

Abstract
Kidney ankyrin repeat–containing proteins (KANK1/2/3/4) belong to a family of scaffold proteins, playing critical roles in cytoskeleton organization, cell polarity, and migration. Mutations in KANK proteins are implicated in cancers and genetic diseases, such as nephrotic syndrome. KANK proteins can bind various target proteins through different protein regions, including a highly conserved ankyrin repeat domain (ANKRD). However, the molecular basis for target recognition by the ANKRD remains elusive. In this study, we solved a high-resolution crystal structure of the ANKRD of KANK1 in complex with a short sequence of the motor protein kinesin family member 21A (KIF21A), revealing that the highly specific target-binding mode of the ANKRD involves combinatorial use of two interfaces. Mutations in either interface disrupted the KANK1-KIF21A interaction. Cellular immunofluorescence localization analysis indicated that binding-deficient mutations block recruitment of KIF21A to focal adhesions by KANK1. In conclusion, our structural study provides mechanistic explanations for the ANKRD-mediated recognition of KIF21A and for many disease-related mutations identified in human KANK proteins.
Keywords: crystal structure, focal adhesion, protein complex, scaffold protein, tumor suppressor gene, KANK, ankyrin repeat
Introduction
Focal adhesions (FAs)4 are sites attached to extracellular matrix for cell adhesion and migration (1). Formation of a functional FA requires the proper assembly and disassembly of numerous FA proteins (focal adhesion kinase, talin, kindlin, vinculin, paxillin, etc.) together with actin filaments and microtubules (2, 3). Recently, kidney ankyrin repeat–containing protein (KANK) family proteins were found to play an important role in bridging microtubules to FAs (4, 5). The KANK family contains four members in vertebrates (KANK1–4) (6), one member in Drosophila (dKANK) (7), and one in Caenorhabditis elegans (VAB-19) (8). All KANK proteins are characterized by an N-terminal 30-residue motif (named KN motif) and a C-terminal ankyrin repeat domain (ANKRD), whereas vertebrate KANKs contain an additional coiled-coil region in the middle (see Fig. 1A) (9). As the most studied KANK family member, KANK1 was first identified as a growth suppressor in human kidney tumor cells (10). Studies suggested that the growth-inhibitory effect of KANK1 is mediated by regulating cytoskeletal actin organization to inhibit cell mobility (11–13). Consistently, deletion and down-regulation of the KANK1 gene have been found in many tumors (9, 14–16) as well as neurological disorders (17, 18). In addition, hereditary mutations in KANKs lead to nephrotic syndrome (NS), a genetic kidney disease, due at least in part to podocyte dysfunction caused by KANK deficiency (7). A KANK2 mutation was identified in patients with keratoderma and woolly hair (19), whereas a genetic variant of KANK4 is associated with corneal dystrophy (20). Several genetic mutations are located in the ANKRD (Fig. 2), suggesting that this domain is important for the functions of the KANK proteins by acting as a target-binding module.
Figure 1.

Biochemical characterization of the KANK1-KIF21A interaction. A, schematic diagrams of domain organizations for KANK1 and KIF21A. The interaction between the ANKRD and the short helical region in KIF21A is indicated by a double-headed arrow. KN, N-terminal 30-residue motif. B, the representative ITC curve showing the strong binding of KANK1_ANKRD to KIF21A_H. C, mapping of the minimal yet sufficient interacting boundaries for both KANK1_ANKRD and KIF21A_H based on ITC analysis shown in Fig. S1. Fitted binding ratios (N) are also indicated. D, gel filtration analysis of the complex formation using 60 μm Trx-KANK1_ANKRD (theoretical molecular mass of 44 kDa), 60 μm Trx-KIF21A_H (18 kDa), and their mixture. Elution volumes of standard size markers are indicated by arrowheads.
Figure 2.

Multiple sequence alignment of the ANKRDs of the KANK family proteins from different species. The species are represented by “h” for human, “m” for mouse, “x” for Xenopus tropicalis, “z” for Danio rerio, and “d” for Drosophila melanogaster. VAB19 is the KANK homologue in C. elegans. Secondary structure elements are labeled above the alignment. Residues that are identical and highly similar in each alignment are shown in red and yellow boxes, respectively. Residues involved in the KANK1-KIF21A interaction are indicated by triangles. Three mutation sites found in genetic diseases are marked by blue circles.
As a scaffold, KANK1 interacts with various target proteins to achieve its functions. KANK1 binds with IRSp53 to negatively regulate cell spreading and lamellipodial development (12, 13). Cortical localization of KANK1 is mediated by association with liprin-β1 (5, 21), and KANK1 is localized to FAs via its binding to talin (4, 5). In particular, KANK1 recruits the cortical microtubule stabilization complex to FAs via a direct interaction between the ANKRD of KANK1 and kinesin family member 21A (KIF21A) (21, 22), which regulates microtubule dynamics during developmental processes, such as axon growth (21, 23). Despite the growing number of known KANK1-binding proteins, the molecular basis governing the target recognition of KANK1 is poorly understood due to a lack of structural information.
Here, we report the crystal structures of the ANKRD of KANK1 both in complex with a 28-residue KIF21A fragment at 1.55-Å resolution and in the apo form at 1.9-Å resolution. The complex structure shows a unique binding mode, mediated by a negatively charged surface patch and a hydrophobic interface of the ANKRD. Mutations disrupting the KANK1-KIF21A interaction lead to the abolished recruitment of KIF21A to FAs. Structural comparison implicates a potential conformational change of the ANKRD upon KIF21A binding. Taken together, our structural, biochemical, and cellular studies of the KANK1-KIF21A interaction provide mechanistic insights into the assembly of the KANK1-mediated complex in connecting microtubules and FAs as well as the effects caused by disease-related mutations in KANKs.
Results
Characterization of the interaction between KANK1 and KIF21A
To obtain insights into the KANK1-mediated target recognition mechanism, we first characterized the binding of KIF21A to KANK1. As the main interacting regions of KANK1 and KIF21A were reported to be the C-terminal ANKRD (KANK1_ANKRD) and a short helical region (KIF21A_H), respectively (Fig. 1A) (21), these two fragments were expressed in Escherichia coli and purified. Consistent with the previous report, KANK1_ANKRD strongly interacts with KIF21A_H with sub-μm binding affinity (Fig. 1, B and C). As indicated by the isothermal titration calorimetry (ITC)–based assay, the binding stoichiometry is close to 1:1. Formation of the KANK1_ANKRD-KIF21A_H heterodimer was confirmed by analytical gel filtration (Fig. 1D).
To identify the minimal binding region of KIF21A, we truncated either N-terminal or C-terminal flanking sequences to KIF21A_H. Based on ITC analysis (Figs. 1C and S1), we determined that the 28-residue region (residues 1142–1169) contains the necessary and sufficient elements for the binding of KIF21A_H to KANK1_ANKRD. To understand how this short sequence in KIF21A can be specifically recognized by KANK1, we sought to solve the KANK1_ANKRD-KIF21A_H complex using crystallography. As extensive trials failed to yield any crystals of the complex, we removed flexible regions on both the N-terminal and C-terminal ends of KANK1_ANKRD without affecting its binding to KIF21A_H (Fig. 1C). By using this KANK1_ANKRD construct (residues 1088–1338), we successfully obtained high-quality crystals for structural study.
Overall structure of the KANK1_ANKRD-KIF21A_H complex
The crystal structure of the KANK1_ANKRD-KIF21A_H complex was determined at 1.55-Å resolution using a molecular replacement method (Table 1). Consistent with our biochemical characterization, the complex structure contains one KANK1_ANKRD molecule and one KIF21A_H molecule in each asymmetric unit. As shown in Fig. 3A, the ANKRD contains five ANK repeats (R1–5), capped by an N-terminal helix bundle (α1–α5; named R0 hereafter) to form an α-solenoid fold (Figs. 2 and 3A). The ANK repeats are similar to each other except for R2, which has a longer sequence and a different conformation at its C-terminal half (Fig. 3B). Thus, the C-terminal helix (α9) of R2 protrudes from the solenoid fold (Fig. 3A).
Table 1.
Statistics of data collection and model refinement
Numbers in parentheses represent the value for the highest-resolution shell.
| Data set |
||
|---|---|---|
| Complex | Apo | |
| Data collection | ||
| Space group | P212121 | P21 |
| Unit cell parameters (Å) | a = 38.0, b = 52.1, c = 136.1 | a = 29.1, b = 70.2, c = 60.7, β = 96.9 |
| Resolution range (Å) | 50–1.55 (1.58–1.55) | 50–1.9 (1.93–1.9) |
| No. of unique reflections | 39,577 (2,005) | 19,134 (949) |
| Redundancy | 5.4 (5.5) | 3.7 (3.5) |
| I/σ | 39.6 (2.7) | 23.8 (1.2) |
| Completeness (%) | 98.2 (100) | 99.8 (99.9) |
| Rmerge (%)a | 5.7 (83.5) | 7.7 (119.9) |
| CC½b | 0.99 (0.69) | 0.99 (0.55) |
| Structure refinement | ||
| Resolution (Å) | 50–1.55 (1.59–1.55) | 50–1.9 (2.0–1.9) |
| Rcryst/Rfree (%)c | 18.3 (24.6)/20.0 (29.8) | 19.3 (31.9)/23.5 (39.5) |
| r.m.s.d. bonds (Å)/angles (°) | 0.012/1.4 | 0.014/1.4 |
| Average B factor | 35.3 | 39.0 |
| No. of atoms | ||
| Protein atoms | 1,990 | 1,774 |
| Water molecules | 121 | 66 |
| Other solvent molecules | 0 | 4 |
| Ramachandran plot | ||
| Favored regions (%) | 98.9 | 97.5 |
| Allowed regions (%) | 1.1 | 2.5 |
| Outliers (%) | 0.0 | 0.0 |
a Rmerge = Σ|Ii − Im|/ΣIi where Ii is the intensity of the measured reflection and Im is the mean intensity of all symmetry-related reflections.
b CC½ is the correlation of one half of randomly chosen observations to the other half.
c Rcryst = Σ‖Fobs| − |Fcalc‖/Σ|Fobs| where Fobs and Fcalc are observed and calculated structure factors, respectively. Rfree = ΣT‖Fobs| − |Fcalc‖/ΣT|Fobs| where T is a test data set of about 5% of the total reflections randomly chosen and set aside prior to refinement.
Figure 3.

Structural characterization of the KANK1-KIF21A interaction. A, ribbon representation of the heterodimeric complex with KANK1_ANKRD in green and KIF21A_H in pink. This color coding scheme is used throughout the remaining figures unless otherwise indicated. B, structural comparison of the five ANK repeats in the ANKRD domain of KANK1. C, surface conservation of the ANKRD. Based on the sequence alignment shown in Fig. 2, identical and similar residues are colored in orange and light orange, respectively. D and E, detailed interactions in site-I (D) and site-II (E). Interface residues are shown in stick representation. Hydrogen bonds and salt bridges are indicated by dashed lines. F, charge potential surface of KANK1 reveals a negatively charged pocket in site-I. G, amino acid sequence alignment of KIF21A_H in vertebrates. Residues directly involved in the site-I and site-II interactions are indicated by triangles.
In the complex structure, the KIF21A_H peptide folds as an extended loop in the N terminus and a short α-helix in the C terminus to interact tightly with the concave side of the ANKRD (Fig. 3A). The five ANK repeats are all involved in forming a highly conserved binding groove using their N-terminal helices and hairpin-like connecting loops (Fig. 3, A and C). Only 15 residues (1154–1168) in KIF21A_H could be successfully traced in the final structural model (Fig. S2). Nevertheless, the binding of KIF21A_H buries a large surface area of ∼900 Å2 on the binding groove of KANK1_ANKRD. This structural finding explains the strong interaction between KIF21A_H and KANK1_ANKRD.
Molecular details of the KANK1_ANKRD-KIF21A_H interaction
Based on the surface properties, the binding groove on the ANKRD can be divided into two parts, site-I and site-II, formed by the repeats R4–5 and R1–3, respectively (Fig. 3, D and E). Site-I contains multiple negatively charged residues, including Glu-1274KANK1, Glu-1284KANK1, Asp-1306KANK1, and Asp-1308KANK1, resulting in a highly negatively charged surface (Fig. 3F). Complementarily, the N-terminal half of KIF21A_H mainly comprises positively charged residues (Fig. 3G). Among these residues, Lys-1154KIF21A, Arg-1156KIF21A, and Arg-1157KIF21A directly form salt bridges with the negatively charged residues on site-I. Although not clearly assigned in the structural model, Lys-1150KIF21A and Lys-1152KIF21A may also contribute to the charge-charge interaction as an N-terminally truncated peptide of KIF21A_H lacking these two residues shows diminished binding affinity (Fig. 1C). Additionally, Arg-1156KIF21A form a hydrogen bond with Ser-1276KANK1 to further strengthen the site-I interaction (Fig. 3D). Unlike site-I, site-II is relatively neutral in its surface charge (Fig. 3F). The site-II interaction is mainly mediated by hydrophobic interactions and hydrogen bonds (Fig. 3E). In detail, the backbone of the KIF21A_H peptide forms hydrogen bonds with the polar residues, Asn-1171KANK1, Asn-1201KANK1, and His-1252KANK1, in site-II, which places the side chains of Leu-1165KIF21A and Leu-1166KIF21A to interact with a hydrophobic surface patch formed by Tyr-1176KANK1, Tyr-1205KANK1, Leu-1210KANK1, Leu-1213KANK1, and Leu-1248KANK1.
To verify the above structural findings, we generated a series of mutations in either KANK1_ANKRD or KIF21A_H designed to disrupt their interaction. As charge complementarity and hydrophobicity are critical for the interactions in site-I and site-II, respectively, we reasoned that a KIF21A_H peptide carrying charge-reversed mutations (e.g. R1156E) or hydrophobic-to-hydrophilic mutations (e.g. L1165Q) would impair its binding to KANK1_ANKRD. Indeed, an ITC-based assay demonstrated that the R1156E variant almost abolishes the binding of KIF21A_H to KANK1_ANKRD, and the L1165Q variant decreases the binding affinity by ∼20-fold (Fig. 4, A and B). Correspondingly, the mutations in site-I and site-II of KANK1_ANKRD (D1306K and L1248Q, respectively) show similar disruptions of binding (Figs. 4C and S1). To further validate this binding mode using the full-length proteins, we performed coimmunoprecipitation experiments by cotransfection of FLAG-tagged KANK1 and GFP-tagged KIF21A in HEK293T cells. Consistent with our findings with the fragments, the binding of KANK1 to KIF21A is disrupted by the R1156E and L1165Q mutations (Fig. 4D). Taken together, our structural and biochemical data demonstrate that KANK1 specifically recognizes KIF21A via a combination of the charge-charge interaction at site-I and the hydrophobic interaction at site-II. Given the high sequence similarity of the interface residues in KIF21A (Fig. 3G), the binding mode found in our complex structure is very likely to be shared by all vertebrate KIF21A homologues.
Figure 4.

The KANK1-KIF21A interaction is disrupted by the mutations in the site-I and site-II interfaces. A and B, ITC curves show that the R1156E and L1165Q mutations in KIF21A_H disrupt binding to site-I and site-II, respectively. C, the dissociation constants of the binding reactions of various mutants of KANK1_ANKRD and KIF21A_H. ITC curves are shown in Fig. S1. D, coimmunoprecipitation analysis of full-length KANK1 and KIF21A in HEK293T cells. IP, immunoprecipitation; IB, immunoblotting.
KANK1 recruits KIF21A to focal adhesions requiring both of the site-I and site-II interactions
Previously, KANK1 was reported to target KIF21A to the cell cortex as well as FAs (5, 21). We therefore used a cellular colocalization assay to confirm the binding mode between KANK1 and KIF21A in cells. As a control, cellular localizations of individual full-length proteins were checked when overexpressed in COS7 cells. KIF21A shows diffuse cellular distribution with modest enrichment at the cell edge (Fig. 5A). In contrast, KANK1 is mainly found in FA as indicated by colocalization with vinculin, although some diffuse localization at the cell edge is also seen (Fig. 5B). Line profile analysis further demonstrated that KANK1 concentrated at the lateral border of FA (Fig. 5C), fully consistent with previous findings (4, 5). We next overexpressed GFP-tagged KIF21A together with Ruby-tagged KANK1 in COS7 cells. As expected, KIF21A colocalized with KANK1 at the FA border (Fig. 5, D and E), indicating that KIF21A was recruited to the FA border by KANK1.
Figure 5.

Recruitment of KIF21A to FAs by KANK1 depends on the site-I and site-II interactions. A and B, COS7 cells transiently transfected with GFP-KIF21A (A) and GFP-KANK1 (B). Anti-vinculin antibody (red) is used to stain FA. Scale bar, 10 μm. C, fluorescence-intensity profiles represent protein localization in the area marked by the white arrow in B. D, cotransfection of GFP-KIF21A and Ruby-KANK1 in COS7 cells showing that KIF21A is recruited by KANK1 to the FA border. However, the recruitment is compromised by the two KIF21A mutants, R1156E and L1165Q, disrupting the site-I and site II interactions, respectively. E, results in D were further confirmed by line profile analysis.
Next, we asked whether recruitment of KIF21A to FA by KANK1 is mediated by site-I and site-II. To address this question, we prepared two mutants (R1156E and L1165Q) of KIF21A, disrupting site-I and site-II binding, respectively (Figs. 3G and 4). By overexpressing the two GFP-KIF21A mutants with Ruby-KANK1 in COS7 cells, we found that neither mutant shows significant enrichment at the FA border as indicated by KANK1 (Fig. 5, D and E). These data indicate that FA localization of KIF21A requires the functional binding interface of the ANKRD; disrupting either the site-I or site-II interaction diminishes the FA targeting of KIF21A.
Conformational differences in the ANKRD
To determine whether the ANKRD undergoes a conformational change upon binding of KIF21A, we determined the structure of KANK1_ANKRD alone (Table 1). Structural comparison indicates that the apo form and the KIF21A-bound ANKRD of KANK1 are essentially identical with an overall r.m.s.d. of 0.5 Å except for the last two ANK repeats (R4 and R5; Fig. 6A). The backbones of the N-terminal helices of R4 and R5 show shifts of 2 and 1 Å, respectively, due at least in part to the charge attraction of Glu-1318KANK1 to Arg-1157KIF21A in the complex (Fig. 5B). This conformational change results in closer packing between R3 and R4 and between R4 and R5, thereby leading to the more compact fold of the ANKRD with bound KIF21A. To test the KIF21A-binding effect on the folding of the ANKRD, we measured the thermal stability of KANK1_ANKRD in the absence or presence of the KIF21A_H peptide using circular dichroism (CD) spectroscopy (Fig. 6C). In line with our prediction, addition of the peptide significantly increased the melting temperature (Tm) of the KANK1_ANKRD protein by ∼2 °C. As the KIF21A_H peptide alone shows little CD signal (Fig. S3), we propose that the thermal stability increase is due to the binding-induced conformational change.
Figure 6.

Structural comparison of the ANKRDs. A, overlap of KANK1_ANKRD in apo form and KIF21A-bound form. Conformational changes in the R4 and R5 repeats are indicated by red circles. B, charge-charge interaction between Glu-1318KANK1 and Arg-1157KIF21A. C, CD-based thermal denaturation analysis of KANK1_ANKRD in the presence and absence of the KIF21A_H peptide. D, overlap of the C-terminal ANK repeats (R3–5) of the ANKRDs in KANK1 and KANK2 (Protein Data Bank code 4HBD) showing the rotational change of the N-terminal region (R0–2). An overall structural comparison of these two ANKRD apo structures is shown in the inset. E, comparison of the R2/3 interfaces of the two structures in D. F, thermal denaturation analysis of KANK2_ANKRD.
We also compared the apo structure of KANK1_ANKRD with the publicly available apo structure of KANK2_ANKRD. The overall structural difference is relatively large (an overall r.m.s.d. of 1.5 Å for 232 aligned residues; Fig. 6D, inset). Interestingly, however, the C-terminal R3–5 regions can be locally superposed with an r.m.s.d. of 0.5 Å for 96 aligned residues (Fig. 6D). Compared with KANK1, the N-terminal R0–2 region in KANK2 shows a ∼6° rotation (Fig. 6D), presumably due to the different packing in the R2/3 interface. As shown in Fig. 6E, Val-754KANK2 is substituted in KANK1 by a methionine (Met-1257KANK1), which has a much longer side chain and thereby forces the convex parts of the R2 and R3 repeats to have a larger distance between each other to avoid steric hindrance. The rotation results in a more compact fold of KANK2_ANKRD, which may increase protein stability. Consistently, the ANKRD of KANK2 shows much higher thermal stability (Fig. 6F). Because Met-1257KANK1 is strictly conserved in vertebrate KANK1/3/4 (Fig. 1), it is likely that KANK3 and KANK4 also adopt the orientation between the N-terminal R0–2 and C-terminal R3–5 regions observed in KANK1 rather than that in KANK2.
Structural implications of KANK mutations in human diseases
As dysfunctions of KANK proteins are associated with cancers (9, 14), we analyzed publicly available mutation data in various cancer samples (http://cancer.sanger.ac.uk/cancergenome/projects/cosmic/).5 Interestingly, somatic cancer mutations appear at higher frequencies in the ANKRD relative to other regions of KANKs (Fig. 7A), suggesting the importance of the ANKRD for KANK functions. By mapping the missense mutations to the ANKRD, we found that four mutation sites are located in the KIF21A-binding groove (Fig. 7B). Based on our structural analysis, we predict that these four mutations would disrupt KIF21A binding by disrupting salt bridges and/or hydrogen bonds or by introducing steric hindrance. Consistent with this hypothesis, the corresponding mutations in KANK1_ANKRD, E1284K, S1276F, N1201D, and Y1176C (corresponding to E738K in human KANK3 and to S1110F, N1035D, and Y1010C in human KANK1, respectively) hamper the binding of KANK1_ANKRD to KIF21A_H (Fig. 4C).
Figure 7.

Disease-related mutations in KANK family members. A, distribution of cancer somatic and genetic mutations in KANKs, including missense, nonsense, deletion, or insertion mutations, which result in changes of protein products. The columns indicate the mutation positions and their reported times according to the COSMIC database (http://cancer.sanger.ac.uk/cancergenome/projects/cosmic/).5 The schematic domain organization is indicated for reference. KN, N-terminal 30-residue motif. B, four somatic mutation sites are mapped to the KANK1_ANKRD structure. The corresponding mutations are also indicated. C, two disease-causing mutation sites are located in the hydrophobic core of KANK1_ANKRD, including the mutations identified in patients with nephrotic syndrome or keratoderma. D, thermal denaturation curves of the wildtype KANK1_ANKRD and its mutants were measured using CD spectroscopy at 222 nm. E, analysis of potential steric clashes in the KANK1_ANKRD-KIF21A_H complex for Ser-1179KANK1 (E1) and three potential side-chain rotamers of the S1179F mutation (E2–E4).
Several mutations in KANKs have been identified in multiple human genetic diseases, such as NS and keratoderma (7, 19). Among these mutations, three sites are located in the ANKRD (Fig. 7A). Because Tyr-1147KANK1 and Ala-1173KANK1 (corresponding to Tyr-801 in KANK4 and Ala-670 in KANK2, respectively) are both involved in forming hydrophobic cores (Fig. 7C), equivalent mutations are expected to destabilize the folding of KANK1_ANKRD. Indeed, the Y1147H and A1173V mutants show much lower Tm values than that of the wildtype protein (Fig. 7D). We also mutated Ser-1179KANK1 to a phenylalanine to mimic the disease-causing mutation (S676F in KANK2). Consistent with the observation that Ser-1179KANK1 is solvent-exposed in KANK1_ANKRD, the thermal stability of the S1179F mutant remains the same as the wildtype protein (Fig. 7D). Although Ser-1179KANK1 is close to site-II of the ANKRD (Fig. 3E), the S1179F mutation does not interfere with the binding of KANK1_ANKRD to KIF21A_H (Fig. 4C). It is likely that the potential clash introduced by the mutated bulky side chain upon KIF21A binding can be tolerated by adopting a specific side-chain conformation (Fig. 7E). Further investigation is thus required to understand the effects of this mutation found in NS patients.
Discussion
Accumulating evidence indicates the important scaffolding functions of KANKs in connecting cytoskeletons to specified membrane regions, such as focal adhesions and the cell cortex (4, 5, 9, 13). In this study, we have solved the crystal structures of the ANKRD of KANK1 and of its complex with the short ANKRD-binding sequence of KIF21A. Cellular recruitment of KIF21A to FAs by KANK1 confirms the regulatory role of KANK1 in the microtubule-FA cross-talk. Our complex structure together with the biochemical and cellular studies reveals the structural basis for the highly specific recognition of KIF21A by the ANK repeats in KANK1, providing a mechanistic explanation of the combinatorial use of the different surface properties of the different ANK repeats as well as the potential effects of several KANK mutations in genetic diseases and cancers. Although the known KANK1-binding targets show varied domain organizations, some of them may share a similar ANKRD-binding mode with that of KIF21A as a short sequence (∼20 residues) is sufficient for high binding affinity. Therefore, the binding sequence pattern identified in KIF21A_H, comprising several positively charged residues plus some hydrophobic residues (Fig. 3G), may assist in studying other ANKRD-mediated interactions in the future.
The ANK repeat is one of the most prevalent structural motifs and is found in numerous proteins as a binding module for mediating specific protein-protein interactions (24, 25). Although most ANK repeats share a well-defined conformation, the N-terminal hairpin-like loops often vary in their sequences (Fig. S4). As the loops are involved in forming the target-binding groove, this sequence variation creates distinct binding environments (e.g. site-I and site-II in the ANKRD) to specifically recognize different parts of the target (e.g. the short binding sequence in KIF21A), providing high binding selectivity and affinity. The combination of multiple sites for target recognition has also been observed in ankyrins that contain 24 ANK repeats (26), indicating that such combinatorial target-binding modes may be a general strategy in ANK repeat–containing proteins.
Despite the high sequence similarity of the ANKRDs in KANKs, KANK4 shows some key differences in the site-I interface residues compared with KANK1/2/3. For example, in KANK4, Glu-1284KANK1 and Asp-1308KANK1 are replaced by a histidine and an alanine, respectively (Fig. 2), resulting in a much less negatively charged surface at site-I (Fig. 8A). Given the importance of the charge-charge interactions in site-I (Figs. 3C and 5D), it is very likely that KANK4 is not capable of interacting with KIF21A. Consistently, our biochemical analysis indicates that KANK4_ANKRD does not form a complex with KIF21A_H (Fig. 8, B and C). As the functional specificities for the four members of the KANK family are poorly understood, our structural analysis provides valuable information for future studies to address isoform specificities.
Figure 8.
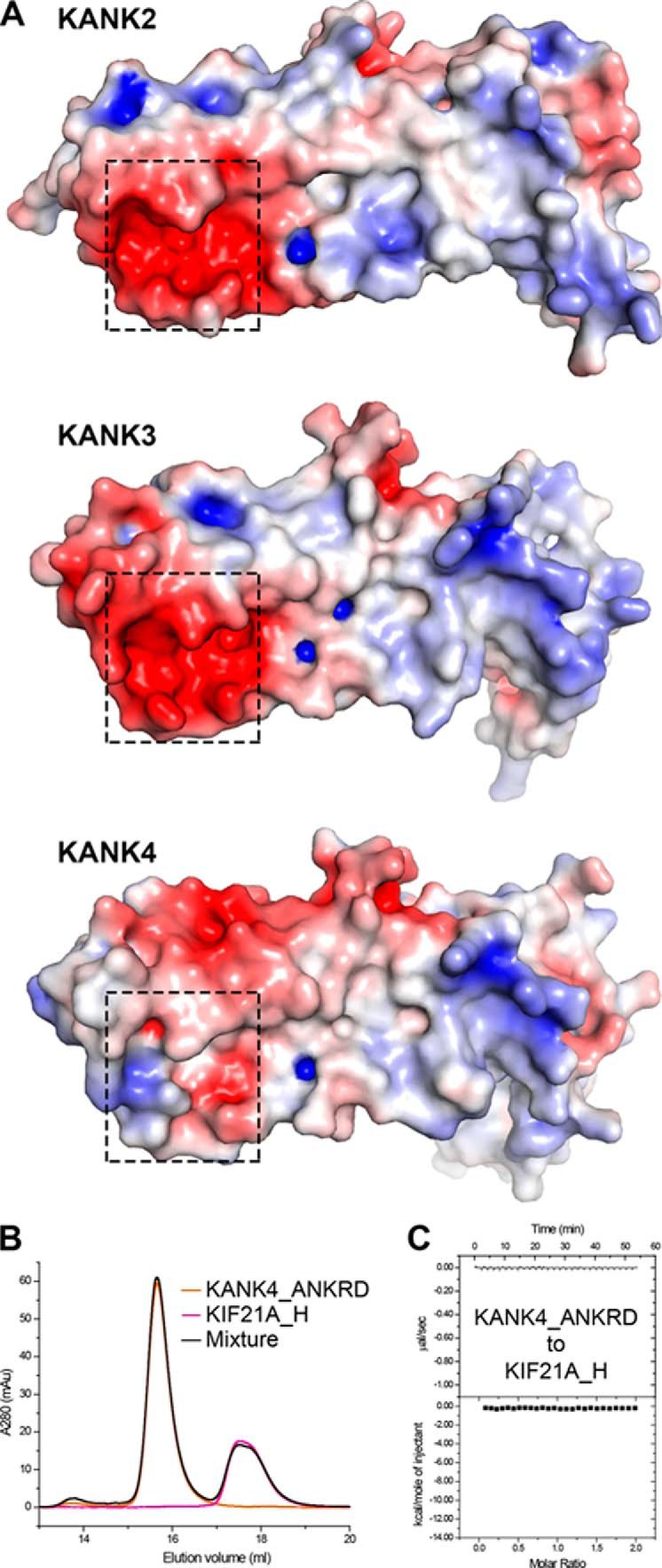
Surface electrostatic potential of the ANKRDs in KANK2/3/4. A, electrostatic potential mapped onto the molecular surface of KANK2_ANKRD (Protein Data Bank code 4HBD) and homology models of the ANKRDs of KANK3 and KANK4. Regions corresponding to site-I are indicated by dashed boxes. The surface analysis clearly shows that site-I is relatively neutral in KANK4. B and C, ITC (B) and gel filtration (C) analysis demonstrate that KANK4_ANKRD cannot bind to KIF21A_H.
Experimental procedures
DNA constructs and site-directed mutagenesis
DNA encoding the ANKRDs of KANK1 and KANK4 and the short H region of KIF21A were PCR-amplified from a mouse cDNA library. All coding sequences were cloned into a home-modified vector, pET32a, with an N-terminal thioredoxin (Trx)-His6 tag for protein expression. The full-length KANK1 and KIF21A constructs for cellular assays were PCR-amplified using plasmids (gifts from Dr. Anna Akhmanova) as template and cloned into mammalian expression vectors containing the GFP, FLAG, or Ruby tag. All point mutations were created using a site-directed mutagenesis kit and confirmed by DNA sequencing.
Protein sample preparation
For the expression of proteins, transformed E. coli C plus cells (Novagen) were grown in LB medium to an A600 nm of 0.8–1.0 at 37 °C. Protein expression was then induced by the addition of isopropyl 1-thio-β-d-galactopyranoside (final concentration, 0.2 mm), and cells were further grown for an additional 14 h at 16 °C. N-terminally Trx-His6-tagged proteins were purified using nickel-affinity chromatography followed by size-exclusion chromatography. For crystallization and CD analysis, the N-terminal tag was cleaved by human rhinovirus 3C protease and removed by size-exclusion chromatography.
Crystallography
To prepare the protein samples for crystallization, the KANK1_ANKRD protein was mixed with synthetic KIF21A_H peptide in a 1:1.2 ratio. Crystals were obtained by the sitting-drop vapor-diffusion method at 16 °C. To set up a sitting drop, 1 μl of concentrated protein solution (20–40 mg/ml) was mixed with 1 μl of crystallization solution containing 0.2 m ammonium formate and 20% (w/v) polyethylene glycol 3350 for the KANK1_ANKRD-KIF21A_H complex and 0.1 m calcium acetate, 0.1 m sodium acetate, pH 4.5, 10% (w/v) polyethylene glycol 4000 for the KANK1 apo form. Before X-ray diffraction experiments, crystals were soaked in the crystallization solution containing an additional 30% glycerol for cryoprotection. Diffraction data were collected at Shanghai Synchrotron Radiation Facility beamlines BL17U1 (27) and BL19U1. Data were processed and scaled using HKL2000 software.
The initial phase of the complex structure was determined by molecular replacement in PHASER (28) using the KANK2_ANKRD structure (Protein Data Bank code 4HBD) as the search model. The KIF21A_H peptide was further built into the model. The model was refined in PHENIX (29). Coot was used for model rebuilding and adjustments (30). In the final stage, an additional Atomic Displacement Parameters refinement with Translation-Libration-Screw-rotation model (TLS) was performed in PHENIX. The model quality was checked by MolProbity (31). The initial phase of the apo structure was determined by molecular replacement using the KANK1_ANKRD structure derived from the complex structure as the search model. The model was refined using the same strategy as for the complex structure. The final refinement statistics are listed in Table 1.
Structural analysis
The binding interface in the complex structure was analyzed using PISA (32). APBS was used to calculate electrostatic potential of the ANKRDs (33). Structural models of the ANKRDs of KANK3 and KANK4 were generated by homologue modeling using SWISS-MODEL (34). All structure figures were prepared using PyMOL.
ITC assay
ITC measurements were carried out on a VP-ITC MicroCal calorimeter (Malvern) at 25 °C. All protein samples were dissolved in 50 mm Tris-HCl buffer, pH 7.5, containing 100 mm NaCl, 1 mm EDTA, and 2 mm DTT. Titrations were performed by injecting a protein solution containing KANK1_ANKRD or its mutants at a concentration of 200–300 μm into a solution containing KIF21A_H or its mutants at a concentration of 20–30 μm. A time interval of 2 min between injections was used to ensure that the titration peak returned to baseline. The titration data were analyzed using the program Origin7.0 and fitted by a one-site binding model.
Analytical gel filtration chromatography
Analytical gel filtration chromatography was carried out on an ÄKTA Pure system (GE Healthcare). Protein samples at a concentration of 40 μm were loaded onto a Superdex 200 Increase 10/300 GL column (GE Healthcare) equilibrated with 50 mm Tris-HCl buffer, pH 7.5, containing 100 mm NaCl, 1 mm EDTA, and 2 mm DTT.
Cell culture and transfection
HEK293T cells and COS7 cells were cultured in DMEM supplemented with 10% FBS, 0.1 mm non-essential amino acids, and 50 units/ml penicillin and streptomycin, respectively, at 37 °C in 5% CO2. Transfection was performed using Lipofectamine 3000 (Thermo Fisher) according to the manufacturer's instructions.
Coimmunoprecipitation assay
Transiently transfected HEK293T cells were lysed in 50 mm Tris buffer, pH 7.5, 100 mm NaCl, 1% Triton X-100. Immunoprecipitation was carried out using 1.5 μg of anti-FLAG antibody conjugated to Protein A/G-Sepharose beads (Sigma), incubated for 1 h at 4 °C, and washed three times in lysis buffer. Bound proteins were resolved by SDS-PAGE and subjected to Western blotting.
Immunofluorescence and confocal microscopy
Cells were transfected with the indicated constructs. One day after transfection, the cells were trypsinized, replated on fibronectin (∼20 μg/ml)-coated coverslips, and cultured for 24 h. After fixation with 4% paraformaldehyde, the cells were stained with anti-vinculin (Abcam) followed by Alexa Fluor 594-conjugated anti-mouse IgG antibody (Invitrogen) and observed under a Leica TCS SP8 confocal microscope. Images were analyzed using Image J software (National Institutes of Health), and image overlays were created with Adobe Photoshop.
CD spectroscopy
CD spectra were recorded using a Chirascan spectrometer (Applied Photophysics, UK). Protein samples of mouse KANK2_ANKRD, KANK1_ANKRD, and its mutants at a concentration of 10 μm were loaded into a quartz cell with 1-mm path length. Thermal melts were recorded at 222 nm in the temperature range from 35 to 56 °C with 1 °C steps. To determine the midpoint of transition (Tm), the thermal denaturation data were fitted with an apparent two-state folding model and plotted as fraction folded versus temperature. Because the KIF21A_H peptide alone is structurally disordered in solution and contributes little to the ellipticity value (Fig. S3), the thermal denaturation profile of the KANK1_ANKRD-KIF21A_H mixture was measured and analyzed using the same strategy as the KANK1_ANKRD sample.
Author contributions
Z. W. conceived the study. W. P., Q. X., and C. M. designed constructs, purified proteins, and performed biochemistry assays. W. P. and Z. W. solved structures. K. S. performed cellular assays. W. P., K. S., K. T., C. Y., and Z. W. analyzed the data. Z. W. supervised the project. K. T. and Z. W. wrote the paper with help from W. P., K. S., and C. Y.
Supplementary Material
Acknowledgments
We thank Dr. Nathan C. Rockwell for critical reading of the manuscript and Dr. Xingqiao Xie for technical assistance. We are grateful to Dr. Anna Akhmanova for generously providing the plasmids encoding full-length KANK1 and KIF21A. We thank the Life Science Research Center, Southern University of Science and Technology for providing facilities. We thank BL19U1 beamline at National Center for Protein Sciences Shanghai and BL17U beamline at Shanghai Synchrotron Radiation Facility for the X-ray beam time.
This work was supported in part by National Natural Science Foundation of China Grants 31770791 and 31570741 (to Z. W.) and 31600612 (to K. T.), Natural Science Foundation of Guangdong Province Grant 2016A030312016, Science and Technology Planning Project of Guangdong Province Grant 2017B030301018, and Shenzhen Science and Technology Innovation Commission Grants JCYJ20160229153100269 and ZDSYS20140509142721429. The authors declare that they have no conflicts of interest with the contents of this article.
This article contains Figs. S1–S4.
The atomic coordinates and structure factors (codes 5YAZ and 5YAY) have been deposited in the Protein Data Bank (http://wwpdb.org/).
Please note that the JBC is not responsible for the long-term archiving and maintenance of this site or any other third party-hosted site.
- FA
- focal adhesion
- KANK
- kidney ankyrin repeat–containing protein
- NS
- nephrotic syndrome
- ANKRD
- ankyrin repeat domain
- ITC
- isothermal titration calorimetry
- r.m.s.d.
- root mean square
- KIF21A
- kinesin family member 21A
- ANK
- ankyrin
- Trx
- thioredoxin.
References
- 1. Burridge K., Fath K., Kelly T., Nuckolls G., and Turner C. (1988) Focal adhesions: transmembrane junctions between the extracellular matrix and the cytoskeleton. Annu. Rev. Cell Biol. 4, 487–525 10.1146/annurev.cb.04.110188.002415 [DOI] [PubMed] [Google Scholar]
- 2. Wehrle-Haller B. (2012) Assembly and disassembly of cell matrix adhesions. Curr. Opin. Cell Biol. 24, 569–581 10.1016/j.ceb.2012.06.010 [DOI] [PubMed] [Google Scholar]
- 3. Stehbens S., and Wittmann T. (2012) Targeting and transport: how microtubules control focal adhesion dynamics. J. Cell Biol. 198, 481–489 10.1083/jcb.201206050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Sun Z., Tseng H.-Y., Tan S., Senger F., Kurzawa L., Dedden D., Mizuno N., Wasik A. A., Thery M., Dunn A. R., and Fässler R. (2016) Kank2 activates talin, reduces force transduction across integrins and induces central adhesion formation. Nat. Cell Biol. 18, 941–953 10.1038/ncb3402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Bouchet B. P., Gough R. E., Ammon Y.-C., van de Willige D., Post H., Jacquemet G., Altelaar A. M., Heck A. J., Goult B. T., and Akhmanova A. (2016) Talin-KANK1 interaction controls the recruitment of cortical microtubule stabilizing complexes to focal adhesions. eLife 5, e18124 10.7554/eLife.18124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Zhu Y., Kakinuma N., Wang Y., and Kiyama R. (2008) Kank proteins: a new family of ankyrin-repeat domain-containing proteins. Biochim. Biophys. Acta 1780, 128–133 10.1016/j.bbagen.2007.09.017 [DOI] [PubMed] [Google Scholar]
- 7. Gee H. Y., Zhang F., Ashraf S., Kohl S., Sadowski C. E., Vega-Warner V., Zhou W., Lovric S., Fang H., Nettleton M., Zhu J. Y., Hoefele J., Weber L. T., Podracka L., Boor A., et al. (2015) KANK deficiency leads to podocyte dysfunction and nephrotic syndrome. J. Clin. Investig. 125, 2375–2384 10.1172/JCI79504 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Ding M., Goncharov A., Jin Y., and Chisholm A. D. (2003) C. elegans ankyrin repeat protein VAB-19 is a component of epidermal attachment structures and is essential for epidermal morphogenesis. Development 130, 5791–5801 10.1242/dev.00791 [DOI] [PubMed] [Google Scholar]
- 9. Kakinuma N., Zhu Y., Wang Y., Roy B. C., and Kiyama R. (2009) Kank proteins: structure, functions and diseases. Cell. Mol. Life Sci. 66, 2651–2659 10.1007/s00018-009-0038-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Sarkar S., Roy B. C., Hatano N., Aoyagi T., Gohji K., and Kiyama R. (2002) A novel ankyrin repeat-containing gene (Kank) located at 9p24 is a growth suppressor of renal cell carcinoma. J. Biol. Chem. 277, 36585–36591 10.1074/jbc.M204244200 [DOI] [PubMed] [Google Scholar]
- 11. Kakinuma N., Roy B. C., Zhu Y., Wang Y., and Kiyama R. (2008) Kank regulates RhoA-dependent formation of actin stress fibers and cell migration via 14-3-3 in PI3K-Akt signaling. J. Cell Biol. 181, 537–549 10.1083/jcb.200707022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Roy B. C., Kakinuma N., and Kiyama R. (2009) Kank attenuates actin remodeling by preventing interaction between IRSp53 and Rac1. J. Cell Biol. 184, 253–267 10.1083/jcb.200805147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Li C.-C., Kuo J.-C., Waterman C. M., Kiyama R., Moss J., and Vaughan M. (2011) Effects of brefeldin A-inhibited guanine nucleotide-exchange (BIG) 1 and KANK1 proteins on cell polarity and directed migration during wound healing. Proc. Natl. Acad. Sci. U.S.A. 108, 19228–19233 10.1073/pnas.1117011108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Guo X., Fan W., Bian X., and Ma D. (2014) Upregulation of the Kank1 gene-induced brain glioma apoptosis and blockade of the cell cycle in G0/G1 phase. Int. J. Oncol. 44, 797–804 10.3892/ijo.2014.2247 [DOI] [PubMed] [Google Scholar]
- 15. Cui Z., Shen Y., Chen K. H., Mittal S. K., Yang J.-Y., and Zhang G. (2017) KANK1 inhibits cell growth by inducing apoptosis though regulating CXXC5 in human malignant peripheral nerve sheath tumors. Sci. Rep. 7, 40325 10.1038/srep40325 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Chen T., Wang K., and Tong X. (2017) In vivo and in vitro inhibition of human gastric cancer progress by upregulating Kank1 gene. Oncol. Rep. 38, 1663–1669 10.3892/or.2017.5823 [DOI] [PubMed] [Google Scholar]
- 17. Lerer I., Sagi M., Meiner V., Cohen T., Zlotogora J., and Abeliovich D. (2005) Deletion of the ANKRD15 gene at 9p24.3 causes parent-of-origin-dependent inheritance of familial cerebral palsy. Hum. Mol. Genet. 14, 3911–3920 10.1093/hmg/ddi415 [DOI] [PubMed] [Google Scholar]
- 18. Vinci G., Chantot-Bastaraud S., El Houate B., Lortat-Jacob S., Brauner R., and McElreavey K. (2007) Association of deletion 9p, 46,XY gonadal dysgenesis and autistic spectrum disorder. Mol. Hum. Reprod. 13, 685–689 10.1093/molehr/gam045 [DOI] [PubMed] [Google Scholar]
- 19. Ramot Y., Molho-Pessach V., Meir T., Alper-Pinus R., Siam I., Tams S., Babay S., and Zlotogorski A. (2014) Mutation in KANK2, encoding a sequestering protein for steroid receptor coactivators, causes keratoderma and woolly hair. J. Med. Genet. 51, 388–394 10.1136/jmedgenet-2014-102346 [DOI] [PubMed] [Google Scholar]
- 20. Afshari N. A., Igo R. P. Jr., Morris N. J., Stambolian D., Sharma S., Pulagam V. L., Dunn S., Stamler J. F., Truitt B. J., Rimmler J., Kuot A., Croasdale C. R., Qin X., Burdon K. P., Riazuddin S. A., et al. (2017) Genome-wide association study identifies three novel loci in Fuchs endothelial corneal dystrophy. Nat. Commun. 8, 14898 10.1038/ncomms14898 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. van der Vaart B., van Riel W. E., Doodhi H., Kevenaar J. T., Katrukha E. A., Gumy L., Bouchet B. P., Grigoriev I., Spangler S. A., Yu K. L., Wulf P. S., Wu J., Lansbergen G., van Battum E. Y., Pasterkamp R. J., et al. (2013) CFEOM1-associated kinesin KIF21A is a cortical microtubule growth inhibitor. Dev. Cell 27, 145–160 10.1016/j.devcel.2013.09.010 [DOI] [PubMed] [Google Scholar]
- 22. Kakinuma N., and Kiyama R. (2009) A major mutation of KIF21A associated with congenital fibrosis of the extraocular muscles type 1 (CFEOM1) enhances translocation of Kank1 to the membrane. Biochem. Biophys. Res. Commun. 386, 639–644 10.1016/j.bbrc.2009.06.109 [DOI] [PubMed] [Google Scholar]
- 23. Cheng L., Desai J., Miranda C. J., Duncan J. S., Qiu W., Nugent A. A., Kolpak A. L., Wu C. C., Drokhlyansky E., Delisle M. M., Chan W.-M., Wei Y., Propst F., Reck-Peterson S. L., Fritzsch B., et al. (2014) Human CFEOM1 mutations attenuate KIF21A autoinhibition and cause oculomotor axon stalling. Neuron 82, 334–349 10.1016/j.neuron.2014.02.038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Michaely P., and Bennett V. (1992) The ANK repeat: a ubiquitous motif involved in macromolecular recognition. Trends Cell Biol. 2, 127–129 10.1016/0962-8924(92)90084-Z [DOI] [PubMed] [Google Scholar]
- 25. Sedgwick S. G., and Smerdon S. J. (1999) The ankyrin repeat: a diversity of interactions on a common structural framework. Trends Biochem. Sci. 24, 311–316 10.1016/S0968-0004(99)01426-7 [DOI] [PubMed] [Google Scholar]
- 26. Wang C., Wei Z., Chen K., Ye F., Yu C., Bennett V., and Zhang M. (2014) Structural basis of diverse membrane target recognitions by ankyrins. eLife 3, e04353 10.7554/eLife.04353 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Wang Q.-S., Yu F., Huang S., Sun B., Zhang K.-H., Liu K., Wang Z.-J., Xu C., Wang S.-S., Yang L.-F., Pan Q.-Y., Li L., Zhou H., Cui Y., Xu Q., et al. (2015) The macromolecular crystallography beamline of SSRF. Nucl. Sci. Tech. 26, 10102–010102 [Google Scholar]
- 28. Storoni L. C., McCoy A. J., and Read R. J. (2004) Likelihood-enhanced fast rotation functions. Acta Crystallogr. D Biol. Crystallogr. 60, 432–438 10.1107/S0907444903028956 [DOI] [PubMed] [Google Scholar]
- 29. Adams P. D., Afonine P. V., Bunkóczi G., Chen V. B., Davis I. W., Echols N., Headd J. J., Hung L. W., Kapral G. J., Grosse-Kunstleve R. W., McCoy A. J., Moriarty N. W., Oeffner R., Read R. J., Richardson D. C., Richardson J. S., et al. (2010) PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 66, 213–221 10.1107/S0907444909052925 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Emsley P., and Cowtan K. (2004) Coot: model-building tools for molecular graphics. Acta Crystallogr. D. Biol. Crystallogr. 60, 2126–2132 10.1107/S0907444904019158 [DOI] [PubMed] [Google Scholar]
- 31. Davis I. W., Leaver-Fay A., Chen V. B., Block J. N., Kapral G. J., Wang X., Murray L. W., Arendall W. B. 3rd, Snoeyink J., Richardson J. S., and Richardson D. C. (2007) MolProbity: all-atom contacts and structure validation for proteins and nucleic acids. Nucleic Acids Res. 35, W375–W383 10.1093/nar/gkm216 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Krissinel E., and Henrick K. (2007) Inference of macromolecular assemblies from crystalline state. J. Mol. Biol. 372, 774–797 10.1016/j.jmb.2007.05.022 [DOI] [PubMed] [Google Scholar]
- 33. Baker N. A., Sept D., Joseph S., Holst M. J., and McCammon J. A. (2001) Electrostatics of nanosystems: application to microtubules and the ribosome. Proc. Natl. Acad. Sci. U.S.A. 98, 10037–10041 10.1073/pnas.181342398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Arnold K., Bordoli L., Kopp J., and Schwede T. (2006) The SWISS-MODEL workspace: a web-based environment for protein structure homology modelling. Bioinformatics 22, 195–201 10.1093/bioinformatics/bti770 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.