

Abstract
The majority of Parkinson’s disease (PD) cases are sporadic with only about 10% of PD patients having a family history of the disease suggesting that this neurodegenerative disorder is the result of both environmental and genetic factors. Both oxidative stress and neuroinflammation are thought to contribute to PD. Previously, we showed that the activation of interleukin 13 receptor alpha 1 (IL-13Rα1) increases the sensitivity of dopaminergic neurons to oxidative damage both in cultured cells and in animals. In this study, we investigated the pathways involved in the IL-13-mediated potentiation of oxidative stress-induced dopaminergic cell death using a combination of cell survival assays and Western blotting with appropriate antibodies. In addition, siRNA was used to examine the role of 4E-BP1 in this cell toxicity paradigm. We show that activation of both the Jak-Stat and PI3 kinase-mTOR pathways play key roles in the promotion of cell death by IL-13 in the presence of mild oxidative stress. The Jak 1/2 inhibitor ruxolitinib, the mTOR inhibitor rapamycin and the PI3 kinase inhibitor LY294002 all prevented the potentiation of cell death by IL-13. Moreover, 4E-BP1, a target of mTOR, appeared to mediate the protective effects of rapamycin. Together, these results indicate that multiple signaling pathways downstream of IL-13Rα1 activation play a role in the toxic effects of IL-13 in dopaminergic neurons in the presence of mild oxidative stress and suggest that any of these pathways might provide potential targets for the treatment of PD.
Keywords: Parkinson’s disease, mTOR, Jak, Akt, 4E-BP1, oxidative stress
Background
Parkinson’s disease (PD) is a chronic, progressive neurodegenerative disease and the second most common after Alzheimer’s disease [1]. In the US, PD affects 1% of the population over age 60 and it is estimated that 60,000 new cases will be diagnosed this year. The clinical symptoms are mainly due to the progressive decrease in dopamine signaling in the basal ganglia that is caused by the loss of the dopaminergic (DA) neurons in the substantia nigra pars compacta (SNc). Although a number of genes have been shown to cause familial PD, these contribute to only ~10% of PD cases with the remainder being sporadic and of unknown origin. While the precise causes of PD still remain to be determined, chronic neuroinflammation is one of the hallmarks of the disease.
Our recent studies on neuroinflammation in the context of PD suggested that the IL-13/IL-13Rα1 system plays a role in disease development and/or the progression [2, 3]. Interestingly, the interleukin 13 receptor alpha 1 (IL-13Rα1) gene lies within the PARK12 locus of PD susceptibility. Moreover, the PARK12 locus is located on the X chromosome which may be related to the observation that PD has a higher incidence in men as compared to women. Even more interesting is the observation that the expression of IL-13Rα1 in the brain is specific to the DA neurons of the ventral tegmental area (VTA) and of the SNc, the area of the brain most affected by PD. Indeed, double immunostaining studies showed that ~80% of the SNc neurons that express the DA marker tyrosine hydroxylase also express IL-13Rα1 [2].
Further evidence for a role for IL-13Rα1 in PD was obtained using the bacterial lipopolysaccharide (LPS) mouse model of PD [2]. We demonstrated that mice that were deficient in IL-13Rα1 were protected from DA neuronal loss when compared to their wild type littermates suggesting that the IL-13/IL-13Rα1 system could exert a neurotoxic effect under certain conditions [2]. Experiments done using the mouse DA nerve cell line MN9D showed that IL-13 alone did not have toxic effects on cells. However, when IL-13 was administered to the cells in the presence of marginally toxic doses of oxidants, it increased cell death in a dose dependent manner. These results suggested that activation of IL-13Rα1 could contribute to the vulnerability of DA neurons under inflammatory conditions when both cytokines and reactive oxygen species (ROS) are produced.
The binding of IL-13 to IL-13Rα1 induces the tyrosine phosphorylation of the receptor [for review see 3] thereby activating its kinase activity which results in the phosphorylation of the downstream substrates of its signaling cascades. IL-13 primarily activates two different intracellular signaling cascades: the Jak-Stat and the phosphatidylinositol 3′-kinase (PI3 kinase) pathways [for review see 4]. Upon IL-13 stimulation of cells, Stat6 is phosphorylated on tyrosine and then forms a homodimer that translocates to the nucleus where it promotes gene transcription [4]. Tyrosine phosphorylation of IRS-2 leads to the activation of PI3 kinase and Akt [4]. However, whether either of these pathways contributes to the potentiation of oxidative stress-induced DA nerve cell death is not known. In this study, we describe experiments designed to determine the signaling pathways activated by IL-13 that contribute to the potentiation of oxidative stress-induced cell death in DA neurons.
Materials and Methods
Materials
IL13 was purchased from Peprotech. Rapamycin, LY294002 and ruxolitinib were purchased from LC Laboratories. Unless otherwise stated, other chemicals and reagents were from Sigma-Aldrich (St. Louis, MO, USA).
Cell culture and treatments
Mouse dopaminergic MN9D cells were grown in high-glucose DMEM containing 10% fetal calf serum as previously published [2]. For toxicity assays, 10,000 cells per well were plated in 96-well plates and grown overnight. The next day, the cells were treated with 1, 5 or 10 ng/ml IL-13 alone or in the presence of 80 μM H2O2 or 2.5 μM t-butylperoxide (tBOOH). In some cases, inhibitors were also included. 24 hr later, cell survival was determined using the MTT assay. None of the concentrations of IL-13 alone had any effect on cell survival. The concentrations of H2O2 and tBOOH were chosen so as to have only a modest effect on cell survival (decrease by 10–15%) as previously described [2]. Human dopaminergic SH-SY5Y cells were grown in high glucose DMEM containing 10% fetal calf serum and plated at 20,000 cells per well in 96-well dishes for the toxicity assays. The next day, the cells were treated with 1, 2.5 or 5 ng/ml IL-13 alone or in the presence of 40 μM H2O2 or 2.5 μM t-butylperoxide (tBOOH). In some cases, rapamycin (100 nM) was also included. 24 hr later, cell survival was determined using the MTT assay. None of the concentrations of IL-13 alone had any effect on cell survival. The concentrations of H2O2 and tBOOH were chosen so as to have only a modest effect on cell survival (decrease by 10–15%).
Transfection
For the siRNA transfections, the MN9D cells were plated in 60 mm dishes at 500,000 cells/dish and grown overnight. The next day, the cells were transfected with 166 pmol 4EBP1 siRNA (#sc-29595) or control siRNA (#sc-37007) (Santa Cruz Biotechnology (Santa Cruz, CA)) using RNAiMAX (Invitrogen) according to the manufacturer’s instructions.
Reactive oxygen species (ROS) measurement
MN9D cells were plated at 10,000 cells/well onto 96-well black walled microtiter plates. The next day, the cells were treated with IL-13 alone or in the presence of H2O2 or tBOOH for the indicated times. The culture medium was then exchanged for 100 μl of phenol red-free Hank’s balanced salt solution containing 10 μM CM-H2DCFDA (Invitrogen). After 30 min, the fluorescence (λ excitation = 495 nm, λ emission = 525 nM) of the cells was measured using a Molecular Devices SpectraMax M3 microplate reader. All the treatments were done in sextuplicate. The CM-H2DCFDA fluorescence in treated cells was normalized to that in control cells not exposed to compounds or oxidant.
Protein Preparation and Western Blotting
For Western blotting, 300,000 MN9D cells were grown overnight in 35 mm dishes prior to the indicated treatments. Total protein extracts were prepared and analyzed by SDS-PAGE and Western blotting as described previously [5]. The primary antibodies used for Western blotting were: rabbit anti-phospho-Akt (ser473) (#9271, 1/1000), rabbit anti-phospho-Stat6 (#9361, 1/1000), rabbit anti-phospho-4E-BP1 (thr37/46) (#2855, 1/10,000), rabbit anti-Akt (#9272, 1/10,000), rabbit anti-Stat6 (#9362, 1/1000), rabbit anti-4E-BP1 (#9644, 1/250,000) and HRP-conjugated rabbit anti-actin (#5125, 1/20,000) from Cell Signaling. Following washing, the Western blots were incubated for 1 hr at room temperature in horseradish peroxidase-goat anti-rabbit or goat anti-mouse (Biorad, Hercules, CA) diluted 1/2500 and developed with the Super Signal reagent (Pierce, Rockford, IL). In all cases, the same membrane was re-probed for actin and/or a parallel membrane was probed with an antibody reacting with the total protein in order to provide a normalization standard. Autoradiographs were scanned and analyzed using a Biorad GS800 scanner. The experiments were repeated a minimum of three times with independent protein samples.
Statistical Analysis
A minimum of three independent experiments were used for statistical analyses which were performed using InStat 3. The results were analyzed for statistically significant differences using either the analysis of variance (ANOVA) test and Tukey’s post test for individual group means comparisons or the t-test, as appropriate.
Results
The simplest explanation for the potentiation of oxidative stress-induced dopaminergic cell death by IL-13 would be that IL-13 increases ROS production from a non-toxic to a toxic level. This would be consistent with reports that IL-13 can rapidly induce NOXs in intestinal epithelial cells [6] and lipoxygenases in monocytes/macrophages cells [7]. To address this question, mouse MN9D dopaminergic nerve cells were treated with increasing doses of IL-13 alone or in the presence of H2O2 or tBOOH. The doses of H2O2 and tBOOH used were based on the results of our earlier study with the MN9D cells [2]. As shown in Figure 1A, treatment of cells with IL-13 alone for 5 or 60 min or 24 hr had no significant effect on ROS levels as measured using CM-H2DCFDA. Short-term treatment of the MN9D cells with H2O2 or tBOOH induced a significant increase in ROS levels (Fig. 1B). However, none of the tested doses of IL-13 further increased these ROS levels. Together, these results indicate that IL-13 does not potentiate oxidative stress-induced cell death by directly increasing intracellular ROS levels.
Figure 1. Effect of IL-13 on ROS Production in MN9D Cells.
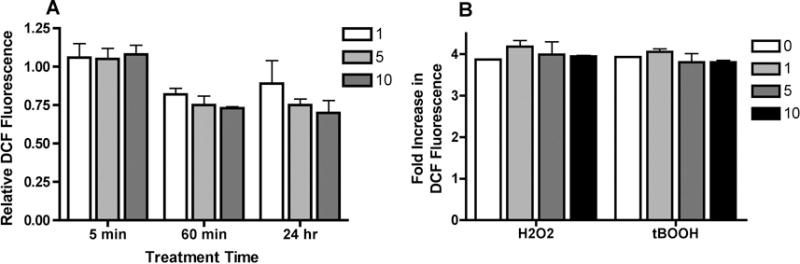
(A) MN9D cells were treated with 1, 5 or 10 ng/ml IL-13 for 5 or 60 min or 24 hr. ROS levels were determined using CM-H2DCFDA as described in Methods. Results are presented as the relative fluorescence compared to untreated cells. All of the experiments were done in sextuplicate and repeated at least three times. (B) MN9D cells were treated with 80 μM H2O2 or 2.5 μM tBOOH alone or in the presence of 1, 5 or 10 ng/ml IL-13. ROS levels were determined using CM-H2DCFDA as described in Methods. Results are presented as the relative fluorescence compared to H2O2- or tBOOH-treated cells. All of the experiments were done in sextuplicate and repeated at least three times.
Since Stat6 phosphorylation is one of the hallmarks of IL-13 receptor activation, we next asked what effect H2O2 or tBOOH have on IL-13-induced Stat6 phosphorylation. As shown in Figure 2, IL-13 rapidly induced the phosphorylation of Stat6 in the MN9D cells and this phosphorylation persisted for at least 4 hr. Neither H2O2 nor tBOOH alone had any effect on Stat6 phosphorylation (Fig. 2) but both brought about a time-dependent decrease in Stat6 phosphorylation in IL-13-treated cells (Fig. 2).
Figure 2. Effects of IL-13 and Oxidative Stress on Stat6 and Akt Phosphorylation.
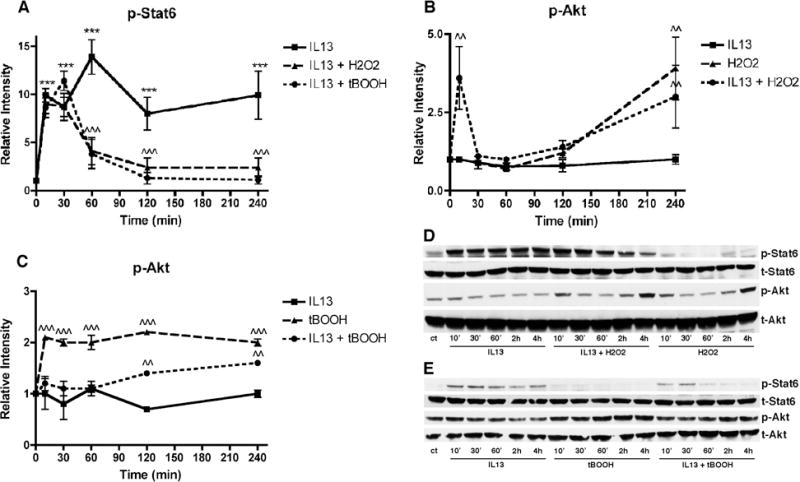
MN9D cells were untreated or treated with 10 ng/ml IL-13, 80 μM H2O2 or 2.5 μM tBOOH or a combination of either IL-13 and H2O2 or IL-13 and tBOOH for the indicated times. Cell lysates were prepared and equal amounts of protein were analyzed by SDS-PAGE and immunoblotting with antibodies to phospho and total Stat6 and phospho and total Akt. (A) – (C) Western blots from three independent experiments similar to the ones shown in (D) and (E) were scanned and quantified. (D) and (E) Representative Western blots. *** p < 0.001 relative to control cells. ^^ p < 0.01; ^^^ p < 0.001 relative to IL-13-treated cells
As indicated in the Introduction, IL-13 has also been reported to induce activation of the PI3 kinase pathway, at least in some types of cells [3, 8]. In the MN9D cells, treatment for 10 min-4 hr with IL-13 did not increase the levels of Akt phosphorylation, a substrate of PI3 kinase (Fig. 2A & B). However, both H2O2 and tBOOH alone increased Akt phosphorylation, albeit with different time courses, and this increase was also seen in the presence of IL13 (Fig. 2). Importantly, the increase in Akt phosphorylation seen in the presence of IL-13 and either H2O2 or tBOOH was most evident after 4 hr of treatment (Fig. 2) and persisted for up to 24 hr of treatment (Fig. 5).
In order to address the roles that both the Jak-Stat and PI3 kinase pathways have in the potentiation of oxidative stress-induced cell death we turned to pathway inhibitors. Ruxolitinib (Rux), an inhibitor of Jak1 and 2 [9], completely blocked IL-13-induced Stat6 phosphorylation at 5–10 μM (Fig. 3A). This concentration of Rux also blocked the potentiation of oxidative stress induced cell death by IL-13 but did not reduce either H2O2 or tBOOH toxicity in the absence of IL-13 (Fig. 3B) suggesting that activation of the Jak-Stat6 pathway was required for the effects of IL-13 in this paradigm. Similar results were obtained with another Jak inhibitor, AZD1480 (not shown).
Figure 3. Effects of the Jak Inhibitor Ruxolitinib on IL-13-mediated Potentiation of Oxidative Stress-Induced Cell Death.
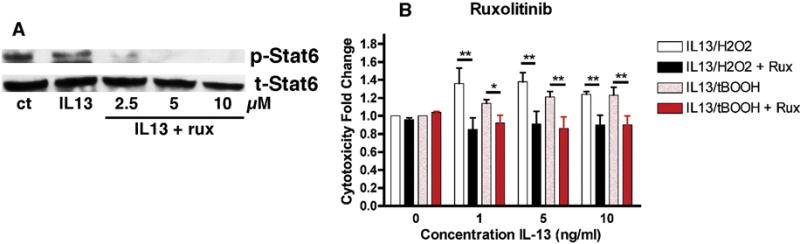
(A) MN9D cells were treated with 1–10 μM ruxolitinib for 60 min prior to the addition of 10 ng/ml IL-13 for 60 min. Cell lysates were prepared and equal amounts of protein were analyzed by SDS-PAGE and immunoblotting with antibodies to phospho and total Stat6. A representative Western blot is shown. Similar results were obtained in 2 independent experiments. (B) MN9D cells grown in 96 well dishes were untreated or treated with 1, 5 or 10 ng/ml IL-13 alone or in the presence of 80 μM H2O2, 2.5 μM tBOOH, 10 μM ruxolitinib or a combination as indicated. Cell survival was measured after 24 hr with the MTT assay. Results are presented as the fold change in cell death as compared to H2O2 or tBOOH alone. The experiments were done in quadruplicate and repeated 4 times. * p < 0.05; ** p < 0.01 relative to no ruxolitinib.
LY294002 is a PI3 kinase inhibitor and rapamycin is an inhibitor of mTOR, one of the targets of the PI3 kinase pathway. Similar to Rux, both LY294002 and rapamycin blocked IL-13-induced potentiation of oxidative stress-induced cell death but did not reduce either H2O2 or tBOOH toxicity in the absence of IL-13 (Fig. 4A and B). These results indicate that activation of the PI3 kinase pathway and specifically, its target mTOR, plays a key role in the potentiation of oxidative stress-induced cell death by IL-13.
Figure 4. Effects of PI3 Kinase Pathway Inhibitors on IL13-mediated Potentiation of Oxidative Stress-Induced Cell Death.
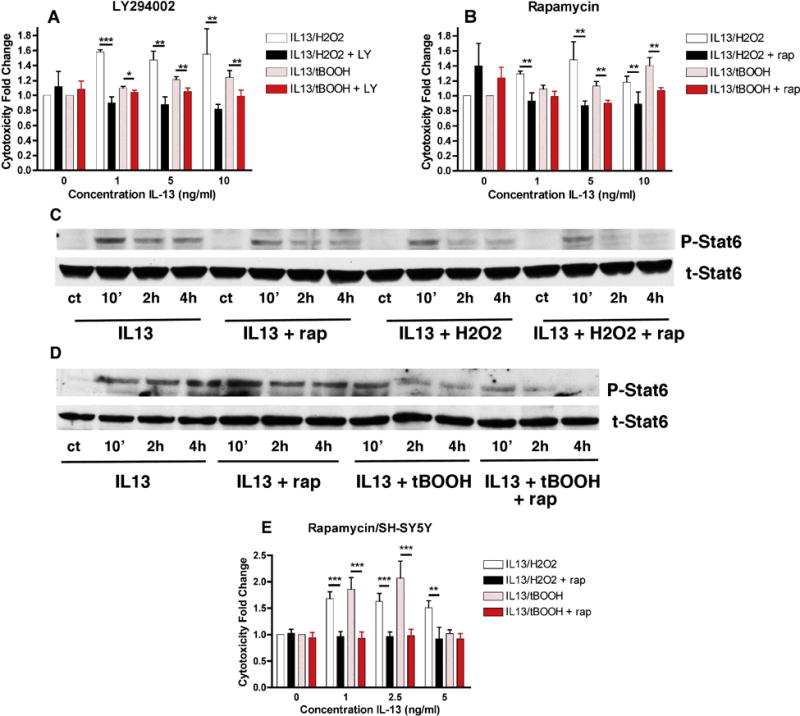
(A) MN9D cells grown in 96 well dishes were untreated or treated with 1, 5 or 10 ng/ml IL-13 alone, in the presence of 80 μM H2O2, 2.5 μM tBOOH or 10 μM LY294002 or as combination as indicated. Cell survival was measured after 24 hr with the MTT assay. Results are presented as the fold change in cell death as compared to H2O2 or tBOOH alone. The experiments were done in quadruplicate and repeated 5 times. (B) MN9D cells grown in 96 well dishes were untreated or treated with 1, 5 or 10 ng/ml IL-13 alone or in the presence of 80 μM H2O2, 2.5 μM tBOOH, 100 nM rapamycin or a combination as indicated. Cell survival was measured after 24 hr with the MTT assay. Results are presented as the fold change in cell death as compared to H2O2 or tBOOH alone. The experiments were done in quadruplicate and repeated 4 times. * p < 0.05; ** p < 0.01; *** p < 0.001 relative to no LY294002 or no rapamycin. (C) and (D) MN9D cells were untreated or treated with 10 ng/ml IL-13 alone or in the presence of 100 nM rapamycin, 80 μM H2O2, 2.5 μM tBOOH or a combination as indicated for 10 min, 2 hr and 4 hr. Cell lysates were prepared and equal amounts of protein were analyzed by SDS-PAGE and immunoblotting with antibodies to phospho and total Stat6. Representative Western blots are shown. Similar results were obtained in 3 independent experiments. (E) Human SH-SY5Y dopaminergic cells grown in 96 well dishes were untreated or treated with 1, 2.5 or 5 ng/ml IL-13 alone or in the presence of 40 μM H2O2, 2.5 μM tBOOH, 100 nM rapamycin or a combination as indicated. Cell survival was measured after 24 hr with the MTT assay. Results are presented as the fold change in cell death as compared to H2O2 or tBOOH alone. The experiments were done in quadruplicate and repeated 4 times. ** p < 0.01; *** p < 0.001 relative to no rapamycin.
As further support for the protective effects of rapamycin against IL-13-induced potentiation of oxidative stress-induced cell death, we tested its effects in the human dopaminergic cell line SH-SY5Y [10]. Similar to the results with the MN9D cells, IL-13 potentiated cell death induced by low concentrations of either H2O2 or tBOOH and this potentiation was blocked by treatment with rapamycin (Fig. 4E).
In order to understand how the mTOR pathway contributes to the potentiation of cell death by IL-13, we decided to focus on rapamycin since this is a well-characterized drug that has been suggested to be beneficial in the context of PD [11, 12]. We first looked at the effects of rapamycin on Stat6 phosphorylation by IL-13 alone and in the presence of H2O2 or tBOOH. Interestingly, rapamycin did not prevent the H2O2- or tBOOH-induced time-dependent decrease in Stat6 phosphorylation (Fig. 4C & D).
Rapamycin has several targets that have been implicated in PD including autophagy [11], REDD1 [13] and 4E-BP1 [14]. Rapamycin induces autophagy and impairments in autophagy are associated with PD [11]. However, the PI3 kinase inhibitor LY294002 has been reported to inhibit autophagy in many but not all studies [15] suggesting that induction of autophagy is unlikely to underlie the protective effects of rapamycin in this model. Moreover, treatment of the MN9D cells with bafilomycin, an autophagy inhibitor, reduced the potentiation of oxidative stress-induced cell death by H2O2 or tBOOH (not shown). Rapamycin can also block the translation of REDD1, a protein that is induced in models of PD and causes the dephosphorylation of Akt. However, as phosphorylation of Akt is increased rather than decreased in the absence of rapamycin in the MN9D cells, REDD1 is also unlikely to be the critical target of rapamycin in this paradigm. Moreover, neither IL-13 alone or in the presence of H2O2 or tBOOH induced changes in the levels of REDD1 (not shown). 4E-BP1 regulates the ratio of cap-independent to cap-dependent mRNA translation [16]. In its hypophosphorylated form, it sequesters eIF4E which mediates binding of the eIF4F complex to the mRNA 5′ cap structure thereby promoting cap-independent mRNA translation. When mTOR is activated, it phosphorylates 4E-BP which promotes its dissociation from eIF4E and stimulates cap-dependent mRNA translation. Modulation of mRNA translation is a key response to stress and impairments in that modulation can have deleterious consequences [16]. Thus, by inhibiting 4E-BP1 phosphorylation, rapamycin promotes cap-independent mRNA translation which is associated with the production of stress-modulating proteins [16]. Treatment with IL-13 alone had little effect on 4E-BP1 phosphorylation after either 8 or 24 hr of treatment (Fig. 5). However, both H2O2 and tBOOH alone increased 4E-BP1 phosphorylation after 24 hr of treatment and this was further increased in the presence of IL-13 (Fig. 5A & B). Importantly, rapamycin greatly reduced both the basal levels of 4E-BP1 phosphorylation and the phosphorylation induced by the combination of IL-13 and either H2O2 or tBOOH (Fig. 5). Interestingly, rapamycin also reduced Akt phosphorylation by the combination of IL-13 and either H2O2 or tBOOH, especially after 24 hr of treatment (Fig. 5).
Figure 5. Effects of Rapamycin on PI3 Kinase Pathway Targets.
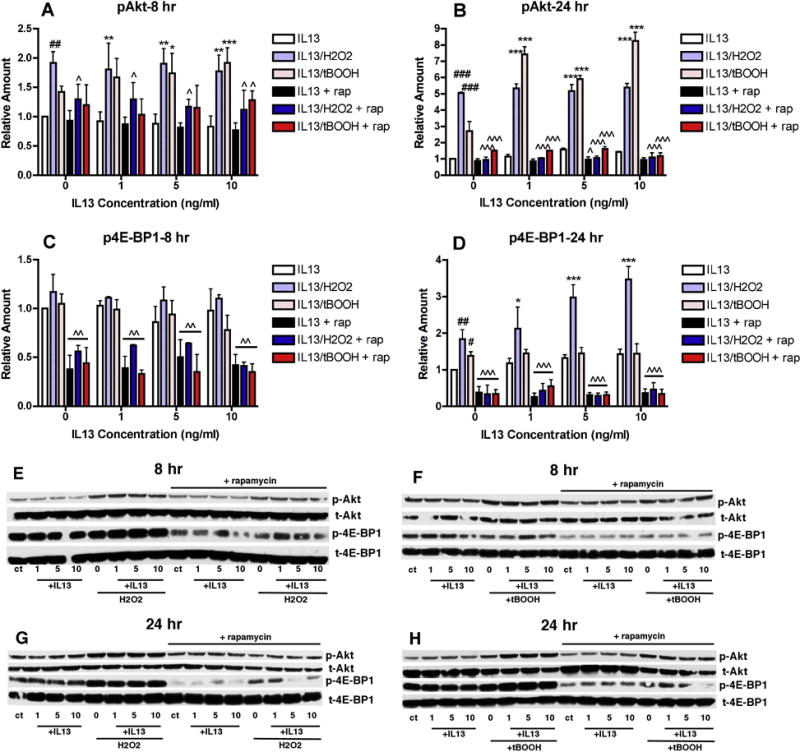
MN9D cells were untreated or treated with 1, 5 or 10 ng/ml IL-13 alone or in the presence of 80 μM H2O2 or 2.5 μM tBOOH in the absence or presence of 100 nM rapamycin as indicated for 8 or 24 hr. Cell lysates were prepared and equal amounts of protein were analyzed by SDS-PAGE and immunoblotting with antibodies to phospho and total Akt and phospho and total 4E-BP1. (A)–(D) Western blots from 3–5 independent experiments similar to the ones shown in (E)–(H) were scanned and quantified. # p < 0.05; ## p < 0.01; ### p < 0.001 relative to control. * p < 0.05; ** p < 0.01; *** p < 0.001 relative to IL-13 alone. ˆ p < 0.05; ˆˆˆ p < 0.001 relative to no rapamycin. (E)–(H) Representative Western blots. Similar results were obtained in 3–5 independent experiments.
To further address the role of 4E-BP1 in the rapamycin-mediated inhibition of the potentiation of oxidative stress-induced dopaminergic cell death by IL-13, 4E-BP1 levels were reduced in the MN9D cells by treatment with siRNA (Fig. 6C). Since phosphorylation of 4E-BP1 reduces its binding to eIF4E, reduction of 4E-BP1 levels should have similar effects to phosphorylation of 4E-BP1 and thereby reduce or eliminate the protective effects of rapamycin if 4E-BP1 is the critical player in the actions of rapamycin in this paradigm. As shown in Figure 6A and B, while control siRNA had no effect on either the IL13-mediated potentiation of oxidative stress-induced cell death or the protection by rapamycin, 4E-BP1 almost completely eliminated the protective effects of rapamycin.
Figure 6. Effect of 4E-BP1 siRNA on Rapamycin-Mediated Protection from IL-13-mediated Potentiation of Oxidative Stress-Induced Cell Death.
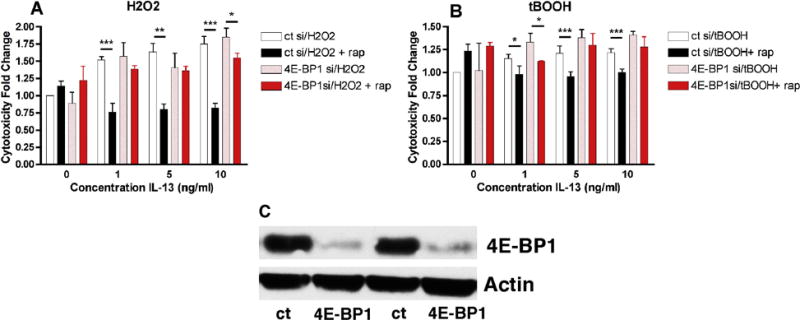
MN9D cells were transfected with control siRNA or 4E-BP1 siRNA as described in Methods. (A) and (B) Transfected MN9D cells grown in 96 well dishes were untreated or treated with 1, 5 or 10 ng/ml IL-13 alone, in the presence of 80 μM H2O2, 2.5 μM tBOOH, 100 nM rapamycin or a combination as indicated. Cell survival was measured after 24 hr with the MTT assay. Results are presented as the fold change in cell death as compared to H2O2 or tBOOH alone. The experiments were done in quadruplicate and repeated 6 times. * p < 0.05; ** p < 0.01; *** p < 0.001 relative to no rapamycin. (C) Cell lysates were prepared and equal amounts of protein were analyzed by SDS-PAGE and immunoblotting with antibodies to phospho and total 4E-BP1. A representative Western blot presenting two independent experiments is shown. Similar results were obtained in 6 independent experiments.
Discussion
The observation that IL-13 potentiated both oxidative stress-induced dopaminergic nerve cell death in vitro and TH-positive neuronal cell loss in the LPS model of PD in vivo [2] not only provided a new pathway for the investigation of PD but also suggested that a better understanding of this pathway could identify additional therapeutic targets for the treatment of the disease. In this manuscript, we show that the PI3 kinase-mTOR pathway plays a key role in this cell death paradigm with inhibition by either the PI3 kinase inhibitor LY294002 or the mTOR inhibitor rapamycin being protective. While a number of studies have shown that rapamycin is neuroprotective in experimental models of neurodegenerative diseases including PD [11], the exact targets appear to vary. Indeed, even in the case of PD models, several different rapamycin targets have been proposed to mediate protection. However, our results strongly suggest that the target most likely to play a protective role against IL-13 potentiation of oxidative stress-induced cell death is 4E-BP1. This conclusion is supported by several observations. First, 4E-BP1 phosphorylation increases in a time- and dose-dependent manner with combinations of IL-13 and either H2O2 or tBOOH. Second, rapamycin reduces this increase in phosphorylation in cells treated with the combination of IL-13 and either H2O2 or tBOOH. Third, siRNA against 4E-BP1 significantly reduces the protective effects of rapamycin in this model. 4E-BP1 phosphorylation/dephosphorylation plays a key role in translational reprogramming [16]. Studies have shown that when cap-dependent translation is reduced by 4E-BP1 hypophosphorylation, there is a selective upregulation of the translation of mRNAs whose products have a role in the response to stress and mitochondrial activity [17]. Indeed, in Drosophila, 4E-BP1 was found to increase the translation of nuclear encoded mitochondrial electron transport chain genes [18]. Since mitochondrial function is compromised in PD [19], this could have a protective effect by helping to maintain the function of the electron transport chain. More recently, skeletal muscle-specific increases in 4E-BP1 in mice were found to directly increase the translation of peroxisome proliferator-activated receptor γ coactivator-1α as well as to enhance respiratory function [20]. Whether or not these or other as yet unidentified targets of 4E-BP1 mediate the protective effects of rapamycin against IL13-mediated potentiation of oxidative stress-induced cell death will be the topic of future studies.
Several previous studies showed that 4E-BP1 hypophosphorylation was protective in genetic models of PD [14, 21]. In flies, overexpression of the Drosophila equivalent of 4E-BP1 suppressed the pathophysiology in Park and Pink mutants [14]. Similar results were obtained with rapamycin. More recently, using cells from mice deficient in PINK1, an impairment in the response to hypoxic stress due to 4E-BP1 hyperphosphorylation was demonstrated [21]. However, our data are the first to show a protective role for 4E-BP1 hypophosphorylation in a non-genetic model of PD and suggest that this might be a target that is worth additional investigation since most cases of PD are sporadic [22].
Our initial results indicated that oxidative stress negatively affected Stat6 activation resulting in a more rapid rate of dephosphorylation. These results suggested that inhibition of IL-13-induced Stat6 phosphorylation might further enhance the potentiation of oxidative stress-induced cell death by IL-13. However, in contrast to these expectations, we found that the Jak1/2 inhibitor ruxolitinib, which completely inhibited IL-13-induced Stat6 phosphorylation, could block the potentiation of oxidative stress-induced cell death. These results indicate that Jak signaling is required for the potentiation of oxidative stress-induced cell death by IL-13. Ruxolitinib is already FDA approved for the treatment of myelofibrosis and is in clinical trials for a variety of inflammatory diseases as well as several cancers [9]. Recently, the Jak1/2 inhibitor AZD1480 was found to be protective in an α-synuclein overexpression model of PD both in vitro and in vivo [23]. Our findings with ruxolitinib support and extend these studies and suggest that Jak inhibitors might be useful therapeutic agents for the treatment of PD resulting from multiple, distinct causes.
In summary, our results show that both the Jak-Stat6 and PI3 kinase-mTOR pathways contribute to the potentiation of oxidative stress-induced cell death in both mouse and human dopaminergic neurons. In contrast to results with other cell types, IL-13 does not stimulate an increase in ROS production in the dopaminergic cells, an observation consistent with our recent in vivo studies on IL-13 and stress [24]. Thus, our results suggest that either Jak1/2 inhibitors or mTOR inhibitors might be useful for treating at least some cases of idiopathic PD.
Highlights.
The Jak-Stat pathway contributes to IL-13 mediated potentiation of cell death.
The PI3 kinase-mTOR pathway contributes to IL-13 mediated potentiation of cell death.
Rapamycin and ruxolitinib can block IL-13 mediated potentiation of cell death.
4E-BP1 phosphorylation is a critical target of rapamycin in this paradigm.
Acknowledgments
None.
Funding
Funding supported by the NIH (NS085155).
Abbreviations
- BSA
bovine serum albumin
- CM-H2DCFDA
5-(and-6)-chloromethyl-2′,7′-dichlorofluorescein diacetate
- mTOR
mammalian target of rapamycin
- MTT
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide
- PI3-kinase
phosphoinositide 3 kinase
- ROS
reactive oxygen species
- Rux
ruxolitinib
- tBOOH
t-butyl hydroxide
- TBS
tris-buffered saline
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Authors’ contributions
PM and BC conceived the project. PM performed the experiments, analyzed the data and wrote the manuscript. BC reviewed the manuscript. Both authors read and approved the final manuscript.
Competing interests
The authors declare that they have no competing interests.
References
- 1.de Lau LM, Breteler MM. Epidemiology of Parkinson’s disease. Lancet Neurol. 2006;5:525–535. doi: 10.1016/S1474-4422(06)70471-9. [DOI] [PubMed] [Google Scholar]
- 2.Morrison BE, Marcondes MC, Nomura DK, Sanchez-Alavez M, Sanchez-Gonzalez A, Saar I, Kim KS, Bartfai T, Maher P, Sugama S, Conti B. IL-13Ralpha1 expression in dopaminergic neurons contributes to their oxidative stress-mediated loss following chronic peripheral treatment with lipopolysaccharide. J Immunol. 2012;189:5498–5502. doi: 10.4049/jimmunol.1102150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mori S, Maher P, Conti B. Neuroimmunology of the Interleukins 13 and 4. Brain Sci. 2016;6:18. doi: 10.3390/brainsci6020018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.McCormick SM, Heller NM. Commentary: Il-4 and IL-13 receptors and signaling. Cytokine. 2015;75:38–50. doi: 10.1016/j.cyto.2015.05.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ehren JL, Maher P. Concurrent regulation of the transcription factors Nrf2 and ATF4 mediates the enhancement of glutathione levels by the flavonoid fisetin. Biochem Pharmacol. 2013;85:1816–1826. doi: 10.1016/j.bcp.2013.04.010. [DOI] [PubMed] [Google Scholar]
- 6.Mandal D, Fu P, Levine AD. REDOX regulation of IL-13 signaling in intestinal epithelial cells: Usage of alternate pathways mediates distinct gene expression patterns. Cell Signal. 2010;22:1485–1494. doi: 10.1016/j.cellsig.2010.05.017. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 7.Bhattacharjee A, Shukla M, Yakubenko VP, Mulya A, Kundu S, Cathcart MK. Il-4 and IL-13 employ discrete signaling pathways for target gene expression in alternatively activate monocytes/macrophages. Free Rad Biol Med. 2013;54:1–16. doi: 10.1016/j.freeradbiomed.2012.10.553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Guo J, Yao H, Lin X, Xu H, Dean D, Zhu Z, Liu G, Sime P. IL-13 induces YY1 through the AKT pathway in lung fibroblasts. PLoS ONE. 2015;10:e0119039. doi: 10.1371/journal.pone.0119039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rask-Andersen M, Zhang J, Fabbro D, Schioth HB. Advances in kinase targeting: current clinical use and clinical trials. Trends Pharmacol Sci. 2014;35:604–620. doi: 10.1016/j.tips.2014.09.007. [DOI] [PubMed] [Google Scholar]
- 10.Xicoy H, Wieringa B, Martens GJM. The SH-SY5Y cell line in Parkinson’s research: a systematic review. Mol Neurodegeneration. 2017;12:10. doi: 10.1186/s13024-017-0149-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bove J, Martinez-Vicente M, Vila M. Fighting neurodegeneration with rapamycin: mechanistic insights. Nature Rev Neurosci. 2011;12:437–452. doi: 10.1038/nrn3068. [DOI] [PubMed] [Google Scholar]
- 12.Liu K, Shi N, Sun Y, Zhang T, Sun X. Therapeutic effects of rapamycin on MPTP-induced Parkinsonism in mice. Neurochem Res. 2013;38:201–207. doi: 10.1007/s11064-012-0909-8. [DOI] [PubMed] [Google Scholar]
- 13.Malagelada C, Jin ZH, Jackson-Lewis V, Przedborski S, Greene LA. Rapamycin protects against neuron disease in in vitro and in vivo models of Parkinson’s disease. J Neurosci. 2010;20:1166–1175. doi: 10.1523/JNEUROSCI.3944-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tain LS, Mortiboys H, Tao RN, Ziviani E, Bandmann O, Whitworth AJ. Rapamycin activation of 4E-BP prevents parkinsonian dopaminergic neuron loss. Nature Neurosci. 2009;12:1129–1136. doi: 10.1038/nn.2372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yang Y, Hu L, Zheng H, Mao C, Hu W, Xiong K, Wang F, Liu C. Application and interpretation of current autophagy inhibitors and activators. Acta Pharmacol Sin. 2013;34:625–635. doi: 10.1038/aps.2013.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Spriggs KA, Bushell M, Willis AE. Translational regulation of gene expression during conditions of cell stress. Mol Cell. 2010;40:228–237. doi: 10.1016/j.molcel.2010.09.028. [DOI] [PubMed] [Google Scholar]
- 17.Nacarelli T, Azar A, Sell C. Aberrant mTOR activation in senescence and aging: A mitochondrial stress response? Exp Gerontol. 2015;68:66–70. doi: 10.1016/j.exger.2014.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zid BM, Rogers AN, Katewa SD, Vargas MA, Kolipinski MC, Lu TA, Benzer S, Kapahi P. 4E-BP extends lifespan upon dietary restriction by enhancing mitochondrial activity in Drosophila. Cell. 2009;139:149–160. doi: 10.1016/j.cell.2009.07.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sarkar S, Raymick J, Imam S. Neuroprotective and therapeutic strategies against Parkinson’s disease: Recent perspectives. Int J Mol Sci. 2016;17:904. doi: 10.3390/ijms17060904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tsai SY, Sitzmann JM, Dastidar SG, Rodriguez AA, Vu SL, McDonald CE, Academia EC, O’Leary MN, Ashe TD, La Spada AR, Kennedy BK. Muscle-specific 4E-BP1 signaling activation improves metabolic parametes during aging and obesity. J Clin Invest. 2015;125:2952–2964. doi: 10.1172/JCI77361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lin W, Wadlington NL, Chen L, Zhuang X, Brorson JR, Kang UJ. Loss of PINK1 attentuates HIF-1alpha induction by preventing 4E-BP1-dependent switch in protein translation under hypoxia. J Neurosci. 2014;34:3079–3089. doi: 10.1523/JNEUROSCI.2286-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Weintraub D, Comella CL, Horn S. Parkinson’s disease. Am J Manag Care. 2008;14:S40–S69. [PubMed] [Google Scholar]
- 23.Qin H, Buckley JA, Liu X, Fox TH, Meares GP, Yu HJ, Yan Z, Harm AS, Li Y, Standaert DG, Benviste EN. Inhibition of the JAK/STAT pathway protects against alpha-synuclein-induced neuroinflammation and dopaminergic degeneration. J Neurosci. 2016;36:5144–5159. doi: 10.1523/JNEUROSCI.4658-15.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mori S, Suguma S, Nguyen W, Michel T, Sanna MG, Sanchez-Alavez M, Cintron-Colon R, Moroncini G, Kakinuma Y, Maher P, Conti B. Lack of interleukin-13 receptor alpha1 delays the loss of dopaminergic neurons during chronic stress. J Neuroinflamm. 2017;14:88. doi: 10.1186/s12974-017-0862-1. [DOI] [PMC free article] [PubMed] [Google Scholar]