

Abstract
The development of novel drugs specifically directed at the ion channels underlying particular features of cardiac action potential (AP) initiation, recovery, and refractoriness would contribute to an optimized approach to antiarrhythmic therapy that minimizes potential cardiac and extracardiac toxicity. Of these, K+ channels contribute numerous and diverse currents with specific actions on different phases in the time course of AP repolarization. These features and their site-specific distribution make particular K+ channel types attractive therapeutic targets for the development of pharmacological agents attempting antiarrhythmic therapy in conditions such as atrial fibrillation. However, progress in the development of such temporally and spatially selective antiarrhythmic drugs against particular ion channels has been relatively limited, particularly in view of our incomplete understanding of the complex physiological roles and interactions of the various ionic currents. This review summarizes the physiological properties of the main cardiac potassium channels and the way in which they modulate cardiac electrical activity and then critiques a number of available potential antiarrhythmic drugs directed at them.
Keywords: potassium channels, repolarization, physiological mechanisms, currents, ion channel, drug target
Introduction
Orderly propagation of cardiac electrophysiological excitation and recovery depends on a normal sequence of cardiac action potential (AP) generation through its component myocytes. The depolarization and repolarization of AP is mediated by multiple, interacting, inward and outward currents mediated by different ion charge carriers dependent on the action of specific membrane ion channels (Figure 1). The initial depolarization phase takes the form of a rapid upstroke and is mainly driven by inward Na+ current (INa) through voltage-gated sodium channels (Nav1.5). The succeeding plateau phase is dominated by inward Ca2+ current (ICa). The resulting entry of extracellular Ca2+ induces release of sarcoplasmic reticular Ca2+ stores, thereby activating excitation–contraction coupling. Repolarization, ultimately returning the membrane to the resting potential, is principally driven by outward current through voltage-gated K+ channels (Kv).1 K+ channel activity is thus a principal determinant of AP duration (APD) as it limits the depolarization duration and therefore both the time course of the Ca2+-mediated contraction and the refractory period. There are numerous and diverse K+ channels types, each with particular kinetic and voltage-dependent properties. These result in numerous and diverse current contributions, each with specific roles at different phases of repolarization. Together these determine the relatively prolonged but finely tuned repolarization time course and the repolarization reserve following recovery of the resting membrane potential. The repolarization reserve refers to the partly overlapping function of these currents, namely, IKr, IKs, and IK1, that gives a limited level of redundancy to the system.2 The kinetics of repolarization varies greatly with cardiac region and species. This reflects variations in the occurrence and density of the different K+ channel subtypes. All these characteristics suggest that explorations of K+ channels may yield a useful group of pharmacological targets for arrhythmic conditions.
Figure 1.
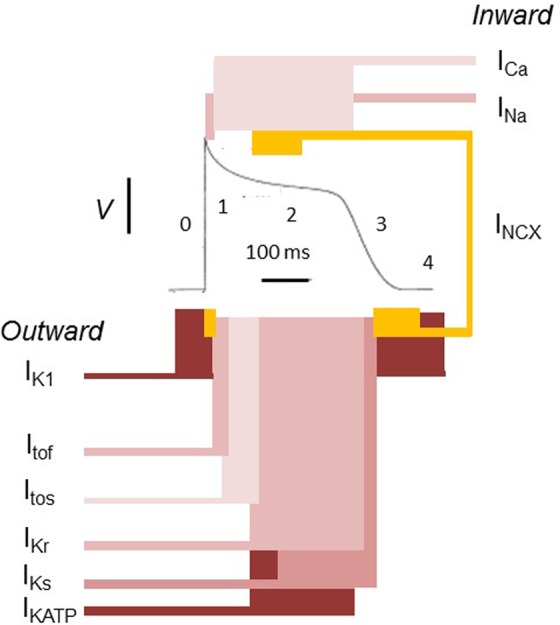
The ventricular action potential as a paradigm for cardiac electrophysiological activity. In the resting state, the voltage of the cell intracellular space is negative to the external environment. This reflects its higher K+ but lower Na+ and Ca2+ concentrations and its lower membrane permeability to Na+ and Ca2+ in comparison to K+. K+ efflux from the cell is then controlled by the inward rectifier K+ channel (IK1). When excitation threshold is reached, a large Na+ influx (INa) into the cell through Na+ channels produces phase 0 depolarization. This is followed by activation of fast and slow transient outward K+ currents (Itof and Itos, respectively) mediating a K+ efflux driving a rapid phase 1 repolarization. There is also an activation of a depolarizing inward Ca2+ current through L-type Ca2+ channels (ICa), which initiates excitation contraction coupling. The reduced membrane K+ permeability due to IK1 rectification combined with ICa maintains the action potential phase 2 plateau phase. Phase 3 repolarization is driven by K+ efflux through the rapid and slow delayed rectifier K+ channels (IKr and IKs, respectively), as well as IK1. At the end of phase 3, the Na+ and Ca2+ that have accumulated in the cells are removed by the Na+, K+ pump, and the Na+, Ca2+ exchanger (NCX). The atrial action potential shows greater contributions to recovery from the ultrarapid delayed rectifier outward currents (IKur) and acetylcholine-activated inward rectifying K+ channel (IKACH). Adapted with permission from Huang.1
Potassium Channels
K+ channels represent the most functionally diverse cardiac ion channel type.3-6 Together, they tightly regulate cardiac repolarization, thus ensuring stable and consistent AP signaling. The different K+ channel types have overlapping functions,2,7 resulting in some degree of functional redundancy,2 which in turn contributes to repolarization reserve. Table 1 summarizes their encoding genes with their chromosomal locations and the structural properties of their pore-forming α- and accessory β-subunits. The α-subunit of different K+ channel types all possess a conserved pore-forming region allowing K+ movement across the plasma membrane down an electrochemical gradient possessing a selective permeability to K+ attributable to a specific structural motif. They may also exhibit gating mechanisms responsive to membrane depolarization and ligand-binding sites whose occupancy could alter channel conformation. Finally, individual monomeric α-subunits may assemble into functional dimers or tetramers due to the presence of one or more subunit-assembly domains.6,8-10 K+ channel α-subunits fall into 3 structural types based on subunit topology (Figure 2). The first has 1 pore-forming region with 6 or 7 transmembrane regions (Figure 2A), the second has 1 pore-forming region and 2 transmembrane regions (Figure 2B), and the third has 2 pore-forming and 4 transmembrane regions (Figure 2C).5,6,10
Table 1.
Molecular Details and Activation Mechanisms of the Cardiac Potassium Channels.2
| Current | Gene | Chromosomal Location | Associated Protein | Type of Subunit |
|---|---|---|---|---|
| I tof | KCND3 | 1p13.2 | Kv4.3 | α |
| KCNIP2 | 10q24.32 | KChIP2 | β | |
| KCNE3 | 11q13.4 | MiRP2 | β | |
| I tos | KCNA4 | 11p14.1 | Kv1.4 | α |
| I Ks | KCNQ1 | 11p15.5-p15.4 | Kv1.7.1/KvLQT1 | α |
| KCNE1 | 21q22.12 | minK | β | |
| AKAP9 | 7q21.2 | AKAP-9 | β | |
| I Kr | KCNH2 | 7q36.1 | Kv11.1/hERG | α |
| KCNE2 | 21q22.11 | MiRP1 | β | |
| I K1 | KCNJ2 | 17q24.3 | Kir2.1/IRK1 | α |
| KCNJ12 | 17p11.2 | Kir2.2/IRK2 | α | |
| I KATP | KCNJ8 | 12p12.1 | Kir6.1 | α |
| KCNJ11 | 11p15.1 | Kir6.2 | α | |
| ABCC9 | 12p12.1 | SUR2A/SUR2Ba | β | |
| I Kur | KCNA5 | 12p12.32 | Kv1.5 | α |
| KCNAB1-B3 | N/A | Kvβ1-3 | β | |
| I KAch | KCNJ3 | 2q24.1 | Kir3.1/GIRK1 | α |
| KCNJ5 | 11q24.3 | Kir3.4/GIRK4 | α |
Abbreviations: IK1, inward rectifier K+ current; IKACH, acetylcholine-activated inward-rectifier K+ current; IKATP, ATP-sensitive K+ current; IKr, rapid component of the delayed rectifier K+ current; IKs, slow component of the delayed rectifier K+ current; IKur, ultrarapid component of the delayed rectifier K+ current; Itof, fast transient outward K+ current; Itos, slow transient outward K+ current.
aSUR2A and SUR2B are splice variant of ABCC9 and considered as cardiac (SUR2A) and vascular (SUR2B) isoforms.
Figure 2.
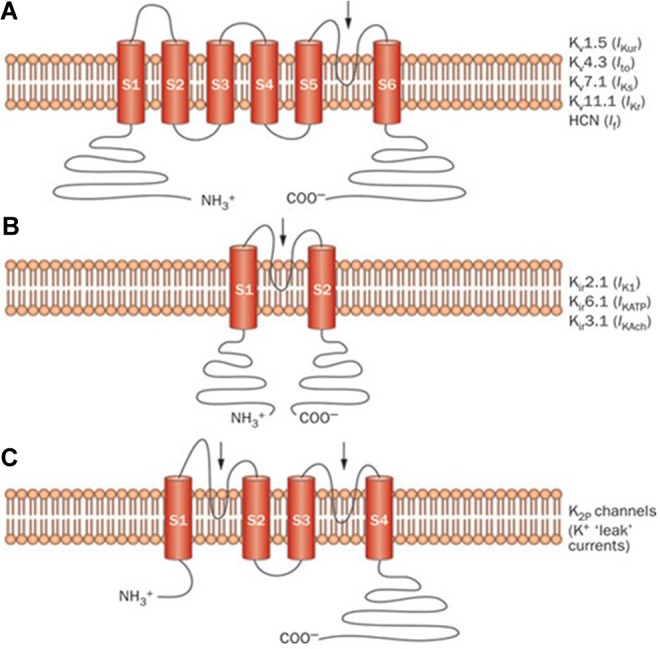
Structure of different cardiac potassium channel species: Schematic representation of selected potassium channel α-subunits. A, The 6-transmembrane 1-pore-region voltage-dependent K+ channel (Kv) α-subunits mediating IKur, Ito, IKs, IKr, and If. B, The 2-transmembrane 1-pore-region inward rectifying K+ channel (Kir) α-subunits mediating IK1, IKATP, and IKAch. C, The 4-transmembrane 2-pore-region K+ channel (K2P) mediating “leak” K+ currents. The arrows indicate the location of the pore-forming region(s). HCN indicates hyperpolarization-activated cyclic nucleotide-gated channel; If, inward rectifier mixed Na+ and K+ “funny” current; IK1, inward rectifier K+ current; IKACH, acetylcholine-activated inward rectifier K+ current; IKATP, ATP-sensitive K+ current; IKr, rapid component of the delayed rectifier K+ current; IKs, slow component of the delayed rectifier K+ current; IKur, ultrarapid component of the delayed rectifier K+ current; Ito, transient outward K+ current. Reprinted with permission from Giudicessi and Ackerman. Macmillan Publishers Ltd, copyright 2012.3
K+ channel β-subunits encompass many molecular groups, such as adenosine triphosphate (ATP)–binding cassette transport-related proteins (eg, sulfonylurea receptors) for inward rectifiers, cytoplasmic proteins (KChIP, KChAP, and Kvβ1-3), and single transmembrane spanning proteins (minK).10 These β-subunits form complexes with the α-subunits and can modify the channel’s functional properties. For example, Kvβ subunits can alter channel trafficking and the kinetics of current activation and inactivation when interacting with Kv1.5.11 More specifically, Kvβ2.1 and Kvβ4.1 behave as chaperone proteins.12 Furthermore, the N-terminus of Kvβ1.2 and Kvβ1.3 has an inactivation domain resembling the inactivation particle of the α-subunit, allowing it to modulate 2channel inactivation.12-14
Cardiac Potassium Currents
Cardiac K+ channels vary in their permeability properties, membrane potential dependence, and their opening or closing activation and inactivation kinetics. The major currents are classified into the transient outward currents, delayed rectifier outward currents, and the inward rectifiers (Figure 3). Advances in electrophysiological and molecular biology techniques have demonstrated additional currents that may fall outside this basic classification. Some brief notes on the major cardiac K+ currents, their role in the cardiac AP, and their functional importance follow.
Figure 3.

Classification of K+ currents: General classification of the main cardiac K+ currents. The relatively new additions to the K+ channel family (Ca2+-activated K+ current [IKCa] and 2-pore domain K+ current [IK2p]) have not been grouped under this scheme. Most K+ currents are grouped according to the direction of their overall rectification property. In some instances, this may vary. With the inward rectifying K+ channels, the name refers to the unusual characteristic whereby net potassium flow is into the cell at potentials lower than the reversal potential where channel conductance is high. As the channel potential becomes more positive, channel conductance decreases. Net ion flow direction reverses at the reversal potential, meaning that net potassium flow is outward at potentials more positive than this. Therefore, at depolarized potentials, potassium loss from the cell is low as conductance through the channel is low.
Transient outward K+ (Ito1) currents
When first described, the transient outward currents (Ito) were attributed to 2 distinct channels, one blocked by 4-aminopyridine (4-AP) and unaffected by extracellular Ca2+ (Ito1) and the other not blocked by 4-AP but sensitive to Ca2+ (Ito2).6 Ito drives the initial rapid repolarization phase of the AP. Regions with shorter APDs, such as the epicardium, right ventricle, and septum, have higher Ito expression. It was later discovered that Ito2 is a Cl- rather than a K+ current.15 Further characterizations subdivided Ito1 into fast (Itof) and slow (Itos) currents (Figure 3). Itof predominates in the atria, whereas both Itof and Itos occur in the ventricles.16 While Itos requires longer recovery times, its classification as “slow” is relative only to Itof. Thus, both Itof and Itos channels activate and inactivate rapidly in comparison to the corresponding processes in other K+ channels.15 Due to differences in the biophysical properties of Itof and Itos, the existence of molecular heterogeneity between these 2 channels has been previously suggested.15
Ultrarapid delayed rectifier currents (IKur)
In addition to Ito, the ultrarapid delayed rectified K+ current (IKur) plays a role in the initial rapid phase 1 AP repolarization. IKur activates rapidly in under 10 milliseconds at voltages in the plateau range and deactivates slowly over the course of the AP.17-19 IKur is the predominant delayed rectifier current for the atria and thus results in the shorter APD seen in the atria compared to the ventricles.10,16,17,19 Where IKur is present, its channels are not evenly distributed over the myocyte surface but instead found at high densities in the intercalated disk.6 This pattern of distribution is often disrupted after cardiac ischemic damage.10 The selective presence of IKur in the atria makes it an interesting target for atria selective therapy, whereby inhibition of IKur would prolong the APD in the atria but not the ventricles.4
Rapid delayed rectifier K+ currents (IKr)
The voltage-gated rapid delayed rectifier outward K+ current (IKr) is critical to phase 3 repolarization. It shows a relatively rapid activation with depolarization. However, its inactivation rate is around 10 times faster than its activation rate due to voltage-dependent C-type inactivation. This renders it relatively nonconducting in phases 1 and 2 of the cardiac AP.20-23 Thus, although termed a delayed rectifier current, it also shows an inward rectification property at positive potentials.22,24 However, with the end of phases 1 and 2, as the membrane potential becomes negative to 0 mV, IKr becomes activated once again, but the deactivation during this phase is much slower. This results in a large outward K+ efflux during phase 3 repolarization.2,10 IKr is found in both human atria and ventricles but is differentially expressed with higher levels in the left atrium and ventricular endocardium.16
Slowly activating delayed rectifier K+ current (IKs)
Cardiac repolarization is also influenced by a third, slowly activating delayed rectifier K+ current (IKs). IKs slowly activates at potentials positive to −20 mV. Unlike IKr, IKs barely inactivates25,26 and consequently accumulates over phase 2 repolarization, significantly influencing phase 3 repolarization.2 This feature of IKs is particularly important during atrial and ventricular APs of long duration. It is also involved in APD shortening during physiological increases in heart rate. An increase in heart rate thus reduces the time required for IKs inactivation. In consequence, more IKs accumulates, leading to a steeper drop in the repolarization rate.27,28 Blocking IKs results in an APD prolongation at increased heart rates.21,28 Inhibition of IKs will increase the vulnerable window for reactivation of voltage-gated Ca2+ channels, thereby increasing the risk of arrhythmic trigger events.2 IKs is found in all cardiac cell types, but its expression is significantly reduced in the mid-myocardial wall; this accounts for the long APD seen in this region.16
Inward rectifier K+ current (IK1)
The inward rectifier K+ current (IK1) functions over a narrow membrane potential range. Its rectifying property results in a marked reduction in IK1 conductance at positive, depolarized, membrane potentials and an increase in IK1 at negative membrane potentials, with the effect of stabilizing the membrane resting potential close to the K+ equilibrium potential (EK).10 The channel mediating IK1 does not show voltage-dependent gating and does not possess a voltage sensor. Nevertheless, IK1 modulation associated with movement of Mg2+ and polyamines results in an indirect sensitivity to voltage.29-32 Between phase 0 and phase 2 of the AP, the membrane potential is more positive than −20 mV, and at this potential, there is no conductance of IK1 as the channel is inhibited by Mg2+ and polyamines. The resulting marked inward rectification property limits the outward current at these positive potentials. This in turn minimizes the inward depolarizing current, which confers energetic efficiency for AP generation as it minimizes changes to ionic gradients that would need to be restored.16 As the potential returns to more negative values (typically around −40 mV), the inhibition by Mg2+ and polyamines is reversed. IK1 conductance then resumes and this contributes to phase 3 cardiac repolarization.31 IK1 occurs in both atria and ventricles and is thereby involved in setting their resting membrane potentials. Channels conducting IK1 are expressed in greater density in the ventricles, making the ventricles less susceptible to pacemaker influence.16
Acetylcholine-activated K+ current (IKACH)
The inwardly rectifying acetylcholine (ACh)-activated K+ current (IKACH) is regulated by G proteins rather than voltage gating. Cardiac parasympathetic nerve endings release ACh, thereby activating M2 muscarinic receptors. This reduces the depolarizing effect of the pacemaker current (If), reducing firing rates of pacemaker cells and in turn reducing heart rate.6 Acetylcholine also opens muscarinic-sensitive IKACH channels allowing the inward rectification of K+. The inward rectifying current shortens the AP and hyperpolarizes the membrane potential.16 Membrane hyperpolarization reduces the rate at which the sinoatrial and atrioventricular (AV) nodes drive pacemaker depolarization in addition to reducing AV conduction velocity.6,33 IKACH is thought to be specific to the atria,2 but there has been a suggestion that it may exist both in the atria and ventricle,16 but with densities 6 times greater in the atria than the ventricles.34
ATP-activated K+ current (IKATP)
The ATP-activated K+ current (IKATP) occurs at both the sarcolemmal (sarc-KATP) and mitochondrial inner membrane (mito-KATP) of cardiomyocytes. The sarc-KATP channels are highly expressed in cardiomyocytes and are composed of Kir6.2 and SUR2A subunits. There may also be contributions from Kir6.1 and SUR1.35 In contrast, although the subunits of mito-KATP channels have been difficult to identify due to the challenge of isolating pure mitochondrial membrane fractions, ROMK2 pore-forming subunits and SUR2 regulatory subunits have been suggested to contribute.36,37
Both channels are controlled by ATP and are thus directly responsive to the cell’s metabolic status, thereby influencing cell membrane potential.6 IKATP is inhibited by physiological intracellular ATP levels, but this reverses with ATP depletion. Thus, under normal energetic circumstances, there is limited IKATP current. However, under both physiological and pathological conditions that reduce ATP, there is increased IKATP current that is essential for adaptation to stress. For example, compared to wild-type controls, mice lacking Kir6.2-containing KATP channels perform less well in acute treadmill exercise testing.38 The increased IKATP has a cardioprotective role in ischemia by shortening the cardiac AP, thus limiting calcium influx into the cytosol.39-41 Specifically, studies have suggested that mito-KATP rather than sarc-KATP channel opening has an energy-modulating property that confers cardioprotection in ischemic hearts.42,43
In some situations, the IKATP-mediated APD shortening and corresponding heterogeneities in repolarization can create a substrate for cardiac reentry arrhythmia. In other situations, KATP channel openers have been described to have antiarrhythmic effects,44-48 and evidence suggests that activation and block of KATP can be pro- or antiarrhythmic depending on the arrhythmogenic mechanism in different animal models.49 For example, selective sarcolemma KATP channel blockers, such as HMR 1883, confer antiarrhythmic effects in the short term,50 although this could be metabolically disadvantageous in the long term due to the abolished adaptive response to stresses. Finally, it is important to note that the channel involved in the conductance of IKATP is also thought to be involved in the regulation of smooth muscle tone and insulin secretion in pancreatic β-cells.6
Other K+ channel family: Ca2+-activated K+ current (IKCa), 2-pore domain K+ current (IK2p), and hyperpolarization-activated cyclic nucleotide-gated channels
Recently, several further currents have been characterized. The Ca2+-activated K+ current, also known as the small conductance Ca2+-activated K+ (SK) current (IKCa), and the 2-pore domain K+ current (IK2p) have attracted considerable physiological and pharmacological interest. IKCa was initially thought to not exist in the human heart.51 However, subsequent studies demonstrated the presence of IKCa, with a higher density in the atria than the ventricle. Various subtypes of Ca2+-activated K+ channels exist in different tissues; the channel subtype conducting the cardiac IKCa is the SK channel.51,52 In neuronal cell, SK channels that are involved in modulating the tonic firing frequency and activation of these channels cause membrane hyperpolarization, thus limiting neuronal AP firing frequency.51 In contrast, cardiac SK channels and consequently IKCa are involved in late AP repolarization, controlling the resting membrane potential in human atria.52 IKCa appears to not play physiologically significant roles in the ventricle.52 IKCa is accordingly of particular pharmacological interest for atrial fibrillation (AF) therapy. Thus, IKCa occurs during late repolarization, when the atrial AP is susceptible to irregular or abnormal excitation such as that resulting from early after-depolarizations (EADs).51
IK2p contributes to the background current, the resting membrane potential, and cellular excitability. The channel involved in the conductance of this current has no voltage dependence, but its activity is modulated by lipids, particularly fatty acids, pH, drugs, particularly local and inhalation anesthetics, and membrane stretch.53,54 These mediators act upon the channel via secondary messenger phosphorylation.55 IK2p is a background current that persists through all phases of the cardiac AP. It thus stabilizes the membrane potential toward Em. IK2p may also prevent the occurrence of EADs, and it may be involved in fine-tuning of Na+ channel availability for phase 0 depolarization.56 The current has been found to occur selectively in the atria and AV node, thereby making it a target for drug development.57-59 Although not entirely new but only recently well characterized, the hyperpolarization-activated cyclic nucleotide-gated (HCN) channel is instrumental in conducting the inward funny current (If) in the heart. The channel is activated by the hyperpolarization of the membrane and is additionally stimulated by intracellular cyclic nucleotides.60,61 The generation of If is attributable to the inward permeability of both Na+ and K+ and occurs at threshold close to the resting membrane potential.62 Although the HCN channel under physiological circumstances conducts both Na+ and K+, the primary sequence of the HCN pore region suggests that it is primarily related to a selective potassium channel.63 In certain pathological conditions such as AF and myocardial infarction, If is increased unusually outside the pacemaker cells, leading to increased propensity to arrhythmia. Thus, targeting the If in such pathological conditions has proven to be therapeutically advantageous.64
Cardiac K+ Channel as Targets for Drug Development
Although there have been significant recent advances in the development and use of cardiological devices and procedures directed at arrhythmic conditions, antiarrhythmic drugs continue to be important whether by themselves or as adjunct therapy to such interventions. These include situations involving acute management of potentially fatal arrhythmic events, particularly where such procedures are contraindicated. Yet progress in antiarrhythmic drug development has been relatively limited. This likely reflects a lack of understanding of cardiac arrhythmic mechanisms. However, recent developments of our understanding of the role of the ion channels in normal AP generation have led to a specific interest into ion channels and their associated currents whose abnormal activity potentially leads to arrhythmia. This would encourage interest in the development of cardiac ion channel activator or blockers directed at modulating the cardiac AP or its refractory period. Introduction of drugs acting specifically on ion channels would optimize the efficacy of therapeutic actions on arrhythmogenic tendency, while minimizing problems arising from potential cardiac and extracardiac toxicity. K+ channels play a vital role in cardiac AP repolarization and thus naturally form potential targets for the development of ion channel-specific antiarrhythmic therapy, such as for AF. However, a limitation of this approach is that arrhythmic conditions, such as AF, are heterogeneous and the efficacy of targeting ion channels varies according to the cause and extent of the arrhythmia.
This is complicated by the fact that in various physiological and pathological conditions, remodeling of K+ channel expression can occur, which can alter the AP and increase the risk of sudden cardiac death.65 For example, AF is maintained and progressed partly due to electrical remodeling, mediating APD shortening.66 Thus, in chronic AF, there is upregulation of IK1, IKs, and IK2P3.1, which offsets the possible downregulation of IKur and Ito.67-69 Nevertheless, the experimental evidence for the reduction in IKur during remodeling is conflicting, as some reports suggest reduced IKur density70,71 and others suggest no change.72,73 It has been suggested that receptor-activated IKACH (rIKACH) mediates AF induced by vagal stimulation, while constitutive IKACH (c IKACH) develops in the time course of AF remodeling.67,74
In physiological cardiac hypertrophy, induced by chronic exercise, for example, there is an increase in IK density.75 This contrasts with pathological cardiac hypertrophy caused by pressure overload where a reduced IK density is noted that was attributable to cellular hypertrophy rather than gene expression changes in Itof and IK1.76 In heart failure, AP prolongation is associated with downregulation of several genes, leading to reduced Itof, IKs, IKr, and IK1.65,77,78 Considering the changes in K+ channel expression in remodeling is clinically important as the sensitivity and efficacy of blocking these channels will change.
Table 2 outlines selected drugs that have been experimentally proven to target different K+ channels, using either native cardiac myocytes or human cell line expression systems. Some of these drugs presently in clinical use have been primarily developed for other ion channels such as the Na+ or Ca2+ cardiac ion channel but have corresponding effects on K+ channels. Several drugs have been proposed to be selective to specific K+ channels, such as A935142, XEN-D0103, and XEN-D0101. However, despite promising experimental findings, many of these drugs have not progressed to clinical use. This may be attributable to limitations associated with experimental studies. Expression systems can often produce off-target effects or nonspecific interactions which may mask the true effect of these drugs. Additionally, expression systems may run the risk of either overexpressing or underexpressing the channel of interest. On the other hand, native cardiac myocytes, while more physiologically representative, may not provide the right platform for the study of specific targets. Additionally, acquisition of viable native cardiac myocytes from a minimally heterogeneous population remains a challenge, and it is widely accepted that channel functions can differ by gender and age. Consequently, while experimental studies may suggest potentially promising options to selectively target K+ channels, the translational capacity of such studies remains limited.
Table 2.
Selected Pharmacological Agents Affecting the Human K+ Channels.
| Current | Pharmacological Agent (Expression System), Reference |
|---|---|
| Activators | |
| I Kr | A-935142 (HEK),79 ICA-105574 (HEK),80 NS1643 (HEK),81 PD-118057 (HEK)82 |
| I Ks | Ephedrine (HEK),83 Tanshinone IIA (HEK)84 |
| I Kca | NS1619 (HEK)85 |
| Blockers | |
| I to | Chromanol 293B (nHVM),86 Flecainidea (nHAM)87 |
| I Kur | Amiodaronea (HEK),88 Bepridil (HEK),88 DPO-1 (nHAM),89 MK-0448 (nHAM),90 NIP-142 (HEK),91 Papaverine (nHAM),92 Pimozide (HEK),93 Sertindole (HEK),94 XEN-D0103 (nHAM)95 |
| I Kr | Cocaine (HEK),96 Fluvoxamine (HEK),97 Ketoconazole (HEK),98 Ketanserin (HEK),99 Ziprasidone (HEK)100 |
| I Ks | HMR 1556 (HEK),101 SKF-96365 (HEK)102 |
| I KACH | NIP-151 (HEK),103 U73122/U73343 (HEK)104 |
| I KATP | 5-Hydroxydecanoate (HEK),105 HMR1098 (HEK)105 |
| Ito, IKur | AVE-0118 (nHAM),106 Acacetin (nHAM),107 Ambasilide (nHAM),108,109 4-aminopyridine (nHAM),87 Diltiazemb (nHAM),110 Docosahexaenoic acid (nHAM),111 Eicosapentaenoic acid (nHAM),111 Nifedipineb (nHAM),110 Quinidinea (nHAM),87 Raloxifene (nHAM),112 U50488 H (nHAM),113 XEN-D0101 (nHAM),114 Vernakalant (RSD1235) (HEK)115 |
| Ito, IKur, IK1 | Propafenonea (nHAM)116 |
| Ito, IKur, IKr, IKs | Clotrimizole (nHAM)117 |
| Ito, IKur, IKr, IKs, IK1 | Azimilide (nHAM)118 |
| IKur, IKr | Cisapride (HEK),119 Verapamilb (nHAM, HEK)120,121 |
| IKr, IKs | Sotalol (nHAM)21,108 |
Abbreviations: nHAM, native human atrial myocyte; HEK, human embryonic kidney; nHVM, native human ventricular myocyte; IK1, inward rectifier K+ current; IKr, rapid component of the delayed rectifier K+ current; IKACH, acetylcholine-activated inward rectifier K+ current; IKATP, ATP-sensitive K+ current;; IKs, slow component of the delayed rectifier K+ current; IKur, ultrarapid component of the delayed rectifier K+ current; Ito, transient outward K+ current; IKCa, small conductance Ca2+-activated K+ (SK) current.
aPrimary Na+ channel blocker.
bPrimary Ca2+ channel blocker.
Furthermore, these activators and blockers often target more than 1 K+ channel species and thus are not entirely specific.10,122 However, a large proportion of these drugs also typically target IKr (known to be present in all cardiac regions) and as such do not constitute ideal candidates for targeted therapy. Nevertheless, mechanisms of cardiac arrhythmia are likely to be region dependent. Drugs that may be antiarrhythmic in some cardiac regions may potentially be pro-arrhythmic in others. Thus, the presence of atrial-specific K+ channels has provided focus on developing drugs that could specifically increase refractory periods, thus preventing atrial reentry arrhythmia, which is the most common mechanism for AF.123
Of ion channels specific to the atrium that might offer specific therapeutic targets, the channel conducting IKur tends to prolong repolarization and effective refractory period (ERP) without altering QT intervals.124,125 The experimental drugs AVE0118 and XEN-D101 are thought to be IKur selective blockers with both prolonging APD in atrial tissue from patients with permanent AF in common with the known IKur blocker 4-AP.106-129 However, a subsequent “first-in-human” study using the highly selective IKur blocker MK-0448 (N-{6-[(1S)-1-(4-fluorophenyl)-2,2-di (pyridine-3-1) ethyl] pyridine2yl} methane sulfonamide) did not reveal any increase in atrial ERP. This led to the conclusion that selective blocking of IKur may have limited clinical value.130 IKACH channels are also atrium specific or at least predominantly occur in the atria and have minimal physiological function in the ventricle.129 Opening of the IKACH channel will lead to shortening of atrial APD and thus increase the likelihood of AF. Therefore, blocking the opening of IKACH channels will prevent such shortening of APD with minimal effect on ventricular APD, in turn reducing the chances of AF. Several drugs block IKACH, but have limited specificity. Nevertheless, selective blocking of IKACH has been experimentally achieved using the compound NTC-801. The compound was found to have selective antifibrillatory properties, achieved by prolonging the atrial ERP.131 Another potential atrial-specific therapeutic target of interest is the IKCa current conducted by SK channels. The selective presence of this current in the atria has recently led to several investigative drugs beings explored. NS8593 is a selective SK channel inhibitor demonstrating significant atrial antiarrhythmic effects in canine and equine experimental models. Experiments using human atrial cardiac myocytes from patients with normal sinus rhythm demonstrated reduction in K+ currents and prolongation in APD. No such changes were observed in intraventricular myocytes.52
Conclusion
There is currently an incomplete understanding of the cellular physiological role of the various cardiac potassium currents and their interacting effects and how dysregulation of their function and expression can provide arrhythmogenic mechanisms. It is thought that the site-specific distribution of some K+ channels could allow targeted therapy to be more spatially selective. However, complex electrical remodeling events that occur in disease states may change channel expression levels to the extent that the selectivity of the drug is hindered, making even this potential therapeutic strategy challenging. Although targeting ion channels responsible for discrete parts of the cardiac AP to modulate the system towards a more physiological state has therapeutic appeal, there are inherent difficulties in developing successful drugs. This is because the ion channels targeted are functionally complex and are interdependent, thus adding a dynamic situation in which function and expression are altered depending on the cell environment. Furthermore, pathophysiological processes of arrhythmic disease may involve functional alterations in 1 or more ion channels. Such single or multiple ion channel functional abnormalities may therefore warrant corresponding use of a single or multichannel activator/blocker approach. However, this approach will only be possible if we are able to identify the specific pathophysiological process affecting individual patients (ie, is this arrhythmic disease related to a single or multichannel abnormality). Thus, although we may be able to develop single or multichannel activators/blockers, actual clinical use will be dependent on a detailed understanding of the exact arrhythmogenic mechanisms affecting individual patients, which thus far is limited. Presently, decisions to use single or multichannel activators/blockers are largely dependent on resolution of clinical signs or the actual arrhythmia rather than a therapeutic approach targeting ion channel functional abnormality. Furthermore, the availability of truly specific ion channel activators/blockers is limited as these agents tend to have off-target actions with corresponding side effects, and this limits the clinical use of selective agents. Focusing on understanding the system at a cellular physiological level through further experimental and computational modeling is needed to enable development of novel insights at a pharmacological level.
Footnotes
Author Contributions: Kamalan Jeevaratnam contributed to conception and design, contributed to interpretation, drafted the manuscript, and critically revised the manuscript. Karan Raj Chadda contributed to interpretation, drafted the manuscript, and critically revised the manuscript. Christopher L-H. Huang contributed to design, contributed to interpretation, drafted the manuscript, and critically revised the manuscript. A. John Camm contributed to conception, contributed to interpretation, and critically revised manuscript. All authors gave final approval and agree to be accountable for all aspects of work ensuring integrity and accuracy
Declaration of Conflicting Interests: The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding: The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: K.J. is funded by the Fundamental Research Grant Scheme (FRGS/2/2014/SKK01/PERDANA/02/1), Ministry of Education, Malaysia and the Research Support Fund, Faculty of Health and Medical Science, University of Surrey. K.R.C. is funded by the Physiological Society, United Kingdom. C. L-H. H. is funded by the Wellcome Trust, Medical Research Council, British Heart Foundation, and McVeigh Benefaction.
ORCID iD: Kamalan Jeevaratnam, DVM, MMedSc, PhD, MRCVS http://orcid.org/0000-0002-6232-388X
References
- 1. Huang CL. Murine electrophysiological models of cardiac arrhythmogenesis. Physiol Rev. 2017;97(1):283–409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Schmitt N, Grunnet M, Olesen SP. Cardiac potassium channel subtypes: new roles in repolarization and arrhythmia. Physiol Rev. 2014;94(2):609–653. [DOI] [PubMed] [Google Scholar]
- 3. Giudicessi JR, Ackerman MJ. Potassium-channel mutations and cardiac arrhythmias—diagnosis and therapy. Nat Rev Cardiol. 2012;9(6):319–332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Wulff H, Castle NA, Pardo LA. Voltage-gated potassium channels as therapeutic targets. Nat Rev Drug Discov. 2009;8(12):982–1001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Coetzee WA, Amarillo Y, Chiu J, et al. Molecular diversity of K+ channels. Ann N Y Acad Sci. 1999;868:233–285. [DOI] [PubMed] [Google Scholar]
- 6. Snyders DJ. Structure and function of cardiac potassium channels. Cardiovasc Res. 1999;42(2):377–390. [DOI] [PubMed] [Google Scholar]
- 7. Roden DM. Taking the “idio” out of “idiosyncratic”: predicting torsades de pointes. Pacing Clin Electrophysiol. 1998;21(5):1029–1034. [DOI] [PubMed] [Google Scholar]
- 8. MacKinnon R. Pore loops: an emerging theme in ion channel structure. Neuron. 1995;14(5):889–892. [DOI] [PubMed] [Google Scholar]
- 9. Bezanilla F, Stefani E. Gating currents. Methods Enzymol. 1998;293:331–352. [DOI] [PubMed] [Google Scholar]
- 10. Tamargo J, Caballero R, Gomez R, Valenzuela C, Delpon E. Pharmacology of cardiac potassium channels. Cardiovasc Res. 2004;62(1):9–33. [DOI] [PubMed] [Google Scholar]
- 11. Uebele VN, England SK, Gallagher DJ, Snyders DJ, Bennett PB, Tamkun MM. Distinct domains of the voltage-gated K+ channel Kv beta 1.3 beta-subunit affect voltage-dependent gating. Am J Physiol. 1998;274(6 pt 1):C1485–C1495. [DOI] [PubMed] [Google Scholar]
- 12. Martens JR, Kwak YG, Tamkun MM. Modulation of Kv channel alpha/beta subunit interactions. Trends Cardiovasc Med. 1999;9(8):253–258. [DOI] [PubMed] [Google Scholar]
- 13. Kurata HT, Fedida D. A structural interpretation of voltage-gated potassium channel inactivation. Prog Biophys Mol Biol. 2006;92(2):185–208. [DOI] [PubMed] [Google Scholar]
- 14. Wang Z, Kiehn J, Yang Q, Brown AM, Wible BA. Comparison of binding and block produced by alternatively spliced Kvbeta1 subunits. J Biol Chem. 1996;271(45):28311–28317. [DOI] [PubMed] [Google Scholar]
- 15. Nerbonne JM, Kass RS. Molecular physiology of cardiac repolarization. Physiol Rev. 2005;85(4):1205–1253. [DOI] [PubMed] [Google Scholar]
- 16. Grant AO. Cardiac ion channels. Circ Arrhythm Electrophysiol. 2009;2(2):185–194. [DOI] [PubMed] [Google Scholar]
- 17. Nattel S, Yue L, Wang Z. Cardiac ultrarapid delayed rectifiers: a novel potassium current family of functional similarity and molecular diversity. Cell Physiol Biochem. 1999;9(4-5):217–226. [DOI] [PubMed] [Google Scholar]
- 18. Snyders DJ, Tamkun MM, Bennett PB. A rapidly activating and slowly inactivating potassium channel cloned from human heart. Functional analysis after stable mammalian cell culture expression. J Gen Physiol. 1993;101(4):513–543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Wang Z, Fermini B, Nattel S. Sustained depolarization-induced outward current in human atrial myocytes. Evidence for a novel delayed rectifier K+ current similar to Kv1.5 cloned channel currents. Circ Res. 1993;73(6):1061–1076. [DOI] [PubMed] [Google Scholar]
- 20. Piper DR, Hinz WA, Tallurri CK, Sanguinetti MC, Tristani-Firouzi M. Regional specificity of human ether-a′-go-go-related gene channel activation and inactivation gating. J Biol Chem. 2005;280(8):7206–7217. [DOI] [PubMed] [Google Scholar]
- 21. Sanguinetti MC, Jurkiewicz NK. Two components of cardiac delayed rectifier K+ current. Differential sensitivity to block by class III antiarrhythmic agents. J Gen Physiol. 1990;96(1):195–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Tseng GN. I(Kr): the hERG channel. J Mol Cell Cardiol. 2001;33(5):835–849. [DOI] [PubMed] [Google Scholar]
- 23. Yellen G. The voltage-gated potassium channels and their relatives. Nature. 2002;419(6902):35–42. [DOI] [PubMed] [Google Scholar]
- 24. Spector PS, Curran ME, Zou A, Keating MT, Sanguinetti MC. Fast inactivation causes rectification of the IKr channel. J Gen Physiol. 1996;107(5):611–619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Jespersen T, Grunnet M, Olesen SP. The KCNQ1 potassium channel: from gene to physiological function. Physiology (Bethesda). 2005;20:408–416. [DOI] [PubMed] [Google Scholar]
- 26. Splawski I, Tristani-Firouzi M, Lehmann MH, Sanguinetti MC, Keating MT. Mutations in the hminK gene cause long QT syndrome and suppress IKs function. Nat Genet. 1997;17(3):338–340. [DOI] [PubMed] [Google Scholar]
- 27. Delpon E, Valenzuela C, Perez O, Casis O, Tamargo J. Propafenone preferentially blocks the rapidly activating component of delayed rectifier K+ current in guinea pig ventricular myocytes. Voltage-independent and time-dependent block of the slowly activating component. Circ Res. 1995;76(2):223–235. [DOI] [PubMed] [Google Scholar]
- 28. Jurkiewicz NK, Sanguinetti MC. Rate-dependent prolongation of cardiac action potentials by a methanesulfonanilide class III antiarrhythmic agent. Specific block of rapidly activating delayed rectifier K+ current by dofetilide. Circ Res. 1993;72(1):75–83. [DOI] [PubMed] [Google Scholar]
- 29. Fakler B, Brandle U, Glowatzki E, Weidemann S, Zenner HP, Ruppersberg JP. Strong voltage-dependent inward rectification of inward rectifier K+ channels is caused by intracellular spermine. Cell. 1995;80(1):149–154. [DOI] [PubMed] [Google Scholar]
- 30. Guo D, Lu Z. Mechanism of IRK1 channel block by intracellular polyamines. J Gen Physiol. 2000;115(6):799–814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Lopatin AN, Nichols CG. Inward rectifiers in the heart: an update on I(K1). J Mol Cell Cardiol. 2001;33(4):625–638. [DOI] [PubMed] [Google Scholar]
- 32. Vandenberg CA. Inward rectification of a potassium channel in cardiac ventricular cells depends on internal magnesium ions. Proc Natl Acad Sci U S A. 1987;84(8):2560–2564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Shieh CC, Coghlan M, Sullivan JP, Gopalakrishnan M. Potassium channels: molecular defects, diseases, and therapeutic opportunities. Pharmacol Rev. 2000;52(4):557–594. [PubMed] [Google Scholar]
- 34. Schram G, Pourrier M, Melnyk P, Nattel S. Differential distribution of cardiac ion channel expression as a basis for regional specialization in electrical function. Circ Res. 2002;90(9):939–950. [DOI] [PubMed] [Google Scholar]
- 35. Zhang H, Flagg TP, Nichols CG. Cardiac sarcolemmal K(ATP) channels: latest twists in a questing tale! J Mol Cell Cardiol. 2010;48(1):71–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Foster DB, Ho AS, Rucker J, et al. Mitochondrial ROMK channel is a molecular component of mitoK(ATP). Circ Res. 2012;111(4):446–454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Grandi E, Sanguinetti MC, Bartos DC, et al. Potassium channels in the heart: structure, function and regulation. J Physiol. 2017;595(7):2209–2228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Zingman LV, Hodgson DM, Bast PH, et al. Kir6.2 is required for adaptation to stress. Proc Natl Acad Sci U S A. 2002;99(20):13278–13283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Ionescu-Ittu R, Abrahamowicz M, Jackevicius CA, et al. Comparative effectiveness of rhythm control vs rate control drug treatment effect on mortality in patients with atrial fibrillation. Arch Intern Med. 2012;172(13):997–1004. [DOI] [PubMed] [Google Scholar]
- 40. Noma A. ATP-regulated K+ channels in cardiac muscle. Nature. 1983;305(5930):147–148. [DOI] [PubMed] [Google Scholar]
- 41. Nichols CG, Lederer WJ. Adenosine triphosphate-sensitive potassium channels in the cardiovascular system. Am J Physiol. 1991;261(6 pt 2):H1675–H1686. [DOI] [PubMed] [Google Scholar]
- 42. Liu Y, Sato T, O’Rourke B, Marban E. Mitochondrial ATP-dependent potassium channels: novel effectors of cardioprotection? Circulation. 1998;97(24):2463–2469. [DOI] [PubMed] [Google Scholar]
- 43. Garlid KD, Paucek P, Yarov-Yarovoy V, et al. Cardioprotective effect of diazoxide and its interaction with mitochondrial ATP-sensitive K+ channels. Possible mechanism of cardioprotection. Circ Res. 1997;81(6):1072–1082. [DOI] [PubMed] [Google Scholar]
- 44. Wolleben CD, Sanguinetti MC, Siegl PK. Influence of ATP-sensitive potassium channel modulators on ischemia-induced fibrillation in isolated rat hearts. J Mol Cell Cardiol. 1989;21(8):783–788. [DOI] [PubMed] [Google Scholar]
- 45. Chi L, Uprichard AC, Lucchesi BR. Profibrillatory actions of pinacidil in a conscious canine model of sudden coronary death. J Cardiovasc Pharmacol. 1990;15(3):452–464. [DOI] [PubMed] [Google Scholar]
- 46. D’Alonzo AJ, Zhu JL, Darbenzio RB, Dorso CR, Grover GJ. Proarrhythmic effects of pinacidil are partially mediated through enhancement of catecholamine release in isolated perfused guinea-pig hearts. J Mol Cell Cardiol. 1998;30(2):415–423. [DOI] [PubMed] [Google Scholar]
- 47. Lepran I, Baczko I, Varro A, Papp JG. ATP-sensitive potassium channel modulators: both pinacidil and glibenclamide produce antiarrhythmic activity during acute myocardial infarction in conscious rats. J Pharmacol Exp Ther. 1996;277(3):1215–1220. [PubMed] [Google Scholar]
- 48. Baczko I, Lepran I, Papp JG. KATP channel modulators increase survival rate during coronary occlusion–reperfusion in anaesthetized rats. Eur J Pharmacol. 1997;324(1):77–83. [DOI] [PubMed] [Google Scholar]
- 49. Baczko I, Husti Z, Lang V, Lepran I, Light PE. Sarcolemmal KATP channel modulators and cardiac arrhythmias. Curr Med Chem. 2011;18(24):3640–3661. [DOI] [PubMed] [Google Scholar]
- 50. Billman GE. The cardiac sarcolemmal ATP-sensitive potassium channel as a novel target for anti-arrhythmic therapy. Pharmacol Ther. 2008;120(1):54–70. [DOI] [PubMed] [Google Scholar]
- 51. Xu Y, Tuteja D, Zhang Z, et al. Molecular identification and functional roles of a Ca(2+)-activated K+ channel in human and mouse hearts. J Biol Chem. 2003;278(49):49085–49094. [DOI] [PubMed] [Google Scholar]
- 52. Skibsbye L, Poulet C, Diness JG, et al. Small-conductance calcium-activated potassium (SK) channels contribute to action potential repolarization in human atria. Cardiovasc Res. 2014;103(1):156–167. [DOI] [PubMed] [Google Scholar]
- 53. Gurney A, Manoury B. Two-pore potassium channels in the cardiovascular system. Eur Biophys J. 2009;38(3):305–318. [DOI] [PubMed] [Google Scholar]
- 54. Lesage F, Lazdunski M. Molecular and functional properties of two-pore-domain potassium channels. Am J Physiol Renal Physiol. 2000;279(5): F793–F801. [DOI] [PubMed] [Google Scholar]
- 55. O’Connell AD, Morton MJ, Hunter M. Two-pore domain K+ channels-molecular sensors. Biochim Biophys Acta. 2002;1566(1-2):152–161. [DOI] [PubMed] [Google Scholar]
- 56. Antzelevitch C, Burashnikov A. Atrial-selective sodium channel block as a novel strategy for the management of atrial fibrillation. J Electrocardiol. 2009;42(6):543–548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Ellinghaus P, Scheubel RJ, Dobrev D, et al. Comparing the global mRNA expression profile of human atrial and ventricular myocardium with high-density oligonucleotide arrays. J Thorac Cardiovasc Surg. 2005;129(6):1383–1390. [DOI] [PubMed] [Google Scholar]
- 58. Gaborit N, Le Bouter S, Szuts V, et al. Regional and tissue specific transcript signatures of ion channel genes in the non-diseased human heart. J Physiol. 2007;582(pt 2):675–693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Limberg SH, Netter MF, Rolfes C, et al. TASK-1 channels may modulate action potential duration of human atrial cardiomyocytes. Cell Physiol Biochem. 2011;28(4):613–624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Wahl-Schott C, Biel M. HCN channels: structure, cellular regulation and physiological function. Cell Mol Life Sci. 2009;66(3):470–494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Sartiani L, Romanelli MN, Mugelli A, Cerbai E. Updates on HCN channels in the heart: function, dysfunction and pharmacology. Curr Drug Targets. 2015;16(8):868–876. [DOI] [PubMed] [Google Scholar]
- 62. Benarroch EE. HCN channels: function and clinical implications. Neurology. 2013;80(3):304–310. [DOI] [PubMed] [Google Scholar]
- 63. Biel M, Schneider A, Wahl C. Cardiac HCN channels: structure, function, and modulation. Trends Cardiovasc Med. 2002;12(5):206–212. [DOI] [PubMed] [Google Scholar]
- 64. Zhao X, Gu T. Dysfunctional hyperpolarization-activated cyclic nucleotide-gated ion channels in cardiac diseases. Braz J Cardiovasc Surg. 2016;31(2):203–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Nass RD, Aiba T, Tomaselli GF, Akar FG. Mechanisms of disease: ion channel remodeling in the failing ventricle. Nat Clin Pract Cardiovasc Med. 2008;5(4):196–207. [DOI] [PubMed] [Google Scholar]
- 66. Heijman J, Voigt N, Nattel S, Dobrev D. Cellular and molecular electrophysiology of atrial fibrillation initiation, maintenance, and progression. Circ Res. 2014;114(9):1483–1499. [DOI] [PubMed] [Google Scholar]
- 67. Dobrev D, Friedrich A, Voigt N, et al. The G protein-gated potassium current I(K, ACh) is constitutively active in patients with chronic atrial fibrillation. Circulation. 2005;112(24):3697–3706. [DOI] [PubMed] [Google Scholar]
- 68. Schmidt C, Wiedmann F, Voigt N, et al. Upregulation of K(2P)3.1 K+ current causes action potential shortening in patients with chronic atrial fibrillation. Circulation. 2015;132(2):82–92. [DOI] [PubMed] [Google Scholar]
- 69. Wiedmann F, Schmidt C, Lugenbiel P, et al. Therapeutic targeting of two-pore-domain potassium (K(2P)) channels in the cardiovascular system. Clin Sci (Lond). 2016;130(9):643–650. [DOI] [PubMed] [Google Scholar]
- 70. Van Wagoner DR, Pond AL, McCarthy PM, Trimmer JS, Nerbonne JM. Outward K+ current densities and Kv1.5 expression are reduced in chronic human atrial fibrillation. Circ Res. 1997;80(6):772–781. [DOI] [PubMed] [Google Scholar]
- 71. Brandt MC, Priebe L, Bohle T, Sudkamp M, Beuckelmann DJ. The ultrarapid and the transient outward K(+) current in human atrial fibrillation. Their possible role in postoperative atrial fibrillation. J Mol Cell Cardiol. 2000;32(10):1885–1896. [DOI] [PubMed] [Google Scholar]
- 72. Workman AJ, Kane KA, Rankin AC. The contribution of ionic currents to changes in refractoriness of human atrial myocytes associated with chronic atrial fibrillation. Cardiovasc Res. 2001;52(2):226–235. [DOI] [PubMed] [Google Scholar]
- 73. Grammer JB, Bosch RF, Kuhlkamp V, Seipel L. Molecular remodeling of Kv4.3 potassium channels in human atrial fibrillation. J Cardiovasc Electrophysiol. 2000;11(6):626–633. [DOI] [PubMed] [Google Scholar]
- 74. Kovoor P, Wickman K, Maguire CT, et al. Evaluation of the role of I(KACh) in atrial fibrillation using a mouse knockout model. J Am Coll Cardiol. 2001;37(8):2136–2143. [DOI] [PubMed] [Google Scholar]
- 75. Yang KC, Foeger NC, Marionneau C, Jay PY, McMullen JR, Nerbonne JM. Homeostatic regulation of electrical excitability in physiological cardiac hypertrophy. J Physiol. 2010;588(pt 24):5015–5032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Marionneau C, Brunet S, Flagg TP, Pilgram TK, Demolombe S, Nerbonne JM. Distinct cellular and molecular mechanisms underlie functional remodeling of repolarizing K+ currents with left ventricular hypertrophy. Circ Res. 2008;102(11):1406–1415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Yang KC, Nerbonne JM. Mechanisms contributing to myocardial potassium channel diversity, regulation and remodeling. Trends Cardiovasc Med. 2016;26(3):209–218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Li GR, Lau CP, Leung TK, Nattel S. Ionic current abnormalities associated with prolonged action potentials in cardiomyocytes from diseased human right ventricles. Heart Rhythm. 2004;1(4):460–468. [DOI] [PubMed] [Google Scholar]
- 79. Su Z, Limberis J, Souers A, et al. Electrophysiologic characterization of a novel hERG channel activator. Biochem Pharmacol. 2009;77(8):1383–1390. [DOI] [PubMed] [Google Scholar]
- 80. Gerlach AC, Stoehr SJ, Castle NA. Pharmacological removal of human ether-a-go-go-related gene potassium channel inactivation by 3-nitro-N-(4-phenoxyphenyl) benzamide (ICA-105574). Mol Pharmacol. 2010;77(1):58–68. [DOI] [PubMed] [Google Scholar]
- 81. Hansen RS, Diness TG, Christ T, et al. Activation of human ether-a-go-go-related gene potassium channels by the diphenylurea 1,3-bis-(2-hydroxy-5-trifluoromethyl-phenyl)-urea (NS1643). Mol Pharmacol. 2006;69(1):266–277. [DOI] [PubMed] [Google Scholar]
- 82. Zhou J, Augelli-Szafran CE, Bradley JA, et al. Novel potent human ether-a-go-go-related gene (hERG) potassium channel enhancers and their in vitro antiarrhythmic activity. Mol Pharmacol. 2005;68(3):876–884. [DOI] [PubMed] [Google Scholar]
- 83. Jing H, Luo L, Li H, et al. Ephedrine controls heart rhythms by activating cardiac I(ks) currents. J Cardiovasc Pharmacol. 2010;55(2):145–152. [DOI] [PubMed] [Google Scholar]
- 84. Sun DD, Wang HC, Wang XB, et al. Tanshinone IIA: a new activator of human cardiac KCNQ1/KCNE1 (I(Ks)) potassium channels. Eur J Pharmacol. 2008;590(1-3):317–321. [DOI] [PubMed] [Google Scholar]
- 85. Kirby RW, Martelli A, Calderone V, McKay NG, Lawson K. Large conductance Ca(2+)-activated K(+) channel (BKCa) activating properties of a series of novel N-arylbenzamides: channel subunit dependent effects. Bioorg Med Chem. 2013;21(14):4186–4191. [DOI] [PubMed] [Google Scholar]
- 86. Bosch RF, Gaspo R, Busch AE, Lang HJ, Li GR, Nattel S. Effects of the chromanol 293B, a selective blocker of the slow, component of the delayed rectifier K+ current, on repolarization in human and guinea pig ventricular myocytes. Cardiovasc Res. 1998;38(2):441–450. [DOI] [PubMed] [Google Scholar]
- 87. Wang Z, Fermini B, Nattel S. Effects of flecainide, quinidine, and 4-aminopyridine on transient outward and ultrarapid delayed rectifier currents in human atrial myocytes. J Pharmacol Exp Ther. 1995;272(1):184–196. [PubMed] [Google Scholar]
- 88. Kobayashi S, Reien Y, Ogura T, Saito T, Masuda Y, Nakaya H. Inhibitory effect of bepridil on hKv1.5 channel current: comparison with amiodarone and E-4031. Eur J Pharmacol. 2001;430(2-3):149–157. [DOI] [PubMed] [Google Scholar]
- 89. Lagrutta A, Wang J, Fermini B, Salata JJ. Novel, potent inhibitors of human Kv1.5 K+ channels and ultrarapidly activating delayed rectifier potassium current. J Pharmacol Exp Ther. 2006;317(3):1054–1063. [DOI] [PubMed] [Google Scholar]
- 90. Loose S, Mueller J, Wettwer E, et al. Effects of IKur blocker MK-0448 on human right atrial action potentials from patients in sinus rhythm and in permanent atrial fibrillation. Front Pharmacol. 2014;5:26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Matsuda T, Masumiya H, Tanaka N, et al. Inhibition by a novel anti-arrhythmic agent, NIP-142, of cloned human cardiac K+ channel Kv1.5 current. Life Sci. 2001;68(17):2017–2024. [DOI] [PubMed] [Google Scholar]
- 92. Choe H, Lee YK, Lee YT, et al. Papaverine blocks hKv1.5 channel current and human atrial ultrarapid delayed rectifier K+ currents. J Pharmacol Exp Ther. 2003;304(2):706–712. [DOI] [PubMed] [Google Scholar]
- 93. Kang J, Wang L, Cai F, Rampe D. High affinity blockade of the HERG cardiac K(+) channel by the neuroleptic pimozide. Eur J Pharmacol. 2000;392(3):137–140. [DOI] [PubMed] [Google Scholar]
- 94. Rampe D, Murawsky MK, Grau J, Lewis EW. The antipsychotic agent sertindole is a high affinity antagonist of the human cardiac potassium channel HERG. J Pharmacol Exp Ther. 1998;286(2):788–793. [PubMed] [Google Scholar]
- 95. Ford J, Milnes J, El Haou S, et al. The positive frequency-dependent electrophysiological effects of the IKur inhibitor XEN-D0103 are desirable for the treatment of atrial fibrillation. Heart Rhythm. 2016;13(2):555–564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Zhang S, Rajamani S, Chen Y, et al. Cocaine blocks HERG, but not KvLQT1+minK, potassium channels. Mol Pharmacol. 2001;59(5):1069–1076. [DOI] [PubMed] [Google Scholar]
- 97. Milnes JT, Crociani O, Arcangeli A, Hancox JC, Witchel HJ. Blockade of HERG potassium currents by fluvoxamine: incomplete attenuation by S6 mutations at F656 or Y652. Br J Pharmacol. 2003;139(5):887–898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Takemasa H, Nagatomo T, Abe H, et al. Coexistence of hERG current block and disruption of protein trafficking in ketoconazole-induced long QT syndrome. Br J Pharmacol. 2008;153(3):439–447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Tang Q, Li ZQ, Li W, et al. The 5-HT2 antagonist ketanserin is an open channel blocker of human cardiac ether-a-go-go-related gene (hERG) potassium channels. Br J Pharmacol. 2008;155(3):365–373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Su Z, Chen J, Martin RL, et al. Block of hERG channel by ziprasidone: biophysical properties and molecular determinants. Biochem Pharmacol. 2006;71(3):278–286. [DOI] [PubMed] [Google Scholar]
- 101. Dong MQ, Lau CP, Gao Z, Tseng GN, Li GR. Characterization of recombinant human cardiac KCNQ1/KCNE1 channels (I(Ks)) stably expressed in HEK 293 cells. J Membr Biol. 2006;210(3):183–192. [DOI] [PubMed] [Google Scholar]
- 102. Liu H, Yang L, Chen KH, et al. SKF-96365 blocks human ether-a-go-go-related gene potassium channels stably expressed in HEK 293 cells. Pharmacol Res. 2016;104:61–69. [DOI] [PubMed] [Google Scholar]
- 103. Hashimoto N, Yamashita T, Tsuruzoe N. Characterization of in vivo and in vitro electrophysiological and antiarrhythmic effects of a novel IKACh blocker, NIP-151: a comparison with an IKr-blocker dofetilide. J Cardiovasc Pharmacol. 2008;51(2):162–169. [DOI] [PubMed] [Google Scholar]
- 104. Klose A, Huth T, Alzheimer C. 1-[6-[[(17beta)-3-Methoxyestra-1,3,5(10)-trien-17-yl]amino]hexyl]-1H-pyrrole-2,5-dione (U73122) selectively inhibits Kir3 and BK channels in a phospholipase C-independent fashion. Mol Pharmacol. 2008;74(5):1203–1214. [DOI] [PubMed] [Google Scholar]
- 105. Liu Y, Ren G, O’Rourke B, Marban E, Seharaseyon J. Pharmacological comparison of native mitochondrial K(ATP) channels with molecularly defined surface K(ATP) channels. Mol Pharmacol. 2001;59(2):225–230. [PubMed] [Google Scholar]
- 106. Christ T, Wettwer E, Voigt N, et al. Pathology-specific effects of the IKur/Ito/IK, ACh blocker AVE0118 on ion channels in human chronic atrial fibrillation. Br J Pharmacol. 2008;154(8):1619–1630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Li GR, Wang HB, Qin GW, et al. Acacetin, a natural flavone, selectively inhibits human atrial repolarization potassium currents and prevents atrial fibrillation in dogs. Circulation. 2008;117(19):2449–2457. [DOI] [PubMed] [Google Scholar]
- 108. Feng J, Wang Z, Li GR, Nattel S. Effects of class III antiarrhythmic drugs on transient outward and ultra-rapid delayed rectifier currents in human atrial myocytes. J Pharmacol Exp Ther. 1997;281(1):384–392. [PubMed] [Google Scholar]
- 109. Koidl B, Flaschberger P, Schaffer P, et al. Effects of the class III antiarrhythmic drug ambasilide on outward currents in human atrial myocytes. Naunyn Schmiedebergs Arch Pharmacol. 1996;353(2):226–232. [DOI] [PubMed] [Google Scholar]
- 110. Gao Z, Sun H, Chiu SW, Lau CP, Li GR. Effects of diltiazem and nifedipine on transient outward and ultra-rapid delayed rectifier potassium currents in human atrial myocytes. Br J Pharmacol. 2005;144(4):595–604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Li GR, Sun HY, Zhang XH, et al. Omega-3 polyunsaturated fatty acids inhibit transient outward and ultra-rapid delayed rectifier K+ currents and Na+ current in human atrial myocytes. Cardiovasc Res. 2009;81(2):286–293. [DOI] [PubMed] [Google Scholar]
- 112. Liu H, Jin MW, Xiang JZ, et al. Raloxifene inhibits transient outward and ultra-rapid delayed rectifier potassium currents in human atrial myocytes. Eur J Pharmacol. 2007;563(1-3):61–68. [DOI] [PubMed] [Google Scholar]
- 113. Xiao GS, Zhou JJ, Cheung YF, Li GR, Wong TM. Effects of U50,488H on transient outward and ultra-rapid delayed rectifier K+ currents in young human atrial myocytes. Eur J Pharmacol. 2003;473(2-3):97–103. [DOI] [PubMed] [Google Scholar]
- 114. Milnes JT, Louis L, Rogers M, Madge DJ, Ford J. The atrial antiarrhythmic drug XEN-D0101 selectively inhibits the human ultra-rapid delayed-rectifier potassium current (IKur) over other cardiac ion channels. Circulation. 2008;118(18):S342–S342. [Google Scholar]
- 115. Fedida D, Orth PM, Chen JY, et al. The mechanism of atrial antiarrhythmic action of RSD1235. J Cardiovasc Electrophysiol. 2005;16(11):1227–1238. [DOI] [PubMed] [Google Scholar]
- 116. Gross GJ, Castle NA. Propafenone inhibition of human atrial myocyte repolarizing currents. J Mol Cell Cardiol. 1998;30(4):783–793. [DOI] [PubMed] [Google Scholar]
- 117. Tian M, Dong MQ, Chiu SW, Lau CP, Li GR. Effects of the antifungal antibiotic clotrimazole on human cardiac repolarization potassium currents. Br J Pharmacol. 2006;147(3):289–297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Chen F, Esmailian F, Sun W, et al. Azimilide inhibits multiple cardiac potassium currents in human atrial myocytes. J Cardiovasc Pharmacol Ther. 2002;7(4):255–264. [DOI] [PubMed] [Google Scholar]
- 119. Rampe D, Roy ML, Dennis A, Brown AM. A mechanism for the proarrhythmic effects of cisapride (Propulsid): high affinity blockade of the human cardiac potassium channel HERG. FEBS Lett. 1997;417(1):28–32. [DOI] [PubMed] [Google Scholar]
- 120. Rampe D, Wible B, Fedida D, Dage RC, Brown AM. Verapamil blocks a rapidly activating delayed rectifier K+ channel cloned from human heart. Mol Pharmacol. 1993;44(3):642–648. [PubMed] [Google Scholar]
- 121. Gao Z, Lau CP, Chiu SW, Li GR. Inhibition of ultra-rapid delayed rectifier K+ current by verapamil in human atrial myocytes. J Mol Cell Cardiol. 2004;36(2):257–263. [DOI] [PubMed] [Google Scholar]
- 122. Li GR, Dong MQ. Pharmacology of cardiac potassium channels. Adv Pharmacol. 2010;59:93–134. [DOI] [PubMed] [Google Scholar]
- 123. Nattel S, Carlsson L. Innovative approaches to anti-arrhythmic drug therapy. Nat Rev Drug Discov. 2006;5(12):1034–1049. [DOI] [PubMed] [Google Scholar]
- 124. Amos GJ, Wettwer E, Metzger F, Li Q, Himmel HM, Ravens U. Differences between outward currents of human atrial and subepicardial ventricular myocytes. J Physiol. 1996;491(pt 1):31–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125. Li GR, Feng J, Yue L, Carrier M, Nattel S. Evidence for two components of delayed rectifier K+ current in human ventricular myocytes. Circ Res. 1996;78(4):689–696. [DOI] [PubMed] [Google Scholar]
- 126. Ford J, Milnes J, Wettwer E, et al. Human electrophysiological and pharmacological properties of XEN-D0101: a novel atrial-selective Kv1.5/IKur inhibitor. J Cardiovasc Pharmacol. 2013;61(5):408–415. [DOI] [PubMed] [Google Scholar]
- 127. Schotten U, de Haan S, Verheule S, et al. Blockade of atrial-specific K+-currents increases atrial but not ventricular contractility by enhancing reverse mode Na+/Ca2+-exchange. Cardiovasc Res. 2007;73(1):37–47. [DOI] [PubMed] [Google Scholar]
- 128. Wettwer E, Hala O, Christ T, et al. Role of IKur in controlling action potential shape and contractility in the human atrium: influence of chronic atrial fibrillation. Circulation. 2004;110(16):2299–2306. [DOI] [PubMed] [Google Scholar]
- 129. Ravens U, Poulet C, Wettwer E, Knaut M. Atrial selectivity of antiarrhythmic drugs. J Physiol. 2013;591(pt 17):4087–4097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130. Pavri BB, Greenberg HE, Kraft WK, et al. MK-0448, a specific Kv1.5 inhibitor: safety, pharmacokinetics, and pharmacodynamic electrophysiology in experimental animal models and humans. Circ Arrhythm Electrophysiol. 2012;5(6):1193–1201. [DOI] [PubMed] [Google Scholar]
- 131. Machida T, Hashimoto N, Kuwahara I, et al. Effects of a highly selective acetylcholine-activated K+ channel blocker on experimental atrial fibrillation. Circ Arrhythm Electrophysiol. 2011;4(1):94–102. [DOI] [PubMed] [Google Scholar]