

Abstract
The transcription factor BCL11A has recently been reported to be a driving force in triple-negative breast cancer (TNBC), contributing to the maintenance of a chemoresistant breast cancer stem cell (BCSC) population. Although BCL11A was shown to suppress γ-globin and p21 and to induce MDM2 expression in the hematopoietic system, its downstream targets in TNBC are still unclear. For its role in transcriptional repression, BCL11A was found to interact with several corepressor complexes; however, the mechanisms underlying these interactions remain unknown. Here, we reveal that BCL11A interacts with histone methyltransferase (PRC2) and histone deacetylase (NuRD and SIN3A) complexes through their common subunit, RBBP4/7. In fluorescence polarization assays, we show that BCL11A competes with histone H3 for binding to the negatively charged top face of RBBP4. To define that interaction, we solved the crystal structure of RBBP4 in complex with an N-terminal peptide of BCL11A (residues 2–16, BCL11A(2–16)). The crystal structure identifies novel interactions between BCL11A and the side of the β-propeller of RBBP4 that are not seen with histone H3. We next show that BCL11A(2–16) pulls down RBBP4, RBBP7, and other components of PRC2, NuRD, and SIN3A from the cell lysate of the TNBC cell line SUM149. Furthermore, we demonstrate the therapeutic potential of targeting the RBBP4–BCL11A binding by showing that a BCL11A peptide can decrease aldehyde dehydrogenase-positive BCSCs and mammosphere formation capacity in SUM149. Together, our findings have uncovered a previously unidentified mechanism that BCL11A may use to recruit epigenetic complexes to regulate transcription and promote tumorigenesis.
Keywords: breast cancer, cancer stem cells, crystal structure, epigenetics, histone chaperone, protein complex, protein–protein interaction, BCL11A, RBBP4, TNBC
Introduction
Although breast cancer research has led to several successful treatments and novel targeted therapies, there still exist the critical issues of recurrence and metastasis. Among the four subtypes of breast cancer, triple-negative breast cancer (TNBC,4 estrogen receptor−/progesterone receptor−/HER2−) has high rates of recurrence and metastasis and thereby has a generally poor prognosis (1, 2). The recent identification of breast cancer stem cells (BCSCs), which are attributed to tumor initiation, chemoresistance, and metastasis, helps explain the aggressive nature of TNBC (3–5). There has been a growing interest in understanding pathways and proteins important for maintaining the BCSC population to develop effective therapies for the prevention and treatment of metastatic breast cancer.
Accumulating evidence has indicated that epigenetic mechanisms are important for the progression of cancer as well as the maintenance of the two BCSC-defining characteristics: self-renewal and multipotency (6–10). Epigenetic complexes may alter the dynamic state of chromatin and regulate access to the genome for DNA-related processes such as repair, transcription, and replication. For example, components of polycomb-repressive complex 2 (PRC2) and nucleosome remodeling and deacetylase (NuRD), two important repressive epigenetic complexes, have been shown to be overexpressed in various types of cancer, including breast cancer, resulting in aberrant silencing of their target genes (11, 12). In BCSCs, epigenetic complexes that have important roles in regulating differentiation, including NuRD and PRC2, may be exploited by oncogenic transcription factors to promote stemness of cancer cells (13–16). Aberrant recruitment of these epigenetic machineries can result in drastic transcriptional changes and gene expression leading to tumorigenesis.
Retinoblastoma-binding proteins 4 and 7 (RBBP4/7) are two histone chaperones that work in tandem with PRC2 and/or NuRD in the cell to facilitate access to the genome and determine cellular identity (17). As members of the WD40 repeat protein family, RBBP4/7 consist of a seven-bladed β-propeller with several interaction surfaces that help them act as keystones for many protein complexes (17). Their role as integral parts of multisubunit epigenetic complexes is to bind to histones H3 and/or H4, allow for nucleosomal association, and expose histones to methylation, acetylation, or deacetylation (18–20).
The top face of RBBP4/7s' β-propeller, otherwise known as the canonical binding site, binds to un-methylated histone H3 as well as several transcription factors, including SALL4 and FOG1 (17, 20–22). The shared binding motif of H3, SALL4, and FOG1 can be used to identify new binding partners of RBBP4/7 and mechanisms by which epigenetic complexes are recruited to target genes. Indeed, we recognized B-cell lymphoma/leukemia 11A (BCL11A), a zinc-finger transcription factor that is necessary for hematopoiesis and regulates developmental globin switching (23), as a potential interacting partner of RBBP4/7.
Typically, the high expression of BCL11A is restricted to fetal brain and germinal center B-cells; however, it was recently demonstrated that BCL11A is up-regulated in TNBC through copy number gains and hypomethylation of the BCL11A locus (24). Furthermore, exogenous overexpression of BCL11A increased the clonogenicity of non-tumorigenic mammary epithelial cells, whereas knockdown of BCL11A resulted in a reduced tumor size and frequency of tumor formation indicating a decrease in BCSCs (24). These data suggest BCL11A has critical functions in BCSCs and TNBC, and hence it is a potential target for TNBC treatment. This led us to question whether BCL11A employs RBBP4/7 to recruit epigenetic complexes and whether one could target this interaction to block the aberrant functions of complexes such as PRC2 and NuRD in cancer.
In this study, we aim to confirm and characterize the interaction between RBBP4 and BCL11A and validate its function in regulating BCSCs. Using a fluorescence polarization (FP) assay, we have shown that a peptide consisting of the BCL11A residues 2–12 (BCL11A(2–12)) is sufficient to bind RBBP4 and compete with a histone H3 peptide for binding to the top face of RBBP4's β-propeller. The crystal structure of RBBP4 in complex with a BCL11A(2–16) peptide shows interactions that extend beyond the top face of RBBP4 and expose new binding sites. Pulldown assays also confirmed that the BCL11A(2–16) peptide is able to interact with RBBP4 and RBBP7 in PRC2, NuRD, and SIN3A complexes in the cell lysate of the TNBC cell line SUM149. Furthermore, blocking the RBBP4–BCL11A interaction by using the BCL11A(2–12) peptide can reduce the BCSC population in SUM149. Together our results reveal a previously unidentified mechanism by which BCL11A recruits epigenetic complexes to regulate transcription and promote tumorigenesis and suggest that the RBBP4/7–BCL11A interaction is a potential therapeutic target. Preventing this interaction could disrupt the aberrant functions of BCL11A, PRC2, and NuRD complexes in the treatment of TNBC.
Results
BCL11A competes with histone H3 for binding to RBBP4
Previous mass spectrometry and pulldown studies have indicated that BCL11A interacts with PRC2 as well as the NuRD and SIN3A histone deacetylase complexes (23, 25). RBBP4/7 are shared among all three complexes, and therefore, we hypothesized that BCL11A may recruit the aforementioned epigenetic machineries through their common core subunit RBBP4/7. In fact, BCL11A shares a similar N-terminal sequence as the transcription factors FOG1 and SALL4, which have been shown to interact with NuRD and specifically RBBP4 (Fig. 1A) (21, 25, 26). It is highly likely this shared motif is a conserved mechanism by which these transcription factors enlist epigenetic complexes to control transcription.
Figure 1.

BCL11A competes with histone H3 for binding to RBBP4. A, sequences of BCL11A, FOG1, SALL4, and histone H3 peptides. All peptides share the RKQ motif thought to be the most important for binding to RBBP4. B, saturation curves of the 5-FAM-labeled H3(1–21) tracer to RBBP4. Tracer was kept constant at 20 nm and incubated with increasing concentrations of purified full-length RBBP4. mP values were measured at the times indicated. Interaction was stable over 24 h. C, Kd values of the 5-FAM-labeled H3(1–21) to either RBBP4 or RBBP7. D, representative competitive binding curves of N-terminal peptides of BCL11A, SALL4A, and scramble peptides to RBBP4 in optimized buffer conditions (50 mm Tris, 50 mm NaCl, 0.1% glycerol (pH 7.5)). Peptides used in competitive binding experiments had a free N-terminal end with a capped amide at the C terminus. Data are presented as mean ± S.D. (n = 3). E, IC50 and Ki values (calculated using the equation from Nikolovska-Coleska et al. (38)) of the peptides tested.
To test our hypothesis, we developed an FP assay with histone H3, a known RBBP4/7-binding partner, as the tracer. A histone H3(1–21) peptide was labeled with 5-carboxyfluorescein (5-FAM) at the C terminus and was incubated at 20 nm with increasing concentrations of full-length RBBP4 (UniProt Q09028). The binding affinity (Kd) of the H3 tracer for RBBP4 was determined to be 0.84 ± 0.08 μm and stable for over 24 h, consistent with published literature (Fig. 1, B and C) (20). Increasing concentrations of salt and/or decreasing the pH of the system resulted in a decreased binding affinity. This indicates the importance of electrostatics in maintaining the interaction between the top face of RBBP4 and its binding partners (Fig. S1).
Competitive binding assays were performed with unlabeled synthetic peptides of BCL11A, SALL4, and histone H3. A scrambled peptide was also tested to determine the specificity of the assay. Although both SALL4(2–12) and BCL11A(2–12) peptides had low inhibitory constants (Ki) of 90 and 268 nm, respectively (Fig. 1, D and E), it is interesting to note that a shorter peptide corresponding to SALL4 amino acids 1–10 resulted in a 160-fold reduced inhibitory potential (Ki = 15 μm) (Fig. 1E). This suggests that histidine at position 11 and a hydrophobic isoleucine or leucine at position 12 may be required to make important contacts with RBBP4. In this platform, a longer peptide of BCL11A(2–16) did not greatly change the Ki value (110 nm versus 268 nm). A scrambled version of BCL11A(2–12) had an IC50 value of >1 mm indicating that a specific sequence order and not the overall charge of the peptide is required for binding to RBBP4. These data also suggest that BCL11A and H3 use the same binding site in PRC2 and NuRD complexes.
Crystal structure of RBBP4 bound to BCL11A(2–16)
To understand the molecular basis for the binding of BCL11A to RBBP4, we solved the crystal structure of RBBP4 bound to BCL11A(2–16) to 2.4 Å (Fig. 2A). Crystallography data and refinement statistics are listed in Table 1. The structure was solved by molecular replacement, and the initial difference electron density map showed clear density for the peptide bound to the negatively charged surface of RBBP4 (Fig. 2, B and C). The conformation of the RBBP4 protein bound to BCL11A(2–16) is identical to the unbound structure, with a root mean square deviation of 0.343 Å between the core atoms of the protein. The BCL11A(2–16) peptide binds in the same pocket as the histone H3 peptide (Fig. 3A), but interestingly the peptide turned 90° and made specific interactions with the side of the β-propeller not seen in previous structures (Figs. 2 and 3) (20).
Figure 2.
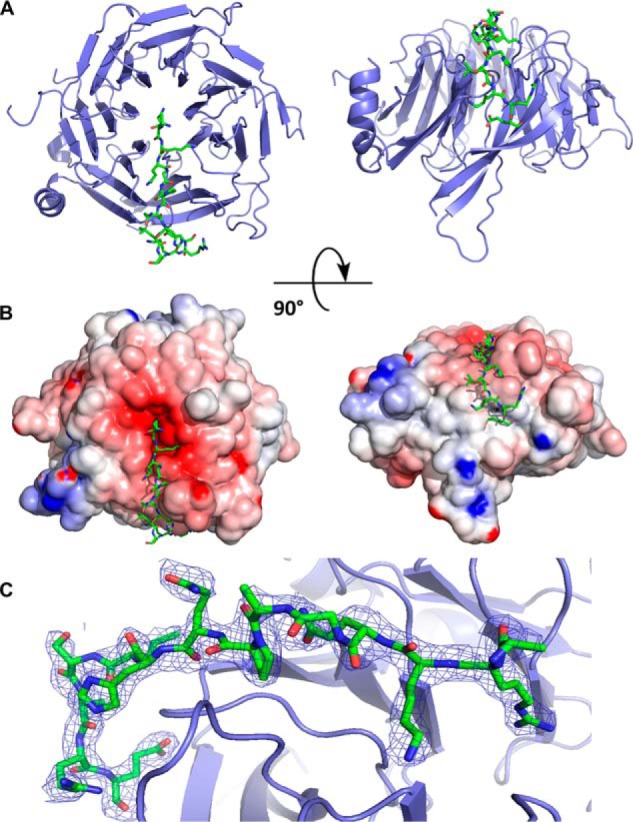
Crystal structure of RBBP4 in complex with BCL11A(2–16) peptide. RBBP4 is shown as a cartoon in blue, and BCL11A(2–16) is shown as sticks in green. A, BCL11A(2–16) is bound to the top face of the RBBP4 β-propeller and then proceeds to wrap down the side. B, electrostatic surface potential representation of RBBP4–BCL11A(2–16). The surface is colored according to the calculated electrostatic potential from −8 kT/e (red) to 8 kT/e (blue). C, 2Fo − Fc electron density map of the BCL11A(2–16) peptide contoured at 1σ is shown as blue mesh.
Table 1.
Crystallography data collection and refinement statistics
| Data collection | RBBP4–BCL11A(2–16) |
|---|---|
| Space group | P212121 |
| Unit cell a, b, c (Å) | 51.301, 85.264, 87.968 |
| Wavelength (Å) | 0.9786 |
| Resolution (Å)a | 2.40 (2.44–2.40) |
| Rsym (%)b | 11.5 (56.6) |
| 〈I/σI〉c | 10 (3) |
| Completeness (%)d | 99.7 (98.7) |
| Redundancy | 8.2 (8.0) |
| Refinement | |
| Resolution (Å) | 2.40 |
| R-factore | 20.1 |
| Rfreef | 24.4 |
| Protein atoms | 2766 |
| Water molecules | 140 |
| Unique reflections | 14917 |
| r.m.s.d.g | |
| Bonds (Å) | 0.010 |
| Angles (°) | 1.08 |
| MolProbity Scoreh | 1.25 |
| Clash Scoreh | 3.01 |
a Statistics for highest resolution bin of reflections are given in parentheses.
b Rsym = ΣhΣj|Ihi − 〈Ih〉|/ΣhΣjIhi, where Ihi is the intensity of observation j of reflection h and 〈Ih〉 is the mean intensity for multiply recorded reflections.
c Intensity signal-to-noise ratio is shown.
d Completeness of the unique diffraction data is shown.
e R-Factor = Σh ‖Fo|−|Fc‖/Σh|Fo|, where Fo and Fc are the observed and calculated structure factor amplitudes for reflection h.
f Rfree is calculated against a 10% random sampling of the reflections that were removed before structure refinement.
g Root mean square deviation of bond lengths and bond angles is shown.
h Data are from Chen et al. (43).
Figure 3.

Comparison of histone H3 and BCL11A peptides bound to RBBP4. A, overlay of histone H3 peptide (shown in yellow) (20) (PDB code 2YBA chains B and C) and the BCL11A peptide (shown in green) bound to the top face of the RBBP4 β-propeller. B, view of the novel interactions of the BCL11A peptide to the side of RBBP4. C, detailed look at the BCL11A- binding site. Hydrogen bonds are depicted as black dashed lines. Carbon atoms are shown in green for the peptide and slate for the protein; nitrogen atoms are blue, and oxygen atoms are red.
The first residue of the BCL11A(2–16) peptide, Ser-2, was disordered, but we observed electron density for the remainder of the BCL11A(2–16) peptide through residue 16. The binding of residues 4–13 of BCL11A is very similar to the binding of residues 2–11 of the histone H3 peptide to RBBP4 (Fig. 3A). The side chain of Arg-4 of BCL11A occupies a similar pocket as Arg-3 of histone H3 and makes a hydrogen bond between NH1 and Glu-231 OE1 of RBBP4 (Fig. 3C). There is also a well-ordered glycerol molecule in the binding pocket, which also interacts with the Arg-4. Similar to Lys-4 of histone H3, the major interaction between RBBP4 and BCL11A is Lys-5, which lies in a highly negatively charged pocket on RBBP4 lined by Glu-179, Glu-126, and the OD1 atom of Asn-128 (Fig. 3C). Gln-6 OE1 makes a hydrogen bond to the backbone carbonyl of Glu-395. Gly-7 and Lys-8 are slightly pushed out and make no interactions with the RBBP4 protein. The side chain of Lys-8 is pointed toward the solvent and disordered. Pro-9 ring packs against the imidazole group of His-71 of RBBP4. The side-chain NE2 of Gln-10 is hydrogen-bonded to the backbone carbonyl of Pro-9 on the peptide. His-11 NE2 is hydrogen-bonded to the OE2 of Glu-41 of RBBP4. The side chain of Leu-12 is pointing toward the solvent, but an important hydrogen bond between the backbone carbonyl of Leu-12 and the backbone amide of Lys-14 forms a tight turn of the peptide. This hydrogen bond is not seen in the H3 structure. In the H3 structure (PDB code 2yba, B chain), the last ordered peptide residue is residue 11; in PDB code 2yba, A chain, the peptide loops out into the solvent. In our structure, Ser-13 is pointed toward the solvent, but the remainder of the peptide makes specific interactions with residues on the side of the β-propeller. The amide of Lys-14 of BCL11A is hydrogen-bonded to the backbone carbonyl of Ala-30 of RBBP4. Arg-15 is bound to Glu-41 of RBBP4, effectively sandwiching this residue between His-11 and Arg-15 of the peptide. The final residue of the peptide Glu-16 is hydrogen-bonded to NE2 of His-38 of RBBP4.
The specific interactions of residues 14–16 of BCL11A on the side face of the histone chaperone RBBP4 may explain the tighter binding of BCL11A to RBBP4 (Ki 110 nm) than H3 to RBBP4 (Kd 348 nm). These interactions, not seen with NURF55/RBBP4 and histone H3 or RBBP4 and FOG1, expose a potential new surface for small molecule inhibitors (20, 21). For example, Glu-16 occupies a small trough-like cavity that may be susceptible to inhibition via small molecules.
BCL11A(2–16) is sufficient to interact with PRC2, NuRD, and SIN3A epigenetic complexes
To assess whether BCL11A(2–16) could interact with epigenetic complexes known to contain RBBP4/7, we performed pulldown experiments with a biotin-labeled BCL11A(2–16) peptide (Fig. 4A). As BCL11A plays a critical role in TNBC and BCSCs, we chose the TNBC cell line SUM149 for these experiments. Ponceau S staining of a nitrocellulose membrane containing the pulldown samples showed that an equal amount of BCL11A(2–16) WT and scramble peptides was used in the experiment (Fig. S2). As shown in Fig. 4B, BCL11A(2–16) was able to bind RBBP4 to a significant degree over scramble control in the SUM149 lysate. In addition, RBBP7 was pulled down by BCL11A(2–16) indicating that BCL11A may bind to both histone chaperones in a similar manner.
Figure 4.

Pulldown assays show that BCL11A(2–16) peptide is sufficient to interact with PRC2, NuRD, and SIN3A epigenetic complexes in SUM149 cell lysate. A, amino acid sequences of the biotin-labeled BCL11A(2–16) WT and scramble (SCR) peptides used in our pulldown studies. B, Western blot analysis of RBBP4, RBBP7, HDAC1, HDAC2, and other components of PRC2 (EZH2 and SUZ12), NuRD (MTA1), SIN3A (SIN3A), and CoREST (CoREST) complexes following BCL11A(2–16) WT or scramble peptide pulldown. Ten micrograms of SUM149 whole-cell lysate (∼4.4% of the lysate used in the pulldown lanes) were loaded as a control for detection of the proteins of interest. C, HDAC activity assay following BCL11A(2–16) WT or scramble peptide pulldown was done using the HDAC fluorometric activity assay kit (Cayman Chemical). To determine the background fluorescence level, the pulldown samples were treated with 1 μm TSA, an HDAC inhibitor, before the assay. Results shown are mean ± S.D. (****, p < 0.0001, one-way analysis of variance). D, HDAC activity (nmol/min/ml) following BCL11A(2–16) WT or scramble peptide pulldown was calculated as described under “Materials and methods.” Results shown are mean ± S.D. (***, p < 0.001, Student's t test).
Because we hypothesized that BCL11A interacts with epigenetic machineries through RBBP4, we checked the levels of various components of these complexes in our pulldown experiment. We confirmed that the enhancer of zeste homolog 2 (EZH2) and the suppressor of zeste 12 (SUZ12), two core subunits of PRC2, were higher in the BCL11A(2–16) than the scramble control pulldown (Fig. 4B). We next examined histone deacetylase 1 (HDAC1) and 2 (HDAC2), two enzymatic components of several histone deacetylase complexes, including NuRD and SIN3A, where RBBP4/7 are also a common component. HDAC1 and HDAC2 were strongly associated with the BCL11A(2–16) peptide and were almost undetectable in the scramble pulldown control (Fig. 4B). The complexes pulled down by BCL11A(2–16) exhibited deacetylase activity, which can be inhibited by trichostatin A (TSA) (Fig. 4C). This result is consistent with other similar studies using SALL4 as the bait (26). Notably, the complexes associated with the BCL11A(2–16) peptide had 16-fold greater HDAC activity compared with scramble peptide control (2.21 versus 0.14 nmol/min/ml) (Fig. 4D).
To distinguish between the several HDAC-containing complexes, we evaluated the presence of the NuRD-specific subunit metastasis-associated protein 1 (MTA1) in the pulldown samples. SIN3A and CoREST, two proteins whose names are shared with their respective histone deacetylase complexes, were also analyzed. As seen in Fig. 4B, MTA1 was strongly detected, and SIN3A was weakly identified in the BCL11A(2–16) pulldown over scramble control. In contrast, CoREST was only detected in the whole-cell lysate. Because the CoREST complex lacks RBBP4 or RBBP7, our results suggest that the N terminus of BCL11A only interacts with epigenetic machineries that contain these two histone chaperones.
Altogether, our results demonstrate that BCL11A interacts with both histone deacetylase complexes (NuRD and SIN3A) and histone methyltransferase complex (PRC2) to dictate transcriptional control.
Disruption of RBBP4–BCL11A interaction decreases ALDH+ BCSC population in TNBC cells SUM149
To demonstrate the functional and biological significance of the RBBP4–BCL11A interaction, we treated SUM149 cells with the BCL11A(2–12) peptide to block this interaction for 72 h. We hypothesized that the peptide would be able to compete with the BCL11A protein to block the RBBP4–BCL11A interaction within the cells and allow us to study the impact of blocking this protein–protein interaction. First, we evaluated the delivery efficiency of a 5-FAM labeled BCL11A(2–12) peptide using the Pep-1/Chariot carrier (27). Using flow cytometry, we found that over 98% of SUM149 cells were successfully transfected (Fig. 5A) when cells were transfected at a confluency of 25%. Treatment at lower cell densities decreased transfection efficiency (data not shown).
Figure 5.

BCL11A peptide decreased the ALDH+ cancer stem cell population in triple-negative breast cancer cells SUM149. A, SUM149 cells were transfected with a 5-FAM-labeled BCL11A(2–12) peptide using Pep-1/Chariot as a carrier. After 24 h, the efficiency of peptide delivery was examined by flow cytometry. B and C, ALDEFLUOR assay of SUM149 cells transfected with BCL11A(2–12) WT or scramble peptide for 72 h. B shows the representative flow cytometry dot plots. C shows relative ALDH+ cells with the average of the scramble peptide-transfected samples being set as 1. Data from four independent experiments are presented as mean ± S.D. (n = 6). BCL11A(2–12) WT peptide decreased ALDH+ cells by ∼50% over scramble peptide. D, ALDEFLUOR assay of SUM149 cells stably expressing BCL11A(1–12) or BCL11A(1–12)K5A. Data from two independent experiments are presented as mean ± S.D. (n = 6). E, mammosphere formation assay of SUM149 cells stably expressing BCL11A(1–12) or BCL11A(1–12)K5A. The results are shown as mean ± S.D. (n = 3). The p values in C–E were calculated by using Student's t test.
Because BCL11A is important for the maintenance of cancer stem cells, we hypothesize that blocking the RBBP4–BCL11A interaction will reduce the aldehyde dehydrogenase-positive (ALDH+) BCSC population (24). We performed an ALDEFLUOR assay, which is a fluorescence-based assay to label cells expressing high levels of ALDH and is a well-established assay for BCSCs. Each ALDEFLUOR-stained sample has a negative control, which is a portion of the cells stained with ALDEFLUOR in the presence of the ALDH inhibitor diethylaminobenzaldehyde (DEAB), to determine the background fluorescence. These DEAB-treated samples are used to set the ALDH+ gates in the flow cytometry analysis. The data showed that treatment of the SUM149 cells with the BCL11A(2–12) peptide decreased the ALDH+ population by ∼50% in comparison with cells treated with a scramble peptide (Fig. 5, B and C).
To further demonstrate the importance of the RBBP4–BCL11A interaction in maintaining BCSC function, we stably expressed BCL11A(1–12) in SUM149 cells using a lentiviral vector. Based on our crystal structure, lysine 5 of BCL11A provides a crucial contact in the interaction with RBBP4 (Fig. 3C), so we changed lysine 5 to alanine (BCL11A(1–12)K5A) and used it as a negative control. As shown in Fig. 5D, expressing BCL11A(1–12) in SUM149 cells decreased the ALDH+ BCSC population by ∼50% compared with BCL11A(1–12)K5A. Furthermore, BCL11A(1–12) also decreased the self-renewal capacity of BCSCs in SUM149 cells as the mammosphere formation assay, an in vitro surrogate assay to evaluate self-renewal ability of BCSCs, showed that a decreased number of mammospheres were formed as compared with the BCL11A(1–12)K5A-expressing SUM149 cells (Fig. 5E). Together, our results support the notion that blocking the interaction between BCL11A and RBBP4 could provide a novel strategy to eliminate BCSCs.
Discussion
BCL11A was recently discovered to be a critical gene in both TNBC and the chemoresistant BCSC population sparking a lot of interest in better understanding the molecular mechanisms by which it operates and how to inhibit its function. Coimmunoprecipitation studies suggest that BCL11A interacts with several histone-modifying and chromatin-remodeling complexes, but how this transcription factor interacts and recruits these epigenetic complexes and whether these interactions are vital for its function as a BCSC regulator remain to be determined (23, 25). Here, we define the interaction between BCL11A and RBBP4, a histone chaperone protein shared by many corepressor complexes. Through RBBP4, BCL11A is capable of recruiting NuRD, PRC2, and SIN3A to initiate transcriptional repression. We further expose this interaction as a potential therapeutic target against BCSCs.
RBBP4 is a WD40 protein with the characteristic seven-bladed β-propeller, which lends itself to multiple interfaces for mediating protein–protein interactions in multisubunit complexes. In chromatin-modifying complexes, the top face of RBBP4 binds the protruding tail of histone H3 to help localize complexes to nucleosomes. We report that BCL11A, specifically its N terminus, utilizes the same site as H3 to interact with RBBP4. This site is shared by many other transcription factors, including PHF6, SALL4A, and FOG1, thus signifying a conserved mechanism by which these transcription factors connect with epigenetic complexes. As they share the same binding pocket, these transcription factors and histone H3 are likely to bind RBBP4 in a mutually exclusive manner. Furthermore, because RBBP4 and RBBP7 share 92% sequence identity, BCL11A and the aforementioned factors may interact with either histone chaperone (17). Indeed, our pulldown studies support the notion that BCL11A can bind both RBBP4 and RBBP7. Specificity of BCL11A to RBBP4 or RBBP7 could therefore be determined by their relative abundances or by other subunits in the epigenetic complexes.
Similar to the other transcription factors that bind to the top face of RBBP4, Arg-4 of BCL11A was inserted into the core of the β-propeller of RBBP4, whereas Lys-5 was anchored to the surface of RBBP4 via hydrogen bonds. Consistent with our structure in which Arg-3 is disordered, this residue is not an important mediator for interactions between RBBP4 and other transcription factors (21, 22). Of note, however, our crystal structure of RBBP4 in complex with BCL11A(2–16) exposes new interactions on the side face of the histone chaperone that are not seen with NURF55/RBBP4 and histone H3 or RBBP4 and FOG1 (20, 21). An intramolecular hydrogen bond (between Leu-12 and Lys-14) creates a 90° turn in the BCL11A peptide allowing for more specific interactions between the BCL11A peptide and the side face of RBBP4. These subtle changes in binding modalities could explain the differences in binding affinity of BCL11A and RBBP4 compared with H3 and RBBP4. Differences in the N-terminal residues (aa 12–16) of FOG1, SALL4, and BCL11A could also be important in determining selectivity between the transcription factors in vivo.
In our FP platform we did not see a difference in the ability of BCL11A(2–12) versus BCL11A(2–16) to compete with H3 for binding to RBBP4; however, previous pulldown studies revealed a drastic decrease in the capacity of shorter peptides, those truncated below the first 15 amino acids, to interact with epigenetic complexes (28). Although the first 12 amino acids of BCL11A are necessary5 and sufficient (our FP experiment) for interacting with RBBP4, our crystal structure indicates the requirement of residues 12–16 to efficiently interact with RBBP4 and endogenous epigenetic complexes, which is consistent with the previous finding. In our pulldown experiment we showed that BCL11A(2–16) is capable of binding components of NuRD, PRC2, and SIN3A but not CoREST, a complex lacking RBBP4/7.
It was recently shown that RBBP4/7 could exist in multiple copies in certain epigenetic complexes. This occurs by either dimerization of the entire complex, such as PRC2, or enhanced recruitment by other subunits (29). For example, the MTA1 subunits of NuRD can each bind two copies of RBBP4/7 (29, 30). This explains the improved efficiency of the BCL11A peptide in pulling down components of the NuRD complex over PRC2 and SIN3A (Fig. 4B). It is interesting to speculate that within NuRD and PRC2, RBBP4 could serve to bind transcription factors and histone H3 at the same time through different RBBP4 molecules. It also suggests that RBBP4 might require contacts with more than one copy of BCL11A for optimal affinity for target genes, i.e. a threshold or dosage constraint that must be met for recruitment of epigenetic complexes. RBBP4/7 may be capable of surveying the surrounding epigenetic landscape and thereby coordinate signals from histone tails and transcription factors in multiple complexes.
From a therapeutic perspective, N-terminal truncations of FOG1 prevent its interaction with NuRD and mitigate its repressive activity suggesting this region is crucial for the function of this transcription factor (31). Similarly, N-terminal missense mutations of BCL11A result in a loss-of-function in vivo (32). Taken together, these studies support our hypothesis that the BCL11A–RBBP4 protein–protein interaction is important in BCL11A's tumorigenic role. When we deliver or stably express a decoy BCL11A N-terminal peptide, we did, in fact, see a decrease in the ALDH+ BCSC population and mammosphere formation capacity. Reducing the ALDH+ BCSC population is imperative for preventing recurrence and improving survival in breast cancer patients as this cell population, although very small, is associated with poor clinical outcome and metastasis (4, 34–36). In our unpublished data,5 knockdown of RBBP4 and/or RBBP7 can also decrease BCSCs. Unfortunately, we were unable to confirm the necessity of RBBP4/7 in the recruitment of epigenetic complexes by BCL11A(2–16) as knockdown of RBBP4/7 resulted in decreased levels of the NuRD, SIN3A, and PRC2 subunits potentially through destabilization of these complexes.5
In summary, BCL11A, through its N terminus, can recruit epigenetic complexes due to its interaction with RBBP4/7 and thereby alter transcription of its target genes. We expose new trough-like cavities on the side of RBBP4 that open avenues for specifically targeting BCL11A. A BCL11A peptide decoy or inhibitor could block recruitment of epigenetic complexes to BCL11A-targeted loci and warrants further investigation for its therapeutic potential in TNBC. Furthermore, such protein–protein interaction inhibitors could be utilized to block association of RBBP4-containing epigenetic complexes to histone H3 and other transcription factors that share the same binding motif as BCL11A. Future studies that focus on genes regulated by BCL11A in TNBC will help delineate the importance of the RBBP4–BCL11A interaction on promoting BCSC stemness and help decipher the impacts of inhibitors of this protein–protein interaction. They will also provide insights into the need for targeting this interaction to inhibit BCL11A's oncogenic function in TNBC.
Materials and methods
Cell culture and reagents
SUM149 cells were cultured in Ham's F-12 medium supplemented with 5% FBS, hydrocortisone (1 μg/ml), insulin (5 μg/ml), and 1× antibiotic-antimycotic (Thermo Fisher Scientific). 293T cells were cultured in Dulbecco's modified Eagle's medium supplemented with 10% FBS and 1× antibiotic-antimycotic. The following antibodies were used in this study: anti-RbAp48 (RBBP4) rabbit polyclonal (ab47456, Abcam; WB 1:2000); anti-RbAp46 (RBBP7) rabbit polyclonal (ab3535, Abcam; WB 1:2000); anti-SUZ12 rabbit monoclonal (no. 3737, Cell Signaling; WB 1:1000); anti-EZH2 rabbit monoclonal (no. 5426, Cell Signaling; WB 1:1000); anti-HDAC1 mouse monoclonal (no. 5356, Cell Signaling; WB 1:1000); anti-HDAC2 mouse monoclonal (no. 5113, Cell Signaling; WB 1:1000); anti-CoREST rabbit monoclonal (no. 14567, Cell Signaling; WB 1:1000); anti-SIN3A rabbit monoclonal (no. 8056, Cell Signaling; WB 1:1000); anti-MTA1 rabbit monoclonal (no. 5647, Cell Signaling; WB 1:1000); goat anti-rabbit IgG, HRP-linked (no. 7074, Cell Signaling; WB 1:2000); and goat anti-mouse IgG, HRP-linked (sc-2005, Santa Cruz Biotechnology).
Protein purification
Cloning, expression, and purification of RBBP4 (UniProt Q09028) was performed similarly to previous reports (21). Briefly, full-length RBBP4 (aa 1–425) was cloned into a pFastBac HT-A vector with both an N-terminal His6 tag and a tobacco etch virus (TEV) cleavage site. Generation of recombinant bacmid and virus was performed using the Bac-to-Bac (Invitrogen) expression system and according to the manufacturer's recommendations. Protein was expressed in Tn5 (High FiveTM; Invitrogen) cells using P3 virus. Infected cells were collected via centrifugation and washed with ice-cold PBS twice, and the pellet was lysed immediately or flash-frozen in liquid nitrogen for later use. All purification steps were performed at 4 °C unless otherwise noted. Infected cells were lysed by sonication in lysis buffer (50 mm Tris (pH 8), 500 mm NaCl, 0.1% Nonidet P-40, 5 mm imidazole, 10% glycerol) with protease inhibitors and Universal Nuclease (PierceTM Thermo Fisher Scientific). The lysate was clarified by centrifugation for 40 min at 12,000 rpm, and supernatant was added to nickel-nitrilotriacetic acid resin (Qiagen). Resin was collected and washed with high-salt buffer (50 mm Tris (pH 8), 500 mm NaCl, 10 mm imidazole, 10% glycerol) followed by low-salt buffer (50 mm Tris (pH 8), 150 mm NaCl, 10 mm imidazole, 10% glycerol). RBBP4 was eluted using the low-salt buffer with 250 mm imidazole. Fractions containing RBBP4 were collected, pooled, and concentrated using an Amicon® Ultra-4 centrifugal filter (EMD Millipore).
Peptide synthesis
Peptides were synthesized as described previously (37). Briefly, all peptides were synthesized manually or with an ABI 433 peptide synthesizer using Fmoc chemistry with Rink amide resin as the solid support with the exception of 5-FAM-labeled H3(1–21) probe that was purchased from Anaspec (catalog no. AS-63824). Either DIC/HOAt or HOBt/HBTU was used as the coupling reagent. Following completion of the peptide, a cleavage mixture composed of TFA/TIF/H2O/thioanisole (19:0.5:1:1 ml) removed the peptide from the resin as well as any side-chain protecting groups. The resulting solution was evaporated, and the crude peptide was precipitated with diethyl ether. Peptides were purified via reverse phase-HPLC (Waters, Sunfire Prep C18, 19 × 50 mm, 5 μm) and confirmed by electrospray ionization-mass spectroscopy. For biotin-labeled peptides, Fmoc-PEG Biotin NovaTagTM Resin (EMD Millipore) was used as solid support. To generate a scramble control, permutations of the original sequence were made.
Fluorescence polarization assay
All fluorescence polarization experiments were conducted in 384-well, black, low volume, round-bottom plates (Corning) using a BioTek Synergy 2 plate reader (Winooski, VT). To each well was added increasing amounts of protein and the 5-FAM-labeled histone H3 N-terminal probe/tracer (20 nm) to a final volume of 20 μl in the assay buffer (50 mm Tris-HCl (pH 7.4), 100 mm NaCl, 0.1% glycerol). The plate was allowed to incubate at room temperature for 5 min to reach equilibrium. The polarization values (mP) were measured at an excitation wavelength at 485 nm and an emission wavelength at 528 nm. An equilibrium-binding isotherm was constructed by plotting the mP values as a function of the protein concentration at a fixed concentration of tracer (20 nm). All experimental data were analyzed using Prism 7.0 software (Graphpad Software, San Diego) and WinNonlin (7.3).
Competitive binding assay
All IC50 values discussed were identified using the competitive binding assay. To a 384-well, black, low volume, round-bottom plate (Corning) was added 10 μl of 2 μm RBBP4 and 4 μl of 100 nm 5-FAM-labeled H3(1–21) probe. Dilutions of test compound (in assay buffer) or peptide were added to the wells to give a final volume of 20 μl and final concentrations of 1 μm RBBP4 and 20 nm probe. All experiments were run in optimized buffer conditions of 50 mm Tris-HCl (pH 7.5), 50 mm NaCl, and 0.1% glycerol. In addition, there were three controls on each plate: blank (buffer only), 100% inhibition (probe only), and 0% inhibition (probe and protein only). Ki values were determined using the previously derived equation (38).
Protein crystallization and structure determination
Prior to crystallization, the N-terminal His6 tag was removed from RBBP4 by TEV cleavage during dialysis against 50 mm Tris (pH 7.5), 150 mm NaCl. The cleaved protein was then purified by gel filtration on a Superdex 75 column in buffer containing 50 mm Tris (pH 7.5) and 150 mm NaCl. The protein was concentrated to 7.7 mg/ml and incubated with a 1.1-fold molar excess of Bcl11a (2–16) overnight at 4 °C. The complex was crystallized from drops containing equal volumes of complex and well solution (25% polyethylene glycol monomethyl ether (Mr 2000) and 0.1 m potassium thiocyanate). Prior to data collection, crystals were cryoprotected in well solution containing 25% glycerol. All data were collected at LS-CAT at the Advanced Photon Source at Argonne National Laboratory on line 21-ID-G equipped with a Mar300 detector. Data were processed and scaled with HKL2000 (39). The structure was solved by molecular replacement with Phaser (CCP4 suite (40)) using a previously solved structure of RBBP4 (PDB 4R7A) as the starting model. The structure was refined using Buster (41) with iterative rounds of fitting in COOT (42). Structures were validated with Molprobity (43). Data refinement and statistics are given in Table 1. The root mean square deviation between core atoms was calculated using secondary structure matching routine in Coot. Electrostatic surface potentials were generated using the Adaptive Poisson-Boltzmann Solver (APBS) plug-in in PyMOL (33). All figures were generated with PyMOL (the PyMOL Molecular Graphics System, Version 1.8.2.3 Schrödinger, LLC).
Pulldown assay
Cell lysate was prepared from SUM149 cells using RIPA buffer (with protease and phosphatase inhibitors added prior to use). Cells were incubated for 30 min on a shaker at 4 °C, and lysate was cleared by centrifugation. Protein concentration was measured using the Bradford assay (Bio-Rad). PierceTM High Capacity streptavidin-agarose resin (catalog no. 20359, Thermo Fisher Scientific) was mixed by inversion and pipetted into two Eppendorf tubes (30 μl each). Resin was washed two times with 500 μl of buffer A (PBS + 0.1% Triton X-100) and centrifuged at 3000 rpm for 5 min to remove the supernatant. To prepare the resin for pulldown, 1 μl (1 mg/ml) of biotinylated peptide (either BCL11A(2–16) WT or scramble) was added to the washed resin in 500 μl of buffer A and incubated for 2 h at 4 °C on a rocking shaker. The beads were washed three times with buffer A and used immediately. 600 μg of SUM149 cell lysate was added to each tube, and the volume was increased to 500 μl with buffer A (plus protease and phosphatase inhibitors). The mixture was incubated overnight at 4 °C on a rocking shaker. The following morning the beads were centrifuged for 5 min at 3000 rpm, and the supernatant was removed. Beads were washed three times with variations of buffer A (increasing detergent and/or salt, plus protease and phosphatase inhibitors). After the final wash, centrifugation, and collection of the beads, 40 μl of 2X× SDS was added to each tube and heated for 5 min at 95 °C. Samples (10 μg of whole-cell lysate and 15 μl of pulldown) were separated and analyzed via SDS-PAGE and Western blotting.
HDAC activity assay
HDAC activity was measured using the HDAC fluorometric activity assay kit (catalog no. 10011563, Cayman Chemical) following the manufacturer's instruction. Briefly, after performing the pulldown as described above, beads were washed three times and then resuspended in 100 μl of HDAC assay buffer (25 mm Tris-HCl (pH 8.0), 137 mm NaCl, 2.7 mm KCl, and 1 mm MgCl2). 10 μl of each sample was added to a well of a black 96-well plate in triplicate. The samples were then diluted further in 140 μl of assay buffer. 10 μl of trichostatin A (final concentration 1 μm) was added to a control well for each sample group to determine the background signal. HDAC substrate (10 μl) was added last to all wells, and the plate was incubated at 37 °C on an orbital shaker for 30 min. Developer was added to each well following incubation. After 15 min at room temperature, fluorescence was read on a BioTek Synergy HT plate reader (excitation 350 nm and emission 450 nm). Fluorescence of the standard wells was plotted as a function of deacetylated substrate concentration to give a standard curve. The deacetylated concentration of the samples was calculated using this equation: deacetylated compound (μm) = ((fluorescence of samples − average fluorescence of TSA-treated samples) − (y-int))/slope. HDAC activity (nmol/min/ml) was calculated taking the deacetylated substrate concentration, divided by the incubation time (30 min), and then multiplying by sample dilution.
Western blotting
Collected samples (15 μl) from the pulldown were run on a 4–15% gradient gel for 5 min at 50 V and then 120 V for 1–1.5 h. Proteins were transferred to a nitrocellulose membrane (0.2 μm) under a constant current of 220 mA. The membrane was blocked with 5% milk in PBST (PBS + 0.1% Tween 20). Primary antibodies were added according to the manufacturer's recommendations in 1% milk in PBST. The following morning, the membrane was washed three times with PBST, and then the corresponding secondary antibody was added to the membrane for 2 h at room temperature. The membrane was washed again using PBST three times before being exposed to enhanced chemiluminescence reagent (PierceTM Thermo Fisher Scientific). Proteins were detected using X-ray development.
Transient transfection
BCL11A (aa 2–12) or scramble peptides were delivered to SUM149 cells using ChariotTM Protein Delivery Reagent (Active Motif) according to the manufacturer's instructions. SUM149 cells were seeded into a six-well plate 24 h prior to treatment. Peptide stocks (2 mm) were diluted in 100 μl of PBS so that the final treatment concentration would be 20 μm. Chariot reagent, diluted 1:10, was further diluted in 100 μl of sterile water. Peptide and Chariot solutions were combined and incubated for 30 min, after which the mixture was overlaid onto cells. 400 μl of serum-free media was added, and the cells were incubated for 2 h at 37 °C before complete media was added. Treated cells were assayed for ALDH activity 72 h post-transfection. Transfection efficiency and conditions were optimized using a 5-FAM-labeled BCL11A(2–12) peptide.
Plasmid construction
An AgeI site was introduced into the 5′-end of turboGFP sequence in the pGIPZ lentiviral vector using site-directed mutagenesis. To replace the turboGFP sequence with BCL11A(1–12), two primers (top, 5′-CCGGCGCCACCATGTCTCGCCGCAAGCAAGGCAAACCCCAGCACTTATGAGC-3′, and bottom, 5′-GGCCGCTCATAAGTGCTGGGGTTTGCCTTGCTTGCGGCGAGACATGGTGGCG-3′) were annealed and ligated into the AgeI and NotI sites of pGIPZ. For BCL11A(1–12)K5A, the primers used are (top) 5′-CCGGCGCCACCATGTCTCGCCGCGCGCAAGGCAAACCCCAGCACTTATGAGC-3′ and (bottom) 5′-GGCCGCTCATAAGTGCTGGGGTTTGCCTTGCGCGCGGCGAGACATGGTGGCG-3′.
Stable expression of BCL11A(1–12) and BCL11A(1–12)K5A in SUM149 cells
The packaging vectors psPAX2 and pMD2.G were co-transfected into 293T cells with pGIPZ lentiviral constructs using the polyethyleneimine method. Lentiviral supernatant was collected 48 h after transfection and added to SUM149 cells with 4 μg/ml Polybrene (Sigma). Forty eight hours later, cells were selected in puromycin (2 μg/ml) for 3 days. The puromycin-resistant cells were collected the next day for ALDEFLUOR and mammosphere formation assays.
ALDH+ assay
ALDH activity was measured by using the ALDEFLUOR kit (StemCell Technologies). Briefly, 2 × 105 cells were resuspended in 800 μl of ALDEFLUOR assay buffer and added to a FACS tube. For negative controls, prepare FACS tubes containing 2 μl of the ALDH inhibitor diethylaminobenzaldehyde (DEAB). Then 4 μl of ALDH substrate was added to the cells and mixed by vortexing immediately. Immediately after mixing, 400 μl of cells were transferred to the tubes containing DEAB and were mixed well. All samples were incubated at 37 °C for 30 min. Cells were washed with 1 ml of PBS containing 2% FBS. After centrifugation and removing the supernatant, cells were resuspended in 300 μl of ALDEFLUOR assay buffer. We analyzed the brightly fluorescent ALDH-expressing cells in the green fluorescence channel (520–540 nm) of a standard flow cytometer. ALDEFLUOR-positive gates were based on the DEAB-treated negative control samples.
Mammosphere formation assay
SUM149 cells stably expressing BCL11A(1–12) or BCL11A(1–12)K5A were plated at a density of 4000 cells per well in an ultra-low attachment six-well plate (Corning) in 2 ml of MammoCult media (STEMCELL Technologies). MammoCult media were prepared according to the manufacturer's instructions. Before resuspending the cells, antibiotic-antimycotic was added to the media. After culturing for 7 days, mammospheres greater than 50 μm in diameter were counted using GelCountTM (Oxford Optronix).
Author contributions
R. R. M., M. C. L., and D. S. conceived and coordinated the study. R. R. M. and M. C. L. wrote the paper. R. R. M., M. C. L., N. O. S., S. L. T., and C. C. L. designed, performed and analyzed the experiments shown in Figs. 1, 4, and 5, A–C. M. C. L. designed vectors for expression of BCL11A(1–12) and BCL11A(1–12)K5A and designed, performed, and analyzed the experiments shown in Fig. 5, D and E. I. J. and A. M. provided technical assistance for plasmid construction and the experiments shown in Fig. 5, D and E. J. L. M. and J. A. S. conducted experiments pertaining to crystallography. All authors reviewed the results and approved the final version of the manuscript.
Supplementary Material
Acknowledgments
We acknowledge the Advanced Photon Source, a United States Department of Energy (DOE) Office of Science User Facility operated for the DOE Office of Science by Argonne National Laboratory under Contract No. DE-AC02–06CH11357. Use of the LS-CAT Sector 21 was supported by the Michigan Economic Development Corp. and the Michigan Technology Tri-Corridor Grant 085P1000817. Research reported in this publication was supported by the NCI, National Institutes of Health under award number P30CA046592 (for flow cytometry).
The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
This article contains Figs. S1 and S2.
The atomic coordinates and structure factors (code 5VTB) have been deposited in the Protein Data Bank (http://wwpdb.org/).
M.-C. Lo, R. R. Moody, C.-C. Lin, N. O. Stevers, S. L. Tinsley, and D. Sun, unpublished data.
- TNBC
- triple-negative breast cancer
- BCSC
- breast cancer stem cell
- ALDH
- aldehyde dehydrogenase
- HDCA
- histone deacetylase
- TEV
- tobacco etch virus
- 5-FAM
- 5-carboxyfluorescein
- DEAB
- diethylaminobenzaldehyde
- FBS
- fetal bovine serum
- PDB
- Protein Data Bank
- WB
- Western blotting
- aa
- amino acid
- 5-FAM
- 5-carboxyfluorescein
- Fmoc
- N-(9-fluorenyl)methoxycarbonyl
- TSA
- trichostatin A
- FP
- fluorescence polarization
- DIC/HOAt
- N,N′-diisopropylcarbodiimide/1-hydroxy-7-azabenzotriazole
- HOBt/HBTU
- hydroxybenzotriazole/2-(1H-benzotriazole-1-yl)-1,1,3,3-tetramethyluronium hexafluorophosphate
- TIF
- triisopropylsilane.
References
- 1. Cetin I., and Topcul M. (2014) Triple negative breast cancer. Asian Pac. J. Cancer Prev. 15, 2427–2431 [DOI] [PubMed] [Google Scholar]
- 2. Lehmann B. D., Bauer J. A., Chen X., Sanders M. E., Chakravarthy A. B., Shyr Y., and Pietenpol J. A. (2011) Identification of human triple-negative breast cancer subtypes and preclinical models for selection of targeted therapies. J. Clin. Invest. 121, 2750–2767 10.1172/JCI45014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Gangemi R., Paleari L., Orengo A. M., Cesario A., Chessa L., Ferrini S., and Russo P. (2009) Cancer stem cells: a new paradigm for understanding tumor growth and progression and drug resistance. Curr. Med. Chem. 16, 1688–1703 10.2174/092986709788186147 [DOI] [PubMed] [Google Scholar]
- 4. Liu S., Cong Y., Wang D., Sun Y., Deng L., Liu Y., Martin-Trevino R., Shang L., McDermott S. P., and Landis M. D., et al. (2013) Breast cancer stem cells transition between epithelial and mesenchymal states reflective of their normal counterparts. Stem Cell Rep. 2, 78–91 10.1016/j.stemcr.2013.11.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Visvader J. E., and Lindeman G. J. (2008) Cancer stem cells in solid tumours: accumulating evidence and unresolved questions. Nat. Rev. Cancer 8, 755–768 10.1038/nrc2499 [DOI] [PubMed] [Google Scholar]
- 6. Deng L., Shang L., Bai S., Chen J., He X, Martin-Trevino R., Chen S., Li X. Y., Meng X., Yu B., Wang X., Liu Y., McDermott S. P., Ariazi A. E., Ginestier C., et al. (2014) MicroRNA100 inhibits self-renewal of breast cancer stem-like cells and breast tumor development. Cancer Res. 74, 6648–6660 10.1158/0008-5472.CAN-13-3710 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Muñoz P., Iliou M. S., and Esteller M. (2012) Epigenetic alterations involved in cancer stem cell reprogramming. Mol. Oncol. 6, 620–636 10.1016/j.molonc.2012.10.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Brown R., Curry E., Magnani L., Wilhelm-Benartzi C. S., and Borley J. (2014) Poised epigenetic states and acquired drug resistance in cancer. Nat. Rev. Cancer 14, 747–753 10.1038/nrc3819 [DOI] [PubMed] [Google Scholar]
- 9. Kagara N., Huynh K. T., Kuo C., Okano H., Sim M. S., Elashoff D., Chong K., Giuliano A. E., and Hoon D. S. (2012) Epigenetic regulation of cancer stem cell genes in triple-negative breast cancer. Am. J. Pathol. 181, 257–267 10.1016/j.ajpath.2012.03.019 [DOI] [PubMed] [Google Scholar]
- 10. Widschwendter M., Fiegl H., Egle D., Mueller-Holzner E., Spizzo G., Marth C., Weisenberger D. J., Campan M., Young J., Jacobs I., and Laird P. W. (2007) Epigenetic stem cell signature in cancer. Nat. Genet. 39, 157–158 10.1038/ng1941 [DOI] [PubMed] [Google Scholar]
- 11. Lai A. Y., and Wade P. A. (2011) Cancer biology and NuRD: a multifaceted chromatin remodelling complex. Nat. Rev. Cancer 11, 588–596 10.1038/nrc3091 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Jene-Sanz A., Váraljai R., Vilkova A. V., Khramtsova G. F., Khramtsov A. I., Olopade O. I., Lopez-Bigas N., and Benevolenskaya E. V. (2013) Expression of polycomb targets predicts breast cancer prognosis. Mol. Cell. Biol. 33, 3951–3961 10.1128/MCB.00426-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Fu J., Qin L., He T., Qin J., Hong J., Wong J., Liao L., and Xu J. (2011) The TWIST/Mi2/NuRD protein complex and its essential role in cancer metastasis. Cell Res. 21, 275–289 10.1038/cr.2010.118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Tong Z. T., Cai M. Y., Wang X. G., Kong L. L., Mai S. J., Liu Y. H., Zhang H. B., Liao Y. J., Zheng F., Zhu W., Liu T. H., Bian X. W., Guan X. Y., Lin M. C., Zeng M. S., et al. (2012) EZH2 supports nasopharyngeal carcinoma cell aggressiveness by forming a co-repressor complex with HDAC1/HDAC2 and Snail to inhibit E-cadherin. Oncogene 31, 583–594 10.1038/onc.2011.254 [DOI] [PubMed] [Google Scholar]
- 15. Cakouros D., Isenmann S., Cooper L., Zannettino A., Anderson P., Glackin C., and Gronthos S. (2012) Twist-1 induces Ezh2 recruitment regulating histone methylation along the Ink4A/Arf locus in mesenchymal stem cells. Mol. Cell. Biol. 32, 1433–1441 10.1128/MCB.06315-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Gupta R. A., Shah N., Wang K. C., Kim J., Horlings H. M., Wong D. J., Tsai M. C., Hung T., Argani P., Rinn J. L., Wang Y., Brzoska P., Kong B., Li R., West R. B., et al. (2010) Long non-coding RNA HOTAIR reprograms chromatin state to promote cancer metastasis. Nature 464, 1071–1076 10.1038/nature08975 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Xu C., and Min J. (2011) Structure and function of WD40 domain proteins. Protein Cell 2, 202–214 10.1007/s13238-011-1018-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Balboula A. Z., Stein P., Schultz R. M., and Schindler K. (2015) RBBP4 regulates histone deacetylation and bipolar spindle assembly during oocyte maturation in the mouse. Biol. Reprod. 92, 105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Kloet S. L., Baymaz H. I., Makowski M., Groenewold V., Jansen P. W., Berendsen M., Niazi H., Kops G. J., and Vermeulen M. (2015) Towards elucidating the stability, dynamics and architecture of the nucleosome remodeling and deacetylase complex by using quantitative interaction proteomics. FEBS J. 282, 1774–1785 [DOI] [PubMed] [Google Scholar]
- 20. Schmitges F. W., Prusty A. B., Faty M., Stützer A., Lingaraju G. M., Aiwazian J., Sack R., Hess D., Li L., Zhou S., Bunker R. D., Wirth U., Bouwmeester T., Bauer A., Ly-Hartig N., et al. (2011) Histone methylation by PRC2 is inhibited by active chromatin marks. Mol. Cell 42, 330–341 10.1016/j.molcel.2011.03.025 [DOI] [PubMed] [Google Scholar]
- 21. Lejon S., Thong S. Y., Murthy A., AlQarni S., Murzina N. V., Blobel G. A., Laue E. D., and Mackay J. P. (2011) Insights into association of the NuRD complex with FOG1 from the crystal structure of an RbAp48.FOG1 complex. J. Biol. Chem. 286, 1196–1203 10.1074/jbc.M110.195842 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Lauberth S. M., and Rauchman M. (2006) A conserved 12-amino acid motif in Sall1 recruits the nucleosome remodeling and deacetylase corepressor complex. J. Biol. Chem. 281, 23922–23931 10.1074/jbc.M513461200 [DOI] [PubMed] [Google Scholar]
- 23. Xu J., Bauer D. E., Kerenyi M. A., Vo T. D., Hou S., Hsu Y.-J., Yao H., Trowbridge J. J., Mandel G., and Orkin S. H. (2013) Corepressor-dependent silencing of fetal hemoglobin expression by BCL11A. Proc. Natl. Acad. Sci. U.S.A. 110, 6518–6523 10.1073/pnas.1303976110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Khaled W. T., Choon Lee S., Stingl J., Chen X., Raza Ali H., Rueda O. M., Hadi F., Wang J., Yu Y., Chin S. F., Stratton M., Futreal A., Jenkins N. A., Aparicio S., Copeland N. G., et al. (2015) BCL11A is a triple-negative breast cancer gene with critical functions in stem and progenitor cells. Nat. Commun. 6, 5987 10.1038/ncomms6987 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Sankaran V. G., Menne T. F., Xu J., Akie T. E., Lettre G., Van Handel B., Mikkola H. K., Hirschhorn J. N., Cantor A. B., and Orkin S. H. (2008) Human fetal hemoglobin expression is regulated by the developmental stage-specific repressor BCL11A. Science 322, 1839–1842 10.1126/science.1165409 [DOI] [PubMed] [Google Scholar]
- 26. Gao C., Dimitrov T., Yong K. J., Tatetsu H., Jeong H. W., Luo H. R., Bradner J. E., Tenen D. G., and Chai L. (2013) Targeting transcription factor SALL4 in acute myeloid leukemia by interrupting its interaction with an epigenetic complex. Blood 121, 1413–1421 10.1182/blood-2012-04-424275 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Morris M. C., Depollier J., Mery J., Heitz F., and Divita G. (2001) A peptide carrier for the delivery of biologically active proteins into mammalian cells. Nat. Biotechnol. 19, 1173–1176 10.1038/nbt1201-1173 [DOI] [PubMed] [Google Scholar]
- 28. Saathoff H., Brofelth M., Trinh A., Parker B. L., Ryan D. P., Low J. K., Webb S. R., Silva A. P., Mackay J. P., and Shepherd N. E. (2015) A peptide affinity reagent for isolating an intact and catalytically active multi-protein complex from mammalian cells. Bioorg. Med. Chem. 23, 960–965 10.1016/j.bmc.2015.01.023 [DOI] [PubMed] [Google Scholar]
- 29. Davidovich C., Goodrich K. J., Gooding A. R., and Cech T. R. (2014) A dimeric state for PRC2. Nucleic Acids Res. 42, 9236–9248 10.1093/nar/gku540 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Millard C. J., Varma N., Saleh A., Morris K., Watson P. J., Bottrill A. R., Fairall L., Smith C. J., and Schwabe J. W. (2016) The structure of the core NuRD repression complex provides insights into its interaction with chromatin. Elife 5, e13941 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Hong W., Nakazawa M., Chen Y. Y., Kori R., Vakoc C. R., Rakowski C., and Blobel G. A. (2005) FOG1 recruits the NuRD repressor complex to mediate transcriptional repression by GATA-1. EMBO J. 24, 2367–2378 10.1038/sj.emboj.7600703 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Dias C., Estruch S. B., Graham S. A., McRae J., Sawiak S. J., Hurst J. A., Joss S. K., Holder S. E., Morton J. E., Turner C., Thevenon J., Mellul K., Sánchez-Andrade G., Ibarra-Soria X., Deriziotis P., et al. (2016) BCL11A haploinsufficiency causes an intellectual disability syndrome and dysregulates transcription. Am. J. Hum. Genet. 99, 253–274 10.1016/j.ajhg.2016.05.030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Baker N. A., Sept D., Joseph S., Holst M. J., and McCammon J. A. (2001) Electrostatics of nanosystems: application to microtubules and the ribosome. Proc. Natl. Acad. Sci. U.S.A. 98, 10037–10041 10.1073/pnas.181342398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Ginestier C., Hur M. H., Charafe-Jauffret E., Monville F., Dutcher J., Brown M., Jacquemier J., Viens P., Kleer C. G., Liu S., Schott A., Hayes D., Birnbaum D., Wicha M. S., and Dontu G. (2007) ALDH1 is a marker of normal and malignant human mammary stem cells and a predictor of poor clinical outcome. Cell Stem Cell 1, 555–567 10.1016/j.stem.2007.08.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Marcato P., Dean C. A., Pan D., Araslanova R., Gillis M., Joshi M., Helyer L., Pan L., Leidal A., Gujar S., Giacomantonio C. A., and Lee P. W. (2011) Aldehyde dehydrogenase activity of breast cancer stem cells is primarily due to isoform ALDH1A3 and its expression is predictive of metastasis. Stem Cells 29, 32–45 10.1002/stem.563 [DOI] [PubMed] [Google Scholar]
- 36. Charafe-Jauffret E., Ginestier C., Iovino F., Tarpin C., Diebel M., Esterni B., Houvenaeghel G., Extra J. M., Bertucci F., Jacquemier J., Xerri L., Dontu G., Stassi G., Xiao Y., Barsky S. H., et al. (2010) Aldehyde dehydrogenase 1-positive cancer stem cells mediate metastasis and poor clinical outcome in inflammatory breast cancer. Clin. Cancer Res. 16, 45–55 10.1158/1078-0432.CCR-09-1630 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Connarn J. N., Assimon V. A., Reed R. A., Tse E., Southworth D. R., Zuiderweg E. R., Gestwicki J. E., and Sun D. (2014) The molecular chaperone Hsp70 activates protein phosphatase 5 (PP5) by binding the tetratricopeptide repeat (TPR) domain. J. Biol. Chem. 289, 2908–2917 10.1074/jbc.M113.519421 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Nikolovska-Coleska Z., Wang R., Fang X., Pan H., Tomita Y., Li P., Roller P. P., Krajewski K., Saito N. G., Stuckey J. A., and Wang S. (2004) Development and optimization of a binding assay for the XIAP BIR3 domain using fluorescence polarization. Anal. Biochem. 332, 261–273 10.1016/j.ab.2004.05.055 [DOI] [PubMed] [Google Scholar]
- 39. Otwinowski Z., and Minor W. (1997) Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 276, 307–326 10.1016/S0076-6879(97)76066-X [DOI] [PubMed] [Google Scholar]
- 40. McCoy A. J., Grosse-Kunstleve R. W., Adams P. D., Winn M. D., Storoni L. C., and Read R. J. (2007) Phaser crystallographic software. J. Appl. Crystallogr. 40, 658–674 10.1107/S0021889807021206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Bricogne G., B., Brandl M., Flensburg C., Keller P., Paciorek W., and Roversi P., S. A., Smart O. S., Vonrhein C., Womack T. O. (2017) BUSTER, Version 2.10.2., Global Phasing Ltd., Cambridge, UK [Google Scholar]
- 42. Emsley P., Lohkamp B., Scott W. G., and Cowtan K. (2010) Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 66, 486–501 10.1107/S0907444910007493 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Chen V. B., Arendall W. B. 3rd., Headd J. J., Keedy D. A., Immormino R. M., Kapral G. J., Murray L. W., Richardson J. S., and Richardson D. C. (2010) MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr. D Biol. Crystallogr. 66, 12–21 10.1107/S0907444909042073 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.