

Abstract
Alzheimer's disease (AD) is a disabling and highly prevalent neurodegenerative condition, for which there are no effective therapies. Soluble oligomers of the amyloid-β peptide (AβOs) are thought to be proximal neurotoxins involved in early neuronal oxidative stress and synapse damage, ultimately leading to neurodegeneration and memory impairment in AD. The aim of the current study was to evaluate the neuroprotective potential of mesenchymal stem cells (MSCs) against the deleterious impact of AβOs on hippocampal neurons. To this end, we established transwell cocultures of rat hippocampal neurons and MSCs. We show that MSCs and MSC-derived extracellular vesicles protect neurons against AβO-induced oxidative stress and synapse damage, revealed by loss of pre- and postsynaptic markers. Protection by MSCs entails three complementary mechanisms: 1) internalization and degradation of AβOs; 2) release of extracellular vesicles containing active catalase; and 3) selective secretion of interleukin-6, interleukin-10, and vascular endothelial growth factor to the medium. Results support the notion that MSCs may represent a promising alternative for cell-based therapies in AD.
Keywords: Alzheimer's disease, catalase, cytokine action, endocytosis, extracellular vesicles, hippocampus, mesenchymal stem cells (MSCs), oxidative stress, synapse, amyloid-β, oligomers
Introduction
Alzheimer's disease (AD)5 is responsible for 50–70% of dementia cases in the elderly, and nearly half of individuals over the age of 85 are at risk of developing the disease (1, 2). AD begins with loss of recent memories and progresses to include loss of consolidated memories and deficits in several cognitive domains, finally depriving patients of their sense of self. The disease poses a great threat to older individuals and their families, becoming a serious socioeconomic problem with increasing longevity. Although considerable insight has been gained into mechanisms of pathogenesis of AD, no treatments are currently available to effectively halt or reverse its progression. Mounting evidence indicates that soluble oligomers of the amyloid-β peptide (AβOs) are the proximal neurotoxins involved in synapse damage and dysfunction leading to memory loss in AD (3–6). Blocking the neuronal impact of AβOs may thus provide effective novel therapies for AD.
Cell therapies have emerged as potential treatments for neurological disorders, including AD (7, 8). Studies employing mesenchymal stem cells (MSCs) from different sources have demonstrated promising results in in vivo AD models (9–12). For example, Lee et al. (13, 14) showed that transplantation of bone marrow MSCs into the hippocampus of the APP/PS1 mouse model of AD reduced Aβ deposition and Tau hyperphosphorylation and reversed learning and spatial memory deficits. However, the mechanisms underlying those neuroprotective actions of MSCs have not been elucidated. It is generally accepted that MSCs do not exert their beneficial actions through direct differentiation into neural tissue, but rather by acting as trophic mediators releasing immune modulatory, proangiogenic, and/or proneurogenic factors (15). Additional mechanisms involved in paracrine signaling promoted by MSCs include the secretion of specific cytokines (16) and the transfer of extracellular vesicles (EVs) or even of healthy mitochondria to cells with impaired mitochondrial function (17–19).
Here, we aimed to investigate the neuroprotective potential of MSCs in an in vitro model of AD, to gain insight into possible mechanisms of cell-to-cell communication (20) that could be exploited in future therapeutic approaches. We demonstrate that MSCs and MSC-derived EVs block oxidative stress and synapse damage induced by AβOs in hippocampal neurons and unveil novel neuroprotective mechanisms of action of MSCs, namely the clearance of extracellular AβOs, selective secretion of cytokines, and the release of active catalase via EVs.
Results
MSCs are resistant to AβOs
We initially evaluated the effects of exposure of MSCs to AβOs (500 nm) by investigating cell viability (Fig. 1, A–C), proliferation (Fig. 1, D–F), oxidative stress (measured by levels of reactive oxygen species (ROS); Fig. 1, G and H), and cellular respiration (via high-resolution respirometry) following exposure to oligomers (Fig. 1I). ROS levels in MSCs were barely detectable (see below). No changes in viability, proliferation rate, intracellular ROS levels, or respiratory parameters of MSCs were detected in AβO-exposed cultures (Fig. 1).
Figure 1.

AβOs do not affect viability, proliferation, respiration, or resistance to oxidative stress of MSCs. Representative photomicrographs show viable (green) or dead (red) MSCs exposed for 72 h to vehicle (Veh) (A) or AβOs (500 nm) (B). Scale bar, 50 μm. Images were acquired on a Zeiss Axiovert 200M microscope with a 10× objective. Cell viabilities are shown in C (n = 3 independent cultures, with triplicate wells in each experimental condition). D and E, Ki67 immunofluorescence (red) and nuclei (DAPI) (blue) in MSCs exposed for 24 h to vehicle (D) or AβOs (500 nm) (E). Scale bar, 50 μm. Images were acquired as described above. F, percentage of proliferative cells, expressed as Ki67-positive cells (n = 3 independent cultures, with triplicate coverslips in each experimental condition). G, representative photomicrograph showing lack of DCF fluorescence in MSCs exposed to AβOs (500 nm) for 24 h; the corresponding bright field image is shown in H (n = 6 independent cultures, with triplicate coverslips in each experimental condition). Scale bar, 100 μm. Images were acquired on a Nikon Eclipse TE300 epifluorescence microscope with a ×20 objective. I, quantification of O2 flow (measured by high-resolution respirometry) under basal culture conditions or after the addition of oligomycin or carbonyl cyanide 4-(trifluoromethoxy)phenylhydrazone (FCCP) in MSCs exposed to vehicle (V) or AβOs (500 nm) (A) for 24 or 48 h (n = 3 independent cultures). In all graphs, data are represented as means ± S.E. (error bars).
MSCs protect neurons against oxidative stress induced by AβOs
To investigate the protective potential of MSCs against neuronal oxidative stress induced by AβOs (21–23), we cocultured MSCs with neurons for 24 h and then exposed the cultures to AβOs (500 nm) or vehicle for 6 h or to H2O2 (100 μm) for 10 min. Representative DCF fluorescence images of vehicle-, AβO-, or H2O2-exposed neurons are shown in Fig. 2, A–D, E–H, or I–L, respectively. The large increases in ROS levels induced by AβOs (Fig. 2M) or H2O2 (Fig. 2N) were fully prevented when neurons were cocultured with MSCs. No evidence of increased cellular oxidative stress was detected in MSCs exposed to AβOs (Fig. 2O) or H2O2 (not shown). In line with previous studies (e.g. see Ref. 24), there was no indication of neuronal death under our experimental conditions (500 nm AβOs, 6 h of exposure) either in the absence or in the presence of MSCs, as revealed by inspection of bright field images corresponding to each DCF fluorescence image (Fig. 2).
Figure 2.

Oxidative stress in hippocampal neurons exposed to AβOs in the absence or presence of MSCs. Photomicrographs showing DCF fluorescence (green) in hippocampal neurons exposed to vehicle (A–D), AβOs (500 nm) for 6 h (E–H), or H2O2 (100 μm) for 10 min (I–L) in the absence or presence of MSCs, as indicated. Scale bar, 100 μm. Images were acquired on a Nikon Eclipse TE300 epifluorescence microscope with a ×20 objective. Corresponding bright field images are shown beside each fluorescence image. M–O, quantification of integrated DCF fluorescence intensity normalized by the total number of cells. Panels show integrated fluorescence for AβO-exposed neurons (M), H2O2-exposed neurons (N), or MSCs cocultured with hippocampal neurons and exposed to vehicle or AβOs, compared with hippocampal neurons alone (O). Data are represented as means ± S.E. (error bars) (n = 6 independent cultures, with triplicate coverslips in each experimental condition); *, p < 0.05; two-way ANOVA followed by Tukey's post hoc test; RU, relative units.
MSCs prevent loss of postsynaptic density protein 95 (PSD-95) and synapses in neurons exposed to AβOs
Double immunolabeling for pre- and postsynaptic markers, synaptophysin and PSD-95, respectively, was used to assess the impact of AβOs on synapses and the possible protective action of MSCs. Compared with vehicle-treated cultures, hippocampal neurons exposed to AβOs presented a significant reduction in levels of PSD-95, and this reduction was blocked by coculture with MSCs (Fig. 3, A–E). Levels of the presynaptic marker, synaptophysin, showed a trend, albeit not statistically significant, of reduction in neurons exposed to AβOs, and this trend was also prevented in the presence of MSCs (Fig. 3F). Quantification of co-localized PSD-95 and synaptophysin puncta, a measure of synapse density, revealed that exposure to AβOs reduced the density of synapses (Fig. 3G) proportionally to the decrease in PSD-95 abundance. Significantly, coculture with MSCs prevented AβO-induced synapse loss.
Figure 3.

Levels of pre- and postsynaptic proteins in hippocampal neurons exposed to AβOs. A–D, representative photomicrographs showing double immunolabeling for presynaptic marker synaptophysin (SYP, red) and postsynaptic marker PSD-95 (green) in hippocampal neurons exposed to vehicle or AβOs (500 nm) for 24 h; nuclei are stained by DAPI (blue). SYP/PSD-95 co-localized puncta (a measure of synapse density) appear in yellow. Scale bar, 10 μm. Images were acquired in a Nikon Eclipse TE300 epifluorescence microscope with a ×63 objective. E–G, quantification of synaptic proteins and synapse density. Data are represented as means ± S.E. (error bars) (n = 4 independent cultures, with triplicate coverslips in each experimental condition); *, p < 0.05; two-way ANOVA followed by Dunnett's post hoc test; n.s., not significant.
MSCs internalize AβOs
Next, we investigated whether protection against neuronal oxidative stress and synapse loss could be related to MSC-mediated clearance of AβOs from the culture medium.
Dot-immunoblotting (using oligomer-sensitive antibody NU4) (25) was used to detect AβO immunoreactivity in the culture medium as described previously (e.g. to detect elevated levels of AβOs in AD brain extracts) (26, 27). Results showed a time-dependent reduction in AβO immunoreactivity in the culture medium of MSCs, suggesting that AβOs were internalized (Fig. 4A, top and middle lanes). Control experiments showed no decrease in AβO immunoreactivity in the medium when oligomers were applied onto gelatin-coated coverslips immersed in Dulbecco's modified Eagle's medium (DMEM)/F-12–filled wells in the absence of cultured MSCs (not shown) or when AβOs were incubated under identical conditions with HEK293 cells instead of MSCs (Fig. 4A, bottom lane). All lanes were quantified by densitometry, as shown in the graph (Fig. 4B).
Figure 4.

MSCs internalize Aβ oligomers. A, dot-blotting analysis (using NU4 oligomer-sensitive antibody) (86) shows a time-dependent reduction in AβO immunoreactivity in the culture medium (top and middle lanes, corresponding to 104 or 105 MSCs/well (lane a or b, respectively). No decrease in AβO immunoreactivity was detected in the presence of 105 HEK293 cells/well instead of MSCs (lane c). B, the bar graph shows quantification by densitometry (using ImageJ version 1.38 software); r.u., relative units. C–H, representative images of AβO labeling (AβOs prepared using Hylite Aβ, as described under “Experimental procedures”; red) in MSCs (labeled with calcein; green) exposed to AβOs. C, MSCs exposed to AβOs for 20 min and imaging carried out in AβO-containing medium; D and E, MSCs were exposed to AβOs for 20 min, followed by exchange of AβO-containing medium by fresh medium and imaging after 3 h (D) or 72 h (E). Internalization of AβOs is evidenced by orthogonal projection analysis of confocal images obtained after 72 h (E). F–H, MSCs were continuously exposed to 500 nm AβOs and imaged after 3 h (F), 24 h (G), or 48 h (H). The kinetics of AβO internalization is accelerated by continuous exposure to AβOs (compare D and F). Arrows in F indicate AβO-containing roughly globular cytoplasmic structures in MSCs, suggesting that AβOs are located within an intracellular compartment after 3 h of exposure (F). D–H, lines indicate orthogonal sections of confocal imaged cells. Scale bar, 50 μm. Images were acquired on a Zeiss LSM510 META confocal microscope using a Plan-Neofluar ×40/1.3 numerical aperture oil differential interference contrast objective.
To directly determine whether MSCs internalized AβOs, we used fluorescence microscopy. Fig. 4C shows that fluorescent HyLite AβOs (prepared using HyLite Aβ, as described under “Methods”) were bound to the surface of MSCs after 20 min of exposure and remained bound 3 h after exchange of the medium containing oligomers for fresh medium (Fig. 4D). Internalization of AβOs by MSCs was evidenced by orthogonal section analysis of confocal images obtained after 72 h (Fig. 4E).
Next, we evaluated the dynamics of internalization by continuously exposing MSCs to 500 nm AβOs for different time intervals and removing the oligomer-containing medium immediately before image acquisition. Under these conditions, AβOs were found to be bound to the external surface of the membrane of MSCs after 1 h of exposure (not shown) and were internalized by ∼75% of the cells 3 h later (Fig. 4F). The presence of AβOs in roughly globular cytoplasmic structures (indicated by arrows in the representative image shown in Fig. 4F) suggests that oligomers were contained within an intracellular compartment in MSCs. After 24 h of exposure to AβOs, the proportion of cells presenting intense oligomer labeling decreased to 50%, and oligomer fluorescence signal was absent or extremely weak in the remaining cells (Fig. 4G). The percentage of cells positive for oligomer signal further decreased to 40% after 48 h of exposure to AβOs (Fig. 4H).
AβOs co-localize with endosomal and lysosomal markers in MSCs
To determine how MSCs process internalized AβOs, we investigated whether oligomers co-localized with endosomal (early endosome antigen 1; EEA1) and lysosomal (lysosome-associated membrane protein 1; LAMP-1) markers. Double immunocytochemistry revealed that AβOs co-localize with both EEA1 (Fig. 5, A–D) and LAMP-1 (Fig. 5, E–L). Treatment of MSCs with the lysosomal protease inhibitor, E64d, resulted in significantly increased AβO immunoreactivity, which almost completely co-localized with LAMP-1 labeling, within ∼100% of the cells after 24 h (Fig. 5, J and K).
Figure 5.

Subcellular localization of internalized Aβ. A–D, double immunofluorescence labeling for early endosome marker EEA1 (green) and Aβ (6E10 monoclonal antibody) in MSCs after exposure to AβOs (500 nm) for 3 h. Note the co-localization of AβOs with EEA1 evidenced by orthogonal projection analysis of confocal images (D). Images were acquired in a Zeiss LSM510 META confocal microscope using a Plan-Apochromat ×20/0.8 numerical aperture M27 objective. Scale bar, 100 μm. E–L, triple immunofluorescence labeling for lysosomal marker LAMP-1 (green), Aβ (6E10 monoclonal antibody; red), and cell mask (blue) in MSCs after exposure to AβOs (500 nm) for 24 h in the absence or presence of the lysosomal cysteine protease inhibitor, E64d. Images were acquired in a Zeiss LSM510 META confocal microscope using a Plan-Neofluar ×40/1.3 numerical aperture oil differential interference-contrast objective. Note that the internalization of AβOs and co-localization with LAMP1 are markedly enhanced in the presence of E64d. Scale bar, 20 μm.
MSCs internalize both AβOs and fibrillar amyloid aggregates
In the course of our study, we found that MSCs internalized polystyrene beads, which were encountered within acidic, LysoTracker-positive compartments in ∼66% of the cells after 3 h of incubation. In line with the results described above, AβOs added to the culture medium were internalized by MSCs and accumulated in the same LysoTracker-positive compartments (Fig. 6, A–D), suggesting that the same internalization pathway was utilized by both beads and AβOs. Because the internalization of polystyrene beads showed that MSCs exhibit endocytic capacity for larger particles, we asked whether MSCs would also be able to internalize and degrade larger amyloid aggregates. This was indeed confirmed for Aβ fibrillar aggregates; after 3 h of exposure of cells to fibrillar Aβ (instead of AβOs), thioflavin S–labeled amyloid aggregates were clearly detected within LAMP-1-positive compartments in MSCs (Fig. 6E).
Figure 6.

MSCs internalize amyloid particles of different sizes. A–D, representative images showing internalization of polystyrene beads and AβOs by MSCs and their localization in acidic, LysoTracker-positive compartments. Images were acquired on a Zeiss LSM510 META confocal microscope using a Plan-Neofluar ×63/1.25 numerical aperture oil objective; scale bar, 10 μm. E, representative image (acquired in the LSM510 META confocal microscope using a Plan Apochromat ×100/1.46 numerical aperture oil M27 objective) showing that fibrillar (thioflavin S–positive) Aβ aggregates are observed within lysosomes (LAMP-1–immunolabeling) after 3 h of exposure of MSCs to Aβ fibrils instead of AβOs. Scale bar, 20 μm.
AβOs stimulate selective release of cytokines from MSCs in coculture with hippocampal neurons
To determine whether MSCs might protect neurons from the toxic impact of AβOs via the release of neuroprotective cytokines, we measured a panel of cytokines in the culture medium of MSCs either alone or in coculture with hippocampal neurons, and exposed or not to AβOs. For all cytokines detected in the multiplex assay we used, exposure to AβOs did not modify cytokine levels in the culture medium of MSCs alone, as illustrated for interleukin (IL)-6, IL-10, and vascular endothelial growth factor (VEGF) (Fig. 7 (A–C), white bars). Furthermore, fractalkine, granulocyte macrophage colony-stimulating factor, IL-1α, IL-1β, IL-4, and tumor necrosis factor-α levels in the culture medium of hippocampal neuronal cultures alone or in coculture with MSCs were not modified by exposure to AβOs. Interestingly, however, levels of IL-6, IL-10, and VEGF in the culture medium of hippocampal neuronal cultures were up-regulated by coculture with MSCs, both in the absence and in the presence of AβOs (Fig. 7). In the case of VEGF, exposure to AβOs further increased the levels secreted to the culture medium compared with vehicle-treated MSC/neuronal cocultures (Fig. 7C).
Figure 7.

Secretion of IL-6, IL-10, and VEGF to the culture medium is increased in neuronal/MSC cocultures and mediates neuroprotection against AβO-induced oxidative stress. A–C, levels of IL-6 (A), IL-10 (B), and VEGF (C) in the culture medium are increased when hippocampal neurons are in coculture with MSCs, either in the absence or presence of AβOs. No changes in cytokine levels are detected when MSCs alone are exposed to AβOs (white bars, normalized by control MSCs in the absence of AβOs). D–H, the addition of antibodies against IL-6 (0.5 μg/ml), IL-10 (1 μg/ml), and VEGF (1 μg/ml) to the culture medium blocks the protection by MSCs against AβO-induced neuronal oxidative stress. D and E, representative DCF fluorescence images from hippocampal neuronal cultures exposed to vehicle or 500 nm AβOs, respectively. F and G, representative DCF fluorescence images from hippocampal neurons cocultured with MSCs and incubated with 500 nm AβOs in the absence or presence of anti-cytokine antibodies, respectively. Images were acquired on a Nikon Eclipse TE300 epifluorescence microscope with a ×20 objective. Scale bar, 100 μm. H, integrated DCF fluorescence intensities in different experimental conditions, normalized by the control group (vehicle-exposed hippocampal neurons in the absence of MSCs). Bars, means ± S.E. (error bars) (n = 2 independent cultures, with triplicate wells per experimental condition). *, p < 0.05; #, p < 0.05; one-way ANOVA followed by Tukey's post hoc test.
To establish whether the release of IL-6, IL-10, and VEGF to the culture medium was mechanistically connected to the protection of neurons against oxidative stress induced by AβOs, we performed experiments in which the three cytokines were blocked by neutralizing antibodies added to the cultured medium. To this end, we cocultured MSCs with neurons for 24 h in the presence of neutralizing antibodies against IL-6 (0.5 μg/ml; R&D Systems), IL-10 (1 μg/ml; BD Pharmingen), and VEGF (1 μg/ml; Avastin®, Roche Applied Science) and then exposed the cultures to AβOs (500 nm) or vehicle for 24 h. Representative DCF fluorescence images of neurons exposed to vehicle, AβOs, AβOs + MSCs, or AβOs + MSCs + cytokine-neutralizing antibodies, respectively, are shown in Fig. 7 (D–G). Consistent with the results described above, the large increase in ROS levels induced by AβOs was fully prevented when neurons were cocultured with MSCs (Fig. 7, E and F). However, neurons cocultured with MSCs in the presence of neutralizing antibodies (Fig. 7G) presented ROS levels similar to those of cultures exposed to AβOs alone (Fig. 7H).
MSC-derived extracellular vesicles protect neurons from oxidative stress via delivery of catalase
We further asked whether the release of EVs by MSCs could constitute an additional protective mechanism against neuronal damage induced by AβOs. To investigate this possibility, we first characterized the EV population secreted by MSCs.
Analysis by transmission electron microscopy (TEM) showed different types of extracellular vesicles exhibiting different morphologies and size distributions. Most of the observed vesicles were small rounded vesicles ranging from ∼30 to 200 nm (Fig. 8, A–C), suggesting that these particles represent exosomes. A second/minor vesicular population with size ranging from ∼200 to 900 nm, consistent with microvesicles, was also detected (Fig. 8, D and E). Analysis of the frequency distribution of vesicle diameter in TEM images (n = 104 vesicles) indicated that most of the vesicles ranged between 30 and 200 nm (Fig. 8F).
Figure 8.

Size distribution and immunological characterization of EVs released by MSCs. A–E, MSC-derived EVs were isolated (see “Experimental Procedures”) and mounted on grids for electron microscopy, negatively stained, and imaged by TEM. Smaller vesicles with diameters ranging between 30 and 200 nm (A–C) and larger vesicles with diameters ranging between 400 and 600 (D–E) nm could be visualized. Vesicles indicated by arrows in A are shown in higher magnification in the inset. F, frequency distribution of vesicle diameter derived from TEM analysis (n = 104 vesicles measured). Most (∼65%) of the vesicles ranged between 30 and 200 nm in diameter. G, dot blot analysis showing that EVs are immunoreactive for exosome-associated tetraspanins CD81 and CD63. H, NanoSight NTA. Size distribution of MSC-derived EVs showed predominance of particles with diameters ranging between 50 and 200 nm. Lines in different colors correspond to quintuplicate analyses from the same EV preparation. Scale bars, 200 nm (A, C, and D); 100 nm (A (inset) and B); and 500 nm (E). I, analysis of EV particle size distribution from forward scatter (FSC) and side scatter (SSC) by flow cytometry. J, microvesicles identified as CD90.1-positive particles. K, overlay of fluorescence histograms (normalized by the respective mode values) in a non-labeled sample (background control, gray curve) and a CD90.1-labeled sample (red curve), showing the presence of 10.1% CD90.1-positive microvesicles in the EV population.
Nanoparticle-tracking analysis (NTA; Fig. 8H) revealed that MSCs in culture secrete EVs (4.810 ± 1.39 particles/ml) with two populations of different sizes, with a mean particle diameter of 168.1 ± 5.3 nm and modal particle diameter of 97.8 ± 3.9 nm. The population exhibiting smaller mean diameter (85.7 ± 3.5 nm) corresponds to exosomes (ranging in dimeter from 30 to 200 nm), and the population exhibiting larger mean diameter (288.6 ± 15.6 nm) constitutes microvesicles (ranging in diameter from 100 to 1,000 nm). The distributions of particle diameters obtained from TEM and NTA analyses were in excellent agreement and indicated that EVs secreted by MSCs and isolated under our conditions comprised predominantly exosomes (consistent with dot blot analysis showing that EVs are immunoreactive for exosome-associated tetraspanins, CD81 and CD63, two typical exosome markers; Fig. 8G), with microvesicles corresponding to about 35% of the total population of secreted vesicles.
The EVs released from MSCs were further characterized by flow cytometry using CD90.1 as a marker of MSC-derived microvesicles (28). Based on a total count of 106 particles detected, 10.1% were CD90.1-positive (Fig. 8, I–K), consistent with EVs comprising predominantly exosomes.
An interesting property of EVs is their stability to cryopreservation. Because EVs have little water, they can be freeze/thawed without significant changes in their physical characteristics (29). Accordingly, NTA results did not indicate any change in particle number when comparing fresh and frozen EVs (not shown). This indicates that EVs can be isolated from MSCs and cryopreserved for future use in neuroprotection.
To determine whether EVs protected neurons against AβO-induced oxidative stress, two different doses of MSC-derived EVs (8 × 107 EV particles or 2.4 × 108 EV particles) were incubated with neurons (50,000 per coverslip) previously exposed to AβOs (500 nm) for 2 h at 37 °C. The lower dose of EVs used corresponds to the amount secreted by the MSCs used in coculture with neurons in the experiments described above. Four hours later, determination of neuronal ROS levels demonstrated that EVs completely restored basal levels of ROS, reversing the deleterious increase induced by AβOs (Fig. 9).
Figure 9.

EV-mediated blockade of AβO-induced oxidative stress in hippocampal cell cultures. A and B, representative DCF fluorescence images from hippocampal neuronal cultures exposed to vehicle or 500 nm AβOs, respectively. C and D, representative DCF fluorescence images of hippocampal neurons exposed to 500 nm AβOs in the presence of 8 × 107 or 2.4 × 108 total EV particles, respectively. Images were acquired on a Zeiss Axiovert 200M microscope with a ×10 objective. Scale bar, 50 μm. E, integrated DCF fluorescence intensities (normalized by cell numbers) in different experimental conditions, normalized by the control group (vehicle-exposed hippocampal neurons in the absence of EVs). EV1, 8 × 107 EV particles; EV2, 2.4 × 108 EV particles. Bars, means ± S.E. (error bars) (n = 3 independent cultures, with triplicate wells per experimental condition). *, p < 0.05; one-way ANOVA followed by Tukey's post hoc test; RU, relative units.
We hypothesized that EVs could exert their protective action against neuronal oxidative stress via secretion of antioxidant enzymes (e.g. catalase). Indeed, we found that MSC-derived EVs harbor significant catalase activity (Fig. 10A). Nonetheless, it is well known that EVs may contain a large number of proteins, miRNAs, and metabolites. To determine whether catalase contained in MSC-derived EVs was directly involved in neuroprotection, we treated EVs with the membrane-permeant specific catalase inhibitor, 3-amino-1,2,4-triazole, prior to the addition of EVs to neuronal cultures. Control measurements confirmed that treatment with 3-amino-1,2,4-triazole fully inhibited catalase activity in EVs (Fig. 10, B and C). Significantly, inhibition of catalase activity abrogated the capacity of MSC-derived EVs to protect neurons against AβO-induced oxidative stress (Fig. 10, D–H).
Figure 10.

Active catalase in EVs mediates protection against AβO-induced neuronal oxidative stress. A, substrate concentration dependence of catalase activity in MSC-derived EVs. O2 production upon the addition of H2O2 to the medium was determined in a high-resolution respirometer (see “Experimental Procedures”). The solid line represents a non-linear regression hyperbolic fit to the data using the equation, VO2 = VO2 max × [H2O2]/([H2O2] + K0.5H2O2). B and C, control EVs or aminotriazole-treated EVs, respectively, were added to the respirometer cell, and successive 80 μm pulses of H2O2 were added (arrows). The increase in O2 concentration in the cell was measured following the catalase-catalyzed chemical reaction, 2 H2O2 ⇔ 2 H2O + O2 (B). Note that no O2 production was detected when aminotriazole-treated EVs were employed, consistent with inactivation of catalase; instead, successive additions of H2O2 generated a reduction in O2 concentration, suggesting O2 consumption due to adduct formation upon lipidic peroxidation (C). D and E, representative DCF fluorescence images from hippocampal neuronal cultures exposed to vehicle or 500 nm AβOs, respectively. F and G, representative DCF fluorescence images of hippocampal neurons exposed to 500 nm AβOs in the presence of control EVs or aminotriazole-treated (inactivated catalase) EVs, respectively. Images were acquired on a Nikon Eclipse TE300 epifluorescence microscope with a ×20 objective. Scale bar, 100 μm. H, integrated DCF fluorescence intensities in different experimental conditions, normalized by vehicle (Veh)-exposed cultures. iEVs, aminotriazole-treated EVs (i.e. containing inactivated catalase). Bars correspond to means ± S.E. (error bars) (n = 3 independent cultures, with triplicate wells per experimental condition). *, p < 0.05; one-way ANOVA followed by Dunnett's post hoc test.
MSC-derived EVs prevent loss of synapses in neurons exposed to AβOs
Finally, we investigated whether MSC-released EVs could protect neurons against synapse damage induced by AβOs. MSC-derived EVs (2.4 × 108 particles) were incubated for 22 h with neurons (50,000 per coverslip) previously exposed to AβOs (500 nm) for 2 h at 37 °C, and cells were fixed for evaluation of synapse integrity (revealed by co-localization of synaptophysin and PSD-95–immunoreactive punctae). As shown in Fig. 11, EVs protected synapses from damage induced by AβOs.
Figure 11.
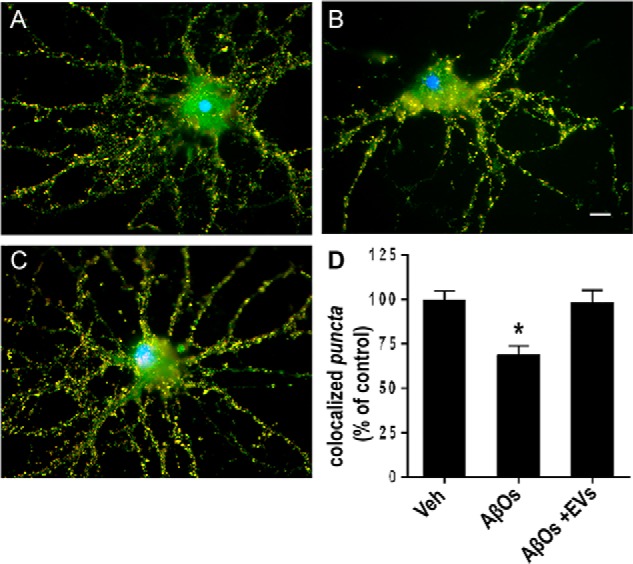
MSC-derived EVs protect hippocampal neurons from synapse damage induced by AβOs. Representative double immunolabeling images for synaptophysin (red) and PSD-95 (green) in cultured hippocampal neurons exposed to vehicle (A), 500 nm AβOs for 24 h (B), or 500 nm AβOs plus EVs (2.4 × 108 particles) (C). EVs were incubated with neuronal cultures for 22 h at 37 °C following initial exposure to AβOs (500 nm) for 2 h. Nuclei are shown in blue (DAPI) and co-localization of pre- and postsynaptic markers is shown in yellow. Images were acquired on a Zeiss Axiovert 200M microscope with a ×63/1.25 numerical aperture oil objective. Scale bar, 10 μm. D, synapse density determined as the number of co-localized synaptophysin/PSD-95 punctae, normalized to vehicle (Veh)-exposed cultures. Bars, means ± S.E. (error bars) (n = 2 independent cultures, with triplicate coverslips per experimental condition). *, p < 0.05; one-way ANOVA followed by Dunnett's post hoc test.
Discussion
Several therapeutic strategies are under investigation to prevent or delay the neurodegenerative process that occurs in AD. Considerable evidence supports a central role of AβOs in triggering harmful events that initiate neuronal oxidative stress and synapse damage leading to cognitive decline in AD (5, 30). The therapeutic potential of MSCs from diverse sources has been investigated with promising results in AD models (31–39). However, the mechanisms by which neuroprotection by MSCs occurs have not been fully elucidated.
We have investigated the neuroprotective potential of MSCs against damage induced by AβOs in hippocampal neurons and possible mechanisms underlying neuroprotection. Results showed that MSCs are resistant to the impact of AβOs in all tested parameters (viability, proliferation, ROS generation, and mitochondrial function), a favorable aspect for possible future use of MSCs as a therapeutic adjuvant or alternative in AD.
We found that MSCs and MSC-derived EVs protect hippocampal neurons against oxidative stress and synapse damage induced by AβOs. In addition, we report mechanisms that may explain, at least in part, neuroprotection by MSCs: 1) their ability to internalize and degrade AβOs; 2) the release of EVs containing the antioxidant enzyme, catalase; and 3) selective secretion of anti-inflammatory and/or trophic cytokines to the medium.
Our findings support a previous report (40) showing that the resistance of human MSCs to death induced by hydrogen peroxide is related to constitutive expression of antioxidant enzymes. In line with this notion, MSCs not only prevented ROS increase induced by AβOs but also reduced basal ROS levels in neurons (Fig. 2). It is possible that exposure to soluble factors secreted by MSCs in the culture medium up-regulates the expression of antioxidant enzymes (e.g. catalase or glutathione peroxidase) in neurons. Alternatively, it is also conceivable that MSC-derived EVs directly supply active catalase to neurons, as suggested by the current findings.
Synapse dysfunction is a central aspect in the pathophysiology of AD, and loss of synapses is directly related to cognitive decline (41). We found that MSCs blocked the reduction in PSD-95 levels and loss of synapses induced by AβOs. Importantly, MSC-derived EVs showed a similar neuroprotective effect on synapse loss. Normal ROS levels are crucial for long-term potentiation and for synaptic strengthening (42). However, at high levels, ROS become detrimental to synapses. Previous studies have shown that AβOs readily promote neuronal oxidative stress (21–23) and inhibit long-term potentiation (43, 44). It is thus possible that protection against neuronal oxidative stress promoted by MSCs and by MSC-derived EVs contributes to the preservation of synapse integrity in AβO-exposed neurons.
We found a progressive and significant reduction in AβO immunoreactivity in the culture medium when MSCs were present (Fig. 4). This suggests that reduced extracellular availability of AβOs can be related to neuroprotection promoted by MSCs. Future identification of binding sites of AβOs in MSCs may enhance application of the endocytic capacity of these cells as a therapeutic tool. For example, cells with higher endocytic capacity could be selected and expanded before transplantation. Previous studies suggest that the binding of AβOs to neurons occurs via a multiprotein receptor complex that probably involves the N-methyl-d-aspartate receptor (21–23), the cellular prion protein (45, 46), the Wnt co-receptor Frizzled (47), the B2 ephrin receptor (48), and the metabotropic glutamate receptor mGluR5 (49, 50). On the other hand, recent evidence indicates that phagocytosis of Aβ by astrocytes is mediated by CD36, CD47, and RAGE receptors (51). Finally, the binding of Aβ to microglia appears to involve Toll-like receptors 2 and 4, CD36, and RAGE (52, 53), and recently it was found that Scara1 is a specific receptor for AβOs in these cells (54). It is, thus, possible that one or more of the above proteins mediates the interaction of AβOs with MSCs.
Our results demonstrate that the endocytic capacity of MSCs is not selective for the size of the aggregates, as MSCs internalize both Aβ oligomers (which typically are a few nanometers in diameter) and much larger Aβ fibrils, as shown by colocalization with EEA1, LysoTracker, and LAMP-1 markers. Analysis of the internalization dynamics of fluorescently labeled AβOs in live cells indicated that ∼75% of MSCs showed internalized Aβ after 3 h, but only 50% of the cells presented oligomer signal after 24 h, decreasing to ∼40% after 48 h of exposure to AβOs (Fig. 4, F, G, and H, respectively). These data suggest that the dynamics or ability to incorporate/clear AβOs differs substantially between cells in the MSC population, with some cells rapidly clearing internalized Aβ, whereas other cells are much slower in doing so. Importantly, in E64d-treated cultures (in which lysosomal degradation was inhibited), almost 100% of cells presented AβOs signals that extensively co-localized with lysosomes (Fig. 5K). Our findings thus indicate that the process of internalization of AβOs is highly dynamic and varies among MSCs cells and that blocking lysosomal degradation using E64d blocks clearance of internalized Aβ. Rapid internalization and degradation by MSCs of both oligomers of a few nanometers in diameter and large Aβ fibrils, leading to a decrease in extracellular levels of AβOs, constitutes a potentially relevant mechanism of protection for therapeutic interventions, because brain accumulation of Aβ in the form of insoluble fibrils and oligomers probably occurs years or even decades before the appearance of clinical symptoms (55, 56). Consequently, the endocytic capacity of MSCs could be therapeutically useful even in the preclinical stage of AD.
Although previous studies have shown the ability of MSCs to reduce amyloid plaques or extracellular levels of Aβ in vivo, the mechanisms revealed in the current study are different from those described in previous reports, which include activation of resident microglia (12–14, 57), increased autophagy in neurons (36), and release of EVs containing active neprilysin (39). Furthermore, given the increasingly recognized role of soluble AβOs in early synapse damage and memory loss in AD, we feel the current results showing protection against the deleterious impact of AβOs represent a contribution to our understanding of the mechanisms by which MSCs may protect neurons from damage in AD.
Several studies have focused on the use of MSC-derived EVs as therapeutic tools, because they have been shown to promote beneficial effects similar to those promoted by their parental cells. Recent reports have shown protection by bone marrow MSC-derived exosomes in different pathological conditions (i.e. by enhancing angiogenesis and neurogenesis after traumatic brain injury or stroke) (59, 60), promoting neurite outgrowth and neural plasticity (61, 62), modulating the immune system (63–65), suppressing fibrosis (66, 67), and preventing apoptosis (68–70). EVs further act as carriers of bioactive molecules, including proteins, lipids, and different types of RNA (e.g. mRNA and miRNAs).
Considering that the molecular signature of EVs differs among different originating cell types (71), and their biological activities depend on the content of molecules they carry, investigation of the complex molecular repertoire of EVs will be crucial for determination of their therapeutic efficacy. For example, it has been reported that MSC-derived EVs from adipose tissue contain enzymatically active neprilysin, an Aβ-degrading enzyme, and are capable of reducing extracellular and intracellular Aβ levels in Neuro-2a cells overexpressing amyloid precursor protein (39). Of note, Kim et al. (72) showed that expression and secretion of galectin-3 (GAL-3) by umbilical cord blood-derived mesenchymal stem cells were up-regulated by Aβ, and coculture with those cells reduced cell death in a GAL3-dependent manner in Aβ42-exposed neurons and SH-SY5Y cells. Another interesting report showed that intracerebrally injected exosomes trapped Aβ on surface glycosphingolipids and transported it into microglia in AD mouse brains, resulting in reduced Aβ pathology (73).
Our results showed that MSC-derived EVs exhibit a neuroprotective effect against AβO-induced oxidative stress similar to that of the originating MSCs and consist of a mixed population of exosomes/microvesicles. Exosome-based CNS delivery of catalase was reported in a recent study in which catalase loaded ex vivo into macrophage-derived exosomes efficiently decreased oxidative stress and increased neuronal survival in in vitro and in vivo models of Parkinson's disease (74). Here, we demonstrate that EVs secreted by MSCs naturally contain and carry endogenous catalase. Another interesting report (75) showed that a single administration of microvesicles derived from human Wharton Jelly mesenchymal stromal cells protects against oxidative stress induced by renal ischemia/reperfusion injury. To our knowledge, the present work is the first to describe the presence of active catalase in MSC-derived EVs and its important role as an antioxidant agent in the AD model. We propose that MSC-derived EVs mediate paracrine mechanisms of neuroprotection against oxidative stress and may represent a novel therapeutic alternative to combat oxidative damage and synapse damage in AD and other neurodegenerative diseases.
Several lines of evidence have shown that inflammation plays a crucial role in AD (e.g. see Refs. 76 and 77). On the other hand, it is known that the release of cytokines and soluble factors is a key component in the paracrine actions of MSCs. Of interest, it was recently described (31) that brain-derived neurotrophic factor–expressing MSCs attenuate the decrease in synaptophysin levels and the presence of aggregation or clustered cell bodies, a sign of degeneration, in neuronal cultures from the 5xFAD mouse model of AD. We found a selective increase in extracellular levels of anti-inflammatory and/or trophic cytokines (IL-6, IL-10, and VEGF) in MSC/neuronal cocultures, even after exposure to AβOs. Interestingly, this effect appears to be mainly related to the cross-talk between MSCs and neurons, because we did not find significant effects of AβOs on IL-6 and IL-10 levels in isolated neuronal or MSC cell cultures. Nonetheless, AβOs potentiated the secretion of VEGF in cocultures compared with levels measured in vehicle-treated cultures. Importantly, we found that blockade of IL-6, IL-10, and VEGF in the culture medium by specific antibodies abrogated the protective action of MSCs against AβO-induced oxidative stress. This indicates that these cytokines play an important role in neuroprotection by MSCs against AβO-induced neuronal damage.
It remains to be determined whether the cytokines indicated above are released only by MSCs or by both neurons and MSCs. Interleukin-6 is a major cytokine in the CNS and can exert opposite actions on neurons in a wide array of pathological conditions, either triggering neuronal survival or causing neuronal degeneration and cell death (78). Although mostly regarded as a pro-inflammatory cytokine, IL-6 also has many regenerative or anti-inflammatory activities, exhibiting a neurotrophin-like behavior (76). IL-6 expression up-regulates glial phagocytic markers in vivo and enhances microglia-mediated phagocytosis of Aβ aggregates in vitro (77). Thus, it is possible that the increase in IL-6 levels in our MSC/neuronal cocultures exerts positive effects in neuroprotection and Aβ clearance.
The association of interleukin-10 polymorphisms with risk of Alzheimer's disease was recently reported (79). It was also reported that IL-10 production in response to amyloid-β differs between AD patients with slow and fast progression of cognitive decline, based on the observation that Aβ-stimulated peripheral blood mononuclear cells from slow decliners present higher IL-10 levels compared with controls, whereas in fast decliners, IL-10 production was abolished (80). These findings suggest that the increase in IL-10 levels found in MSC/neuronal cocultures may represent an important component of the neuroprotective action of MSCs, by neutralizing the cytotoxic inflammatory process induced by AβOs and down-regulating the synthesis of pro-inflammatory cytokines, as proposed previously (81).
There is evidence that the blood–brain barrier may be compromised in AD (82, 83), which might permit peripherally infused MSCs to enter the CNS and establish direct contact with neurons. Further, our group and others have shown that MSCs are able to cross the blood–brain barrier and migrate to lesion areas in the brain (84, 85). Nonetheless, even if MSCs circulating in the brain bloodstream did not come into direct contact with neurons, we feel our in vitro results support the notion that the molecular cross-talk between cells could still occur (as observed through the Millicell membrane in our experiments).
In summary, our findings suggest that MSCs may represent a promising alternative for future therapeutic interventions, especially during the initial phase of AD, by releasing EVs, trophic factors, and cytokines and, importantly, by promoting Aβ clearance, thus preventing harmful events triggered by AβOs. Results further provide insight into possible neuroprotective mechanisms of action that could be activated and/or enhanced in MSCs, enabling the implementation of new therapeutic approaches for neurodegenerative diseases.
Experimental procedures
Ethical considerations
All procedures were approved by and followed the guidelines of the institutional animal care and utilization committee of the Federal University of Rio de Janeiro (Protocol IBCCF 076).
Materials
The sources of antibodies, culture media and supplements, amyloid peptide, antibiotics, probes, transwell system, enzymes, inhibitors, instruments, and software are mentioned below. Salts, buffers, Me2SO, paraformaldehyde, and H2O2 were of the highest purity available.
MSC cultures
Bone marrow cells were obtained from tibias and femurs from male Wistar rats (250–300 g). After removal of the epiphyses, the diaphyses were inserted into 1-ml polypropylene tips and immediately placed in 15-ml tubes. After centrifugation at 300 × g for 1 min, the bone marrow–containing pellets were suspended and dissociated in DMEM/F-12 (Invitrogen) supplemented with 10% fetal bovine serum (Invitrogen), 100 units/ml penicillin (Sigma-Aldrich), and 100 μg/ml streptomycin (Sigma-Aldrich). The suspensions were plated in 75-cm2 flasks, supplemented with the same medium, and maintained in a 5% CO2 atmosphere at 37 °C. After 24 h, non-adherent cells were removed by washing with PBS, and the medium was changed every 2–3 days. Cells were grown until ∼90% confluence, trypsinized (0.25% trypsin plus 1 mm EDTA; Invitrogen), and plated again at a density of 7 × 103 cells/cm2. After 3–4 passages, the cultures were highly enriched with MSCs, as revealed by flow cytometry and tissue differentiation (86).
Neuronal cultures
Hippocampi from 18-day-old rat embryos were dissected and cultured as described previously (25, 87) with minor modifications. Briefly, cells were plated on glass coverslips previously coated with 0.1 mg/ml poly-l-lysine and cultured in Neurobasal medium supplemented with B27 at 37 °C in a humidified 5% CO2 atmosphere for 18–21 days before use.
Cocultures of hippocampal neurons and MSCs
Cocultures were established in two different ways, depending on the type of experiment. In both conditions, intercellular communication between neurons and MSCs occurred via factors released into the culture medium shared by both cell types, without direct physical contact between cells. For oxidative stress assays, neurons and MSCs were plated on different glass coverslips, which were placed side by side inside a Petri dish during coculture. For analysis of synaptic integrity, the MSCs were plated onto the porous membranes (12-mm diameter, 1-μm pore size) of a transwell system (Millicell®, product code PIRP 12R 48, Millipore). The membranes were then transferred to the chamber in which neurons had previously been layered. In both coculture systems, MSCs were plated at a ratio of ∼1:10 with respect to neurons. After 24 h of coculture, a freshly prepared solution of AβOs (500 nm final concentration) or vehicle was added to the culture medium for 6 h (for oxidative stress measurements) or 24 h (for analysis of synaptic integrity).
MSC viability assay
The viability of MSCs was assessed using the Live/Dead kit (Life Technologies, Inc.). Live cells were identified by green calcein fluorescence, and dead cells were identified by red propidium iodide fluorescence. The percentage of live MSCs was expressed relative to the total number of cells in each well. Twelve images were analyzed per experimental condition (carried out in triplicate wells in each of three independent experiments using different MSCs cultures) on a Zeiss Axiovert 200M microscope.
MSC proliferation analysis
MSCs were plated onto glass coverslips (13-mm diameter) in 24-well plates (3 × 104 cells/well) containing 500 μl of supplemented DMEM/F-12. After 24 h, the medium was replaced by fresh medium, and cells were incubated with AβOs (500 nm) or vehicle for 24 h. After fixation with 4% paraformaldehyde (w/v) for 15 min, cultures were permeabilized with 0.1% (v/v) Triton X-100 for 5 min at room temperature, and nonspecific sites were blocked with 10% goat serum albumin (v/v) for 1 h. Immunoreaction was carried out overnight with rabbit anti-Ki67 primary antibody (1:100; Abcam) followed by washing with PBS and incubation with Cy3-conjugated secondary antibody (goat anti-rabbit IgG; 1:400; Jackson ImmunoResearch) for 2 h at room temperature. Nuclei were counterstained with DAPI. Fifteen different fields were imaged per coverslip (3–4 coverslips/experimental condition) in three independent experiments using different MSC cultures. Ki67 is a nuclear protein expressed in all phases of the cell cycle, with the exception of G0 and the initial period of G1 (88). Thus, quantification of the number of Ki67 positive (Ki67+) cells allowed determination of the percentage of proliferative cells.
Respiration measurements
MSCs were plated on adherent plastic dishes (Petri dishes, 96 × 21 mm) containing 10 ml of supplemented DMEM/F-12 (106 cells/dish). After 24 h, the medium was completely replaced by fresh medium, and the cells were incubated with AβOs (500 nm) or vehicle for 24 h. Cells were then trypsinized (0.25% trypsin plus 1 mm EDTA; Invitrogen), quantified, and centrifuged at 300 × g for 5 min. MSC-containing pellets (corresponding to 2 × 106 cells/experimental group) were dissociated in 200 μl of supplemented DMEM/F-12 and added to 1.8 ml of the same solution (at 37 °C) contained in the chamber of a high-resolution respirometer (Oroboros Oxygraph-O2k, Oroboros Instruments). Measurement of O2 consumption (expressed in pmol of O2/s/106 cells) was carried out in basal conditions, after the addition of oligomycin (2 μg/ml; Sigma-Aldrich) to inhibit the F0F1-ATP synthase or after successive additions of 1 μm carbonyl cyanide 4-(trifluoromethoxy)phenylhydrazone (Sigma-Aldrich) to uncouple mitochondrial respiration.
Formation of ROS
ROS formation was evaluated in live hippocampal neurons and MSCs using, respectively, 2 or 4 μm CM-H2DCFDA (Life Technologies) as described previously (21). Probe fluorescence was analyzed using ImageJ software (National Institutes of Health) (89) as described (21). Twelve images were analyzed per experimental condition in each of triplicate wells in six independent experiments using different cocultures and AβO preparations and were combined to allow quantitative estimates of changes in ROS levels. Assays using EVs secreted by MSCs and hippocampal neurons were performed in three independent cultures under identical experimental conditions.
Isolation of EVs secreted by MSCs
MSCs were cultured for 24 h in DMEM/F-12 supplemented with 100 units/ml penicillin and 100 μg/ml streptomycin, without fetal bovine serum. EV isolation was performed as described previously (90) with minor modifications. The supernatants were collected and centrifuged at 2,000 × g for 20 min to remove cellular debris and, subsequently, at 100,000 × g for 2 h at 4 °C (Optima L-90K ultracentrifuge; Beckman Coulter). The pellet containing EVs was resuspended in PBS and stored at −80 °C. Nanoparticle tracking analysis using NanoSight LM10 (Malvern) was performed to determine the size and number of EVs. Each MSC secreted an average of 16,000 EVs, and the number of EVs used in cocultures with neurons was ∼8 × 107, corresponding to the total number of EVs released by ∼5,000 MSCs. This proportion of EVs to neurons corresponded to the original number of MSCs plated for coculture with ∼50,000 neural cells/well. In some experiments, a 3-fold higher EV dose (2.4 × 108 EVs) was employed.
TEM and morphometric analysis of MSC-derived EVs
Isolated EVs were deposited on glow-discharged, Formvar-coated copper grids, fixed with 4% paraformaldehyde (w/v), and stained with a mixture of 9 parts 2% methyl cellulose and 1 part 4% uranyl acetate mixed just before use. Samples were imaged on a Tecnai Spirit electron microscope (FEI Co., Eindhoven, The Netherlands) operating at 120 kV and coupled to a “2,000×2,000”-pixel CCD camera. Vesicle diameter measurements were made using ImageJ software, and data were plotted on a distribution graph using GraphPad Prism software.
Dot-immunoblotting for exosomal markers
The presence of exosomes in the EV population secreted by MSCs was confirmed by dot-immunoblotting for exosomal markers (91). EVs (20 μg of protein) were homogenized in 200 μl of radioimmune precipitation assay buffer with protease inhibitors. Samples were immobilized on nitrocellulose membranes, and the membranes were blocked in 3% BSA in TBS-T. After three successive 10-min washes with TBS-T, the membranes were incubated overnight with anti-CD81 (sc-7637, Santa Cruz Biotechnology) or anti-CD63 (sc-5275; Santa Cruz Biotechnology) monoclonal antibodies (1:100 dilution in blocking solution). After three successive 10-min washes with TBS-T, the membranes were incubated with fluorescent anti-mouse secondary antibody (LI-COR) diluted in blocking solution (1:10,000). Membranes were washed three times for 10 min with TBS-T, and immunoreactivity (fluorescence intensity) was detected in an Odyssey imaging system.
Flow cytometry analysis of EVs
To characterize the microvesicles present in the EV population secreted by MSCs, 25 μg of EVs were incubated with blocking solution (PBS containing 0.5% bovine serum albumin) for 30 min at 4 °C and labeled with phycoerythrin-conjugated anti-CD90.1 (BD Pharmingen, catalog no. 551401; 1:50 dilution) for 30 min in the dark at 4 °C. The EVs were centrifuged at 100,000 × g for 60 min and resuspended in 200 μl of PBS for data acquisition. Unlabeled EVs were used as a background (negative) control. Flow cytometry analysis was performed on an Accuri C6 flow cytometer (BD Biosciences). Appropriate FSC-H and fluorescence thresholds were selected to improve the identification of small particles and decrease noise events. For detection, we used a 585/40 band pass filter to capture the CD90.1-phycoerythrin fluorescence signal. Samples were loaded and run at a stable rate, and 106 events were recorded at a low flow rate (100 events/s). Data were acquired using BD Accuri software C6 (BD Biosciences) and analyzed with FlowJo version 10.1 (TreeStar).
Catalase detection
In this experiment, the protein content of EVs was quantified using the Pierce BCATM protein assay kit (Thermo Fisher Scientific). One mg of EV protein was suspended in PBS in the chamber of the high-resolution respirometer described above. After temperature equilibration (37 °C), the suspension received successive H2O2 pulses (corresponding to 80, 200 (2 pulses), and 400 μm peroxide), and O2 production resulting from catalase-mediated breakdown of H2O2 was quantified using DataLab version 4 software coupled to the respirometer. The rate of catalase activity (dO2/dt, in pmol of O2 × (mg of EV protein)−1 × min−1) was calculated from the linear phase of O2 formation after each pulse of H2O2. To confirm the role of catalase in O2 production, 1 mm KCN (an inhibitor of the enzyme) was added.
To confirm that protection by EVs against AβO-induced neuronal oxidative stress was related to the presence of active catalase within the vesicles, we incubated EVs with 1 mm 3-amino-1,2,4-triazole, a specific and irreversible catalase inhibitor (92), at 37 °C for 30 min. After treatment, EVs were washed two times with PBS and centrifuged (100,000 × g for 2 h at 4 °C) before the addition to neuronal cultures (see “Results”).
Preparation of AβOs
AβOs were prepared as described previously (21). Briefly, the peptide was dissolved to 1 mm in hexafluoro-2-propanol and stored in aliquots as a dried film at −80 °C after solvent evaporation. The film was resuspended in Me2SO to a final concentration of 5 mm. The solution was diluted to 100 μm in ice-cold PBS and left at 4 °C overnight. The preparation was centrifuged at 14,000 × g for 10 min at 4 °C to remove any insoluble aggregates, and the supernatant containing soluble AβOs was transferred to clean tubes and stored at 4 °C. Protein concentration was determined using the BCA assay (Pierce). Oligomer solutions were used within 48 h of preparation. Routine characterization of oligomer preparations was performed by size-exclusion chromatography and, occasionally, by Western blotting using anti-oligomer NU4 antibody (93). Preparations consistently comprised a mixture of soluble oligomeric species, including dimers, trimers, tetramers, and higher molecular mass oligomers of 50–180 kDa, ranging in diameter from 1.5 to 3.5 nm.
Fluorescently labeled AβOs
Fluorescently labeled AβOs were prepared by combining HiLyte Fluor 647-labeled Aβ(1–42) (Anaspec) and Aβ(1–42) at a 1:4 molar ratio. Preparations were characterized by HPLC size exclusion chromatography using a GPC-100 column (Eprogen) and by Western immunoblotting using oligomer-sensitive NU4 antibody (93). Protein concentration was determined with the BCA kit (Pierce). The fluorescently labeled AβOs were used at 300 or 500 nm (expressed as Aβ monomer concentration).
Aβ amyloid fibril formation
Synthetic Aβ(1–42) was freshly dissolved from the lyophilized powder in a 50% (v/v) solution of trifluoroethanol in PBS. Aβ fibrils were prepared by dilution of small aliquots from the stock solution into PBS (resulting in ≤0.5% residual trifluoroethanol) to a final concentration of 100 μm Aβ, exactly as described previously (94). Amyloid fibrils formed using this protocol have been previously characterized by staining samples with 1% uranyl acetate and examination on a Jeol 1200-EX transmission electron microscope (94).
For internalization assays (see below), 5 μl of the fibrillar suspension (obtained from an original 100 μm Aβ solution) were resuspended in 500 μl of DMEM/F-12 under intense stirring.
Treatment of hippocampal cells and MSCs with AβOs
Hippocampal neuronal cultures or MSC cultures were exposed to vehicle (2% (v/v) Me2SO in PBS) or freshly prepared AβOs (300 or 500 nm final concentration, as indicated under “Results”) and were further incubated at 37 °C for different times (ranging between 5 min and 72 h, as indicated).
Aβ internalization by MSCs
To investigate AβO internalization by MSCs, cultures were exposed to fluorescently labeled AβOs (HyLite AβOs). We first investigated whether AβOs were able to bind to the surface of living MSCs and then investigated the time course of their removal from the culture medium and their fate within cells.
In the first series of experiments, MSCs (3 × 104 cells) were plated on coverslips deposited inside Petri dishes suitable for video microscopy containing supplemented DMEM/F-12. After 24 h, the medium was removed and replaced by a fresh solution of calcein (Life Technologies) in DMEM/F-12 (1:2,000 dilution), and after 6 min at 37 °C, the cells were washed three times with PBS before incubation with HyLite AβOs (300 nm) in DMEM/F-12 for 20 min. After replacing the medium by AβO-free medium, the MSCs were maintained at 37 °C, and HyLite AβOs fluorescence was followed for 72 h by confocal microscopy (LSM510 META).
In the second series of experiments, MSCs were treated with calcein as above, continuously exposed to HyLite AβOs (500 nm), and imaged at various time intervals for 48 h. The AβOs-containing medium was removed just before image acquisition. One hundred cells were analyzed in random fields to minimize damage to cells by the laser beam during the time course analyses.
Aiming to determine the intracellular localization of AβOs in MSCs, double labeling experiments for subcellular markers and internalized Aβ were performed. We first investigated whether internalized AβOs were found inside early endosomes. Cells were incubated with AβOs (500 nm) for 3 h and fixed with 4% (w/v) paraformaldehyde for 10 min. Next, cells were probed for EEA1 and Aβ using rabbit anti-EEA1 (Santa Cruz Biotechnology; 1:100 dilution) and mouse anti-β-amyloid 1–16 (6E10) (Covance; 1:100), respectively. Secondary antibodies were goat anti-rabbit Alexa 488 (Life Technologies; 1:2,000) and goat anti-mouse Alexa 555 (Life Technologies; 1:2,000). Images were acquired using 3–4 coverslips with MSCs obtained from different cultures.
To assess whether internalized AβOs were located inside lysosomes, cells were treated as above with 500 nm E64d, a cysteine protease inhibitor (Sigma-Aldrich), for 5 min before the addition of 500 nm AβOs. After 24 h, cells were labeled with CellMaskTM deep red plasma membrane stain (Life Technologies; 1:1,500 dilution of the 5 mg/ml stock solution) for 10 min at 37 °C and fixed with 4% (w/v) paraformaldehyde for 10 min. Next, cells were probed for LAMP-1 and Aβ using rabbit anti-LAMP-1 (Abcam; 1:100 dilution) and 6E10 antibodies, respectively. Secondary antibodies were goat anti-rabbit Alexa 488 (Life Technologies; 1:2,000) and goat anti-mouse Alexa 555 (Life Technologies; 1:2,000). Images were acquired using 3–4 coverslips with MSCs obtained from different cultures.
To investigate whether the endocytic capacity of MSCs was restricted to small Aβ oligomers or could include larger particles, MSCs were processed as above and incubated for 1 h at 37 °C in supplemented DMEM/F-12 containing 50 nm LysoTracker® (Invitrogen) (a marker of acidic compartments). The medium was then replaced by medium containing fluorescent polystyrene beads (500-nm diameter; Sigma-Aldrich; 1:200 dilution of the 10% (solids) stock suspension in DMEM/F-12). Three hours later, the medium was replaced by fresh supplemented DMEM/F-12 containing 500 nm AβOs, incubated at 37 °C for 24 h, and fixed with 4% (w/v) paraformaldehyde before probing for β-amyloid peptide as above.
In yet another experimental approach, MSCs were submitted to the initial general process above, labeled with CellMaskTM deep red plasma membrane stain (Life Technologies; 1:1,500 dilution of the 5 mg/ml stock solution) for 10 min at 37 °C, washed with PBS, and incubated for 3 h with 500 μl of supplemented DMEM/F-12 containing 1 μm Aβ fibrils (see above) labeled with thioflavin S. Cells were fixed and probed for LAMP-1 using anti-rabbit Alexa 555 as secondary antibody. Confocal images (Zeiss LSM510 META) were acquired using 10 coverslips from three different MSC cultures.
Dot-immunoblotting for AβO immunoreactivity in the conditioned medium
To assess the amount of AβOs present in the culture medium of MSCs after different exposure times, we used a dot-immunoblotting technique. MSCs were plated at two densities (104 or 105 cells/well) in 24-well plates containing 500 μl of supplemented DMEM. After 24 h, the medium was replaced, and cells were incubated with AβOs (500 nm) or vehicle for different times. The medium was removed and homogenized, and 3-μl aliquots were spotted onto Hybond ECL nitrocellulose membranes (Amersham Biosciences) and stained with Ponceau red (0.1% (w/v) Ponceau S in 5% (v/v) acetic acid) to verify the efficiency of protein adsorption. Membranes were washed with Tris-buffered saline supplemented with 0.1% (v/v) Tween 20 (TBS-T), and nonspecific sites were blocked with 3% (w/v) skimmed milk solution or 3% (w/v) BSA under gentle stirring for 1 h at room temperature, incubated overnight at 4 °C with anti-AβOs mouse monoclonal antibody (NU4; 1 μg/ml diluted 1:2,000 in blocking solution; a kind gift from Dr. William L. Klein) (93). After three successive 10-min washes with TBS-T, the membranes were incubated for 2 h at room temperature with secondary antibody coupled to horseradish peroxidase (Invitrogen), diluted in blocking solution (1:10,000). Finally, the membranes were washed three times for 10 min with TBS-T, developed with SuperSignal West Femto maximum sensitivity substrate (Pierce) (1:1 in TBS-T), and exposed on Kodak films, which were scanned at high resolution (600 dpi) and quantified by densitometry using Image J version 1.38 software. As a control, HEK293 cells were plated in 24-well plates (105 cells/well) and incubated with AβOs (500 nm) for different times (see “Results”).
Cytokine level analysis in neurons and MSCs cocultures
Hippocampal neurons were plated (106 cells/well) and cultured as described previously. The same amount of neuronal cells was cocultured with MSCs (105) in a transwell system for 24 h, as described above. Cultures without MSCs were used as controls. After this period, AβOs (500 nm final concentration or an equivalent volume of vehicle) were added to the neuronal cultures alone or to neuronal/MSC cocultures, and cultures were maintained under identical conditions for an additional period of 24 h. Culture media from each experimental group (wells in triplicate) were pooled and used in a multiplex system (Luminex® 100/200TM) for simultaneous detection of cytokines. The RECTTMAZ-65K kit (Merck Millipore) was used for the detection of nine cytokines: IL-1α, IL-1β, IL-4, IL-6, IL-10, chemokine fractalkine, tumor necrosis factor-α, VEGF, and granulocyte macrophage colony-stimulating factor.
Immunocytochemistry and analysis of synapse density
To evaluate synapse integrity, we double-immunolabeled cultures for PSD-95 and presynaptic protein synaptophysin. After fixation with 4% (w/v) paraformaldehyde for 15 min, cultures were permeabilized with 0.1% (v/v) Triton X-100 for 5 min at room temperature, and nonspecific sites were blocked with 10% (v/v) goat serum albumin (Sigma-Aldrich) for 1 h before immunoreactions. Primary antibodies used were rabbit anti-PSD-95 (1:1,000; Cell Signaling Technology) and mouse anti-synaptophysin (1:1,000; Chemicon International). After overnight incubation with primary antibodies, cells were washed with PBS and incubated with secondary antibodies (Alexa Fluor 555–conjugated goat anti-mouse IgG (1:2,000) and Alexa Fluor 488–conjugated goat anti-rabbit IgG (1:2,000); Invitrogen) for 2 h at room temperature. Nuclei were counterstained with DAPI (Sigma-Aldrich). To analyze synapse density, neurons separated by a distance equivalent to at least two cell body diameters were selected. After immunostaining, cells were imaged on a Zeiss Axiovert 200M microscope equipped with an Apotome slider. Co-localization of synaptic proteins was analyzed as described under “Puncta analysis.”
Puncta analysis
Neurons located at least two cell body diameters away from neighboring neurons were randomly selected. Green (PSD-95) and red (synaptophysin) channel images were acquired, aligned, and quantified following digital cell body removal using the Puncta Analyzer plug-in in ImageJ, as described (58). In each experiment, at least 50 neurons were analyzed per experimental condition. Experiments were performed in triplicate coverslips, and results represent means from experiments performed with two (for experiments with EVs) or four (for experiments with MSCs) independent neuronal cultures, as described in the figure legends.
Statistical analyses
Results were analyzed using one- or two-way ANOVA, followed by Tukey or Dunnett post hoc tests, as indicated in the corresponding figure legends. p < 0.05 values were considered indicators of statistically significant difference. All data were analyzed using GraphPad Prism version 4.0.
Author contributions
R. M. O. and S. T. F. conceived and coordinated the study, and analyzed all results. M. A. G., L. M. S., L. R. P. C., A. V.-S., and R. B. L. wrote the initial version of the manuscript. R. M.-O., S. T. F., and A. V. critically revised the paper. F. G. D. F. participated in study design, analysis, and interpretation of data. M. A. G. and L. M. S. performed and analyzed the experiments shown in Figs. 1–11, and M. A. G. prepared the figures. A. V. and H. J. V. B. designed, performed, and analyzed the experiment shown in Fig. 1I. A. G. and L. R. P. C. designed, performed, and analyzed experiments shown in Figs. 9 and 10. A. B. R. and A. V. performed and analyzed experiments shown in Fig. 7. A. P. C. A. L. and N. L. C. S. designed and analyzed experiments shown in Figs. 5 and 6. A. V.-S., L. R. P. S., R. B. L., V. H. S. M., C. V. B., C. A. A.-S., and L. C. S. participated in the establishment and maintenance of cell cultures and acquisition of data shown in Figs. 1 and 4. L. R. P. C., V. B. S., T. H. K. B., C. L. A., and N. L. C. S. designed, performed, and analyzed experiments shown in Fig. 8. All authors reviewed the results and approved the final version of the manuscript.
Acknowledgments
Imaging of Fig. 5 (A–D) was performed at the Institute of Biophysics Carlos Chagas Filho Light Microscopy Facility-PLAMOL. We thank Adriana Campos de Carvalho, André Batista, Antonio Campos de Carvalho, Amanda Souza, Cristiane Arantes, Daianne Torres, Danielle Cozachenco, Dominique Nicolaci, Eduardo Ferreira, Elizabeth Giestal, Fabiana Evaristo, Federica Collino, Felipe Marins, Grasielle Kincheski, Helen Melo, Humberto Muzi Filho, Jarlene Lopes, Jordano Brito-Moreira, Júlia de Deus, Juliana Tiemi, Leandro Coelho, Ligia Castro, Luís Santos, Maíra Oliveira, Mariângela Viana, Michael Rivadeneira, Milena Peclat, Mychael Lourenço, Natalia Silva, Pedro Coelho, Rachel Raxidi, Rafael Lindoso, Regina Goldenberg, Ricardo Lima-Filho, Rudimar Frozza, Sara Farias, Suelen Sério, Taina Panta, and Teresa Puig for assistance; Camilla Bayer and Marcelo Santiago for assistance and suggestions in image acquisition; and Dany Reznik for editing the manuscript.
This work was supported by grants from Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq), Coordenação de Aperfeiçoamento de Pessoal de Nível Superior (CAPES), Fundação Carlos Chagas Filho de Amparo à Pesquisa do Estado do Rio de Janeiro (FAPERJ), National Institute for Translational Neuroscience, and Departamento de Ciência e Tecnologia do Ministério da Saúde (DECIT/MS) (to S. T. F., R. M. O., F. G. D. F., A. V., A. G., N. L. C. S., and A. P. C. A. L.). The authors declare that they have no conflicts of interest with the contents of this article.
- AD
- Alzheimer's disease
- AβO
- soluble oligomers of the amyloid-β peptide
- EEA1
- early endosome antigen 1
- MSC
- mesenchymal stem cell
- EV
- extracellular vesicle
- DMEM
- Dulbecco's modified Eagle's medium
- IL
- interleukin
- VEGF
- vascular endothelial growth factor
- DCF
- 2′,7′-dichlorofluorescein
- ROS
- reactive oxygen species
- TEM
- transmission electron microscopy
- NTA
- nanoparticle-tracking analysis
- CNS
- central nervous system
- LAMP-1
- lysosome-associated membrane protein 1
- PSD-95
- postsynaptic density protein 95
- ANOVA
- analysis of variance.
References
- 1. Anand R., Gill K. D., and Mahdi A. A. (2014) Therapeutics of Alzheimer's disease: past, present and future. Neuropharmacology 76, 27–50 10.1016/j.neuropharm.2013.07.004 [DOI] [PubMed] [Google Scholar]
- 2. Imtiaz B., Tolppanen A.-M., Kivipelto M., and Soininen H. (2014) Future directions in Alzheimer's disease from risk factors to prevention. Biochem. Pharmacol. 88, 661–670 10.1016/j.bcp.2014.01.003 [DOI] [PubMed] [Google Scholar]
- 3. Mucke L., and Selkoe D. J. (2012) Neurotoxicity of amyloid β-protein: synaptic and network dysfunction. Cold Spring Harb. Perspect. Med. 2, a006338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Ferreira S. T., Lourenco M. V., Oliveira M. M., and De Felice F. G. (2015) Soluble amyloid-β oligomers as synaptotoxins leading to cognitive impairment in Alzheimer's disease. Front. Cell Neurosci. 9, 191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Ferreira S. T., and Klein W. L. (2011) The Aβ oligomer hypothesis for synapse failure and memory loss in Alzheimer's disease. Neurobiol. Learn. Mem. 96, 529–543 10.1016/j.nlm.2011.08.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Haass C., and Selkoe D. J. (2007) Soluble protein oligomers in neurodegeneration: lessons from the Alzheimer's amyloid β-peptide. Nat. Rev. Mol. Cell Biol. 8, 101–112 10.1038/nrm2101 [DOI] [PubMed] [Google Scholar]
- 7. Liste I., García-García E., Bueno C., and Martínez-Serrano A. (2007) Bcl-XL modulates the differentiation of immortalized human neural stem cells. Cell Death Differ. 14, 1880–1892 10.1038/sj.cdd.4402205 [DOI] [PubMed] [Google Scholar]
- 8. Bissonnette C. J., Lyass L., Bhattacharyya B. J., Belmadani A., Miller R. J., and Kessler J. A. (2011) The controlled generation of functional basal forebrain cholinergic neurons from human embryonic stem cells. Stem Cells 29, 802–811 10.1002/stem.626 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Simard A. R., Soulet D., Gowing G., Julien J. P., and Rivest S. (2006) Bone marrow-derived microglia play a critical role in restricting senile plaque formation in Alzheimer's disease. Neuron 49, 489–502 10.1016/j.neuron.2006.01.022 [DOI] [PubMed] [Google Scholar]
- 10. Wu Q.-Y., Li J., Feng Z.-T., and Wang T.-H. (2007) Bone marrow stromal cells of transgenic mice can improve the cognitive ability of an Alzheimer's disease rat model. Neurosci. Lett. 417, 281–285 10.1016/j.neulet.2007.02.092 [DOI] [PubMed] [Google Scholar]
- 11. Lee J. K., Schuchman E. H., Jin H. K., and Bae J. (2012) Soluble CCL5 derived from bone marrow-derived mesenchymal stem cells and activated by amyloid β ameliorates Alzheimer's disease in mice by recruiting bone marrow-induced microglia immune responses. Stem Cells 30, 1544–1555 10.1002/stem.1125 [DOI] [PubMed] [Google Scholar]
- 12. Kim J.-Y., Kim D. H., Kim J. H., Lee D., Jeon H. B., Kwon S.-J., Kim S. M., Yoo Y. J., Lee E. H., Choi S. J., Seo S. W., Lee J. I., Na D. L., Yang Y. S., Oh W., and Chang J. W. (2012) Soluble intracellular adhesion molecule-1 secreted by human umbilical cord blood-derived mesenchymal stem cell reduces amyloid-β plaques. Cell Death Differ. 19, 680–691 10.1038/cdd.2011.140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Lee J. K., Jin H. K., and Bae J. (2009) Bone marrow-derived mesenchymal stem cells reduce brain amyloid-β deposition and accelerate the activation of microglia in an acutely induced Alzheimer's disease mouse model. Neurosci. Lett. 450, 136–141 10.1016/j.neulet.2008.11.059 [DOI] [PubMed] [Google Scholar]
- 14. Lee H. J., Lee J. K., Lee H., Shin J. W., Carter J. E., Sakamoto T., Jin H. K., and Bae J. S. (2010) The therapeutic potential of human umbilical cord blood-derived mesenchymal stem cells in Alzheimer's disease. Neurosci. Lett. 481, 30–35 10.1016/j.neulet.2010.06.045 [DOI] [PubMed] [Google Scholar]
- 15. Caplan A. I., and Dennis J. E. (2006) Mesenchymal stem cells as trophic mediators. J. Cell Biochem. 98, 1076–1084 10.1002/jcb.20886 [DOI] [PubMed] [Google Scholar]
- 16. Montemurro T., Viganò M., Ragni E., Barilani M., Parazzi V., Boldrin V., Lavazza C., Montelatici E., Banfi F., Lauri E., Giovanelli S., Baccarin M., Guerneri S., Giordano R., and Lazzari L. (2016) Angiogenic and anti-inflammatory properties of mesenchymal stem cells from cord blood: soluble factors and extracellular vesicles for cell regeneration. Eur. J. Cell Biol. 95, 228–238 10.1016/j.ejcb.2016.04.003 [DOI] [PubMed] [Google Scholar]
- 17. Spees J. L., Olson S. D., Whitney M. J., and Prockop D. J. (2006) Mitochondrial transfer between cells can rescue aerobic respiration. Proc. Natl. Acad. Sci. U.S.A. 103, 1283–1288 10.1073/pnas.0510511103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Plotnikov E. Y., Khryapenkova T. G., Galkina S. I., Sukhikh G. T., and Zorov D. B. (2010) Cytoplasm and organelle transfer between mesenchymal multipotent stromal cells and renal tubular cells in co-culture. Exp. Cell Res. 316, 2447–2455 10.1016/j.yexcr.2010.06.009 [DOI] [PubMed] [Google Scholar]
- 19. Collino F., Deregibus M. C., Bruno S., Sterpone L., Aghemo G., Viltono L., Tetta C., and Camussi G. (2010) Microvesicles derived from adult human bone marrow and tissue specific mesenchymal stem cells shuttle selected pattern of miRNAs. PLoS One 5, e11803 10.1371/journal.pone.0011803 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Lindoso R. S., Sandim V., Collino F., Carvalho A. B., Dias J., da Costa M. R., Zingali R. B., and Vieyra A. (2016) Proteomics of cell-cell interactions in health and disease. Proteomics 16, 328–344 10.1002/pmic.201500341 [DOI] [PubMed] [Google Scholar]
- 21. De Felice F. G., Velasco P. T., Lambert M. P., Viola K., Fernandez S. J., Ferreira S. T., and Klein W. L. (2007) Aβ oligomers induce neuronal oxidative stress through an N-methyl-d-aspartate receptor-dependent mechanism that is blocked by the Alzheimer drug memantine. J. Biol. Chem. 282, 11590–11601 10.1074/jbc.M607483200 [DOI] [PubMed] [Google Scholar]
- 22. De Felice F. G., Vieira M. N. N., Bomfim T. R., Decker H., Velasco P. T., Lambert M. P., Viola K. L., Zhao W.-Q., Ferreira S. T., and Klein W. L. (2009) Protection of synapses against Alzheimer's-linked toxins: insulin signaling prevents the pathogenic binding of Aβ oligomers. Proc. Natl. Acad. Sci. U.S.A. 106, 1971–1976 10.1073/pnas.0809158106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Decker H., Jürgensen S., Adrover M. F., Brito-Moreira J., Bomfim T. R., Klein W. L., Epstein A. L., De Felice F. G., Jerusalinsky D., and Ferreira S. T. (2010) N-Methyl-d-aspartate receptors are required for synaptic targeting of Alzheimer's toxic amyloid-β peptide oligomers. J. Neurochem. 115, 1520–1529 10.1111/j.1471-4159.2010.07058.x [DOI] [PubMed] [Google Scholar]
- 24. De Felice F. G., Wu D., Lambert M. P., Fernandez S. J., Velasco P. T., Lacor P. N., Bigio E. H., Jerecic J., Acton P. J., Shughrue P. J., Chen-Dodson E., Kinney G. G., and Klein W. L. (2008) Alzheimer's disease-type neuronal tau hyperphosphorylation induced by Aβ oligomers. Neurobiol. Aging. 29, 1334–1347 10.1016/j.neurobiolaging.2007.02.029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. De Felice F. G., Vieira M. N. N., Saraiva L. M., Figueroa-Villar J. D., Garcia-Abreu J., Liu R., Chang L., Klein W. L., and Ferreira S. T. (2004) Targeting the neurotoxic species in Alzheimer's disease: inhibitors of Aβ oligomerization. FASEB J. 18, 1366–1372 10.1096/fj.04-1764com [DOI] [PubMed] [Google Scholar]
- 26. Gong Y., Chang L., Viola K. L., Lacor P. N., Lambert M. P., Finch C. E., Krafft G. A., and Klein W. L. (2003) Alzheimer's disease-affected brain: Presence of oligomeric Aβ ligands (ADDLs) suggests a molecular basis for reversible memory loss. Proc. Natl. Acad. Sci. U.S.A. 100, 10417–10422 10.1073/pnas.1834302100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Lacor P. N., Buniel M. C., Chang L., Fernandez S. J., Gong Y., Viola K. L., Lambert M. P., Velasco P. T., Bigio E. H., Finch C. E., Krafft G. A., and Klein W. L. (2004) Synaptic targeting by Alzheimer's-related amyloid β oligomers. J. Neurosci. 24, 10191–10200 10.1523/JNEUROSCI.3432-04.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. L Ramos T., Sánchez-Abarca L. I., Muntión S., Preciado S., Puig N., López-Ruano G., Hernández-Hernández Á., Redondo A., Ortega R., Rodríguez C., Sánchez-Guijo F., and del Cañizo C. (2016) MSC surface markers (CD44, CD73, and CD90) can identify human MSC-derived extracellular vesicles by conventional flow cytometry. Cell Commun. Signal. 14, 2 10.1186/s12964-015-0124-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Sokolova V., Ludwig A.-K., Hornung S., Rotan O., Horn P. A., Epple M., and Giebel B. (2011) Characterisation of exosomes derived from human cells by nanoparticle tracking analysis and scanning electron microscopy. Colloids Surfaces B Biointerfaces 87, 146–150 10.1016/j.colsurfb.2011.05.013 [DOI] [PubMed] [Google Scholar]
- 30. Ferreira S. T., Vieira M. N. N., and De Felice F. G. (2007) Soluble protein oligomers as emerging toxins in Alzheimer's and other amyloid diseases. IUBMB Life 59, 332–345 10.1080/15216540701283882 [DOI] [PubMed] [Google Scholar]
- 31. Song M. S., Learman C. R., Ahn K. C., Baker G. B., Kippe J., Field E. M., and Dunbar G. L. (2015) In vitro validation of effects of BDNF-expressing mesenchymal stem cells on neurodegeneration in primary cultured neurons of APP/PS1 mice. Neuroscience 307, 37–50 10.1016/j.neuroscience.2015.08.011 [DOI] [PubMed] [Google Scholar]
- 32. Danielyan L., Beer-Hammer, Stolzing A., Schäfer R., Siegel G., Fabian C., Kahle P., Biedermann T., Lourhmati A., Buadze M., Novakovic A., Proksch B., Gleiter C. H., Frey W. H., and Schwab M. (2014) Intranasal delivery of bone marrow-derived mesenchymal stem cells, macrophages, and microglia to the brain in mouse models of Alzheimer's and Parkinson's disease. Cell Transplant. 23, S123–S139 [DOI] [PubMed] [Google Scholar]
- 33. Xie Z. H., Liu Z., Zhang X. R., Yang H., Wei L. F., Wang Y., Xu S. L., Sun L., Lai C., Bi J. Z., and Wang X. Y. (2016) Wharton's Jelly-derived mesenchymal stem cells alleviate memory deficits and reduce amyloid-β deposition in an APP/PS1 transgenic mouse model. Clin. Exp. Med. 16, 89–98 10.1007/s10238-015-0375-0 [DOI] [PubMed] [Google Scholar]
- 34. Yang H., Xie Z., Wei L., Yang H., Yang S., Zhu Z., Wang P., Zhao C., and Bi J. (2013) Human umbilical cord mesenchymal stem cell-derived neuron-like cells rescue memory deficits and reduce amyloid-β deposition in an AβPP/PS1 transgenic mouse model. Stem Cell Res. Ther. 4, 76 10.1186/scrt227 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Garcia K. O., Ornellas F. L. M., Matsumoto Martin P. K., Patti C. L., Mello L. E., Frussa-Filho R., Han S. W., and Longo B. M. (2014) Therapeutic effects of the transplantation of VEGF overexpressing bone marrow mesenchymal stem cells in the hippocampus of murine model of Alzheimer's disease. Front. Aging Neurosci. 6, 30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Shin J. Y., Park H. J., Kim H. N., Oh S. H., Bae J. S., Ha H. J., and Lee P. H. (2014) Mesenchymal stem cells enhance autophagy and increase β-amyloid clearance in Alzheimer disease models. Autophagy 10, 32–44 10.4161/auto.26508 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Lee H. J., Lee J. K., Lee H., Carter J. E., Chang J. W., Oh W., Yang Y. S., Suh J. G., Lee B. H., Jin H. K., and Bae J. (2012) Human umbilical cord blood-derived mesenchymal stem cells improve neuropathology and cognitive impairment in an Alzheimer's disease mouse model through modulation of neuroinflammation. Neurobiol. Aging 33, 588–602 10.1016/j.neurobiolaging.2010.03.024 [DOI] [PubMed] [Google Scholar]
- 38. Naaldijk Y., Jäger C., Fabian C., Leovsky C., Blüher A., Rudolph L., Hinze A., and Stolzing A. (2017) Effect of systemic transplantation of bone marrow-derived mesenchymal stem cells on neuropathology markers in APP/PS1 Alzheimer mice. Neuropathol. Appl. Neurobiol. 43, 299–314 10.1111/nan.12319 [DOI] [PubMed] [Google Scholar]
- 39. Katsuda T., Tsuchiya R., Kosaka N., Yoshioka Y., Takagaki K., Oki K., Takeshita F., Sakai Y., Kuroda M., and Ochiya T. (2013) Human adipose tissue-derived mesenchymal stem cells secrete functional neprilysin-bound exosomes. Sci. Rep. 3, 1197 10.1038/srep01197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Valle-Prieto A., and Conget P. A. (2010) Human mesenchymal stem cells efficiently manage oxidative stress. Stem Cells Dev. 19, 1885–1893 10.1089/scd.2010.0093 [DOI] [PubMed] [Google Scholar]
- 41. Terry R. D., Masliah E., Salmon D. P., Butters N., DeTeresa R., Hill R., Hansen L. A., and Katzman R. (1991) Physical basis of cognitive alterations in Alzheimer's disease: synapse loss is the major correlate of cognitive impairment. Ann. Neurol. 30, 572–580 10.1002/ana.410300410 [DOI] [PubMed] [Google Scholar]
- 42. Serrano F., and Klann E. (2004) Reactive oxygen species and synaptic plasticity in the aging hippocampus. Ageing Res. Rev. 3, 431–443 10.1016/j.arr.2004.05.002 [DOI] [PubMed] [Google Scholar]
- 43. Lambert M. P., Barlow A. K., Chromy B. A., Edwards C., Freed R., Liosatos M., Morgan T. E., Rozovsky I., Trommer B., Viola K. L., Wals P., Zhang C., Finch C. E., Krafft G. A., and Klein W. L. (1998) Diffusible, nonfibrillar ligands derived from Aβ1–42 are potent central nervous system neurotoxins. Proc. Natl. Acad. Sci. U.S.A. 95, 6448–6453 10.1073/pnas.95.11.6448 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Walsh D. M., Klyubin I., Fadeeva J. V., Cullen W. K., Anwyl R., Wolfe M. S., Rowan M. J., and Selkoe D. J. (2002) Naturally secreted oligomers of amyloid β protein potently inhibit hippocampal long-term potentiation in vivo. Nature 416, 535–539 10.1038/416535a [DOI] [PubMed] [Google Scholar]
- 45. Laurén J., Gimbel D. A., Nygaard H. B., Gilbert J. W., and Strittmatter S. M. (2009) Cellular prion protein mediates impairment of synaptic plasticity by amyloid-β oligomers. Nature 457, 1128–1132 10.1038/nature07761 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Gimbel D. A., Nygaard H. B., Coffey E. E., Gunther E. C., Laurén J., Gimbel Z. A., and Strittmatter S. M. (2010) Memory impairment in transgenic Alzheimer mice requires cellular prion protein. J. Neurosci. 30, 6367–6374 10.1523/JNEUROSCI.0395-10.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Magdesian M. H., Carvalho M. M. V. F., Mendes F. A., Saraiva L. M., Juliano M. A., Juliano L., Garcia-Abreu J., and Ferreira S. T. (2008) Amyloid-β binds to the extracellular cysteine-rich domain of Frizzled and inhibits Wnt/β-catenin signaling. J. Biol. Chem. 283, 9359–9368 10.1074/jbc.M707108200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Cissé M., Halabisky B., Harris J., Devidze N., Dubal D. B., Sun B., Orr A., Lotz G., Kim D. H., Hamto P., Ho K., Yu G.-Q., and Mucke L. (2011) Reversing EphB2 depletion rescues cognitive functions in Alzheimer model. Nature 469, 47–52 10.1038/nature09635 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Renner M., Lacor P. N., Velasco P. T., Xu J., Contractor A., Klein W. L., and Triller A. (2010) Deleterious effects of amyloid β oligomers acting as an extracellular scaffold for mGluR5. Neuron. 66, 739–754 10.1016/j.neuron.2010.04.029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Salazar S. V., and Strittmatter S. M. (2017) Cellular prion protein as a receptor for amyloid-β oligomers in Alzheimer's disease. Biochem. Biophys. Res. Commun. 483, 1143–1147 10.1016/j.bbrc.2016.09.062 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Jones R. S., Minogue A. M., Connor T. J., and Lynch M. A. (2013) Amyloid-β-induced astrocytic phagocytosis is mediated by CD36, CD47 and RAGE. J. Neuroimmune Pharmacol. 8, 301–311 10.1007/s11481-012-9427-3 [DOI] [PubMed] [Google Scholar]
- 52. Ledo J. H., Azevedo E. P., Beckman D., Ribeiro F. C., Santos L. E., Razolli D. S., Kincheski G. C., Melo H. M., Bellio M., Teixeira A. L., Velloso L. A., Foguel D., De Felice F. G., and Ferreira S. T. (2016) Cross-talk between brain innate immunity and serotonin signaling underlies depressive-like behavior induced by Alzheimer's amyloid-β oligomers in mice. J. Neurosci. 36, 12106–12116 10.1523/JNEUROSCI.1269-16.2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Noda M., and Suzumura A. (2012) Sweepers in the CNS: microglial migration and phagocytosis in the Alzheimer disease pathogenesis. Int. J. Alzheimers Dis. 2012, 891087 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Frenkel D., Wilkinson K., Zhao L., Hickman S. E., Means T. K., Puckett L., Farfara D., Kingery N. D., Weiner H. L., and El Khoury J. (2013) Scara1 deficiency impairs clearance of soluble amyloid-β by mononuclear phagocytes and accelerates Alzheimer's-like disease progression. Nat. Commun. 4, 2030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Goedert M., and Spillantini M. G. (2006) A century of Alzheimer's disease. Science 314, 777–781 10.1126/science.1132814 [DOI] [PubMed] [Google Scholar]
- 56. De Felice F. G. (2013) Alzheimer's disease and insulin resistance: translating basic science into clinical applications. J. Clin. Invest. 123, 531–539 10.1172/JCI64595 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Nikolic W. V., Hou H., Town T., Zhu Y., Giunta B., Sanberg C. D., Zeng J., Luo D., Ehrhart J., Mori T., Sanberg P. R., and Tan J. (2008) Peripherally administered human umbilical cord blood cells reduce parenchymal and vascular β-amyloid deposits in Alzheimer mice. Stem Cells Dev. 17, 423–439 10.1089/scd.2008.0018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Christopherson K. S., Ullian E. M., Stokes C. C., Mullowney C. E., Hell J. W., Agah A., Lawler J., Mosher D. F., Bornstein P., and Barres B. A. (2005) Thrombospondins are astrocyte-secreted proteins that promote CNS synaptogenesis. Cell 120, 421–433 [DOI] [PubMed] [Google Scholar]
- 59. Zhang Y., Chopp M., Meng Y., Katakowski M., Xin H., Mahmood A., and Xiong Y. (2015) Effect of exosomes derived from multipluripotent mesenchymal stromal cells on functional recovery and neurovascular plasticity in rats after traumatic brain injury. J. Neurosurg. 122, 856–867 10.3171/2014.11.JNS14770 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Doeppner T. R., Herz J., Görgens A., Schlechter J., Ludwig A.-K., Radtke S., de Miroschedji K., Horn P. A., Giebel B., and Hermann D. M. (2015) Extracellular vesicles improve post-stroke neuroregeneration and prevent postischemic immunosuppression. Stem Cells Transl. Med. 4, 1131–1143 10.5966/sctm.2015-0078 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Xin H., Li Y., Buller B., Katakowski M., Zhang Y., Wang X., Shang X., Zhang Z. G., and Chopp M. (2012) Exosome-mediated transfer of miR-133b from multipotent mesenchymal stromal cells to neural cells contributes to neurite outgrowth. Stem Cells 30, 1556–1564 10.1002/stem.1129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Xin H., Li Y., Cui Y., Yang J. J., Zhang Z. G., and Chopp M. (2013) Systemic administration of exosomes released from mesenchymal stromal cells promote functional recovery and neurovascular plasticity after stroke in rats. J. Cereb. Blood Flow Metab. 33, 1711–1715 10.1038/jcbfm.2013.152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Blazquez R., Sanchez-Margallo F. M., de la Rosa O., Dalemans W., Alvarez V., Tarazona R., and Casado J. G. (2014) Immunomodulatory potential of human adipose mesenchymal stem cells derived exosomes on in vitro stimulated T cells. Front. Immunol. 5, 556 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Chi Y., Jin Y., He Z., and Yu T. (2014) Detection of cytokines in supernatant from hematopoietic stem/progenitor cells co-cultured with mesenchymal stem cells and endothelial progenitor cells. Cell Tissue Bank. 15, 397–402 10.1007/s10561-013-9404-y [DOI] [PubMed] [Google Scholar]
- 65. Zhang B., Yin Y., Lai R. C., Tan S. S., Choo A. B., and Lim S. K. (2014) Mesenchymal stem cells secrete immunologically active exosomes. Stem Cells Dev. 23, 1233–1244 10.1089/scd.2013.0479 [DOI] [PubMed] [Google Scholar]
- 66. Choi M., Ban T., and Rhim T. (2014) Therapeutic use of stem cell transplantation for cell replacement or cytoprotective effect of microvesicle released from mesenchymal stem cell. Mol. Cells 37, 133–139 10.14348/molcells.2014.2317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. He J., Wang Y., Sun S., Yu M., Wang C., Pei X., Zhu B., Wu J., and Zhao W. (2012) Bone marrow stem cells-derived microvesicles protect against renal injury in the mouse remnant kidney model. Nephrology 17, 493–500 10.1111/j.1440-1797.2012.01589.x [DOI] [PubMed] [Google Scholar]
- 68. Tan C. Y., Lai R. C., Wong W., Dan Y. Y., Lim S. K., and Ho H. K. (2014) Mesenchymal stem cell-derived exosomes promote hepatic regeneration in drug-induced liver injury models. Stem Cell Res. Ther. 5, 76 10.1186/scrt465 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Farinazzo A., Turano E., Marconi S., Bistaffa E., Bazzoli E., and Bonetti B. (2015) Murine adipose-derived mesenchymal stromal cell vesicles: in vitro clues for neuroprotective and neuroregenerative approaches. Cytotherapy 17, 571–578 10.1016/j.jcyt.2015.01.005 [DOI] [PubMed] [Google Scholar]
- 70. Zhou Y., Xu H., Xu W., Wang B., Wu H., Tao Y., Zhang B., Wang M., Mao F., Yan Y., Gao S., Gu H., Zhu W., and Qian H. (2013) Exosomes released by human umbilical cord mesenchymal stem cells protect against cisplatin-induced renal oxidative stress and apoptosis in vivo and in vitro. Stem Cell Res. Ther. 4, 34 10.1186/scrt194 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Villarroya-Beltri C, Baixauli F., Gutiérrez-Vázquez C., Sánchez-Madrid F., and Mittelbrunn M. (2014) Sorting it out: regulation of exosome loading. Semin. Cancer Biol. 28, 3–13 10.1016/j.semcancer.2014.04.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Kim J.-Y., Kim D. H., Kim D.-S., Kim J. H., Jeong S. Y., Jeon H. B., Lee E. H., Yang Y. S., Oh W., and Chang J. W. (2010) Galectin-3 secreted by human umbilical cord blood-derived mesenchymal stem cells reduces amyloid-β42 neurotoxicity in vitro. FEBS Lett. 584, 3601–3608 10.1016/j.febslet.2010.07.028 [DOI] [PubMed] [Google Scholar]
- 73. Yuyama K., Sun H., Sakai S., Mitsutake S., Okada M., Tahara H., Furukawa J., Fujitani N., Shinohara Y., and Igarashi Y. (2014) Decreased amyloid-β pathologies by intracerebral loading of glycosphingolipid-enriched exosomes in Alzheimer model mice. J. Biol. Chem. 289, 24488–24498 10.1074/jbc.M114.577213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Haney M. J., Klyachko N. L., Zhao Y., Gupta R., Plotnikova E. G., He Z., Patel T., Piroyan A., Sokolsky M., Kabanov A. V., and Batrakova E. V. (2015) Exosomes as drug delivery vehicles for Parkinson's disease therapy. J. Control. Release 207, 18–30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Zhang G., Zou X., Miao S., Chen J., Du T., Zhong L., Ju G., Liu G., and Zhu Y. (2014) The anti-oxidative role of micro-vesicles derived from human Wharton-Jelly mesenchymal stromal cells through NOX2/gp91(phox) suppression in alleviating renal ischemia-reperfusion injury in rats. PLoS One 9, e92129 10.1371/journal.pone.0092129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Erta M., Quintana A., and Hidalgo J. (2012) Interleukin-6, a major cytokine in the central nervous system. Int. J. Biol. Sci. 8, 1254–1266 10.7150/ijbs.4679 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Chakrabarty P., Jansen-West K., Beccard A., Ceballos-Diaz C., Levites Y., Verbeeck C., Zubair A. C., Dickson D., Golde T. E., and Das P. (2010) Massive gliosis induced by interleukin-6 suppresses Aβ deposition in vivo: evidence against inflammation as a driving force for amyloid deposition. FASEB J. 24, 548–559 10.1096/fj.09-141754 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Gadient R. A., and Otten U. H. (1997) Interleukin-6 (IL-6)–a molecule with both beneficial and destructive potentials. Prog. Neurobiol. 52, 379–390 10.1016/S0301-0082(97)00021-X [DOI] [PubMed] [Google Scholar]
- 79. Vargas-Alarcón G., Juárez-Cedillo E., Martínez-Rodríguez N., Fragoso J. M., García-Hernández N., and Juárez-Cedillo T. (2016) Association of interleukin-10 polymorphisms with risk factors of Alzheimer's disease and other dementias (SADEM study). Immunol. Lett. 177, 47–52 10.1016/j.imlet.2016.07.011 [DOI] [PubMed] [Google Scholar]
- 80. Asselineau D., Benlhassan K., Arosio B., Mari D., Ferri E., Casati M., Gussago C., Tedone E., Annoni G., Mazzola P., Piette F., Belmin J., Pariel S., Bornand A., Beaudeux J. L., Doulazmi M., Mariani J., and Bray D. H. (2015) Interleukin-10 production in response to amyloid-β differs between slow and fast decliners in patients with Alzheimer's disease. J. Alzheimers Dis. 46, 837–842 10.3233/JAD-142832 [DOI] [PubMed] [Google Scholar]
- 81. Heiskanen M., Kähönen M., Hurme M., Lehtimäki T., Mononen N., Juonala M., Hutri-Kähönen N., Viikari J., Raitakari O., and Hulkkonen J. (2010) Polymorphism in the IL10 promoter region and early markers of atherosclerosis: the cardiovascular risk in young Finns study. Atherosclerosis 208, 190–196 10.1016/j.atherosclerosis.2009.06.032 [DOI] [PubMed] [Google Scholar]
- 82. Brkic M., Balusu S., Van Wonterghem E., Gorlé N., Benilova I., Kremer A., Van Hove I., Moons L., De Strooper B., Kanazir S., Libert C., and Vandenbroucke R. E. (2015) Amyloid β oligomers disrupt blood-CSF barrier integrity by activating matrix metalloproteinases. J. Neurosci. 35, 12766–12778 10.1523/JNEUROSCI.0006-15.2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. van de Haar H. J., Burgmans S., Jansen J. F. A., van Osch M. J. P., van Buchem M. A., Muller M., Hofman P. A. M., Verhey F. R. J., and Backes W. H. (2016) Blood-brain barrier leakage in patients with early Alzheimer disease. Radiology 281, 527–535 10.1148/radiol.2016152244 [DOI] [PubMed] [Google Scholar]
- 84. Moraes L., Vasconcelos-dos-Santos A., Santana F. C., Godoy M. A., Rosado-de-Castro P. H., Jasmin, Azevedo-Pereira R. L., Cintra W. M., Gasparetto E. L., Santiago M. F., and Mendez-Otero R. (2012) Neuroprotective effects and magnetic resonance imaging of mesenchymal stem cells labeled with SPION in a rat model of Huntington's disease. Stem Cell Res. 9, 143–155 10.1016/j.scr.2012.05.005 [DOI] [PubMed] [Google Scholar]
- 85. Tang X., Kim J., Zhou L., Wengert E., Zhang L., Wu Z., Carromeu C., and Muotri A. R., Marchetto M. C., Gage F. H., Chen G. (2016) KCC2 rescues functional deficits in human neurons derived from patients with Rett syndrome. Proc. Natl. Acad. Sci. U.S.A. 113, 751–756 10.1073/pnas.1524013113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. de Vasconcelos Dos Santos A., da Costa Reis J., Diaz Paredes B., Moraes L., Jasmin, Giraldi-Guimarães A., and Mendez-Otero R. (2010) Therapeutic window for treatment of cortical ischemia with bone marrow-derived cells in rats. Brain Res. 1306, 149–158 10.1016/j.brainres.2009.09.094 [DOI] [PubMed] [Google Scholar]
- 87. Vieira M. N. N., Forny-Germano L., Saraiva L. M., Sebollela A., Martinez A. M. B., Houzel J. C., De Felice F. G., and Ferreira S. T. (2007) Soluble oligomers from a non-disease related protein mimic Aβ-induced tau hyperphosphorylation and neurodegeneration. J. Neurochem. 103, 736–748 10.1111/j.1471-4159.2007.04809.x [DOI] [PubMed] [Google Scholar]
- 88. Scholzen T., and Gerdes J. (2000) The Ki-67 protein: from the known and the unknown. J. Cell. Physiol. 182, 311–322 10.1002/(SICI)1097-4652(200003)182:3%3C311::AID-JCP1%3E3.0.CO%3B2-9 [DOI] [PubMed] [Google Scholar]
- 89. Abràmoff M. D., Magalhães P. J., and Ram S. J. (2004) Image processing with ImageJ. Biophotonics Int. 11, 36–42 [Google Scholar]
- 90. Lindoso R. S., Collino F., Bruno S., Araujo D. S., Sant'Anna J. F., Tetta C., Provero P., Quesenberry P. J., Vieyra A., Einicker-Lamas M., and Camussi G. (2014) Extracellular vesicles released from mesenchymal stromal cells modulate miRNA in renal tubular cells and inhibit ATP depletion injury. Stem Cells Dev. 23, 1809–1819 10.1089/scd.2013.0618 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Kowal E. J. K., Ter-Ovanesyan D., Regev A., and Church G. M. (2017) Extracellular vesicle isolation and analysis by Western blotting. Methods Mol. Biol. 1660, 143–152 10.1007/978-1-4939-7253-1_12 [DOI] [PubMed] [Google Scholar]
- 92. Margoliash E., and Schejter A. (1962) Kinetics of the irreversible inhibition of catalase by 3-amino-1,2,4-triazole in the presence of hydrogen peroxide and catalase-hydrogen peroxide complex I hydrogen donors. J. Biol. Chem. 237, 2359–2363 [PubMed] [Google Scholar]
- 93. Lambert M. P., Velasco P. T., Chang L., Viola K. L., Fernandez S., Lacor P. N., Khuon D., Gong Y., Bigio E. H., Shaw P., De Felice F. G., Krafft G. A., and Klein W. L. (2007) Monoclonal antibodies that target pathological assemblies of Aβ. J. Neurochem. 100, 23–35 10.1111/j.1471-4159.2006.04157.x [DOI] [PubMed] [Google Scholar]
- 94. De Felice F. G., Houzel J.-C. C., Garcia-Abreu J., Louzada P. R. Jr., Afonso R. C., Meirelles M. N., Lent R., Neto V. M., and Ferreira S. T. (2001) Inhibition of Alzheimer's disease β-amyloid aggregation, neurotoxicity, and in vivo deposition by nitrophenols: implications for Alzheimer's therapy. FASEB J. 15, 1297–1299 10.1016/j.cell.2004.12.020 [DOI] [PubMed] [Google Scholar]