

Abstract
Histone deacetylase 6 (HDAC6), a class IIb HDAC, plays an important role in many biological and pathological processes. Previously, we found that ERK1, a downstream kinase in the mitogen-activated protein kinase signaling pathway, phosphorylates HDAC6, thereby increasing HDAC6-mediated deacetylation of α-tubulin. However, whether HDAC6 reciprocally modulates ERK1 activity is unknown. Here, we report that both ERK1 and -2 are acetylated and that HDAC6 promotes ERK1 activity via deacetylation. Briefly, we found that both ERK1 and -2 physically interact with HDAC6. Endogenous ERK1/2 acetylation levels increased upon treatment with a pan-HDAC inhibitor, an HDAC6-specific inhibitor, or depletion of HDAC6, suggesting that HDAC6 deacetylates ERK1/2. We also noted that the acetyltransferases CREB-binding protein and p300 both can acetylate ERK1/2. Acetylated ERK1 exhibits reduced enzymatic activity toward the transcription factor ELK1, a well-known ERK1 substrate. Furthermore, mass spectrometry analysis indicated Lys-72 as an acetylation site in the ERK1 N terminus, adjacent to Lys-71, which binds to ATP, suggesting that acetylation status of Lys-72 may affect ERK1 ATP binding. Interestingly, an acetylation-mimicking ERK1 mutant (K72Q) exhibited less phosphorylation than the WT enzyme and a deacetylation-mimicking mutant (K72R). Of note, the K72Q mutant displayed decreased enzymatic activity in an in vitro kinase assay and in a cellular luciferase assay compared with the WT and K72R mutant. Taken together, our findings suggest that HDAC6 stimulates ERK1 activity. Along with our previous report that ERK1 promotes HDAC6 activity, we propose that HDAC6 and ERK1 may form a positive feed-forward loop, which might play a role in cancer.
Keywords: enzyme mechanism; histone acetylase; histone deacetylase (HDAC); histone deacetylase 6 (HDAC6); mitogen-activated protein kinase (MAPK), extracellular signal-regulated kinase1/2 (ERK1/2); acetylation;
Introduction
Histone deacetylases (HDACs)3 and histone acetyltransferases (HATs) are the enzymes that regulate core histones and non-histone proteins by deacetylation and acetylation, respectively (1). HATs acetylate proteins via adding acetyl groups to lysine residues, whereas HDACs catalyze a reverse reaction by removing the acetyl group from lysine residues. Until now, a total of 18 HDACs were identified in humans and grouped into four classes based on their sequence similarity to yeast orthologs (2). Class I HDACs are homologous to yeast-reduced potassium dependence 3 (Rpd3) and include HDACs 1–3 and 8. Class II HDACs are homologous to yeast histone deacetylase 1 (HdaI) and are further divided into class IIa and class IIb. Class IIa contains HDACs 4, 5, 7, and 9, and class IIb includes HDACs 6 and 10. HDAC11 is the only member in class IV. The deacetylase activity of HDAC classes I, II, and IV is zinc-dependent. Class III HDACs, also knowns as sirtuins, are homologous to yeast silent information regulator 2 (Sir2), and the deacetylase activity of this class is oxidized nicotinamide adenine dinucleotide (NAD+)-dependent.
HDAC6 belongs to class IIb HDACs. Its structure is quite unique among all HDACs, in that it contains two functional deacetylase domains in tandem and a zinc finger domain in the C terminus (2, 3). HDAC6 participates in numerous biological and pathological processes, such as cell migration, DNA damage response, and oncogenesis, through modulating its substrates (4–7). For example, HDAC6 deacetylates cytoskeleton proteins and their associated proteins, such as α-tubulin and cortactin, to regulate cell mobility (4, 5). HDAC6 also deacetylates and ubiquitinates the DNA mismatch repair protein MSH2, to regulate MutSα homeostasis, DNA mismatch repair, and DNA damage response (6). In addition, HDAC6 deacetylates cell-signaling regulators K-Ras and β-catenin, leading to altered oncogenic activity and nuclear localization, respectively (8, 9).
Mitogen-activated protein kinases (MAPKs) are a conserved family of serine/threonine protein kinases connected to various essential cellular processes (10). To date, a total of 14 MAPKs have been isolated in humans, all of which fall into the seven classes that follow: the extracellular signal-regulated kinase class, p38 class, c-Jun N-terminal kinase (JNKs) class, and ERK5 class belong to the conventional group of MAPKs, all of which have been studied extensively; and the Nemo-like kinase (NLK) class, ERK3/4 class, and ERK7 class belong to the atypical group of MAPKs, all of which have been studied inadequately (10, 11). In general, the MAPK pathway contains at least three tiers: a MAPK kinase kinase (MAP3K); a MAPK kinase (MAP2K); and a MAPK. These MAPK pathways participate in transducing signals from the surface to the interior of the cell. Being triggered by extracellular stimuli, the first tier MAP3Ks are activated to phosphorylate MAP2Ks, which subsequently phosphorylate MAPKs. These MAPK pathways have their own unique primary kinases in different tiers, but they also share some minor activators (10, 11). All MAPKs, except ERK3/4 and NLK, contain a conserved Thr-Xaa-Tyr motif in their kinase domain. Phosphorylation of both Thr and Tyr residues in this motif is a critical step for MAPK activation (10).
Human ERK1 (also known as MAPK3 or p44 MAPK) and ERK2 (also known as MAPK1 or p42 MAPK) are 84% identical in sequence; they share many functions (12). Thus, they are often referred to as ERK1/2. Among these MAPKs, ERK1/2 are associated with cell proliferation, cell growth, cell mobility, and cell survival (13), and the Ras-Raf (MAP3K)–MEK (MAP2K)–ERK1/2 (MAPK) signal transduction cascade can be activated by growth factors, osmotic stress, and cytokines (11, 14, 15). To date, more than 160 substrates of ERK1/2 have been discovered from the nucleus and cytosol to the cell membrane (16).
Post-translational modifications (PTMs) have long been documented as a critical means of regulating ERK1/2 activity. Compared with the decades of studies of ERK1/2 phosphorylation, especially at the Thr-202/Tyr-204 sites in ERK1 and Thr-185/Tyr-187 in ERK2 of the Thr-Xaa-Tyr motif, the studies of other PTMs of ERK1/2, including acetylation, methylation, and ubiquitination, are just emerging. Recently, two lysine sites at the ERK1 C terminus, Lys-302 and Lys-361, have been revealed to be tri-methylated, and methylation of ERK1 enhances its phosphorylation (17). Arg-309 of ERK1 has also been reported as a methylation site via a proteome-wide analysis, yet the function of this site is not clear (18). Ubiquitination of ERK1/2 has been reported from three different proteome-wide analyses, but the role of ubiquitination in ERK1/2 still remains elusive (19–21). Likewise, one ERK1 acetylation site has been identified by SILAC assays, but the function of this site still remains to be determined (22). Lately, several reports have shown that when cell lines, including A549, MB361, BT474, MV4-11, PC-3, SKBR-3, HN-9, and SQ20B, were treated with HDAC6 inhibitors, the level of phosphorylated ERK1/2 decreased (23–26), suggesting that acetylation of ERK1/2 compromises their activities and that HDAC6 inhibition may down-regulate ERK1/2 activities. However, the mechanisms underlying this observation are not clear.
Our previous study showed that ERK1 phosphorylates HDAC6 at its Ser-1035 site, and phosphorylation of this site increases HDAC6's activity toward α-tubulin and stimulates cell migration (27). In this study, we have determined that ERK1/2 are acetylated proteins and that ERK1/2 are novel substrates of HDAC6. Both ERK1/2 show the ability to physically interact with HDAC6 in vitro. Also, both CBP and p300 acetylate ERK1/2. One novel acetylation site, Lys-72, was identified in ERK1 via mass spectrometry analysis, and the acetylation-mimicking mutant of ERK1 exhibits reduced kinase activity, suggesting that the acetylation status of ERK1 plays an important role in regulating ERK1 enzymatic activity.
Results
ERK1/2 interact with HDAC6 directly
We previously showed that ERK1/2 interact with HDAC6 endogenously (27). However, how these proteins interact with each other was unknown. To determine whether ERK1/2 interact with HDAC6 directly or through other proteins, we performed in vitro GST pulldown assays with bacterially-purified HDAC6 and ERK1/2. As shown in Fig. 1A, GST-HDAC6, but not GST, efficiently pulled down His-ERK1. Similarly, as shown in Fig. 1B, GST-HDAC6 also pulled down His-ERK2. Therefore, ERK1/2 physically interact with HDAC6.
Figure 1.

ERK1/2 interact with HDAC6 physically. A, His-ERK1 binds to GST-HDAC6. The GST pulldown assays were performed with indicated proteins as described under the “Experimental procedures.” The proteins pulled down by glutathione-agarose were resolved on SDS-PAGE followed by Western blot analyses using the anti-ERK1/2 antibody (upper panel). Bacterially purified proteins, GST, GST-HDAC6, and His-ERK1, were stained by Coomassie Blue (middle and lower panels). B, His-ERK2 binds to GST-HDAC6. The GST pulldown assays and Coomassie Blue staining were conducted as described in A. IB, immunoblot.
Inhibition or depletion of HDAC6 increases ERK1/2 acetylation
Previously, we demonstrated that ERK1 phosphorylates HDAC6 at the Ser-1035 site (27). Given the fact that HDAC6 is a deacetylase, we interrogated HDAC6 to see if it deacetylates ERK1, in other words, whether ERK1 is a substrate of HDAC6. To this end, we set out to determine whether ERK1 is acetylated. Before using the anti-acetylated lysine (AcK) antibodies to examine ERK1 acetylation, we tested two commercial anti-AcK antibodies from Cell Signaling Technology, Inc. As shown in Fig. 2, both antibodies specifically recognized acetylated BSA but not non-acetylated BSA. We then used these two antibodies in the following experiments. To determine whether ERK1 is acetylated, mammalian expression vector GST-ERK1 was transfected into HEK293T cells. Then the transfected cells were treated with 0, 50, 100, 200, 400, or 600 ng/ml pan-HDAC inhibitor trichostatin A (TSA) 12 h prior to harvest. As shown in Fig. 3A, after normalizing with the total ERK1, the level of acetylated GST-ERK1 was increased as the dosage of TSA was increased. 600 ng/ml TSA increased the level of acetylated GST-ERK1 by 4-fold as compared with a vehicle. We then treated the GST-ERK1-transfected HEK293T cells with 600 ng/ml TSA at 0, 2, 4, 8, 12, and 24 h. As shown in Fig. 3B, the level of acetylated GST-ERK1 increased as the TSA treatment time increased. At the 24-h time point, the longest time point, the level of acetylated GST-ERK1 is nearly 2-fold as compared with that at the 0-h time point. To ensure that TSA is functional (TSA inhibits classes I, II, and IV HDACs, including HDAC6), we also detected the level of acetylated α-tubulin, which is a well-known HDAC6 substrate (4). Using a similar approach, we have shown that ERK2 is acetylated upon TSA treatment (Fig. 3, C and D). Taken together, TSA increases ERK1/2 acetylation in a dose- and time-dependent manner.
Figure 2.

Characterization of the anti-acetylated lysine antibodies. Increasing doses of non-acetylated BSA and acetylated BSA were resolved on SDS-PAGE. Two commercial anti-acetylated lysine antibodies as indicated were used for Western blot analyses. Both short exposures and long exposures were shown. IB, immunoblot.
Figure 3.

Inhibition of HDACs by TSA increases ERK1 and ERK2 acetylation. A, TSA increases ERK1 acetylation in a dose-dependent manner. Left panels, HEK293T cells were transfected with GST-ERK1 followed by treatment with TSA for 12 h with indicated concentrations. GST-ERK1 proteins were then isolated from HEK293T cells as described under the “Experimental procedures.” Western blot analyses were performed with the indicated antibodies. Right panel, acetylated GST-ERK1 and total GST-ERK1 bands were quantified by densitometry using the software ImageLabTM from Bio-Rad. The value of untreated acetylated GST-ERK1 band normalized against the total GST-ERK1 band was designated as 1, and the value of the other normalized treated and acetylated GST-ERK1 bands were indicated as a fold change relative to the untreated one. A bar graph was used to show relative intensity of untreated and treated acetylated GST-ERK1. Four independent experiments were performed. B, TSA increases ERK1 acetylation in a time-dependent manner. Left panels, HEK293T cells were transfected with GST-ERK1 followed by treatment with 600 ng/ml TSA at indicated time points. GST-ERK1 proteins were then isolated from HEK293T cells as described under the “Experimental procedures.” Western blot analyses were performed with the indicated antibodies. Right panel, bar graph was drawn as described in A. Five independent experiments were performed. C and D, TSA increases ERK2 acetylation in a dose- and time-dependent manner. C and D, experiments were performed the same as described in A and B, respectively. C, four independent experiments were performed. D, three independent experiments were performed. Student's t tests were performed with *, p < 0.05; **, p < 0.01. Error bars, S.D. IB, immunoblot.
Next, we set out to determine whether specific inhibition of HDAC6 would increase ERK1/2 acetylation. We treated GST-ERK1- or GST-ERK2-transfected HEK293T cells with an HDAC6-specific inhibitor, ACY-1215, at the concentrations of 0, 0.5, 1, 2, 4, and 6 μg/ml for 12 h. As shown in Fig. 4, ACY-1215 increased the level of acetylated GST-ERK1 and GST-ERK2 by 9- and 4-fold, respectively.
Figure 4.

Inhibition of HDAC6 by ACY-1215 increases ERK1 and ERK2 acetylation. A, ACY-1215 increases ERK1 acetylation in a dose-dependent manner. The experiments were performed the same as described in Fig. 3A, except that ACY-1215 was used for the treatment instead of TSA. B, ACY-1215 increases ERK2 acetylation in a dose-dependent manner. The experiments were performed the same as described in A, except that GST-ERK2 was used for transfection instead of GST-ERK1. A, four independent experiments were performed. B, six independent experiments were performed. Student's t tests were performed with *, p < 0.05; **, p < 0.01; ***, p < 0.001. Error bars, S.D. IB, immunoblot.
To determine whether endogenous ERK1/2 are acetylated, we treated 293T cells with the pharmacological inhibitor TSA or ACY-1215 and examined the level of acetylation of ERK1/2. As shown in Fig. 5, both inhibitors increased the acetylation levels of endogenous ERK1/2, suggesting that inhibition of HDAC6 increases the acetylation of endogenous ERK1/2. To further confirm the role of HDAC6 in regulating ERK1/2 deacetylation, we compared the acetylation level of endogenous ERK1/2 in HDAC6 wildtype and HDAC6 knockout MEFs. As shown in Fig. 6A, the acetylation of endogenous ERK1/2 increased significantly in HDAC6 knockout MEFs as compared with HDAC6 wildtype MEFs. Likewise, the level of endogenous ERK1/2 increased in HDAC6 knockdown A549 cells as compared with that of control A549 cells (Fig. 6B). These results validate that inhibition or depletion of HDAC6's enzymatic activity is responsible for the increase of ERK1/2 acetylation.
Figure 5.

Inhibition of HDACs by TSA and inhibition of HDAC6 by ACY-1215 increase endogenous ERK1 and ERK2 acetylation. A, TSA increases endogenous ERK1/2 acetylation. Left panels, HEK293T cells were treated with a vehicle or 600 ng/ml TSA for 24 h. The anti-AcK antibody was used to immunoprecipitate (IP) acetylated ERK1/2. The immunoprecipitates were resolved on SDS-PAGE, and the anti-ERK1/2 Western blot analysis was performed. 0.5% of whole-cell lysate was used as the input. Western blot analyses were performed using anti-ERK1/2, anti-acetyl–α-tubulin, or α-tubulin antibodies as indicated. Right panel, bar graph was used to show relative intensity of endogenous ERK1 and ERK2. Three independent experiments were performed. B, ACY-1215 increases endogenous ERK1/2 acetylation. The experiments were performed as described in A except that ACY-1215 was used for the treatment instead of TSA. Four independent experiments were performed. Student's t tests were performed with *, p < 0.05; **, p < 0.01. Error bars, S.D. IB, immunoblot.
Figure 6.

Depletion or knockdown of HDAC6 increases endogenous ERK1/2 acetylation. A, knockout of HDAC6 increases ERK1/2 acetylation. Left panels, anti-AcK antibody was used to immunoprecipitate acetylated ERK1/2 in HDAC6 wildtype or HDAC6 knockout MEFs. The immunoprecipitates were resolved on SDS-PAGE, and the anti-ERK1/2 Western blot analysis was performed. 0.5% of whole-cell lysate was used as the input. Western blot analyses were performed using anti-ERK1/2, anti-HDAC6, anti-acetyl–α-tubulin, or α-tubulin antibodies as indicated. Right panel, bar graph was used to show relative intensity of endogenous ERK1 and ERK2. Five independent experiments were performed. B, knockdown of HDAC6 increases ERK1/2 acetylation. The experiments were conducted the same as A except that A549 control and A549 HDAC6-KD pair were used to replace the wildtype and HDAC6KO MEFs pair. Four independent experiments were performed. Student's t tests were performed with *, p < 0.05; **, p < 0.01. Error bars, S.D. IB, immunoblot.
CBP and p300 acetylate ERK1/2 in vivo and in vitro
To determine which HAT acetylates ERK1/2, we tested five HATs, which belong to the following three families: Gcn5-related N-acetyltransferase family (PCAF); MYST family (TIP60 and HBO1); and p300/CBP family (p300 and CBP) (28–30). These HATs were co-expressed with GST-ERK1 in 293T cells. When GST-ERK1 was co-expressed with CBP, the acetylation level of GST-ERK1 was much higher than that with empty vector and other HATs (Fig. 7A). p300 also weakly acetylated ERK1 (Fig. 7A). To further confirm the results, we co-expressed the increasing amounts of CBP or p300 with GST-ERK1. As shown in Fig. 7, B and C, CBP and p300 acetylated GST-ERK1 in a dose-dependent manner. To further confirm CBP and p300's effect on endogenous ERK1, we overexpressed p300 in HEK293T cells and tested the acetylation level of endogenous ERK1. As shown in Fig. 7D, exogenous p300 increased the acetylation of endogenous ERK1. To eliminate the potential influence of endogenous HATs and HDACs in the cells on ERK1 acetylation, we executed in vitro acetylation assays to confirm that CBP is an ERK1 acetyltransferase. As shown in Fig. 7E, recombinant CBP indeed acetylated bacterially purified GST-ERK. Similarly, we have shown that both CBP and p300 promote ERK2 acetylation in cells and that CBP acetylated ERK2 in vitro (Fig. 8, A–D).
Figure 7.

ERK1 is acetylated by CBP and p300 in vivo and in vitro. A, CBP and p300 acetylate ERK1. GST-ERK1 was co-transfected with each of the indicated HAT plasmids into HEK293T cells. GST-ERK1 was pulled down by glutathione-agarose and then the bead-bound proteins were resolved on SDS-PAGE followed by Western blot analyses with anti-acetyl-lysine antibodies. The membrane was then stripped and reblotted with anti-ERK1/2 antibodies. The input was subjected to Western blot analyses with indicated antibodies. B, CBP acetylates ERK1 in a dosage-dependent manner. GST-ERK1 was co-transfected with an increasing amount of HA-CBP plasmids as indicated into HEK293T cells. GST-ERK1 was further pulled down by glutathione-agarose, and then the bead-bound ERK1 was subjected to the anti-acetyl-lysine Western blot analysis. The membrane was then stripped and reprobed with anti-ERK1/2 antibodies. The input was subjected to Western blot analyses with the indicated antibodies. C, p300 acetylates ERK1 in a dosage-dependent manner GST-ERK1 was co-transfected with an increasing amount of HA-p300 plasmids. The experiments were performed as described in B. D, p300 acetylates endogenous ERK1/2. HEK293T cells were transfected with empty vector or HA-p300. The immunoprecipitation assays were carried out with anti-AcK antibodies as described under the “Experimental procedures.” The immunoprecipitates (IP) were subjected to the anti-AcK Western blot analysis. 0.5% of the whole-cell lysate was used as input, which was subjected to Western blot analyses with indicated antibodies. E, recombinant CBP acetylates ERK1 in vitro. Bacterially expressed GST-ERK1 and the catalytic domain of CBP were subjected to in vitro acetylation assays as described under the “Experimental procedures.” An equal amount of GST-ERK1 was incubated with or without 2 μg of the recombinant CBP catalytic domain, and the reactions were analyzed by the anti-AcK Western blot analysis (upper panel). After transfer, the membrane was stained with Ponceau S to confirm the amount and purity of GST-ERK1 (lower panel). A–C, acetylated GST-ERK1 bands were quantified by the software Image LabTM from Bio-Rad and normalized by the total GST-ERK1 bands. The values of the empty vector-transfected Ac-GST-ERK1 bands normalized by total GST-ERK1 were designated as 1, and the density of other HAT-transfected Ac-GST-ERK1 bands was indicated as a fold change relative to the corresponding empty vector transfected bands. D, upper panel, the ERK1/2 bands were quantified by densitometry, and the empty vector-transfected ERK1/2 bands were designated as 1. The density of the HA-p300-transfected ERK1/2 bands were quantified relative to the empty vector-transfected ones. IB, immunoblot.
Figure 8.
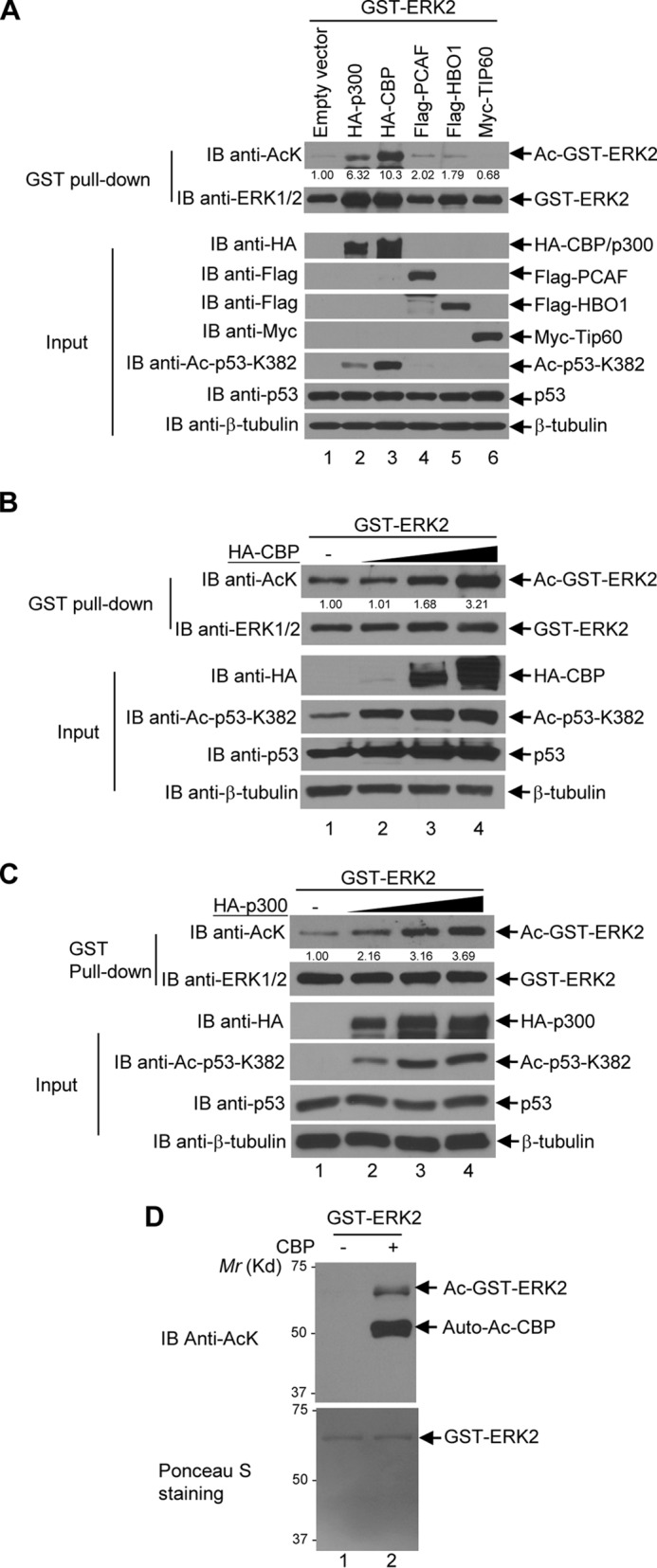
ERK2 is acetylated by CBP and p300 in vivo and in vitro. A, CBP and p300 acetylate ERK2. GST-ERK2 was co-transfected with each of the indicated HAT plasmids into HEK293T cells. GST-ERK2 was pulled down by glutathione-agarose, and then the bead-bound proteins were resolved on SDS-PAGE followed by Western blot analyses with anti-acetyl-lysine antibodies. The membrane was then stripped and reblotted with anti-ERK1/2 antibodies. The input was subjected to Western blot analyses with the indicated antibodies. B, CBP acetylates ERK2 in a dosage-dependent manner. GST-ERK2 was co-transfected with an increasing amount of HA-CBP plasmids as indicated into HEK293T cells. GST-ERK2 was further pulled down by glutathione-agarose, then the bead-bound ERK2 was subjected to anti-acetyl-lysine Western blot analysis. The membrane was then stripped and reprobed with anti-ERK1/2 antibodies. The input was subjected to Western blot analyses with the indicated antibodies. C, p300 acetylates ERK2 in a dosage-dependent manner GST-ERK2 was co-transfected with an increasing amount of HA-p300 plasmids in HEK293T cells. The experiments were performed as described in B. D, recombinant CBP acetylates ERK2 in vitro. Bacterially expressed GST-ERK2 and catalytic domain of CBP were subjected to in vitro acetylation assays as described under the “Experimental procedures.” Equal amount of GST-ERK2 was incubated with or without 2 μg of the recombinant CBP catalytic domain, and the in vitro acetylation reactions were analyzed by the anti-AcK Western blot analysis (upper panel). For the Western blot analysis, after transfer, the membrane was stained with Ponceau S to confirm the amount and purity of GST-ERK2 (lower panel). A–C, the acetylated GST-ERK2 bands were quantified as described in Fig. 7. IB, immunoblot.
HDAC6 deacetylates ERK1 in vivo and in vitro
Because of the low basal acetylation level of ERK1 and to confirm whether HDAC6 deacetylates ERK1, we co-expressed CBP to increase the ERK1 acetylation level. As shown in Fig. 9, A and B, as expected, both CBP and p300 increased acetylation of GST-ERK1, whereas overexpression of HDAC6 reduced CBP- or p300-mediated ERK1 acetylation. Then we tested whether HDAC6 is able to deacetylate in vitro acetylated GST-ERK1 purified from bacteria. As shown in Fig. 9C, HDAC6 purified from 293T cells could efficiently deacetylate acetylated ERK1 in vitro, suggesting that ERK1 is a bona fide substrate of HDAC6.
Figure 9.

HDAC6 deacetylates ERK1 in vivo and in vitro. A, HDAC6 decreases CBP-induced ERK1 acetylation in vivo. GST-ERK1 was transfected into HEK293T cells alone, with CBP, or with CBP and HDAC6. GST-ERK1 proteins were purified as described under the “Experimental procedures” followed by the anti-AcK Western blot analysis. The membrane was then stripped and then reprobed with anti-ERK1/2 antibodies. The input was subjected to Western blot analyses with indicated antibodies. B, HDAC6 decreases p300-induced ERK1 acetylation in vivo. The experiments were performed as described in A, except that HA-p300 was used to replace HA-CBP. C, HDAC6 deacetylates ERK1 in vitro. Left panels, acetylated bacterially purified GST-ERK1 protein was incubated with or without F-HDAC6 purified from 293T cells as described under the “Experimental procedures” followed by the anti-AcK Western blot analysis. The procedure for generating acetylated GST-ERK1 was described under the “Experimental procedures.” The membrane was stripped and reblotted with the anti-ERK1/2 antibody. Right panel, relative intensity of acetylated GST-ERK1 band was quantified by densitometry against the total ERK1 band shown in a bar graph. This experiment was repeated three times. Student's t tests were performed with ***, p < 0.001. Error bars, S.D. IB, immunoblot.
Acetylation of ERK1 reduces ERK1's enzymatic activity
We next examined whether acetylation of ERK1 affects its enzymatic activity. Because HDAC6 deacetylates ERK1, we then hypothesize that in the absence of HDAC6, ERK1 acetylation would be increased, which may alter ERK1's enzymatic activity. To test this hypothesis, the ERK1 plasmid was transfected into wildtype 293T cells or HDAC6KO 293T cells. Then ERK1 was isolated from these two type of cells, and ERK1's kinase activity was examined using recombinant ELK1 as a substrate. ELK1 is a member of Ets transcription factor family, and several serine and threonine sites of ELK1 can be phosphorylated by ERKs (31). Among these sites, the phosphorylation status of ELK1 Ser-383 is pivotal for ELK1 transcriptional activation (32). Because of this reason, we used ELK1 as the substrate to execute non-radioactive kinase assays to measure the ability of ERK1 to phosphorylate the Ser-383 site in ELK1. As shown in Fig. 10A, ERK1 purified from HDAC6KO cells displayed significantly lower activity than that from wildtype cells. Then we also directly acetylated ERK1 using CBP and investigated the enzymatic activity of vehicle-incubated ERK1 versus CBP-incubated ERK1. As shown in Fig. 10B, CBP-acetylated ERK1 harbored lower enzymatic activity. In summary, we concluded that acetylation of ERK1 decreases its enzymatic activity.
Figure 10.

Acetylated ERK1 exhibits decreased enzymatic activity. A, GST-ERK1 exhibits lower enzymatic activity in 293T-HDAC6KO cells than in 293T wildtype cells. Left panels, GST-ERK1 was transfected into 293T wildtype cells and 293T-HDAC6KO cells. Then the GST-ERK1 protein was purified by GST pulldown followed by kinase assays using recombinant ELK1 as a substrate as described under the “Experimental procedures.” Anti-p-ELK(S383) Western blot was performed. The membrane was then stripped and reprobed with the anti-ELK1 antibody. Anti-ERK1/2, anti-HDAC6, anti-acetyl–α-tubulin, and anti-α-tubulin Western blots were also performed. Right panel, pELK1 bands were quantified by densitometry against total ELK1 bands. The results are shown in a bar graph. The same experiments were repeated five times. B, acetylated ERK1 exhibits decreased enzymatic activity toward ELK1. Left panels, in vitro kinase assays were performed with either vehicle-incubated GST-ERK1 or CBP-incubated GST-ERK1. GST-ERK1 protein was purified from 293T cells. Recombinant ELK1 fusion protein purified from Escherichia coli containing ELK1 residues 307–428 was used as a substrate. The kinase assays were performed as described under the “Experimental procedures.” Anti-pELK1(S383) Western blot was used as a readout for ERK1 activity (upper panel). The membrane was stripped and reprobed with the anti-ELK1 antibody. The anti-AcK Western blot was performed to examine the acetylation status of ERK1 (3rd panel). The membrane was stripped and reprobed with the anti-ERK1/2 antibody. Right panel, relative intensity of ELK1(S383) phosphorylation was plotted as a bar graph. The experiments were repeated five times. Student's t tests were performed with **, p < 0.01; ***, p < 0.001. Error bars, S.D. IB, immunoblot.
ERK1 lysine 72 is acetylated
To detect the acetylation site of ERK1, we prepared samples for mass spectrometry analyses. GST-ERK1 and CBP were co-expressed in HEK293T cells for 36 h. Cells were harvested and lysed in lysis buffer. GST-ERK1 was then pulled down by glutathione-agarose and resolved on SDS-PAGE. The SDS-polyacrylamide gel was stained by Coomassie Blue, and the specific bands were excised. Samples were further digested with chymotrypsin and Lys-C endoproteinase sequentially and subjected to LC-tandem mass spectrometry analysis. Lys-72 was identified as a novel acetylation site of ERK1 (Fig. 11A). Lys-72 is located on the β3-strand of ERK1's N-lobe and is very close to the glycine-rich loop (Fig. 11B). To show the conservation of Lys-72 in ERK1, ERK1 sequences from human to nematode were compared by the T-Coffee alignment program. The alignment results showed that this mass spectrometry-identified ERK1 Lys-72 was highly conserved among mammals and even in zebrafish (Danio rerio), Drosophila, and Caenorhabditis elegans (Fig. 11C), indicating that Lys-72 plays an important role in ERK1 function.
Figure 11.

Lysine 72 site of ERK1 is acetylated. A, Lys-72 site of ERK1 is acetylated. The doubly charged peptide was detected with a mass-to-charge ratio 646.3179, which represents an error of −3.8 ppm. The tandem mass spectrum matched the following sequence, KISPFEHQTY, indicating that the Lys-72 (highlighted in red) was acetylated; the detection of b2 is consistent with this localization. The assignment was made with Sequest with Xcorr score of 2.1 and ΔCN score of 0.19. B, diagram of ERK1's domain structure. The numbers showed the amino acid residues of ERK1. Lysine 72, detected by mass spectrometry as an acetylated site, is indicated in a red circle. Thr-202 and Tyr-204 of the Thr-Xaa-Tyr motif, whose phosphorylation status is critical for ERK1 activity, are shown as blue triangles. The kinase domain is highlighted in light gray. Other important motifs and regions are indicated as well. C, stretch of ERK1 amino acids shows the conservation of Lys-72 in different species from mammals to nematode. The mass spectrometry-detected acetylation site Lys-72 is highlighted in red.
Acetylation mimetic mutant of ERK1 abolishes ERK1 kinase activity toward ELK1
To test whether acetylation status of Lys-72 in ERK1 affects its kinase activity, Lys-72 was mutated to glutamine (an acetylation mimetic mutant, K72Q) or arginine (a deacetylation mimetic mutant, K72R), and the resulting mutants were tested by their phosphorylation status in cells followed by kinase assays. As shown in Fig. 12A, K72Q, but not K72R, displayed a reduced level of phosphorylation as compared with wildtype ERK1, implying that the K72Q mutant exhibits a diminished enzymatic activity. To confirm this notion, the kinase assay using the most thoroughly studied ERK1 substrate, ELK1, was performed. As shown in Fig. 12B, the acetylation mimetic mutant, ERK1 (K72Q), displayed a significantly reduced kinase activity toward ELK1 as compared with the deacetylation mimetic mutant, ERK1 (K72R), and the wildtype of ERK1.
Figure 12.

Acetylation-mimicking mutant of ERK1(K72Q) decreases the ERK1 kinase activity. A, acetylation-mimicking mutant of ERK1(K72Q) displays decreased ERK1 phosphorylation in HEK293T cells. The GST-tagged dominant-negative, wildtype, K72Q, or K72R ERK1 was transfected into HEK293T cells, and GST-tagged proteins were purified as described under the “Experimental procedures.” Glutathione-agarose– bound proteins were analyzed by the anti-phospho-ERK1/2 Western blot analysis. The membrane was then stripped and reprobed with anti-ERK1/2 antibodies. B, K72Q mutant of ERK1 exhibits a decreased kinase activity toward ELK1. Dominant-negative, wildtype, K72Q, or K72R GST-ERK1 was transfected into HEK293T cells. The GST-tagged proteins were purified as described under the “Experimental procedures.” The glutathione-agarose–bound proteins were subjected to non-radioactive in vitro kinase assays using recombinant ELK1 as a substrate. The phosphorylation of ELK1 was measured by anti-phospho-ELK1 (Ser-383) Western blot analysis. The anti-ERK1/2 Western blot analysis was also performed. C, acetylation-mimicking mutant of ERK1(K72Q) exhibits decreased luciferase activity. HeLa cells were transfected with indicated plasmids. Luciferase reporter assays were performed with the dual-luciferase reporter kit (Promega) as described under the “Experimental procedures.” The expression of those plasmids was examined by anti-ELK1, anti-HA, anti-ERK1/2, and anti-β-actin Western blot analyses as indicated. This experiment was repeated three times. Student's t tests were performed with *, p < 0.05; ***, p < 0.001, ns, not significant. Error bars, S.D. D, potential structural effects of Lys-72 mutation and acetylation. Ribbon diagram of ERK1 structure (Protein Data Bank code 2ZOQ) and a close-up view of Lys-72 interaction (right panel). Lys-72 and its interacting residues (Asp-117 and Tyr-119) are depicted by sticks with their carbon atoms colored in gray and cyan, respectively. Residues Thr-202 and phosphorylated Tyr-204 indicate the location of the activation loop. The ATP-binding site is indicated by binding of the inhibitor 5-iodotubercidin (5ID). Hydrogen bonds are illustrated as red broken lines. IB, immunoblot.
To further demonstrate the impact of ERK1-K72 acetylation in vivo, we monitored activity of ELK1 in a reporter assay. The luciferase construct, (ELK1)2-TATA-Luc, being used in this assay was a kind gift from Dr. Manohar Ratnam. In this construct, the cis-element preferred by ELK1 was placed as two tandem repeat elements upstream of a minimal TATA-dependent Firefly luciferase promoter (33). Wildtype, K72Q, and K72R of ERK1 were examined by their ability to activate ELK1-mediated transcription by luciferase assays in HeLa cells. The pRL plasmid encoding Renilla luciferase was also transfected in HeLa cells together with the above constructs. The Renilla reading was then used to normalize the Firefly luciferase reading. As shown in Fig. 12C, the ELK1-dependent promoter activity is significantly lower in acetylation mimetic mutant ERK1(K72Q)-transfected cells than in wildtype or deacetylation mimetic mutant ERK1(K72R)-transfected cells, indicating that acetylation at Lys-72 site reduces ERK1's activity to activate ELK1-mediated transcription.
To determine how acetylation/deacetylation of Lys-72 regulates ERK1 kinase activity, we examined ERK1's crystal structure. Lys-72 is located near the ATP-binding site and stabilizes one wall of the ATP-binding site via intramolecular contacts. In particular, Lys-72 forms a salt bridge with Asp-117 and links to Tyr-119 with a hydrogen bond. Acetylation mimetic Lys-72 would break the contacts to both Asp-117 and Tyr-119 and might change the conformation of the ATP-binding site leading to reduced enzymatic activity of ERK1 (Fig. 12D).
Discussion
In this study, we have demonstrated that the acetylation/deacetylation status of Lys-72 in ERK1 regulates its enzymatic activity. For the first time, we have revealed that ERK1 and ERK2 are acetylated proteins and are novel substrates of the deacetylase HDAC6 and the acetyltransferases CBP and p300. We have also discovered that Lys-72 of ERK1 is a novel acetylation site. Furthermore, we have shown that the ERK1 Lys-72 acetylation-mimicking mutant (K72Q) displayed reduced kinase activity as compared with the wildtype and deacetylation-mimicking mutant (K72R). Overall, our results suggest that HDAC6/CBP and p300 govern ERK1's kinase activity via deacetylation/acetylation of Lys-72.
Although we have provided strong evidence that HDAC6 deacetylates ERK1/2, we cannot rule out the possibility that other class I, II, and IV HDACs can deacetylate ERK1/2. In addition, we tested whether class III HDACs, also called sirtuins, can deacetylate ERK1/2. We found that the sirtuin inhibitor, nicotinamide, increased ERK2, but not ERK1, acetylation (data not shown), suggesting that one or multiple sirtuins may deacetylate ERK2. Further investigations are warranted to study whether other HDACs and sirtuins regulate ERK1/2.
In addition, we have identified a conserved lysine, Lys-72, which is adjacent to a critical ATP-binding site, Lys-71, as a novel acetylation site in ERK1, and the acetylation status of Lys-72 significantly decreases ERK1's enzymatic activity toward a well-known ERK1 substrate, ELK1. According to the structural analysis, the acetylation mimetic mutant of ERK1(K72Q), but not the deacetylation mimetic mutant ERK1(K72R), would block the formation of the salt bridges to Asp-117 and Tyr-119, leading to decreased stability of the β3-strand, diminished ATP binding, and reduced ERK1 kinase activity. Our study is the first to report that the acetylation/deacetylation of a conserved lysine in subdomain II of ERK1 could influence its enzymatic activity. It would be interesting to speculate whether the acetylation/deacetylation of Lys-53 in ERK2, which is equivalent to Lys-72 in ERK1, regulates ERK2's kinase activity, although Lys-53 has not been identified as an acetylation site yet.
More than 2 decades ago, it was demonstrated that Lys-71 within subdomain II is critical for ATP binding (34, 35). Substitution of Lys to Arg at this site therefore abolishes ERK1 kinase activity (35). Interestingly, we found that Lys-71 can be acetylated by mass spectrometry analysis (data not shown). It was expected that the replacement of Lys with any other amino acid would ablate ERK1 kinase activity. Because of this reason, Lys to Arg (the deacetylation mimetic mutation) or Lys to Glu (the acetylation mimetic mutation) substitution of Lys-71 would not tell us how deacetylation/acetylation regulates ATP binding and ERK1 enzymatic activity. Future studies using a special tRNA synthetase capable of binding Nϵ-acetyl-lysine to synthesize ERK1 with acetylated Lys-72 may elucidate the role of the acetylation of this site in ERK1 function.
However, it is intriguing that acetylation of Lys-53, a homologous site of ERK1's Lys-71, in p38 augments p38's kinase activity (36). Moreover, Lys-52 in ERK2 and Lys-55 in JNK1 and JNK2 are also homologous to ERK1's Lys-71 (36, 37), but whether these sites are acetylated remains to be determined. It is tempting to hypothesize that the acetylation/deacetylation of the two conserved lysines in subdomain II (in the case of ERK1, Lys-71 and Lys-72), which either bind to ATP or form salt bridges to affect ATP binding, is a strategy employed by HATs and HDACs to fine-tune the enzymatic activities of MAPKs.
As the main moderator in the downstream of the MAPK pathway, ERK1/2 are emerging as alternative targets, especially when inhibitors of the upstream kinases become resistant to patients (38). There are several ERK1/2-specific inhibitors being used to combat the resistance to EGFR, Raf, or MEK inhibitors in clinical trials. HDAC6-specific inhibitors are also being tested in clinical trials. Most of these trials were conducted with other anti-cancer drugs (33, 39).4–11 The combination of HDAC inhibitors and ERK1/2 pathway inhibitors have shown synergistic cell killing (40–44). Here, we show that inhibition of HDAC6 down-regulates ERK1's enzymatic activity, suggesting that the combination of HDAC6 inhibitors and ERK1/2 inhibitors may be a promising strategy to overcome the resistance to EGFR, Raf, or MEK inhibitors.
Experimental procedures
Antibodies
Anti-acetylated lysine mouse mAb (AcK-103) (catalog no. 9681), anti-acetylated lysine rabbit polyclonal antibody (catalog no. 9441), anti-acetyl–p53 (Lys-382) (catalog no. 2525), anti-p44/42 (ERK1/2) (catalog no. 9102), anti-phospho-p44/42 (ERK1/2) (Thr-202/Tyr-204) (catalog no. 9101), anti-ELK-1 (catalog no. 9182), anti-α-tubulin (catalog no. 2125), anti-α-tubulin (catalog no. 2146), and anti-β-actin (catalog no. 4967) antibodies were purchased from Cell Signaling Technology, Inc. The anti-FLAG M2 antibody (F1804), anti-FLAG® M2 affinity gel antibody (A2220), and anti-Myc antibody (C3956) were purchased from Sigma. Anti-p53 (sc-126), anti-p-ELK-1(B4) (sc-8406), and anti-HDAC6 (sc-11420) antibodies were purchased from Santa Cruz Biotechnology. The anti-HA antibody (16B12) was purchased from Covance.
Chemicals and reagents
Q5® high-fidelity DNA polymerase (catalog no. M0491S), BamHI (catalog no. R0136S), NotI (catalog no. R0189S), XhoI (catalog no. R0146S), SalI (catalog no. R0138S), and SpeI (catalog no. R0133S) restriction enzymes were purchased from New England Biolabs. Protease inhibitor mixture (catalog no. 11836170001) was purchased from Roche Applied Science. Protein G-agarose (catalog no. 15920-010) and Western blotting substrates (catalog no. 32106) were purchased from Thermo Fisher Scientific. Glutathione-agarose (catalog no. G4510) and trichostatin A (catalog no. T8552) were purchased from Sigma. Ni2+-NTA-agarose (catalog no. 635659) was purchased from Clontech. ELK-1 fusion protein (catalog no. 9184) was purchased from Cell Signaling Technology, Inc. Recombinant CBP protein (catalog no. BML-SE452-0100) was purchased from Enzo Life Sciences. ACY-1215 was purchased from Selleckchem. Dual-Luciferase® reporter assay system was purchased from Promega.
Plasmids construction
pGEX-4T-1-HDAC6 was generated by PCR using HA-HDAC6-F (5) as the template and the following primers: GST-HD6-F 5′-CCCGTCGACTCATGACCTCAACCGGCCAGGA-3′ (SalI) and GST-HD6-R 5′-TGCGGCCGCTTAGTGTGGGTGGGGCATATC-3′ (NotI). The PCR product was inserted into the SalI and NotI site of the pGEX-4T-1 vector to generate pGEX-4T-1-HDAC6. pLEX-GST-ERK1 was generated from the pGEX-ERK1 plasmids described in Williams et al. (27). Briefly, ERK1 cDNA was isolated from the pDONR223-MAPK3 (Addgene plasmid 23509) vector by PCR using the following primers: ERK1-F 5′-CCCGGATCCATGGCGGCGGCGGCGGCTCAG-3′ (BamHI) and ERK1-R 5′-GGGCTCGAGCTAGGGGGCCTCCAGCACTCC-3′ (XhoI). The PCR product was then inserted into the BamHI and XhoI sites into the pGEX-4T-1 vector to generate pGEX-ERK1. GST-ERK1 cDNA was isolated by PCR using pGEX-ERK1 as the template and the following primers: GST-SpeI 5′-CCCACTAGTATGTCCCCTATACTAGGTTATTG-3′ (SpeI) and ERK1-R 5′-GGGCTCGAGCTAGGGGGCCTCCAGCACTCC-3′ (XhoI). The PCR product was then inserted into the SpeI and XhoI sites of the pLEX-MCS vector to generate pLEX-GST-ERK1. The pLEX-GST-DN-ERK1 plasmid was described in Williams et al. (27). The mammalian expression GST-tagged ERK2 construct pLEX-GST-ERK2 was generated as follows. Human wildtype ERK2 cDNA was amplified from pDONR223-MAPK1 (Addgene plasmid 23498) by PCR, and the PCR product was inserted into pGEX-4T-1 between SalI and NotI sites to construct pGEX-4T-1-GST-ERK2. The primers used were 5′-GCGGTCGACTTATGGCGGCGGCGGCGGCGGGC-3′ (SalI) and 5′-CGCGCGGCCGCTCAAGATCTGTATCCTGGCTGGAATC-3′ (NotI). pGEX-4T-1-GST-ERK2 was then used as a template for PCR using the following primers: 5′-CCCACTAGTATGTCCCCTATACTAGGTTATTG-3′ (SpeI) and 5′-CGCGCGGCCGCTCAAGATCTGTATCCTGGCTGGAATC-3′ (NotI) to generate GST-ERK2 cDNA, which was further subcloned into the pLEX-MCS vector (Thermo Fisher Scientific, catalog no. OHS4735) between SpeI and NotI sites to generate pLEX-GST-ERK2.
Then we generated the mammalian expression K72Q and K72R mutants of ERK1 in the pLEX-MCS vector as described below. The GST-ERK1 cDNA was generated by PCR using pGEX-4T-1-ERK1 as a template and the following primers: 5′-CCCACTAGTATGTCCCCTATACTAGGTTATTG-3′ (SpeI) and 5′-GGGCTCGAGCTAGGGGGCCTCCAGCACTCC-3′ (XhoI). Then the PCR product was inserted between SpeI and XhoI sites to generate pcDNA3.1/Hygro-GST-ERK1. The pcDNA3.1/Hygro-GST-ERK1 (K72Q) and pcDNA3.1/Hygro-GST-ERK1 (K72R) plasmids were made by site-directed mutagenesis using the following primers: K72Q, 5′-GTGGCCATCAAGCAGATCAGCCCCTTC-3′ and 5′-GATGGCCACGCGAGTCTTGCGCA-3′; K72R, 5′-GTGGCCATCAAGAGGATCAGCCCCTTC-3′ and 5′-GATGGCCACGCGAGTCTTGCGCA-3′. The PCR cycle for site-directed mutagenesis was as follows: 95 °C 5 min, 95 °C 3 min, 55 °C 1 min, 72 °C 6 min for 16 cycles and finally 72 °C 10 min. Then the pcDNA3.1/Hygro-GST-ERK1 (K72Q) and pcDNA3.1/Hygro-GST-ERK1 (K72R) plasmids were cut with SpeI and XhoI to isolate the cDNA fragments of GST-ERK1(K72Q) and GST-ERK1(K72R), which were subcloned into SpeI and XhoI sites of the pLEX-MCS vector to generate pLEX-GST-ERK1(K72Q) and pLEX-GST-ERK1(K72R), respectively. To generate the His6-tagged ERK1, pET-ERK1, the cDNA of ERK1 was transferred from pGEX-4T-1-ERK1 (27) to the pET28a vector by digesting with BamHI and XhoI to generate pET28a-ERK1. To generate pET28a-ERK2, the cDNA of ERK2 was excised from the pGEX-4T-1-GST-ERK2 vector as described above. Then the insert was subcloned into the pET28a vector via SalI and NotI sites. pCMV-ELK1 purchased from Origene (SC116858) contains untagged human ELK1 (member of ETS oncogene family (ELK1), transcript variant 2, NM_005229.2). The construct was made by inserting the cDNA of ELK1 into the NotI site of the pCMV6-XL4 vector.
Cell culture and cell lines
HEK293T cells were grown in Dulbecco's modified Eagle's medium (DMEM) with 10% bovine calf serum. HDAC6 wildtype and HDAC6 knockout mouse embryonic fibroblasts (MEFs) were cultured in DMEM with 10% fetal bovine serum. HDAC6 control and HDAC6 knockdown A549 cells were cultured in RPMI 1640 medium with 10% fetal bovine serum. All cell lines were cultured in the medium with penicillin (100 units/ml) and streptomycin (100 g/ml) and in the incubators with 5% CO2 at 37 °C.
Establishment of HDAC6 stable knockdown A549 cells and HDAC6 knockout 293T cells
A549 scramble and HDAC6 knockdown stable cell lines were clonally selected by 0.5 μg/ml puromycin. Briefly, A549 cells were transiently transfected with control vector pRS (catalog no. TR20003) or shRNA vector against HDAC6 (recognize sequence 5-AGGTCTACTGTGGTCGTTACATCAATGGC-3′, tube ID: TI349960, ORIGENE). Twenty-four hours after transfection, cells were split to duplicate plates of 1:20 in RPMI 1640 medium containing 0.5 μg/ml puromycin. Puromycin was replenished every 2 days to maintain a sufficient level of selection pressure. The well-isolated single clones were transferred into 24-well plates. The knockdown effect was verified by Western blot analysis using anti-HDAC6 antibodies.
HDAC6 knockout 293T cells were created using CRISPR/Cas9 (clustered regularly interspaced short palindromic repeats) method. Briefly, the guide RNA targeting HDAC6 exon 5 (5′-GAAAGGACACGCAGCGATCT-3′) was selected and inserted into the LentiCRISPRv2 vector (Addgene plasmid 52961) to generate the HDAC6KO vector that also expressed the codon-optimized Cas9 protein as well as the puromycin resistance gene. The 293T cells infected with the HDAC6-KO virus were selected for stable clones using puromycin at 1 μg/ml. The HDAC6-knockout clones were screened by anti-HDAC6 (H-300) Western blot analysis.
Immunoprecipitation and immunoblotting
For immunoprecipitation assays, cells were lysed in lysis buffer (25 mm Tris-HCl, pH 7.4, 150 mm NaCl, 1 mm EDTA, 1% Nonidet P-40, 5% glycerol and protease inhibitor mixture (11836170001, Roche Applied Science)). Then the lysates were first pre-cleared with protein G-agarose for 30 min at 4 °C with rotation and then incubated with interested antibodies overnight at 4 °C on a rocker, followed by protein G-agarose incubation for 4 h at 4 °C with rotation. The samples were washed by washing buffer (TBS with 0.5% Triton X-100) four times and subjected to further analyses. For immunoblotting, the samples were resolved on SDS-PAGE and transferred to nitrocellulose membranes. The membranes were blocked by 5% nonfat milk in TBST (0.1% Tween 20 in TBS (20 mm Tris, pH 7.5, 150 mm NaCl)) for 1 h at room temperature. Then the membranes were incubated with a first antibody overnight at 4 °C and washed with TBST three times. Membranes were further incubated with an appropriate secondary antibody that was conjugated with horseradish peroxidase for 2–4 h at room temperature and washed with TBST three times. Proteins on the membranes were detected by Western blotting substrates and exposed to the X-ray films. To re-blot the same membrane with a different antibody, the membrane would be stripped with stripping buffer (2% SDS, 62.5 mm Tris-HCl, pH 6.8, and 0.8% β-mercaptoethanol) for 45 min at 50 °C. The membrane would then be re-blocked by 5% non-fat milk in TBST and subjected to the above immunoblotting procedures.
Purification of GST-tagged ERK proteins from 293T cells
pLEX-GST-ERK1, pLEX-GST-ERK2, pLEX-GST-ERK1(K72Q), or pLEX-GST-ERK1 (K72R) was transfected into 293T cells. Cells were lysed in lysis buffer (25 mm Tris-HCl, pH 7.4, 150 mm NaCl, 1 mm EDTA, 1% Nonidet P-40, 5% glycerol) followed by incubation with glutathione-agarose at 4 °C overnight. The agarose beads were spun down and washed four times with cold wash buffer (TBS with 0.5% Triton X-100). The agarose-bound GST proteins were subjected to the assays that follow.
Purification of His-ERK1/2 proteins
BL21 bacteria harboring His-ERK1 or His-ERK2 were grown in the log phase and induced with isopropyl β-d-1-thiogalactopyranoside (IPTG) at 4 h. The cell pellets were lysed in bacteria lysis buffer (10 mm Tris-HCl, pH 8.0, 150 mm NaCl, 1 mm EDTA, 5 mm DTT, 1.5% N-lauroylsarcosine, and 1% Triton X-100). The mixtures were further sonicated in appropriate conditions until the mixtures were clear. The mixtures were centrifuged at 10,000 × g at 4 °C for 5 min, and the supernatants were incubated with Ni2+-NTA-agarose (catalog no. 635659, Clontech) at 4 °C overnight. The agarose was washed four times with cold washing buffer (TBS with 0.5% Triton X-100) after incubation. His-ERK1 and His-ERK2 were further eluted by 250 mm imidazole from Ni2+-NTA-agarose for next assays.
GST pulldown assay
BL21 bacteria harboring GST or GST-HDAC6 were grown in the log phase and induced with IPTG at 4 h. The cell pellets were resuspended in bacteria lysis buffer (10 mm Tris-HCl, pH 8.0, 150 mm NaCl, 1 mm EDTA, 5 mm DTT, 1.5% N-lauroylsarcosine, and 1% Triton X-100) and sonicated in appropriate conditions until the mixtures were clear. The mixtures were centrifuged at 10,000 × g at 4 °C for 5 min, and the supernatants were incubated with glutathione-agarose to isolate bead-bound GST or GST-HDAC6. The purified His-ERK1 or His-ERK2, described under the “Experimental Procedures,” was incubated with either glutathione-agarose–bound GST or GST-HDAC6 for 30 min at 4 °C on a rotator. After incubation, the glutathione-agarose beads were spun down and washed four times with cold washing buffer (TBS with 0.5% Triton X-100). The samples were then subjected to Western blot analyses.
Non-radioactive in vitro kinase assay
Wildtype, dominant-negative K72Q or K72R mutant ERK1 proteins were overexpressed in 293T cells, and the cells were lysed in lysis buffer (25 mm Tris-HCl, pH 7.4, 150 mm NaCl, 1 mm EDTA, 1% Nonidet P-40, 5% glycerol and protease inhibitor mixture (11836170001, Roche Applied Science)). These GST fusion ERK1 proteins were purified as described under the “Experimental Procedures.” GST fusion proteins were washed once by 1× kinase buffer (5 mm Tris, pH 7.5, 5 mm β-glycerol phosphate, 2 mm DTT, 0.1 mm Na3VO4, 10 mm MgCl2) before the reaction. All glutathione-agarose–bound proteins were incubated with 250 ng of ELK1 (9184, Cell Signaling Technology, Inc.), 200 μm ATP, and 1× kinase assay buffer for 30 min at 30 °C. The reactions were stopped by adding 5× SDS sample loading buffer and heating for 5 min at 100 °C. The samples were subjected to Western blot analyses, and the anti-phospho-Ser-383–ELK1 antibody was used for phospho-ELK1 detection.
In vitro acetylation assay
Bacterial GST-ERK1 or GST-ERK2 protein was purified by glutathione-agarose as described in the GST pulldown assay. The glutathione-agarose–bound GST-ERK1 or GST-ERK2 protein was mixed with 2 μg of recombinant CBP proteins (BML-SE452–0100, Enzo Life Sciences), 100 nm acetyl-CoA in 1× acetylation buffer (50 mm Tris-HCl, pH 8.0, 10% glycerol, 0.1 mm EDTA, 1 mm DTT, 10 mm sodium butyrate, and protease inhibitor mixture), and the mixtures were incubated at 30 °C for 60 min. Then the agarose beads were washed with washing buffer (TBS with 0.5% Triton X-100) three times, and the reactions were terminated by adding 5× SDS sample loading buffer and boiling at 100 °C for 5 min. The samples were then subjected to Western blot analyses.
Samples preparation for mass spectrometry
GST-ERK1 and HA-CBP were co-expressed in 293T cells for 36 h. Cells were harvested and lysed in lysis buffer. GST-ERK1 was then pulled down by glutathione-agarose and analyzed by SDS-PAGE. The gel was stained by Coomassie Blue, and the specific bands were excised. The excised gels were further digested with chymotrypsin and Lys-C endoproteinase sequentially and subjected to mass spectrometry analysis.
LC-MS/MS analysis
A nanoflow ultra-high-performance-liquid chromatograph (RSLC, Dionex, Sunnyvale, CA) coupled to an electrospray bench top orbitrap mass spectrometer (Q-Exactive plus, Thermo Fisher Scientific, San Jose, CA) was used for tandem mass spectrometry peptide sequencing experiments. The sample was first loaded onto a pre-column (2-cm × 100-μm inner diameter packed with C18 reversed-phase resin, 5 μm, 100 Å) and washed for 8 min with aqueous 2% acetonitrile and 0.04% trifluoroacetic acid. The trapped peptides were eluted onto the analytical column (C18, 75-μm inner diameter × 50 cm, 2 μm, 100 Å, Dionex, Sunnyvale, CA). The 90-min gradient was programmed as follows: 95% solvent A (2% acetonitrile + 0.1% formic acid) for 8 min, solvent B (90% acetonitrile + 0.1% formic acid) from 5 to 38.5% in 60 min, then solvent B from 50 to 90% B in 7 min and held at 90% for 5 min, followed by solvent B from 90 to 5% in 1 min and re-equilibrate for 10 min. The flow rate on analytical column was 300 nl/min. Sixteen tandem mass spectra were collected in a data-dependent manner following each survey scan. Both MS and MS/MS scans were performed in Orbitrap to obtain accurate mass measurement using 60 s exclusion for previously sampled peptide peaks.
Data analysis
Sequest (45) and Mascot (46) searches were performed against the Swiss-Prot human database. Two trypsin missed cleavages were allowed, and the precursor mass tolerance was 10 ppm. MS/MS mass tolerance was 0.05 Da. Dynamic modifications included carbamidomethylation (Cys), oxidation (Met), and acetylation (Lys). Both MASCOT and SEQUEST search results were summarized in Scaffold 4.4.
Luciferase reporter assay
Briefly, HeLa cells were seeded in 24-well plates overnight prior to transfection. The cells were transfected with plasmids of (ELK1)2-TATA-Luc (33), pCMV-ELK1 (Origene), HA-MEK (S218D/S222D), GST-ERK1-WT, GST-ERK1-K72Q, or GST-ERK1-K72R as shown in Fig. 12C. The pRL plasmid encoding Renilla luciferase gene was also transfected. 24 h post-transfection, cells were lysed with 100 μl of 1× lysis buffer (Dual-Luciferase reporter, E1910/Promega). 10 μl of lysates was taken out to mix with 50 μl of LARII and 50 μl of “STOP & Glo” for firefly and Renilla reading, sequentially, in a white opaque 96-well plate using a BioTek Synergy 2 Multi-Mode reader.
In vitro deacetylation assay
F-HDAC6 was overexpressed in 293T cells and purified with anti-FLAG® M2 affinity gel by following the vendor's instruction. Purified F-HDAC6 was resolved on SDS-PAGE and stained by Coomassie Blue to verify the amount and purity. Purified GST-ERK1 was first acetylated by CBP in vitro, and then the acetylated GST-ERK1 was incubated with or without F-HDAC6 in the 1× deacetylation buffer (20 mm Tris, pH 8.0, 150 mm NaCl, and 10% glycerol) at 30 °C for 2 h. The reaction was stopped by adding 5× SDS-PAGE sample buffer and boiled in 95 °C for 10 min.
Author contributions
J.-Y. W. performed most of the experiments. S. X. helped make some constructs. M. Z. did the luciferase reporter assays. B. F., H. H., O. K. K., and Y. Z. did the mass spectrometry analyses. Z. Y. did the structural analysis of ERK1. W. B. and G. B. provided the critical reagents and helped design the experiments. J.-Y. W. and X. M. Z. wrote the manuscript. All authors reviewed the results and approved the final version of the manuscript.
Acknowledgments
We thank Victoria Izumi at the H. Lee Moffitt Cancer Center Proteomics Facility and Dr. Paul Stemmer at Wayne State University Proteomics Core Facility for the mass spectrometry analysis; Dr. Edward Seto for the HBO1 plasmid; Dr. Tso-Pang Yao for the CBP construct; Dr. Manohar Ratnam and Dr. Rayna Rosati for the plasmids of pCMV-ELK1 and (ELK1)2-TATA-Luc and assistance with the luciferase reporter assays; Dr. Jie Wu for HA-MEK(S218/222D) plasmid and discussion; and Joshua Haakenson for proofreading the manuscript.
This work was supported in part by National Institutes of Health Grant R01CA164147 and by Karmanos Cancer Institute start-up funds (to X. M. Z.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Liu, J. (2016) https://clinicaltrials.gov/ct2/show/NCT02661815. A phase 1b study of paclitaxel and ricolinostat for the treatment of gynecological cancer. (Please note that the JBC is not responsible for the long-term archiving and maintenance of this site or any other third party–hosted site.)
Kalinsky, K. (2015) https://clinicaltrials.gov/ct2/show/NCT02632071. ACY-1215 + Nab-paclitaxel in metastatic breast cancer. (Please note that the JBC is not responsible for the long-term archiving and maintenance of this site or any other third party hosted site.)
Celgene (2014) https://clinicaltrials.gov/ct2/show/NCT02189343. Phase 1b study evaluating ACY-1215 (ricolinostat) in combination with pomalidomide and dexamethasone in relapsed or relapsed-and-refractory multiple myeloma. (Please note that the JBC is not responsible for the long-term archiving and maintenance of this site or any other third party hosted site.)
Amengual, J. (2014) https://clinicaltrials.gov/ct2/show/NCT02091063. ACY-1215 for relapsed/refractory lymphoid malignancies. (Please note that the JBC is not responsible for the long-term archiving and maintenance of this site or any other third party hosted site.)
Acetylon Pharmaceuticals Incorporated (2014) https://clinicaltrials.gov/ct2/show/NCT02088398. Alternative 10 mg/ml liquid formulation of ACY 1215 (ricolinostat) in healthy subjects. (Please note that the JBC is not responsible for the long-term archiving and maintenance of this site or any other third party hosted site.)
Celgene (2013) https://clinicaltrials.gov/ct2/show/NCT01997840. ACY-1215 (ricolinostat) in combination with pomalidomide and low-dose dex in relapsed-and-refractory multiple myeloma. (Please note that the JBC is not responsible for the long-term archiving and maintenance of this site or any other third party hosted site.)
Celgene (2012) https://clinicaltrials.gov/ct2/show/NCT01583283. Study of ACY-1215 in combination with lenalidomide, and dexamethasone in multiple myeloma. (Please note that the JBC is not responsible for the long-term archiving and maintenance of this site or any other third party hosted site.)
Celgene (2011) https://clinicaltrials.gov/ct2/show/NCT01323751. Study of ACY-1215 alone and in combination with bortezomib and dexamethasone in multiple myeloma. (Please note that the JBC is not responsible for the long-term archiving and maintenance of this site or any other third party hosted site.)
- HDAC
- histone deacetylase
- ERK1/2
- extracellular signal-regulated kinase 1/2
- MAPK
- mitogen-activated protein kinase
- JNK
- c-Jun N-terminal kinase
- NLK
- Nemo-like kinase
- PTM
- post-translational modification
- MEF
- mouse embryonic fibroblast
- HAT
- histone acetyltransferase
- Rpd3
- reduced potassium dependency 3
- MAP3K
- MAPK kinase kinase
- MAP2K
- MAPK kinase
- TSA
- trichostatin A
- EGFR
- epidermal growth factor receptor
- GST
- glutathione S-transferase
- CBP
- cAMP-response element-binding protein-binding protein
- IPTG
- isopropyl β-d-1-thiogalactopyranoside
- DMEM
- Dulbecco's modified Eagle's medium
- Ni2+-NTA
- Ni2+-nitrilotriacetic acid
- MEK
- MAPK/ERK kinase.
References
- 1. Yang X. J., and Seto E. (2007) HATs and HDACs: from structure, function and regulation to novel strategies for therapy and prevention. Oncogene 26, 5310–5318 10.1038/sj.onc.1210599 [DOI] [PubMed] [Google Scholar]
- 2. Yang X. J., and Seto E. (2008) The Rpd3/Hda1 family of lysine deacetylases: from bacteria and yeast to mice and men. Nat. Rev. Mol. Cell Biol. 9, 206–218 10.1038/nrm2346 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Seto E., and Yoshida M. (2014) Erasers of histone acetylation: the histone deacetylase enzymes. Cold Spring Harb. Perspect. Biol. 6, a018713 10.1101/cshperspect.a018713 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Hubbert C., Guardiola A., Shao R., Kawaguchi Y., Ito A., Nixon A., Yoshida M., Wang X. F., and Yao T. P. (2002) HDAC6 is a microtubule-associated deacetylase. Nature 417, 455–458 10.1038/417455a [DOI] [PubMed] [Google Scholar]
- 5. Zhang X., Yuan Z., Zhang Y., Yong S., Salas-Burgos A., Koomen J., Olashaw N., Parsons J. T., Yang X. J., Dent S. R., Yao T. P., Lane W. S., and Seto E. (2007) HDAC6 modulates cell motility by altering the acetylation level of cortactin. Mol. Cell 27, 197–213 10.1016/j.molcel.2007.05.033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Zhang M., Xiang S., Joo H. Y., Wang L., Williams K. A., Liu W., Hu C., Tong D., Haakenson J., Wang C., Zhang S., Pavlovicz R. E., Jones A., Schmidt K. H., Tang J., et al. (2014) HDAC6 deacetylates and ubiquitinates MSH2 to maintain proper levels of MutSα. Mol. Cell 55, 31–46 10.1016/j.molcel.2014.04.028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Lee Y. S., Lim K. H., Guo X., Kawaguchi Y., Gao Y., Barrientos T., Ordentlich P., Wang X. F., Counter C. M., and Yao T. P. (2008) The cytoplasmic deacetylase HDAC6 is required for efficient oncogenic tumorigenesis. Cancer Res. 68, 7561–7569 10.1158/0008-5472.CAN-08-0188 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Yang M. H., Laurent G., Bause A. S., Spang R., German N., Haigis M. C., and Haigis K. M. (2013) HDAC6 and SIRT2 regulate the acetylation state and oncogenic activity of mutant K-RAS. Mol. Cancer Res. 11, 1072–1077 10.1158/1541-7786.MCR-13-0040-T [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Li Y., Zhang X., Polakiewicz R. D., Yao T. P., and Comb M. J. (2008) HDAC6 is required for epidermal growth factor-induced β-catenin nuclear localization. J. Biol. Chem. 283, 12686–12690 10.1074/jbc.C700185200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Cargnello M., and Roux P. P. (2011) Activation and function of the MAPKs and their substrates, the MAPK-activated protein kinases. Microbiol. Mol. Biol. Rev. 75, 50–83 10.1128/MMBR.00031-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Johnson G. L., and Lapadat R. (2002) Mitogen-activated protein kinase pathways mediated by ERK, JNK, and p38 protein kinases. Science 298, 1911–1912 10.1126/science.1072682 [DOI] [PubMed] [Google Scholar]
- 12. Lloyd A. C. (2006) Distinct functions for ERKs? J. Biol. 5, 13 10.1186/jbiol46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Roskoski R. (2012) ERK1/2 MAP kinases: Structure, function, and regulation. Pharmacol. Res. 66, 105–143 10.1016/j.phrs.2012.04.005 [DOI] [PubMed] [Google Scholar]
- 14. Raman M., Chen W., and Cobb M. H. (2007) Differential regulation and properties of MAPKs. Oncogene 26, 3100–3112 10.1038/sj.onc.1210392 [DOI] [PubMed] [Google Scholar]
- 15. Boulton T. G., Yancopoulos G. D., Gregory J. S., Slaughter C., Moomaw C., Hsu J., and Cobb M. H. (1990) An insulin-stimulated protein kinase similar to yeast kinases involved in cell cycle control. Science 249, 64–67 10.1126/science.2164259 [DOI] [PubMed] [Google Scholar]
- 16. Yoon S., and Seger R. (2006) The extracellular signal-regulated kinase: multiple substrates regulate diverse cellular functions. Growth Factors 24, 21–44 10.1080/02699050500284218 [DOI] [PubMed] [Google Scholar]
- 17. Vougiouklakis T., Sone K., Saloura V., Cho H. S., Suzuki T., Dohmae N., Alachkar H., Nakamura Y., and Hamamoto R. (2015) SUV420H1 enhances the phosphorylation and transcription of ERK1 in cancer cells. Oncotarget 6, 43162–43171 10.18632/oncotarget.6351 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Larsen S. C., Sylvestersen K. B., Mund A., Lyon D., Mullari M., Madsen M. V., Daniel J. A., Jensen L. J., and Nielsen M. L. (2016) Proteome-wide analysis of arginine monomethylation reveals widespread occurrence in human cells. Sci. Signal. 9, rs9 10.1126/scisignal.aaf7329 [DOI] [PubMed] [Google Scholar]
- 19. Kim W., Bennett E. J., Huttlin E. L., Guo A., Li J., Possemato A., Sowa M. E., Rad R., Rush J., Comb M. J., Harper J. W., and Gygi S. P. (2011) Systematic and quantitative assessment of the ubiquitin-modified proteome. Mol. Cell 44, 325–340 10.1016/j.molcel.2011.08.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Mertins P., Qiao J. W., Patel J., Udeshi N. D., Clauser K. R., Mani D. R., Burgess M. W., Gillette M. A., Jaffe J. D., and Carr S. A. (2013) Integrated proteomic analysis of post-translational modifications by serial enrichment. Nat. Methods 10, 634–637 10.1038/nmeth.2518 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Wagner S. A., Beli P., Weinert B. T., Nielsen M. L., Cox J., Mann M., and Choudhary C. (2011) A proteome-wide, quantitative survey of in vivo ubiquitylation sites reveals widespread regulatory roles. Mol. Cell. Proteomics 10, M111.013284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Hornbeck P. V., Zhang B., Murray B., Kornhauser J. M., Latham V., and Skrzypek E. (2015) PhosphoSitePlus, 2014: mutations, PTMs and recalibrations. Nucleic Acids Res. 43, D512–D520 10.1093/nar/gku1267 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Chuang M. J., Wu S. T., Tang S. H., Lai X. M., Lai H. C., Hsu K. H., Sun K. H., Sun G. H., Chang S. Y., Yu D. S., Hsiao P. W., Huang S. M., and Cha T. L. (2013) The HDAC inhibitor LBH589 induces ERK-dependent prometaphase arrest in prostate cancer via HDAC6 inactivation and down-regulation. PLoS One 8, e73401 10.1371/journal.pone.0073401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Inks E. S., Josey B. J., Jesinkey S. R., and Chou C. J. (2012) A novel class of small molecule inhibitors of HDAC6. ACS Chem. Biol. 7, 331–339 10.1021/cb200134p [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Kim I. A., No M., Lee J. M., Shin J. H., Oh J. S., Choi E. J., Kim I. H., Atadja P., and Bernhard E. J. (2009) Epigenetic modulation of radiation response in human cancer cells with activated EGFR or HER-2 signaling: potential role of histone deacetylase 6. Radiother. Oncol. 92, 125–132 10.1016/j.radonc.2009.03.008 [DOI] [PubMed] [Google Scholar]
- 26. Tien S. C., and Chang Z. F. (2014) Oncogenic Shp2 disturbs microtubule regulation to cause HDAC6-dependent ERK hyperactivation. Oncogene 33, 2938–2946 10.1038/onc.2013.241 [DOI] [PubMed] [Google Scholar]
- 27. Williams K. A., Zhang M., Xiang S., Hu C., Wu J. Y., Zhang S., Ryan M., Cox A. D., Der C. J., Fang B., Koomen J., Haura E., Bepler G., Nicosia S. V., Matthias P., et al. (2013) Extracellular signal-regulated kinase (ERK) phosphorylates histone deacetylase 6 (HDAC6) at serine 1035 to stimulate cell migration. J. Biol. Chem. 288, 33156–33170 10.1074/jbc.M113.472506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Marks P., Rifkind R. A., Richon V. M., Breslow R., Miller T., and Kelly W. K. (2001) Histone deacetylases and cancer: causes and therapies. Nat. Rev. Cancer 1, 194–202 10.1038/35106079 [DOI] [PubMed] [Google Scholar]
- 29. Lee K. K., and Workman J. L. (2007) Histone acetyltransferase complexes: one size doesn't fit all. Nat. Rev. Mol. Cell Biol. 8, 284–295 10.1038/nrm2145 [DOI] [PubMed] [Google Scholar]
- 30. Roth S. Y., Denu J. M., and Allis C. D. (2001) Histone acetyltransferases. Annu. Rev. Biochem. 70, 81–120 10.1146/annurev.biochem.70.1.81 [DOI] [PubMed] [Google Scholar]
- 31. Gille H., Kortenjann M., Thomae O., Moomaw C., Slaughter C., Cobb M. H., and Shaw P. E. (1995) ERK phosphorylation potentiates Elk-1-mediated ternary complex formation and transactivation. EMBO J. 14, 951–962 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Marais R., Wynne J., and Treisman R. (1993) The SRF accessory protein Elk-1 contains a growth factor-regulated transcriptional activation domain. Cell 73, 381–393 10.1016/0092-8674(93)90237-K [DOI] [PubMed] [Google Scholar]
- 33. Patki M., Chari V., Sivakumaran S., Gonit M., Trumbly R., and Ratnam M. (2013) The ETS domain transcription factor ELK1 directs a critical component of growth signaling by the androgen receptor in prostate cancer cells. J. Biol. Chem. 288, 11047–11065 10.1074/jbc.M112.438473 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Hanks S. K., Quinn A. M., and Hunter T. (1988) The protein kinase family: conserved features and deduced phylogeny of the catalytic domains. Science 241, 42–52 10.1126/science.3291115 [DOI] [PubMed] [Google Scholar]
- 35. Robbins D. J., Zhen E., Owaki H., Vanderbilt C. A., Ebert D., Geppert T. D., and Cobb M. H. (1993) Regulation and properties of extracellular signal-regulated protein kinases 1 and 2 in vitro. J. Biol. Chem. 268, 5097–5106 [PubMed] [Google Scholar]
- 36. Pillai V. B., Sundaresan N. R., Samant S. A., Wolfgeher D., Trivedi C. M., and Gupta M. P. (2011) Acetylation of a conserved lysine residue in the ATP binding pocket of p38 augments its kinase activity during hypertrophy of cardiomyocytes. Mol. Cell. Biol. 31, 2349–2363 10.1128/MCB.01205-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Robinson M. J., Harkins P. C., Zhang J., Baer R., Haycock J. W., Cobb M. H., and Goldsmith E. J. (1996) Mutation of position 52 in ERK2 creates a nonproductive binding mode for adenosine 5′-triphosphate. Biochemistry 35, 5641–5646 10.1021/bi952723e [DOI] [PubMed] [Google Scholar]
- 38. Samatar A. A., and Poulikakos P. I. (2014) Targeting RAS-ERK signalling in cancer: promises and challenges. Nat. Rev. Drug Discov. 13, 928–942 10.1038/nrd4281 [DOI] [PubMed] [Google Scholar]
- 39. Mevissen T. E., and Komander D. (2017) Mechanisms of deubiquitinase specificity and regulation. Annu. Rev. Biochem. 86, 159–192 10.1146/annurev-biochem-061516-044916 [DOI] [PubMed] [Google Scholar]
- 40. Zhang L., Zhang Y., Mehta A., Boufraqech M., Davis S., Wang J., Tian Z., Yu Z., Boxer M. B., Kiefer J. A., Copland J. A., Smallridge R. C., Li Z., Shen M., and Kebebew E. (2015) Dual inhibition of HDAC and EGFR signaling with CUDC-101 induces potent suppression of tumor growth and metastasis in anaplastic thyroid cancer. Oncotarget 6, 9073–9085 10.18632/oncotarget.3268 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Carson R., Celtikci B., Fenning C., Javadi A., Crawford N., Carbonell L. P., Lawler M., Longley D. B., Johnston P. G., and Van Schaeybroeck S. (2015) HDAC inhibition overcomes acute resistance to MEK inhibition in BRAF-mutant colorectal cancer by downregulation of c-FLIPL. Clin. Cancer Res. 21, 3230–3240 10.1158/1078-0432.CCR-14-2701 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Ahn M. Y., Ahn J. W., Kim H. S., Lee J., and Yoon J. H. (2015) Apicidin inhibits cell growth by downregulating IGF-1R in salivary mucoepidermoid carcinoma cells. Oncol. Rep. 33, 1899–1907 10.3892/or.2015.3776 [DOI] [PubMed] [Google Scholar]
- 43. Yu C., Friday B. B., Lai J. P., McCollum A., Atadja P., Roberts L. R., and Adjei A. A. (2007) Abrogation of MAPK and Akt signaling by AEE788 synergistically potentiates histone deacetylase inhibitor-induced apoptosis through reactive oxygen species generation. Clin. Cancer Res. 13, 1140–1148 10.1158/1078-0432.CCR-06-1751 [DOI] [PubMed] [Google Scholar]
- 44. Bahr J. C., Robey R. W., Luchenko V., Basseville A., Chakraborty A. R., Kozlowski H., Pauly G. T., Patel P., Schneider J. P., Gottesman M. M., and Bates S. E. (2016) Blocking downstream signaling pathways in the context of HDAC inhibition promotes apoptosis preferentially in cells harboring mutant Ras. Oncotarget 7, 69804–69815 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Eng J. K., McCormack A. L., and Yates J. R. (1994) An approach to correlate tandem mass spectral data of peptides with amino acid sequences in a protein database. J. Am. Soc. Mass Spectrom. 5, 976–989 10.1016/1044-0305(94)80016-2 [DOI] [PubMed] [Google Scholar]
- 46. Perkins D. N., Pappin D. J., Creasy D. M., and Cottrell J. S. (1999) Probability-based protein identification by searching sequence databases using mass spectrometry data. Electrophoresis 20, 3551–3567 10.1002/(SICI)1522-2683(19991201)20:18%3C3551::AID-ELPS3551%3E3.0.CO%3B2-2 [DOI] [PubMed] [Google Scholar]