapatinib in treatment-refractory, KRAS codon 12/13 wild-type, HER2-positive metastatic colorectal cancer (HERACLES): a proof-of-concept, multicentre, open-label, phase 2 trial. Lancet Oncol. 2016;17:738–746. doi: 10.1016/S1470-2045(16)00150-9. [DOI] [PubMed] [Google Scholar]
- 35.Kaufman B, et al. Olaparib monotherapy in patients with advanced cancer and a germline BRCA1/2 mutation. J Clin Oncol. 2015;33:244–250. doi: 10.1200/JCO.2014.56.2728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Le DT, et al. Mismatch-repair deficiency predicts response of solid tumors to PD-1 blockade. Science. 2017 doi: 10.1126/science.aan6733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hyman DM, et al. The efficacy of larotrectinib (LOXO-101), a selective tropomyosin receptor kinase (TRK) inhibitor, in adult and pediatric TRK fusion cancers. J Clin Oncol. 2017;35(suppl) abstr LBA2501. [Google Scholar]
- 38.Wahl RL, Jacene H, Kasamon Y, Lodge MA. From RECIST to PERCIST: Evolving considerations for PET response criteria in solid tumors. J Nucl Med. 2009;50(Suppl. 1):122S–150S. doi: 10.2967/jnumed.108.057307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zehir A, et al. Mutational landscape of metastatic cancer revealed from prospective clinical sequencing of 10,000 patients. Nat Med. 2017;23:703–713. doi: 10.1038/nm.4333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Cheng DT, et al. Memorial Sloan Kettering-Integrated Mutation Profiling of Actionable Cancer Targets (MSK-IMPACT): A Hybridization Capture-Based Next-Generation Sequencing Clinical Assay for Solid Tumor Molecular Oncology. J Mol Diagn. 2015;17:251–64. doi: 10.1016/j.jmoldx.2014.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chang MT, et al. Identifying recurrent mutations in cancer reveals widespread lineage diversity and mutational specificity. Nat Biotechnol. 2016;34:155–63. doi: 10.1038/nbt.3391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Chang MT, et al. Accelerating discovery of functional mutant alleles in cancer. Cancer Discov. doi: 10.1158/2159-8290.CD-17-0321. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kanehisa M, Goto S. KEGG: kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000;28:27–30. doi: 10.1093/nar/28.1.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Chakravarty D, et al. OncoKB: A Precision Oncology Knowledge Base. JCO Precis Oncol. 2017 Jul; doi: 10.1200/PO.17.00011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Shen R, Seshan VE. FACETS: allele-specific copy number and clonal heterogeneity analysis tool for high-throughput DNA sequencing. Nucleic Acids Res. 2016;44:e131. doi: 10.1093/nar/gkw520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Carter SL, et al. Absolute quantification of somatic DNA alterations in human cancer. Nat Biotechnol. 2012;30:413–21. doi: 10.1038/nbt.2203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.McGranahan N, et al. Clonal status of actionable driver events and the timing of mutational processes in cancer evolution. Sci Transl Med. 2015;7:283ra54. doi: 10.1126/scitranslmed.aaa1408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Niu B, et al. MSIsensor: microsatellite instability detection using paired tumor-normal sequence data. Bioinformatics. 2014;30:1015–6. doi: 10.1093/bioinformatics/btt755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Alexandrov LB, et al. Signatures of mutational processes in human cancer. Nature. 2013;500:415–21. doi: 10.1038/nature12477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Middha et al. submitted JCO
- 51.Zhou X, et al. Exploring genomic alteration in pediatric cancer using ProteinPaint. Nat Genetics. 2016;48:4–6. doi: 10.1038/ng.3466. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All datasets generated during and/or analysed during the current study, including patient-level clinical data as well as all sequencing data have been deposited and are publically available in the cBioPortal for Cancer Genomics under the accession code “SUMMIT, Nature, 2018” (http://www.cbioportal.org/study?id=summit_2018). All figure source data are also provided at www.nature.com/nature.