

Abstract
Most FDA-approved adjuvants for infectious agents boost humoral but not cellular immunity, and have poorly-understood mechanisms. Stimulator of interferon genes (STING, also known as MITA, MPYS, or ERIS) is an exciting adjuvant target due to its role in cyclic dinucleotide (CDN)-driven anti-viral immunity; however, a major hindrance is STING’s cytosolic localization which requires intracellular delivery of its agonists. As a result, STING agonists administered in a soluble form have elicited suboptimal immune responses. Delivery of STING agonists via particle platforms has proven a more successful strategy, but the opportunity for improved formulations and bioactivity remains. In this study we evaluated the adjuvant activity of the potent STING agonist, CDN 3′3′-cGAMP (cGAMP), encapsulated in acid-sensitive acetalated dextran (Ace-DEX) polymeric microparticles (MPs) which passively target antigen-presenting cells for intracellular release. This formulation was superior to all particle delivery systems evaluated and maintained its bioactivity following a sterilizing dose of gamma irradiation. Compared to soluble cGAMP, the Ace-DEX cGAMP MPs enhanced type-I interferon responses nearly 1000-fold in vitro and 50-fold in vivo, caused up to a 104-fold boost in antibody titers, increased Th1-associated responses, and expanded germinal center B cells and memory T cells. Furthermore, the encapsulated cGAMP elicited no observable toxicity in animals and achieved protective immunity against a lethal influenza challenge seven months post-immunization when using CDN adjuvant doses up to 100-fold lower than previous reports. For these reasons, Ace-DEX MP-encapsulated cGAMP represents a potent vaccine adjuvant of humoral and cellular immunity.
Keywords: STING, cGAMP adjuvant, influenza vaccine, microparticle, acetalated dextran, electrospray
Graphical abstract
1. Introduction
The influenza virus is a significant source of morbidity and mortality, with 3 to 5 million severe cases worldwide each year [1], and it remains a considerable economic burden with an estimated annual cost of $87.1 billion in the United States alone [2]. The primary strategy for control and prevention of influenza is annual vaccination, but current influenza vaccines frequently suffer from relatively low efficacy [3] and short-lived (3 to 4 months) antibody responses [4], highlighting the need for improved formulations.
Influenza vaccines typically consist of killed or inactivated virus produced in eggs. This approach, however, has several significant drawbacks, including the lengthy chicken egg-based vaccine production process (6 to 8 months), limitation to using strains that grow well in eggs, and necessity of inactivation procedures that can decrease vaccine potency [3, 5]. Furthermore, recent evidence has demonstrated that passaging viruses through eggs also can induce mutations that decrease protection against circulating strains of virus [6].
Subunit protein-based formulations (e.g., FDA-approved Flublok®) have emerged as an alternative. This type of formulation circumvents some of the shortcomings of egg-based vaccines and allows relatively quick (< 2 months) production of recombinant viral proteins, enabling a more rapid response to emerging strains [5]. Protein antigens, however, often suffer from poor immunogenicity, necessitating delivery of high doses of recombinant protein or the addition of a vaccine adjuvant. Although traditional adjuvants (e.g., alum, squalene-based emulsions) have proven to be effective inducers of potent Th2-biased responses and humoral immunity, they have notable drawbacks. They often fail to induce significant Th1-responses that drive protective immunity against the influenza virus [7] and/or their mechanism of action is poorly understood [8]. As a result, there is a need for novel adjuvants for influenza vaccines that are capable of inducing balanced Th1/Th2 immunity and acting through well-defined mechanisms.
Among the many adjuvant candidates in development [9], considerable interest has been generated over the potential use of interferons (IFNs) [10]. IFNs are produced in response to infection and cancer [11], and can be broadly categorized by type, with type-I IFNs (e.g., IFN-α, IFN-β) binding to the IFN-α/β receptor (IFNAR). Transient expression of type-I IFNs can enhance activation of dendritic cells (DCs) [12–14], leading to enhanced priming of T cells [15]. Type-I IFNs also drive B cell activation and Ig class-switching [16–18]. Consequently, these IFNs are potent enhancers of both cellular and humoral immunity.
As an adjuvant, exogenous type-I IFNs have many critical drawbacks, including relatively short in vivo half-lives and the potential formation of anti-IFN neutralizing antibodies [19, 20]. An alternative to exogenous application is to stimulate endogenous type-I IFN production via innate immune receptors, the central one being the recently described stimulator of interferon genes (STING [21]; also known as MITA [22], MPYS [23], or ERIS [24]). Cyclic dinucleotides (CDNs), which are produced by microbes or mammalian cells, bind to STING. One robust STING-activating CDN is 3′3′ cyclic GMP-AMP (cGAMP), first identified in Vibrio cholera [25]. After cGAMP binds to STING, IRF3 and NFκB are activated, which drives expression of type-I IFNs and pro-inflammatory cytokines [26].
Although cGAMP and other CDNs represent an exciting novel class of vaccine adjuvants in animals models [27, 28], CDN delivery is still faced with the formidable physiological plasma membrane barrier that separates extracellular CDNs from their cognate cytosolic STING receptor. This obstacle has previously been overcome via the in vivo delivery of high doses of soluble CDNs (5–140 μg/dose) [29–37]. Parenteral delivery is one of the most common administration routes for clinical vaccines, but recent work has indicated that soluble CDNs injected via the intramuscular (i.m.) route does not augment strong vaccine-mediated immunity due to rapid drug diffusion away from the injection site [37]. Alternative approaches for in vivo parenteral CDN vaccine adjuvant delivery include formulating them with cell-penetrating peptides [38] or designing an adenovirus vector that induces CDN production [39]. Delivery vehicles can also be used for in vivo parenteral CDN administration and offer many advantages, such as targeted delivery and dose-sparing [40]. Liposomes and polymeric hydrogel particles, for instance, have been used to encapsulate CDNs as either a vaccine adjuvant [41, 42] or cancer immunotherapeutic [41, 43–45]. While these studies lay important groundwork which shows enhanced bioactivity of encapsulated CDNs compared to their soluble form, the formulation processes can result in relatively low CDN encapsulation efficiencies (e.g., 2 to 47% [41–45]). Furthermore, some of these vehicles can have limited long-term stability [40] and are fabricated by batch techniques [46, 47]. To address these outstanding issues, and identify a platform for efficient CDN encapsulation and delivery, we have developed an electrohydrodynamic spraying (electrospray) formulation of cGAMP-loaded acetalated dextran (Ace-DEX) polymeric microparticles (MPs) for intracellular delivery of a STING agonist.
Organic soluble Ace-DEX is formed via a one-step synthesis, where the pendant hydroxyl groups of FDA-approved water soluble dextran homopolysaccharide are converted into acetal groups. Unlike dextran, Ace-DEX’s solubility enables it to be processed into polymeric MPs using fabrication methods such as electrospray. Ace-DEX is an attractive biomaterial due to its biocompatibility, tunable biodegradability, ease of synthesis, and stability at elevated temperatures [48–51]. We have previously used electrospray to formulate Ace-DEX MPs encapsulating subunit vaccine components [52, 53]. Electrospray is a continuous method which facilitates ease of scalability [54] and efficient adjuvant encapsulation [52, 53]. Furthermore, electrosprayed Ace-DEX MPs can passively target antigen-presenting cells (APCs) based on size [55], and once phagocytosed, the MPs’ acid sensitivity results in rapid intracellular release of their payload in the phagolysosome’s acidic environment [48–50].
To evaluate our newly developed electrospray Ace-DEX cGAMP MP formulation, we compared this formulation’s bioactivity to several other particulate platforms, assessed its in vitro bioactivity in multiple APCs, evaluated in vivo adjuvant activity through measurement of antibody and cellular responses, as well as evaluated its ability to provide long-term protection against a lethal influenza challenge when formulated into a recombinant influenza protein vaccine.
2. Materials and methods
Additional detailed materials and methods can be found in supplementary information.
2.1 Reagents for Ace-DEX Synthesis and Ace-DEX cGAMP MP Fabrication
All materials used for polymer synthesis and MP fabrication were purchased from Sigma Aldrich (St. Louis, MO), unless otherwise indicated. Vaccine grade 3′3′-cGAMP was purchased from Invivogen (San Diego, CA).
2.2. Ace-DEX Synthesis
Ace-DEX was synthesized according to Kauffman et al. using dextran (average molecular weight of 70 kDa) from Leuconostoc mesenteroides [50]. After rapidly hydrolyzing the polymer in 10% v/v deuterium chloride in deuterium oxide, its relative cyclic acetal coverage was determined to be 40 ± 3% using 1H-NMR spectroscopy (Inova 400 MHz spectrometer; Varian Medical Systems, Palo Alto, CA) [50].
2.3. Electrospray Microparticle Fabrication and Characterization
ES cGAMP MPs (Ace-DEX or poly(lactic-co-glycolic acid) [PLGA], 85:15, 50–75 kDa, ester-terminated) were fabricated by a coaxial electrohydrodynamic spraying method (Figure S1) similar to Gallovic et al. [53]. A stainless steel plate (McMaster Carr, Elmhurst, IL) used as the MP collection surface was UV-treated for sterilization (1 hr) and incubated at high heat (260 °C, 45 min) to inactivate any potential endotoxin contamination [56]. cGAMP (varying concentrations dissolved in molecular grade water) and Ace-DEX (in an ethyl acetate/butanol/ethanol co-solvent) were driven through the core and shell of the coaxial needle (Ramé-Hart Instrument Co., Succasunna, NJ), respectively, using two separate syringe pumps (Scientific Lab Supply, Acton, MA). The needle and egg phosphatidylcholine (egg PC; Avanti Polar Lipids, Alabaster, AL)-coated plate, located 15 cm from each other, were charged (−7 and +2.5 kV, respectively) using high voltage power sources (Gamma High Voltage Research, Inc., Ormond Beach, FL), and aerosolized MPs were collected on the plate. Blank MPs were fabricated by the same process using pure molecular grade water in the inner phase. In order to determine cGAMP loading capacity in the MPs, cGAMP was first extracted from the MPs using 50% by volume dichloromethane with water. After vigorous vortexing, the mixture was centrifuged at 20,000 × g (4 °C, 60 min) to separate the water and dichloromethane phases. The amount of cGAMP was then quantified using high performance liquid chromatography (HPLC) with an isocratic mobile phase (80% water, 20% methanol by volume) operating at 0.6 mL/min through an Aquasil C18 (150 mm length, 4.6 mm inner diameter, 5 μm pore size; Thermo Fisher Scientific, Waltham, MA). The drug was detected at a UV-absorbance wavelength of 256 nm. A standard curve of soluble cGAMP alone was subjected to the same extraction conditions as the MPs. The MPs’ cGAMP loading capacity (Equation 1) and cGAMP encapsulation efficiency (Equation 2) was determined following background correction with complementary extracted blank MPs. The experimental cGAMP loading capacity is the actual amount of cGAMP encapsulated within the microparticles, as determined by HPLC, while the theoretical cGAMP loading capacity is the theoretical amount of cGAMP used in the microparticle formulation preparation.
| Equation 1 |
| Equation 2 |
The MPs were characterized according to the following methods. The endotoxin content was measured per Gallovic et al. [57] using a Pierce™ LAL Chromogenic Endotoxin Quantitation Kit. All MPs had an endotoxin content of less than 0.25 EU/mg, within the recommended level for preclinical subunit vaccine formulations [58]. For microscopic characterization, MPs were placed on carbon tape attached to aluminum pin stubs (Ted Pella, Inc., Redding, CA) and sputter-coated with 10 nm of AuPd using a Sputter Coater 108 Auto attached to a Thickness Monitor MTM-10 (Cressington Scientific Instruments, Hertfordshire, United Kingdom). Electron micrographs were acquired using an S-4700 scanning electron microscope (Hitachi High Technologies America, Schaumburg, IL) operating at an accelerating voltage of 2.0 kV. For particle size distribution and zeta potential measurements, each particle formulation was suspended separately in deionized water, and their hydrodynamic size (z-average, nm) and zeta potential (kV) were acquired using a Zetasizer Nano Z (Malvern Instruments Ltd, Malvern, United Kingdom). The reported size and zeta potential data are the mean of three values determined by a minimum of ten readings each.
2.4. In vitro analysis
Murine bone marrow derived dendritic cells (BMDCs) were obtained from C57BL/6 mice and cultured as described previously [59]. BMDCs for all experiments were treated with MPs suspended in supplemented RPMI media (10% inactivated fetal bovine serum [FBS], 100 U/mL penicillin, 100 μg/mL streptomycin, 50 μM 2-mercaptoethanol, 1X non-essential amino acids, and 2 mM L-glutamine). For the 28 day release experiment, MPs were suspended in RPMI media containing just 10% inactivated FBS, 100 U/mL penicillin, and 100 μg/mL streptomycin, but subsequent BMDC treatment with MPs remaining at each time point used the fully supplemented media described above. Primary murine peritoneal macrophages were isolated from the peritoneal space of C57BL/6 mice and treated with MPs suspended in RPMI media containing 10% inactivated FBS, 100 U/mL penicillin, and 100 μg/mL streptomycin. Human DCs were generated from patients enrolled in a clinical trial approved by the University of North Carolina (UNC) Institutional Review Board (Study #05-2860). These cells were provided as de-identified samples prior to use in the described study. The human cells were treated with MPs suspended in the same supplemented RPMI media used for the murine BMDC experiments. All cells were treated as described in figure legends, and cytokine levels produced in all in vitro murine and human cell experiments were subsequently assessed by ELISA or Luminex.
2.5. Immunization and in vivo assessment of immune response
All studies were conducted in accordance with National Institutes of Health’s guidelines for the care and use of laboratory animals and approved by the Institutional Animal Care and Use Committee at UNC. All animals, aged between 8 and 15 weeks, were maintained in a specific pathogen-free facility at UNC. Age and sex matched C57BL/6 mice were obtained from Jackson Laboratory (Bar Harbor, ME). Mice were immunized as indicated in figure legends via intramuscular injection of 50 μL. All antigens were delivered as soluble protein, and mixed with particle formulations immediately prior to immunization. Antibody responses were assessed via ELISA, and cellular responses were assessed via ELISA, ELISPOT, and flow cytometry.
2.6 Injection Site Cytokines
Mice were injected i.m. with 50 μL containing 0.2, 1, or 10 μg cGAMP delivered either in soluble form or 0.8–1 mg of Ace-DEX MPs. PBS or blank MP controls were included as well. Six hours later mice were euthanized and quadriceps were collected and homogenized in 4 μL of PBS containing 2 μg of protease inhibitor per mg of muscle tissue. This homogenate was then centrifuged at 12,000 × g for 10 minutes, supernatants were collected, and cytokine production was assessed by ELISA.
2.7 Influenza Infection and Animal Monitoring
C57BL/6 mice were immunized as described above. Mice were infected intranasally at post-immunization time points indicated in figure legends with influenza strain A/Puerto Rico/8/1934 H1N1. Animals were monitored daily for body weight and condition to determine survival.
2.8 Flow Cytometry
Lymph nodes and spleens were harvested from vaccinated mice. These organs were then processed into a single cell suspension after being passed through a 40 μM strainer, lysed with ammonium-chloride-potassium (ACK) solution, and washed in Hank’s balanced salt solution (HBSS) containing 3% FBS before staining and fixation in 1% paraformaldehyde. Immune cell populations were identified by flow cytometry using an LSRII (BD Biosciences, San Jose, CA) and analyzed by FlowJo software (Tree Star, Ashland, OR). Lymph nodes or splenocytes were stained with anti-mouse CD3-FITC and anti-mouse CD44-APC purchased from eBioscience (San Diego, CA). Additionally, cells were stained for anti-mouse CD4-APC-Cy7, anti-mouse CD62L PeCy7, anti-mouse CD8-Pacific Blue, anti-mouse CD95-PE, anti-mouse CD19-PECy7, and anti-mouse GL-7 Pacific Blue (Biolegend, San Diego, CA).
2.9 Statistical Analysis
Statistical analyses for antibody titers were performed in R using the Wilcoxon rank sum test. All remaining statistical analyses were performed with GraphPad Prism Version 6. Analysis of groups was performed as indicated in figures. All data points were included in the analyses, and no outliers were excluded in calculations of means or statistical significance. All box plots are displayed in a min-to-max format.
3. Results and discussion
3.1. Ace-DEX MPs are an efficient and stable platform for cGAMP delivery
In an effort to generate an efficient and potent STING-targeting delivery carrier we encapsulated cGAMP in multiple particulate platforms: electrospray (ES) Ace-DEX MPs, ES PLGA MPs, emulsion (Em) Ace-DEX MPs, and liposomes. A characterization of each particle formulation was performed (Table 1).
Table 1.
Characterization of cGAMP delivery vehicles.
| Formulation | Hydrodynamic Diameter (μm) | Polydispersity Index | Zeta Potential (mV) | cGAMP Loading Capacity (%) | Encapsulation Efficiency (%) |
|---|---|---|---|---|---|
| Electrospray Ace-DEX MPs | 1.54 ± 0.47 | 0.56 | −32.0 ± 0.7 | 0.52 | 89.7 |
| Electrospray PLGA MPs | 2.89 ± 0.69 | 0.71 | −27.1 ± 1.7 | 0.30 | 70.2 |
| Emulsion Ace-DEX MPs | 0.77 ± 0.03 | 0.22 | −34.5 ± 0.3 | 0.41 | 41.0 |
| Emulsion PLGA MPs | N/A* | N/A* | N/A* | 0 | 0 |
| Liposome | 0.46 ± 0.04 | 0.66 | +25.5 ± 1.4 | 1.37 | 30.4 |
Characterization not performed because cGAMP was not reliably encapsulated.
cGAMP loading capacity was quantified by HPLC. Hydrodynamic diameter and zeta potential were determined using a Zetasizer Nano Z (data reported as mean ± SEM, n = 3). Ace-DEX = acetalated dextran, PLGA = poly(lactic-co-glycolic acid), MP = microparticle.
Both of the ES MPs were micron-sized, with the PLGA MPs being larger as a result of the conditions required to electrospray each of the polymers [60]. The polydispersity indices (PDIs) for these MPs were relatively high, suggesting the presence of a mixed population of MP sizes and/or MP aggregation. The Em particles were sub-micron sized, which is consistent with previous reports using a double emulsion (via homogenization) followed by solvent evaporation to fabricate Ace-DEX MPs [53, 61, 62]. The lower PDI for this particle set suggests a more narrow size distribution and potentially less MP aggregation. The liposomes demonstrated some aggregation, contributing to their higher than expected hydrodynamic diameter and PDI. In contrast to the negative zeta potential of the polymeric particles, the liposomes were positively charged as a result of the DOTAP lipid component. cGAMP loading capacity was matched as closely as possible between polymeric MP platforms, since this property can have an effect on in vitro activation by adjuvant-loaded polymeric MPs [52]. The ES MPs had much higher encapsulation efficiencies (EEs) than their respective emulsion MPs. Hydrophilic small molecule cargo like cGAMP can easily be lost to the continuous outer water phase of a double emulsion, while the electrospray system consists of a continuous air phase, enhancing retention of the CDN. The less than 100% cGAMP encapsulation efficiency for the ES Ace-DEX MPs was likely the result of satellite particles lost to the ambient environment, while the ES PLGA MPs’ even lower EE was due in part to a less stable spray regime, leading to suboptimal collection of cGAMP-loaded MPs. cGAMP could not be reliably encapsulated within PLGA MPs using the double emulsion procedure. This is consistent with previous studies that attempted to encapsulate other hydrophilic cargo within PLGA MPs using similar emulsion methods [63, 64]. Compared to other reported CDN delivery vehicles (polymeric hydrogel particles (47%) [42] or liposomes (2 to 35%) [41, 43–45]), electrospray was able to achieve a significantly superior CDN EE (90%). This multi-fold increase in EE has promising manufacturing implications for using this electrospray platform to formulate hydrophilic, small molecule CDNs.
Although there were some differences in chemical properties between each formulated vehicle that could lead to variable in vitro murine BMDC activation, a proof-of-concept study was performed to compare the potency of each particulate platform. In this experiment, BMDCs were treated with identical doses of soluble cGAMP, cGAMP encapsulated within the various platforms, or cGAMP delivered using Lipofectamine, a micelle-based transfection platform. Type-I IFN and IL-6 production was assayed 6 hours later (Figure 1A and B). All of the polymer and liposome formulations significantly enhanced cGAMP-mediated IFN-β and IL-6 production over soluble and transfection controls, supporting the hypothesis that particulate delivery of cGAMP enhances its activity. This is consistent with previous reports where c-di-GMP was delivered via liposomes [41]. ES Ace-DEX cGAMP MPs, whose electron micrograph is displayed in Figure 1C, proved to be the most potent vehicle for induction of type-I IFN and IL-6, illustrating drastically improved type-I IFN production over ES PLGA cGAMP MPs and liposomes. This improved efficacy may be attributed to the polymer’s acid-sensitivity, resulting in rapid MP degradation, triggered release of the cargo within the acidic environment of the phagolysosome, and subsequent endosomal escape [65]. Although PLGA is attractive because of its biocompatibility and biodegradability, it is very slow-degrading compared to Ace-DEX and shows minimal release of its payload within acidic environments [66]. This could explain why Ace-DEX MPs outperformed PLGA MPs made through similar methods. In support of this hypothesis, antigen cross-presentation, which also relies on intracellular delivery of payload, is significantly enhanced via antigen encapsulation in Ace-DEX MPs relative to PLGA MPs [49]. Finally, degrading PLGA MPs produce acidic byproducts that could be potentially detrimental to vaccine outcomes [67]. Because of the ES Ace-DEX MP platform’s numerous advantages, they were used in all further experiments.
Figure 1. Evaluation of cGAMP delivery platforms.

(A–B) Bone marrow derived dendritic cells (BMDCs) from C57BL/6 mice were treated with 1 μg/mL soluble cGAMP, or an equivalent dose of cGAMP delivered in electrospray (ES) acetalated dextran microparticles (ES Ace-DEX MPs), ES poly(lactic-co-glycolic acid) microparticles (ES PLGA MPs), emulsion acetalated dextran microparticles (Em Ace-DEX MPs), SoyPC-DOTAP liposomes (Liposomes), or Lipofectamine 3000 transfection reagent (Transfection). Supernatants were collected 6 hours later and assayed for secreted IFN-β and IL-6. (C) Representative electron micrograph of Ace-DEX cGAMP MPs. (D–E) ES Blank MPs or ES Ace-DEX cGAMP MPs were incubated in pH-neutral RPMI media containing 10% inactivated fetal bovine serum, 100 U/mL penicillin, and 100 μg/mL streptomycin for 28 days. MPs still intact at 28 days (Figure S3B) were then used to treat BMDCs as either blank MPs (equivalent MP dose) or 1 μg/mL cGAMP in ES Ace-DEX MPs for 6 hours, and (D) IFN-β and (E) IL-6 levels were measured (all data represent BMDCs cultured from 3–4 individual mice ± SEM. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001).
A notable drawback to using MPs is the inability to sterilize the formulation via terminal 0.2 μm filtration, a common technique available for nanoparticulate formulations. While one option is to fabricate MPs in stringent aseptic cleanroom conditions, alternative terminal sterilization techniques are often preferred [68]. Other common terminal sterilization options such as ethylene oxide gas or e-beam treatment have substantial drawbacks (e.g., toxic residues or poor material penetration), whereas gamma irradiation circumvents these problems [68]. Here we have provided a proof-of-concept that Ace-DEX cGAMP MPs can be terminally sterilized using a sterilizing 25 kGy γ-irradiation dose without impacting the MPs’ immunological activity (Figure 2). MP structural integrity was not detrimentally affected (Figure S2).
Figure 2. cGAMP microparticles can be sterilized through gamma irradiation.
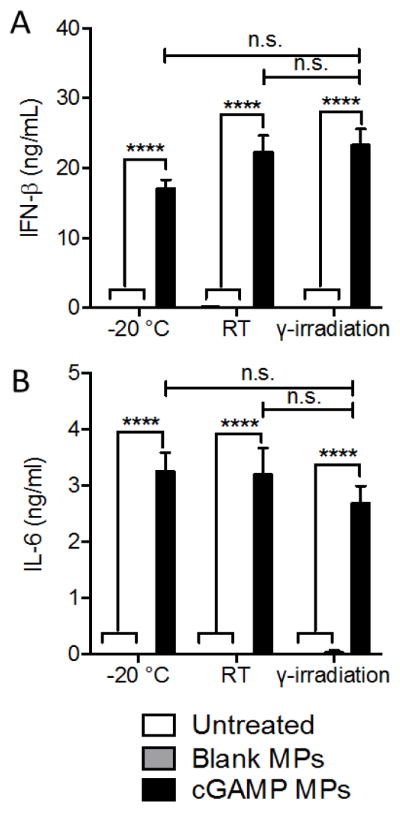
Blank and cGAMP loaded ES Ace-DEX microparticles (Blank MPs and cGAMP MPs, respectively) were incubated for 39 hours at −20°C, room temperature or 25 kGy gamma irradiation at RT. Bone marrow derived dendritic cells from C57BL/6 mice were then treated with 1 μg/mL cGAMP MPs stored at each condition, as well as blank MPs and untreated controls for 6 hours at 37 °C. Supernatants were collected and analyzed by ELISA for (A) IFN-β and (B) IL-6 (n=3 batches of particles. ****p < 0.0001).
To assess long-term encapsulated cGAMP stability under physiological conditions, ES Ace-DEX cGAMP MPs were incubated for 28 days in pH-neutral media at 37 °C, and cGAMP release was assessed (Figure S3A). After 28 days, particles remained intact (Figure S3B), and the remaining encapsulated cGAMP was still capable of stimulating IFN-β and IL-6 production in BMDCs (Figure 1D and E). These data demonstrate that after an initial burst release, the remaining encapsulated cGAMP was stable over 28 days at neutral pH and retained significant bioactivity. Future work will focus on improving drug retention within MPs at neutral pH to unlock the untapped potential of the platform.
3.2. Ace-DEX cGAMP MPs are dose-sparing and non-toxic, and enhance immune activation in vitro and in vivo
In order to assess the bioactivity of the ES Ace-DEX cGAMP MPs (hereafter referred to as cGAMP MPs), several primary APCs from murine and human donors were treated with this formulation. Murine BMDCs were treated with either soluble cGAMP, cGAMP MPs, or blank ES Ace-DEX MP controls (hereafter referred to as blank MPs). Soluble cGAMP did not induce inflammatory cytokines or type-I IFN at a concentration lower than 5 μg/mL cGAMP (Figure S4). cGAMP MPs, on the other hand, resulted in significantly enhanced responses (without inducing cell death), and provided 100 and 1000-fold dose-sparing compared to soluble cGAMP for pro-inflammatory cytokines and type-I IFN responses, respectively (Figure S4). Soluble cGAMP and blank MPs also did not induce detectable quantities of pro-inflammatory (TNF and IL-6). cGAMP MPs resulted in rapidly induced and sustained responses for all three cytokines (Figure S5). Similar results were obtained with primary murine peritoneal macrophages, where cGAMP MPs induced dramatically more cytokines than a Lipofectamine transfection reagent (Figure S6). Furthermore, despite the relatively rapid diffusion of cGAMP out of the MPs (Figure S2), a significant advantage was observed when using cGAMP encapsulation within MPs compared to cGAMP mixed with blank MPs (Figure S7). To evaluate human cells response to the formulated adjuvant, human-derived DCs from six individual donors were treated with cGAMP MPs and exhibited significantly enhanced production of type-I IFN, TNF, IL-6, and chemotactants (MIP-1α, IP-10 and RANTES), compared to soluble cGAMP (Figure S8). These results are consistent with other work demonstrating CDN activation of human DCs [69], and is of critical importance since a previously developed small molecule STING agonist, DMXAA, performed well in murine models but failed late-stage clinical trials. It was later determined that DMXAA could not activate STING in human cells [70].
Intramuscular (i.m.) administration of blank MPs or cGAMP MPs (with doses up to 20 μg cGAMP) in a preclinical mouse model resulted in no significant weight loss or increases in serum liver enzyme activity (Figure 3A and B). In addition, no mortality, deterioration in body condition, drop in body temperature, infection site reactogenicity, or detectable levels of serum IL-6, which is indicative of systemic inflammation, were observed (Figure S9). These results are highly significant as vaccines are routinely administered to healthy patients, and therefore, adjuvants are held to a particularly high safety standard. Very favorable safety profiles and a lack of adverse events in animal models, as reported here, are consequently critical characteristics for any adjuvant candidate warranting further development.
Figure 3. Microparticle delivery of cGAMP is safe and enhances cytokine responses in vivo.

(A–B) C57BL/6 mice were injected i.m. with cGAMP Ace-DEX microparticles (cGAMP MPs) at a final dose of 20, 10, 1, or 0.1 μg of cGAMP. A dose of blank MPs equivalent to 10 μg cGAMP MPs was also injected (Blank MPs). Weight loss (A) and ALT liver enzyme activity in serum (B) was assessed. (n=4 ± SD, all data are non-significant). (C–D) C57BL/6 mice were injected i.m. with the indicated dose of cGAMP either as soluble or within cGAMP MPs. Alternatively, mice were injected with PBS or blank MPs (Vehicle Ctrl). Six hours later the injected muscle tissue was harvested, and (C) IFN-β and (D) IL-6 concentrations were assessed in tissue homogenates by ELISA (n=5 mice ± SD, **p < 0.01, ****p < 0.0001).
Local immune activation in muscle tissue was assessed six hours after injection with either soluble cGAMP or cGAMP MPs with various drug loading capacities (Figure 3C and D). As with in vitro studies, cGAMP MPs profoundly enhanced local type-I IFN and IL-6 responses in vivo, achieving at least 10-fold and 50-fold dose-sparing, respectively, compared to soluble cGAMP. This is similar to local production of type-I IFNs resulting from adenoviral induction of c-di-GMP [39] and intradermal injection of 2′,3′-cGAMP [37]. It should be noted that although soluble CDN displayed very little bioactivity in vitro, relatively greater bioactivity was observed in vivo. These results may point to a yet uncharacterized mechanism which enhances soluble CDN bioactivity in vivo that is not captured in any of the in vitro models performed here. Type-I IFN and cytokine responses induced by cGAMP were, however, dramatically enhanced when encapsulated in Ace-DEX MPs, demonstrating that soluble CDNs represent a suboptimal adjuvant formulation.
In order to drive a strong and safe immune response, CDNs should ideally reach the lymph nodes without disseminating systemically. While some nanoparticulate delivery vehicles can drain directly from injection sites to lymph nodes [71], larger MPs may require active trafficking by immune cells. In order to assess MP trafficking in vivo, fluorescently-labeled blank Ace-DEX MPs were injected i.m. to mimic a vaccine administration route. Organs were collected at various time points up to 1 week post injection, and accumulation of fluorescently labeled particles was quantified using the IVIS imaging system. A time dependent accumulation of particles was observed in the draining inguinal lymph node, proximal to the injection site, which peaked 72 h post injection (Figure S10). This is in close agreement with our previous findings [72]. No accumulation of particles was observed in any other organs (lungs, spleen, liver, heart, brain, and blood), demonstrating that the injected particles trafficked efficiently to the lymph nodes and did not disseminate systemically. These data may explain why local, but not systemic inflammatory responses were observed following i.m. administration of cGAMP MPs (Figure S9D), contributing to a favorable safety profile.
3.3. Ace-DEX cGAMP MPs induce a potent humoral and cellular response to a model antigen independent of MP dose
We next assessed the adjuvant activity of cGAMP MPs in vivo using the model antigen ovalbumin (OVA). Although relatively non-inflammatory in vitro, polymeric particles, without inclusion of pathogen-associated molecular patterns, can exert adjuvant activity by inducing humoral immunity when combined with antigens in vivo [73–76]. This could be the result of antigen adsorbing to particle surfaces, leading to increased internalization of antigen by APCs [73, 74]. Thus, in order to begin to dissect out the individual contributions of the cGAMP cargo and polymeric delivery vehicle, Ace-DEX MPs were formulated with various cGAMP loading capacities, allowing for delivery of a fixed cGAMP dose (0.2 μg) in varying amounts of Ace-DEX MPs (0.02 to 1.0 mg). Equivalent doses of blank MPs were also tested. Other mice were immunized with either soluble OVA alone, or soluble OVA delivered with a conventional adjuvant (alum) or soluble cGAMP. While the OVA plus a low concentration of soluble cGAMP did not induce substantial OVA-specific total IgG levels over OVA alone, OVA plus cGAMP encapsulated in Ace-DEX MPs significantly enhanced these titers by 104 to 106-fold (Figure 4A). Antigen-specific total IgG titers, equivalent to OVA alone or OVA plus soluble cGAMP, were observed with higher doses (greater than or equal to 0.1 mg) of blank MPs co-delivered with soluble OVA, but waned at the low 0.02 mg MP dose. In the presence of soluble OVA, the cGAMP MP formulation at this low MP dose (0.2 μg cGAMP in 0.02 mg MP) yielded a greater than 106-fold increase in titers compared to the corresponding blank MPs and 103-fold increase compared to soluble cGAMP. OVA plus cGAMP MPs resulted in titers that were similar to OVA plus the conventional strong humoral adjuvant alum, independent of the MP dose, which suggests that cGAMP MPs have substantially higher adjuvant activity than the blank MP vehicle alone.
Figure 4. cGAMP microparticles enhance humoral and cellular immune responses.

C57BL/6 mice were injected i.m. on days 0 and 21 with PBS, or soluble ovalbumin (OVA, 10 μg) either alone or combined with blank microparticles (Blank MPs; 1.0–0.02 mg), Alhydrogel 2% (Alum, 1:1 by volume), soluble cGAMP (0.2 μg), or cGAMP MPs (0.2 μg cGAMP in 1.0–0.02 mg MP). Serum was collected on day 28 and assayed for OVA-specific (A) total IgG titers, (B) IgG1 titers, and (C) IgG2c titers (n=6–10 mice ± SEM pooled from two separate experiments, *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001). Alternatively, mice were injected i.m. on days 0, 21, and 35 with PBS, or soluble ovalbumin (10 μg) either alone (No Adjuvant) or in combination with Blank MPs (1 mg), Alum (1:1 by volume), soluble cGAMP (0.2 μg), or cGAMP MPs (0.2 μg in 1 mg MP). Splenocytes were harvested on day 42 and restimulated with CD8 restricted OVA peptide (SIINFEKL, 10 μg/mL) for 36 h. (D) IFN-γ or (F) IL-2 specific T cells were quantified by ELISPOT. Alternatively, splenocytes were re-stimulated using whole OVA protein (10 μg/mL) for 36 h. Supernatants were evaluated by ELISA for (E) IFN-γ or (G) IL-2 (n=3–5 mice ± SEM, representative of two individual experiments. *p < 0.05, **p < 0.01, ****p < 0.0001).
The impact of cGAMP MPs on antibody isotype skewing between Th2-associated IgG1 (Figure 4B) and Th1-associated IgG2c (Figure 4C) was next assessed. While IgG1 titers closely reflected total IgG levels described above, IgG2c titers were detected only when OVA was delivered with soluble cGAMP, cGAMP MPs, or the highest dose of blank MPs. As expected, the Th2-polarizing alum-adjuvanted formulation demonstrated no detectable IgG2c titers at all. At all particle doses, OVA plus cGAMP MPs induced significantly higher anti-OVA IgG2c titers than blank MPs. At the medium dose of particles (0.1 mg) plus OVA, blank MPs did not induce any IgG2c, while cGAMP MPs enhanced IgG2c titers 103-fold. The current results demonstrate that encapsulation of the cGAMP adjuvant within Ace-DEX MPs further enhances humoral adjuvant activity, as well as promotes a balance of both Th1 and Th2-associated IgG isotypes not achieved with OVA plus the blank MP vehicle alone or the conventional alum adjuvant.
To assess the impact of cGAMP MPs on cellular immunity, mice were immunized with either OVA alone or OVA in combination with blank MPs, 0.2 μg of soluble or Ace-DEX MP-encapsulated cGAMP, or alum. On day 42 (following three immunizations), splenocytes were stimulated with a CD8 restricted OVA peptide (SIINFEKL) (Figure 4D and 4F) or whole OVA protein (Figure 4E and 4G). T cell responses were assessed by IFN-γ and IL-2 ELISPOT (Figure 4D and 4F), as well as by ELISA (Figure 4E and 4G). IFN-γ and IL-2 positive spots were significantly increased in cGAMP MP-treated splenocytes compared to all other groups, while soluble cGAMP induced a small increase in all of these measurements. Similar results were observed with total cytokine levels following re-stimulation with whole protein. These findings are consistent with previous studies which found enhanced cellular responses following immunization with soluble CDN-adjuvanted model antigens [27–29, 77]. A notable difference between the current findings and these reports is that cGAMP MPs induced cellular responses at significantly lower doses of CDN than the soluble CDN studies.
Other studies have also reported an enhancement in OVA antigen-specific humoral and cellular responses with CDN-loaded delivery vehicles. When tested in a murine OVA vaccine model, polymeric hydrogel particles encapsulating cGAMP exhibited a very modest effect on antibody production over soluble cGAMP (< 20%) [42]. This is in contrast to the current data where cGAMP MPs demonstrated a nearly 104-fold antigen-specific antibody enhancement over soluble cGAMP (Fig. 3A). While cellular responses were not assessed in the polymeric hydrogel particle study, Hanson et al. [41] used multi-lamellar liposomes to deliver c-di-GMP and demonstrated comparable enhancement in OVA-specific cellular responses. It is important to note that these responses were achieved at a 25-fold higher dose of CDN than the current study.
3.4. Ace-DEX cGAMP MP vaccination generates a strong influenza-specific antibody response
We next tested cGAMP MPs in a viral antigen model. In this study, we used a fixed MP dose and varied the cGAMP dose. As MP dose did not impact cGAMP MPs’ efficacy in the model OVA vaccine (Figure 4A–C), 1 mg of MPs was used per injection to allow evaluation of a broad range of cGAMP loading capacities (0.02 to 1%). Mice were immunized with a hemagglutinin (HA) protein from influenza strain A/Puerto Rico/8/1934 H1N1 (PR8). This protein was produced by Protein Sciences, the same company that markets the unadjuvanted subunit HA Flublok® vaccine. Adsorption of the HA antigen to both blank and cGAMP loaded Ace-DEX MPs was characterized prior to injection (Table S1). Antigen adsorption did not vary significantly with the addition of cGAMP to the formulation. The HA antigen was then delivered either alone, or adjuvanted with blank MPs, indicated doses of cGAMP encapsulated within MPs, soluble cGAMP, or alum. Following a boost on day 21, HA antibody titers were assessed on day 28 (Figure 5). Total HA-specific IgG was induced by both soluble and cGAMP MPs in a dose dependent fashion (Figure 5A). cGAMP MPs increased antibody titers between 9 and 41-fold over soluble cGAMP and between 60 and 600-fold over the conventional vaccine adjuvant alum. They also generated total IgG titers significantly greater than the blank MP control. Notably, all mice receiving adjuvanted HA had detectable anti-HA antibody titers.
Figure 5. cGAMP microparticles enhance influenza-specific humoral immune response.

C57BL/6 mice were injected i.m. on days 0 and 21 with PBS, or soluble hemagglutinin (HA, 1 μg) either alone or combined with blank microparticles (Blank MPs), soluble cGAMP (Sol. cGAMP, 10–0.2 μg), cGAMP MPs (10–0.2 μg cGAMP in 1 mg MPs), or Alhydrogel 2% (Alum, 1:1 by volume). Serum was collected on day 28 and assayed for HA-specific (A) total IgG titer, (B) IgG1 titer, (C) IgG2c titer, (D) IgG2c:IgG1 isotype skewing, and (E) neutralizing titers (n=12–13 mice ± SEM, *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001).
Similar trends noted in the OVA studies were also observed with the anti-HA IgG1 (Figure 5B) and IgG2c (Figure 5C) titers. Soluble cGAMP and alum favored Th2-associated IgG1 isotypes, whereas cGAMP MPs profoundly promoted Th1-associated IgG2c production approaching 105-fold over soluble cGAMP and 103-fold over alum (Figure 5C). A comparison of the IgG2c:IgG1 ratio allows for a relative measure of Th1 and Th2 skewing where a ratio of 1 represents a balanced Th1/Th2 response, a ratio greater than 1 indicates a Th1 skewed response, and a ratio less than 1 indicates Th2 skewing. This analysis revealed that cGAMP MPs favored a balanced Th1:Th2 response and greater Th1-skewing at the 10 μg encapsulated-cGAMP dose, in contrast to Th2-polarization observed with soluble cGAMP and alum (Figure 5D).
These data are consistent with previous studies demonstrating that CDN adjuvants can increase anti-HA titers [30, 32–37], and in particular Th1-biased IgG subtype antibody levels [34–37]. There are, however, critical differences between the experimental design of the current study and several of the previous ones. These prior studies have focused only on mucosal CDN delivery and subsequent mucosal antibodies through intranasal, intratracheal, or sublingual administration [30, 33, 35, 36]. While these needle-free routes offer the opportunity for enhanced mucosal immunity, they do not mimic the parenteral administration routes used for the vast majority of clinical flu vaccines. Indeed, the Centers for Disease Control and Prevention recommended against immunizing patients with FluMist®, the only FDA-approved intranasally (i.n.)-administered influenza vaccine, for the 2016–17 flu season due to the poor efficacy (~3%) reported since 2014 [78]. In the context of a more clinically relevant i.m. delivery route, it is critical to note that both previous work [34, 37], as well the results reported here, demonstrate that soluble CDN does not result in skewing toward Th1-biased anti-influenza IgG titers. These results may reflect rapid diffusion of the unencapsulated CDN away from the injection site following immunization [37]. On the other hand, when CDN was encapsulated within Ace-DEX MPs, and administered i.m. using at least a 25 to 100-fold lower dose than previous reports [34–37], either balanced Th1:Th2 responses or Th1 skewing was observed.
In order to assess the functional neutralizing capacity of antibodies elicited by each formulation, neutralizing titers were assessed against the PR8 influenza virus (Figure 5E). Immunization with the HA antigen alone, a formulation representative of FDA-approved Flublok®, did not result in any detectable neutralizing titers. HA adjuvanted with blank MPs or alum also failed to induce neutralizing antibodies enhanced over the un-immunized PBS control group. While no significant difference was observed between soluble and encapsulated cGAMP, all cGAMP-loaded MP groups did produce high levels of neutralizing antibodies that were significantly greater than the levels exhibited with either antigen alone or antigen adjuvanted with alum. This is similar to another study where viral neutralizing titers were similarly enhanced versus antigen alone when adjuvanting with a soluble CDN [36].
3.5. Ace-DEX cGAMP MPs expand germinal center B cell, and memory T cell populations
cGAMP MPs (10 μg cGAMP) significantly increased germinal center B cell population in the draining (inguinal) lymph nodes 14 days post-boost compared to alum and blank MPs. In the spleen, total populations of central memory CD4 and CD8 T cells were significantly expanded following immunization using HA adjuvanted with cGAMP MPs compared to soluble cGAMP (Figure 6). These results are consistent with previous reports which demonstrate that CDN-based adjuvants can enhance influenza virus-specific CD4+ and/or CD8+ T cells, and favor Th1 or Th17-biased cellular responses [32–37]. These reports did not, however, examine whether influenza-specific T cells displayed a memory phenotype, which is a critical aspect of a durable cellular response. Our findings demonstrate a notable expansion of both CD4+ and CD8+ memory populations. While the lack of known T cell epitopes in PR8 HA prevented demonstration that these populations were influenza-specific, the data obtained from OVA studies (Figure 4) confirmed the generation of antigen-specific and Th1-polarized T cells. Together these experiments demonstrate that cGAMP MPs induce robust cellular memory responses.
Figure 6. cGAMP microparticles expand germinal center B cells and central memory T cells, and protect against lethal influenza challenge.

(A–C) C57BL/6 mice were injected i.m. on days 0 and 21 with PBS, or soluble hemagglutinin (HA, 1 μg) either alone or combined with blank microparticles (Blank MPs), soluble cGAMP (Sol. cGAMP, 0.2 μg), cGAMP MPs (0.2 μg cGAMP in 1 mg MPs), or Alhydrogel 2% (Alum, 1:1 by volume). (A) Lymph nodes were collected on day 35 and analyzed for total germinal center B cells (CD19+GL7+CD95+). (B–C) Spleens were collected and analyzed for total central memory CD4+ and CD8+ T cells (CD4/CD8+CD62hiCD44hi) (n=6–10 mice ± SD pooled from two individual experiments, *p < 0.05, **p < 0.01). (D–E) Alternatively, mice were immunized as above and challenged one month post-boost. Animal survival (D) and weight loss (E) were monitored for 14 days post challenge. The last recorded weight of deceased animals was used to calculate group averages at subsequent time points. (n=12–13 ± SD, *p < 0.05).
3.6. Ace-DEX cGAMP MP vaccination protects against lethal influenza challenge
To assess whether a cGAMP MP vaccine is protective against influenza infection, mice were vaccinated with a subset of the above groups and then challenged one month later with a lethal dose of PR8 influenza virus. Survival and weight loss were monitored for 14 days (Figure 6D and E). Soluble HA adjuvanted with cGAMP MPs demonstrated nearly complete protection (12 of 13 mice survived) against this highly virulent H1N1 influenza virus. This lethal challenge killed greater than 90% of unvaccinated mice and greater than 75% of animals vaccinated with the unadjuvanted HA formulation. Soluble cGAMP demonstrated modest 50% protection that was significantly lower than the protection offered by cGAMP MPs. Alum also provided a lower level of protection than cGAMP MPs. In addition to increased survival, cGAMP MP vaccinated animals displayed the least weight loss.
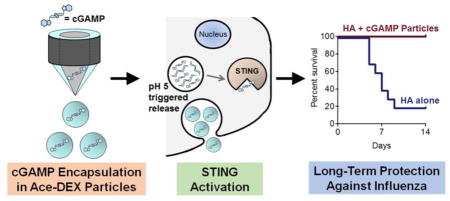
A common issue with existing influenza vaccines is relatively short-lived protection (3 to 4 months) [4]. In order to evaluate the long-term protection afforded with the vaccine formulations of interest in this study, animals were immunized per the short-term study, and then HA-specific IgG and neutralizing antibody titers were monitored for 4 months (Figure 7A and B). Alum, soluble cGAMP, and cGAMP MPs provided sustained levels of HA-specific IgG over time, with cGAMP MPs providing the highest overall titers. Similarly, serum collected from mice that were immunized with alum or cGAMP MP adjuvants resulted in sustained virus neutralizing activity, while the neutralizing capacity of serum from mice immunized with soluble cGAMP began to wane by 4 months post-immunization. In order to assess whether mice were protected over the timeframe of a clinical flu season, they were challenged with a lethal dose of A/Puerto Rico/8/34 H1N1 influenza virus seven months post-immunization. Mice immunized with cGAMP MPs maintained body weight post-infection, showing significantly less weight loss than all other groups tested (Figure 7C). Similarly, of the groups tested, only cGAMP MPs provided complete protection (Figure 7D). Moreover, similar protection was conferred by soluble cGAMP and unadjuvanted HA as before (Figure 6D). Furthermore, the protection afforded by the cGAMP MP-adjuvanted formulation was significantly superior to the alum group. Together these results demonstrate that immunization with a subunit influenza protein vaccine adjuvanted with cGAMP MPs provided long-term protection against a lethal influenza infection.
Figure 7. cGAMP MPs provide long term protection against lethal influenza challenge.

8 week old female C57BL/6 mice were immunized with PBS, or soluble hemagglutinin (HA, 1 μg) from strain A/Puerto Rico/8/1934/H1N1 either alone or combined with soluble cGAMP (HA + Sol cGAMP, 0.2 μg), cGAMP microparticles (HA + cGAMP MPs, 0.2 μg cGAMP in 1 mg MP) or Alhydrogel 2% (HA + Alum, 1:1 by volume). A boost was administered 21 days later. (A) Total HA specific IgG endpoint titers (B) and virus neutralizing titers were assessed over 4 months. Seven months post-immunization mice were infected intranasally with 2,000 ffu of A/Puerto Rico/8/1934/H1N1. (C) Survival, (D) weight loss, and (E) disease score were assessed daily for 14 days. The last recorded weight and disease score for deceased animals were used to calculate group averages at subsequent time points (n=7–10 ± SD. *p < 0.05, **p < 0.01).
While many of the previously reported CDN-adjuvanted influenza vaccine studies did not evaluate protection against an infection challenge [32–34, 36], there have been at least three studies where a challenge was performed following immunization. The first of these studies challenged with a sub-lethal dose of strain A/Puerto Rico/8/1934/H1N1 influenza virus and demonstrated that mice immunized i.n. with relatively high doses of nucleoprotein (10 μg) and a bacterially derived CDN (c-di-AMP; 10 μg) prevented weight loss compared to controls [35]. Similar protection was observed with the cGAMP MP-adjuvanted group in the current study, which was in the context of a more difficult to protect lethal infection model, and at a 50-fold lower CDN dose. In another more pertinent study, mice were immunized i.n. with a vaccine composed of 1.5 μg of a plant-derived HA combined with 5 μg of soluble c-di-GMP [30]. These immunizations conferred 80% protection against a lethal H5N1 influenza challenge using a CDN dose 25-fold higher than was used in the current study. Furthermore, while providing an important proof-of-principle for protection following immunization with a CDN-adjuvanted influenza vaccine, these two previously reported studies relied on intranasal vaccine administration, which is not commonly used in the clinic. As discussed earlier, the CDC recommended against immunizing with i.n.-administered FluMist® for the 2016–17 flu season due to poor vaccine efficacy over the past few years [78]. In one of the few studies that has explored i.m. delivery of a CDN-adjuvanted influenza vaccine, 0.3 μg HA content of an inactivated, monovalent seasonal vaccine combined with 20 μg of 2′3′-cGAMP, a host-derived CDN, was used [37]. This vaccine provided only ~15% protection against a lethal influenza challenge, and the authors concluded that i.m. delivered CDNs had suboptimal effectiveness as a vaccine adjuvant. In the current study, a related CDN (3′3′-cGAMP) was used and considerably higher protection (~50%) at much lower doses (100-fold) was observed. Critical differences in the choice of antigen (recombinant protein vs. inactivated virus) and dose of HA (1 μg vs. 0.3 μg) could account for this discrepancy.
Another important observation is that none of the previously reported work demonstrated long-lasting protection against infection. The maximum amount of time between immunization and challenge in the three studies was two months. In contrast, the current work has demonstrated that cGAMP MPs protect robustly against lethal influenza infection for at least seven months following a clinically relevant i.m. immunization using 25 to 100-fold lower CDN adjuvant doses. The current results are highly significant, because they demonstrate that cGAMP MP-adjuvanted immunization can achieve long-term memory over a timeframe of a clinical flu season.
Although our findings indicate very promising protection, neutralizing titers were not strongly correlated with weight loss, or days survived post challenge, suggesting that immune mechanisms other than direct antibody-mediated virus neutralization contributed to protection induced by cGAMP MPs. Similar findings have been reported by others [79–81], and antibody dependent cell-mediated cytotoxicity (ADCC) has emerged as an important component of a protective response to influenza infection [79, 80, 82]. This suggests that immune mechanisms contributing to cGAMP MP-mediated protection against influenza are likely multifaceted.
4. Conclusions
Encapsulation of cGAMP within Ace-DEX MPs by electrospray is a highly efficient and potentially scalable system for the production of potent type-I IFN, as well as pro-inflammatory cytokine responses, both in vitro and in vivo. We demonstrate that these particles produce balanced Th1/Th2-mediated humoral and cellular immune responses, and provide significant dose-sparing compared to soluble cGAMP. Finally, we provide a proof-of-principle that HA adjuvanted with Ace-DEX cGAMP MPs can provide long term protection in a mouse model of lethal influenza infection using a CDN adjuvant dose up to 100-fold lower than previously reported. Together these results demonstrate that Ace-DEX cGAMP MPs represent an effective vaccine adjuvant that warrants further development and investigation.
Supplementary Material
Acknowledgments
This work was supported by U19 AI109784 (to JPYT, GDS and EMB), UC6-AI058607 (to GDS), UNC internal funds (to KMA), T32-HL007106 (to BMJ), and T32-AI007151 (to RDJ). The following reagents were obtained through the NIH Biodefense and Emerging Infectious Research Repository, NIAID, NIH: Antisera Panel to Isolated Antigens of Influenza Virus, NR-10208. Madin-Darby Canine Kidney (MDCK) Cells, London Line, FR-58, was obtained through the Influenza Reagent Resource, Influenza Division, WHO Collaborating Center for Surveillance, Epidemiology and Control of Influenza, Centers for Disease Control and Prevention, Atlanta, GA, USA. The authors would like to thank the Chapel Hill Analytical and Nanofabrication Laboratory (CHANL) for their assistance with obtaining scanning electron micrographs. CHANL is a member of the North Carolina Research Triangle Nanotechnology Network (RTNN), which is supported by the National Science Foundation (Grant ECCS-1542015) as part of the National Nanotechnology Coordinated Infrastructure (NNCI). The authors would also like to acknowledge the valuable technical assistance of: Dr. Willie June Brickey, Kimberly Parks, Kristina Riebe, Melissa Samo, and Christopher Sample.
Footnotes
In addition to supporting figures, a detailed explanation of materials and methods is available in supplementary information.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.T.W.H. Organization. Influenza, Seasonal Fact Sheet. 2016. [Google Scholar]
- 2.Molinari NA, Ortega-Sanchez IR, Messonnier ML, Thompson WW, Wortley PM, Weintraub E, Bridges CB. The annual impact of seasonal influenza in the US: measuring disease burden and costs. Vaccine. 2007;25:5086–5096. doi: 10.1016/j.vaccine.2007.03.046. [DOI] [PubMed] [Google Scholar]
- 3.Osterholm MT, Kelley NS, Sommer A, Belongia EA. Efficacy and effectiveness of influenza vaccines: a systematic review and meta-analysis. The Lancet infectious diseases. 2012;12:36–44. doi: 10.1016/S1473-3099(11)70295-X. [DOI] [PubMed] [Google Scholar]
- 4.Kissling E, Valenciano M, Larrauri A, Oroszi B, Cohen JM, Nunes B, Pitigoi D, Rizzo C, Rebolledo J, Paradowska-Stankiewicz I, Jimenez-Jorge S, Horvath JK, Daviaud I, Guiomar R, Necula G, Bella A, O’Donnell J, Gluchowska M, Ciancio BC, Nicoll A, Moren A. Low and decreasing vaccine effectiveness against influenza A(H3) in 2011/12 among vaccination target groups in Europe: results from the I-MOVE multicentre case-control study. Euro surveillance: bulletin Europeen sur les maladies transmissibles = European communicable disease bulletin. 2013;18 doi: 10.2807/ese.18.05.20390-en. [DOI] [PubMed] [Google Scholar]
- 5.Yang LP. Recombinant trivalent influenza vaccine (flublok(®)): a review of its use in the prevention of seasonal influenza in adults. Drugs. 2013;73:1357–1366. doi: 10.1007/s40265-013-0103-6. [DOI] [PubMed] [Google Scholar]
- 6.Raymond DD, Stewart SM, Lee J, Ferdman J, Bajic G, Do KT, Ernandes MJ, Suphaphiphat P, Settembre EC, Dormitzer PR, Del Giudice G, Finco O, Kang TH, Ippolito GC, Georgiou G, Kepler TB, Haynes BF, Moody MA, Liao HX, Schmidt AG, Harrison SC. Influenza immunization elicits antibodies specific for an egg-adapted vaccine strain. Nat Med. 2016;22:1465–1469. doi: 10.1038/nm.4223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bungener L, Geeraedts F, Ter Veer W, Medema J, Wilschut J, Huckriede A. Alum boosts TH2-type antibody responses to whole-inactivated virus influenza vaccine in mice but does not confer superior protection. Vaccine. 2008;26:2350–2359. doi: 10.1016/j.vaccine.2008.02.063. [DOI] [PubMed] [Google Scholar]
- 8.Awate S, Babiuk LA, Mutwiri G. Mechanisms of action of adjuvants. Frontiers in immunology. 2013;4:114. doi: 10.3389/fimmu.2013.00114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.O’Hagan DT, Fox CB. New generation adjuvants–from empiricism to rational design. Vaccine. 2015;33:B14–B20. doi: 10.1016/j.vaccine.2015.01.088. [DOI] [PubMed] [Google Scholar]
- 10.Arico E, Belardelli F. Interferon-alpha as antiviral and antitumor vaccine adjuvants: mechanisms of action and response signature. Journal of interferon & cytokine research: the official journal of the International Society for Interferon and Cytokine Research. 2012;32:235–247. doi: 10.1089/jir.2011.0077. [DOI] [PubMed] [Google Scholar]
- 11.Ivashkiv LB, Donlin LT. Regulation of type I interferon responses. Nature reviews Immunology. 2014;14:36–49. doi: 10.1038/nri3581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Diamond MS, Kinder M, Matsushita H, Mashayekhi M, Dunn GP, Archambault JM, Lee H, Arthur CD, White JM, Kalinke U, Murphy KM, Schreiber RD. Type I interferon is selectively required by dendritic cells for immune rejection of tumors. The Journal of experimental medicine. 2011;208:1989–2003. doi: 10.1084/jem.20101158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Montoya M, Schiavoni G, Mattei F, Gresser I, Belardelli F, Borrow P, Tough DF. Type I interferons produced by dendritic cells promote their phenotypic and functional activation. Blood. 2002;99:3263–3271. doi: 10.1182/blood.v99.9.3263. [DOI] [PubMed] [Google Scholar]
- 14.Parlato S, Santini SM, Lapenta C, Di Pucchio T, Logozzi M, Spada M, Giammarioli AM, Malorni W, Fais S, Belardelli F. Expression of CCR-7, MIP-3beta, and Th-1 chemokines in type I IFN-induced monocyte-derived dendritic cells: importance for the rapid acquisition of potent migratory and functional activities. Blood. 2001;98:3022–3029. doi: 10.1182/blood.v98.10.3022. [DOI] [PubMed] [Google Scholar]
- 15.Welsh RM, Bahl K, Marshall HD, Urban SL. Type 1 interferons and antiviral CD8 T-cell responses. PLoS pathogens. 2012;8:e1002352. doi: 10.1371/journal.ppat.1002352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Alsharifi M, Lobigs M, Regner M, Lee E, Koskinen A, Mullbacher A. Type I interferons trigger systemic, partial lymphocyte activation in response to viral infection. Journal of immunology (Baltimore, Md: 1950) 2005;175:4635–4640. doi: 10.4049/jimmunol.175.7.4635. [DOI] [PubMed] [Google Scholar]
- 17.Braun D, Caramalho I, Demengeot J. IFN-alpha/beta enhances BCR-dependent B cell responses. International immunology. 2002;14:411–419. doi: 10.1093/intimm/14.4.411. [DOI] [PubMed] [Google Scholar]
- 18.Le Bon A, Thompson C, Kamphuis E, Durand V, Rossmann C, Kalinke U, Tough DF. Cutting edge: enhancement of antibody responses through direct stimulation of B and T cells by type I IFN. Journal of immunology (Baltimore, Md: 1950) 2006;176:2074–2078. doi: 10.4049/jimmunol.176.4.2074. [DOI] [PubMed] [Google Scholar]
- 19.Salmon P, Le Cotonnec JY, Galazka A, Abdul-Ahad A, Darragh A. Pharmacokinetics and pharmacodynamics of recombinant human interferon-beta in healthy male volunteers. J Interferon Cytokine Res. 1996;16:759–764. doi: 10.1089/jir.1996.16.759. [DOI] [PubMed] [Google Scholar]
- 20.Sorensen PS, Ross C, Clemmesen KM, Bendtzen K, Frederiksen JL, Jensen K, Kristensen O, Petersen T, Rasmussen S, Ravnborg M, Stenager E, Koch-Henriksen NG Danish Multiple Sclerosis Study. Clinical importance of neutralising antibodies against interferon beta in patients with relapsing-remitting multiple sclerosis. Lancet. 2003;362:1184–1191. doi: 10.1016/S0140-6736(03)14541-2. [DOI] [PubMed] [Google Scholar]
- 21.Ishikawa H, Barber GN. STING is an endoplasmic reticulum adaptor that facilitates innate immune signalling. Nature. 2008;455:674–678. doi: 10.1038/nature07317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhong B, Yang Y, Li S, Wang YY, Li Y, Diao F, Lei C, He X, Zhang L, Tien P, Shu HB. The adaptor protein MITA links virus-sensing receptors to IRF3 transcription factor activation. Immunity. 2008;29:538–550. doi: 10.1016/j.immuni.2008.09.003. [DOI] [PubMed] [Google Scholar]
- 23.Jin L, Hill KK, Filak H, Mogan J, Knowles H, Zhang B, Perraud AL, Cambier JC, Lenz LL. MPYS is required for IFN response factor 3 activation and type I IFN production in the response of cultured phagocytes to bacterial second messengers cyclic-di-AMP and cyclic-di-GMP. Journal of immunology (Baltimore, Md: 1950) 2011;187:2595–2601. doi: 10.4049/jimmunol.1100088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sun W, Li Y, Chen L, Chen H, You F, Zhou X, Zhou Y, Zhai Z, Chen D, Jiang Z. ERIS, an endoplasmic reticulum IFN stimulator, activates innate immune signaling through dimerization. Proceedings of the National Academy of Sciences of the United States of America. 2009;106:8653–8658. doi: 10.1073/pnas.0900850106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Davies BW, Bogard RW, Young TS, Mekalanos JJ. Coordinated regulation of accessory genetic elements produces cyclic di-nucleotides for V. cholerae virulence. Cell. 2012;149:358–370. doi: 10.1016/j.cell.2012.01.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Diner EJ, Burdette DL, Wilson SC, Monroe KM, Kellenberger CA, Hyodo M, Hayakawa Y, Hammond MC, Vance RE. The innate immune DNA sensor cGAS produces a noncanonical cyclic dinucleotide that activates human STING. Cell reports. 2013;3:1355–1361. doi: 10.1016/j.celrep.2013.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Dubensky TW, Jr, Kanne DB, Leong ML. Rationale, progress and development of vaccines utilizing STING-activating cyclic dinucleotide adjuvants. Therapeutic advances in vaccines. 2013;1:131–143. doi: 10.1177/2051013613501988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Li XD, Wu J, Gao D, Wang H, Sun L, Chen ZJ. Pivotal roles of cGAS-cGAMP signaling in antiviral defense and immune adjuvant effects. Science (New York, NY) 2013;341:1390–1394. doi: 10.1126/science.1244040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gray PM, Forrest G, Wisniewski T, Porter G, Freed DC, DeMartino JA, Zaller DM, Guo Z, Leone J, Fu TM, Vora KA. Evidence for cyclic diguanylate as a vaccine adjuvant with novel immunostimulatory activities. Cellular immunology. 2012;278:113–119. doi: 10.1016/j.cellimm.2012.07.006. [DOI] [PubMed] [Google Scholar]
- 30.Major D, Chichester JA, Pathirana RD, Guilfoyle K, Shoji Y, Guzman CA, Yusibov V, Cox RJ. Intranasal vaccination with a plant-derived H5 HA vaccine protects mice and ferrets against highly pathogenic avian influenza virus challenge. Human vaccines & immunotherapeutics. 2015;11:1235–1243. doi: 10.4161/21645515.2014.988554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Skrnjug I, Guzman CA, Rueckert C. Cyclic GMP-AMP displays mucosal adjuvant activity in mice. PloS one. 2014;9:e110150. doi: 10.1371/journal.pone.0110150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Madhun AS, Haaheim LR, Nøstbakken JK, Ebensen T, Chichester J, Yusibov V, Guzman CA, Cox RJ. Intranasal c-di-GMP-adjuvanted plant-derived H5 influenza vaccine induces multifunctional Th1 CD4+ cells and strong mucosal and systemic antibody responses in mice. Vaccine. 2011;29:4973–4982. doi: 10.1016/j.vaccine.2011.04.094. [DOI] [PubMed] [Google Scholar]
- 33.Neuhaus V, Chichester JA, Ebensen T, Schwarz K, Hartman CE, Shoji Y, Guzmán CA, Yusibov V, Sewald K, Braun A. A new adjuvanted nanoparticle-based H1N1 influenza vaccine induced antigen-specific local mucosal and systemic immune responses after administration into the lung. Vaccine. 2014;32:3216–3222. doi: 10.1016/j.vaccine.2014.04.011. [DOI] [PubMed] [Google Scholar]
- 34.Pedersen GK, Ebensen T, Gjeraker IH, Svindland S, Bredholt G, Guzmán CA, Cox RJ. Evaluation of the sublingual route for administration of influenza H5N1 virosomes in combination with the bacterial second messenger c-di-GMP. PLoS One. 2011;6:e26973. doi: 10.1371/journal.pone.0026973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sanchez MV, Ebensen T, Schulze K, Cargnelutti D, Blazejewska P, Scodeller EA, Guzmán CA. Intranasal delivery of influenza rNP adjuvanted with c-di-AMP induces strong humoral and cellular immune responses and provides protection against virus challenge. PLoS One. 2014;9:e104824. doi: 10.1371/journal.pone.0104824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Svindland SC, Pedersen GK, Pathirana RD, Bredholt G, Nøstbakken JK, Jul-Larsen Å, Guzmán CA, Montomoli E, Lapini G, Piccirella S, Jabbal-Gill I, Hinchcliffe M, Cox RJ. A study of Chitosan and c-di-GMP as mucosal adjuvants for intranasal influenza H5N1 vaccine. Influenza Other Respir Viruses. 2013;7:1181–1193. doi: 10.1111/irv.12056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wang J, Li P, Wu MX. Natural STING agonist as an “ideal” adjuvant for cutaneous vaccination. The Journal of investigative dermatology. 2016 doi: 10.1016/j.jid.2016.05.105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yildiz S, Alpdundar E, Gungor B, Kahraman T, Bayyurt B, Gursel I, Gursel M. Enhanced immunostimulatory activity of cyclic dinucleotides on mouse cells when complexed with a cell-penetrating peptide or combined with CpG. Eur J Immunol. 2015;45:1170–1179. doi: 10.1002/eji.201445133. [DOI] [PubMed] [Google Scholar]
- 39.Koestler BJ, Seregin SS, Rastall DP, Aldhamen YA, Godbehere S, Amalfitano A, Waters CM. Stimulation of innate immunity by in vivo cyclic di-GMP synthesis using adenovirus. Clin Vaccine Immunol. 2014;21:1550–1559. doi: 10.1128/CVI.00471-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Irvine DJ, Hanson MC, Rakhra K, Tokatlian T. Synthetic Nanoparticles for Vaccines and Immunotherapy. Chemical reviews. 2015;115:11109–11146. doi: 10.1021/acs.chemrev.5b00109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hanson MC, Crespo MP, Abraham W, Moynihan KD, Szeto GL, Chen SH, Melo MB, Mueller S, Irvine DJ. Nanoparticulate STING agonists are potent lymph node-targeted vaccine adjuvants. The Journal of clinical investigation. 2015;125:2532–2546. doi: 10.1172/JCI79915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lee E, Jang HE, Kang YY, Kim J, Ahn JH, Mok H. Submicron-sized hydrogels incorporating cyclic dinucleotides for selective delivery and elevated cytokine release in macrophages. Acta biomaterialia. 2016;29:271–281. doi: 10.1016/j.actbio.2015.10.025. [DOI] [PubMed] [Google Scholar]
- 43.Miyabe H, Hyodo M, Nakamura T, Sato Y, Hayakawa Y, Harashima H. A new adjuvant delivery system ‘cyclic di-GMP/YSK05 liposome’ for cancer immunotherapy. Journal of controlled release: official journal of the Controlled Release Society. 2014;184:20–27. doi: 10.1016/j.jconrel.2014.04.004. [DOI] [PubMed] [Google Scholar]
- 44.Nakamura T, Miyabe H, Hyodo M, Sato Y, Hayakawa Y, Harashima H. Liposomes loaded with a STING pathway ligand, cyclic di-GMP, enhance cancer immunotherapy against metastatic melanoma. Journal of controlled release: official journal of the Controlled Release Society. 2015;216:149–157. doi: 10.1016/j.jconrel.2015.08.026. [DOI] [PubMed] [Google Scholar]
- 45.Koshy ST, Cheung AS, Gu L, Graveline AR, Mooney DJ. Liposomal Delivery Enhances Immune Activation by STING Agonists for Cancer Immunotherapy. Advanced Biosystems. 2017 doi: 10.1002/adbi.201600013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Duong AD, Collier MA, Bachelder EM, Wyslouzil BE, Ainslie KM. One Step Encapsulation of Small Molecule Drugs in Liposomes via Electrospray-Remote Loading. Molecular pharmaceutics. 2016;13:92–99. doi: 10.1021/acs.molpharmaceut.5b00528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Johansen P, Martinez Gomez JM, Gander B. Development of synthetic biodegradable microparticulate vaccines: a roller coaster story. Expert Rev Vaccines. 2007;6:471–474. doi: 10.1586/14760584.6.4.471. [DOI] [PubMed] [Google Scholar]
- 48.Bachelder EM, Beaudette TT, Broaders KE, Dashe J, Frechet JM. Acetal-derivatized dextran: an acid-responsive biodegradable material for therapeutic applications. Journal of the American Chemical Society. 2008;130:10494–10495. doi: 10.1021/ja803947s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Broaders KE, Cohen JA, Beaudette TT, Bachelder EM, Frechet JM. Acetalated dextran is a chemically and biologically tunable material for particulate immunotherapy. Proceedings of the National Academy of Sciences of the United States of America. 2009;106:5497–5502. doi: 10.1073/pnas.0901592106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kauffman KJ, Do C, Sharma S, Gallovic MD, Bachelder EM, Ainslie KM. Synthesis and characterization of acetalated dextran polymer and microparticles with ethanol as a degradation product. ACS applied materials & interfaces. 2012;4:4149–4155. doi: 10.1021/am3008888. [DOI] [PubMed] [Google Scholar]
- 51.Kanthamneni N, Sharma S, Meenach SA, Billet B, Zhao JC, Bachelder EM, Ainslie KM. Enhanced stability of horseradish peroxidase encapsulated in acetalated dextran microparticles stored outside cold chain conditions. International journal of pharmaceutics. 2012;431:101–110. doi: 10.1016/j.ijpharm.2012.04.043. [DOI] [PubMed] [Google Scholar]
- 52.Duong AD, Sharma S, Peine KJ, Gupta G, Satoskar AR, Bachelder EM, Wyslouzil BE, Ainslie KM. Electrospray encapsulation of toll-like receptor agonist resiquimod in polymer microparticles for the treatment of visceral leishmaniasis. Molecular pharmaceutics. 2013;10:1045–1055. doi: 10.1021/mp3005098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Gallovic MD, Schully KL, Bell MG, Elberson MA, Palmer JR, Darko CA, Bachelder EM, Wyslouzil BE, Keane-Myers AM, Ainslie KM. Acetalated Dextran Microparticulate Vaccine Formulated via Coaxial Electrospray Preserves Toxin Neutralization and Enhances Murine Survival Following Inhalational Bacillus Anthracis Exposure. Advanced healthcare materials. 2016;5:2617–2627. doi: 10.1002/adhm.201600642. [DOI] [PubMed] [Google Scholar]
- 54.Almeria B, Fahmy TM, Gomez A. A multiplexed electrospray process for single-step synthesis of stabilized polymer particles for drug delivery. Journal of controlled release: official journal of the Controlled Release Society. 2011;154:203–210. doi: 10.1016/j.jconrel.2011.05.018. [DOI] [PubMed] [Google Scholar]
- 55.Kohane DS. Microparticles and nanoparticles for drug delivery. Biotechnology and bioengineering. 2007;96:203–209. doi: 10.1002/bit.21301. [DOI] [PubMed] [Google Scholar]
- 56.Tsuji K, Harrison SJ. Dry-heat destruction of lipopolysaccharide: dry-heat destruction kinetics. Applied and environmental microbiology. 1978;36:710–714. doi: 10.1128/aem.36.5.710-714.1978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Gallovic MD, Montjoy DG, Collier MA, Do C, Wyslouzil BE, Bachelder EM, Ainslie KM. Chemically modified inulin microparticles serving dual function as a protein antigen delivery vehicle and immunostimulatory adjuvant. Biomaterials science. 2016;4:483–493. doi: 10.1039/c5bm00451a. [DOI] [PubMed] [Google Scholar]
- 58.Brito LA, Singh M. Acceptable levels of endotoxin in vaccine formulations during preclinical research. Journal of pharmaceutical sciences. 2011;100:34–37. doi: 10.1002/jps.22267. [DOI] [PubMed] [Google Scholar]
- 59.Lutz MB, Kukutsch N, Ogilvie AL, Rossner S, Koch F, Romani N, Schuler G. An advanced culture method for generating large quantities of highly pure dendritic cells from mouse bone marrow. Journal of immunological methods. 1999;223:77–92. doi: 10.1016/s0022-1759(98)00204-x. [DOI] [PubMed] [Google Scholar]
- 60.Chakraborty S, Liao IC, Adler A, Leong KW. Electrohydrodynamics: a facile technique to fabricate drug delivery systems. Advanced drug delivery reviews. 2009;61:1043–1054. doi: 10.1016/j.addr.2009.07.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Schully K, Bell M, Prouty A, Gallovic M, Gautam S, Peine K, Sharma S, Bachelder EM, Pesce J, Elberson M. Evaluation of a biodegradable microparticulate polymer as a carrier for Burkholderia pseudomallei subunit vaccines in a mouse model of melioidosis. International journal of pharmaceutics. 2015;495:849–861. doi: 10.1016/j.ijpharm.2015.09.059. [DOI] [PubMed] [Google Scholar]
- 62.Schully KL, Sharma S, Peine KJ, Pesce J, Elberson MA, Fonseca ME, Prouty AM, Bell MG, Borteh H, Gallovic M. Rapid vaccination using an acetalated dextran microparticulate subunit vaccine confers protection against triplicate challenge by bacillus anthracis. Pharmaceutical research. 2013;30:1349–1361. doi: 10.1007/s11095-013-0975-x. [DOI] [PubMed] [Google Scholar]
- 63.Hennig D, Schubert S, Dargatz H, Kostenis E, Fahr A, Schubert US, Heinzel T, Imhof D. Novel insights into appropriate encapsulation methods for bioactive compounds into polymers: a study with peptides and HDAC inhibitors. Macromol Biosci. 2014;14:69–80. doi: 10.1002/mabi.201300213. [DOI] [PubMed] [Google Scholar]
- 64.Peine KJ, Bachelder EM, Vangundy Z, Papenfuss T, Brackman DJ, Gallovic MD, Schully K, Pesce J, Keane-Myers A, Ainslie KM. Efficient delivery of the toll-like receptor agonists polyinosinic:polycytidylic acid and CpG to macrophages by acetalated dextran microparticles. Mol Pharm. 2013;10:2849–2857. doi: 10.1021/mp300643d. [DOI] [PubMed] [Google Scholar]
- 65.Murthy N, Xu M, Schuck S, Kunisawa J, Shastri N, Frechet JM. A macromolecular delivery vehicle for protein-based vaccines: acid-degradable protein-loaded microgels. Proceedings of the National Academy of Sciences of the United States of America. 2003;100:4995–5000. doi: 10.1073/pnas.0930644100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Walter E, Dreher D, Kok M, Thiele L, Kiama SG, Gehr P, Merkle HP. Hydrophilic poly(DL-lactide-co-glycolide) microspheres for the delivery of DNA to human-derived macrophages and dendritic cells. Journal of controlled release: official journal of the Controlled Release Society. 2001;76:149–168. doi: 10.1016/s0168-3659(01)00413-8. [DOI] [PubMed] [Google Scholar]
- 67.Jiang W, Gupta RK, Deshpande MC, Schwendeman SP. Biodegradable poly(lactic-co-glycolic acid) microparticles for injectable delivery of vaccine antigens. Advanced Drug Delivery Reviews. 2005;57:391–410. doi: 10.1016/j.addr.2004.09.003. [DOI] [PubMed] [Google Scholar]
- 68.Desai KGH, Kadous S, Schwendeman SP. Gamma irradiation of active self-healing PLGA microspheres for efficient aqueous encapsulation of vaccine antigens. Pharmaceutical research. 2013;30:1768–1778. doi: 10.1007/s11095-013-1019-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Karaolis DK, Means TK, Yang D, Takahashi M, Yoshimura T, Muraille E, Philpott D, Schroeder JT, Hyodo M, Hayakawa Y, Talbot BG, Brouillette E, Malouin F. Bacterial c-di-GMP is an immunostimulatory molecule. J Immunol. 2007;178:2171–2181. doi: 10.4049/jimmunol.178.4.2171. [DOI] [PubMed] [Google Scholar]
- 70.Conlon J, Burdette DL, Sharma S, Bhat N, Thompson M, Jiang Z, Rathinam VA, Monks B, Jin T, Xiao TS, Vogel SN, Vance RE, Fitzgerald KA. Mouse, but not human STING, binds and signals in response to the vascular disrupting agent 5,6-dimethylxanthenone-4-acetic acid. J Immunol. 2013;190:5216–5225. doi: 10.4049/jimmunol.1300097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Moyer TJ, Zmolek AC, Irvine DJ. Beyond antigens and adjuvants: formulating future vaccines. The Journal of clinical investigation. 2016;126:799–808. doi: 10.1172/JCI81083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Chen N, Peine KJ, Collier MA, Gautam S, Jablonski KA, Guerau-de-Arellano M, Ainslie KM, Bachelder EM. Co-Delivery of Disease Associated Peptide and Rapamycin via Acetalated Dextran Microparticles for Treatment of Multiple Sclerosis. Advanced Biosystems. 2017;1:1700022-n/a. [Google Scholar]
- 73.Galloway AL, Murphy A, DeSimone JM, Di J, Herrmann JP, Hunter ME, Kindig JP, Malinoski FJ, Rumley MA, Stoltz DM, Templeman TS, Hubby B. Development of a nanoparticle-based influenza vaccine using the PRINT technology. Nanomedicine. 2013;9:523–531. doi: 10.1016/j.nano.2012.11.001. [DOI] [PubMed] [Google Scholar]
- 74.Metz SW, Tian S, Hoekstra G, Yi X, Stone M, Horvath K, Miley MJ, DeSimone J, Luft CJ, de Silva AM. Precisely Molded Nanoparticle Displaying DENV-E Proteins Induces Robust Serotype-Specific Neutralizing Antibody Responses. PLoS Negl Trop Dis. 2016;10:e0005071. doi: 10.1371/journal.pntd.0005071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Zhang W, Wang L, Liu Y, Chen X, Liu Q, Jia J, Yang T, Qiu S, Ma G. Immune responses to vaccines involving a combined antigen–nanoparticle mixture and nanoparticle-encapsulated antigen formulation. Biomaterials. 2014;35:6086–6097. doi: 10.1016/j.biomaterials.2014.04.022. [DOI] [PubMed] [Google Scholar]
- 76.Kipper MJ, Wilson JH, Wannemuehler MJ, Narasimhan B. Single dose vaccine based on biodegradable polyanhydride microspheres can modulate immune response mechanism. Journal of Biomedical Materials Research Part A. 2006;76:798–810. doi: 10.1002/jbm.a.30545. [DOI] [PubMed] [Google Scholar]
- 77.Ebensen T, Libanova R, Schulze K, Yevsa T, Morr M, Guzmán CA. Bis-(3′,5′)-cyclic dimeric adenosine monophosphate: strong Th1/Th2/Th17 promoting mucosal adjuvant. Vaccine. 2011;29:5210–5220. doi: 10.1016/j.vaccine.2011.05.026. [DOI] [PubMed] [Google Scholar]
- 78.ACIP votes down use of LAIV for 2016–2017 flu season. Centers for Disease Control and Prevention; https://www.cdc.gov/media/releases/2016/s0622-laiv-flu.html. [Google Scholar]
- 79.Impagliazzo A, Milder F, Kuipers H, Wagner MV, Zhu X, Hoffman RM, van Meersbergen R, Huizingh J, Wanningen P, Verspuij J, de Man M, Ding Z, Apetri A, Kukrer B, Sneekes-Vriese E, Tomkiewicz D, Laursen NS, Lee PS, Zakrzewska A, Dekking L, Tolboom J, Tettero L, van Meerten S, Yu W, Koudstaal W, Goudsmit J, Ward AB, Meijberg W, Wilson IA, Radosevic K. A stable trimeric influenza hemagglutinin stem as a broadly protective immunogen. Science (New York, NY) 2015;349:1301–1306. doi: 10.1126/science.aac7263. [DOI] [PubMed] [Google Scholar]
- 80.Yassine HM, Boyington JC, McTamney PM, Wei CJ, Kanekiyo M, Kong WP, Gallagher JR, Wang L, Zhang Y, Joyce MG, Lingwood D, Moin SM, Andersen H, Okuno Y, Rao SS, Harris AK, Kwong PD, Mascola JR, Nabel GJ, Graham BS. Hemagglutinin-stem nanoparticles generate heterosubtypic influenza protection. Nature medicine. 2015;21:1065–1070. doi: 10.1038/nm.3927. [DOI] [PubMed] [Google Scholar]
- 81.Henry Dunand CJ, Leon PE, Huang M, Choi A, Chromikova V, Ho IY, Tan GS, Cruz J, Hirsh A, Zheng NY, Mullarkey CE, Ennis FA, Terajima M, Treanor JJ, Topham DJ, Subbarao K, Palese P, Krammer F, Wilson PC. Both Neutralizing and Non-Neutralizing Human H7N9 Influenza Vaccine-Induced Monoclonal Antibodies Confer Protection. Cell host & microbe. 2016;19:800–813. doi: 10.1016/j.chom.2016.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Jegaskanda S, Reading PC, Kent SJ. Influenza-specific antibody-dependent cellular cytotoxicity: toward a universal influenza vaccine. The Journal of Immunology. 2014;193:469–475. doi: 10.4049/jimmunol.1400432. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.