

Abstract
The strains serotyped as M71 from group A Streptococcus are common causes of pharyngeal and skin diseases worldwide. Here we characterize the genome of a unique non-invasive M71 human isolate, NS53. The genome does not contain structural rearrangements or large-scale gene gains/losses, but encodes a full set of non-truncated known virulence factors, thus providing an ideal reference for comparative studies. However, the NS53 genome showed incongruent phenotypic implications from distinct genotypic markers. NS53 is characterized as an emm pattern D and FCT (fibronectin-collagen-T antigen) type-3 strain, typical of skin tropic strains, but is phylogenetically close to emm pattern E strains with preference for both skin and pharyngeal infections. We propose that this incongruence could result from recombination within the emm gene locus, or, alternatively, selection has been against those genetic alterations. Combined with the inability to select for CovS switching, a process is indicated whereby NS53 has been pre-adapted to specific host niches selecting against variations in CovS and many other genes. This may allow the strain to attain successful colonization and long-term survival. A balance between genetic variations and fitness may exist for this bacterium to form a stabilized genome optimized for survival in specific host environments.
Keywords: Streptococcus pyogenes, genetic markers, virulence factors, invasive, phylogenetic structure
The authors discuss the incongruent genotype:phenotype correlation in Streptococcus.
INTRODUCTION
Group A Streptococcus (GAS) causes a wide range of human diseases with diverse colonization sites and clinical manifestations, ranging from relatively mild pharyngitis to life-threatening invasive diseases (Schwartz, Facklam and Breiman 1990). The versatility of this microbe has been associated with its highly plastic genetic repertoire, including diverse virulence factors, extensive recombination mechanisms and finely tuned regulatory systems, which, in combination, facilitate the response and adaptation to challenging host niche environments (Cole et al.2011).
Numerous efforts have been made to correlate genetic markers with their phenotypic traits in order to allow insights into the mechanisms underlying its pathogenesis and adaptation to human host niches. The emm locus encodes the emm gene and other emm-like genes, e.g. fcR, enn and fbaA, responsible for cell surface binding of host proteins, e.g. immunoglobulins and fibronectin. These genes are under the regulation of the multigene activator protein (Mga) (Ribardo and McIver 2006). Based on the chromosomal organization of the emm-like genes in the mga locus and their 3΄ sequence similarities, GAS strains are categorized into one of five different emm patterns (A–E) (Svensson, Sjobring and Bessen 1999). The emm pattern A–C strains have been shown to have a predilection for throat or pharyngeal infections, pattern D strains for skin infections and pattern E strains are generalists, causing both throat and skin infection (Bessen et al.1996).
Pattern D GAS strains also uniquely encode a direct human plasma plasminogen (hPg)-binding M-protein (PAM), and, employing the fibrinolytic system, exhibit enhanced virulence in skin infections (McKay et al.2004). Sequence comparisons and epidemiological surveillance have revealed that the presence of PAM is strongly linked with secretion of the subcluster 2 isoform of streptokinase, SK2b, the GAS secreted-hPg activator that shows optimal activity when hPg is bound to PAM (Kalia and Bessen 2004). Therefore, the co-evolution of PAM and SK2b in GAS genomes strongly suggests a preference for skin infection, along with a high probability of invasiveness of the carrier strain (Zhang et al.2012). Additionally, the fibronectin-collagen-T antigen (FCT) binding locus, responsible for surface binding of GAS to skin and pharyngeal epithelial cells, plays an important role in initial adherence and colonization of GAS to host tissues (Kreikemeyer et al.2011). Based on gene compositions and sequence divergences, the FCT locus is classified into at least nine subtypes, from FCT-1 to FCT-9, and these subtypes exhibit high variability of genes in the FCT locus, likely linked to the specificity in host adherence and adaptation (Falugi et al.2008).
Due to the emergence of whole-genome information of GAS strains, it is possible to perform genetic characterizations at single nucleotide resolutions using genome-wide single nucleotide polymorphisms (SNPs). Our previous study, based on genome-wide SNPs, demonstrated an association between genetic variations and tissue-specific diseases in GAS (Bao et al.2016b).
In the current work, we present a genomic study of a serotype M71 GAS isolate, NS53, isolated from a patient with an uncomplicated skin infection. This strain exhibits a relatively intact genomic backbone without structural rearrangements or truncated virulence genes. Interestingly, this strain exhibits incongruent phenotypic implications from distinct genotypic markers, such as emm patterns, FCT types and genome-wide polymorphisms. The current study of the NS53 expands our knowledge of GAS genetics and provides a relatively intact reference strain for future comparative studies.
MATERIALS AND METHODS
Bacterial strain and growth measurements
GAS strain NS53 was originally isolated from skin of a febrile patient with a non-invasive infection in Australia and was provided by MJ Walker (Queensland, Australia). The genomic DNA was extracted using a Qiagen (Valencia, CA) Mini-DNA kit.
Whole-genome sequencing and assembly
Whole-genome sequencing of NS53 was performed with a combined strategy of the Roche (Branford, CT) 454 GS FLX+ and Illumina (San Diego, CA) Miseq systems. Two genomic libraries containing 10-kb paired ends and 500-bp shotgun inserts were constructed and sequenced by these two systems, respectively. De novo genome assembly was accomplished using Newbler v2.9 (454 Life Sciences) and MIRA3 (Chevreux et al.2004). Gap closure was conducted by PCR and standard Sanger sequencing (Applied Biosystems, Foster City, CA).
Genome annotation
Coding sequence predictions and annotations were performed using RAST (Overbeek et al.2014), Glimmer 3.0 (Delcher et al.1999), and BLAST searches against GenBank, and Clusters of Orthologous Groups (COG) databases (von Mering et al.2003). The circular genome map was constructed with CGView Server (Grant and Stothard 2008).
Pan-genome and core genome analysis
The estimation of pan-genome and core genome was conducted as described by Medini et al. (2005). Homologous clusters used for subsequent analyses were determined by OrthoMCL (Li, Stoeckert and Roos 2003). For pan-genome analyses, starting with a single genome, genomes were added in a randomized order without replacement at each fixed number of genomes. Statistical analyses of the core genome followed a similar procedure. The sizes of pan-genome and core genome were calculated as a function of the number of genomes sequentially included. Power-law regression and exponential curve fit models were used in the pan-genome and core genome analysis, respectively.
Phylogenomic analysis
SNPs detected in the core genome were concatenated for each genome, and the tree was inferred based on the maximum-likelihood method implemented in MEGA (Tamura et al.2013). The genetic distance was estimated as the number of substitutions per site using the maximum composite-likelihood method. The SNPs were detected using the variant ascertainment algorithm (Nusbaum et al.2009).
Quantitative real-time (qRT-PCR) analysis of virulence genes
Bacteria were cultured overnight at 37°C with 5% CO2 in the THY broth. The cells were collected from single colonies grown to logarithm phase (OD600nm = 0.5–0.6). Total RNA was extracted using RNeasy Mini Kit (QIAGEN, Valencia, CA) and were reverse transcribed to cDNA using the iScript cDNA Synthesis Kit (Bio-Rad Laboratories, Inc.). The qPCR reactions were performed with 12.5 μl of 2 × SYBR green PCR master mix (Applied Biosystems). The gene, plr (gapdh), was used as a control. Sequences of primers used are provided in Table S1 (Supporting Information).
Mouse survival assays
C57BL/6 mice and hPg transgenic mice hPgTg were injected subcutaneously with 1.2 × 107 CFU of GAS/mouse. The mice were observed for 10 days for survival status, and the survival differences were plotted by Kaplan-Meier curves and evaluated using the log-rank test. All animal experiments were performed after approval of the University of Notre Dame Institutional Animal Care and Use Committee.
Mouse passage in NS53 and CovS mutant screening
Single colonies of GAS NS53 grown to logarithm phases were used for subcutaneous injection in a cohort of three hPgTg mice with a dose of 1.2 × 107 CFU of GAS/mouse. Mice were sacrificed at 3-day post-infection and the skin wounds, spleens and blood were collected for bacteria recovery. Four rounds of passages were performed. For preliminary screening of CovS mutants, the recovered GAS colonies were plated on THY milk/agar plates and observed for the lytic zone on the plates based on the SpeB-proteolytic phenotype. In order to accurately identify covS mutations, the genotyping of the covS gene in GAS gDNAs was further performed using primers covering the full length of covS (primers are provided in Table S1).
Nucleotide sequence accession number
The complete genome sequence of M71 NS53 has been deposited in Genbank with accession number CP015238.
RESULTS
Overview of the genomic properties of the strain NS53
The genome of GAS strain NS53 is a single circular chromosome of 1 765 123 bp with an average G+C content of 38.4% (Fig. 1). A total of 1723 open reading frames, 44 tRNA genes and 5 rRNA operons were predicted. Based on the 5΄-terminal sequence of the M-protein, NS53 is an M71 serotype. The sequence of NS53 exhibits synteny with other GAS genomes except at the phages, mga locus, FCT locus and short mobile elements (Fig. S1, Supporting Information). No integrative conjugative elements (ICE) or functional phages were identified. Only three short phage remnants (of length 8.6–17.5 kb) were found in the NS53 genome.
Figure 1.

Circular representation of the genome of GAS strain NS53. Data are shown from the outermost to innermost circles. Circle 1 indicates the genome scale in nucleotides. Circles 2 and 3 display the ORFs encoding in the forward and reverse strand, respectively. The colors of the ORFs indicate the COG functional categories. Circles 4 and 5 represent the RNA genes and short mobile elements (IS) in the forward and reverse strand, respectively. Circles 6 and 7 show the GC content and GC skew (G-C)/(G+C), respectively. The red bars outside the circle represent the locations and lengths of phage remnants. The COG categories are as follows: (A) RNA processing and modification; (B) chromatin structure and dynamics; (C) energy production and conversion; (D) cell cycle control and mitosis; (E) amino acid metabolism and transport; (F) nucleotide metabolism and transport; (G) carbohydrate metabolism and transport; (H) coenzyme metabolism; (I) lipid metabolism; (J) translation; (K) transcription; (L) replication and repair; (M) cell wall/membrane/envelop biogenesis; (N) cell motility; (O) post-translational modifications, protein turnover and chaperone; (P) inorganic ion transport and metabolism; (Q) secondary structure; (T) signal transduction; (U) intracellular trafficking, secretion and vesicular transport; (V) defense mechanisms; (W) extracellular structure; (Y) nuclear structure; (Z) cytoskeleton; (R) General function prediction only; (S) function unknown.
Prophage elements and phage-associated virulent genes
Whereas no functional prophages were present in the genome of NS53, three short prophage remnants, designated ΦNS53.1, ΦNS53.2 and ΦNS53.3, with sizes from 8.6to 17.5 kb were identified (Fig. S1). Both ΦNS53.1 and ΦNS53.3 share high similarities with an extensively decayed phage, Φ370.4, from an M1 strain SF370 (with similarities >97%, covering >50% of its length) (Ferretti et al.2001). Phage Φ370.4 is broadly present in GAS genomes and is integrated at the same att site by interrupting the co-regulated mismatch repair genes, mutS and mutL (Scott et al.2008). Only ΦNS53.3 is integrated at the same site as Φ370.4, whereas ΦNS53.1 is inserted between genes encoding a histidine triad family protein and a tyrosyl-tRNA synthetase, without an apparent att site. This implies that ΦNS53.1 and ΦNS53.3 may have arisen from a common ancestor, wherein distinct subsequent evolutionary processes have occurred. Another phage remnant ΦNS53.2 comprises only 8.6 kb, but encodes the virulence gene speA4, a variant of streptococcal pyrogenic exotoxin (speA). Phage ΦNS53.2 is virtually identical to the phage Φ10394.2 in an M6 GAS strain MGAS10394 (Banks et al.2004). The sequences in the upstream and downstream regions of ΦNS53.2 are also highly similar to those of Φ10394.2. The characteristics of ΦNS53.2 suggest that this phage may have been inherited by clonal transmission within specific lineages, or acquired by horizontal transfer from specific isolates. Taken together, NS53 manifests itself as a GAS strain with phage paucity, in contrast with other GAS genomes that contain functional phage elements.
Genetic markers of NS53 for tissue-specific infection
The emm pattern and FCT type have been established as important genetic markers for tissue-specific infections and host cell adhesion (Bessen and Lizano 2010). The gene composition of the mga locus and sequence comparison indicates NS53 to be an emm pattern D strain with the gene arrangement mga-fcR-emm-enn-scpA-fbaA (Fig. 2). Unlike other pattern D strains that produce PAM, NS53 does not encode PAM, but encodes an M-protein lacking the hPg-binding module A repeats (Fig. S2, Supporting Information). Amino-acid sequence alignments of the β-domain of the hPg activator, SK, revealed a SK cluster 1 allele, SK1 (Fig. S2) (Kalia and Bessen 2004). This indicates that NS53 likely does not employ hPg sequestering or activation for its survival, at least directly via binding to M-protein, as happens with pattern D GAS strains. Flow cytometric analysis confirmed the absence of hPg binding of NS53 cells via M-protein. As with other emm pattern D strains, NS53 also possesses the FCT-3 locus, with the gene organization, nra-cpa-fctA-srtB-fctB-msmR-prtF2 (Fig. S3, Supporting Information). Unlike those emm pattern D/FCT-3 strains, which are skin tropic, GAS strains with serotype M71 caused both skin and throat infections (Steer et al.2009). These data indicate that M71 strains have a pattern D-like genotype, but exhibit a pattern E-like phenotype.
Figure 2.

Emm pattern designation of NS53 based on sequence comparisons. (A) Multiple sequence comparison of the genes in the mga locus from distinct GAS strains. The sequence similarities are shown in gradient scale from 66% to 100%. NS53 exhibits the same gene organization in the mga locus as the pattern D strains, except that pam is replaced by emm71 at the corresponding location. The gene fcR in AP53 is a pseudogene truncated by a missense point mutation, indicated with a kink in the middle of the gene. (B) Sequence alignments of the conserved C-terminus of M-protein or PAM. The functional domains in the conserved regions are indicated. The C-terminal sequences of M-protein in NS53 are more homologous to the PAM protein in the pattern D strains (similarity > 99%) than the M-protein in the pattern E strains (similarity ∼ 50%). From this evidence, NS53 is designated as an emm pattern D strain.
Assessment of virulence phenotypes and lack of phenotype switching in NS53
The two-component system, CovRS, regulating the transcription of diverse virulence genes in GAS, is one of the deterministic factors for invasiveness and lethality of GAS due to its role in remodeling the transcriptome of GAS (Graham et al.2002). Inactivated CovS is known to switch the phenotype of GAS isolates from low to high virulence. Sequence comparisons showed that NS53 encodes an intact CovRS system, consistent with a non-invasive phenotype. Furthermore, mouse-passage experiments using mice models showed the lack of CovS switching in the recovered bacteria by examining the speB-suppressed phenotypes and covS genotyping (see Materials and Methods section). We next used qRT-PCR to assess the expression properties of essential virulence genes regulated by CovRS (including mga locus genes, streptokinase ska, capsule synthesis genes hasA and exotoxin B speB) in NS53 and compared them with those in the CovS-inactivated invasive strain, AP53/CovS–, and an isogenic non-invasive mutant complemented with intact CovS (AP53/CovS+). Among the genes we tested, NS53 shows a typical expression pattern for a CovS+ strain, largely comparable with that of the non-invasive AP53/CovS+ (Fig. 3A). The only unusual feature is the very low expression levels of fbaA in NS53, as a characteristic of this strain, which would diminish its adherence to host fibronectin and perhaps contribute to the low virulence of this strain. The genotypes and expression patterns of key virulence genes are also reflected in a mouse survival study, where NS53 shows little-to-no virulence in hPgTg mice (Fig. 3B).
Figure 3.
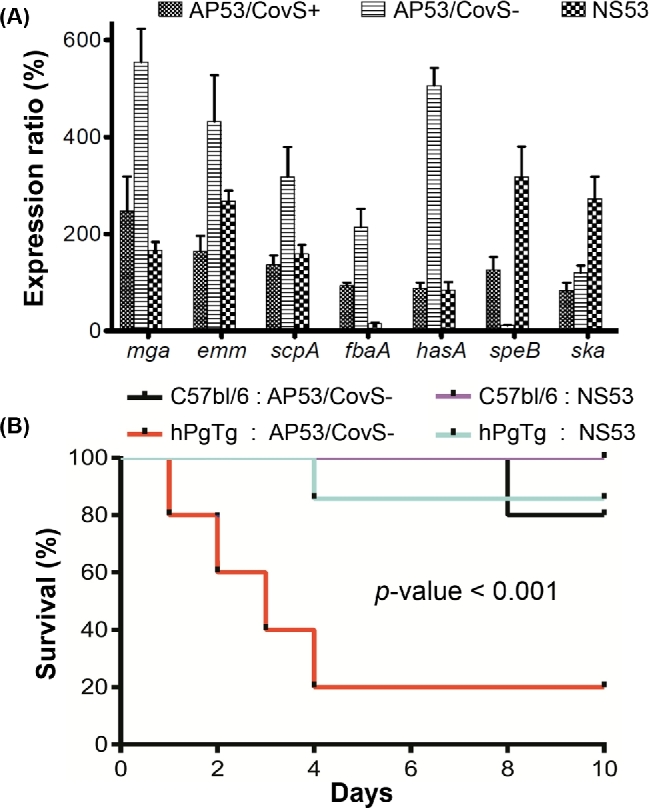
Assessment of the virulence genotype and phenotype of NS53 and in vitro growth in comparison with a serotype M53 strain AP53 capable of selecting for CovS switching (AP53/CovS– and AP53/CovS+). (A) qRT-PCR analysis of transcription levels of key virulence genes, i.e. mga locus genes, hasA, speB and ska. The qRT-PCR assays were performed for logarithm-phase grown cells of NS53, the invasive strain AP53/CovS– and the isogenic non-invasive mutant complemented with the intact covS gene, AP53/CovS+. The expression levels are expressed as ratios relative to that for AP53/CovS+. NS53 shows comparable expression patterns with the non-invasive strain AP53/CovS+. (B) Survival study of GAS in C57BL/6 male mice models and the transgenic mice hPgTg containing human plasminogen. The mice were injected subcutaneously with 1.2 × 107 CFU of GAS cells, and monitored for 10 days. NS53 exhibits significantly lower lethality than the invasive strain AP53/CovS– (P-value < 0.001). The differences between survival curves were evaluated using log-rank test (n = 5–7 mice).
A putative virulence gene encoding a protective antigen was identified in NS53
We cataloged a set of known virulence factors and performed comparisons between NS53 and other GAS genomes (Fig. 4). The virulence genes identified in NS53 are commonly found in GAS genomes, but are not ubiquitous. NS53 is among the three genomes that encode the full set of intact virulence genes that we cataloged (Fig. 4).
Figure 4.

Catalog of a set of known virulence genes among the known GAS genomes. The genes were compared with a non-redundant reference set of virulence genes and the sequence similarities in amino acids are shown in gradient scale. The pseudogenes, which encode premature truncated proteins, were considered to be absent from the relevant strains. One of the GAS strains, M44 STAB901, was removed from the comparison due to its low-sequence quality. Most of the virulence genes show high conservation across distinct genomes, while several ligand-binding-related genes are mosaic including ska, ideS, graB, endoS, sclA and sclB. The strains encoding the full set of intact virulence genes are indicated with black arrows, including M71 NS53, M59 MGAS1882 and M101 NGAS638.
Besides the common virulence genes, a putative virulence gene, spa, encoding an alternative streptococcal protective antigen, was uniquely identified in NS53. Only a distant homolog with amino-acid similarity of 41% was found in a serotype M18 strain, MGAS8232. Additional orthologs (41%–96% similarities) were also identified in several other GAS serotypes without full-genome information (M15, M36, M65, M67, M74 and M78). Like M-like proteins in GAS, Spa is a surface protein containing a signal peptide, a cell-wall-anchoring motif at its C-terminus and α-helical binding domain in the central region (Fig. S4A, Supporting Information) (McLellan et al.2001). However, Spa does not exhibit significant sequence similarities to M-like proteins in GAS, but is phylogenetically related with an M-like protein, Sem, present in Streptococcus equi (S. equi) subsp. equi and Szm in Streptococcus equi subsp. zooepidemicus by sharing C-terminal conserved binding domains (Fig. S4B–D) (Meehan et al.2009).
The phylogenetic tree constructed based on the conserved C-terminal region of Spa shows that two GAS strains, M18 MGAS8232 and M36 ss873, cluster together with the S. equi branch, while other GAS strains, including NS53, cluster in a separate branch (Fig. 5A). We propose that spa from GAS strains in the two branches may have been acquired from different origins with distinct recombination mechanisms. First, multiple sequence comparisons show that the sequences flanking spa in M18 MGAS8232 are more similar to those from the S. equi strains (with similarity ∼95%) than to GAS strains (with <75% similarity over 20% of the length). By identifying the break points of the divergent regions, we found that M18 MGAS8232 probably have acquired spa and its flanking sequences, of length ∼ 5 kb, from the donors of S. equi by replacing a segment of ∼2.9 kb via gene conversion (Fig. 5B). This replacement is further supported by the GC content profile (Fig. S5A, Supporting Information). The M18 strain MGAS8232 exhibits a similar GC content to S. equi strains in spa and its flanking regions, but to other GAS strains in the regions beyond the replaced range (Fig. S5A). Second, spa and its flanking sequences in NS53 are located at a different locus in the genome, and may have been inserted between the genes nusG and nga via a transposon element IS1562 downstream of spa (Fig. 5C). We were unable to identify the possible donors of spa encoded by NS53 based on sequence comparisons or GC contents. The inserted fragment shows sequence divergence and a different GC content profile from that acquired by MGAS8232 (Fig. S5B), indicating its distinct and yet unknown evolutionary origin.
Figure 5.

Phylogenetic location and genetic origin of spa from GAS strains by comparison with other Streptococcus species. (A) The phylogenetic construction shows that the Spa protein in GAS strains is phylogenetically close to the M-proteins Szm from S. equi subsp. zooepidemicus and Sem from S. equi subsp. equi. The phylogenetic topology was constructed based on the C-terminal conserved sequences of Spa using maximum-likelihood method with 1000 bootstraps. The following synonyms for species names are used: Szoo for Streptococcus equi subsp. zooepidemicus, Sequi for Streptococcus equi subsp. equi and Spyo for Streptococcus pyogenes. The synonyms for strain names are also used: M18–8232 for M18 MGAS8232, M71-NS53 for M71 NS53, M53-AP53 for M53 AP53, M6–10394 for M6 MGAS10394, M1-SF370 for M1 SF370, and M12–9429 for M12 MGAS9429. Most GAS strains cluster in one single branch, including NS53. Two GAS strains M18 MGAS8232 and M36 ss873 cluster in a separate branch with S. equi. (B) The gene organization and break-point identification show that spa and its flanking sequences of length ∼ 5 kb from M18 MGAS8232 may have been acquired from donors of S. equi via gene conversion by replacing a fragment of length ∼ 2.9 kb containing the gene encoding the collagen-binding protein SclA. The double arrows indicate the break points, where the replacement may have occurred. (C) The spa and its flanking sequences of length ∼ 5.9 kb in NS53 may have been inserted between the gene nusG and nga probably via a transposon element IS1562 downstream spa.
Phylogenomic analysis of NS53 and other GAS strains based on genome-wide core SNPs
In order to obtain insights of the genome-wide phylogenetic location of NS53 compared with other GAS strains, we identified the core genome and pan-genome by fitting the growth curves of gene numbers by sequential addition of each new genome. The number of genes from core genome and pan-genome were estimated to be 1160 and 3541, respectively (Fig. S6, Supporting Information). We further identified 73 387 SNPs in the core genome, and inferred the core genome phylogeny based on the detected SNPs (Fig. 6A). Interestingly, NS53, characterized as emm pattern D/FCT-3, is not clustered with the isolates with emm pattern D and FCT-3, previously identified to have predisposition for skin infection (i.e. M53, M83, M101 and M80), but is grouped with an emm pattern E strain, M114 NGAS322 and more closely phylogenetically related with several other generalist strains associated with both skin and throat infection (i.e. M44 and M59). The clustering pattern is further revealed by the pairwise genetic distance comparison, where NS53 is closer to the generalist strains (M114, M44 and M59 strains) than to the skin infection-associated strains (M53, M83, M101 NGAS638, M80 ATCC19615, M3 strains and M14 HSC5) or the throat specialists (M18 MGAS8232, M5 Manfredo, M23ND and M6 strains) (Fig. 6B). The deviation of NS53 from skin tropic strains is also reflected by the low proportion of skin infection-specific SNPs present in NS53. Previously, we identified 895 SNPs and 221 non-synonymous SNPs significantly associated with skin infection using a genome-wide association study (Bao et al.2016b). Among them, only 206 (23.0%) and 41 (18.6%) were found in NS53, respectively. However, we also note that NS53 is not significantly as close as M114 NGAS322 to other generalist strains based on genetic distances, indicating its multifaceted genotypes (Fig. 6C).
Figure 6.

The phylogenetic relationship of NS53 with other GAS strains. (A) The phylogenetic tree of all considered GAS genomes based on the core genome SNPs. The tree was inferred using maximum-likelihood method with 1000 bootstraps. The isolates associated with skin and throat infection are clustered in separate branches (highlighted in blue and green, respectively). NS53 is clustered tightly with an emm pattern E strain M114 NGAS322 (indicated in red), and closely related with several other strains associated with both skin and throat infection (highlighted in magenta). (B,C) Genetic distances of NS53 (B) and M114 NGAS322 (C) with the strains associated with both skin and throat infection, strains associated with skin infection and those associated with throat infection. The low genetic distance between NS53 and NGAS322 is indicated with the black arrow in (B). The genetic distances were estimated as number of substitutions per site using the maximum composite-likelihood method. The differences between genetic distances were evaluated using the unpaired t-test.
These analyses raise an important issue regarding to the nature of the evolutionary forces that drive the incongruence between the serotyping classification and the genome-wide phylogeny, as revealed by NS53. We propose two possible explanations. First, strains, such as NS53, may have undergone serotype switching via recombination in the emm gene locus considering the generally high recombination rates that occur at this locus. Alternatively, it is possible that selection mechanisms acting on NS53 have been against the polymorphisms common to other skin tropic isolates. Similar incongruences were also observed for M3 and M14 GAS isolates (Bao et al.2016b). These observations reiterate the multifaceted correlation between genotypes and phenotypes of GAS, and single genetic markers may not be sufficient for precise prediction of the phenotypes of individual strains.
DISCUSSION
In this study, we present a comparative genomic study of non-invasive serotype M71 GAS strain, NS53 and other epidemic or invasive isolates. NS53 exhibits many unique genomic characteristics with seemingly less intact genomic backbones, making it a valuable reference for future comparative studies.
NS53 possesses a relatively small genome compared to other GAS strains, without structural rearrangements, integrated functional phages or ICEs. The absence of functional phages is rare in GAS and is only otherwise found in an M59 strain MGAS15252. Because MGAS15252 is an epidemic strain and caused severe infections in animal models, we are unable to establish a robust correlation between the paucity of phage elements and infection severity. NS53 may have never had the evolutionary pressure to acquire any functional phages, and/or alternatively, it has suffered from extensive phage decay.
The catalog of the virulence genes prompted us to prefer the former possibility. NS53 maintained all the key regulatory genes (i.e. covRS, mga, rgg, rocA, rivR and fasBCA system) (Sarkar and Sumby 2017) and virulence factors without being interrupted by mutations or segmental deletions. NS53 is one of the few strains encoding the full set of intact virulence genes that we identified. Interestingly, we note that some of the key virulence genes, previously considered to be essential for lethality of GAS, are lacking or are truncated in some strains. For example, the capsule biosynthesis operon hasABC is absent in an M4 strain MGAS10750. Previous studies also reported the absence of hasABC in a series of serotype M4 and M22 strains (Flores et al.2012; Turner et al.2015), probably due to the pressure exerted by the catalytic enzyme, hyaluronate lyase (HylA), which is only active in these two types of strains (Henningham et al.2014), and in epidemic M89 strains, proposed to be related with increased adherence and colonization in the hosts (Turner et al.2015; Zhu et al.2015). This implies that some of the virulence genes are inactivated due to selective pressures or conferred advantages in specific bacterial and host settings. NS53 does not seem to acquire those kinds of inactivations in any known virulence genes.
With the lack of functional phages and intact virulence genes, the non-invasive isolate, NS53, appears to have interacted with host niches with selections against extensive gene gain and loss or other alterations in virulence factors. An exception is the acquisition of the putative protective antigen spa with unknown donors. Genetic alterations are frequently observed in GAS genomes and usually confer advantages for enhanced survival or virulence. In this regard, NS53 provides a relatively ‘clean’ genetic background without being significantly influenced by genetic variations. This strain may contribute as an ideal reference for evolutionary and comparative studies in GAS, which are obscured in many cases by the gene gain and loss, structural rearrangements or interrupting mutations.
NS53 is characterized to be an emm pattern D/FCT-3 strain. Previously, we proposed that the emm pattern D strains with skin tropicity have subtype FCT-3 (Bao et al.2016a). Based on this correlation, serotype M71 strains should be probably more disposed to cause skin infection. However, serotype M71 isolates were reported to cause both skin and pharyngeal diseases with equivalent capacity (Steer et al.2009). Similar incongruence between genotypic markers and phenotypic traits is also observed for serotype M59, which was classified as emm pattern D, but demonstrated tissue tropism for both skin and throat (Fittipaldi et al.2012). Both M71 and M59 strains contain an emm pattern E-like gene organization in the mga locus, but encode pattern D-like C-terminal sequences in their M-proteins. Furthermore, the genome-wide phylogeny analysis indicates that strain NS53 is clustered together with an emm pattern E strain M114 NGAS322, and is phylogenetically closely related with several generalist strains causing both skin and throat infection. We propose two possible explanations for the inconsistency. First, this discrepancy could be related to the concept that many surface proteins, including M-proteins, encoded by this organism, may have undergone frequent recombination, causing serotype alterations. Second, the specific host niches of NS53 may have the selections against the variations shared by other pattern D strains, and probably also against high virulence. The unfavorable selections for the variations were also reiterated by our animal passage experiments, where we were unable to obtain the NS53 mutants with CovS switching. Combined with the observations that inactivations were not identified in the known virulence genes, the data point towards a scenario, wherein this bacterium has been pre-adapted to the specific host niches selecting against variations in covS and many other virulence genes. This may in turn allow NS53 to achieve successful colonization and survival. A balance between genetic variations and fitness may exist for the bacteria to form a stabilized genome in a specific host environment.
Meanwhile, it should also be noted that the final outcome of the balance between virulence and fitness may have also been influenced by many other factors, such as the changing environments from natural hosts to growth media or growth media to animal models, which could compromise the evolution of virulence (Mikonranta et al.2015).
Supplementary Material
Supplementary data are available at FEMSPD online.
SUPPLEMENTARY DATA
Supplementary data are available at FEMSPD online.
FUNDING
This work is supported by NIH grant HL013423.
Conflict of interest. None declared.
REFERENCE
- Banks DJ, Porcella SF, Barbian KD et al. . Progress toward characterization of the group A Streptococcus metagenome: complete genome sequence of a macrolide-resistant serotype M6 strain. J Infect Dis 2004;190:727–38. [DOI] [PubMed] [Google Scholar]
- Bao Y-J, Liang Z, Mayfield JA et al. . Genomic characterization of a pattern D Streptococcus pyogenes emm53 isolate reveals a genetic rationale for invasive skin tropicity. J Bacteriol 2016a;198:1712–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bao Y-J, Shapiro BJ, Lee SW et al. . Phenotypic differentiation of Streptococcus pyogenes populations is induced by recombination-driven gene-specific sweeps. Sci Rep 2016b;6:36644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bessen DE, Lizano S. Tissue tropisms in group A streptococcal infections. Future Microbiol 2010;5:623–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bessen DE, Sotir CM, Readdy TL et al. . Genetic correlates of throat and skin isolates of group A Streptococci. J Infect Dis 1996;173:896–900. [DOI] [PubMed] [Google Scholar]
- Chevreux B, Pfisterer T, Drescher B et al. . Using the miraEST assembler for reliable and automated mRNA transcript assembly and SNP detection in sequenced ESTs. Genome Res 2004;14:1147–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cole JN, Barnett TC, Nizet V et al. . Molecular insight into invasive group A streptococcal disease. Nat Rev Microbiol 2011;9:724–36. [DOI] [PubMed] [Google Scholar]
- Delcher AL, Harmon D, Kasif S et al. . Improved microbial gene identification with GLIMMER. Nucleic Acids Res 1999;27:4636–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falugi F, Zingaretti C, Pinto V et al. . Sequence variation in Group A Streptococcus pili and association of pilus backbone types with Lancefield T serotypes. J Infect Dis 2008;198:1834–41. [DOI] [PubMed] [Google Scholar]
- Ferretti JJ, McShan WM, Ajdic D et al. . Complete genome sequence of an M1 strain of Streptococcus pyogenes. P Natl Acad Sci USA 2001;98:4658–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fittipaldi N, Beres SB, Olsen RJ et al. . Full-genome dissection of an epidemic of severe invasive disease caused by a hypervirulent, recently emerged clone of Group A Streptococcus. Am J Pathol 2012;180:1522–34. [DOI] [PubMed] [Google Scholar]
- Flores AR, Jewell BE, Fittipaldi N et al. . Human disease isolates of serotype M4 and M22 Group A Streptococcus lack genes required for hyaluronic acid capsule biosynthesis. mBio 2012;3:e00413–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graham MR, Smoot LM, Migliaccio CAL et al. . Virulence control in Group A Streptococcus by a two-component gene regulatory system: global expression profiling and in vivo infection modeling. P Natl Acad Sci USA 2002;99:13855–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grant JR, Stothard P. The CGView Server: a comparative genomics tool for circular genomes. Nucleic Acids Res 2008;36:W181–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henningham A, Yamaguchi M, Aziz RK et al. . Mutual exclusivity of hyaluronan and hyaluronidase in invasive group A Streptococcus. J Biol Chem 2014;289:32303–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalia A, Bessen DE. Natural selection and evolution of streptococcal virulence genes involved in tissue-specific adaptations. J Bacteriol 2004;186:110–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kreikemeyer B, Gámez G, Margarit I et al. . Genomic organization, structure, regulation and pathogenic role of pilus constituents in major pathogenic Streptococci and Enterococci. Int J Med Microbiol 2011;301:240–51. [DOI] [PubMed] [Google Scholar]
- Li L, Stoeckert CJ, Roos DS. OrthoMCL: identification of ortholog groups for eukaryotic genomes. Genome Res 2003;13:2178–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKay FC, McArthur JD, Sanderson-Smith ML et al. . Plasminogen binding by Group A streptococcal isolates from a region of hyperendemicity for streptococcal skin infection and a high incidence of invasive infection. Infect Immun 2004;72:364–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLellan DGJ, Chiang EY, Courtney HS et al. . Spa contributes to the virulence of type 18 Group A Streptococci. Infect Immun 2001;69:2943–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medini D, Donati C, Tettelin H et al. . The microbial pan-genome. Curr Opin Genet Dev 2005;15:589–94. [DOI] [PubMed] [Google Scholar]
- Meehan M, Lewis MJ, Byrne C et al. . Localization of the equine IgG-binding domain in the fibrinogen-binding protein (FgBP) of Streptococcus equi subsp. equi. Microbiology 2009;155:2583–92. [DOI] [PubMed] [Google Scholar]
- Mikonranta L, Mappes J, Laakso J et al. . Within-host evolution decreases virulence in an opportunistic bacterial pathogen. BMC Evol Biol 2015;15:165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nusbaum C, Ohsumi TK, Gomez J et al. . Sensitive, specific polymorphism discovery in bacteria using massively parallel sequencing. Nat Methods 2009;6:67–, 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Overbeek R, Olson R, Pusch GD et al. . The SEED and the Rapid Annotation of microbial genomes using Subsystems Technology (RAST). Nucleic Acids Res 2014;42:D206–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ribardo DA, McIver KS. Defining the Mga regulon: comparative transcriptome analysis reveals both direct and indirect regulation by Mga in the group A streptococcus. Mol Microbiol 2006;62:491–508. [DOI] [PubMed] [Google Scholar]
- Sarkar P, Sumby P. Regulatory gene mutation: a driving force behind group A Streptococcus strain- and serotype-specific variation. Mol Microbiol 2017;103:576–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartz B, Facklam RR, Breiman RF. Changing epidemiology of group A streptococcal infection in the USA. Lancet 1990;336:1167–71. [DOI] [PubMed] [Google Scholar]
- Scott J, Thompson-Mayberry P, Lahmamsi S et al. . Phage-associated mutator phenotype in group A streptococcus. J Bacteriol 2008;190:6290–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steer AC, Law I, Matatolu L et al. . Global emm type distribution of group A streptococci: systematic review and implications for vaccine development. Lancet Infect Dis 2009;9:611–6. [DOI] [PubMed] [Google Scholar]
- Svensson MD, Sjobring U, Bessen DE. Selective distribution of a high-affinity plasminogen-binding site among group A Streptococci associated with impetigo. Infect Immun 1999;67:3915–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamura K, Stecher G, Peterson D et al. . MEGA6: Molecular Evolutionary Genetics Analysis version 6.0. Mol Biol Evol 2013;30:2725–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turner CE, Abbott J, Lamagni T et al. . Emergence of a new highly successful acapsular Group A Streptococcus clade of genotype emm89 in the United Kingdom. mBio 2015;6:e00622–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Mering C, Huynen M, Jaeggi D et al. . STRING: a database of predicted functional associations between proteins. Nucleic Acids Res 2003;31:258–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Liang Z, Hsueh HT et al. . Characterization of streptokinases from Group A Streptococci reveals a strong functional relationship that supports the coinheritance of plasminogen-binding M protein and cluster 2b streptokinase. J Biol Chem 2012;287:42093–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu L, Olsen RJ, Nasser W et al. . Trading capsule for increased cytotoxin production: contribution to virulence of a newly emerged clade of emm89 Streptococcus pyogenes. mBio 2015;6:e01378–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary data are available at FEMSPD online.