

Abstract
Copper is required for the activity of cytochrome c oxidase (COX), the terminal electron-accepting complex of the mitochondrial respiratory chain. The likely source of copper used for COX biogenesis is a labile pool found in the mitochondrial matrix. In mammals, the proteins that transport copper across the inner mitochondrial membrane remain unknown. We previously reported that the mitochondrial carrier family protein Pic2 in budding yeast is a copper importer. The closest Pic2 ortholog in mammalian cells is the mitochondrial phosphate carrier SLC25A3. Here, to investigate whether SLC25A3 also transports copper, we manipulated its expression in several murine and human cell lines. SLC25A3 knockdown or deletion consistently resulted in an isolated COX deficiency in these cells, and copper addition to the culture medium suppressed these biochemical defects. Consistent with a conserved role for SLC25A3 in copper transport, its heterologous expression in yeast complemented copper-specific defects observed upon deletion of PIC2. Additionally, assays in Lactococcus lactis and in reconstituted liposomes directly demonstrated that SLC25A3 functions as a copper transporter. Taken together, these data indicate that SLC25A3 can transport copper both in vitro and in vivo.
Keywords: copper, cytochrome c oxidase (complex IV), mitochondria, copper transport, membrane transport
Introduction
Mitochondrial dysfunction contributes to the pathogenesis of heart failure, neurodegenerative disorders, myopathies, and diabetes (1). Mitochondria are dynamic, double membrane-bound organelles with a semi-permeable outer membrane that allows exchange of metabolites between the cytosol and the intermembrane space (IMS).5 In contrast, the inner membrane (IM) that separates the IMS and the matrix is tightly sealed. Thus, numerous transporters are required to provide the matrix with a diverse range of substrates that are necessary to support metabolism, the biogenesis of iron-sulfur clusters, and the assembly of the electron transport chain (ETC) required for oxidative phosphorylation (2).
Cytochrome c oxidase (COX) is the terminal electron-accepting complex of the ETC. Mammalian COX contains 14 major subunits, two of which bind three redox centers required for electron transfer (3). The catalytic core consists of the mitochondrially-encoded subunits COX1, COX2, and COX3. COX2 binds the binuclear CuA site required for accepting electrons from cytochrome c. These electrons are then transferred to the cofactors of COX1, first to heme a and then to the heme a3–CuB site where oxygen is bound. COX biogenesis requires >25 accessory proteins known as COX assembly factors (1). The overwhelming majority of ETC defects that underlie mitochondrial dysfunction and human disease is caused by pathogenic mutations in accessory factors (1). At least nine of these factors facilitate the insertion of the copper cofactors that are essential for the catalytic competence of the COX holoenzyme (4). In yeast, it has been demonstrated that the copper used for COX assembly comes from the matrix, and this matrix copper pool is conserved in mammals (5, 6). However, how copper traverses the tightly sealed IM for its storage in the matrix and how copper is mobilized from the matrix to the IMS where it is then used by these COX assembly factors remain major unanswered mechanistic questions.
Biogenesis of the COX complex occurs in a modular pathway with different assembly proteins exchanging into relatively transient complexes to promote the incorporation of structural subunits or cofactor insertion. Because the copper and heme cofactors in COX1 are largely buried within the core structure of the enzyme, their early insertion is a necessity. COX1 translation and insertion into the IM are mediated by a suite of specific translational activators and chaperones that form a complex known as MITRAC (7). Once COX1 is inserted into the IM, SURF1 adds the heme cofactor (8), and COX11 facilitates the formation of the CuB site (9, 10). The copper-binding cysteinyl sulfurs of COX11 are reduced via an interaction with COX19 (11), and the copper required for CuB site maturation is then donated to COX11 by COX17 (9). Like COX1, specific factors promote the translation and membrane insertion of COX2. After membrane insertion, COX2 is bound by a complex composed of COX20, SCO1, SCO2, and COA6, which facilitates the maturation of its binuclear CuA site (12–14). CuA site formation requires that copper is transferred from COX17 to SCO1, which subsequently catalyzes its insertion into COX2 (9). SCO2 appears to be essential for this latter copper transfer step because it acts as an oxidoreductase on the cysteinyl thiolates of COX2 (15, 16).
Copper recruitment to the mitochondrion is independent of the presence and activity of COX (17). In yeast, biochemical depletion of the matrix copper pool results in a COX defect, suggesting that this pool is redistributed to the IMS for maturation of the holoenzyme (5). The mitochondrial carrier family (MCF) protein Pic2 acts as an importer of copper into the matrix (18). The human genome encodes for more than 50 MCF proteins that are involved in translocation of substrates across the IM, including various TCA intermediates and nucleoside di- and triphosphates for energy metabolism (19). MCF proteins have a basic structure consisting of three repeats of ∼100 amino acids that contain two transmembrane helices connected by a loop with a short α-helix. The transmembrane helices contain a conserved PX(D/E)XX(R/K) motif that is a signature of all MCF proteins (20) and allows for the formation of salt bridges that are thought to determine the requirement for counter substrates, co-substrates, and the directionality of transport (20). In general, MCF function has been conserved between yeast and mammals. The closest homolog of Pic2 in mammals is SLC25A3 (49% identity and 65% similarity with Pic2). SLC25A3 is an established phosphate carrier in mammals, but in yeast Mir1 (41% identity and 61% similarity to SLC25A3; 40% identity and 59% similarity to Pic2) instead of Pic2 is the major phosphate transporter (21, 22).
Mammals have two isoforms of SLC25A3 as a result of alternative splicing of exon 3. The two isoforms differ by 13 amino acids between residues 54 and 80. Each isoform is expressed in a cell–type-specific manner, with heart and skeletal muscle expressing SLC25A3-A and all other tissues expressing SLC25A3-B (23). SLC25A3-B has an ∼3-fold higher rate of phosphate transport than SLC25A3-A in liposomes after purification from Escherichia coli (23). Patients with mutations in the third exon that are specific to the a-isoform of SLC25A3 present with multisystem disorders characterized by muscle hypotonia, lactic acidosis, and hypertrophic cardiomyopathy (24, 25). In this report, we use cell culture systems and in vitro transport studies to demonstrate a role for SLC25A3 in mitochondrial copper transport and to establish that this transport function directly impinges upon copper delivery to COX during holoenzyme assembly.
Results
SLC25A3 depletion in human and mouse cells causes a COX deficiency that is suppressed by copper supplementation
To investigate the role of SLC25A3 in mitochondrial copper distribution, we focused on the copper requirement for COX assembly in multiple mammalian cell types. We observed a COX deficiency in HEK293 cells in which siRNA knockdown reduced SLC25A3 abundance to ∼25% that in cells treated with a scrambled siRNA control (Fig. 1, A and B). Although this was the only cell type in which we were reliably able to detect endogenous SLC25A3 levels with commercially available antibodies, application of various SLC25A3 siRNA oligonucleotides consistently produced a COX deficiency in multiple cell types. Knockdown of SLC25A3 in wildtype human fibroblasts resulted in a COX deficiency that was rescued by supplementing the culture medium with copper bound to an ionophore (CuATSM) that freely crosses biological membranes but was not rescued by supplementation with phosphate (Fig. 1C). Addition of supplemental copper increased whole-cell copper levels ∼6-fold, whereas supplementation with phosphate increased the cellular phosphorus content by ∼50-fold as assessed by inductively coupled plasma-optical emission spectroscopy (ICP-OES) (data not shown). Because HEK293 cells and human fibroblasts express the SLC25A3-B isoform, we performed the same siRNA knockdown experiment in the murine cardiac muscle cell line HL-1, which predominantly expresses the SLC25A3-A isoform, and once again we observed a COX deficiency (21 ± 3% of control COX, n = 3). Finally, we used the C2C12 skeletal muscle cell line and found that treatment with an Slc25a3-A-specific siRNA also resulted in a significant COX deficiency in myoblasts, which express both the A- and B-isoforms (26) (37 ± 3% of control COX, n = 3), and in differentiated myotubes which exclusively express the A-isoform (26) (43 ± 4% of control COX, n = 3). Taken together, these data suggest that SLC25A3-A and SLC25A3-B are required for COX assembly in human and murine cells.
Figure 1.
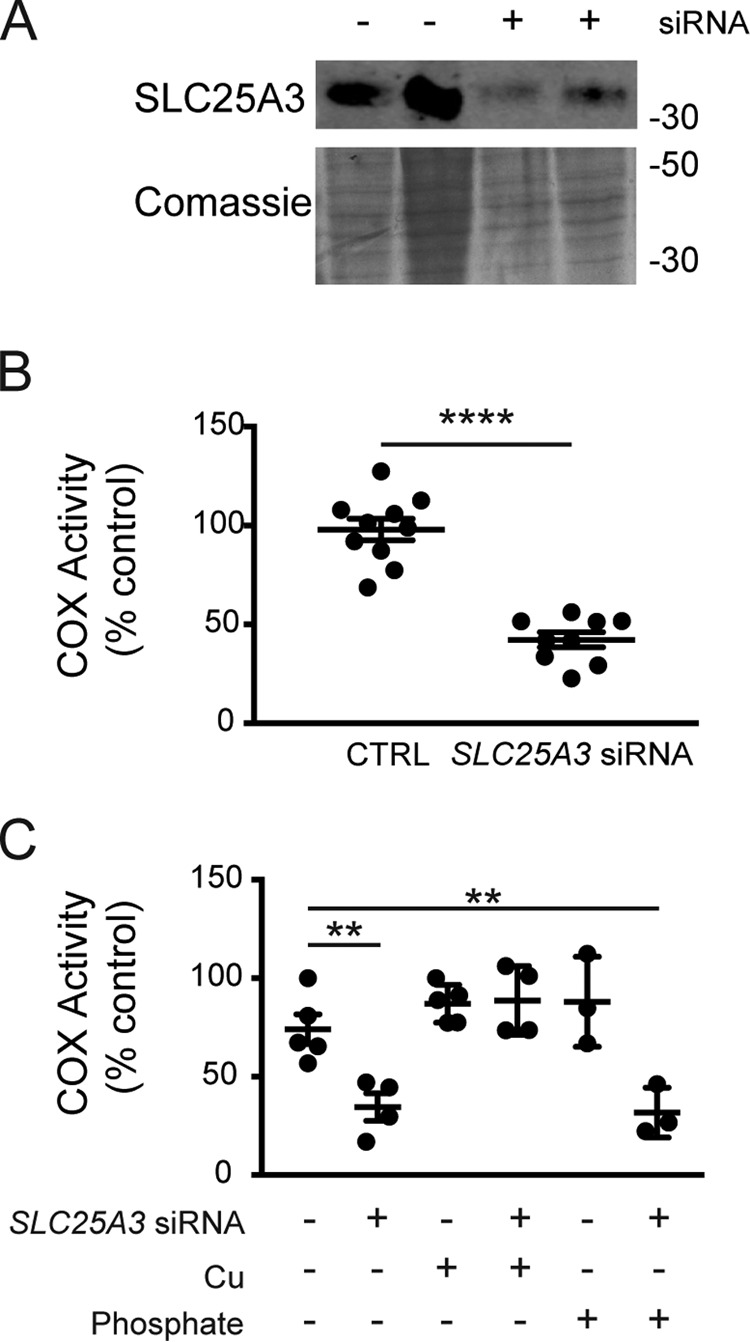
A, immunoblot analysis of SLC25A3 in HEK293 cells treated with negative control siRNA (−) or SLC25A3 siRNA (+). B, cytochrome c oxidase (COX) activity in HEK293 cells treated with negative control siRNA (CTRL) or SLC25A3 siRNA (n = 10, ****, p < 0.0001 based on Student's t test). C, COX activity in control human fibroblasts treated with SLC25A3 siRNA (+) (n = 4) or negative control siRNA (−) (n = 5) grown in basal media or media supplemented with either 50 μm CuATSM (n = 4–5) or 50 mm phosphate (n = 3). **, p < 0.01 based on Student's t test. All data are expressed as a percentage of activity in negative control cells grown in basal medium.
Deletion of Slc25a3 in mouse embryonic fibroblasts results in COX and copper homeostasis defects
To confirm that the loss of SLC25A3 affects mitochondrial copper handling, we immortalized primary mouse embryonic fibroblasts (MEFs) in which the first and second exons of Slc25a3 are floxed (27), then isolated, and amplified mock-treated (Slc25a3FLOX) and Cre-treated (Slc25a3−/−) clonal lines. Immunoblot analysis showed decreased levels of COX subunits 1 and 4 and reduced COX activity in the Slc25a3−/− relative to the isogenic Slc25a3FLOX MEFs (Fig. 2A). The COX deficiency was rescued by retroviral expression of a Slc25a3-B cDNA (Fig. 2B) or by supplementing the media with copper (Fig. 2B).
Figure 2.

A, immunoblot analysis of COX subunit abundance in immortalized Slc25a3 MEFs treated with buffer alone (flox) or Cre recombinase (KO). GAPDH served as an internal loading control. flox-1, flox-2, KO-1, and KO-2 denote independent clonal isolates whose genotype was confirmed by PCR analysis (data not shown). B, COX activity in Slc25a3−/− MEFs alone (n = 8) or transduced with an Slc25a3-B cDNA (Slc25a3-B) (n = 6) cultured in standard medium (NA) or Slc25a3−/− MEFs in standard media supplemented with 50 μm CuATSM (n = 4). ****, p < 0.0001 based on Student's t test. All data are expressed as a percentage of matched Slc25a3FLOX MEFs. C, immunoblot analysis of copper homeostasis and control proteins in Slc25a3FLOX (flox-1) and Slc25a3−/− (KO-1) cells alone (NA) or transduced with retrovirus containing Slc25a3-B cDNA. D, total SOD activity in Slc25a3FLOX (flox) and Slc25a3−/− (KO) MEFs (n = 6, ****, p < 0.0001 based on Student's t test). Cyanide-resistant activity was also measured and was not significantly different in flox versus KO MEFs (data not shown).
Previous studies of pathogenic mutations in SCO1 and SCO2 revealed that perturbing the function of either mitochondrial protein can also affect the stability and localization of other proteins crucial to copper homeostasis and, by extension, total cellular copper levels (28). We therefore assessed the steady-state levels of copper homeostasis proteins in this mutant background. Slc25a3−/− cells had reduced levels of the cytosolic copper enzyme SOD1 and its chaperone CCS, which were restored by retroviral expression of a Slc25a3-B cDNA (Fig. 2C). Consistent with the decreased levels of CCS and SOD1, SOD activity was reduced by ∼60% in these cells (Fig. 2D). The observed decrease in activity was not due to loss of SOD2 as cyanide-resistant SOD activity (∼15% of total attributed to SOD2, data not shown) was equal in both Slc25a3 backgrounds. In contrast to SOD1 and CCS, we did not detect a change in the abundance of the copper exporter ATP7A, the high affinity copper importer CTR1, or the COX assembly protein SCO1 (Fig. 3A). However, measurement of the total metal content of Slc25a3−/− MEFs cells showed a statistically significant decrease in total copper (−26% of control, p = 0.005, n = 3) and calcium (−37% of control, p = 0.01, n = 3) (Fig. 3B). These data indicate that, in MEFs, the loss of SLC25A3 affects cellular copper homeostasis.
Figure 3.
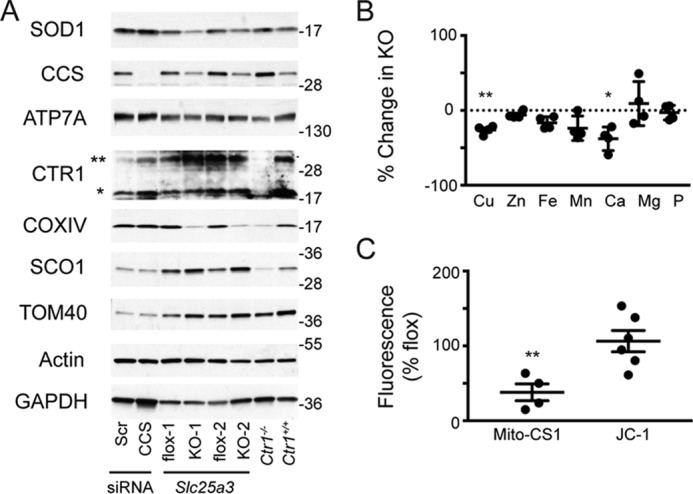
A, immunoblot analysis of copper-related proteins and controls in Slc25a3FLOX (flox-1 and flox-2) and Slc25a3−/− (KO-1 and KO-2) MEFs. Floxed MEFs treated with scrambled (Scr) or CCS siRNA as well as Ctr1+/+ and Ctr1−/− MEFs were used as controls and to validate antibody specificity. ** on the immunoblot indicates the mature, glycosylated CTR1, and * is the cleaved form of the protein. B, ICP-OES analysis of metal content of Slc25a3−/− MEFs expressed as percentage of Slc25a3FLOX MEFs. Changes in both copper (n = 4, **, p < 0.001 based on Student's t test) and calcium (n = 4, *, p < 0.05 based on Student's t test) were statistically significant. All other elements are n = 4, p > 0.05 based on Student's t test. C, whole-cell fluorescence of Slc25a3−/− MEFs (KO) as a percentage of Slc25a3FLOX loaded with the copper-dependent dye Mito-CS1 (n = 4, **, p < 0.01 based on Student's t test) or the mitochondrial membrane potential dependent dye JC-1 (n = 6, p > 0.05 based on Student's t test) expressed as a percentage of matched Slc25a3FLOX MEFs.
To directly demonstrate that loss of SLC25A3 affects mitochondrial copper levels, we quantified the exchangeable copper in the matrix with mitochondrially targeted copper sensor 1 (Mito-CS1) (29), and we found that matrix copper was reduced by 65% in Slc25a3−/− MEFs (Fig. 3C). Consistent with previous investigations (27), mitochondrial membrane potential as assessed by JC-1 staining was unchanged in the Slc25a3−/− MEFs (Fig. 3C). The reduced signal of Mito-CS1 was not a function of reduced organelle content as TOM40 levels (Fig. 2C, 3A) and the total MitoTracker Green signal (data not shown) were equivalent in Slc25a3−/− and Slc25a3FLOX MEFs. Taken together, these data support a role for SLC25A3 in COX assembly and the maintenance of the mitochondrial copper pool.
Heterologous expression of SLC25A3 in yeast and Lactococcus lactis facilitates copper transport
The MCF protein Pic2 is a mitochondrial copper transporter in budding yeast (18). To test for the copper transport function of SLC25A3 in yeast, we expressed the human SLC25A3-A cDNA under the control of the constitutive ADH1 promoter in a pic2Δ background. SLC25A3-A restored mitochondrial copper uptake in isolated mitochondria (Fig. 4A) and rescued the COX defect in these cells under copper limitation (Fig. 4B). These data suggest that SLC25A3 can function as a copper transporter, particularly because pic2Δ yeast do not exhibit any phosphate defects and increased phosphate levels do not suppress the copper defects in this mutant (18).
Figure 4.
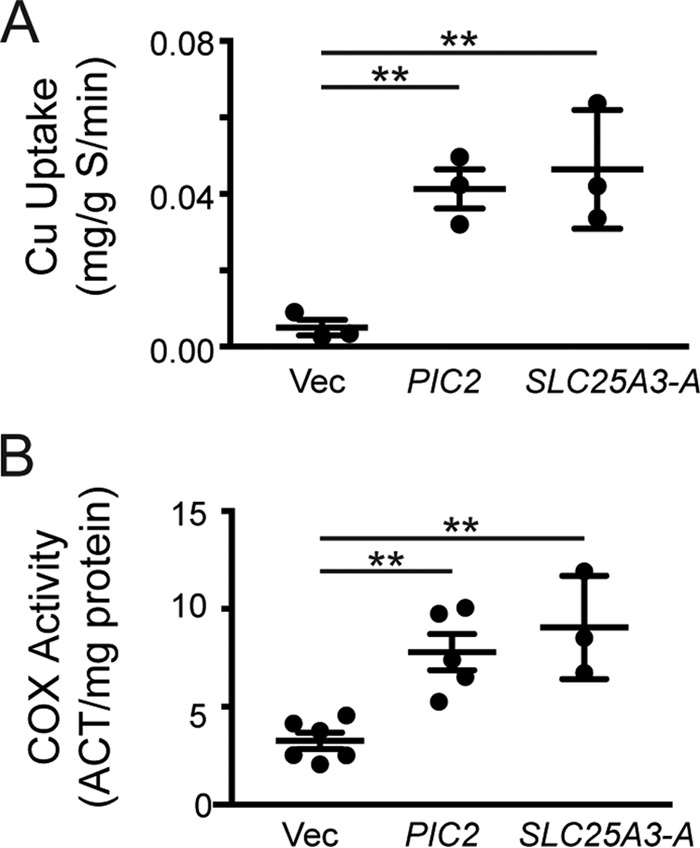
A, ICP-OES analysis of copper uptake into mitochondria isolated from pic2Δ yeast expressing empty vector (Vec) (n = 3) or PIC2 (n = 3, **, p < 0. 01 based on Student's t test) or human SLC25A3-A (n = 3, **, p < 0.01 based on Student's t test). B, COX activity in mitochondria isolated from pic2Δ yeast expressing empty vector (Vec) (n = 6) or PIC2 (n = 5, **, p < 0.01 based on Student's t test) or human SLC25A3-A (n = 3, **, p < 0.01 based on Student's t test).
MCF proteins are known to insert into the cytoplasmic membrane of L. lactis (30). Expression of human SLC25A3-A in L. lactis resulted in the time-dependent uptake of copper, as assessed by ICP-OES (Fig. 5, A and B). As an additional assessment of copper transport, we used silver in place of copper as silver is more toxic to L. lactis and is widely used to demonstrate copper transport. In this assay, expression of a functional copper transporter increases the silver sensitivity of L. lactis (18, 31). Silver was more toxic to L. lactis expressing human SLC25A3-A when compared with L. lactis harboring an empty expression vector, as indicated by a lack of growth at 24 h in silver-containing medium (Fig. 5C). To confirm that SLC25A3-A expressed in this system could also function as a phosphate transporter, we used arsenate as a toxic mimetic of phosphate. Cells expressing SLC25A3-A were unable to grow in increasing arsenate when compared with cells expressing empty vector (Fig. 5D). Finally, we tested the growth of L. lactis expressing SLC25A3-A, SLC25A3-B, or the empty vector in the concentration of silver or arsenate that inhibited growth by at least 50% in SLC25A3-A (Fig. 5, C and D) and observed a significant growth defect for both isoforms of the transporter (Fig. 5, E and F).
Figure 5.
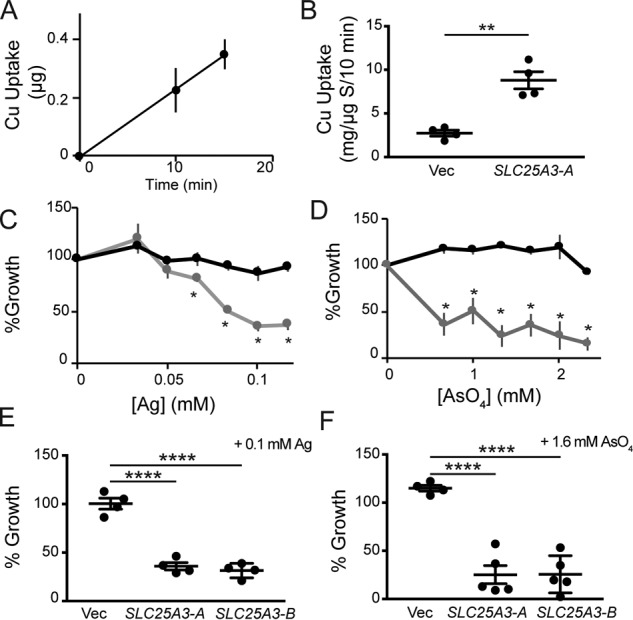
A, copper uptake over time in L. lactis expressing human SLC25A3-A monitored by ICP-OES. Uptake or association with L. lactis expressing an empty vector is subtracted at each time point. B, initial rate of copper uptake by L. lactis harboring an empty vector (Vec) (n = 4) or vector containing the human SLC25A3-A cDNA (n = 4, **, p < 0.01 based on Student's t test). C, growth of L. lactis expressing human SLC25A3-A (gray line, gray circles) or empty vector (black line, black circle) in media with increasing silver concentrations. Each data point represents the average of four independent cultures (n = 4, *, p < 0.05 based on Student's t test relative to empty vector at each silver concentration). D, growth of L. lactis expressing human SLC25A3-A (gray line, gray circles) or empty vector (black line, black circle) in media with increasing arsenate concentrations. Each data point represents the average of four independent cultures (n = 4, *, p < 0.05 based on Student's t test relative to empty vector at each arsenate concentration). E, growth of cells expressing empty vector (Vec) (n = 4) or vector containing the cDNA from human SLC25A3-A (n = 4, ****, p < 0.0001 based on Student's t test) or human SLC25A3-B (n = 4, ****, p < 0.0001 based on Student's t test) in the presence of 100 μm silver. F, L. lactis growth represented as in E in the presence of 1.6 mm arsenate (n = 4–5, ****, p < 0.0001 based on Student's t test).
SLC25A3 purified from E. coli transports copper when reconstituted into liposomes
To directly test copper transport by human SLC25A3-A and SLC25A3-B, we expressed each cDNA with a His6 tag in E. coli, purified the protein from inclusion bodies, and reconstituted it into liposomes, a method that has been used extensively to assess MCF substrate transport and specificity (23, 32–36). Reconstituted SLC25A3-A (Fig. 6A) and SLC25A3-B (Fig. 6B) both transport copper into liposomes without internal counter substrates, suggesting they act as unidirectional copper transporters. The kinetic parameters for both reconstituted proteins suggest an apparent transport affinity of 15 μm and a specific activity of 25 mmol of copper/min/g under our standard conditions in the absence of internal substrates (Fig. 6, A and B). SLC25A3 proteins did not appear to transport other metals that were tested (calcium, zinc, magnesium, and iron), and none of these metals affected copper transport when added externally at a 10-fold molar excess (Fig. 6C). However, the initial rate of copper transport was inhibited by the addition of a 10-fold excess of arsenate to the uptake assay in SLC25A3-A (Fig. 6D) and SLC25A3-B (Fig. 6E). To test whether SLC25A3 can interact with the fluorescent anionic copper complex found in the mitochondrial matrix, we used fluorescence anisotropy. We observed a stabilization of the rotational time of the ligand with increasing concentrations of human SLC25A3-A that is suggestive of an interaction (Fig. 6F). Taken together, these data show that SLC25A3 is capable of transporting copper in the absence of additional accessory factors.
Figure 6.

Copper uptake into proteoliposomes reconstituted with purified human SLC25A3-A (K½, 15 ± 3 μm; Vmax, 25 ± 0.8 mmol/g/min) and control vesicles (without reconstituted protein) (A) or human SLC25A3-B (K½, 11 ± 2 μm; Vmax, 30 ± 1.1 mmol/g/min) (n = 6, average with standard deviation as error bars and apparent kinetics taken from curve fitting) (B) is shown. C, initial rate of uptake of copper, zinc, calcium, magnesium, and iron expressed as a percentage of rate with copper alone (open bars) by SLC25A3-A. Initial rate of uptake of copper was in the presence of 10-fold excess of zinc, calcium, magnesium, and iron expressed as a percentage of rate of copper alone (black bars). The rate of copper uptake into proteoliposomes containing human SLC25A3-A (D) or human SLC25A3-B (E) in the presence of 10-fold excess of arsenate (n = 4–5, ***, p < 0.001 based on Student's t test) is shown. F, fluorescence anisotropy of the mitochondrial anionic copper complex isolated from yeast and recombinant human SLC25A3-A in liposomes. Equal amounts of control liposome without protein were assayed and subtracted from each point.
Discussion
Copper delivery and insertion into COX during its assembly occurs in the IMS, and it requires at least nine copper-binding assembly factors (1). However, we have a limited understanding of how copper is recruited and delivered to these ancillary proteins. In this report, we demonstrate an unexpected role for SLC25A3 in mitochondrial copper transport. SLC25A3 is a phosphate transporter, and our findings raise the possibility that phosphate levels could control copper availability or transport. However, three independent observations support a direct role for SLC25A3 as a mitochondrial copper importer. First, purified and reconstituted SLC25A3 is able to transport copper into proteoliposomes, and its expression in a heterologous system facilitates copper uptake. Second, copper supplementation suppresses the COX deficiency phenotype in Slc25a3−/− cells. Third, depletion or deletion of Slc25a3 decreases total mitochondrial copper levels. Therefore, we suggest that although secondary effects of phosphate on copper homeostasis may contribute to the COX defect, this phenotype is primarily due to loss of the copper transport function of SLC25A3. How this transporter can mediate the transport of both phosphate and copper ions (or the chelated form of copper that is found in the matrix of yeast and mammalian cells (5, 17)) remains to be established through mutagenesis studies. One likely possibility is that SLC25A3 transports copper ions that are chelated in anionic complexes (e.g. copper chloride in vitro or in the anionic copper complex observed in vivo (5)). Significant precedent exists in the literature for the transport of metal–anion complexes in eukaryotic systems, including transport of copper into a number of epithelial cells via anion transporters (37, 38). We previously reported an interaction between the copper chelate found in the mitochondrial matrix and Pic2 from yeast (31), and we observed a similar interaction here with recombinant human SLC25A3-A using fluorescence anisotropy. Therefore, we speculate that this anionic copper chelate is the main source of copper transported by SLC25A3 in vivo.
Patients with mutations in the exon 3a isoform of SLC25A3 present with multisystem disorders characterized by muscle hypotonia, lactic acidosis, and hypertrophic cardiomyopathy (24, 25). These patients show phenotypes consistent with decreased ATP production from limiting phosphate availability. In the initial case report by Mayr et al. (24), ETC enzyme activity was measured and considered to be normal. However, the data presented suggest that loss of SLC25A3 in muscle results in a mild, isolated COX deficiency, because the enzyme activity in the patient was lower than even the lowest reference control (patient value with range for controls in parentheses): for COX, 81 (90–281) (24). It may be that high copper levels present at birth make it possible to partially compensate for loss of SLC25A3 function by redistributing copper to the peripheral tissues, thereby allowing for the partial rescue of COX activity. Such a scenario would be reminiscent of the compensation observed in the myocardium of the heart-specific knockout of Ctr1 (39–41). There is no information regarding the copper status of SLC25A3 patients, and additional experiments will be required to address this possibility.
The fact that the loss of an importer of copper correlates with a decrease in COX activity suggests that matrix copper is used as a source of copper for the maturation of the CuA and CuB sites of the holoenzyme (5). Although we have genetically and biochemically manipulated the matrix copper pool in Saccharomyces cerevisiae to affect COX activity, this is the first demonstration of a genetic manipulation in mammalian cells that affects matrix copper levels and impacts COX activity. Thus, the pathway of copper import into the matrix prior to redistribution to the IMS for COX assembly in yeast appears to be conserved in mammalian cells. The phenotypes observed in L. lactis expressing either the A or B isoform of SLC25A3 upon exposure to both silver and arsenate suggest that the transporter is working as an importer. However, it remains possible that these transporters can also export matrix copper and that our current assays lack the necessary components either in small molecules or interacting protein partners to stimulate this activity. SLC25A3 has been identified in multiple complexes in the IM, including the ATP synthasomes and respirasomes (42–44). The physical association of the respiratory chain complexes with other MCFs, most notably ANT1, is known to significantly affect both nucleotide transport rates and oxidative phosphorylation (44). Therefore, further work is needed to elucidate the other components of this pathway involving SLC25A3, including the identity of possible metabolic products that could serve as modulators of transport and other transporters required to redistribute copper to the IMS.
Mitochondria generate and transduce a signal that can regulate the activity of the copper import and export machinery and therefore cellular copper concentrations (28, 45, 46). The integrity of this signaling pathway is compromised by mutations in SCO1 and SCO2 (28, 45, 46). SCO1 and SCO2 patients present with clinically heterogeneous forms of fatal disease that affect liver, heart, or brain function (47–54). Affected tissues in SCO patients are both COX- and copper-deficient, and both deficiencies appear to be key components of the underlying tissue specificity of disease (46, 55). Although the levels of SCO1 are unchanged in the Slc25a3−/− MEFs, the redox state of the copper-binding CXXXC motif of this protein in the absence of SLC25A3 is unknown, and this is an important component of the mitochondrial signal that controls cellular copper homeostasis. However, it should be noted that the deletion of Slc25a3 in MEFs results in a modest decrease in total copper when compared with the SCO cell lines. Here, the decrease in cellular copper is accompanied by a decrease in calcium content. The connection between these changes is not obvious; however, SLC25A3 has been implicated as indirectly regulating the mitochondrial permeability transition pore in response to calcium, and SLC25A3 mutants are desensitized to mitochondrial calcium overload (27).
Strikingly, manipulation of SLC25A3 causes a significant decrease in the activity of whole-cell SOD1. The observed decrease in total cellular copper is not expected to be sufficient to explain the reduction in SOD1 activity. Although we did not examine the levels of SOD1 in the IMS, the total decrease in its abundance far exceeds the amount of protein found in this compartment. We have previously observed a decrease in SOD1 activity in multiple models with SCO mutations (28, 46, 55). However, in these models the cellular copper content along with CCS abundance are both dramatically reduced, giving rise to the loss of SOD1 activity. CCS levels are normally elevated under copper deficiency as we observed here upon deletion of Ctr1 (56). These data collectively suggest a novel role for the mitochondrial copper-handling machinery in regulating or controlling cytosolic copper levels or bioavailability. In yeast, cytosolic SOD1 is known to transmit signals from oxygen and glucose to repress respiration via an interaction with the yeast homolog of casein kinase 1-γ (57). The interaction between these two proteins promotes the stability of the kinase and requires the activity of SOD1 to convert superoxide to hydrogen peroxide (57). In the absence of SOD1 respiration rates increase. The interaction between SOD1 and casein kinase 1-γ is also observed in mammalian cells (57). Therefore, under mitochondrial copper deficiency this mechanism could be designed to alter respiration rates, which raises the intriguing possibility that mitochondrial copper status is functionally coupled to glucose metabolism through SOD1.
The identification of SLC25A3 as a copper importer is a fundamental advance in our basic understanding of copper homeostasis. It adds yet another piece to the complicated COX assembly process that is implicated and disrupted in numerous metabolic diseases.
Experimental procedures
Cell culture conditions
HEK293 cells maintained in high-glucose DMEM containing sodium pyruvate, 50 μg/ml uridine, and 15% fetal bovine serum at 37 °C at an atmosphere of 5% CO2 were treated with siRNA for SLC25A3 (Human Silencer Select no. s10429 and Human Stealth RNAi no. hss107924 from Thermo Fisher Scientific) or negative control (Stealth RNAi negative control no. 452001, Thermo Fisher Scientific). All oligonucleotides were transfected into cells at 60–80% confluence using the RNAi Max protocol at 8 nm final concentration (Thermo Fisher Scientific) and allowed to grow for 36–48 h at 37 °C at an atmosphere of 5% CO2. HL-1 cells were maintained in Claycombs medium (58) at 37 °C at an atmosphere of 5% CO2 and were treated with siRNA as described above. Mouse C2C12 cells were maintained in high-glucose DMEM containing sodium pyruvate, 50 μg/ml uridine, and 10% fetal bovine serum at 37 °C at an atmosphere of 5% CO2. RNA silencing was performed as above using a mouse siRNA (Mouse Silencer select no. s71565 from Thermo Fisher Scientific). C2C12 myoblasts were differentiated into myotubes by switching the cells to serum-free medium. Differentiation was assessed by visual inspection of the culture over time, and siRNAs were re-applied every 48 h.
MEFs were isolated from Slc25a3FLOX/FLOX embryos and immortalized by retroviral transduction with telomerase and the E7 gene of the human papillomavirus (59, 60). Recombinant Cre recombinase was purified from E. coli, and it or buffer alone were added to Slc25a3FLOX/FLOX MEFs to generate Slc25a3−/− or Slc25a3FLOX/FLOX alleles, respectively (61). Clonal Slc25a3FLOX/FLOX and Slc25a3−/− MEF lines were then isolated and maintained in high-glucose DMEM containing sodium pyruvate, 50 μg/ml uridine, 0.1 mm mercaptoethanol, and 15% fetal bovine serum at 37 °C at an atmosphere of 5% CO2. Mouse Slc25a3-b cDNA was amplified from RNA and cloned into a Gateway-modified retroviral expression vector (62). The fidelity of this construct was confirmed by sequencing, and retrovirus was produced with the Phoenix amphotrophic packaging cell line (45) and used to transduce MEFs.
ATSM was synthesized as described previously (63). Briefly, 5 g of 4-methyl-3-thiosemicarbazide was added to 100 ml of ethanol (100 ml), heated to 60 °C, followed by the addition of 2 ml of diacetyl (2,3-butadione). ATSM was precipitated by addition of concentrated sulfuric acid, then collected by filtration, and washed with methanol. CuATSM was made by dissolving 4 g of ATSM in 100 ml of methanol, followed by the dropwise addition of copper chloride dissolved in water, and stirring the mixture for 30 min. CuATSM was precipitated by adding water; the product was collected via filtration and washed with methanol and water. CuATSM was diluted in DMSO (0.75 g/15 ml) and then added to the culture medium at 50 μm final concentration. Phosphate was supplemented into the medium at a concentration of 50 mm.
Yeast strains
The yeast strain used was BY4741 pic2Δ (MATa, leu2Δ, met15Δ, ura3Δ, his3Δ, PIC2::KANMX). Cultures were grown in 1% yeast extract, 2% peptone with 1% glucose medium at 30 °C for 24–36 h. Preparation and fractionation of mitochondria were performed as described previously (17). The mitochondrial copper uptake rate was obtained by incubation of isolated mitochondria with copper for 0–4 min at room temperature, centrifugation, and then acid digestion followed by ICP-OES (PerkinElmer Life Sciences 7300-DV) as described previously (18).
Expression of recombinant proteins
SLC25A3-A and SLC25A3-B human cDNAs were purchased from Life Technologies, Inc., and then amplified by PCR for subcloning into the E. coli expression vector pHis parallel 1. The fidelity of the construct was verified by sequencing. BL21(DE3) E. coli was transformed with the vector, and after isolation of single colonies with inducible protein expression, 1-liter cultures were grown to an absorbance at 600 nm of 0.6–0.8, and protein expression was induced with isopropyl β-d-1-thiogalactopyranoside for 2 h. Inclusion bodies were isolated in a manner similar to that described by Palmieri (19). Briefly, cells were resuspended in potassium phosphate buffer (140 mm NaCl, 2.7 mm KCl, 8.3 mm K2HPO4, 1.8 mm KH2PO4, pH 7.5) and disrupted by sonication. Insoluble material was collected by centrifugation at 18,500 × g. Insoluble material was resuspended in potassium phosphate buffer, pH 7.5, and loaded onto a stepwise 40, 53, and 70% sucrose gradient. Samples were centrifuged at 18,500 × g for 1 h, and inclusion bodies were isolated as a gray-colored band at the interface between the 53 and 70% layers. Presence of the protein of interest was confirmed by immunoblot and Coomassie staining. To load protein into liposomes, inclusion bodies were solubilized in 6 m urea or 1% Sarkosyl, then mixed with egg yolk phospholipids purified by the Folch extraction method, and dialyzed overnight in 25 mm Tris-HCl buffer, pH 7.2. The dialyzed mixture was sonicated (in the presence of 1 mm PhenGreen), then purified by loading the vesicles in 35% sucrose under 20% sucrose, and centrifuged at 18,500 × g for 60 min. The final proteoliposomes were isolated from the top of the sucrose layer and diluted into 25 mm Tris-HCl buffer, pH 7.2, and the protein concentration was determined by the Bradford assay. Equal protein concentrations were diluted in 25 mm Tris-HCl, pH 7.2, and loaded into a 96-well plate. A copper ascorbate stock solution was created from 10 mm copper sulfate solution and 10 mm ascorbate, and then the stock solution was diluted in 25 mm Tris-HCl buffer, pH 7.2, at concentrations ranging from 0 to 5 mm to yield working solutions for uptake assays. Assays were started by delivering copper-ascorbate with a multichannel pipette and monitoring the decrease in fluorescence over time in a Cytation 3 plate reader (BioTek). All reactions contained trace amounts of chloride and sulfate present from the initial preparation of buffers and reagents. Initial rates of copper uptake were calculated over the linear portion of the curve. Copper uptake is expressed as change in PhenGreen fluorescence/min/mg of protein. Arsenate competition studies were performed by making the copper-ascorbate stock solution with arsenate added at 10-fold excess of copper.
Expression and uptake in L. lactis
L. lactis cells transformed with vector (pNZ8148) alone or pNZ8148 (MoBiTec) carrying the human SLC25A3-A or SLC25A3-B gene were grown overnight at 30 °C in M17 medium with 0.5% glucose and 10 μg/ml chloramphenicol. Cells were diluted into fresh medium at an A600 of 0.1, grown to an A600 of 0.4, and induced using 1 ng/ml nisin for 5 h or overnight. Copper uptake was assayed in L. lactis using whole cells resuspended in 2 μm copper-sulfate salts dissolved in water. Cells were incubated for different time points at room temperature, removed by centrifugation, and washed in water, and total metals were measured by ICP-OES. Uptake was reported as the increase in copper over time. To determine silver toxicity in L. lactis strains containing vector, or human SLC25A3-A, or human SLC25A3-B, cells were grown in a 96-well plate containing M17 medium plus 1 ng/ml nisin and increasing concentrations of silver (0–250 μm) or arsenate (0–2.5 mm) (31). Controls containing M17 without nisin or M17 plus silver or arsenate without nisin were included. Absorbance at 600 nm was used to assess growth after 24 h. Percent growth was quantified by comparing to the optical density of the same genotype in nisin alone.
Elemental analysis
Samples were digested in 40% nitric acid by boiling for 1 h in capped acid-washed tubes, diluted in ultra-pure, metal-free water, and analyzed by ICP-OES (PerkinElmer Life Sciences, Optima 7300DV) versus acid-washed blanks. Concentrations were determined from a standard curve constructed with serial dilutions of two commercially available mixed metal standards (Optima). Blanks of nitric acid with and without “metal spikes” were analyzed to ensure reproducibility.
Immunoblot and activity assays
This study used monoclonal antibodies raised against GAPDH, COX1, and COX4 subunits of COX (Abcam) and polyclonal antisera raised against SOD1 (Enzo), CTR1 (custom order Genscript, VSIRYNSMPVPGPNGTILC), CCS (Joe Prohaska, University of Minnesota), ATP7A (Mick Petris, University of Missouri), SCO1 (in-house, keyhole limpet hemocyanin-coupled peptide CEKMIEVVEEIDSIPSLPNLT), and SLC25A3 (Invitrogen and Abcam). COX activity was determined by monitoring the decrease in absorbance at 550 nm of chemically reduced cytochrome c in the presence of cell or mitochondrial extracts (64). Superoxide dismutase (SOD) activity was measured using a xanthine oxidase-linked assay kit (Sigma Life Science), and absorbance was measured on a Cytation 3 plate reader (BioTek). All activities were normalized to protein concentration and then converted to percentage of maximum control value.
Fluorescence measurements
Mito-CS1 was synthesized as described (29). Reactivity with copper was measured by titrating the freshly diluted probe with copper. 4,6-Diamidino-2-phenylindole (DAPI), Mitotracker Green, JC-1, and Phen Green SK were purchased from Life Technologies, Inc. Cells were harvested from the plate with addition of trypsin for 15 min. Equal numbers of cells were loaded with dyes by incubation for 15 min at 37 °C, then washed by centrifugation with Dulbecco's PBS (Corning), and then read at appropriate wavelengths in a 96-well plate format using a Cytation 3 plate reader (BioTek).
Fluorescence anisotropy was performed using mitochondrial anionic copper complex (CuL) isolated from yeast and recombinant SLC25A3-A reconstituted into liposomes (31). Equal amounts of control liposome without protein were assayed and subtracted from each point to account for increase viscosity. CuL was isolated as described previously (5, 17, 31) and then diluted to give a fluorescence intensity of ∼30 using an excitation at 320 nm and monitoring emission at 400 nm. SLC25A3 protein incorporated into liposomes was added in 1–5-μl increments. Protein concentrations were determined by Bradford assay. Anisotropy was measured using a PerkinElmer Life Sciences LS55 spectrofluorimeter.
Data analysis
All results are presented as mean with errors bars of standard error of the mean. p values <0.05 were considered significant as assessed by Student's t test. *, p < 0.05; **, p < 0.01; ***, p < 0.001; ****, p < 0.0001. Curve fitting for calculation of half-maximal transport (K½) and Vmax was done using KaleidaGraph Michaelis-Menten curve fitting of concentration versus uptake and/or values from linear regression on a Lineweaver-Burk plot. Conversion of relative fluorescence to copper concentration in vesicle was calculated by using titration of PhenGreen in the same buffer conditions at the time of analysis in the absence of vesicles.
Author contributions
A. B., K. E. V., M. K. M., M. G. G., A. C. R., A. T. M., S. E. C., X. Z., C. B. P., J. Q. K., S. C. D., S. C. L., and P. A. C. investigation; A. B., K. E. V., S. C. L., and P. A. C. writing-review and editing; K. E. V., S. C. L., and P. A. C. conceptualization; K. E. V., S. C. L., and P. A. C. writing-original draft; S. C. D. and P. A. C. methodology; S. C. L. and P. A. C. formal analysis; S. C. L. and P. A. C. funding acquisition; S. C. L. and P. A. C. data curation; S. C. L. and P. A. C. supervision; P. A. C. project administration.
Acknowledgments
We thank Chistopher J. Chang (University of California, Berkeley) for helpful discussions and Jeffery D. Molkentin (Cincinnati Children's Hospital Medical Center) for generously providing the original Slc25a3 MEFs that were immortalized for analysis in this study.
This work was supported in part by National Science Foundation Grant MCB1158497 (to P. A. C.), National Institutes of Health Grants R01GM120211 (to P. A. C. and S. C. L.) and R01GM079465 (to Christopher J. Chang (University of California-Berkeley) for work performed by S. C. D.), and Canadian Institutes of Health Research (to S. C. L.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
This article was selected as one of our Editors' Picks.
- IMS
- intermembrane space
- IM
- inner membrane
- COX
- cytochrome c oxidase; cytochrome c oxidase
- SOD
- superoxide dismutase
- MEF
- mouse embryonic fibroblast
- ETC
- electron transport chain
- MCF
- mitochondrial carrier family
- ICP-OES
- inductively coupled plasma-optical emission spectroscopy
- DMEM
- Dulbecco's modified Eagle's medium
- ATSM
- diacetylbis(4-methylthiosemicarbazonato)copper.
References
- 1. Diaz F. (2010) Cytochrome c oxidase deficiency: patients and animal models. Biochim. Biophys. Acta 1802, 100–110 10.1016/j.bbadis.2009.07.013 [DOI] [PubMed] [Google Scholar]
- 2. Palmieri F. (2004) The mitochondrial transporter family (SLC25): physiological and pathological implications. Pflugers Arch. 447, 689–709 10.1007/s00424-003-1099-7 [DOI] [PubMed] [Google Scholar]
- 3. Tsukihara T., Aoyama H., Yamashita E., Tomizaki T., Yamaguchi H., Shinzawa-Itoh K., Nakashima R., Yaono R., and Yoshikawa S. (1996) The whole structure of the 13-subunit oxidized cytochrome c oxidase at 2.8 A. Science 272, 1136–1144 10.1126/science.272.5265.1136 [DOI] [PubMed] [Google Scholar]
- 4. Jett K. A., and Leary S. C. (September 29, 2017) Building the CuA site of cytochrome c oxidase: a complicated, redox-dependent process driven by a surprisingly large complement of accessory proteins. J. Biol. Chem. 293, 10.1074/jbc.R117.816132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Cobine P. A., Pierrel F., Bestwick M. L., and Winge D. R. (2006) Mitochondrial matrix copper complex used in metallation of cytochrome oxidase and superoxide dismutase. J. Biol. Chem. 281, 36552–36559 10.1074/jbc.M606839200 [DOI] [PubMed] [Google Scholar]
- 6. Baker Z. N., Cobine P. A., and Leary S. C. (2017) The mitochondrion: a central architect of copper homeostasis. Metallomics 9, 1501–1512 10.1039/C7MT00221A [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Mick D. U., Dennerlein S., Wiese H., Reinhold R., Pacheu-Grau D., Lorenzi I., Sasarman F., Weraarpachai W., Shoubridge E. A., Warscheid B., and Rehling P. (2012) MITRAC links mitochondrial protein translocation to respiratory-chain assembly and translational regulation. Cell 151, 1528–1541 10.1016/j.cell.2012.11.053 [DOI] [PubMed] [Google Scholar]
- 8. Smith D., Gray J., Mitchell L., Antholine W. E., and Hosler J. P. (2005) Assembly of cytochrome-c oxidase in the absence of assembly protein Surf1p leads to loss of the active site heme. J. Biol. Chem. 280, 17652–17656 10.1074/jbc.C500061200 [DOI] [PubMed] [Google Scholar]
- 9. Horng Y. C., Cobine P. A., Maxfield A. B., Carr H. S., and Winge D. R. (2004) Specific copper transfer from the Cox17 metallochaperone to both Sco1 and Cox11 in the assembly of yeast cytochrome c oxidase. J. Biol. Chem. 279, 35334–35340 10.1074/jbc.M404747200 [DOI] [PubMed] [Google Scholar]
- 10. Carr H. S., George G. N., and Winge D. R. (2002) Yeast Cox11, a protein essential for cytochrome c oxidase assembly, is a Cu(I)-binding protein. J. Biol. Chem. 277, 31237–31242 10.1074/jbc.M204854200 [DOI] [PubMed] [Google Scholar]
- 11. Bode M., Woellhaf M. W., Bohnert M., van der Laan M., Sommer F., Jung M., Zimmermann R., Schroda M., and Herrmann J. M. (2015) Redox-regulated dynamic interplay between Cox19 and the copper-binding protein Cox11 in the intermembrane space of mitochondria facilitates biogenesis of cytochrome c oxidase. Mol. Biol. Cell 26, 2385–2401 10.1091/mbc.E14-11-1526 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Bourens M., Boulet A., Leary S. C., and Barrientos A. (2014) Human COX20 cooperates with SCO1 and SCO2 to mature COX2 and promote the assembly of cytochrome c oxidase. Hum. Mol. Genet. 23, 2901–2913 10.1093/hmg/ddu003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Stroud D. A., Maher M. J., Lindau C., Vögtle F. N., Frazier A. E., Surgenor E., Mountford H., Singh A. P., Bonas M., Oeljeklaus S., Warscheid B., Meisinger C., Thorburn D. R., and Ryan M. T. (2015) COA6 is a mitochondrial complex IV assembly factor critical for biogenesis of mtDNA-encoded COX2. Hum. Mol. Genet. 24, 5404–5415 10.1093/hmg/ddv265 [DOI] [PubMed] [Google Scholar]
- 14. Pacheu-Grau D., Bareth B., Dudek J., Juris L., Vögtle F. N., Wissel M., Leary S. C., Dennerlein S., Rehling P., and Deckers M. (2015) Cooperation between COA6 and SCO2 in COX2 maturation during cytochrome c oxidase assembly links two mitochondrial cardiomyopathies. Cell Metab. 21, 823–833 10.1016/j.cmet.2015.04.012 [DOI] [PubMed] [Google Scholar]
- 15. Leary S. C., Sasarman F., Nishimura T., and Shoubridge E. A. (2009) Human SCO2 is required for the synthesis of CO II and as a thiol-disulphide oxidoreductase for SCO1. Hum. Mol. Genet. 18, 2230–2240 10.1093/hmg/ddp158 [DOI] [PubMed] [Google Scholar]
- 16. Morgada M. N., Abriata L. A., Cefaro C., Gajda K., Banci L., and Vila A. J. (2015) Loop recognition and copper-mediated disulfide reduction underpin metal site assembly of CuA in human cytochrome oxidase. Proc. Natl. Acad. Sci. U.S.A. 112, 11771–11776 10.1073/pnas.1505056112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Cobine P. A., Ojeda L. D., Rigby K. M., and Winge D. R. (2004) Yeast contain a non-proteinaceous pool of copper in the mitochondrial matrix. J. Biol. Chem. 279, 14447–14455 10.1074/jbc.M312693200 [DOI] [PubMed] [Google Scholar]
- 18. Vest K. E., Leary S. C., Winge D. R., and Cobine P. A. (2013) Copper import into the mitochondrial matrix in Saccharomyces cerevisiae is mediated by Pic2, a mitochondrial carrier family protein. J. Biol. Chem. 288 23884–23892 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Palmieri L., Runswick M. J., Fiermonte G., Walker J. E., and Palmieri F. (2000) Yeast mitochondrial carriers: bacterial expression, biochemical identification and metabolic significance. J. Bioenerg. Biomembr. 32, 67–77 10.1023/A:1005564429242 [DOI] [PubMed] [Google Scholar]
- 20. Robinson A. J., Overy C., and Kunji E. R. (2008) The mechanism of transport by mitochondrial carriers based on analysis of symmetry. Proc. Natl. Acad. Sci. U.S.A. 105, 17766–17771 10.1073/pnas.0809580105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Takabatake R., Siddique A. B., Kouchi H., Izui K., and Hata S. (2001) Characterization of a Saccharomyces cerevisiae gene that encodes a mitochondrial phosphate transporter-like protein. J. Biochem. 129, 827–833 10.1093/oxfordjournals.jbchem.a002926 [DOI] [PubMed] [Google Scholar]
- 22. Hamel P., Saint-Georges Y., de Pinto B., Lachacinski N., Altamura N., and Dujardin G. (2004) Redundancy in the function of mitochondrial phosphate transport in Saccharomyces cerevisiae and Arabidopsis thaliana. Mol. Microbiol. 51, 307–317 10.1046/j.1365-2958.2003.03810.x [DOI] [PubMed] [Google Scholar]
- 23. Fiermonte G., Dolce V., and Palmieri F. (1998) Expression in Escherichia coli, functional characterization, and tissue distribution of isoforms A and B of the phosphate carrier from bovine mitochondria. J. Biol. Chem. 273, 22782–22787 10.1074/jbc.273.35.22782 [DOI] [PubMed] [Google Scholar]
- 24. Mayr J. A., Merkel O., Kohlwein S. D., Gebhardt B. R., Böhles H., Fötschl U., Koch J., Jaksch M., Lochmüller H., Horváth R., Freisinger P., and Sperl W. (2007) Mitochondrial phosphate-carrier deficiency: a novel disorder of oxidative phosphorylation. Am. J. Hum. Genet. 80, 478–484 10.1086/511788 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Mayr J. A., Zimmermann F. A., Horváth R., Schneider H. C., Schoser B., Holinski-Feder E., Czermin B., Freisinger P., and Sperl W. (2011) Deficiency of the mitochondrial phosphate carrier presenting as myopathy and cardiomyopathy in a family with three affected children. Neuromuscul. Disord. 21, 803–808 10.1016/j.nmd.2011.06.005 [DOI] [PubMed] [Google Scholar]
- 26. Seifert E. L., Gál A., Acoba M. G., Li Q., Anderson-Pullinger L., Golenár T., Moffat C., Sondheimer N., Claypool S. M., and Hajnóczky G. (2016) Natural and induced mitochondrial phosphate carrier loss: differential dependence of mitochondrial metabolism and dynamics and cell survival on the extent of depletion. J. Biol. Chem. 291, 26126–26137 10.1074/jbc.M116.744714 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Kwong J. Q., Davis J., Baines C. P., Sargent M. A., Karch J., Wang X., Huang T., and Molkentin J. D. (2014) Genetic deletion of the mitochondrial phosphate carrier desensitizes the mitochondrial permeability transition pore and causes cardiomyopathy. Cell Death Differ. 21, 1209–1217 10.1038/cdd.2014.36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Leary S. C., Cobine P. A., Kaufman B. A., Guercin G. H., Mattman A., Palaty J., Lockitch G., Winge D. R., Rustin P., Horvath R., and Shoubridge E. A. (2007) The human cytochrome c oxidase assembly factors SCO1 and SCO2 have regulatory roles in the maintenance of cellular copper homeostasis. Cell Metab. 5, 9–20 10.1016/j.cmet.2007.04.002 [DOI] [PubMed] [Google Scholar]
- 29. Dodani S. C., Leary S. C., Cobine P. A., Winge D. R., and Chang C. J. (2011) A targetable fluorescent sensor reveals that copper-deficient SCO1 and SCO2 patient cells prioritize mitochondrial copper homeostasis. J. Am. Chem. Soc. 133, 8606–8616 10.1021/ja2004158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Monné M., Chan K. W., Slotboom D. J., and Kunji E. R. (2005) Functional expression of eukaryotic membrane proteins in Lactococcus lactis. Protein Sci. 14, 3048–3056 10.1110/ps.051689905 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Vest K. E., Wang J., Gammon M. G., Maynard M. K., White O. L., Cobine J. A., Mahone W. K., and Cobine P. A. (2016) Overlap of copper and iron uptake systems in mitochondria in Saccharomyces cerevisiae. Open Biol. 6, 150223 10.1098/rsob.150223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Marobbio C. M., Punzi G., Pierri C. L., Palmieri L., Calvello R., Panaro M. A., and Palmieri F. (2015) Pathogenic potential of SLC25A15 mutations assessed by transport assays and complementation of Saccharomyces cerevisiae ORT1 null mutant. Mol. Genet. Metab. 115, 27–32 10.1016/j.ymgme.2015.03.003 [DOI] [PubMed] [Google Scholar]
- 33. Marobbio C. M., Agrimi G., Lasorsa F. M., and Palmieri F. (2003) Identification and functional reconstitution of yeast mitochondrial carrier for S-adenosylmethionine. EMBO J. 22, 5975–5982 10.1093/emboj/cdg574 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Fiermonte G., Dolce V., David L., Santorelli F. M., Dionisi-Vici C., Palmieri F., and Walker J. E. (2003) The mitochondrial ornithine transporter. Bacterial expression, reconstitution, functional characterization, and tissue distribution of two human isoforms. J. Biol. Chem. 278, 32778–32783 10.1074/jbc.M302317200 [DOI] [PubMed] [Google Scholar]
- 35. Cavero S., Vozza A., del Arco A., Palmieri L., Villa A., Blanco E., Runswick M. J., Walker J. E., Cerdán S., Palmieri F., and Satrústegui J. (2003) Identification and metabolic role of the mitochondrial aspartate-glutamate transporter in Saccharomyces cerevisiae. Mol. Microbiol. 50, 1257–1269 10.1046/j.1365-2958.2003.03742.x [DOI] [PubMed] [Google Scholar]
- 36. Catalina-Rodriguez O., Kolukula V. K., Tomita Y., Preet A., Palmieri F., Wellstein A., Byers S., Giaccia A. J., Glasgow E., Albanese C., and Avantaggiati M. L. (2012) The mitochondrial citrate transporter, CIC, is essential for mitochondrial homeostasis. Oncotarget 3, 1220–1235 10.18632/oncotarget.714 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Kaplan J. H., and Maryon E. B. (2016) How mammalian cells acquire copper: an essential but potentially toxic metal. Biophys. J. 110, 7–13 10.1016/j.bpj.2015.11.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Zimnicka A. M., Ivy K., and Kaplan J. H. (2011) Acquisition of dietary copper: a role for anion transporters in intestinal apical copper uptake. Am. J. Physiol. Cell Physiol. 300, C588–C599 10.1152/ajpcell.00054.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Allen K. J., Buck N. E., Cheah D. M., Gazeas S., Bhathal P., and Mercer J. F. (2006) Chronological changes in tissue copper, zinc and iron in the toxic milk mouse and effects of copper loading. Biometals 19, 555–564 10.1007/s10534-005-5918-5 [DOI] [PubMed] [Google Scholar]
- 40. Kim B. E., Turski M. L., Nose Y., Casad M., Rockman H. A., and Thiele D. J. (2010) Cardiac copper deficiency activates a systemic signaling mechanism that communicates with the copper acquisition and storage organs. Cell Metab. 11, 353–363 10.1016/j.cmet.2010.04.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Pyatskowit J. W., and Prohaska J. R. (2008) Copper deficient rats and mice both develop anemia but only rats have lower plasma and brain iron levels. Comp. Biochem. Physiol. C Toxicol. Pharmacol. 147, 316–323 10.1016/j.cbpc.2007.11.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Chen C., Ko Y., Delannoy M., Ludtke S. J., Chiu W., and Pedersen P. L. (2004) Mitochondrial ATP synthasome: three-dimensional structure by electron microscopy of the ATP synthase in complex formation with carriers for Pi and ADP/ATP. J. Biol. Chem. 279, 31761–31768 10.1074/jbc.M401353200 [DOI] [PubMed] [Google Scholar]
- 43. Ko Y. H., Delannoy M., Hullihen J., Chiu W., and Pedersen P. L. (2003) Mitochondrial ATP synthasome. Cristae-enriched membranes and a multiwell detergent screening assay yield dispersed single complexes containing the ATP synthase and carriers for Pi and ADP/ATP. J. Biol. Chem. 278, 12305–12309 10.1074/jbc.C200703200 [DOI] [PubMed] [Google Scholar]
- 44. Lu Y. W., Acoba M. G., Selvaraju K., Huang T. C., Nirujogi R. S., Sathe G., Pandey A., and Claypool S. M. (2017) Human adenine nucleotide translocases physically and functionally interact with respirasomes. Mol. Biol. Cell 28, 1489–1506 10.1091/mbc.E17-03-0195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Leary S. C., Cobine P. A., Nishimura T., Verdijk R. M., de Krijger R., de Coo R., Tarnopolsky M. A., Winge D. R., and Shoubridge E. A. (2013) COX19 mediates the transduction of a mitochondrial redox signal from SCO1 that regulates ATP7A-mediated cellular copper efflux. Mol. Biol. Cell 24, 683–691 10.1091/mbc.E12-09-0705 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Hlynialuk C. J., Ling B., Baker Z. N., Cobine P. A., Yu L. D., Boulet A., Wai T., Hossain A., El Zawily A. M., McFie P. J., Stone S. J., Diaz F., Moraes C. T., Viswanathan D., Petris M. J., and Leary S. C. (2015) The mitochondrial metallochaperone SCO1 is required to sustain expression of the high-affinity copper transporter CTR1 and preserve copper homeostasis. Cell Rep. 2015, S2211–S1247 [DOI] [PubMed] [Google Scholar]
- 47. Horváth R., Freisinger P., Rubio R., Merl T., Bax R., Mayr J. A., Shawan Müller-Höcker J., Pongratz D., Moller L. B., Horn N., and Jaksch M. (2005) Congenital cataract, muscular hypotonia, developmental delay and sensorineural hearing loss associated with a defect in copper metabolism. J. Inherit. Metab. Dis. 28, 479–492 10.1007/s10545-005-0479-x [DOI] [PubMed] [Google Scholar]
- 48. Jaksch M., Horvath R., Horn N., Auer D. P., Macmillan C., Peters J., Gerbitz K. D., Kraegeloh-Mann I., Muntau A., Karcagi V., Kalmanchey R., Lochmuller H., Shoubridge E. A., and Freisinger P. (2001) Homozygosity (E140K) in SCO2 causes delayed infantile onset of cardiomyopathy and neuropathy. Neurology 57, 1440–1446 10.1212/WNL.57.8.1440 [DOI] [PubMed] [Google Scholar]
- 49. Jaksch M., Ogilvie I., Yao J., Kortenhaus G., Bresser H. G., Gerbitz K. D., and Shoubridge E. A. (2000) Mutations in SCO2 are associated with a distinct form of hypertrophic cardiomyopathy and cytochrome c oxidase deficiency. Hum. Mol. Genet. 9, 795–801 10.1093/hmg/9.5.795 [DOI] [PubMed] [Google Scholar]
- 50. Jaksch M., Paret C., Stucka R., Horn N., Müller-Höcker J., Horvath R., Trepesch N., Stecker G., Freisinger P., Thirion C., Müller J., Lunkwitz R., Rödel G., Shoubridge E. A., and Lochmüller H. (2001) Cytochrome c oxidase deficiency due to mutations in SCO2, encoding a mitochondrial copper-binding protein, is rescued by copper in human myoblasts. Hum. Mol. Genet. 10, 3025–3035 10.1093/hmg/10.26.3025 [DOI] [PubMed] [Google Scholar]
- 51. Joost K., Rodenburg R., Piirsoo A., van den Heuvel B., Zordania R., and Ounap K. (2010) A novel mutation in the SCO2 gene in a neonate with early-onset cardioencephalomyopathy. Pediatr. Neurol. 42, 227–230 10.1016/j.pediatrneurol.2009.10.004 [DOI] [PubMed] [Google Scholar]
- 52. Papadopoulou L. C., Sue C. M., Davidson M. M., Tanji K., Nishino I., Sadlock J. E., Krishna S., Walker W., Selby J., Glerum D. M., Coster R. V., Lyon G., Scalais E., Lebel R., Kaplan P., et al. (1999) Fatal infantile cardioencephalomyopathy with COX deficiency and mutations in SCO2, a COX assembly gene. Nat. Genet. 23, 333–337 10.1038/15513 [DOI] [PubMed] [Google Scholar]
- 53. Leary S. C., Antonicka H., Sasarman F., Weraarpachai W., Cobine P. A., Pan M., Brown G. K., Brown R., Majewski J., Ha K. C., Rahman S., and Shoubridge E. A. (2013) Novel mutations in SCO1 as a cause of fatal infantile encephalopathy and lactic acidosis. Hum. Mutat. 34, 1366–1370 10.1002/humu.22385 [DOI] [PubMed] [Google Scholar]
- 54. Valnot I., Osmond S., Gigarel N., Mehaye B., Amiel J., Cormier-Daire V., Munnich A., Bonnefont J. P., Rustin P., and Rötig A. (2000) Mutations of the SCO1 gene in mitochondrial cytochrome c oxidase deficiency with neonatal-onset hepatic failure and encephalopathy. Am. J. Hum. Genet. 67, 1104–1109 10.1016/S0002-9297(07)62940-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Baker Z. N., Jett K., Boulet A., Hossain A., Cobine P. A., Kim B.-E., El Zawily A. M., Lee L., Tibbits G. F., Petris M. J., and Leary S. C. (2017) The mitochondrial metallochaperone SCO1 maintains CTR1 at the plasma membrane to preserve copper homeostasis in the murine heart. Hum. Mol. Genet. 26, 4617–4628 10.1093/hmg/ddx344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. West E. C., and Prohaska J. R. (2004) Cu,Zn-superoxide dismutase is lower and copper chaperone CCS is higher in erythrocytes of copper-deficient rats and mice. Exp. Biol. Med. 229, 756–764 10.1177/153537020422900807 [DOI] [PubMed] [Google Scholar]
- 57. Reddi A. R., and Culotta V. C. (2013) SOD1 integrates signals from oxygen and glucose to repress respiration. Cell 152, 224–235 10.1016/j.cell.2012.11.046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Claycomb W. C., Lanson N. A. Jr., Stallworth B. S., Egeland D. B., Delcarpio J. B., Bahinski A., and Izzo N. J. Jr. (1998) HL-1 cells: a cardiac muscle cell line that contracts and retains phenotypic characteristics of the adult cardiomyocyte. Proc. Natl. Acad. Sci. U.S.A. 95, 2979–2984 10.1073/pnas.95.6.2979 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Yao J., and Shoubridge E. A. (1999) Expression and functional analysis of SURF1 in Leigh syndrome patients with cytochrome c oxidase deficiency. Hum. Mol. Genet. 8, 2541–2549 10.1093/hmg/8.13.2541 [DOI] [PubMed] [Google Scholar]
- 60. Lochmüller H., Johns T., and Shoubridge E. A. (1999) Expression of the E6 and E7 genes of human papillomavirus (HPV16) extends the life span of human myoblasts. Exp. Cell Res. 248, 186–193 10.1006/excr.1999.4407 [DOI] [PubMed] [Google Scholar]
- 61. Anand R., Wai T., Baker M. J., Kladt N., Schauss A. C., Rugarli E., and Langer T. (2014) The i-AAA protease YME1L and OMA1 cleave OPA1 to balance mitochondrial fusion and fission. J. Cell Biol. 204, 919–929 10.1083/jcb.201308006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Antonicka H., Mattman A., Carlson C. G., Glerum D. M., Hoffbuhr K. C., Leary S. C., Kennaway N. G., and Shoubridge E. A. (2003) Mutations in COX15 produce a defect in the mitochondrial heme biosynthetic pathway, causing early-onset fatal hypertrophic cardiomyopathy. Am. J. Hum. Genet. 72, 101–114 10.1086/345489 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Williams J. R., Trias E., Beilby P. R., Lopez N. I., Labut E. M., Bradford C. S., Roberts B. R., McAllum E. J., Crouch P. J., Rhoads T. W., Pereira C., Son M., Elliott J. L., Franco M. C., Estévez A. G., et al. (2016) Copper delivery to the CNS by CuATSM effectively treats motor neuron disease in SOD(G93A) mice co-expressing the Copper-Chaperone-for-SOD. Neurobiol. Dis. 89, 1–9 10.1016/j.nbd.2016.01.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Capaldi R. A., Marusich M. F., and Taanman J. W. (1995) Mammalian cytochrome-c oxidase: characterization of enzyme and immunological detection of subunits in tissue extracts and whole cells. Methods Enzymol. 260, 117–132 10.1016/0076-6879(95)60134-1 [DOI] [PubMed] [Google Scholar]