

Abstract
Mounting evidence suggests that prolonged exposure to general anesthesia (GA) during brain synaptogenesis damages the immature neurons and results in long-term neurocognitive impairments. Importantly, synaptogenesis relies on timely axon pruning to select axons that participate in active neural circuits formation. This process is in part dependent on proper homeostasis of neurotrophic factors, in particular brain-derived neurotrophic factor (BDNF).
We set out to examine how GA may modulate axon maintenance and pruning and focused on the role of BDNF. We exposed post-natal day (PND) 7 mice to ketamine using a well-established dosing regimen known to induce significant developmental neurotoxicity. We performed morphometric analyses of the infrapyramidal bundle (IPB) since IPB is known to undergo intense developmental modeling and as such is commonly used as a well-establish model of in vivo pruning in rodents.
When IPB remodeling was followed from PND10 until PND65 we noted a delay in axonal pruning in ketamine-treated animals when compared to controls; this impairment coincided with ketamine-induced downregulation in BDNF protein expression and maturation suggesting two conclusions: a surge in BDNF protein expression ‘signals’ intense IPB pruning in control animals and, ketamine-induced downregulation of BDNF synthesis and maturation could contribute to impaired IPB pruning.
We conclude that the combined effects on BDNF homeostasis and impaired axon pruning may in part explain ketamine-induced impairment of neuronal circuitries formation.
Keywords: Infrapyramidal bundle, Synaptogenesis, Brain-derived neurotrophic factor, Mossy fibers, General anesthesia, Immature brain
Introduction
Advances in modern medicine enable us to care for sick and premature children, but force us to rely on extensive and frequent use of sedatives and general anesthetics during painful interventions. Unfortunately, recent discoveries show that exposure to sedation and general anesthesia (GA) during critical stages of brain development (i.e. synaptogenesis), may be damaging to immature neurons by causing extensive apoptotic cell death ultimately resulting in long-term neurocognitive and behavioral impairments [1-7].
Synaptogenesis involves two equally important regressive events: 1) naturally occurring neuronal death by apoptosis to eliminate neurons that are not appropriately connected with their targets [8, 9] and, 2) axon pruning to select axons that participate in active neural circuits [8, 9]. We know that GA exacerbates neuronal apoptosis, thus causing wide-spread deletion of developing neurons in vulnerable brain regions. However, it is not yet clear whether or how GA impairs selection of appropriate axons and elimination of redundant axons, two balancing forces necessary for the formation and fine-tuning of neuronal networks. Considering the protracted nature of cognitive and behavioral impairments that appear to worsen over a lifetime [1, 10], we set out to examine whether an early exposure to GA causes long-term impairments of proper axon maintenance and pruning in neurons that survive the initial apoptotic ‘attack’.
It seems that axon pruning manifests through at least two major phases: one, more robust and related to interplay of major neutrotrophic factors and later phase, dominated by locally produced and secreted neuronal growth factors. Hence, to begin to understand the mechanisms of anesthesia-induced modulation of axon maintenance and pruning we focused on the role of neurotrophic factors. Brain-derived neurotrophic factor (BDNF) was of particular interest for this study, because the disturbances in BDNF expression and function have been shown to impair axon pruning. For example, sympathetic neuron targets are inappropriately innervated in BDNF(+/-) knockout mice [11] whereas BDNF deprivation in neuronal cultures impairs axonal growth, causes extensive axonal degeneration and impairs axon competition [11]. Given that GA exposure during critical stages of synaptogenesis causes perturbation in BDNF regulation and impairment of Trk-pathway activation [12, 13] while decreasing neuronal activity, we hypothesize that GA impairs the axon selection and pruning, which may explain long-term defects in neuronal networking and may in turn be the culprit for reported functional impairments.
We used an early exposure to ketamine as a well-established model of anesthesia-induced developmental neurotoxicity in mice [14] and developmental modeling of infrapyramidal bundle as well-establish model of in vivo pruning [15]. We report that an early exposure to ketamine delays axonal pruning; this impairment coincides with ketamine-induced downregulation in BDNF expression and maturation suggesting that ketamine-induced modulation of BDNF synthesis and maturation could at least in part contribute to the impairment of neuronal circuitries formation.
Materials and Methods
Animals
We used 7-day-old (PND7) CD-1 mice (Harlan Laboratories, Indianapolis, IN) for all experiments. We chose this age because 1) it is when rodents are most vulnerable to GA-induced developmental neurotoxicity [16] and, 2) it falls before developmental pruning of the IPB begins [15]. Our ketamine anesthesia protocol was as follows: experimental mouse pups were exposed to 6h of ketamine anesthesia and controls were exposed to 6h of mock anesthesia (vehicle) injected I.M. During anesthesia, pups were carefully monitored. After the administration of anesthesia, mice were reunited with their mothers until sacrifice (from P8 until P65). The weaning was done at P21 using the standard protocol. At the desired age mice were divided randomly into two groups: one group for assessing expression of pro- and the mature form of BDNF using the Western blotting technique and the second group for morphometric studies of IPB development. Our randomization process was designed to provide each group with roughly equal representation of pups from each dam.
The experiments were approved by the Animal Use and Care Committees at the University of Colorado the Office of Animal Resources (OLAR), Aurora, Colorado and the Animal Use and Care Committees of the University of Virginia, Charlottesville, Virginia. The experiments were done in accordance with the Public Health Service's Policy on Humane Care and Use of Laboratory Animals. Efforts were made to minimize the number of animals used while being able to conduct meaningful statistical analyses.
Anesthesia administration
To achieve general anesthesia state, we used a ketamine protocol known to cause significant developmental neurotoxicity in PND7 mice whereby mouse pups received a total of four doses of ketamine, at 75 mg/kg, IM every 90 minutes so that the loss of righting reflex and lack of response to tail pinch could be maintained for 6 hours [14]. For control animals, saline was administered using the same volume and administration schedule. During entire anesthesia procedure, animals were kept away from their mother and were housed in standard, tightly closed mice cages, with free air flow through air filters. Animals were kept in close proximity to each other in the cages, so they could preserve, even under anesthesia, important olfactory cues and stimuli, necessary to bust and sustain their metabolic output. During the experiment, we carefully monitored animals and measured environmental temperature in their breeding cages. We established that the ambient temperature in the breeding cage was around 37.0±1°C. Considering that animals at this age are very sensitive to change in body temperature, they were kept under constant ambient temperature maintained with heating pads conveniently set up around the cages. The ambient temperature was assessed at frequent time intervals using the thermometer.
Western blot studies
For BDNF protein quantification, we dissected the hippocampus immediately after the brains were removed from the individual pups using a dissecting scope (10× magnification). Tissue was collected on ice and was snap-frozen in liquid nitrogen immediately. The protein concentration of the lysates was determined with the Total Protein kit using the Bradford method (Cayman Chemical, Ann Arbor, MI). Approximately 10-25 μg of total protein was heat- denatured, and subjected to sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) through 4-20% Tris-glycine polyacrylamide gradient gels (BioRad, Hercules, CA). Separated proteins were transferred to polyvinylidene difluoride (PVDF) membrane (Millipore, Billerica, MA), blocked at room temperature for 1h in 3% bovine serum albumin (BSA) followed by incubation at 4°C overnight with primary antibody, anti-BDNF (1:500, Alomone Labs, Jerusalem, Israel), and anti-β-actin antibody (1:10000 Sigma Aldrich, USA) as a loading control.
Membranes were incubated for 1h at room temperature with horseradish peroxidase (HRP)-conjugated secondary antibodies - goat anti-rabbit or goat anti-mouse IgG (1:10000, Santa Cruz, Dallas, TX). Three washes with 0.3% Tween-20 in Tris-buffered saline were performed between all steps. Immunoreactivity was detected using enhanced chemiluminescence substrate (Super Signal west Femto; Thermo Scientific, UT). Images were captured using GBOX (Chemi XR 5, Syngene, MD) and gels were analyzed densitometrically with the computerized image analysis program ImageQuant 5.0 (GE Healthcare, Life Sciences, Piscataway, NJ).
Histological Preparation
Mice were deeply anesthetized with 2% isoflurane and immediately perfused with 4% paraformaldehyde in 0.1 M phosphate buffer (at pH 7.4). Brains were extracted and immersed in fresh 4% paraformaldehyde and incubated at 4°C for additional 2-3 days before being embedded in agar. Briefly, brain coronal sections (50μm thickness) were cut using vibratome. Tissue sections were blocked with 1× TBS contains normal goat serum 5%, 1% BSA and 0.1% triton X-100 for 1h at room temperature before incubated with primary antibodies against calbindin (anti-calbindin D-28K antibody, 1:1000; Gene Tex, CA, USA) overnight at 4°C. Free floating sections were then washed three times with TBS, and then incubated with corresponding HRP-conjugated secondary antibodies (1:200) at room temperature for 2h. Tissue sections were mounted on glass slides and air dried. For detection, we used DAB Peroxidase substrate kit (Vector Laboratories) following manufacturer's instructions.
Histological Morphometric Assessment
The morphometric analyses of IBP developmental shortening (from PND10 until PND65 in both control and ketamine-treated mice) were performed using coronal hippocampal slices (50μm) cut from bregma -1.34mm to -2.30mm (as determined using a mouse brain atlas). The images were scanned at 20× magnification using an Aperio Scanscope XT digital slide scanner (Aperio Technologies Inc., Vista, CA) at University of Virginia, Charlottesville, VA and at University of Colorado, Aurora, CO. The hippocampal area in digital sections (.svs file) was extracted at 600μm scale to convert to a .tiff file and was spatially calibrated using 1000 μm2 grid prior to quantifying using Image-Pro Plus 7.0 software (Media Cybernetics, MD). The morphometric approach used to evaluate so called ‘normalized length of IPB’, which takes into consideration individual variability and developmental growth of hippocampus. The IPB length was approximated from the tip of the inferior blade of the dentate granule cell layer (“a”). The length of CA3 was approximated from the tip of the inferior blade to the apex of the curvature of the CA3 pyramidal cell layer (“b”). Normalized IPB length was taken as a ratio between “a” and “b”. The values from serial sections (n=3-6 serial sections per animal from 6-7 animals per age group) were averaged to provide a single data point and is presented as normalized IPB length. The results from different age groups were statistically analyzed by t test and between both groups by Two-way ANOVA using Graph Pad Prism 5.01 software (Graph Pad, CA). The experimenters were blinded to the experimental condition.
Statistical analysis
Comparisons among groups were made using one-way and two-way ANOVAs followed by Tukey's post hoc test. Using the standard version of GraphPad Prism 5.01 software (Media Cybernetics, Inc, Bethesda, MD), we considered p<0.05 to be statistically significant. All data are presented as mean ±SD or mean ±SEM. The sample sizes reported throughout the Results and in the Figure Legends were based on previous experience.
Results
To begin to understand the effects of general anesthesia on neuronal circuitries remodeling we focused on well-established infrapyramidal bundle (IPB) model of in vivo axon connectivity. The IPB is formed from the axons of granule cells in the dentate gyrus projecting to pyramidal cells mostly in region CA3 (Fig. 1). During normal development, the IPB undergoes a progressive decrease in size due to tightly controlled axon selection and pruning, a process necessary to assure proper circuitry formation in developing hippocampus [15].
Figure 1. Schematic representation of the infrapyramidal bundle (IPB).
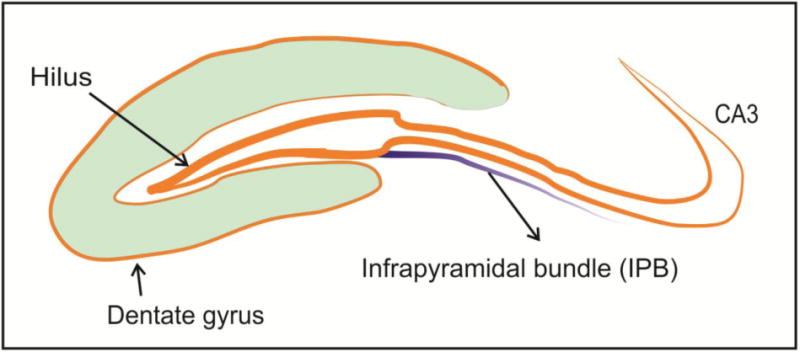
The IPB is formed from the axons of granule cells in the dentate gyrus projecting to pyramidal cells in region CA3. During normal development, the IPB undergoes progressive decrease in size due to tightly controlled axon selection and pruning.
The IPB fails to undergo proper shortening in ketamine-treated mice
To begin to understand whether an early GA exposure impairs axon pruning we examined the IPB length in CD-1 mice that were exposed to either saline (vehicle control) or ketamine at postnatal day (PND)7 (Fig. 2A). We chose this age because 1) it is when rodents are most vulnerable to GA-induced developmental neurotoxicity [16] and, 2) it falls immediately before developmental pruning of the IPB begins [15]. As stated earlier, a total of four doses of ketamine, at 75 mg/kg, IM were administered every 90 minutes so that the loss of righting reflex and lack of response to tail pinch could be maintained for 6 hours. The IPB length was measured at different age points -when IPB length is about maximal (PND10); the IPB length begins to decrease (PNDs 20 and 30) and the IPB length reaches its final length (PNDs 40 and 65) [15]. The morphometric approach used to evaluate so called ‘normalized length of IPB’, which takes into consideration individual variability and developmental growth of hippocampus, is shown in Fig. 2B. The IPB length was measured from the tip of the inferior blade of the dentate granule cell layer (“a”). The length of CA3 was measured from the tip of the inferior blade to the apex of the curvature of the CA3 pyramidal cell layer (“b”). Normalized IPB length was taken as a ratio between “a” and “b” [15].
Figure 2. The timeline of the experimental design and the IPB morphometry in mice.
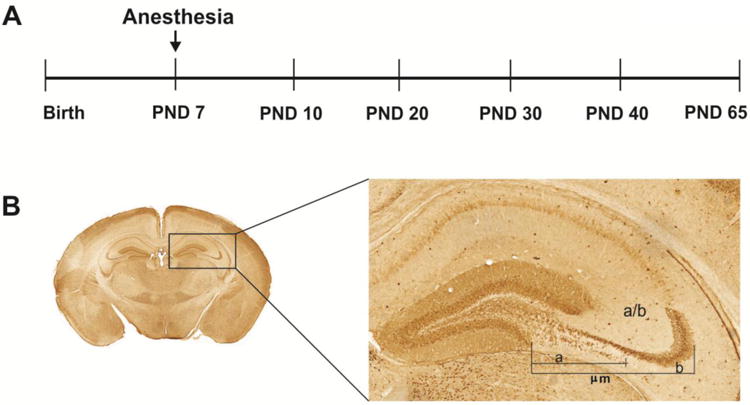
A Ketamine exposure occurred at post-natal day (PND) 7 when synaptogenesis is at the peak in mice. The IPB length was measured at different age points - when IPB length is about maximal (PND10); IPB length begins to decrease (PNDs 20 and 30) and IPB length reaches its final length (PNDs 40 and 65).
B. The morphometric approach used to evaluate so called ‘normalized length of IPB’, which takes into consideration individual variability and developmental growth of hippocampus. The IPB length was approximated from the tip of the inferior blade of the dentate granule cell layer (“a”). The length of CA3 was approximated from the tip of the inferior blade to the apex of the curvature of the CA3 pyramidal cell layer (“b”). Normalized IPB length was taken as a ratio between “a” and “b”.
As shown in Fig. 3, control animals underwent substantial shortening of the IPB from PND20 until the PND65 compared to PND10 (shortening from 20 to 80%, respectively; see table for pairwise comparison to PND10) whereas ketamine-treated ones maintained the IPB length close to PND10 level until the PND30 (p=0.572), resulting in a significantly longer IPB throughout the experimental time line [two-way ANOVA main effect on treatment (F (1, 56)=9.247); (**, p<0.01)] (n=6-7 pups per data point) suggesting a rightward shift with ketamine treatment (Fig. 3A). Representative microphotographs from the control and experimental animals are depicted in Fig. 3B (black line underlines the IBP length. As stated earlier, we used calbindin staining (with anti-calbindin D-28K antibody) to label mossy fibers in the IPB since it labels long unmyelinated axons [17].
Figure 3. Ketamine exposure impairs IPB pruning in young mice.
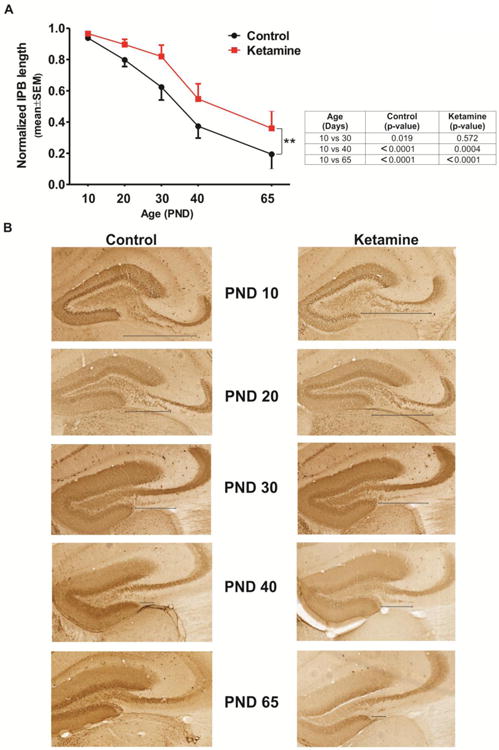
A Control animals underwent significant shortening of the IPB from PND20 until the PND65 compared to PND10, whereas ketamine-treated ones maintained the IPB length at PND10 level until the PND30 (see the Table depicting within-the-group comparisons). The IPB remained longer throughout the experimental time line in ketamine-treated animals suggesting a rightward shift with ketamine treatment [two-way ANOVA main effect on treatment (F (1, 56) = 9.247); (**, p<0.01)] (n=6-7 pups per data point).
B. Representative microphotographs from the control and experimental animals are depicted from PND10 to PND65. To label mossy fibers in the IPB we used calbindin staining (with anti-calbindin D-28K antibody) since it labels long unmyelinated axons (magnification ×20). Note a delay in the IPB shortening in ketamine-treated animals (right panels) when compared to controls (left panels).
BDNF protein expression is down-regulated in ketamine-treated mice
To begin to decipher the mechanism of ketamine-induced delay in the IPB pruning, we assessed BDNF protein expression changes during early stages of brain development. We measured protein levels of mature (Fig. 4A; n=3-4) and pro- (Fig. 4B; n=3-4) forms of BDNF in saline- and ketamine-treated mice. The treatment was performed at PND7 and the Western blot analyses of hippocampal tissue were performed in frequent intervals (until PND30) to capture the BDNF changes during the initial phase of IPB pruning. As shown in Fig. 4A, the mature BDNF levels rise steadily in both control and ketamine-treated mice over the course of first 20 postnatal days, peak around PND22, and slowly decline thereafter in controls (PND22 vs. PND 19, ††, p<0.01 and PND22 vs. PND26, †, p<0.05). Ketamine-treated animals exhibit a much less robust increase in BDNF levels throughout with a lower BDNF levels when compared to age-matched controls. Note that there is over a twofold decrease in BDNF expression in the ketamine group on PND22 as compared with age-matched controls (***, p<0.001). Interestingly, pro-BDNF levels appeared to be somewhat elevated in ketamine-treated animals shortly after the treatment (Fig. 4B). However, starting from PND13 there was a precipitous decline in their pro-BDNF levels that was significant on PND22 as compared with age-matched controls (*, p<0.05).
Figure 4. Ketamine exposure impairs BDNF homeostasis in young mice.
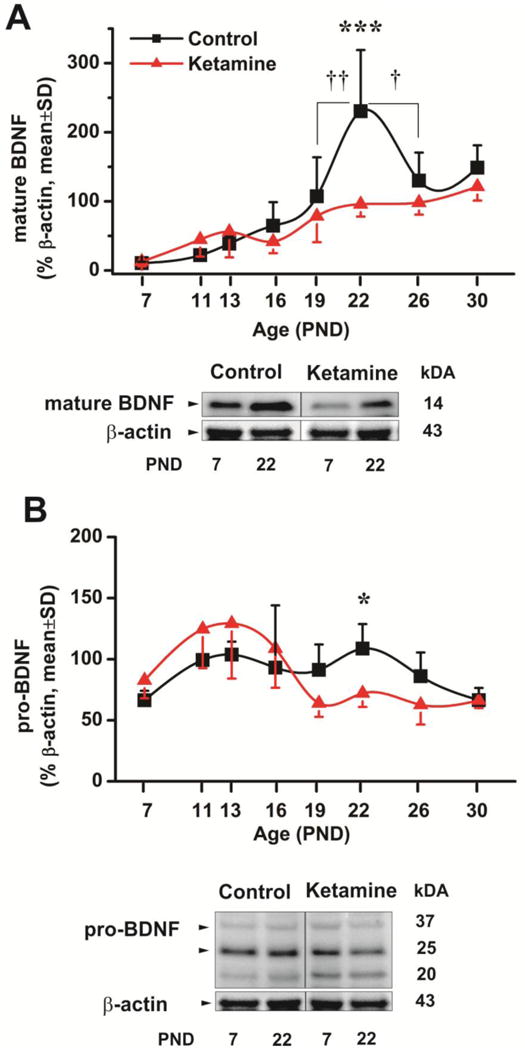
A Mature BDNF protein expression was examined during the early stage of brain development in saline and ketamine-treated mice. The treatment was performed at PND7 and the Western blot analyses of hippocampus were performed in frequent intervals as indicated (until PND30). The mature BDNF levels rise steadily in both control and ketamine-treated mice over the course of first 20 postnatal days, peak around PND22, and slowly decline thereafter in controls (PND22 vs. PND 19, ††, p<0.01 and PND22 vs. PND26, †, p<0.05). Ketamine-treated animals exhibit much less robust increase in BDNF levels throughout with a lower BDNF levels when compared to age-matched controls. Note that there is over a 2-fold decrease in BDNF expression on PND22 in the ketamine group as compared with age-matched controls (***, p<0.001)(n=3-4 animals per data point).
B. Pro-BDNF protein expression was examined during the early stage of brain development in saline and ketamine-treated mice. The treatment was performed at PND7 and the Western blot analyses of CA3 and dentate gyrus were performed in frequent intervals (until PND30). Pro-mature BDNF levels appeared to be somewhat elevated (although not significantly) in ketamine-treated animals shortly after the treatment. However, starting from PND13 there was a precipitous decline in their pro-BDNF levels found to be significant on PND22 as compared with age-matched controls (*, p<0.05) (n=3-4 animas per data point).
The impairment of BDNF homeostasis corresponds with the timing of intense IPB pruning
To assess how the changes in mature BDNF levels correspond to the timeline of the IPB pruning we superimposed the IPB pruning curve on the mature (Fig. 5A) and pro-BDNF expression curves (Fig. 5B) and discovered that around the time when intense IPB pruning is normally initiated (the time course of normal IPB pruning is superimposed with a blue dashed line) there is a peak in BDNF expression followed by a precipitous decline in control animals suggesting that under normal circumstance, an increase in pro- and mature BDNF provides a homeostatic ‘signal’ for intense IPB pruning (around PND22; marked with shaded rectangles).
Figure 5. The peak of BDNF protein expression coincides with the initiation of intense IPB pruning in young mice.
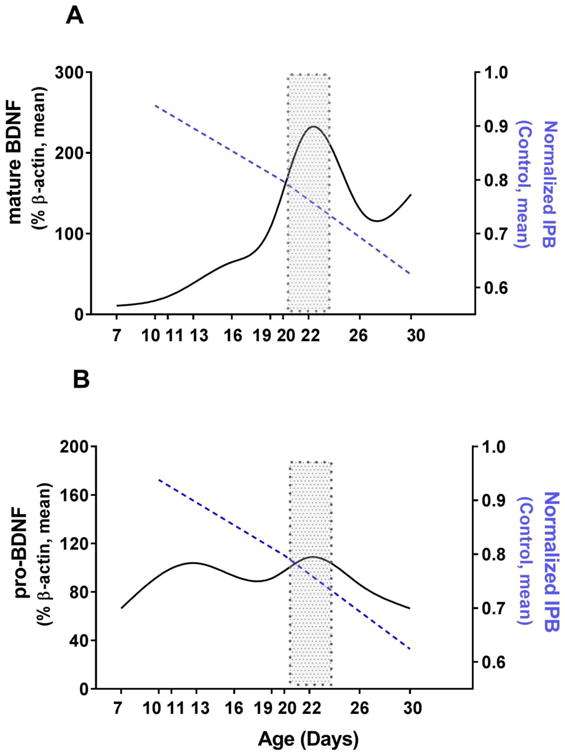
A When the IPB pruning curve (dashed blue line) was superimposed on the mature BDNF protein expression curve in control animals, it showed that a substantial peak in BDNF expression occurs around the time when intense IPB pruning is normally initiated.
B. When the IPB pruning curve (dashed blue line) was superimposed on the pro-BDNF protein expression curve in control animals, it showed that an increase in pro-BDNF expression occurs around the time when intense IPB pruning is normally initiated.
Discussion
We show in this study that ketamine exposure during the critical stages of mouse brain development impairs timely IPB pruning. Since both pro- and mature BDNF forms exhibit a significant decline at the time when intense IBP shortening begins (around PND20) we propose that ketamine-induced impairment in developmental axon pruning could be, at least in part, BDNF-dependent. We further hypothesize that disturbance in axonal selection and pruning may result in faulty formation of functional neuronal networks among the remaining (‘normal’) neurons. This notion remains to be confirmed in future mechanistic and functional studies.
The importance of neuronal activity in regulating developmental axon competition is well-established. For example, developing rat sympathetic eye projecting neurons initially extend axon collaterals to two different eye sections, but then axon elimination occurs, so that any individual neuron ultimately only projects to one section [18]. If, however, the activity is disturbed during this critical time period, axon selection does not occur, thus affecting the innervations of an eye. Although the effect of general anesthesia on neuronal activity in vast brain circuitries is complex and not well understood, it is clear that a substantial decrease in neuronal activity has to occur to induce the state of unconsciousness, amnesia, and insensitivity to pain- three key features of the general anesthesia state. Hence, GA-induced neuronal inhibition may be the cause of improper axon selection manifested as delayed pruning.
The role of neurotrophic factors in developmental axon competition is becoming more appreciated. We, along with others have previously reported that the exposure to GA causes profound disturbances in homeostasis of the neurotrophic factor, BDNF. This, in turn, leads to the inhibition of Trk-B- dependent pathways, either directly or indirectly via p75NTR signaling, ultimately resulting in neuronal death [12, 13] since both Trk and p75NTR receptors modulate the activation of major survival pathways for neurons [19, 20]. However, after a decade of intense investigation we question whether the detrimental effects of GA on BDNF signaling have consequences beyond inducing neuronal death to include compromising the development and function of the remaining surviving neurons by impairing not only current but also future connections, maintenance of neuronal circuits and general plasticity of dentate gyrus-CA3 communications. We base this hypothesis on data presented herein which suggests the impairment of proper and timely axon pruning, a crucial process during normal development that allows removal of exuberant or misguided axon branches while maintaining other appropriate connections of the same neuron. If, indeed, GA impairs axon selection during critical stages of synaptogenesis, this effect may account for long-lasting impairment of neuronal networking and circuitry formation [21].
We find that the time course of this impairment correlates with GA-induced down-regulation of BDNF protein expression in developing hippocampus in vivo. Although our work was not focused on studying Trk-mediated signaling, based on presently available knowledge, the basic mechanism suggests that active axons secrete BDNF enabling extensive activation of p75NTR receptors located on a ‘losing’ (less active) axon. p75NTR activation inhibits Trk-mediated signaling, which is essential for axon maintenance, thus promoting degeneration and pruning of losing axons [11, 22]. When BDNF levels are downregulated, this timely activation of p75NTR receptors is impaired resulting in faulty axon pruning similar to the one we report herein.
Our findings indicate that GA impairs the normal progressive decrease in the infrapyramidal bundle (IPB) of mossy fiber projections (IMF) in the hippocampus, an important event in the formation of proper circuitry between the dentate gyrus and the CA3 region, a neuronal circuitry that plays a crucial role in learning and memory [23]. We focused on this well-established IPB model of in vivo axon connectivity because during normal development, the IPB undergoes a progressive decrease in size due to tightly controlled axon selection and pruning, a process necessary to assure proper circuitry formation in developing hippocampus [15]. The size of the IPB correlates inversely with performance in a variety of cognitive tasks, i.e. the longer the IPB, the more learning and memory development (in particular spatial learning) [23] are impaired, suggesting that subtle disturbances in hippocampal circuitries could have detrimental long-term effects on cognitive development. Because spatial learning is impaired in animals exposed to ketamine [24] (intravenous anesthetics used in pediatric medicine) and because our data suggest that ketamine compromises IPB pruning, we propose a temporal association between ketamine-induced impairment of BDNF homeostasis and disturbances in normal development of IPB. It remains to be determined whether this temporal association could explain the impairment in cognitive functioning previously reported. This notion could be considered based on the observation that BDNF is critical for cognitive development [25], it modulates the strength of existing synaptic connections and it assists in the formation of new synaptic contacts [26, 27]. In addition, pro- and mature forms of BDNF can induce long-term potentiation and depression [28], known to be impaired after an early exposure to anesthesia [1]. A decrease greater than twofold in BDNF expression in the ketamine group around the time of intense IPB pruning approaches a reduction in BDNF previously reported to be sufficient to eliminate the competitive advantage of active neurons, thus resulting in impaired pruning [29]. It is noteworthy that somewhat elevated pro-BDNF levels in ketamine-treated animals we report herein could be an attempt to compensate for a decrease in mature BDNF.
In conclusion, we report that ketamine exposure during critical stages of mammalian brain development lead to an impaired BDNF homeostasis and delayed pruning of axons known to be critically important for proper cognitive development. We suggest that disturbance in axonal selection and pruning may lead to a faulty formation of functional neuronal networks among the remaining (‘normal’) neurons thus resulting in an impaired synaptic neurotransmission we and others have previously reported [1, 21]. Further studies of neuronal circuitry formations vis-à-vis the studies of axonal selection and pruning are needed to make final determination.
Acknowledgments
This study was supported by the grants R0144517 (NIH/NICHD), R0144517-S (NIH/NICHD), R01 GM118197 (NIH/NIGMS), R21 HD080281 (NIH/NICHD), John E. Fogarty Award 007423-128322 (NIH) and March of Dimes National Award, USA (to Vesna Jevtovic-Todorovic). Vesna Jevtovic-Todorovic was an Established Investigator of the American Heart Association. We thank Jonathan Park for his assistance with the morphometric analysis of the IPB pruning.
Footnotes
Conflicts of Interest: The authors declare no competing interests.
References
- 1.Jevtovic-Todorovic V, Hartman RE, Izumi Y, Benshoff ND, Dikranian K, Zorumski CF, Olney JW, Wozniak DF. Early exposure to common anesthetic agents causes widespread neurodegeneration in the developing rat brain and persistent learning deficits. J Neurosci. 2003;23(3):876–882. doi: 10.1523/JNEUROSCI.23-03-00876.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Loepke AW, Istaphanous GK, McAuliffe JJ, 3rd, Miles L, Hughes EA, McCann JC, Harlow KE, Kurth CD, Williams MT, Vorhees CV, Danzer SC. The effects of neonatal isoflurane exposure in mice on brain cell viability, adult behavior, learning, and memory. Anesth Analg. 2009;108(1):90–104. doi: 10.1213/ane.0b013e31818cdb29. [DOI] [PubMed] [Google Scholar]
- 3.Paule MG, Li M, Allen RR, Liu F, Zou X, Hotchkiss C, Hanig JP, Patterson TA, Slikker W, Jr, Wang C. Ketamine anesthesia during the first week of life can cause long-lasting cognitive deficits in rhesus monkeys. Neurotoxicol Teratol. 2011;33(2):220–230. doi: 10.1016/j.ntt.2011.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rizzi S, Carter LB, Ori C, Jevtovic-Todorovic V. Clinical anesthesia causes permanent damage to the fetal guinea pig brain. Brain Pathol. 2008;18(2):198–210. doi: 10.1111/j.1750-3639.2007.00116.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Fredriksson A, Pontén E, Gordh T, Eriksson P. Neonatal exposure to a combination of N-methyl-D-aspartate and gamma-aminobutyric acid type A receptor anesthetic agents potentiates apoptotic neurodegeneration and persistent behavioral deficits. Anesthesiology. 2007;107(3):427–436. doi: 10.1097/01.anes.0000278892.62305.9c. [DOI] [PubMed] [Google Scholar]
- 6.Viberg H, Pontén E, Eriksson P, Gordh T, Fredriksson A. Neonatal ketamine exposure results in changes in biochemical substrates of neuronal growth and synaptogenesis, and alters adult behavior irreversibly. Toxicology. 2008;249(2-3):153–159. doi: 10.1016/j.tox.2008.04.019. [DOI] [PubMed] [Google Scholar]
- 7.Kodama M, Satoh Y, Otsubo Y, Araki Y, Yonamine R, Masui K, Kazama T. Neonatal desflurane exposure induces more robust neuroapoptosis than do isoflurane and sevoflurane and impairs working memory. Anesthesiology. 2011;115(5):979–991. doi: 10.1097/ALN.0b013e318234228b. [DOI] [PubMed] [Google Scholar]
- 8.Bishop DL, Misgeld T, Walsh MK, Gan WB, Lichtman JW. Axon branch removal at developing synapses by axosome shedding. Neuron. 2004;44:651–661. doi: 10.1016/j.neuron.2004.10.026. [DOI] [PubMed] [Google Scholar]
- 9.Luo L, O'Leary DD. Axon retraction and degeneration in development and disease. Annu Rev Neurosci. 2005;28:127–156. doi: 10.1146/annurev.neuro.28.061604.135632. [DOI] [PubMed] [Google Scholar]
- 10.Wilder RT, Flick RP, Sprung J, et al. Early exposure to anesthesia and learning disabilities in a population-based birth cohort. Anesthesiology. 2009;110:796–804. doi: 10.1097/01.anes.0000344728.34332.5d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Singh KK, Park KJ, Hong EJ, Kramer BM, Greenberg ME, Kaplan DR, Miller FD. Developmental axon pruning mediated by BDNF-p75NTR–dependent axon degeneration. Nature Neuroscience. 2008;11:649–658. doi: 10.1038/nn.2114. [DOI] [PubMed] [Google Scholar]
- 12.Lu LX, Yon JH, Carter LB, Jevtovic-Todorovic V. General anesthesia activates BDNF-dependent neuroapoptosis in the developing rat brain. Apoptosis. 2006;11:1603–15. doi: 10.1007/s10495-006-8762-3. [DOI] [PubMed] [Google Scholar]
- 13.Pearn ML, Hu Y, Niesman IR, Patel HH, Drummond JC, Roth DM, Akassoglou K, Patel PM, Head BP. Propofol neurotoxicity is mediated by p75 neurotrophin receptor activation. Anesthesiology. 2012;116:352–61. doi: 10.1097/ALN.0b013e318242a48c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Young C, Jevtovic-Todorovic V, Qin YQ, Tenkova T, Wang H, Labruyere J, Olney JW. Potential of ketamine and midazolam, individually or in combination, to induce apoptotic neurodegeneration in the infant mouse brain. Br J Pharmacol. 2005;146(2):189–97. doi: 10.1038/sj.bjp.0706301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bagri A, Cheng HJ, Yaron A, Pleasure SJ, Tessier-Lavigne M. Stereotyped pruning of long hippocampal axon branches triggered by retraction inducers of the semaphorin family. Cell. 2003;113:285–99. doi: 10.1016/s0092-8674(03)00267-8. [DOI] [PubMed] [Google Scholar]
- 16.Yon JH, Daniel-Johnson J, Carter LB, Jevtovic-Todorovic V. Anesthesia induces neuronal cell death in the developing rat brain via the intrinsic and extrinsic apoptotic pathways. Neuroscience. 2005;135:815–27. doi: 10.1016/j.neuroscience.2005.03.064. [DOI] [PubMed] [Google Scholar]
- 17.Liu XB, Low LK, Jones EG, Cheng HJ. Stereotyped axon pruning via plexin signaling is associated with synaptic complex elimination in the hippocampus. J Neurosci. 2005;25:9124–34. doi: 10.1523/JNEUROSCI.2648-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lawrence JM, Black IB, Mytilineou C, Field PM, Raisman G. Decentralization of the superior cervical ganglion in neonates impairs the development of the innervations of the iris. A quantitative ultrastructural study. Brain Res. 1979;168:13–19. doi: 10.1016/0006-8993(79)90124-0. [DOI] [PubMed] [Google Scholar]
- 19.Majdan M, Miller FD. Neuronal life and death decisions: functional antagonism between the Trk and p75 neurotrophin receptors. Int J Dev Neurosci. 1999;17:153–161. doi: 10.1016/s0736-5748(99)00016-7. [DOI] [PubMed] [Google Scholar]
- 20.Miller FD, Kaplan DR. Neurotrophin signaling pathways regulating neuronal apoptosis. Cell Mol Life Sci. 2001;58:1045–1053. doi: 10.1007/PL00000919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mintz CD, Barrett KM, Smith SC, Benson DL, Harrison NL. Anesthetics interfere with axon guidance in developing mouse neocortical neurons in vitro via a γ-aminobutyric acid type A receptor mechanism. Anesthesiology. 2013;118:825–33. doi: 10.1097/ALN.0b013e318287b850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Singh KK, Miller FD. Activity regulates positive and negative neurotrophin-derived signals to determine axon competition. Neuron. 2005;45:837–845. doi: 10.1016/j.neuron.2005.01.049. [DOI] [PubMed] [Google Scholar]
- 23.Crusio WE, Schwegler H. Learning spatial orientation tasks in the radial-maze and structural variation in the hippocampus in inbred mice. Behav Brain Funct. 2005;1(1):3–11. doi: 10.1186/1744-9081-1-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Fredriksson A, Archer T, Alm H, Gordh T, Eriksson P. Neurofunctional deficits and potentiated apoptosis by neonatal NMDA antagonist administration. Behav Brain Res. 2004;153:367–76. doi: 10.1016/j.bbr.2003.12.026. [DOI] [PubMed] [Google Scholar]
- 25.Lu Y, Christian K, Lu B. BDNF: a key regulator for protein synthesis-dependent LTP and long-term memory? Neurobiol Learn Mem. 2008;89(3):312–23. doi: 10.1016/j.nlm.2007.08.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Thoenen H. Neurotrophins and neuronal plasticity. Science. 1995;270:593–8. doi: 10.1126/science.270.5236.593. [DOI] [PubMed] [Google Scholar]
- 27.Lu B, Figurov A. Role of neurotrophins in synapse development and plasticity. Rev Neurosci. 1997;8:1–12. doi: 10.1515/revneuro.1997.8.1.1. [DOI] [PubMed] [Google Scholar]
- 28.Woo NH, Teng HK, Siao CJ, Chiaruttini C, Pang PT, Milner TA, Hempstead BL, Lu B. Activation of p75NTR by proBDNF facilitates hippocampal long-term depression. Nat Neurosci. 2005;8:1069–77. doi: 10.1038/nn1510. [DOI] [PubMed] [Google Scholar]
- 29.Chen ZY, Ieraci A, Teng H, Dall H, Meng CX, Herrera DG, Nykjaer A, Hempstead BL, Lee FS. Sortilin controls intracellular sorting of brain-derived neurotrophic factor to the regulated secretory pathway. J Neurosci. 2005;25:6156–6166. doi: 10.1523/JNEUROSCI.1017-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]