

Graphical abstract

1. INTRODUCTION
Graphene is composed of single-atom thick sheets of sp2 bonded carbon atoms that are arranged in a perfect two-dimensional (2D) honeycomb lattice. Because of this structure, graphene is characterized by a number of unique and exceptional structural, optical, and electronic properties.1 Specifically, these extraordinary properties include, but are not limited to, a high planar surface area that is calculated to be 2630 m2 g−1,2 superior mechanical strength with a Young’s modulus of 1100 GPa,3 unparalleled thermal conductivity (5000 W m−1 K−1),4 remarkable electronic properties (e.g., high carrier mobility [10 000 cm2 V−1 s−1] and capacity),5 and alluring optical characteristics (e.g., high opacity [~97.7%] and the ability to quench fluorescence).6 As such, it should come as no surprise that graphene is currently, without any doubt, the most intensively studied material for a wide range of applications that include electronic, energy, and sensing outlets.1c Moreover, because of these unique chemical and physical properties, graphene and graphene-based nanomaterials have attracted increasing interest, and, arguably, hold the greatest promise for implementation into a wide array of bioapplications.7
In the last several years, numerous studies have utilized graphene in bioapplications ranging from the delivery of chemotherapeutics for the treatment of cancer8 to biosensing applications for a host of medical conditions9 and even for the differentiation and imaging of stem cells.10 While promising and exciting, recent reports have demonstrated that the combination of graphene with nanomaterials such as nanoparticles, thereby forming graphene–nanoparticle hybrid structures, offers a number of additional unique physicochemical properties and functions that are both highly desirable and markedly advantageous for bioapplications when compared to the use of either material alone (Figure 1).11 These graphene–nanoparticle hybrid structures are especially alluring because not only do they display the individual properties of the nanoparticles, which can already possess beneficial optical, electronic, magnetic, and structural properties that are unavailable in bulk materials, and of graphene, but they also exhibit additional advantageous and often synergistic properties that greatly augment their potential for bioapplications.
Figure 1.
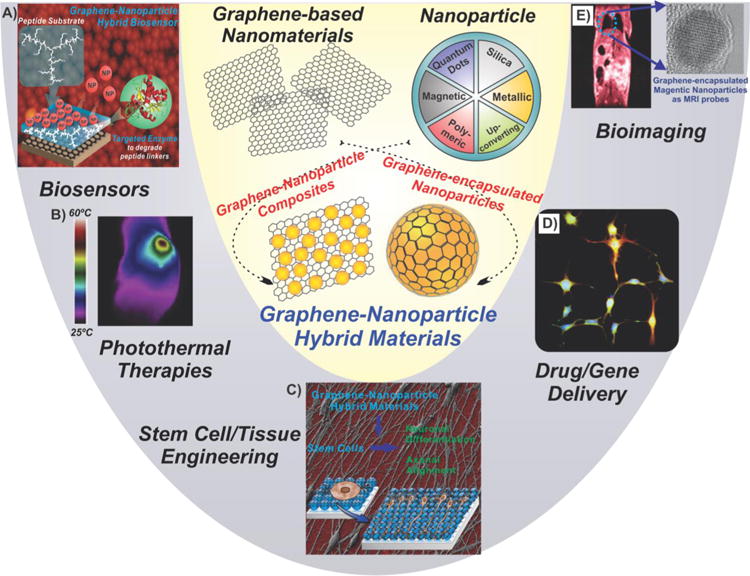
Graphene nanoparticle hybrids exist in two forms, as graphene–nanoparticle composites and graphene-encapsulated nanoparticles, and can be used for various bioapplications including biosensors, photothermal therapies, stem cell/tissue engineering, drug/gene delivery, and bioimaging. Panel (A) reprinted with permission from ref 110. Copyright 2012 Wiley. Panel (B) reprinted with permission from ref 211. Copyright 2013 Elsevier. Panel (C) reprinted with permission from ref 244. Copyright 2013 Wiley.
In general, these graphene–nanoparticle hybrid materials can be categorized into two classes (Figure 2). They can exist as (1) graphene–nanoparticle composites, where the nanoparticles decorate or are grown on graphene sheets (Figure 2A and B), or (2) graphene-encapsulated nanoparticles wherein the nanoparticle surface is wrapped or coated with graphene (Figure 2C and D). In particular, graphene–nanoparticle hybrid structures can be synthesized by combining graphene or its derivatives, graphene oxide (GO) and reduced graphene oxide (rGO), with various types of nanoparticles including, but not limited to, quantum dots (QDs), metal (e.g., noble metal and magnetic), metal oxide, or silica nanoparticles (SiNPs), depending on the final functional properties that are desired. By combining these unique and robust materials, a striking synergy can often be achieved. For instance, the decoration of metal or metal oxide nanoparticles on graphene surfaces has been shown to have implications on the charge transfer behavior in graphene through the modification of the local electronic structure.12 As a result, this type of composite material can show improved performance as catalysts. On the other hand, in sensing applications, the combination of nanoparticles, which have excellent conductivity and catalytic properties, with graphene materials allows for the enhancement of achievable sensitivity and selectivity over either graphene or nanoparticle-based sensors alone.11a,13 Finally, by combining graphene with nanoparticles, it is possible to deliver cargo (e.g., small molecule drugs, nucleic acids, etc.) to target cells (e.g., cancer cells or stem cells) while enabling complementary multifunctionalities such as imaging (e.g., surface-enhanced Raman spectroscopy [SERS] or magnetic resonance imaging [MRI]) and hyperthermia (e.g., using near-infrared [NIR] radiation).14
Figure 2.

Graphene–nanoparticle hybrid structures. Panels (A) and (B) show graphene-nanoparticle composites wherein nanoparticles are decorated on the surface of graphene sheets. Panel (A) reprinted with permission from ref 146. Copyright 2011 Elsevier. Panel (B) reprinted with permission from ref 102. Copyright 2010 American Chemical Society. Panels (C) and (D) show graphene-encapsulated nanoparticles wherein nanoparticles are wrapped by graphene. Panel (C) reprinted with permission from ref 112. Copyright 2012 American Chemical Society. Panel (D) reprinted with permission from ref 104. Copyright 2010 Wiley.
In this Review, we will systematically discuss graphene–nanoparticle hybrid materials in terms of their synthesis, characterization, and implementation to a host of bioapplications. While there are already numerous excellent reviews that cover graphene synthesis, its chemical and physical properties, and its bioapplications,1,15 graphene–nanoparticle hybrid materials and their implementation into bioapplications is a relatively new area that has not been comprehensively reviewed. Moreover, graphene–nanoparticle hybrid materials are gaining significant traction in the field of graphene research, where, in 2013, approximately 20% of all papers published on graphene involved the synthesis and application of graphene–nanoparticle hybrid materials (Figure 3). Thus, we present the first complex work covering all modes and methods for the synthesis of graphene–nanoparticle hybrid materials, its unique chemical and physical properties, and, most importantly, its utilization for bioapplications. Specifically, we will focus on graphene–nanoparticle hybrid materials for biosensing, multifunctional drug delivery, imaging, as well as for the control of stem cell differentiation with particular emphasis on the advantages and differences that are conferred by the use of graphene–nanoparticle hybrid materials over conventional methods in each area. We will also discuss trends, future directions, and any controversies that exist in the field. As a result, we hope that this Review will inspire interest from various disciplines and highlight an up and coming field wherein graphene–nanoparticle hybrid structures can bring significant advantages to a wide variety of bioapplications.
Figure 3.
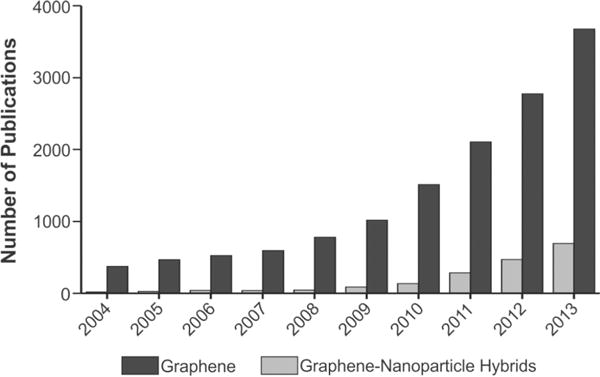
The number of publications utilizing graphene–nanoparticle hybrid materials is increasing (numbers were obtained from PubMed).
2. SYNTHESIS AND CHARACTERIZATION OF GRAPHENE–NANOPARTICLE HYBRID MATERIALS
Because of the many unique and advantageous properties of graphene and its derivatives, GO and rGO, a significant amount of effort has been invested in utilizing these materials either by themselves, or in combination with other interesting nanomaterials such as nanoparticles. As mentioned previously, graphene–nanoparticle hybrid materials can be broadly categorized into two main classes on the basis of their structural morphology. They can exist as (1) graphene–nanoparticle composites, where nanoparticles are decorated or grown on sheets of graphene or its derivatives, and (2) graphene-encapsulated nanoparticles wherein nanoparticles are wrapped by graphene or its derivatives. The main difference between these two classes can be found in the relative size ratio that exists between the diameter of the nanoparticles and the lateral dimensions of the graphene sheets. Specifically, when the diameter of the nanoparticles is in the range of a few nanometers to about a hundred nanometers, the nanoparticles are generally small in comparison to the graphene sheets and can easily be decorated onto the sheets, thereby forming graphene–nanoparticle composites. On the other hand, when the size of the nanoparticles is larger and becomes more comparable with the graphene sheets, the small 2D sheets can be wrapped around the nanoparticles resulting in graphene-encapsulated nanoparticles. In this section, we will begin by briefly describing the primary methods used to produce graphene, GO, and rGO. This will be followed by an in-depth analysis of the various methods that have been developed for the synthesis of graphene–nanoparticle hybrid structures, with particular emphasis on the properties and characteristics that result from these different procedures.
2.1. Graphene and Graphene Oxide
Graphene sheets can be obtained using a number of procedures; however, there are three primary methods, which include: (1) mechanical exfoliation, (2) chemical vapor deposition (CVD) onto metal or Si substrates, and (3) the chemical, electrochemical, or thermal reduction of GO (Figure 4). For the mass-production of graphene, the CVD method is the most effective and can be used to fabricate large areas of graphene while limiting the number of defects.16 Consequently, graphene that is obtained using the CVD method can have a carrier mobility that reaches values as high as ~2000–4000 cm2 V−1 s−1.17 On the other hand, mechanical exfoliation using the Scotch tape method or by rubbing lithographically patterned pillars with “tipless” atomic force microscopy (AFM) cantilevers can be used to generate very high-quality graphene with a carrier mobility that reaches ~10 000 cm2 V−1 s−1 at room temperature.18 Using this method, the physical limit of the graphene sheets that are produced has lateral dimensions on the order of tens to hundreds of micrometers. However, mechanical exfoliation has poor reproducibility and is not amenable to large-scale fabrication.1c Finally, the reduction of GO has also been demonstrated to be a relatively economical and facile technique for the production of graphene and will be explained in more detail below; however, it has been reported that the quality of these graphene (rGO) sheets, in terms of electrical conductivity, is generally lower than that produced using the other two methods described.19 As such, overall, the reduction method is a better fit for the mass-production of small graphene sheets, while the CVD method is more efficient for the mass-production of high-quality graphene. Hence, the application for which graphene is being synthesized must first be considered before the appropriate synthesis method can be selected (Figure 4).
Figure 4.
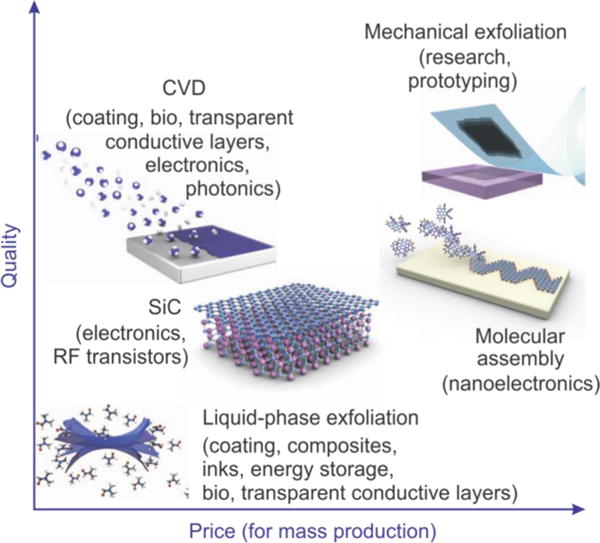
Methods for the mass-production of graphene. There are several choices depending on the particular application, each with differences in terms of size, quality (e.g., presence of defects and impurities), and price. Reprinted with permission from ref 1c. Copyright 2012 Nature.
On the other hand, GO has many distinct characteristics that greatly differentiate it from those of graphene due to the presence of numerous oxygen-containing hydroxyl and epoxy groups on the basal plane, along with smaller quantities of carboxyl, carbonyl, phenol, lactone, and quinone that are present at the edges,20 which collectively act to inhibit electron transfer (Figure 5A and B). Specifically, GO films are hydrophilic and typically insulating with an energy gap in the electron density of states and a sheet resistance of about 1012 Ω sq−1 or higher.21 However, similar to graphene, GO exhibits excellent electronic, thermal, electrochemical, and mechanical properties and is flexible, transparent, and also biocompatible due to its hydrophilic nature. In terms of its fabrication, GO is most commonly produced using the Brodie, Staudenmaier, and Hummer methods or some variation of these methods.19b,22 All three methods involve the oxidation of graphite to various levels resulting in the formation of hydrophilic groups on the surface. In particular, a combination of potassium chlorate (KClO3) and nitric acid (HNO3) is typically used to oxidize graphite in the Brodie and Staudenmaier methods, while potassium permanganate (KMnO4) and sulfuric acid (H2SO4) are used in the Hummer method.23 After the oxidation process, the resulting product is then exfoliated to obtain one- or multilayered GO sheets with ultrasonication being the most commonly used procedure.
Figure 5.
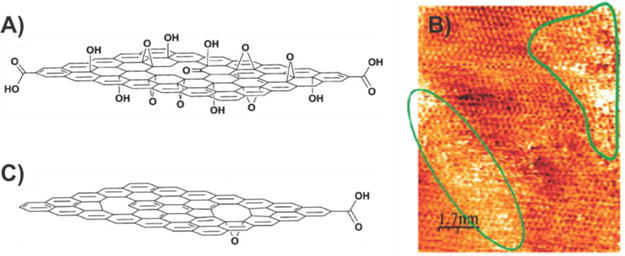
Structure of graphene oxide and reduced graphene oxide. (A) Chemical structure of GO. (B) Scanning tunneling microscope (STM) image of a GO monolayer on a highly oriented pyrolytic graphite substrate. Oxidized regions are marked by green contours. Panel (B) reprinted with permission from ref 5b. Copyright 2007 American Chemical Society. (C) Chemical structure of rGO. Panels (A) and (C) reprinted with permission from ref 24. Copyright 2010 Wiley.
Finally, as mentioned above, rGO can be obtained from GO through chemical, electrochemical, or thermal reduction, which removes the oxygen-containing branches from the basal planes and edges of the GO sheets (Figure 5C).22,24 As a consequence, the electrical conductivity of GO (as well as its thermal stability) can be restored close to the levels found in graphite. Specifically, the electrical conductivity of rGO has been reported to fall within the range of 200–42 000 Sm−1.19a,25 However, this depends significantly on the parameters used during reduction, including the type of reducing agent, duration of reduction, temperature, annealing time, and annealing temperature. Moreover, while the specific capacitance of rGO is significantly higher than that of GO, the transparency and dispersibility of rGO are significantly reduced when compared to pure GO sheets. To form rGO from GO, hydrazine, hydroquinone, ascorbic acid (AA), and sodium borohydride are commonly used to remove the hydroxyl groups from GO. Electrochemical tools can also be used to fabricate rGO. For instance, this can be accomplished using either acidic (e.g., sulfuric acid) or nonacidic solutions such as Na-phosphate-buffered saline (PBS), K-PBS, NaOH, and KCl in the presence of a constant reduction potential or sweeping potential.26 Finally, thermal reduction is also a well-known method for the removal of oxygen functional groups from the surface of GO. In this method, oxygen functional groups are removed in the form of water, carbon dioxide, and carbon monoxide by placing the GO sample in a preheated furnace at 1000–1100 °C for 30–45 s in the absence of air.27 However, chemical and electrochemical methods tend to be preferred as they can be performed under more mild conditions.
2.2. Graphene–Nanoparticle Composites
Graphene–nanoparticle composites, wherein sheets of graphene, GO, or rGO, are decorated with nanoparticles that are a few nanometers to a couple hundred nanometers in diameter,28 can be obtained by anchoring various types of nanoparticles to the surface of graphene sheets through both in situ (e.g., growing the nanoparticles on the graphene surface) and ex situ (e.g., attaching premade nanoparticles to the graphene surface) methods. GO and rGO are especially promising templates for this purpose as the presence of defects and oxygen functional groups on their surfaces allows for the nucleation, growth, and attachment of various metal (e.g., Au,29 Ag,30 Pt,31 etc.) and metal oxide nanoparticles (e.g., Fe3O4,32 TiO2, 33 ZnO,34 SnO2,35 Cu2O,36 MnO2,37 NiO,38 etc.). The resulting graphene–nanoparticle composites are then able to offer numerous unique and advantageous properties for various applications depending on the particular characteristics possessed by the nanoparticles used to form the composites. For example, graphene–nanoparticle composites can confer excellent catalytic activity, enhancements in mass transport, and a significantly higher effective surface area.20b As such, recent efforts in this area have not only focused on methods to form graphene–nanoparticle composites while preserving the excellent properties of graphene but also on precisely tuning the physicochemical features that are present. In this section, we will give a comprehensive review of the different methods that are available for the preparation of graphene–nanoparticle composites including various in situ (e.g., reduction, hydrothermal, and electrochemical) and ex situ methods (Table 1). We will specifically focus on the distinct characteristics and properties that each technique imparts on the resulting graphene–nanoparticle composite.
Table 1.
Summary of the Different Methods Used To Prepare Graphene–Nanoparticle Composites
| method | typical NPs | key characteristics | key refs |
|---|---|---|---|
| reduction | metal NPs, especially noble metals | (1) one-pot synthesis; (2) highly efficient and easy to perform; (3) can be difficult to control the size and morphology of the NPs; however, can be addressed by using microwave-facilitated reduction | 30, 39–55 |
| hydrothermal | metal oxide NPs, QDs | (1) can create NPs with high crystallinity and narrow size distribution; (2) high temperature and long reaction times can partially or completely reduce GO on its own | 56–76 |
| electrochemical | metal NPs, especially noble metals | (1) simple, fast, and green technique; (2) low cost, easy to miniaturize and automate, and is highly stable and reproducible; (3) pulse current can be adjusted to control density, size, and morphology of NPs; also allows for simultaneous reduction of GO | 78–84 |
| ex situ | inorganic NPs, especially noble metals | (1) NPs are synthesized in advanced; this allows for precise control of the size, shape, and density of the NPs that are used to form hybrids; (2) ex situ methods include covalent or noncovalent interactions such as van der Waals interactions, hydrogen bonding, π–π stacking, or electrostatic interactions | 86–103 |
2.2.1. Reduction Methods To Form Graphene–Nanoparticle Composites
Graphene–metal nanoparticle composites are most frequently fabricated via the reduction of metallic salts (e.g., HAuCl4, AgNO3, and K2PtCl4) using chemical agents such as ethylene glycol, sodium citrate, and sodium borohydride.30,39 More specifically, the negatively charged functional groups that exist on the surface of GO allow for the nucleation of positively charged metallic salts, resulting in the growth of metal nanoparticles on the GO surface. By utilizing this method to form rGO–metal nanoparticle composites, it is possible to preserve the excellent electrical properties of graphene. Moreover, by controlling the density of oxygen-containing groups on the GO and rGO surface, one can easily tune the density of nanoparticles formed on the resulting graphene–nanoparticle composites. In general, to form graphene–metallic nanoparticle composites in situ via reduction, a one-step method is used wherein the metal precursor and GO sheets are mixed in an aqueous solution and then reduced simultaneously (Figure 6). This reaction is similar to conventional nanoparticle synthesis methods and follows three steps: (1) reduction, (2) nucleation, and (3) nanoparticle growth. More specifically, the functionalities that exist on the GO and rGO surface, such as alcohols, carbonyl groups, and acids, are responsible for the attachment of free metal ions through electrostatic interactions. Subsequently, the addition of a reducing agent promotes the reduction of the attached metal ions, thereby enabling the growth of metal nanoparticles on the GO and rGO surfaces.40 While this method is highly efficient and easy to perform, the size and morphology of the metal nanoparticles on the resulting composite can be difficult to control, resulting in samples that are decorated with nanoparticles that have a wide size distribution.41
Figure 6.
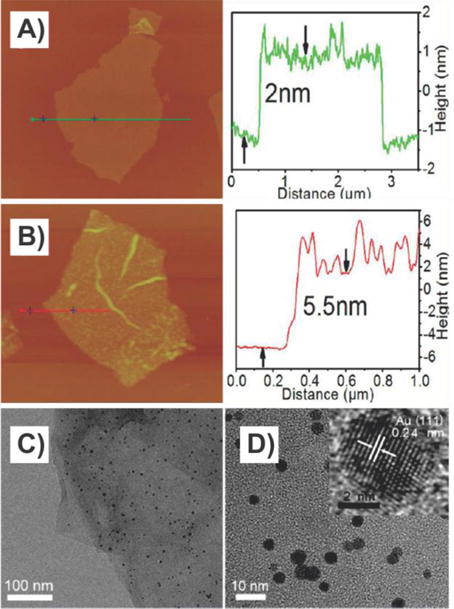
Graphene–nanoparticle composite composed of GO sheets decorated with AuNPs. (A) AFM image of a single GO sheet and (B) a GO sheet decorated with 3.5 nm AuNPs. The corresponding curves on the right side show the thicknesses of the GO sheet and the GO/AuNP sheet. (C and D) TEM images of the GO/AuNP sheet with different magnifications. Inset of (D) shows a high-resolution TEM image of a single AuNP. Reprinted with permission from ref 41. Copyright 2013 American Chemical Society.
To date, the reduction technique has been used primarily for the preparation of graphene–noble metal nanoparticle composites. Noble metal nanoparticles, especially gold (AuNP) and silver nanoparticles (AgNPs), are among the most extensively studied nanomaterials and have led to the development of numerous biotechniques and applications including diagnostics, imaging, drug delivery, and other therapeutics.42 Noble metal nanoparticles are of particular interest because of their unique and unusual properties such as high biocompatibility and optical properties (e.g., surface plasmon resonance), which can easily be tuned to the desired wavelength according to their shape (nanoparticles, nanoshells, nanorods, etc.), size, and composition.43 In addition, when combined with graphene as a composite, graphene–noble metal nanoparticle hybrids are able to exhibit SERS as well as enhanced catalytic potential.44 To this end, numerous methods have been used to fabricate graphene–AuNP composites, which are currently the most commonly prepared and utilized graphene–nanoparticle composite. Briefly, graphene–AuNP composites can be attained by mixing HAuCl4 precursor with exfoliated GO and sodium citrate, resulting in gold precursors anchoring to the surface of GO via electrostatic interaction. Afterward, the gold precursors are reduced using NaBH4 to form AuNPs.45 Similarly, graphene–AgNP composites can be obtained by mixing GO with AgNO3, followed by reduction with NaBH4.46 Finally, graphene decorated with platinum or palladium nanoparticles has also been produced by mixing graphene with chloroplatinic acid (H2PtCl6) or tetrachloropalladic acid (H2PdCl4), followed by reduction with ethylene glycol, respectively.47 In terms of the characteristics that are imparted by the formation of graphene–noble metal nanoparticle composites, Subrahmanyam et al. recently studied the interaction between noble metal nanoparticles and graphene utilizing Raman spectroscopy.47a Specifically, they reported that the decoration of graphene with noble metal nanoparticles results in electronic interactions that give rise to significant changes in the ionization energies of the metals as well as in their charge-transfer interaction and, subsequently, the Raman spectrum of the graphene sheets.
On the other hand, graphene–nanoparticle composites containing bimetallic nanoparticle hybrids can also be obtained utilizing a two-step reduction process. By fabricating such a structure, not only can synergism be achieved between the different nanoparticle species that are present but also between the bimetallic nanoparticles and graphene.48 For example, the reduction of H2PdCl4 by formic acid, followed by the addition of K2PtCl4 and reduction by AA, yielded a graphene–PtPd bimetallic nanoparticle composite structure (Figure 7).49 The resulting bimetallic hybrid composite showed much higher catalytic activity than conventional graphene–platinum nanoparticle (PtNP) hybrid structures and PtPd bimetallic catalysts. This can be attributed to a combination of the increased surface area of Pt in bimetallic nanoparticles, which is an essential factor in improving catalytic activity, as well as their better dispersion on graphene nanosheets that have a high surface area.50 Similarly, Yang et al. described the preparation of another bimetallic nanoparticle hybrid wherein GO was decorated with PtCo bimetallic nanoparticles.48b In this case, ethylene glycol-functionalized GO sheets were loaded with PtCo bimetallic nanoparticles by the addition of H2PtCl6 and CoCl2 at room temperature, followed by reduction with NaBH4. Similar to the graphene–PtPd bimetallic nanoparticle composites, the resulting graphene–PtCo bimetallic nanoparticle composite also exhibited good stability, resistance to degeneration, and an especially high catalytic activity as compared to other PtNPs and graphene–PtNP composites, presumably for the same reasons as mentioned for graphene PtPd composites.
Figure 7.
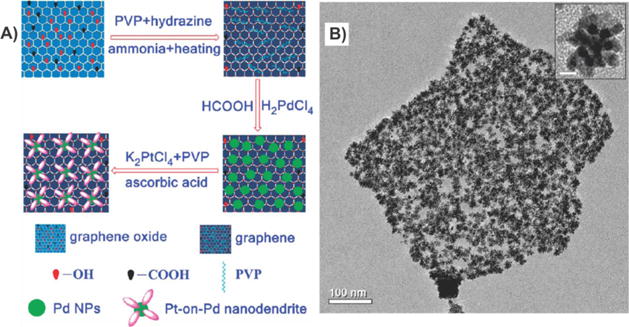
Graphene–bimetallic nanoparticle composites. (A) Procedure to fabricate graphene nanosheet/Pt-on-Pd bimetallic nanodendrite hybrids. (B) TEM images of the graphene–bimetallic nanoparticle composites. Inset of (B) shows the Pt-on-Pd bimetallic nanodendrites at a higher magnification. Reprinted with permission from ref 49. Copyright 2010 American Chemical Society.
Finally, as an alternative to the traditional reduction methods used to prepare graphene–nanoparticle composites, microwaves can also be used as a source of energy that facilitates the process.51 For instance, graphene and its derivatives were decorated with metal (e.g., Au,51 Ag,52 and Pt53) and metal oxide (e.g., Co3O4 and MnO2) nanoparticles,54 in the presence or absence of reducing agents and stabilizing molecules, with the help of rapid microwave irradiation. The main advantage of microwave irradiation is the uniform and rapid heating of the reaction mixture, thereby reducing the barrier to reduction, nucleation, and ion incorporation.55 Hence, nanoparticles with a very small size and narrow size distribution can be obtained.
2.2.2. Hydrothermal Methods To Form Graphene–Nanoparticle Composites
The hydrothermal method is also commonly used to synthesize inorganic nanoparticles that have a high crystallinity and narrow size distribution on graphene sheets. Moreover, this method allows for the formation of nanoparticles on graphene without the need for postannealing and calcination.11b In general, the process involves the use of high temperatures and pressures, which induce the growth of nanocrystals and, at the same time, reduce GO to rGO. However, while the high temperature and long reaction times can partially or completely reduce GO on its own, in most cases, reducing agents are added to ensure the complete reduction of GO.11b,56
Graphene–metal oxide nanoparticle composites (e.g., ZnO,57 TiO2,58 Fe3O4,59 SnO2,60) are, by far, the most common hybrids synthesized using the hydrothermal method. Specifically, metal oxide nanoparticles can provide the graphene–metal oxide nanoparticle hybrids with a number of advantages such as a higher capacitance, which depends on the nanoparticle structure, size, and crystallinity, while suppressing agglomeration and the restacking of graphene.61 Moreover, graphene–metal oxide nanoparticle composites result in enhanced electron conductivity, shortened ion paths, and a significant increase in the available surface area when compared to graphene alone, which altogether lead to higher electrochemical activity.11c For example, in 2012, Park et al. reported the one-pot synthesis of rGO–SnO2 nanoparticle composites, wherein the resulting composites exhibited outstanding cycling performance and could be used as an electrode.60b In their study, a hydrothermal synthesis assisted by hydrazine, which promoted the complete reduction of GO to rGO, was utilized. The resulting composites exhibited a first discharge capacity of 1662 mA h g−1, which rapidly stabilized and remained at 626 mA h g−1 even after 50 cycles when cycled at a current density of 100 mA g−1, whereas the capacity of pure SnO2 nanoparticles decreases continuously. This could be attributed to the lack of aggregation in the rGO-supported SnO2 composites and the uncontrolled aggregation of the pure SnO2 nanoparticles. On the other hand, Ren et al. reported the synthesis of graphene–magnetic nanoparticle (MNP) composites. In particular, these graphene–MNP composites exhibited excellent electrical conductivity and mechanical strength while possessing the magnetic properties of the attached MNPs.62 In this case, a one-step hydrothermal method was performed, wherein anhydrous FeCl3 provided the source of iron, and ethylene glycol (or a bisolvent of diethylene glycol and ethylene glycol) was used as the reductant and solvent. Consequently, Fe3O4 nanoparticles with a diameter of 7 nm were densely and uniformly deposited on the rGO sheets. Moreover, the reduction of GO by this process was comparable to that achieved by conventional methods. For example, the D/G intensity ratio of rGO in the aforementioned hybrids was 2.30:1, which is similar to the ratio typically obtained for pristine graphene (2.45:1), indicating the recovery of the sp2 domain in the carbon network.62
In addition, various chalcogenide QDs such as CdS,63 ZnS,64 Cu2S,65 MoS2,66 Sn3S4,67 and CdTe68 have been successfully immobilized on graphene utilizing hydrothermal methods. These semiconductor nanostructures have attracted intense interest due to their fundamental importance as well as their enormous potential in optoelectronic, magnetic, and catalytic applications.69 Specifically, for biological applications, QDs provide a high quantum yield (0.1–0.8 [visible], 0.2–0.7 [NIR] vs 0.5–1.0 [visible], 0.05–0.25 [NIR] for organic dyes),70 high molecular extinction coefficients (~10–100× that of organic dyes),71 broad absorption with narrow symmetric photoluminescence spectra (full-width at half-maximum ~25–40 nm) spanning from ultraviolet (UV) to NIR, high resistance to photobleaching, and exceptional resistance to photo- and chemical degradation.72 Moreover, because of their size-tunable fluorescence emission and the broad excitation spectra, QDs have a significant advantage over molecular dyes. However, these semiconductor nanoparticles are limited by their tendency to aggregate, resulting in a reduction in the surface area that is available for subsequent applications. In terms of the fabrication of rGO–sulfide nanocomposites, the sulfur sources often act as a reducing agent for GO. Zhang et al. recently reported the synthesis of graphene–CdS nanoparticle composites wherein a facile one-step hydrothermal approach was utilized to simultaneously form CdS nanoparticles and reduce GO (Figure 8).73 By combining these two excellent materials in a single composite, the graphene–CdS nanoparticle composites were able to serve as promising visible-light-driven photocatalysts, whose excellent photoactivity could be attributed to the integrative effect of the enhanced light absorption intensity, high electron conductivity of graphene, which significantly prolonged the lifetime of photogenerated electron–hole pairs, and its significant influence on the morphology and structure of the samples (e.g., the density and size of the nanoparticles could be controlled during synthesis).
Figure 8.
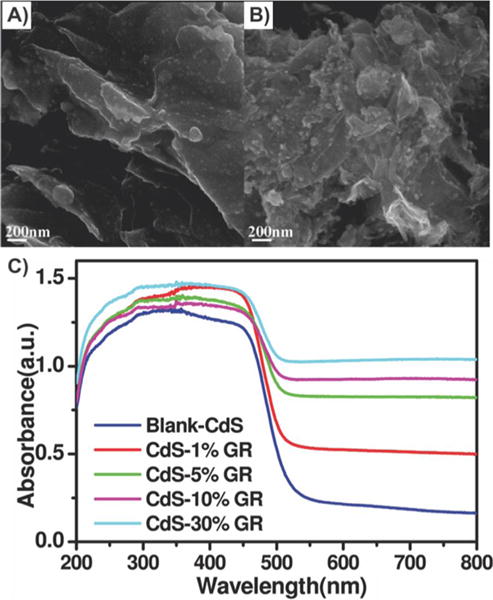
Graphene–quantum dot composites. SEM images of the as-prepared samples of (A) CdS–5% graphene, (B) CdS–30% graphene, and (C) UV–vis diffuse reflectance spectra of the samples of blank-CdS and Cd-GR nanocomposites with different weight addition ratios. Reprinted with permission from ref 73. Copyright 2011 American Chemical Society.
Finally, there have also been some reports utilizing hydrothermal methods for the preparation of rGO–noble metal nanoparticle composites (Figure 9).74 In the case of graphene–AuNPs, a solution of HAuCl4·3H2O and NaOH was mixed with GO.75 Subsequently, the solution was sonicated at a frequency of 40 kHz and heated to 180 °C. The resulting graphene–AuNP composites had AuNPs with a narrow size distribution in the range of 2 or 18 nm, with and without sonication, respectively. Graphene–Pt or –Pd nanoparticle composites can also be prepared by a similar method using H2PtCl6·6H2O and PdCl2, respectively.75 Moreover, graphene–bimetallic nanoparticle hybrid composites have been reported using the hydrothermal method. For instance, platinum–ruthenium nanoparticles with a mean size of 2.17 nm were decorated on graphene nanosheets, and it was found that the size and morphology of these nanoparticles could easily be controlled by modifying the synthesis temperature and the initial materials used.76
Figure 9.
Hydrothermal methods to synthesize graphene–AgNP composites. (A) (1) GO is adsorbed on the APTES-modified SiOx substrate. (2) GO is reduced, and rGO is obtained. (3) Growth of Ag particles by heating the rGO substrate in 0.1 M AgNO3 at 75 °C for 30 min. (4) Growth of AgNPs by heating the GO substrate in 0.1 M AgNO3 at 75 °C for 30 min. (B) Tapping mode AFM topographic image and height profile of a single layer of GO adsorbed on an APTES-modified SiOx substrate. (C) SEM image of Ag particles grown on a rGO surface. Reprinted with permission from ref 74b. Copyright 2009 American Chemical Society.
2.2.3. Electrochemical Methods To Form Graphene–Nanoparticle Composites
Electrochemical deposition is a simple, fast, and green technique that can be used to form graphene–nanoparticle composites while preventing the contamination of the synthesized materials. In addition, it is low cost, easy to miniaturize and automate, and is highly stable and reproducible.77 As such, by utilizing electrochemical deposition, the size and shape of the nanoparticles that are deposited can be precisely controlled by simply altering the conditions of electrochemical deposition. In particular, electrochemical deposition methods have been developed extensively for the fabrication of graphene–inorganic nanoparticle composites with the vast majority of composites formed using this method being noble metals such as Au,78 Ag,79 Pt,80 as well as bimetallic metals.
In a typical electrochemical deposition experiment, there are three steps wherein (1) the graphene sheets are first assembled onto an electrode, (2) the graphene-coated electrode is then immersed in an electrolytic solution containing metallic precursors, and (3) a potential is applied. For the formation of graphene–AgNP composites, Golsheikh and co-workers recently reported an electrochemical deposition method wherein the resulting nanoparticles fell within a very narrow size distribution with a mean size of 20 nm.79 In this case, a solution containing silver–ammonia [Ag(NH3)2OH] and GO was exposed to cyclic voltammetry (CV), which was performed using a three-electrode system that consisted of an indium tin oxide (ITO) working electrode, a platinum foil counter electrode, and a saturated calomel electrode (SCE) reference electrode (scanning between −1.5 and 0 V at a rate of 25 mV s−1). Fisher et al. also utilized electrochemical tools to fabricate graphene–PtNP composites wherein PtNPs were decorated on multilayered graphene petal nanosheets (MGPNs).80b According to their report, a three-electrode system, wherein the MGPNs acted as the working electrode, Pt gauze as the auxiliary electrode, and Ag/AgCl as the reference electrode, was dipped in a solution containing H2PtCl6·6H2O and Na2SO4. Utilizing this method, the density, size, and morphology of the PtNPs could be precisely controlled by simply adjusting the intensity of the pulse current. In particular, this allowed for the simultaneous reduction of GO and H2PtCl6 to rGO and PtNPs, respectively, without the need for any other reagents or thermal treatment.
As for graphene–nanoparticle composites composed of non-noble metal nanoparticles, Wu et al. recently deposited Cu nanoparticles on rGO and, more interestingly, investigated the mechanism underlying their electrochemical deposition (Figure 10).81 In their study, the nucleation of Cu on rGO was achieved using an electrolyte solution containing CuSO4 in a conventional three-electrode system, wherein the rGO electrode, a Pt mesh, and an Ag/AgCl electrode were used as the working, counter, and reference electrodes, respectively (Figure 10A). To fundamentally study the nucleation of Cu on rGO via electrochemical deposition, the authors used CV, Tafel plots, and chronoamperometry. From CV, it was inferred that Cu deposition on rGO electrodes initiated at a more positive potential of 0.105 V (vs Ag/AgCl) as compared to that found for glassy carbon and pencil graphite. On the other hand, the Tafel plot confirmed that the rate-determining step for Cu deposition on rGO was mass transport and, finally, that nucleation on rGO occurred either instantaneously or progressively depending on the initial concentration of the electrolyte (e.g., instantaneously at higher concentrations [50 mM] and progressively at lower concentrations [10 mM]).
Figure 10.
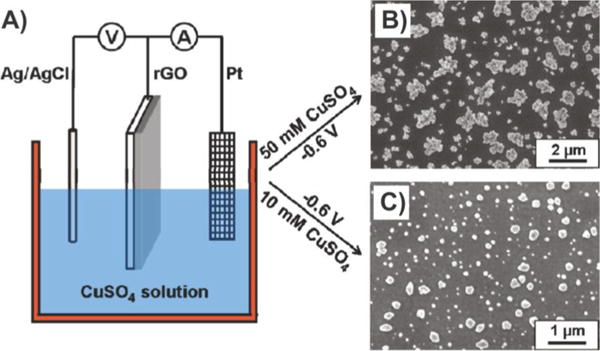
Electrochemical deposition of Cu nanoparticles on rGO. (A) Electrochemical experiments were performed by an electrochemical workstation (CHI600C, CH Instrument Inc., U.S.) in a conventional three-electrode electrochemical cell. The rGO electrode, a Pt mesh, and an Ag/AgCl (3 M NaCl) electrode were used as the working, counter, and reference electrodes, respectively. (B) SEM image of Cu electrodeposited on a rGO electrode in 50 mM CuSO4 solution at −0.6 V. (C) SEM image of Cu electrodeposited on a rGO electrode in 10 mM CuSO4 solution at −0.6 V. Reprinted with permission from ref 81. Copyright 2011 American Chemical Society.
Last, although the vast majority of research efforts has concentrated on the electrochemical deposition of metal nanoparticles on graphene sheets, there have also been several reports of the electrochemical deposition of metal oxide nanoparticles onto graphene. For instance, Wu et al. deposited Cl-doped n-type Cu2O nanoparticles, which are abundant and nontoxic nanoparticles that have a direct band gap of ca. 2.0 eV,82 on rGO electrodes resulting in a carrier concentration of up to 1 × 1020 cm−3.83 Specifically, to deposit Cl–Cu2O nanoparticles on rGO, a solution of CuSO4, CuCl2, and lactic acid was added to a three-electrode system where the rGO electrode, a Pt mesh, and SCE were used as the working, counter, and reference electrodes, respectively. The deposition used a potentiostatic process (potential of −0.4 V, charge density of 2 C cm−2) at a temperature of 60 °C. Similarly, ZnO nanorods could also be deposited on rGO films using a solution containing ZnCl2 and KCl as the supporting electrolyte in a conventional three-electrode system where an rGO-polyethylene terephthalate electrode, a Pt mesh, and a SCE were used as the working, counter, and reference electrodes, respectively.84
2.2.4. Ex Situ Methods To Form Graphene–Nanoparticle Composites
Graphene–nanoparticle composites can also be produced by the ex situ assembly of nanoparticles onto the graphene surface. In this method, the nanoparticles are synthesized in advance and then later attached to the surface of the graphene sheets via linking agents that can utilize either covalent or noncovalent interactions including van der Waals interactions, hydrogen bonding, π–π stacking, or electrostatic interactions. Although this method requires more time and steps to complete, it can offer a number of advantages when compared to in situ growth. For instance, ex situ methods result in a significantly narrower size distribution as well as better control over the size, shape, and density of the nanoparticles that decorate the graphene sheets while utilizing self-assembly.85
For the covalent attachment of nanoparticles, GO rather than rGO is preferred due to the large amount of oxygen-containing groups on its surface, which can facilitate linkage with other functional groups. A variety of nanoparticles have been attached to graphene using this method. Fan et al. covalently attached MNPs to GO by first modifying the Fe3O4 nanoparticles with tetraethyl orthosilicate and (3-aminopropyl)triethoxysilane (APTES), thereby introducing amino groups on its surface.86 Next, these amino groups were reacted with the carboxylic groups on the surface of GO with the aid of 1-ethyl-3-(3-(dimethylamino)propyl)carbodiimide (EDC) and N-hydroxysuccinimide (NHS), resulting in the formation of GO-Fe3O4 nanoparticle composites. Moreover, they demonstrated that the GO could subsequently be reduced to rGO using NaBH4 as the reducing agent. Similarly, cadmium sulfide (CdS) QDs have been immobilized covalently on GO nanosheets.87 In this case, amino-functionalized CdS QDs were first prepared by modification of the kinetic trapping method.88 Next, GO nanosheets were aceylated with thionyl chloride to introduce acyl chloride groups on its surface. To covalently bond the QDs to the GO nanosheets, an amidation reaction between the amino groups on the QDs and the acyl chloride groups on the GO surface was performed. Finally, noble metal nanoparticles such as AuNPs have also been covalently attached to graphene. Specifically, Ismaili and co-workers demonstrated the light-activated covalent formation of AuNPs on rGO.89 In this study, 4 nm AuNPs were modified with a 3-aryl-3-(trifluoromethyl)-diazirine functionality. Correspondingly, upon irradiation with wavelengths above 300 nm and in the presence of rGO, the terminal diazirine group lost nitrogen to generate a reactive carbene that could then undergo addition or insertion reactions with the functional groups on graphene leading to covalent linkage.
Alternatively, nanoparticles can be attached to graphene sheets via noncovalent bonds including van der Waals interactions, hydrogen bonding, π–π stacking, and electrostatic interactions. Among these noncovalent bonds, π–π stacking and electrostatic interactions have been the most widely used. For π–π stacking, generally, aromatic compounds are attached to the nanoparticle surface, which enables their attachment to graphene via π–π stacking. For example, derivatives of the pyrene molecule as well as pyrene-functionalized block copolymers have provided an effective way for the noncovalent functionalization of carbon nanomaterials including graphene.90 In particular, pyrene groups have the ability to interact strongly with the basal plane of graphene via π–π stacking. For example, 1-pyrenebutyric acid (PBA) is one of the simplest pyrene-containing molecules that are attached to a carboxyl group. Resultantly, graphene sheets functionalized with PBA become negatively charged allowing for the attachment of positively charged nanoparticles via electrostatic interaction.91 The use of pyrene-containing molecules has been reported for various nanoparticles. Examples include PBA, which was used to form graphene–AuNP hybrids,29c and pyrene-grafted poly(acrylic acid), which was used to form graphene–CdSe nanoparticle hybrids.92 Pyridine is another aromatic structure that has also seen significant use in anchoring nanoparticles such as Au44b,93 and CdSe94 nanoparticles to the basal planes of GO/rGO sheets via π–π stacking. Importantly, pyrene- or pyridine-modified graphene sheets have a high loading capacity for nanoparticles, and the amount of nanoparticle that assembles on the graphene sheets can easily be modulated by controlling the feeding weight ratio of both components.29c
DNA molecules, which contain both purine and pyrimidine bases, have also been used to mediate the fabrication of graphene–nanoparticle composites. In particular, DNA is able to interact with graphene via π–π stacking interactions as well as the surface binding model wherein DNA electrostatically interacts with graphene basal planes, which is similar to what is observed between single-stranded DNA (ssDNA) and CNTs.95 For instance, Liu et al. developed a strategy wherein thiolated DNA oligos (d(GT)29SH) were first adsorbed onto GO sheets (DNA–GO) and then reduced by hydrazine to obtain DNA–rGO sheets.96 Consequently, the addition of a large excess of 6 nm AuNPs to a solution containing either DNA–GO or DNA–rGO resulted in the formation of GO–AuNP and rGO–AuNP composites, respectively. Similarly, Wang et al. fabricated GO–AuNP and GO–AgNP composites by first functionalizing AuNPs or AgNPs with DNA via didentate capping ligands and then assembling them onto GO via π–π stacking interactions.97
As mentioned previously, electrostatic interactions are also commonly used to modify graphene with various nanoparticles as it provides a facile and scalable method to form composite structures in a controlled manner while avoiding conglomeration. GO and rGO have an inherent negative charge as a result of the ionization of the oxygen functional groups on their surface. As such, they can be decorated with positively charged inorganic nanoparticles through electrostatic interactions. For instance, graphene–metal oxide nanoparticle composites (e.g., rGO–Fe3O4 nanoparticle composites98 and GO–MnO2 nanoparticle composites99) have been formed by mixing positively charged metal oxide nanoparticles with negatively charged graphene nanosheets.98 Similarly, graphene–noble metal and other inorganic nanoparticles have also been prepared in this way. To this end, Lu et al. noncovalently decorated GO sheets with positively charged aerosol Ag nanocrystals that were synthesized from an arc plasma source using an electrostatic force-directed assembly technique.100 Moreover, reports have decorated graphene with APTES-modified Si nanoparticles.101 Specifically, while the amine functional groups of APTES can function to reduce GO and form covalent bonds with rGO, APTES can also help disperse rGO and Si nanoparticles due to polar–polar interactions.
Finally, Deng and co-workers utilized a novel nontoxic synthetic method wherein bovine serum albumin (BSA) was utilized for the fabrication of graphene–nanoparticle composites that could be composed of various types of nanoparticles (Au, Pt, Pd, Ag, and polystyrene beads).102 In this study, the use of BSA not only effectively reduced GO to rGO, but also acted as a stabilizer to induce the attachment of nanoparticles onto the graphene surface (Figure 11A). Specifically, BSA–GO/rGO conjugates were first obtained via the adsorption of BSA onto the basal planes of GO/rGO. Afterward, the nanoparticles were mixed in a solution containing the BSA–GO/rGO conjugates overnight to form the final graphene–nanoparticle composites. Moreover, the density of nanoparticles on the graphene–nanoparticle composites could be controlled by simply changing the concentration of BSA and NaCl during assembly (Figure 11B–D). Similarly, Wang et al. recently reported an inexpensive and unique green synthetic method for the production of Ag–GO nanocomposites that utilized glucose as both the reducing and the stabilizing agent, eliminating the need for toxic reduction agents.103 In particular, Wang and co-workers demonstrated that AgNPs could be directly reduced from silver ions on GO in a glucose solution.
Figure 11.
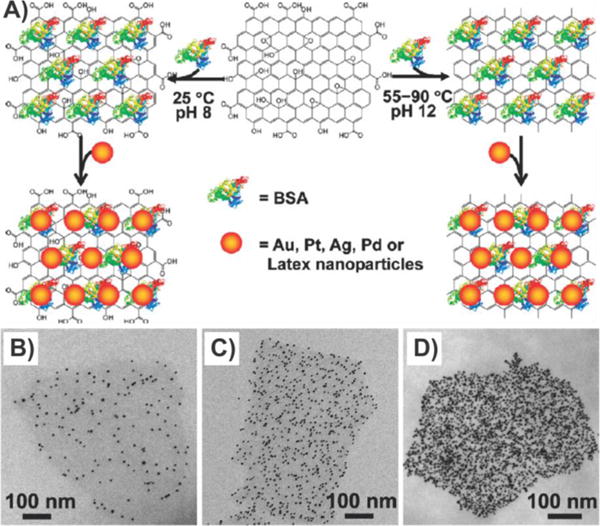
Protein-induced reduction and decoration of GO for the assembly of multiple nanoparticles. (A) General scheme depicting the BSA protein-based decoration and reduction of GO, leading to a general nanoplatform for nanoparticle assembly. (B) TEM images of AuNP decorated BSA–GO with well-controlled densities of AuNPs. (B and C) AuNP densities were varied by increasing the concentrations of BSA from 0.5 mg/mL (B) to 20 mg/mL (C), during the preparation of BSA–GO. NaCl was omitted for the samples in (B) and (C). (D) AuNP density was further increased (in comparison with (C)) by adding 0.1 M NaCl to the assembly system as in (C). Reprinted with permission from ref 102. Copyright 2010 American Chemical Society.
2.3. Graphene-Encapsulated Nanoparticles
Because of the flexible and 2D sheet-like nature of graphene and its derivatives, these sheets can easily be used to wrap or encapsulate nanoparticles that range in diameter from 100 nm to several hundreds of nanometers and even micrometers (Figure 12). rGO sheets are the most frequently utilized carbon material for the encapsulation of nanoparticles due to their slightly hydrophilic nature and the ease with which small fractions of rGO can be fabricated. In particular, methods used to fabricate graphene-encapsulated nanoparticles typically utilize noncovalent bonds. For instance, the most frequently used method to encapsulate nanoparticles with rGO consists of endowing the surface of the nanoparticle with a positive charge (e.g., by coating with APTES), resulting in the strong attachment of rGO via electrostatic interaction.101b,104 By controlling the size of cracked rGO, a variety of nanomaterials such as polymer as well as inorganic, metals, and metal oxide nanoparticles can be encapsulated by graphene/rGO to enhance their properties as well as to obtain additional advantages.
Figure 12.
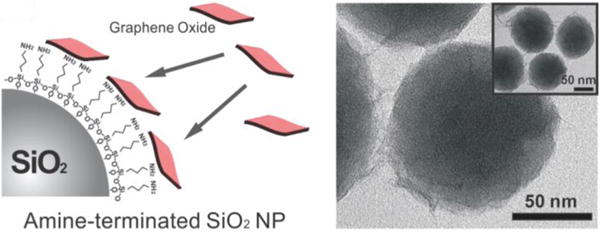
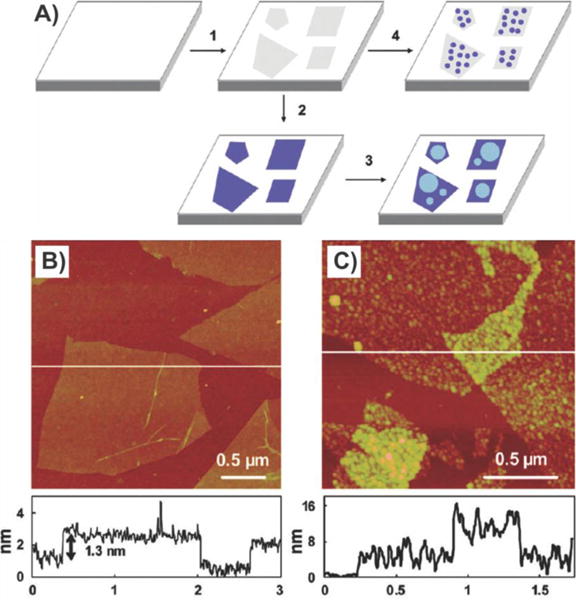
Fabrication process for graphene-coated NPs. Schematic diagram of GO assembly on amine-functionalized NPs and TEM image of NPs coated with GO (inset: zoomed-out TEM image of NPs coated with GO). Reprinted with permission from ref 101b. Copyright 2011 Wiley.
In terms of its benefits, the encapsulation of nanoparticles with graphene endows similar enhancements in electrical, electrochemical, and optical properties that were observed for graphene–nanoparticle composites. However, due to the characteristically strong negative charge of rGO, the encapsulation of small nanoparticles with rGO also results in the suppression of aggregation, which is a major issue in many nanoparticle-based bioapplications.96,105 Moreover, because of the high degree of contact that exists between graphene and the encapsulated nanoparticle, which is significantly greater than that seen in graphene–nanoparticle composites, graphene-encapsulated nanoparticles are very stable, thereby limiting the degree of exfoliation of the nanoparticles from graphene or vice versa.11c
Numerous reports have demonstrated the encapsulation of metal oxide nanoparticles with graphene. For example, Yang et al. reported rGO-encapsulated cobalt oxide nanoparticles (Co3O4). These rGO-encapsulated Co3O4 nanoparticles exhibited a very high reversible capacity (1000 mA h g−1) over 130 cycles, which was superior to normal cobalt oxide nanoparticles used for capacitors.104 In particular, using alternating current impedance measurements (30 cycles), Nyquist plots were obtained wherein the diameter of the semicircle for rGO-encapsulated Co3O4 electrodes in the high–medium frequency region was much smaller than that of bare Co3O4 electrodes. This suggested that the rGO-encapsulated Co3O4 electrodes possessed lower contact and charge-transfer impedances. Feng and co-workers also reported graphene-encapsulated TiO2 nanospheres for efficient photocatalysis due to their high specific surface area (133 m2 g−1). Specifically, the resulting hybrid material was much more efficient at decomposing rhodamine B (91%) than normal TiO2 (65%) due to the presence of graphene, which was beneficial for the separation of photogenerated electrons and holes.106 Similarly, the performance of tin oxide (SnO2) nanoparticles was also improved by encapsulation of individual aggregates with graphene, resulting in excellent performance including a charge capacity of 700 mA h g−1 at the current density of 0.1 A g−1 and 423 mA h g−1 after a 10-fold increase in the current density to 1 A g−1 in the 0.005–2 V voltage window.107 Finally, Lin and coworkers recently created rGO-encapsulated amine-functionalized Fe3O4 MNPs that were used to support Pt catalysts.108 Specifically, after the Fe3O4 nanoparticles were functionalized with APTES and coated with rGO, PtNPs were uniformly anchored by a polyol reduction reaction and the GO was simultaneously reduced to rGO. Resultantly, the electrochemical activity of the catalyst for methanol oxidation was significantly improved. The authors claimed that this was due to the accessibility of the PtNPs on the graphene surface and the greatly enhanced electronic conductivity of the underlying rGO-encapsulated Fe3O4 nanoparticles.
Others have focused on encapsulating metal nanoparticles with graphene. For instance, Zhang et al. recently reported an interesting material, a “graphene-veiled gold nanostructure”. In this study, they used graphene as a passivation nanosheet to prevent metal–molecule chemical interactions and to control the spatial resolution of molecules to achieve sensitive SERS signals from analytes of interest.16 Kawasaki and co-workers have also reported graphene-encapsulated cobalt nanomagnets, wherein the cobalt nanoparticles were first functionalized with benzylamine groups.109 These 30 nm graphene-encapsulated nanoparticles had a high specific surface area of 15 m2 g−1 and a high strength saturation magnetization of 158 emu g−1, which led to efficient extraction of analytes by magnetic separation for surface-assisted laser desorption/ionization mass spectrometry (affinity SALDI-MS) analysis.
In addition to the above examples, our group recently reported a method to convert nonconducting silicon oxide nanoparticles into conducting rGO-encapsulated nanoparticles, which could then be used as the “bridging-material” in a field-effect transistor (FET)-based biosensor.101b,110 Specifically, SiO2 nanoparticles were functionalized with APTES thereby imparting them with a positive surface charge, which allowed for encapsulation with rGO via electrostatic interaction (Figure 12). In doing so, we were able to prevent aggregation while maintaining a high electrical conductivity and enhanced surface area for the detection of cancer markers. Similarly, Zhou et al. also encapsulated Si nanoparticles with rGO via electrostatic interaction, again using APTES.111 As a consequence of encapsulation, the Si nanoparticles exhibited less aggregation and destruction than pristine SiNPs and acted as an outstanding electrode, exhibiting high reversible capacity (902 mA h g−1 after 100 cycles at 300 mA g−1). On the other hand, pristine SiNPs exhibited an initial discharge capacity of 3220 mA h g−1, which dropped to 13 mA h g−1 after only 50 cycles.
Finally, while electrostatic interaction has been the most commonly used method to form graphene-encapsulated nanoparticles, Luo et al. reported an innovative method to fabricate graphene-encapsulated nanoparticles via a facile and scalable, capillary-driven aerosol droplet method. Specifically, in a typical experiment, Si nanoparticles in an aqueous suspension were directly added to a dispersion of micrometer-sized GO sheets. Nebulization of the colloidal mixture resulted in the formation of aerosol droplets, which were blown through a preheated tube furnace at 600 °C with a N2 carrier gas (Figure 13A). As a result, during the process of evaporation, the amphiphilic GO sheets migrated to the surface of the droplets to form a shell, and as the droplets evaporated further, the GO shell collapsed forming a “crumpled” shell around the Si nanoparticles (Figure 13B).112
Figure 13.
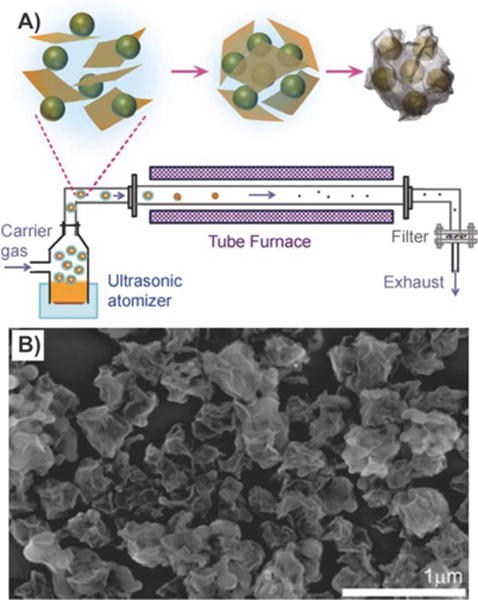
Crumpled graphene-encapsulated Si nanoparticles. (A) Schematic drawing illustrating aerosol-assisted capillary assembly of crumpled-graphene-wrapped Si nanoparticles. Aqueous dispersion of GO and Si particles was nebulized to create a mist of aerosol droplets, which were passed through a preheated tube furnace. During evaporation, GO sheets first migrated to the surface of the droplets and then tightly wrapped the Si particles upon complete evaporation. (B) SEM image showing the crumpled capsules of graphene-wrapped Si. Reprinted with permission from ref 112. Copyright 2012 American Chemical Society.
3. GRAPHENE–NANOPARTICLE HYBRID MATERIALS FOR BIOSENSING APPLICATIONS
Biosensors are analytical devices that utilize biological sensing elements to detect and/or quantify a particular target analyte or family of analytes. As such, biosensors are applicable to and are important for virtually every conceivable analytical task in the biomedical field, which can range from applications in medical diagnostics to drug discovery, food safety, environmental monitoring, and defense.
In general, biosensors are composed of two fundamental elements: a receptor and a transducer. The receptor consists of any material, either organic or inorganic, that can interact with a target analyte or family of analytes. On the other hand, the transducer converts the recognition event that occurs between the analyte and the receptor (e.g., the binding of an enzyme to its substrate, binding between an antibody and its target protein, or reduction/oxidation of an electroactive biomolecule by the sensing electrode) into a measurable signal that can come in many forms including, but not limited to, electronic, electrochemical, and optical signals. In terms of its performance, biosensors are evaluated on the basis of sensitivity to the target(s), limit of detection (LOD), linear and dynamic ranges, reproducibility or precision of its response, and selectivity.113 Other parameters that are often compared and useful include the sensor’s response time (e.g., the amount of time needed for the sensor response to reach 95% of its final value after introduction of the analyte(s)), operational and storage stability, ease of use, and portability. Moreover, the ideal biosensor should be reusable, thereby allowing for several consecutive measurements to be made.
Graphene–nanoparticle hybrids are particularly well-suited for biosensing applications. As mentioned previously, graphene possesses numerous unique and advantageous physicochemical properties including an extremely high surface area, excellent electrical properties, high mechanical strength, advantageous optical properties (e.g., transparent and can quench fluorescence), and is relatively easy to functionalize and mass produce. As such, there has been significant effort invested in utilizing this material for the development of biosensors.9a,114 Moreover, nanoparticles have also been widely investigated in the field of biosensing due to the exquisite sensitivity that nanomaterials can offer for this type of application.115 Specifically, because of the diameter of nanoparticles (e.g., 1–100 nm scale), these nanomaterials can display unique physical and chemical features (e.g., quantum size effect, surface effect, and macro-quantum tunnel effect). As such, nanoparticles can be used to enhance achievable sensitivities by amplifying the obtained signal as well as increasing the available surface area for analyte binding.
By combining these two excellent and unique modalities as graphene–nanoparticle hybrids, a number of advantageous properties are attained for biosensing applications. In particular, it has been observed that graphene acts as an excellent material with which to immobilize nanoparticles and enhance their stability (e.g., preventing aggregation). Moreover, the combination of graphene with nanoparticles can increase the available surface area for analyte binding as well as improve their electrical conductivity and electron mobility, thereby enhancing the achievable sensitivity and selectivity.11a In particular, the field of graphene–nanoparticle hybrid materials for biosensing applications can be generally divided into three classes based on the underlying mechanism of detection. These classes include (1) electronic, (2) electrochemical, and (3) optical sensors, with each class having its own advantages and disadvantages. As such, in this section, we will give a comprehensive review of recent work that has been conducted on the development of graphene–nanoparticle hybrid biosensors. In particular, we will focus primarily on the use of graphene–nanoparticle hybrid materials in electronic (e.g., FET), electrochemical, and optical biosensors, with emphasis on how they compare to current gold standards and their achievable sensitivities and selectivities for various biomolecules (Table 2).
Table 2.
Summary of the Different Detection Mechanisms Discussed
| type | common NPs | LOD | advantages | key refs |
|---|---|---|---|---|
| FET | Au, Pt, SiO2 | aM–pM | (1) enhanced surface area for detection; (2) can preserve the electrical properties of graphene by conjugating the probe on the nanoparticle | 124, 127, 128, 130 |
| electrochemical | Au, Pt, Cu | pM–μM | (1) immobilization of biomolecules; (2) catalyze electrochemical reactions; (3) act as a reactant | 137, 140, 143–146, 148–152 |
| FRET | QD, UCNP, Au, Ag | pM–nM | (1) GO is superquenching, which can result in double-quenching (e.g., with Au or Ag) | 156, 162, 163, 165 |
| SERS | Au | nM | (1) dual enhancement of Raman signals via chemical and electromagnetic enhancement | 174–179, 181 |
3.1. Electronic Sensors
As compared to the other methods that are available for biosensing applications, nanomaterial-based electronic biosensing offers significant advantages, such as high achievable sensitivities, high spatial resolution for localized detection, easy miniaturization, facile integration with standard semiconductor processing, and label-free, real-time detection that can be achieved in a nondestructive manner.116 In particular, these electronic sensors primarily utilize the principle of FETs to convert the biological recognition event to a measurable electronic signal. In a standard FET device, current flows along a semiconductor path (the channel) that connects two electrodes (the source and the drain). The conductance of the channel between the source and the drain is then switched on and off by a third electrode (the gate) that is capacitively coupled to the device through a thin dielectric layer.116,117 Specifically, in FET-based biosensors, the channel is in direct contact with the sensing sample, which enhances the achievable sensitivity as any single biological event that occurs at the channel surface could result in a variation in the surface potential thereby modulating the channel conductance.118
Currently, FET sensors that are composed of Si nanowires or carbon nanotubes (CNTs) are the most heavily investigated.118,119 FET sensors that utilize either of these materials exhibit exceptional performance with their achievable LOD falling in the range of picomolar (pM) to femtomolar (fM); however, the achievable sensitivity of devices that use these materials is limited by the rarity of binding events that occur between the probe and its target molecule due to the scarcity of available binding sites on the surface of the materials.116 Moreover, the use of Si nanowires is expensive. On the other hand, while CNT sensors represent a significantly cheaper option, the reproducibility of CNT-based devices in terms of their fabrication and electrical properties is considered a significant limiting factor.120 As such, graphene-based materials have a major advantage in FET sensing applications in that graphene has an extremely high surface-to-volume ratio, which increases the likelihood of binding events. In particular, graphene-based FET biosensors are able to compete with CNT and Si nanowire-based FET sensors with an ultrasensitive LOD down to a similar (pM to fM) and potentially lower range.121 More specifically, because of this high surface-to-volume ratio, any analytes that adsorb onto the graphene surface could potentially alter its electronic properties (e.g., the conductivity can be altered when an analyte is adsorbed due to doping or a change in the carrier mobility of graphene). In addition, by utilizing graphene–nanoparticle hybrid materials it is possible to further push this limit to the attomolar (aM) range for biomolecule detection by utilizing the synergism that occurs in these unique structures, wherein the combination of two materials results in additional surface area for analyte binding as well as signal amplification and enhanced electrical conductivity. For instance, Zhang et al. determined that the covalent linkage of AuNPs to GO could enhance the electronic conductivity when compared to GO alone.122 Similarly, Dinh and colleagues reported that the formation of rGO–AgNPs decreased the sheet resistance from 10.93 kΩ sq−1 (for rGO) to 270 Ω sq−1.123
Demonstrations utilizing graphene–nanoparticle hybrid-based FET biosensors have focused on exploiting variations of a single mechanism. Specifically, studies have shown that by conjugating the detection probe (e.g., antibody) to the nanoparticle and then using these nanoparticle–probe conjugates to form graphene–nanoparticle composites, one can preserve the superb electrical properties of graphene. In these cases, the formation of the graphene–nanoparticle hybrid generally occurs via electrostatic interaction and van der Waals binding. As such, as long as detection probe conjugation does not significantly affect the charge of the nanoparticles, graphene–nanoparticle composites can be formed without any steric hindrance. One popular FET-based biosensing application that has utilized this concept is for the detection of proteins. Protein detection is particularly important as proteins play an essential role in all biological functions. As such, they are at the center of almost all pathological conditions, and the majority of disease markers are composed of proteins. Chen et al. reported the first graphene–AuNP hybrid sensor for the detection of proteins.124 In this case, thermally reduced GO sheets (TRGO) (e.g., a few layers with a thickness of 3–6 nm) were decorated with 20 nm AuNPs, which were covalently conjugated to anti-immunoglobulin G (IgG) antibodies (Figure 14A). Upon introduction of the target protein (e.g., IgG), FET and direct current was measured resulting in a LOD of approximately 13 pM (Figure 14B), which is among the best LODs when compared to carbon nanomaterial-based protein sensors including CNTs,125 graphene, and GO.126 This sensor also showed excellent selectivity when exposed to samples containing mismatched protein such as immunoglobulin M (IgM) or horseradish peroxidase (HRP). In particular, when 0.8 mg mL−1 IgM and 0.2 mg mL−1 HRP were introduced to the sensor using the exact procedure as that used for IgG, the sensor response was 15.3% and 12.4%, respectively, which was significantly lower than that from the complementary IgG (68.0%). Last, it was observed that binding of the IgGs to their anti-IgGs resulted in local geometric deformations and an increase in the number of scattering centers across the sheet, thereby reducing the mobility of holes and, subsequently, the conductivity of the TRGO sheets.
Figure 14.
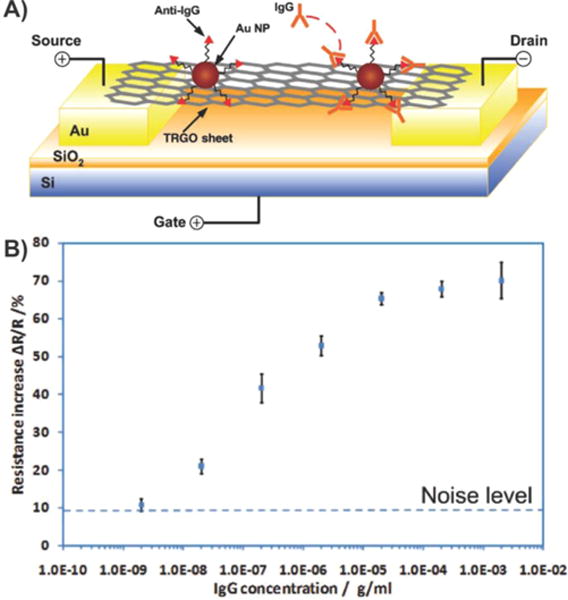
Protein detection using thermally reduced graphene oxide (TRGO) sheets decorated with gold nanoparticle–antibody conjugates. (A) Schematic of a TRGO FET. Anti-IgG is anchored to the TRGO sheet surface through AuNPs and functions as a specific recognition group for IgG binding. The electrical detection of protein binding is accomplished by FET and direct current measurements. (B) Sensor sensitivity (relative resistance change, %) versus IgG concentration. Dashed line represents the noise level from the buffer solution. Reprinted with permission from ref 124. Copyright 2010 Wiley.
Besides preserving the excellent electrical properties of graphene, graphene–nanoparticle composites also exhibit additional advantages such as increasing available surface area for the binding of target analyte, enhanced stability, and also amplified transduction signals.127 For example, Kwon et al. reported a novel liquid-ion gated FET using large-scale graphene micropattern nanohybrids decorated with closely packed conducting polymer nanoparticles for the detection of HIV.127a Specifically, this closely packed nanoparticle array was composed of 20 nm carboxylated polypyrrole nanoparticles that were covalently modified with HIV-2 gp36 antigen and provided an enlarged surface area and stable sensing geometry. Therefore, the authors could detect the HIV biomarker at concentrations as low as 1 pM, which is better than any biosensor that has been reported for this particular purpose. Moreover, this biosensor exhibited excellent mechanical flexibility and durability. On the other hand, Kim and coworkers demonstrated that, in addition to preserving the superb electrical properties of graphene and increasing available surface area, graphene–nanoparticle hybrids could also be designed to amplify the transduction signal, thereby further increasing the achievable LOD by a full order of magnitude.127b In this work, the authors fabricated a FET biosensor that had networked channels of chemically reduced GO nanosheets, which were modified with aptamers specific for the detection of anthrax toxin (e.g., protective antigen) (Figure 15A). Briefly, in their design, the source/drain electrodes were formed on a networked film composed of rGO nanosheets using a shadow mask to prevent the deposition of polymeric residues during photolithography. Next, passivation of the electrodes was achieved with a 200 nm thick Al2O3 layer and direct pasting with PDMS, which minimized local work function modulation and isolated the leakage current between the electrode and electrolytes. In this way, Kim et al. achieved an ultralow LOD of 12 aM in 10 μM PBS. Furthermore, by utilizing secondary aptamer-conjugated AuNPs, they were able to achieve an even lower LOD of 1.2 aM (Figure 15B). This was attributed to the ability of the secondary aptamer-conjugated AuNPs to further amplify the transduction signal. As for the achievable selectivity, the authors exposed their sensor to a PBS solution containing carcinoembryonic antigen (CEA). The results showed no shift in voltage and no change in current, indicating that no CEA binding occurred.
Figure 15.
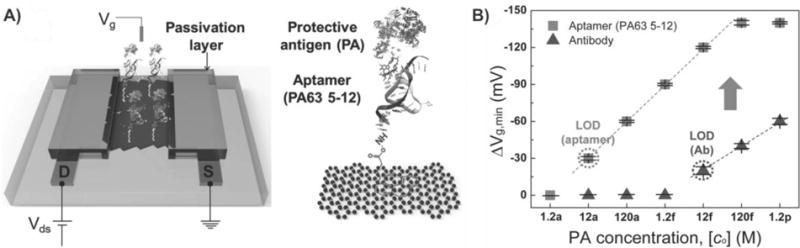
Electronic graphene–nanoparticle composite sensor utilizing aptamers to detect anthrax toxin. (A) Schematic illustration of the aptamer-immobilized graphene FET for detection of protective antigen (PA). (B) Vg,min shift (ΔVg,min) versus PA concentration in PBS solutions with different probe molecules. The ΔVg,min value was obtained by calculating the difference in the charge neutrality point, Vg,min, as a reference for the device with no binding of PA. Reprinted with permission from ref 127b. Copyright 2013 Wiley.
As a variation of the above-mentioned mechanism, encapsulating nanoparticles with graphene can also enhance the surface-to-volume ratio that is available for the capture of target analyte in FET sensors while enhancing stability. Recently, our group developed an rGO encapsulated nanoparticle-based FET sensor for the sensitive and selective detection of proteins (Figure 16).101b In particular, we sought to detect human epidermal growth factor receptor 2 (HER2) and epidermal growth factor receptor (EGFR), which are both known to be overexpressed in breast cancers.128 To this end, individual silicon oxide nanoparticles (100 nm diameter) functionalized with APTES were coated with a thin layer of rGO (5 nm thick) due to the electrostatic interaction that could occur between the negatively charged GO sheets and the positively charged silicon oxide nanoparticles. Arrays of rGO nanoparticles (rGO-NP) were then patterned to form channels between gold electrodes, which occurred through a self-assembly process upon centrifugation of the device with a solution containing rGO–NPs (Figure 16A). Finally, the rGO–NPs were functionalized with monoclonal antibodies against HER2 or EGFR (Figure 16B). Specifically, this was accomplished using a well-established process where the rGO surface was functionalized with 4-(pyren-1-yl)butanal via π–π stacking. Next, the aldehyde groups were coupled to the amine groups of the HER2 or EGFR antibodies through reductive amination and unreacted aldehyde groups were blocked using ethanolamine. In this way, we were able to preserve the electrical properties of the rGO by not conjugating the antibodies directly to the rGO surface while increasing the available surface area available for detection over rGO alone. Using this device, we were able to achieve a detection limit as low as 1 pM for HER2 and 100 pM for EGFR (Figure 16D–F). In addition, we demonstrated the highly selective nature of our biosensor in the presence of other proteins such as BSA (50 μg mL−1), which did not induce a change in conductance.
Figure 16.
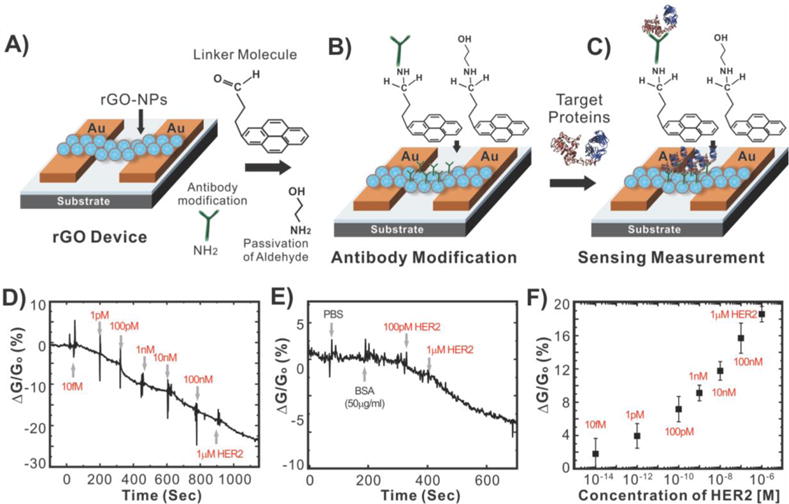
Real-time detection of cancer marker, HER2, using a graphene-encapsulated nanoparticle-based FET biosensor. (A) The preparation of the rGO–NP device. (B) Surface functionalization of rGO for immobilizing the antibody. (C) Measuring conductance of the devices when the target protein is introduced. (D) The sensitivity of the biosensor (relative conductance change, %) in response to the concentration of HER2 with VDS (voltage drain to source) = 1 V and Vg (gate voltage) = 0 V. (E) The selectivity of the biosensor in response to PBS buffer, BSA with 50 μg mL−1, and HER2 (100 pM and 1 μM). (F) Sensor sensitivity (relative conductance change, %) as a function of the HER2 concentration with VDS = 1 V and Vg = 0 V. All experiments were performed multiple times (sample number, n = 30) to collect statistical data (with error bars) and confirm the reproducibility and robustness of the biosensing system. Reprinted with permission from ref 101b. Copyright 2011 Wiley.
Finally, besides proteins, the detection of specific nucleic acids (e.g., DNA or RNA) has garnered significant attention as it can be utilized for various bioapplications including, but not limited to, pathogen identification, the recognition of genetic mutations, and forensic analysis.129 For instance, Yin and coworkers reported a PtNP-decorated rGO FET where a thiolated DNA probe was attached to the PtNPs via Pt–S bonding.130 Specifically, a large, continuous, few-layer thick film of GO was fabricated via the Langmuir–Blodgett method and subsequently reduced with high temperature annealing in an Ar/H2 atmosphere at 1000 °C. To form graphene–nanoparticle composites, the PtNPs were directly synthesized on the rGO film by immersion of the rGO in an ethanolic solution of PtCl4 followed by light irradiation. It was found that this graphene–nanoparticle composite-based FET, when modified with probe DNA, was able to detect the real-time hybridization of target DNA in PBS with a calculated detection limit of 2.4 nM. Moreover, the sensor exhibited good selectivity. For instance, when 1 μM of noncomplementary DNA was added to the sensing chamber, there was no obvious change in the conductance.
Overall, the performance of hybrid electronic sensors depends strongly on graphene morphology (e.g., wrinkles, folds, number of layers), the number of graphene sheets, the level of graphene reduction, and the interface that exists between the nanoparticles and graphene, which can all influence the electrical properties of the device. In particular, the morphology (e.g., wrinkles, folds, number of layers) of graphene should be free of defects and, as such, hinges on the synthesis method used. On the other hand, the nanoparticle–graphene interface is highly susceptible to modulations by adsorbed species, and the Schottky barrier of the interface has been shown to significantly change device conductance.124 In particular, smaller sized nanoparticles can be distributed more uniformly on graphene materials and provide more contact area than bigger sized nanoparticles, resulting in an improvement in electrical properties.123
3.2. Electrochemical Sensors
Electrochemical sensors are, by far, the largest group of sensors and provide an especially attractive means with which to analyze the content of a biological sample due to the direct conversion of a biological recognition event to an electrical signal. A typical electrochemical sensor consists of a sensing (or working) electrode that has a biological recognition element and a counter electrode that are separated by a layer of electrolytes. Electrochemical biosensors can be divided into two main categories based on the nature of their biological recognition process: (1) affinity-based sensors and (2) catalytic sensors. Affinity sensors rely on the selective binding interaction that occurs between a biological component such as an antibody, enzyme, nucleic acid, or a receptor (e.g., immunosensor or DNA hybridization biosensor) and its target analyte, which results in the production of a measurable electrical signal. On the other hand, catalytic sensors generally incorporate nanoparticles or enzymes that recognize the analyte of interest and produce electroactive species. The amount of analyte that is either reduced or oxidized at the sensing electrode would then correlate with the concentration of the target analyte present. In particular, various forms of voltammetry (e.g., linear sweep, differential pulse, squarewave, stripping) and amperometry are commonly used for the electrochemical detection of biomolecules.131
Graphene is an ideal material for electrochemical biosensors as it is an excellent conductor of electrical charge.9a Moreover, because of its high surface area, graphene can facilitate a large number of defects and thus electroactive sites.132 The electrochemical behavior of graphene is also excellent and comparable to other carbon-based materials including CNTs and graphite where recent reports have even demonstrated that graphene-based electrochemical sensors have superior performance when compared to CNTs due to the presence of more sp2-like planes and edge defects on the surface of graphene.133
While graphene exhibits great promise, graphene–nanoparticle hybrid structures have recently gained increasing attention for their applicability to electrochemical sensing. In particular, various types of nanoparticles, including metal nanoparticles such as Au and Pt, metal oxide nanoparticles, and semiconductor nanoparticles, are already widely used for electrochemical sensing applications.115a,134 These nanoparticles can have different roles in electrochemical sensing platforms; for example, they can function to (1) immobilize biomolecules, (2) catalyze electrochemical reactions, or (3) act as a reactant. As such, by incorporating graphene–nanoparticle hybrid structures, one can impart unique and advantageous properties to electrochemical biosensors resulting in the exhibition of the advantages provided by the individual nanoparticle and graphene components as well as synergy from the hybrid. For example, graphene sheets that are decorated with nanoparticles can help overcome the poor utilization coefficient of aggregated nanoparticles.48d In certain cases, by decorating graphene with nanoparticles, one can also efficiently improve the electron transfer that occurs between the analyte and the electrode.135 Finally, similar to the strategy that was used in some hybrid FET sensors, instead of immobilizing the sensing biomolecules directly to graphene, which is often difficult and can negatively affect electrical properties, graphene–nanoparticle hybrids can be formed wherein the sensing biomolecules are first immobilized on the nanoparticles prior to decoration onto graphene. As a result, this can enhance the achievable sensitivity of graphene–nanoparticle hybrid electrochemical sensors.
In the following subsections, we will focus on graphene–nanoparticle hybrid structures for the immobilization of biomolecules and the catalysis of electrochemical reactions. Emphasis will be placed on the different biological applications that they can be used for and the advantages and disadvantages that they provide. Finally, a comparison between graphene–nanoparticle hybrid-based electrochemical sensors and electrochemical sensors that use other more established materials will also be provided throughout.
3.2.1. Immobilization of Biomolecules
Because of its large specific surface area, graphene–nanoparticle hybrids are advantageous for the immobilization of biomolecules. Moreover, the excellent electrical properties of graphene significantly improve the electronic and ionic transport capacity of the resulting hybrid electrochemical sensor, thereby enhancing achievable sensitivities and measurement ranges. Specifically, efforts to apply graphene–nanoparticle hybrids to the immobilization of biomolecules have primarily focused on utilizing enzymes, antibodies, and DNA with enzymes being the most common.
There have been numerous reports demonstrating the graphene–nanoparticle hybrid-based immobilization of enzymes for electrochemical detection. Specifically, the mechanism of action is based on utilizing enzymes that catalyze redox reactions. In this case, when the immobilized enzyme catalyzes a redox reaction of the target analyte, a direct electron transfer from the enzyme to the electrode occurs, which provides an amperometric signal that is proportional to the concentration of analyte.136 For example, Shan et al. gave the first report of an electrochemical biosensor based on a graphene–AuNP nanocomposite for the detection of glucose.137 In this instance, the enzyme glucose oxidase, which reduces O2 into H2O2 in the presence of glucose, was immobilized in thin films consisting of a graphene/AuNP/chitosan nanocomposite on a gold electrode. Specifically, chitosan was an ideal candidate for the immobilization of bioactive molecules onto the electrodes as it has excellent biocompatibility and film-forming ability.138 The resulting composite film had a LOD of 180 μM for glucose. Moreover, the sensor exhibited good reproducibility (4.7% standard deviation for six successive measurements) and an amperometric response to glucose with a linear range from 2 to 14 mM, which is a suitable range for the clinic as the blood glucose level of normal people range from 4 to 6 mM. In terms of the selectivity, glucose (2.5–7.5 mM) could be reproducibly detected in real blood samples. All of this was a result of the synergistic effect that occurred in the graphene–AuNP hybrid structure, where the presence of graphene in the nanocomposite film improved the electronic and ionic transport capacity, resulting in a considerable enhancement in its electrocatalytic activity toward H2O2 when compared to AuNPs alone.139
Another enzymatic electrochemical sensor utilizing hybrid structures for the detection of glucose was reported by Gunasekaran and co-workers.140 In this work, a green, simple, fast, and controllable approach was developed in which a novel nanocomposite consisting of electrochemically reduced GO (ERGO) and gold–palladium (1:1) nanoparticles (AuPdNPs) was synthesized in the absence of reducing agents. Bimetallic nanoparticles are advantageous, especially for electrocatalysis, due to their ability to enhance electrocatalytic activity, improve biocompatibility, promote electron transfer, and poison resistance.141 In particular, Pd is one of the most frequently used electrocatalysts to facilitate reduction reactions involving oxygen, while the introduction of Au into the hybrid offers many appealing properties such as improved biocompatibility and provides an excellent surface for biofunctionalization with biomolecules that contain primary amine groups.142 Glucose oxidase was then immobilized as a model enzyme to detect O2 consumption that occurs during the enzymatic reaction of glucose oxidase in the presence of glucose. While quantifying the electrochemical properties of the ERGO–AuPdNP nanocomposites using electrochemical impedance spectroscopy, it was determined that the charge transfer resistance drastically decreased when ERGO was functionalized with metal nanoparticles. (49.0, 108.9, and 39.6 Ω for Au, Pd, and AuPd nanoparticles, respectively). This result demonstrated that AuPdNPs had a better electron-transfer interface with the electrode surface, the electrolyte solution, and also between the electroactive sites of the immobilized enzyme and electrode when compared to pure metal nanoparticles. Importantly, the resulting ERGO–AuPdNP nanocomposite-based electrochemical sensor exhibited excellent biocompatibility and had a LOD of 6.9 μM, a linear range up to 3.5 mM, and a sensitivity of 266.6 μA mM−1 cm−2. Last, in terms of its selectivity, the device could be used to measure glucose in clinical blood samples, where its detection ability was only ±10% different from a clinical Roche Modular Chemistry Analyzer.
The immobilization of other biomolecules such as DNA and antibodies has also been demonstrated with great success. In the case of antibody immobilization (e.g., immunosensor), a trace antibody is labeled with an electroactive species, such as an enzyme, metal nanoparticle, or QD. As such, when a potential is applied, the concentration of the analyte can be quantified by measuring the resulting current at the electrode.131 Using this principle, Qi et al. recently developed a sensitive Pd-rGO-based label-free electrochemical immunosensor for the detection of the hepatocellular carcinoma biomarker alpha fetoprotein (AFP).143 Specifically, a glassy carbon electrode (GCE) was first coated with Pd–rGO, where the Pd nanoparticles were 4–8 nm in diameter (Figure 17A). Afterward, the anti-AFP antibody solution was added onto the electrode surface. The mechanism of the resulting immuno-sensor was based on the formation of antigen antibody immunocomplexes on the surface of the electrode, which was coated with Pd–rGO. The decrease in the resulting amperometric current of H2O2 was then directly proportional to the concentration of AFP present. As such, the LOD of the immunosensor for AFP was 5 pg mL−1 and was found to have a linear range from 0.01 to 12 ng mL−1 (Figure 17B). They were also able to use this immunosensor to determine AFP in clinical serum samples and demonstrated that the devices offered good stability, biocompatibility, as well as a large surface area. In terms of its selectivity, the device was exposed to several biomolecules found in serum, including BSA (60 000 000 ng mL−1), L-lysine (30 000 ng mL−1), ascorbic acid (2000 ng mL−1), and dopamine (150 ng mL−1), in the presence and absence of 1 ng mL−1 AFP. It was found that the peak current in these cases differed by less than 6%. Similarly, Lu et al. developed a facile and sensitive electrochemical immunosensor for the detection of human chorionic gonadotrophin (hCG), which is secreted from the placenta during pregnancy and from patients with gestational trophoblastic diseases. In this device, they constructed an electrochemical immunosensor using AuNP decorated CNT–graphene composites.144 As a result of the larger surface area available to capture the primary antibody and, subsequently, the analyte of interest as well as improvements in the electron transmission rate, the as-prepared immunosensor had an exceptional linear response range from 0.005 to 500 mIU mL−1 with a low LOD of 0.0026 mIU mL−1 and also proved sensitive, selective, stable, and reproducible for the detection of hCG from human serum samples. Moreover, the immunosensor maintained a relative standard deviation of only 1.12–2.45% when hCG was detected in the presence of interfering biomolecules such as CEA, AFP, cancer antigen 125, prostate-specific antigen, and BSA, further demonstrating its selectivity.
Figure 17.
Label-free electrochemical graphene–palladium nanoparticle nanocomposite-based immunosensor for alpha fetoprotein. (A) Schematic illustration for the preparation of Pd nanoparticles conjugated with rGO. (B) Differential pulse voltammetric response for the modified electrode toward different concentrations of AFP in 0.01 M phosphate buffer (pH 7.4); pulse period, 0.2 s; amplitude, 50 mV. (C) Calibration curves of Pd–rGO modified immunosensor to different concentrations of AFP, error bar = relative standard deviation (n = 5). Reprinted with permission from ref 143. Copyright 2014 Elsevier.
Last, as mentioned previously, the accurate and rapid detection of DNA has received significant attention due to its important role in the diagnosis of genetic diseases including Alzheimer’s disease and various cancers. As such, effort has been invested in the development of DNA-based electrochemical sensors (e.g., genosensor), which utilize a mechanism similar to that stated above for immunosensors. To this end, Lin and co-workers reported an electrochemical DNA sensor wherein the captured DNA was directly immobilized on the surface of a graphene-modified GCE through π–π stacking (Figure 18A).145 AuNPs modified with oligonucleotide probes were then cohybridized on the GCE surface in a sandwich assay format for the detection of the targeted DNA sequence. By utilizing graphene–nanoparticle composite structures, Lin et al. demonstrated enhanced analytical performance with a linear range from 200 pM to 500 nM, and a low LOD of 72 pM due to the higher electron transfer resistance and peak current of the composite structure as compared to graphene alone. Moreover, the biosensor could discriminate the target complementary sequence from single-base pair mismatches (Figure 18B). On the other hand, Du et al. utilized graphene–mesoporous silica–AuNP hybrids (GSGHs) for the ultra-sensitive and selective detection of DNA using strand-displacement DNA polymerization and parallel-motif DNA triplex as a dual amplification system.146 In this system, GSGH was synthesized through self-assembly, and the device was fabricated via layer-by-layer assembly on an ITO electrode (Figure 19A and B). In terms of the sensing mechanism, in the first stage, the target DNA strand acted as a trigger to initiate a polymerase chain reaction (PCR). This process resulted in strand migration along the template and production of a complementary strand that would then displace the target DNA, allowing the target strand to be reused. Next, the sensing interface was incubated with the duplex product resulting in the formation of triplexes between the duplex DNA and the single-stranded acceptor DNA attached on the sensor surface. This duplex blocked the electrode surface with a partially covered adsorption layer leading to a sharply decreased differential pulse voltammetry signal (Figure 19C). In this way, a very low LOD of 6.6 pM was achieved, which is 4 orders of magnitude lower than those of graphene-based DNA sensors coupled with fluorescent assays (100 pM to 1 nm) and 5 orders of magnitude lower than those of FET-based assays (1 and 2 nM) due to the combination of the advantages from the graphene–nanoparticle composites with those from the dual amplification system (Figure 19E and F). This system also demonstrated excellent selectivity wherein the addition of mismatched DNAs containing single nucleotide polymorphisms resulted in only a 6.3–22.2% decrease in the original signal (vs a 66.7% decrease for original target DNA).
Figure 18.
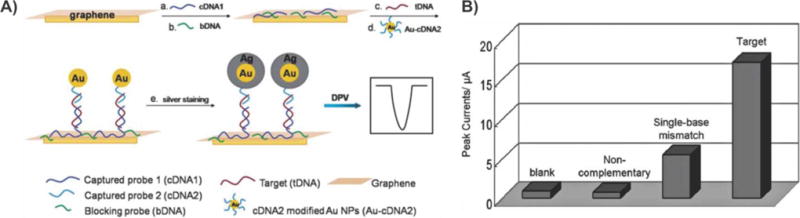
Electrochemical DNA sensor. (A) Schematic diagram. In a typical experiment, captured probe 1 (cDNA1) was adsorbed on the surface of a graphene-modified glassy carbon electrode directly (a), followed by a further adsorption of blocking probe (bDNA) (b). Different target sequences (c) and AuNPs-modified oligonucleotide probes (d) were then cohybridized to the target-active substrates in buffer solution. After silver staining (e) on AuNPs tags as a signal amplification method, a subsequent differential pulse voltammetry (DPV) technique was applied as a detection means for deposited silver. The magnitude of the anodic peak current, which corresponds to the oxidation of silver particles, reflected the amount of complementary target oligonucleotides bound to the GCE-GR/cDNA1 surface. (B) DPV responses of the electrochemical DNA sensor in the blank, noncomplementary sequence, single-base mismatch sequence, and complementary sequence. Electrolyte: 0.1 M KNO3. Pulse amplitude: 50 mV. Pulse period: 0.2 s. Silver staining time: 10 min. Reprinted with permission from ref 145. Copyright 2011 Royal Society of Chemistry.
Figure 19.
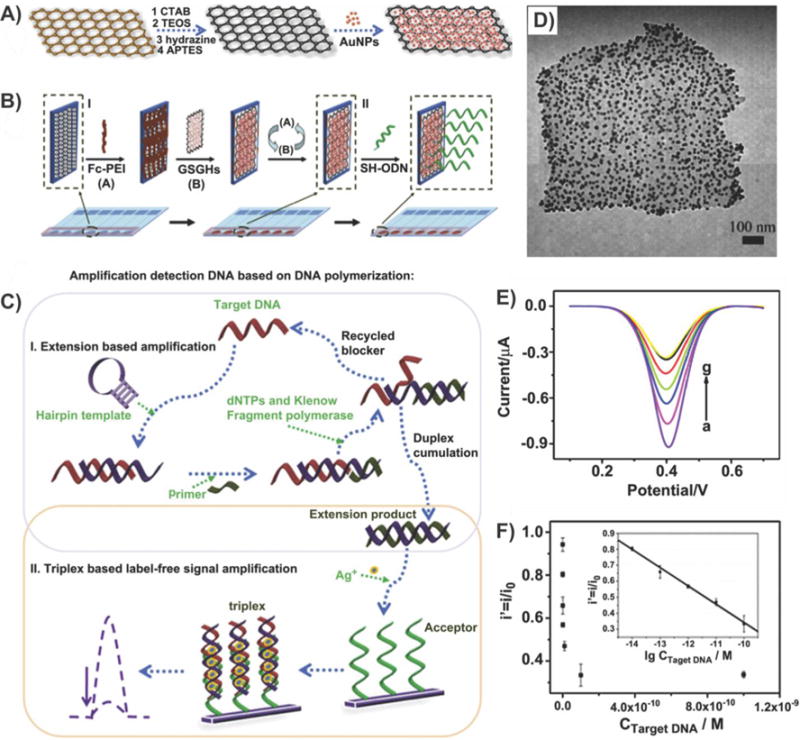
Detection of DNA using a novel parallel-motif DNA triplex system and graphene–mesoporous silica–gold nanoparticle hybrids. (A) Schematic procedure for the synthesis of graphene–mesoporous silica–gold nanosheets. (B) Illustration of the procedure for preparing the electrochemical sensing interface. (C) The procedure of strand-displacement DNA polymerization amplification and parallel-motif DNA triplex amplification. (D) TEM image of graphene–mesoporous silica hybrids. (E) The differential pulse voltammetric response to target DNA with different concentrations: (a) 0 M, (b) 10−14 M, (c) 10−13 M, (d) 10−12 M, (e) 10−11 M, (f) 10−10 M, and (g) 10−9 M. (F) The effect of probe currents on different concentrations of target DNA (from 10−14 to 10−9 M). Inset: A good linear detection range from 10−14 to 10−10 M was obtained. Reprinted with permission from ref 146. Copyright 2011 Elsevier.
3.2.2. Catalysis of Electrochemical Reactions
Sensors that utilize biomolecules such as enzymes, antibodies, and DNA have been widely used in electrochemical sensors as well as other sensor types, allowing users to achieve excellent sensitivity and selectivity. However, biomolecules are also associated with potential limitations such as poor long-term stability, high costs, and may require complicated immobilization procedures. Therefore, there has been increasing interest in the development of enzyme-free sensors as they are cheaper to produce, more stable, simpler to fabricate, and highly reproducible.113,147
For this purpose, there has been significant interest in nonenzymatic electrochemical sensors that utilize catalysis to directly detect electroactive biomolecules at the electrode surface. Graphene–nanoparticle hybrids, wherein metal nanoparticles (e.g., Pt, Au) are used, exhibit excellent catalytic properties for this purpose with the majority of such sensors being applied for the detection of neurotransmitters (e.g., AA, dopamine (DA), and uric acid [UA]), glucose and other carbohydrates, and hydrogen peroxide. In particular, the common detection principle in this case relies on amperometric detection, wherein the analyte of interest is oxidized or reduced (usually oxidized) at the sensing electrode (e.g., anode) when a potential is applied. In terms of the advantages that graphene–nanoparticle hybrids hold for this application, the introduction of metal nanoparticles into electrochemical sensors can decrease overpotentials that are typically seen with many electrochemical reactions. Moreover, the graphene–nanoparticle hybrids have advantages that enhance the overall performance of such sensors as graphene provides an excellent supporting material to facilitate the immobilization and stabilization of nanoparticles on the electrode surface. Moreover, as mentioned previously, graphene–nanoparticle hybrids show high conductivity and enhanced electron transfer over nanoparticles alone and thus can greatly increase the achievable sensitivity and selectivity.
The detection of neurotransmitters such as AA, DA, and UA is a particularly significant application for biosensors. DA is an especially important neurotransmitter in the central nervous system. Low levels of DA have been shown to cause neurological disorders such as schizophrenia and Parkinson’s disease. As such, there is an urgent need for the accurate and rapid determination of DA in bodily fluids. However, this issue is confounded by the existence of AA and UA, which have oxidation potentials similar to those of DA but coexist with DA at concentrations that are several orders of magnitude higher. To address this issue and accurately detect DA and distinguish it from AA and UA, Sun et al. developed a graphene-PtNP-modified GCE.148 Specifically, size-selected PtNPs with a mean diameter of 1.7 nm were self-assembled onto the graphene surface and incorporated as the working electrode in a standard three-electrode cell, where a silver/silver chloride and platinum electrode were used as the reference and counter electrodes, respectively. Measurements were then taken via CV and differential pulse voltammetry. Using this method, the graphene-PtNP-modified GCE was able to simultaneously detect the aforementioned three analytes via their oxidation peaks. In particular, according to the CV curves, the overlapping voltammetric peaks could be resolved into three, well-defined oxidation peaks at approximately 40 mV (AA), 225 mV (DA), and 369 mV (UA). In comparison to GCE and graphene alone, the hybrid electrode provided a significantly enhanced voltammetric peak separation and higher currents for all three analytes. Similarly, for PtNPs alone, peaks potentials were observed at 70 mV (AA), 190 mV (DA), and 330 mV (UA) due to the catalytic properties of Pt, however, with very low currents. Consequently, detection limits as low as 0.15, 0.03, and 0.05 μM are achieved for AA, DA, and UA, respectively. As such, the authors claimed that this electrode system could differentiate well between AA, DA, and UA due to the graphene support, which allowed for enhanced dispersion of PtNPs and thereby full utilization of their catalytic properties for the detection of these three neurotransmitters.
Besides neurotransmitters, there is also significant interest in the development of nonenzymatic biosensors for glucose and other carbohydrates as they are important in many areas such as clinical diagnostics (e.g., diabetes), medicine, and in the food industry. To this end, Luo et al. developed a novel enzyme-free glucose sensor based on graphene–Cu nanoparticle composites, which was synthesized by electrodepositing Cu nanoparticles on a graphene sheet-modified GCE.149 In particular, Cu was chosen as a sensing material on the basis of its wide usage in the amperometric detection of carbohydrates like glucose. As a result of its hybrid structure, the graphene–Cu nanoparticle electrode showed much better electrocatlytic properties for glucose oxidation and detection as compared to unmodified graphene electrodes and Cu/GC electrodes, as well as many other nonenzymatic glucose sensors that have been reported in the literature. Specifically, the authors found that when studying the electrodes with electrochemical impedance spectroscopy, the graphene–Cu nanoparticle electrodes had the lowest electron transfer resistance value (22 Ω) followed by Cu electrodes (24 Ω) and graphene electrodes (28 Ω). This demonstrated the graphene–Cu nanoparticle electrode’s superior electrochemical activity, which the authors hypothesized was due to the synergy that existed between graphene and Cu, where the Cu nanoparticles act as an electron mediator in the electron transfer process. As such, the authors reported that the device was characterized by a linear response up to 4.5 mM and a very fast response time (<2 s). Importantly, the device could achieve a low LOD of 0.5 μM for glucose, which is superior to other devices reported for glucose sensing. Finally, in the presence of interfering species such as fructose, lactose, sucrose, AA, UA, and DA, the device exhibited a minimal change in current (0.2–4%) and could still respond strongly to glucose.
Similarly, Chen and co-workers have demonstrated electrodes utilizing graphene–copper nanoparticle150 and graphene–nickel nanoparticle151 hybrid structures for the enhanced electrochemical detection of carbohydrates when compared to nanoparticle systems alone (Figure 20). In these studies, copper and nickel were chosen on the basis of their wide usage in the amperometric detection of carbohydrates and their strong electrocatalytic activity with regard to the oxidation of carbohydrates in alkaline solutions, respectively. In the case of the graphene–copper nanoparticle hybrid sensor, the hybrid structures were packed into fused silica capillaries to form microdisc electrodes (Figure 20A and B).150 In the presence of 2.0 mM glucose, CV indicated that the peak current for the oxidation of glucose on the graphene–copper nanoparticle composite packed electrode was much higher than that on the copper nanoparticle electrode alone (+0.65 V). In particular, the authors suggested that the deposition of copper nanoparticles on graphene sheets with high specific surface area resulted in higher elecrocatalytic activity due to improved electron transduction. To demonstrate its performance, the graphene–copper nanoparticle composite packed electrode was coupled with a CE system as an end-column amperometric detector for the separation of carbohydrates such as fructose, glucose, lactose, mannitol, and sucrose. The novel detection electrodes were able to successfully separate and detect a mixture of the five carbohydrates even when used for real samples such as milk and fruits after CE separation, demonstrating its selectivity and sensitivity. Specifically, the sensitivity and detection limit of the sensor were determined to be between 45.61 and 85.96 nA mM−1, and 0.87 and 1.64 μM, respectively (Figure 20D).
Figure 20.
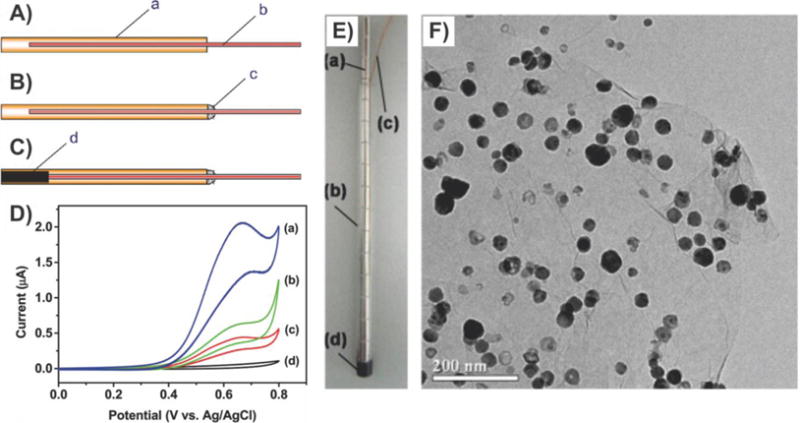
Electrochemical detection of carbohydrates. (A–C) Schematics illustrating the fabrication process of a graphene–copper nanoparticle composite paste electrode. (D) Cyclic voltammograms at (a) a graphene–copper nanoparticle composite paste electrode, (b) a copper nanoparticle paste electrode, (c) a graphite–copper nanoparticle composite paste electrode, and (d) a graphene paste electrode in 75 mM NaOH aqueous solution containing 2 mM glucose. Scan rate, 50 mV/s. The content of paraffin oil in the four pastes was 25% (w/w). Panels (A)–(D) reprinted with permission from ref 150. Copyright 2012 American Chemical Society. (E) Photograph of a magnetic electrode and (F) TEM image of graphene–NiNP hybrid: (a) NdFeB magnet; (b) glass tube; (c) copper wire; and (d) graphite–epoxy composite. Panels (E) and (F) reprinted with permission from ref 151. Copyright 2013 Elsevier.
The graphene–nickel hybrid sensor was prepared via a single-step far-infrared-assisted reduction method, wherein GO and nickel(II) ions were reduced in a hydrazine-containing solution (Figure 20E and F).151 The graphene–nanoparticle hybrid structures were then loaded onto the surface of a magnetic electrode and used to detect the same carbohydrates including glucose, fructose, lactose, mannose, and sucrose. Using this system, the peak currents were found to be significantly stronger than those of Ni nanoparticles alone for the same reasons as described for the graphene–copper nanoparticle hybrids. However, in comparison to the graphene–Cu nanoparticle hybrid, the graphene–Ni nanoparticle hybrid was much easier to load on to the magnetic electrode due to the ferromagnetic properties of nickel and could be fabricated easily and at a low cost. The authors also observed that the graphene–Ni nanoparticle hybrid sensor had superior performance in comparison to the graphene–Cu nanoparticle hybrid sensor. In particular, this sensor exhibited a good linearity from 0.001 to 1 mM and a sensitivity of 42.15 nA mM−1. Moreover, the detection limit was estimated to be 474 nM, and the sensor exhibited good reproducibility (relative standard deviation of 4.3% over nine measurements) as well as selectivity (current response to 0.1 mM UA, DA, and AA was 5.7%, 7.2%, and 5.1% of the response to 1 mM glucose). Overall, the nickel-based hybrid sensor was superior to the copper-based sensor due to the fact that nickel exhibits a much stronger electrocatalytic activity toward the oxidation of carbohydrates in alkaline solutions.151 However, in both cases, the presence of graphene greatly enhanced the strength of the electrochemical signal when compared to either nanoparticles (e.g., copper or nickel) alone due to the high surface area and excellent electrical properties of graphene.
Finally, the concept of utilizing graphene–nanoparticle hybrid structures for the catalysis of electrochemical reactions has also been extended to the detection of hydrogen peroxide (H2O2) in living cells. For instance, Jiang and co-workers recently fabricated an electrochemical sensor that integrated graphene with AuNPs and poly(toluidine blue O) (PTBO) films (Figure 21).152 The rationale behind this choice of nanomaterial was based on the fact that AuNPs have an excellent catalytic ability for H2O2 and incorporation of AuNPs could improve the electron transfer between the analyte and electrode.153 On the other hand, PTBO provided the ideal matrix to enclose the AuNP–graphene hybrid structures. Specifically, a layer-by-layer approach was used to deposit AuNPs (80 nm) and PTBO film onto rGO. The resulting sensor was characterized by a H2O2 sensitivity of 24.52 μA mM−1 cm−2 and a LOD of 0.2 μM. Furthermore, exposure to DA, AA, UA, and glucose yielded little current response and did not affect the sensor’s response to H2O2. As a potential explanation for these observations, the authors noted that the low applied potential (−0.3 V) as well as the design of the electrode helped endow the sensor with its high sensitivity and selectivity. To detect H2O2 in living cells, the cells were separated from the culture medium and resuspended in deoxygenated PBS. H2O2 generation was then stimulated by the addition of AA resulting in the efflux of H2O2 from cells, which was detected and quantified. In particular, H2O2 levels were determined for K562 (human leukemia), PC12 (rat adrenal medulla pheochromocytoma), HepG2 (human hepatocarcinoma), L02 (human embryo liver), and RSC (rat synovial) cells (Figure 21B). The results indicated that a higher efflux of H2O2 occurred in tumor cells versus normal cells suggesting that there was a decline in the enzymatic ROS-scavenging mechanisms in tumor cells, as AA should have induced equal production and distribution of H2O2 in tumor and normal cells.
Figure 21.
Evaluating oxidative stress in tumor cells. (A) Schematic of the LBL assembly of RGO–Au–PTBO modified GCE used for detecting H2O2 efflux from cells stimulated with AA. (B) Amount of H2O2 released by PC12, RSC, L02, HepG2, and K562 cell lines stimulated by 4 μM AA. The values are expressed as means ± SD of at least three independent measurements. Reprinted with permission from ref 152. Copyright 2013 Elsevier.
3.3. Optical Sensors
The deoxidation of GO to rGO results in the formation of a material that is not only electrically conductive but also optically transparent with a transparency of ~97.7% and a transmittance that linearly decreases with the number of layers.154 As such, besides their excellent electronic and electrochemical properties, graphene–nanoparticle hybrid materials that utilize graphene or rGO possess a number of advantageous optoelectronic properties as well, which can be utilized for optical sensing applications. In particular, optical sensors that incorporate graphene nanomaterials fall into two categories: (1) fluorescence-based and (2) SERS-based biosensors. In the case of fluorescence-based biosensors, GO and rGO have the unique property in that not only are they fluorescent over a broad range of wavelengths (e.g., visible to NIR range with their maximum intensity being located between 500 and 800 nm)155 but they also have the ability to quench fluorescent molecules such as dyes and fluorescent nanomaterials (QDs and upconversion nanoparticles [UCNPs]).156 As such, graphene–nanopaticle composites composed of fluorescent nanomaterials are immensely useful for the development of fluorescence resonance energy transfer (FRET)-based and nanometal surface energy transfer (NSET)-based biosensors, which can enable the quantitative analysis of molecular dynamics in living cells on the single cell level. On the other hand, graphene exhibits remarkable Raman scattering properties, which are related to its unique electron and phonon structure.157 This Raman scattering can be greatly enhanced by the inclusion of metal nanoparticles (e.g., AuNPs and AgNPs) as graphene–metal nanoparticle hybrids, resulting in exceptional SERS effects, which can be used to detect the presence of individual biomolecules.29b In the following subsections, we will describe the use of graphene–nanoparticle hybrid materials for optical sensing. In particular, we will focus on fluorescence-based and SERS-based biosensors.
3.3.1. Fluorescence-Based Sensors
FRET is a type of fluorescence phenomenon that can occur when two fluorescent molecules are sufficiently close to one another (typically 1–10 nm). In this phenomenon, the energy from one fluorophore flows to the other via a nonradiative transfer.158 Specifically, the first fluorophore, which is typically denoted as the “donor”, is initially excited by an external optical source. Instead of emitting a photon, the excited donor transfers energy to the neighboring fluorophore, which is termed the “acceptor”. This leads to (1) the emission of fluorescence at the acceptor’s characteristic wavelength and also (2) quenching of the donor fluorophore. Because of the fact that the intensity of FRET depends strongly on the distance between the two fluorophores and their relative orientation as a donor or acceptor, FRET can be used to study, detect, and quantify interactions that occur between two molecules. The donor and acceptor fluorophores can be brought together in many different configurations. For instance, one fluorophore can be attached to a substrate while the other is attached to its binding site, and once the analyte of interest binds, FRET would be initiated. Alternatively, two fluorophores can be attached to a single protein and FRET can be initiated if and when the protein alters its conformation. Finally, the fluorophores can be linked to induce FRET, and once the linkage is cleaved (e.g., by an enzyme), FRET will cease to occur. As such, FRET is an excellent biosensing technique that can be superior to other optical detection methods in terms of its selectivity and sensitivity.159 Moreover, FRET sensing is a nondestructive method that can be used to detect molecular interactions within single living cells.
Graphene and GO sheets were recently found to exhibit super quenching capabilities as energy acceptors for fluorescent dyes, whose efficiency is estimated to reach up to 30 nm, thereby providing greater flexibility for the construction of fluorescence sensors based on FRET. Similar to the quenching effect seen with AuNPs, the mechanism underlying the superquenching effect of graphene and GO follows the NSET mechanism. Therefore, due to the superior-quenching ability of graphene (e.g., the rate of this long-range resonance energy transfer depends on d−4, where d is the distance, whereas traditional FRET depends on d−6),160 it can be coupled with other fluorophores such as fluorescent nanoparticles (e.g., QDs and UCNPs) for use as FRET-based biosensors with enhanced capabilities.
In recent years, the use of anti-Stokes fluorophores such as UCNPs, which can be excited in the NIR, have successfully circumvented issues with autofluorescence and the scattering of light, which typically arise when utilizing FRET in biological tissue. In particular, rare earth-containing UCNPs are able to emit high-energy photons under NIR excitation, which results from a nonlinear optical upconversion process, where the sequential absorption of two or more photons results in the emission of a single photon at a shorter wavelength.161 As such, the use of UCNPs has made it possible to utilize FRET-based sensors directly on biological samples, wherein the use of graphene–UCNP hybrids has even greater advantages due to the superquenching properties of graphene, which greatly enhance the range of achievable FRET. As an example, Zhang et al. utilized graphene–UCNP hybrids for glucose sensing.162 In their system, they synthesized 50 nm water-soluble NaYF4:Yb,Er UCNPs modified with poly(acrylic acid), which were then conjugated with concanavalin A (conA) via EDC coupling. On the other hand, the GO sheets were functionalized with chitosan, which also occurred via EDC coupling. In terms of the underlying mechanism, conA and chitosan were able to form tight bonds, which brought the UCNPs and GO into appropriate proximity to induce FRET (81% degree of quenching). However, in the presence of glucose, the FRET process was inhibited because of competition between glucose and chitosan for ConA. By utilizing this system, Zhang and coworkers were able to achieve a LOD of 0.025 μM even in the presence of serum. Specifically, the FRET process in the presence of serum was found to be nearly the same (with minor differences in the slope), suggesting the selectivity of the sensor.
Similarly, Wu et al. utilized BaYF5:Yb,Er and BaYF5:Yb,Tm UCNPs that were functionalized with aptamers against ochratoxin A (OTA) and fumonisin B1 (FB1), respectively.156c In particular, OTA and FB1 are mycotoxins, which are a group of chemical substances that are produced by some fungal species and can cause illness or even death. As such, strong π–π stacking between the aptamers and the sp2 atoms of GO could be used to initiate FRET, and, in the presence of OTA and FB1, strong binding of the aptamers to the analytes resulted in the release of the UCNPs, thereby decreasing FRET (Figure 22). By utilizing this mechanism, the authors achieved a linear range from 0.05 to 100 ng mL−1 for OTA and 0.1 to 500 ng mL−1 for FB1 and a detection limit of 0.02 and 0.1 ng mL−1 for OTA and FB1, respectively. In terms of selectivity, aflatoxins B1, B2, G1, and G2, fumonisin B2, and zearalenone, which are all homologues of other mycotoxins found in foods, were evaluated. It was determined that only FB1 and OTA could induce a dramatic fluorescence enhancement at their corresponding peaks, whereas the other analogues could not.
Figure 22.
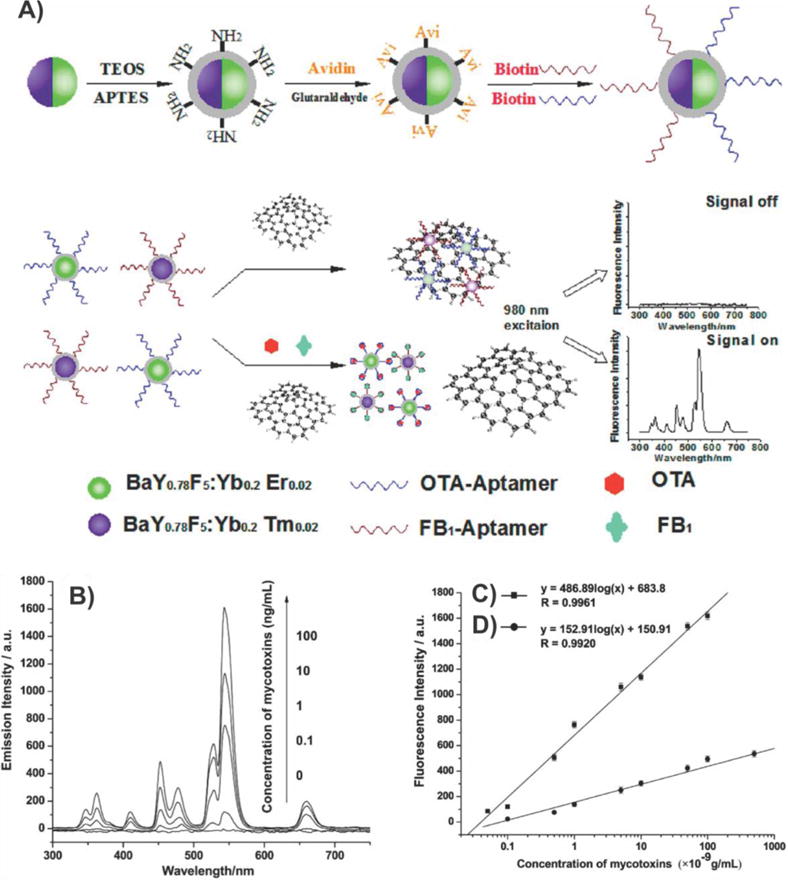
Multiplexed fluorescence resonance energy transfer aptasensor. (A) Schematic illustration of the multiplexed upconversion fluorescence resonance energy transfer between aptamers-UCNPs and GO for FB1 and OTA detection. (B) Upconversion fluorescence spectra of the multiplexed UCNPs-GO FRET aptasensor in the simultaneous presence of 0–100 ng mL−1 FB1 and OTA. (C) A standard curve of the fluorescence intensity versus OTA concentration and (D) FB1 concentrations measured by this developed method. Reprinted with permission from ref 156c. Copyright 2012 American Chemical Society.
QDs also exhibit a number of advantageous properties that make it an attractive nanomaterial for the formation of graphene–nanoparticle hybrids for FRET-based biosensors. In particular, QDs are inorganic and, as such, exhibit good resistance to photobleaching. Moreover, they exhibit a narrow emission band, broad absorption spectra, and have a size-tunable emission.69 Dong et al. gave the first report of efficient FRET between QDs and GO for biosensing applications.156b Specifically, CdTe QDs that were conjugated with either a molecular beacon (MB), which is a single-stranded oligonucleotide hybridization probe with a stem-and-loop structure in which the loop contains a probe sequence that is complementary to a target sequence and the annealing of self-complementary 5′ and 3′ ends forms a stem, or aptamer, were used to detect DNA concentration and sequence (e.g., via MB) as well as proteins such as thrombin (e.g., via aptamer) (Figure 23). By utilizing this structure, a linear range was obtained from 50 to 1500 nM and a LOD of 0.5 and 50 nM was achieved for thrombin and ssDNA, respectively, which is highly sensitive and selective when compared to the other fluorescence or FRET-based methods (Figure 23B).156b Specifically, the MB-QD probe was able to differentiate between the perfectly complementary target, single-base mismatched strand (signal was 40% of the perfectly complementary target), and three-base mismatched strand (signal was 15% of that perfectly complementary target). Moreover, the quenching efficiency was much higher for MB-QDs than the typical efficiency seen in MB-based detection, which helped improve the achievable sensitivity and dynamic range.
Figure 23.
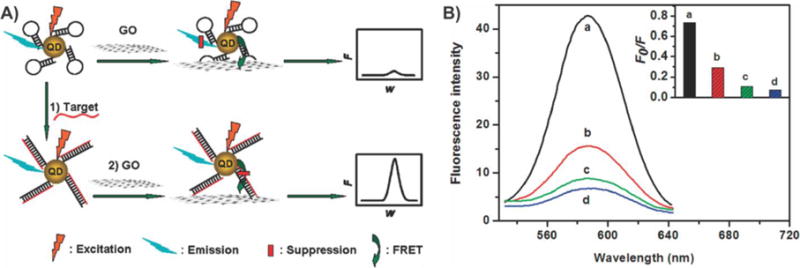
FRET between QDs and GO to sense biomolecules. (A) Schematic representation of GO-induced fluorescence quenching of MB-QDs and biosensing mechanism. (B) Fluorescence emission spectra of MB-QDs (50 nM) after incubation with (a) target (800 nM), (b) single-base mismatch strand (800 nM), (c) three-base mismatch strand (800 nM), and (d) no target and then addition of GO (0.1 μg/mL) for 5 min. Inset: Fluorescence intensity ratio F0/F for four cases. Reprinted with permission from ref 156b. Copyright 2010 American Chemical Society.
Similarly, Li et al. reported a novel “turn-on” fluorescent sensor that utilized a QD/aptamer-GO hybrid to detect lead(II) ions.163 However, it should be noted that this concept could also be readily applied to the detection of biomolecules. Specifically, hybrids were formed between the aptamer-functionalized CdSe/ZnS QDs, which had a characteristic fluorescence emission at 569 nm, and GO via π–π stacking. In the presence of Pb2+, these complexes were capable of changing their structural conformation from a one-dimensional structure to a G-quadruplex/Pb2+ complex, leading to the detachment of the QD/aptamer complex from the surface of GO. This detachment then allowed for monitoring using fluorescence microscopy. In this way, the authors achieved a linear response range of 0.1–10 nM and a LOD of 90 pM (0.019 parts per billion) with the authors attributing this high sensitivity to the super quenching capabilities of GO, the significant difference in the affinity of the aptamer toward Pb2+ and GO, as well as the low background signal due to the “turn-on” configuration. Moreover, when comparing the performance of this sensor to previous reports utilizing fluorescence, colorimetric, and electrochemical devices, this hybrid sensor exhibited a LOD that was 1–3 orders of magnitude better.164 Finally, the sensor exhibited excellent selectivity toward Pb2+ wherein exposure to other metal ions such as Ag+, Ca2+, Cd3+, CO2+, Cu2+, Fe3+, Hg2+, K+, and Ni2+ did not induce any evident change in fluorescence.
Last, researchers have combined GO with noble metal nanoparticles to induce a double-quenching effect that resulted in an increase in the achievable signal-to-noise ratio and thereby amplified the achievable sensitivity. For example, Qu et al. reported a DNA-silver nanocluster–GO nanohybrid material for the detection of multiple nucleic acid targets (Figure 24).165 In this biosensor, Ag nanoclusters were functionalized with a ssDNA reporter that had a cytosine-rich DNA sequence (C12) resulting in highly fluorescent Ag nanoclusters. These DNA–silver nanoclusters were then added to a solution of GO resulting in complex formation via π–π stacking and fluorescence quenching. Once the target sequence was introduced, the high binding affinity between the reporter DNA and the target sequence resulted in the release of the DNA–silver nanocluster and induced strong fluorescence that correlated with the concentration of target sequence DNA present. Using this mechanism, the authors could detect multiple target nucleic acids with a high sensitivity (LOD: 1 nM), which was attributed to the high achievable signal-to-noise ratio resulting from the high quenching efficiency of GO. In terms of the selectivity, the sensor could discriminate between perfectly complementary target, one-base mismatched target (88.4% of the perfect target’s signal), two-based mismatched target (67.4% of the perfect target’s signal), and noncomplementary target (22% of the perfect target’s signal). Chen and co-workers also reported a similar detection platform that utilized AuNP-functionalized graphene for the detection of lead ions. This sensor was found to function in the concentration range of 50–1000 nM and had a LOD of 10 nM.166
Figure 24.
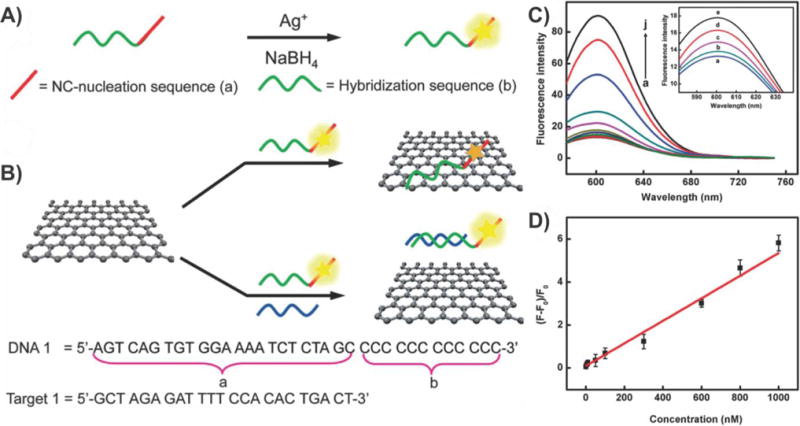
DNA-templated silver nanoclusters–GO hybrids for the detection of multiple nucleic acids. (A) Schematic representation of the preparation of silver nanoclusters on DNA scaffolds in aqueous solution. (B) Schematic illustration of the assay for label-free DNA detection using AgNCs–GO nanohybrid materials. (C) Fluorescence emission spectra of P1 (1 mM) upon addition of T1 with different concentrations: (a) control; (b) 1 nM; (c) 5 nM; (d) 10 nM; (e) 50 nM; (f) 100 nM; (g) 300 nM; (h) 600 nM; (i) 800 nM; and (j) 1000 nM, which was then quenched with GO. Inset: A zoomed-in view of the data for low concentrations of target (a–e). (D) Linear relationship between (F − F0)/F0 (relative fluorescence intensity, where F0 and F are the fluorescence intensities without and with the presence of target DNA, respectively) and the concentration of target DNA (1–1000 nM). Error bars were obtained from three parallel experiments. Reprinted with permission from ref 165. Copyright 2012 Royal Society of Chemistry.
3.3.2. SERS-Based Sensors
SERS is a surface-sensitive technique that allows for the enhancement of Raman scattering by molecules attached to or in close proximity of metal nanostructures and is one of the best techniques available for molecular analysis with sensitivities that can reach as low as the detection of single molecules.167 In particular, SERS is a phenomenon that can amplify normally weak Raman signals by many orders of magnitude and occurs due to a combination of chemical enhancement, which is mainly related to the charge transfer that occurs between the metal nanostructure and the analyte molecules, with electromagnetic enhancement (e.g., induced by the surface electron oscillation in the structure).168 As such, SERS-based sensors have been used in a variety of detection applications ranging from physics and engineering to biology and medicine.169
To achieve high electric field intensities and to acquire significant enhancement in achievable sensitivities, it has become a common practice to pattern nanoparticles/nano-structures composed of noble metals (e.g., Cu, Ag, or Au) on substrates (e.g., silicon wafer). Enhancement is primarily caused by amplified, light-induced electric fields that exist on the surface of the patterned metallic nanoparticles. Specifically, when the incident light is in resonance with the oscillations of conducting electrons in a metallic nanoparticle, all of the electrons are collectively driven to oscillate in an optical phenomenon known as localized surface plasmon resonance (LSPR).170 This LSPR phenomenon is then responsible for the strong scattering and absorption of light that is typically observed when utilizing metallic nanoparticles. This phenomenon is also responsible for the generation of enhanced E-fields on the surface of the nanoparticles at sites that are known as “hotspots”. Consequently, molecules within these hotspots experience enormous enhancement in their Raman cross-section, thereby potentially allowing for the detection of single molecules.171
Interestingly, recent studies have found that graphene and GO also have the ability to enhance Raman signals via a chemical enhancement mechanism, which is independent of the Raman enhancement caused by noble metal nanoparticles.172 In particular, it has been shown that mechanically exfoliated graphene sheets can induce Raman enhancement with a maximal enhancement of 17-fold, due to electron transfer that occurs between graphene and the molecules adsorbed on its surface.173 Therefore, by using graphene or GO, which possesses numerous active oxygen sites that can enhance graphene–metal nanoparticle/molecule binding, it can be expected that the combination of graphene materials and metal nanoparticles would act synergistically to further enhance SERS when compared to using either graphene or metal nanoparticles alone (e.g., dual-enhancement of Raman signals by hybrid materials via chemical and electromagnetic enhancement). Using this strategy, graphene–nanoparticle hybrid materials have been developed to successfully detect a variety of biomolecules. For example, He et al. developed a SERS-active substrate based on AuNP decorated CVD graphene and used it for the multiplexed detection of DNA.174 Specifically, large films of graphene (120 μm × 120 μm) were generated and AuNPs (20 to 50 nm) were formed spontaneously on the film by immersion in HAuCl4 wherein direct redox occurred between the metal ions and the graphene sheets. To detect DNA, the AuNPs were functionalized with thiolated DNA probes (up to two different DNA probes were used). Following the exposure of the device to the target DNA and the addition of a reporter DNA labeled with Cy3, which resulted in the formation of a sandwich composed of capture/target/reporter DNA, multiplex detection of DNA was achieved with a LOD of 10 pM (Figure 25). On the other hand, addition of uncomplementary DNA at the same concentration resulted in little to no signal. Moreover, it was shown that the graphene–AuNP substrate enhanced Raman signals significantly more than SiO2/Si and graphene–SiO2/Si substrates alone due to the coupled surface plasmon resonance absorption of AuNPs on the graphene film.
Figure 25.
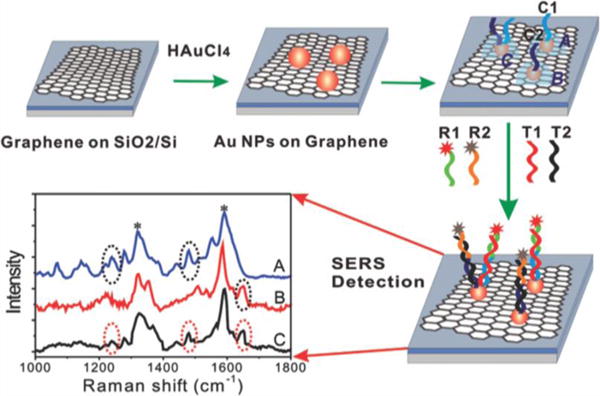
Graphene–AuNP-based SERS platform for multiplex DNA detection. The large-sized graphene films were grown by CVD on copper foils and then transferred onto SiO2/Si substrate. Graphene film was then decorated with AuNPs by immersing it into HAuCl4 solution. The detection of target and multiplex (two targets) DNA by Au-G–SiO2/Si substrate-based SERS sensor. SERS spectra obtained in the absence of target DNA (black, C) and in the presence of 1 nM non target DNA (red, B) and complementary target DNA (blue, A). Reprinted with permission from ref 174. Copyright 2012 American Chemical Society.
A number of studies have also utilized graphene–AgNP hybrids for SERS biosensing. For example, Ren et al. developed a GO–AgNP hybrid structure that could function as a Raman-enhancing material to detect folic acid (FA) in water, as FA has been reported to play a significant role in diseases such as cancer, heart attack, and mental degeneration.175 In particular, GO–AgNP hybrids were obtained via self-assembly, wherein GO sheets were first functionalized with poly-(diallyldimethylammonium chloride) (PDDA). Because of the cationic nature of PDDA, the GO–AgNP hybrids could capture negatively charged FA molecules resulting in a sensor that exhibited a LOD of 9 nM and a linear response between 9 and 180 nM even when mixed with serum proteins. Importantly, despite depending on electrostatic interaction as its sensing mechanism, the unique SERS spectra of FA could be distinguished from serum proteins. Moreover, it was actually found that the SERS spectra of serum were quite similar to that in water with comparable intensities and the addition of a few peaks. As with the report by He et al., the authors reported that the Raman signals of FA were much higher on GO–AgNP hybrid structures than on normal AgNPs due to the presence of strong electrostatic interactions. GO–AgNP hybrids have also been applied to monitor prohibited colorants in food,176 wherein the GO–AgNP hybrids were used to detect mixtures of up to four prohibited red (e.g., allura red, ponceau, amaranth, and erythrosine) or yellow (e.g., lemon yellow, sunset yellow, orange II, and chrysoidin) colorants by their characteristic peaks with a LOD of 10−5 M. Similarly, GO–AgNP hybrids that were prepared via a one-pot method, where tannic acid, a water-soluble, phenolic hydroxyl-rich compound, was used as the reducing agent, were used to detect H2O2 and glucose without the need for glucose oxidase at a LOD of 7 and 100 μM, respectively.177 Finally, Long et al. recently reported a disposable biosensor composed of a GO–AgNP composite on a screen-printed electrode that was capable of monitoring different polar antibiotics (e.g., MT, AT, 6-AA, and PG) in situ with a LOD of 1 nM (Figure 26).178 In this case, SERS was combined with electrophoretic preconcentration (EP), a process that draws charged analytes toward SERS substrates through electrostatic force, to increase the concentration of analyte in the enhancement regions of the graphene–nanoparticle hybrid device. By combining SERS with EP, the antibiotics could be selectively adsorbed on the GO–AgNP sensor by controlling the applied potential in the EP process, and the SERS spectra of a mixture of different analytes could be obtained and distinguished within a 10 min time frame without any preseparation.
Figure 26.
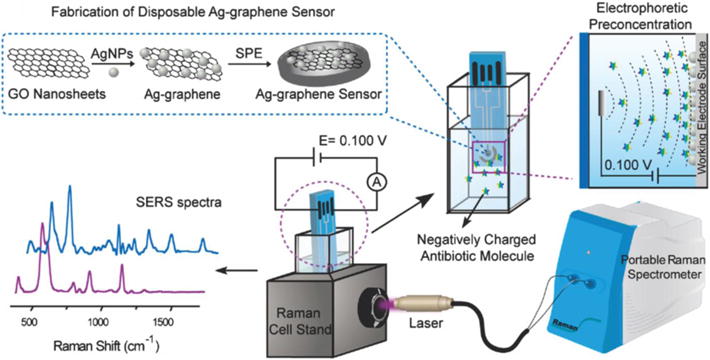
Graphene–AgNP sensor based on electrophoretic preconcentration and SERS. Schematic representation of a disposable Ag–graphene sensor for the detection of polar antibiotics in water. The magnification insets show the fabrication of Ag–graphene sensors and the electrophoretic preconcentration process of polar antibiotics. The distribution of antibiotics molecules is sketched for the case of a negatively charged analyte. At a given potential, most of the negatively charged antibiotics are concentrated onto the positively charged printed electrode, due to the generated electric field between the working electrode and the counter electrode. In SERS experiments, the laser comes vertically from the side view of the spectroelectrochemical cell and is focused on the Ag–graphene sensor. SPE = screen-printed electrodes. Reprinted with permission from ref 178. Copyright 2013 Elsevier.
Last, GO nanocomposites have also been applied for live cell-based SERS applications. Liu et al. recently reported a graphene–AuNP hybrid for SERS in which gold nanostructures were grown intracellularly.179 Specifically, this process was assisted by polyvinylpyrrolidone (PVP)-functionalized GO, which acted as an activator for AuNP (IGAuNPs) biosynthesis. In terms of its mechanism of action, PVP is a nonionic, water-soluble, and nontoxic polymer surfactant that can be employed as a stabilizing and coordinating agent for the synthesis of metal nanoparticles.180 As such, by delivering trace amounts of PVP into the cell via functionalization of GO, not only did PVP serve as a biocompatible stabilizer for GO, but it also provided a template to coordinate the reduction of gold ions via intracellular redox systems, thereby forming 100 nm graphene–AuNP hybrids termed PVP/GO/IGAuNPs (Figure 27A and B).179 The distribution of PVP/GO/IGAuNs in the cells (A549, 4T1, HeLa cells) then allowed for the sensitive monitoring of intracellular chemical compositions including proteins, nucleic acids, lipids, and carbohydrates within 15 h. Moreover, the hybrids could be used to monitor the intracellular composition of the different cellular compartments including the cytoplasm, nucleoplasm, and even the nucleus using SERS (Figure 27C and D). Specifically, a comparison between the SERS spectra of GO/PVP/IGAuNPs and IGAuNPs alone showed that the hybrid structure results in a 5 times larger Raman enhancement, possibly due to the formation of IGAuNP aggregates on GO. As such, the authors stated that the device showed potential for cancer detection. Similarly, Liu and co-workers also reported GO–AgNPs, which enabled very rapid cancer cell probing and imaging with a detection time of 0.06 s per pixel.181 In this case, the GO–AgNP composites were synthesized via an in situ reduction process that also used PVP as the reductant and stabilizer.182 For their studies, Raman images of HeLa cells were taken after 8 h of incubation with 1 ug mL−1 GO or GO–AgNPs. The Raman signals of GO were almost undetectable in the cells incubated with GO alone. However, the hybrids showed remarkable enhancement (~48.4-fold enhancement). Finally, selective labeling of cancer cells could be achieved by covalently functionalizing the GO–AgNPs with FA.
Figure 27.
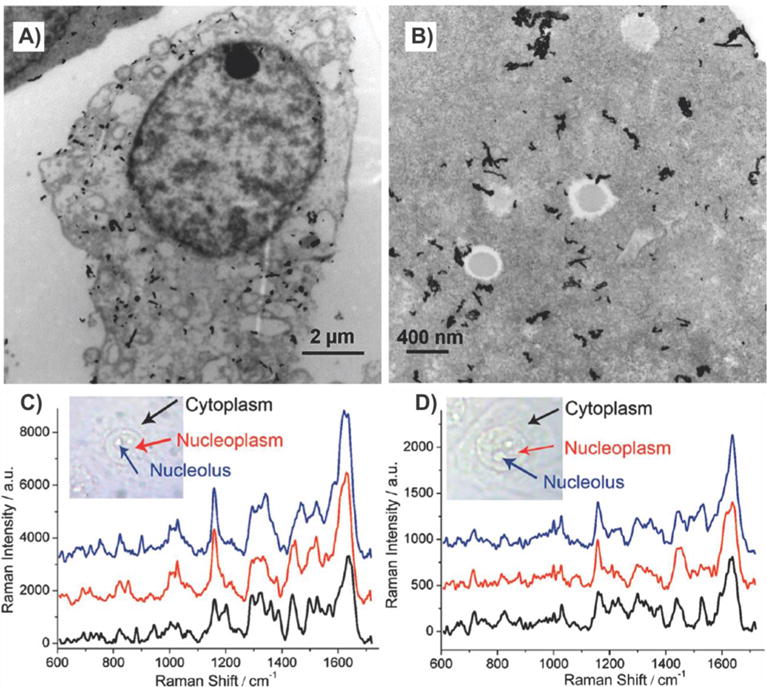
Intracellularly grown GO–AuNP hybrids for SERS. (A and B) TEM images of A549 cells containing GO/PVP/IGAuNs nanocomposites. (C, GO/PVP/IGAuNs; and D, IGAuNs) SERS spectra of A549 cells collected from the regions corresponding to the cytoplasm, nucleoplasm, and nucleolus. Reprinted with permission from ref 179. Copyright 2012 American Chemical Society.
4. CELLULAR INTERACTIONS WITH GRAPHENE–NANOPARTICLE HYBRID MATERIALS
Nanoparticles possess a number of distinctive properties that make them useful for applications in cellular biology. For instance, their small size (e.g., 5–100 nm diameter) allows for easy delivery into cells by crossing the plasma membrane.183 Moreover, the surface chemistry can be adjusted such that the nanoparticles selectively interact with individual biomolecules found either on the cell membrane or within the cytoplasm, specific normal/diseased tissues, or bodily fluids (e.g., blood, interstitial fluids, etc.). As such, numerous studies have reported the synthesis of nanoparticles with different inorganic and organic compositions, including noble metal, magnetic, polymeric, semiconducting, upconversion, and silica nanoparticles.184 The ability to tune the properties of these nanoparticles has permitted scientists and clinicians to effectively utilize nanoparticles for various cellular applications including bioimaging, drug delivery, and tissue engineering.180,185 At the same time, graphene-based nanomaterials exhibit unique physicochemical, thermal, mechanical, and optical properties, which have also made them attractive for biomedical applications.186 Therefore, in the last five years, researchers have successfully combined the intrinsic properties of nanoparticles and graphene-based nanomaterials within single graphene–nanoparticle hybrid platforms to make remarkable advances in a variety of cellular applications. In this section, we will highlight the recent cellular applications of graphene–nanoparticle hybrid structures, mainly focusing on the areas of bioimaging, photothermal therapies (PTTs), drug/gene delivery, multifunctional therapies, and stem cell/tissue engineering.
4.1. Bioimaging
Understanding complex biological systems entails learning how individual components within living cells form, how they are organized, how they interact, and how they assemble to provide structure and function. The need to visualize and, in turn, assess these different processes within living cells, tissues, and whole organisms has undoubtedly necessitated the development of sophisticated bioimaging techniques. In addition to acquiring a better understanding of how normal biological processes occur, bioimaging can even enable the monitoring of abnormal processes caused by cancer and disease, thus underscoring its importance in medicine. At the most basic level, bioimaging encompasses techniques to visualize biological processes in living cells or animals using specialized imaging probes. However, with clinical applications hampered by poor sensitivity, specificity, and targeting, there is a constant need to develop more advanced bioimaging probes and imaging modalities.187 To this end, graphene–nanoparticle hybrid materials have shown great promise as the next generation of imaging probes. We will briefly review the three main imaging modalities in which these hybrid nanostructures have made the most impact, specifically (1) MRI, (2) Raman spectroscopy, and (3) fluorescence imaging.
4.1.1. Magnetic Resonance Imaging (MRI)
When it comes to investigating tissues within the body and monitoring the progression of cancer or disease, MRI is the most widely used medical imaging technique. MRI takes advantage of tissue contrast that is generated from nuclear magnetic resonance (NMR) signals received from hydrogen nuclei located in different physiological environments/tissues throughout an organism. Among other factors, the spin–lattice longitudinal relaxation time (T1) and the spin–spin transverse relaxation time (T2) are crucial in determining the MRI signal intensity, wherein the magnitude of the MRI signal increases with decreasing relaxation times. However, the inherent difference in the relaxation times between two given biological tissues tends to be insufficient to obtain a detectable signal. As a result, contrast agents bearing paramagnetic or superparamagnetic properties (e.g., from metals belonging to lanthanide and transition metal series) were developed to achieve enhanced contrast.
MRI contrast agents work by reducing the T1 and/or T2 relaxation times of the protons in the vicinity of the metal-containing agent, in turn making areas in the image brighter or darker for better detection. Initially, GdIII was established as one of the best contrast agents due its large magnetic moment, but it has issues with uptake in extravascular space and toxicity in its ionic form.188 In recent years, MNPs, such as Fe2O3 and Fe3O4, have proven to be excellent MRI contrast agents, offering sufficient sensitivity for T2-weighted imaging.189 Moreover, the formation of aggregates of MNPs has been reported to increase relaxivity rates (r2) and, in turn, improve MRI contrast.190 However, the uncontrolled aggregation of iron oxide NPs (IONPs) can cause precipitation of the NPs and thus reduce the time of circulation in the blood due to reticuloendothelial clearance.191 In this regard, graphene-based nanomaterials were initially employed to serve as a scaffold with which to anchor IONPs, wherein IONP clustering on graphene sheets would enhance MRI contrast and the chemical functionalities of the graphene sheets would improve the physiological stability of the aggregated MNPs. Early work in this area focused on developing efficient methodologies to prepare these graphene–MNP composites.192 For example, Cong et al. were among the first to report the synthesis and MRI imaging capabilities of rGO sheets decorated with monodisperse Fe3O4 NPs.193 In this case, the composites were prepared using a simple in situ method, wherein rGO sheets were reacted with an iron precursor, iron(III) acetylacetoante (Fe(acac)3). Using TEM, it was then determined that all of the MNPs were coated on the rGO sheets with no aggregated or free particles. Moreover, by adjusting the initial weight ratio of Fe(acac)3:rGO sheets, the coverage density of the MNPs and, in turn, the magnetic properties of the composites were finely tuned. For instance, saturation magnetization values of 19.6, 51.7, and 64.8 emu g−1 were achieved with initial weight ratios of 1:1, 2:1, and 4:1, respectively. While these composites were only shown to provide contrast in T2-weighted MR imaging (performed in 0.5% agarose gels), in vitro cellular imaging using GO–MNP composites as contrast agents was later demonstrated by Chen et al. using composites of aminodextran-coated Fe3O4 nanoparticles and GO sheets.194 The T2 relaxivity of the GO–Fe3O4 composites (76 Fe mM−1 s−1) was found to be higher than that of Fe3O4 nanoparticles (21 Fe mM−1 s−1), indicating that enhanced MRI intensities can be achieved due to the formation of hybrid structures wherein the Fe3O4 NPs formed aggregates on the GO sheets. In addition, the GO–Fe3O4 composites remained stable under physiological conditions, and delivery to HeLa cells allowed for the detection of as few as 1000 cells mL−1 (Fe concentration of 20 μg mL−1). However, there was a significant drop in the MR relaxation rate of the Fe3O4–GO composites to 31 mM−1 s−1 upon incubation with/delivery into cells. While this is to be expected, possibly due to the slower rate of diffusion for water inside the cell, when the composites are confined in the endosomes, a further enhancement of T2 relaxivity would be desirable to apply these hybrids for in vivo MRI cellular labeling.
To this end, recent studies have demonstrated a method to further enhance the T2 relaxivity by assembling nanoparticles possessing varying morphologies and composition onto GO sheets. One such study by Chen et al. reported the in situ growth of β-FeOOH nanorods (longitudinal length of 20 nm and transverse diameter of 3 nm) onto polyethyelene glycol-coated (PEGylated) GO sheets.195 These GO-PEG-β-FeOOH composites exhibited an ultrahigh transverse relaxivity (r2) of 303.81 mM−1 s−1, which was 60 times higher than those achieved by previous β-FeOOH-based MRI contrast agents (Figure 28).196 These nanocomposites produced an exceptional enhancement in T2-weighted MR imaging for in vitro HeLa cell cultures, as well as for in vivo studies in mice. Specifically, a significant darkening of the liver was observed in the T2-weighted images about 2–4 h after intravenous administration of the nanocomposites, which was further confirmed with TEM images that showed uptake of the composites into the mouse liver. Overall, these studies provide remarkable evidence for the benefits of combining MNPs with GO sheets to enhance the capabilities of MRI contrast agents via the controlled formation of MNP aggregates on the graphene sheets.
Figure 28.
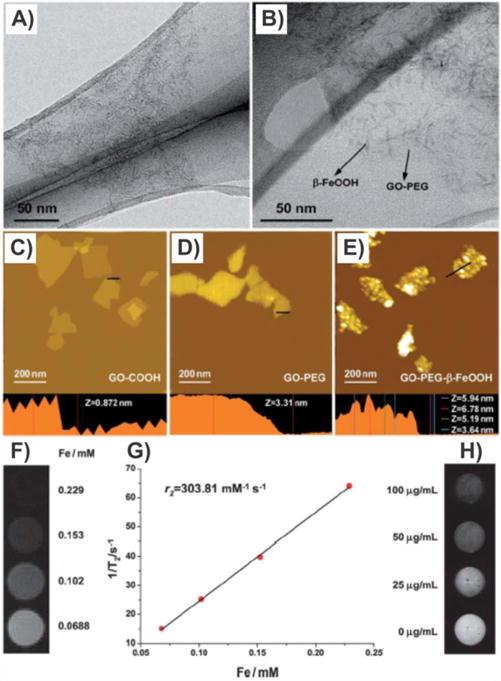
In situ growth of β-FeOOH nanorods on GO for in vivo MRI and cancer therapy. (A and B) TEM images of β-FeOOH nanorods on GO. AFM images of (C) GO-COOH, (D) GO-PEG, and (E) GO-PEG-β-FeOOH. (F) T2-weighted MR images of GO-PEG-β-FeOOH. (G) Plot of 1/T2 versus Fe concentration in GO-PEG-β-FeOOH. The slope indicates the specific relaxivity (r2). (H) T2-weighted MR images of HeLa cells after 4 h incubation with different concentrations of GO-PEG-β-FeOOH. Reprinted with permission from ref 195. Copyright 2013 Royal Society of Chemistry.
4.1.2. Raman Spectroscopy
Raman spectroscopy is an alternative mode of imaging that relies on the intrinsic structure and properties of graphene-based nanomaterials. Raman spectroscopy is widely used to characterize GO due to its characteristic fingerprint in the Raman spectra (D band at ~1350 cm−1 and G band at ~1600 cm−1). However, the introduction of noble metals, such as AuNPs and AgNPs, onto GO sheets has been found to promote surface-enhanced Raman scattering (SERS), which can be used to improve the Raman intensity and sensitivity of GO considerably.44b Resultantly, a number of studies have exploited this feature to utilize GO–noble metal composites as imaging probes in biological cells. For example, employing a traditional in situ synthetic approach as seen with MNPs, 20 nm AuNPs were formed on GO sheets via the reduction of AuCl4− with citrate to obtain GO–AuNP hybrids (Figure 29A).197 A 4-fold enhancement of the Raman spectra was observed for the GO–AuNP hybrids when compared to the original GO under the same test conditions, due to the surface enhancement effect of the AuNPs. Furthermore, when Raman imaging was performed following delivery of the GO–AuNP hybrids into HeLa 229 cells, it was observed that cells incubated with the GO–AuNP hybrids exhibited significantly enhanced Raman signals when compared to control cells incubated with only GO and only AuNPs (Figure 29B–I). Finally, rapid Raman imaging and high-resolution Raman images were acquired using a low laser power (3.5 mW) and short acquisition time (1 s). In terms of the mechanism of uptake, a simple study suggested that the GO–AuNP hybrids were internalized by an energy-dependent process such as endocytosis. These findings were further supported by a more detailed study performed by Huang et al., wherein SERS was used to demonstrate that internalization occurred through the clathrin-mediated energy-dependent endocytosis route.198
Figure 29.
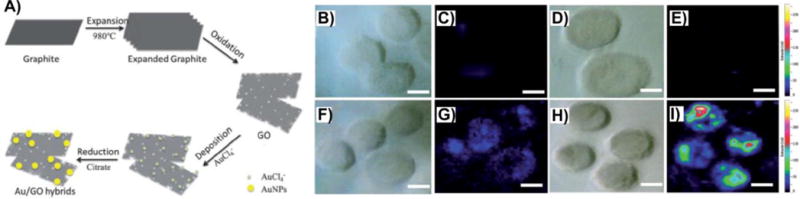
GO–AuNP hybrid-based SERS probe for cancer cell imaging. (A) Scheme for the preparation of GO and Au nanoparticle decorated GO (Au/GO hybrids). Optical (B, D, F, H) and Raman (C, E, G, I) images of HeLa 229 cells. Cells were incubated in medium without GO (B and C), in medium with Au nanoparticles (D and E), in medium with GO (F and G), and Au/GO hybrids (H and I). Scale bars: 10 μm. Reprinted with permission from ref 197. Copyright 2012 Royal Society of Chemistry.
In contrast to intracellular delivery, these hybrids can also be used as highly sensitive SERS probes to achieve targeted labeling of the cellular surface membrane. In one such study, GO–AgNP composites were synthesized using an in situ reduction method for HeLa cell labeling.181 By varying the weight ratio between AgNO3 and GO, an optimal Raman enhancement was found with a ratio of 192. The GO–AgNPs were initially delivered without any modifications, resulting in the distribution of the hybrids almost exclusively in the cytoplasm. However, the authors also demonstrated targeted cellular delivery by covalently binding a FA molecule to GO hybrid structure via amide bond formation. In particular, FA is known to target the folate receptor (FR), which is overexpressed in the vast majority of cancers (e.g., epithelial, ovarian, breast, lung, and brain tumors) while having limited expression in healthy tissues and organs.199 As such, it is suitable for HeLa cell targeting.200 After incubating the FA-GO-AgNPs with FR-positive cells (HeLa) and FR-negative cells (A549), the intensities of Raman signals acquired from HeLa cell were 2 orders of magnitude higher than that of A549 cells. Moreover, extensive accumulation of the hybrids was found on the plasma membrane of the FR-positive HeLa cells after 4 h, with moderate signals found in the cytoplasm.
4.1.3. Fluorescence Imaging
Fluorescence-based techniques are widely used in the life sciences for imaging and diagnostics. However, fluorescence-based detection methods tend to have several limitations, including photobleaching, degradation of the chemical dye compound, and background autofluorescence from cells or tissues.201 Nevertheless, a number of studies employing GO–nanoparticle hybrids for fluorescence imaging have been conducted, which address several of these limitations. Sreejith et al. fabricated a GO-wrapped mesoporous silica nanoparticle (MSNP) for the encapsulation of squaraine dyes, a class of zwitterionic dye that exhibits photophysical properties in the NIR region.202 While suitable for numerous bioapplications, squaraine dyes tend to lose their photophysical properties in aqueous medium due to aggregation and nucleophilic attack of the squaryl rings. Therefore, sequestering the dye in MSNPs and then further capping the pores with GO ensured the minimal loss of the dye from leakage, improved the stability of the dye by preventing nucleophilic attack from thiol-containing molecules generally found in the cytoplasm (e.g., cysteine, glutathione), and allowed for fluorescence imaging in cancer cells.
While dyes are conventionally used for fluorescence imaging, semiconductor QDs overcome many of the limitations of traditional dyes and allow for tunable emissions.203 Recently, hybrid nanocomposites of zinc-doped AgInS (AIZS) QDs and GO were synthesized for in vitro imaging of NIH/3T3 cells.204 Freshly prepared hydrophobic AIZS nanoparticles were assembled onto oleylamine-modified GO, followed by PEGylation of the nanocomposites to improve colloidal stability in aqueous medium (Figure 30A). In doing so, the authors reported that they could enhance the solubility and stability over AIZS nanoparticles alone. Specifically, the AIZS–GO nanocomposites dispersed well in water with no observable aggregation. Moreover, varying the doping amount of zinc in the AIZS nanoparticles allowed for the preparation of nanocomposites with green, yellow, orange, and red colors (Figure 30B). These nanocomposites possessed a diameter of about 130 nm under physiological conditions and exhibited no obvious cytotoxicity in NIH/3T3 cells (1 to 100 ug mL−1). The nanocomposites also showed strong photoluminescence, wherein confocal imaging revealed that the nanocomposites accumulated in the cytoplasm around the nuclei (Figure 30C).
Figure 30.
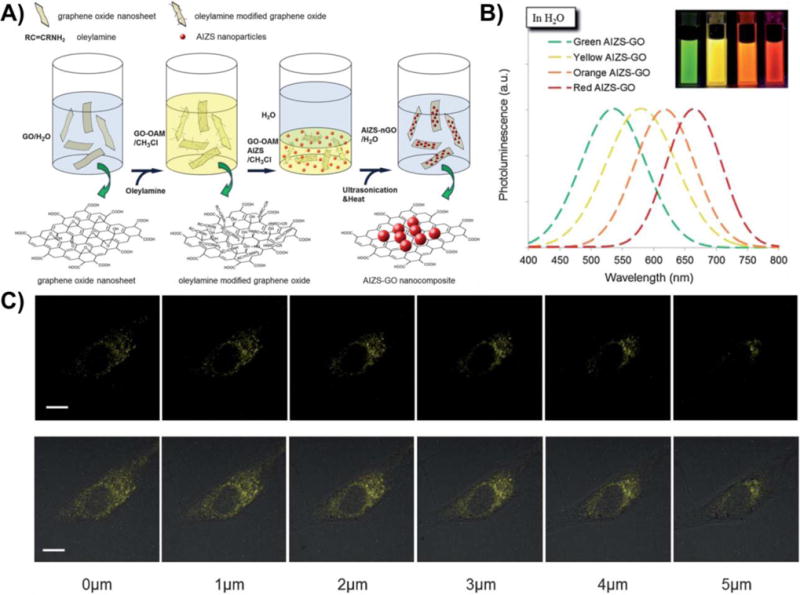
GO–AIZS NP composites for cellular imaging. (A) Schematic illustration of the procedure for synthesizing fluorescent zinc-doped AgInS2 quantum dot decorated GO (AIZS–GO) nanocomposites. (B) The photoluminescence spectra of AIZS–GO nanocomposites suspended in water. (C) Confocal laser scanning microscope (CLSM) images (top row) and merged images (bottom row) of one typical NIH/3T3 cell tagged with yellow color emitting AIZS–GO–PEG nanocomposites at different cross sections (1 μm intervals). Scale bars: 10 μm. Reprinted with permission from ref 204. Copyright 2013 Royal Society of Chemistry.
4.2. Photothermal Therapies
Temperature is an important factor that determines the viability and functionality of biological entities ranging from cells to entire organisms. While an increase in temperature above the normal body temperature (37 °C) is normally a sign of illness, the controlled increase in temperature of targeted tissues has been reported to have numerous therapeutic benefits in patients with cancer or disease.205 The clinically relevant temperature range for thermal treatments has been reported to be between 41 and 48 °C, a range that is referred to as hyperthermia.206 Moderate levels of hyperthermia appear to be adequate for inducing protein denaturation and aggregation, while reducing cellular resistance against orthogonal cancer treatments (e.g., chemotherapy, radiation, etc.). On the other hand, short thermal treatments above 48 °C for even several minutes (or long exposures in the clinically relevant range for several hours) can cause excessive necrosis and ablation, leading to severe and irreversible damage. As a result, the development of novel tools and techniques for controlled heating in localized areas has been a major focus for advancing thermal therapies. In regard to such thermal therapies, PTT has gained tremendous attention, wherein photoabsorbing agents generate heat upon exposure to optical energy. Among the various types of nanomaterials available for this purpose, graphene-based structures exhibit strong optical absorbance in the NIR range, which makes it an especially effective PTT agent. In addition, modification of the graphene surface to further incorporate inorganic nanoparticles allows for the generation of hybrid materials with a greater dynamic range of heating, enhanced temperature control, and the incorporation of imaging modalities as well as other functionalities.
In one study, rGO–IONP composites were applied for in vivo PTT in a 4T1 murine breast tumor mouse model.207 In particular, the nanocomposite was prepared by the hydrothermal reaction of GO and iron chloride hexahydrate, resulting in the distribution of IONPs (8–10 nm) on the rGO surface.208 Further grafting with a PEG-based moiety imparted stability in physiological solutions, as well as a significant reduction in the mean hydrodynamic diameter from >200 to ~50 nm. The resulting rGO-IONP-PEG exhibited greater NIR absorbance than the as-made GO at the same GO concentration, underscoring the advantage of a graphene–nanoparticle hybrid for inducing photothermal effects. Intravenous injection of 125I labeled rGO-IONP-PEG in mice bearing 4T1 tumors revealed primary accumulation in the liver and spleen as expected (~12–15% injected dose [ID] g−1), in addition to passive accumulation in the tumor site (~5% ID g−1). In vivo photothermal studies were then conducted by intravenously injecting tumor-bearing mice with rGO-IONP-PEG and then irradiating the tumors after 48 h with 808 nm laser (0.5 W cm−2) for 5 min. The surface temperature of the tumors in nanocomposite injected mice increased to 48 °C after laser exposure, as compared to 39 °C in untreated mice after laser exposure. Moreover, the tumors were ablated after photo-irradiation without regrowth over 40 days, whereas the control groups showed rapid growth and eventually led to death within 20 days.
In another study, IONPs were replaced with Au plasmonic nanostructures to form GO–Au nanocomposites.209 Given that Au nanostructures absorb NIR light and convert the electronic oscillations at the particle surface to heat, the combination with GO was observed to enhance photothermal effects. In contrast to assembling nanoparticle structures on GO, Au nanoshells (AuNS) (150 nm in diameter) and Au nanorods (AuNR) (10 nm width, aspect ratio = 3.5) were coated with rGO (Figure 31A–D). The photothermal performance of the AuNS and AuNR with and without rGO was studied in the dry state and solution state under NIR illumination (808 nm, 3.0 W cm−2). In the dry state for both particle types, rGO coating resulted in an approximately 2.9-fold increase in the temperature after photoirradiation. The solution state showed a similar trend, with a temperature change of 42.3 ± 0.2 °C for rGO–AuNS and 47.5 ± 0.2 °C for rGO–AuNR over 5 min, both of which were greater than the rGO and Au nanostructures alone (Figure 31E,F). To explain this enhancement, the authors suggested that the AuNS and AuNR could serve as both photothermal sources through nonradiative decay as well as local nanoantennae to enhance the optical energy absorption of graphene at select plasmon frequencies.210 Finally, it was reported that human umbilical vein endothelial cells (HUVECs) exposed to the nanocomposites showed an irradiation time-dependent decrease in cell viability, wherein 1 min of irradiation led to a sharp decrease in cell viability for RGO–AuNS treated (to 23%) and RGO–AuNR treated (to 33%) conditions (Figure 31).
Figure 31.
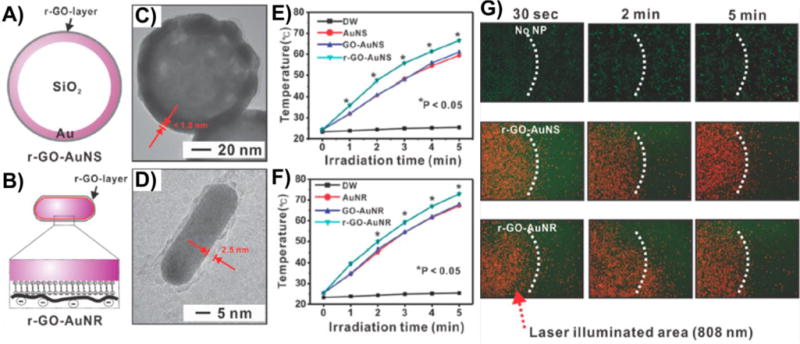
Plasmonic nanoparticles coated with rGO for photothermal therapy. Schematic representation of (A) rGO coated Au nanoparticles (rGO–AuNS) and (B) rGO coated Au nanorods. HR-TEM image of (C) rGO–AuNS and (D) rGO–AuNR. (E and F) Solution temperature changes over time upon irradiation (3.0 W cm−2, 808 nm) of solutions of coated and uncoated nanoparticles and distilled water (DW). (G) Photothermal cell killing with rGO–AuNS and rGO–AuNR, measured by live/dead assay after 30 s, 2 min, and 5 min of irradiation (3.0 W cm−2, 808 nm). The dotted white line represents the boundary of the irradiated area. Reprinted with permission from ref 209. Copyright 2013 American Chemical Society.
Further advancing upon these hybrid platforms, Shi et al. generated a dual-nanostructured composite by combining both IONPs and Au nanostructures on GO.211 In this case, a layer of Au nanostructures was first synthesized on GO–IONPs, and then the overall hybrid was functionalized with PEG (Figure 32A). These GO-IONP-Au-PEG nanocomposites allowed for efficient PTT as well as dual-modal MRI (magnetic IONPs) and X-ray computed tomography (CT) imaging (X-ray absorbance from Au). In vitro studies in 4T1 breast cancer cells showed that almost all cancer cells were killed after incubation with the hybrids and exposure to an 808 nm laser (2 W cm−2 for 5 min). Mice bearing 4T1 tumors were then intratumorally injected with GO-IONP-Au-PEG to assess in vivo imaging and PTT. T2-weighted MR and CT images showed a clear contrast in the tumor of the hybrid-injected mice. Tumor-bearing mice were then injected with either GO-PEG or GO-IONP-Au-PEG at the same dose of GO (50 ug mL−1), and subjected to an 808 nm laser at 0.75 W cm−2 for 5 min as controls. IR thermal images revealed that the surface temperature of the tumors injected with the nanocomposites rapidly increased to about 55 °C within 5 min of laser irradiation (as compared to 45 °C for GO-PEG and 38 °C for PBS only) (Figure 32B). Measurements of the tumor volume after irradiation showed effective ablation in the nanocomposite condition, whereas other control conditions (saline and GO-PEG, with and without irradiation) showed a continuous increase in tumor volume.
Figure 32.
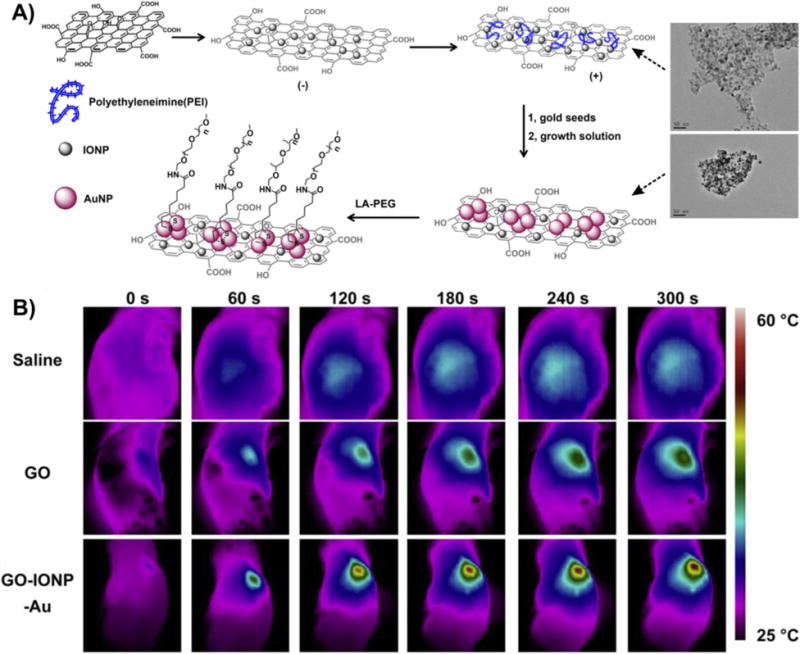
GO–AuNP composite for dual bioimaging and photothermal therapy. (A) Schematic illustration of GO-IONP-Au nanocomposite synthesis. (B) IR thermal images of tumor-bearing mice injected with saline, GO-PEG, or GO-IONP-Au-PEG under laser irradiation (808 nm, 0.75 W/cm2). Reprinted with permission from ref 211. Copyright 2013 Elsevier.
Last, to monitor the increase in temperature and the progress of PTT, Hu et al. designed QD-tagged reduced GO (QD–rGO) nanocomposites.212 In this case, highly fluorescent CdSe/ZnS core–shell QDs, capped with a surfactant (TOPO) and a self-assembled monolayer of 11- mercaptoundecanoic acid (MUA), were assembled on 260 nm small rGO (S-rGO) and 38 nm ultrasmall rGO (US-rGO). FA was further tethered to the composites to enhance uptake in FR-expressing cells. As compared to the QD–S-rGO composites, the QD–US-rGO exhibited higher fluorescence for cell imaging as well as greater cellular uptake. Interestingly, increasing the time of exposure to an 808 nm laser (2 W cm−2) not only resulted in cellular death, but there was a simultaneous loss of QD fluorescence. The loss in fluorescence was speculated to be due to the degradation of the MUA coating under excessive heating, which in turn reduced the QD protection and led to oxidation. In this way, the QD–rGO composite provided orthogonal functionalities, in which the cell death occurred from the local heating of rGO and the progress of the PTT was monitored and quantitatively estimated by measuring the decrease in QD fluorescence intensity.
4.3. Delivery of Drugs and Genes
The delivery of small molecule drugs and biomacromolecules (e.g., proteins, peptides, genes) to specific tissues of interest has been a fundamental approach in medicine. As compared to the conventional method of oral administration or intravenous injection, significant effort has recently been dedicated to enhancing bioavailability, achieving targeted delivery, more effectively solubilizing drugs/biomolecules of interest, and protecting drugs/biomolecules from enzymatic degradation.213 In this regard, nanomaterial-based delivery platforms have revolutionized drug delivery systems, providing benefits from both the effective delivery and the imaging standpoints.214 Since the discovery of graphene in 2004, there has been increasing interest in loading graphene-based nanomaterials for the delivery of drugs/genes due to graphene’s high specific surface area and unique modes of complex formation including π–π stacking, electrostatic interactions, and hydrophobic interactions.186 More recently, the well-controlled modification of graphene materials with nanoparticles to form graphene–nanoparticle hybrids has been applied to improve their utility for both in vitro cancer cell assays and in vivo chemotherapies. In particular, as with the other cellular applications mentioned so far, the formation of graphene–nanoparticle hybrids can prevent aggregation and provide additional functionalities over graphene or nanoparticles alone. Additionally, graphene–nanoparticle hybrids can greatly improve the drug loading capacity over graphene materials alone due to increases in overall surface area. To this end, early work focused on optimizing synthetic procedures to achieve high drug loading of poorly soluble drugs while maintaining maximal potency. For example, Yang et al. formed a GO–Fe3O4 hybrid to investigate the binding of the anticancer agent doxorubicin (DOX).215 The hybrids were prepared via chemical deposition, followed by conjugation of DOX via sonication and overnight stirring resulting in a drug loading capacity as high as 1.08 mg mg−1. Recent work with a hybrid containing IONPs, AuNPs, and GO further improved the DOX loading capacity to 6.05 mg mg−1, which is significantly higher than GO alone (2.35 mg mg−1).216 While a range of drug loading capacities have been demonstrated, the maximum achievable loading is heavily dependent on the synthetic conditions employed to prepare the hybrids, the chemical composition of individual components (e.g., GO, RGO, Fe3O4, Au, etc.), and the structure/composition of the small molecule drug.
While attaining sufficient drug loading is a key criterion for achieving a therapeutic effect, the effective delivery of the hybrid nanocarrier into the desired cancer cell/tissue is equally important. In one study, gold nanocluster (GNC)–rGO nanocomposites were loaded with DOX to investigate the effect on hepatocarcinoma (HepG2) cells.217 Specifically, GNCs ranging between 2 and 3 nm in diameter were noncovalently attached onto the rGO surface resulting in GNC–rGO nanocomposites that were less toxic than GNCs alone (IC50 values: 1.36 μg mL−1 for GNC–rGO versus 0.36 μg mL−1 for GNCs). Next, DOX was loaded onto the GNC–rGO nanocomposites at a 91% drug loading efficiency, where the concentration of DOX was 0.22 mg mL−1 and that of GNC–rGO was 1 mg mL−1. The authors found that the DOX-loaded GNC–rGO inhibited HepG2 cell growth more strongly than DOX and GNC–rGO alone, which suggested that DOX was more effectively transported into the cell by the GNC–rGO nanocomposite than when used alone. Moreover, while DOX treatment alone led to DOX molecules resting on the cellular membrane, the DOX-loaded GNC–rGO allowed DOX to be well-distributed inside the cells, thus suggesting the importance of such a composite vehicle to enhance membrane permeability. In a similar study, NGO sheets were wrapped onto the surface of individual AuNPs and then noncovalently bound with DOX for delivery into HeLa cells (Figure 33A).218 This methodology generated Au@NGO composites with a hydrodynamic size of 133.5 nm in culture media, a size range that allowed for facile cellular delivery. The intrinsic SERS signal from the Au@NGO further allowed for Raman imaging, which confirmed the efficient delivery and distribution of the nanocomposites in the cytoplasm. Interestingly, sustained release of the drug was observed in the DOX-loaded Au@ NGO composite condition, wherein the red fluorescence (DOX) within the nucleus gradually increased over 48 h and became higher than that seen in the cytoplasm (Figure 33B). In contrast, treatment with an equivalent amount of DOX showed fast nuclear accumulation within only 2 h, with no further enhancement in fluorescence over time. Moreover, DOX-loaded Au@NGO induced a significant decrease in HeLa cell viability when compared to free DOX (Figure 33C). This work suggests that while treatment with free DOX led to a burst dosing profile, the Au@NGO composite allowed for sustained release within the cancer cells, showing promise for long-term therapeutic efficiency.
Figure 33.
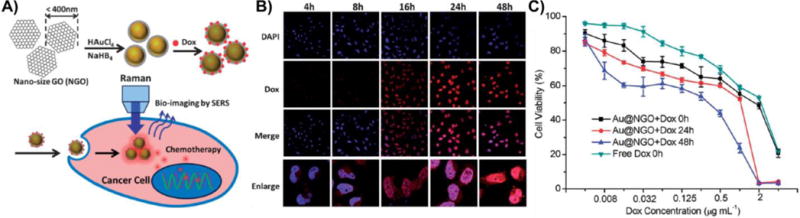
GO wrapped AuNPs for intracellular Raman imaging and drug delivery. (A) Illustrative mechanism of SERS-based bioimaging and anticancer drug delivery by using Au@NGO. (B) Confocal laser scanning microscopy (CLSM) images of HeLa cells treated with DOX-loaded Au@ NGO (Au@NGO + DOX, 25 μg mL−1) for different times. (C) MTT cytotoxicity assay of HeLa cells after being treated with DOX-loaded Au@ NGO (Au@NGO + DOX) and free DOX. Reprinted with permission from ref 218. Copyright 2013 Royal Society of Chemistry.
Targeted delivery of therapeutic agents is also highly desirable because it can allow for higher bioavailability of the agent at its site of action, while reducing side effects.219 As compared to passive transfection into cancer cells, a number of studies have attempted to chemically or physically modify the drug-loaded hybrid nanocomposite to enhance cellular internalization. Fan et al. prepared a graphene-CNT-MNP (Fe3O4) nanocomposite to investigate the delivery of the anticancer drug 5-fluorouracil (5-FU) into HepG2 cells.220 While the high specific surface area of graphene allowed for higher drug loading than graphene-based drug carriers alone and the IONPs imparted superparamagentic behavior to the nanocomposite, the incorporation of CNTs was found to enhance transportation of the graphene-CNT-Fe3O4 hybrid across the cell membrane. TEM images comparing magnetic CNT nanocomposites (CNT–Fe3O4) and magnetic graphene nanocomposites (graphene–Fe3O4) showed distribution of the CNT–Fe3O4 nanocomposites in the cell cytoplasm but graphene–Fe3O4 only outside of the cell. The study further showed dose-dependent in vitro cell toxicity studies using the graphene-CNT-Fe3O4 for fluorouracil (5-FU) drug delivery, providing further evidence for improved cellular delivery. On the other hand, to achieve a more targeted drug delivery, Chen et al. designed a QD-graphene-based hybrid composite that was conjugated with the transferrin (Trf) ligand.221 Specifically, Trf ligands are known to bind with high affinity to Trf receptors, which are expressed on the plasma membrane of various types of cancer cells.222 DOX-graphene-QD-Trf (loading capacity 1.4 mg mg−1) was incubated in a Trf positive cell line (HeLa) and a Trf negative cell line (HEK293). The DOX-graphene-QD-Trf exhibited increased toxicity to HeLa cells as compared to the HEK293 cells, while the DOX-graphene-QD and DOX-graphene-QD-Trf showed a similar effect on HEK293 cell viability. This effect was attributed to the specific Trf receptor recognition, which suggested that the ligand-specific conjugation of the nanocomposites could selectively increase drug cytotoxicity. Moreover, the QDs (CdTe–CdS nanocrystal clusters) allowed for fluorescence imaging, thus providing the capability to track and monitor drug delivery.
In addition to targeted therapies, remotely triggerable drug delivery systems have become popular because they enable the user to adjust dosing regimens on-demand,223 based on a patient’s physiological response. In a recent study, He et al. describe a photocontrolled release system by incorporating a photoacid generator (PAG) into DOX-loaded, GO-capped MSNPs (Figure 34A,B).224 In particular, PAG can generate strong acid via illumination with UV or near-UV light.225 As such, by assembling GO as nanogates on the MSNPs with acid-labile boroester linkers, the PAG was used to generate a locally high concentration of H+ upon illumination. This, in turn, induced the hydrolysis of the boroester linkers to open the pores (by releasing the GO cap) and release the loaded DOX. The nanocomposites were further conjugated with folate, which allowed for targeted delivery to cancer cells overexpressing FRs. Selective cell death from the DOX-loaded nanocomposites was indicated by the significant cell death of approximately 80% in HeLa cells (high expression of FRs), but a negligible change in cell viability of L02 cells (low expression of FRs) (Figure 34C).
Figure 34.
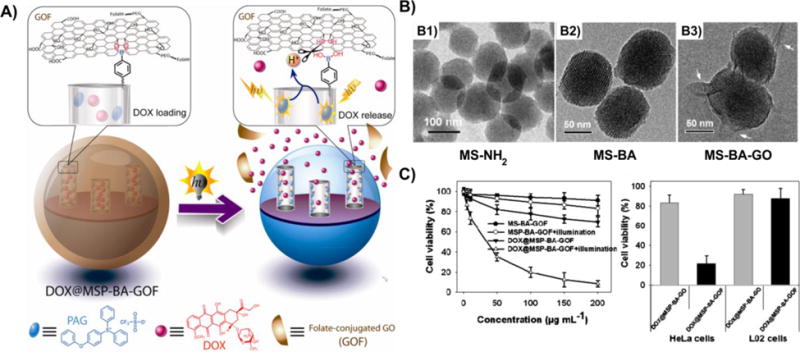
Remote-controlled drug release from GO-capped mesoporous silica to cancer cells by photoinduced pH-jump activation. (A) Schematic illustration of DOX@MS-BA-GOF as a drug delivery system for remote light control of drug release. (B) TEM images of amine-terminated mesoporous silica (b1), boronic acid-grafted MS (b2), and GO-capped MS-BA (b3). (C) Viability of HeLa cells after being incubated with different nanoparticles (MS-BA-GOF, MSP-BA-GOF, and DOX@MSP-BA-GOF). Cytotoxicity of DOX@MSP-BA-GOF and DOX@MSP-BA-GO incubated with HeLa cells and L02 cells for 24 h. Reprinted with permission from ref 224. Copyright 2014 American Chemical Society.
While the majority of studies on graphene-based nanocomposites for drug delivery applications have employed inorganic nanoparticles for complexation with GO, Wang et al. were among the first to demonstrate the use of a soft nanoparticle in this regard, by adsorbing zwitterionic dioleoyl-sn-glycero-3-phosphocholine (DOPC) liposomes onto NGO.226 The rationale for using DOPC liposomes was to adsorb multiple molecules on the same surface with little intermolecular competition, thus allowing for highly effective drug loading and controlled release. The nanoscale heterogeneity of the GO surface proved to be ideal, wherein DOX was loaded using the well-established hydrophobic interactions that occurs with the basal aromatic surface. On the other hand, the DOPC liposomes were speculated to adsorb using hydrogen bonding and hydration forces with the peripheral carboxyl groups. As such, DOPC liposomes may be adsorbed on GO at different sites from DOX, thereby avoiding competition. Interestingly, the DOPC/NGO complex not only exhibited improved aqueous dispersion and colloidal stability, but also allowed for an increased loading capacity of DOX (~500%) as compared to loading on only GO (~300%), which indicated that the DOPC liposomes did not interfere with the drug loading. In addition, it was also found that while free DOPC liposomes were not internalized by HeLa cells, the DOPC/NGO complex had high colloidal stability and readily entered the cells. Finally, the DOPC liposomes were simultaneously loaded with a calcein dye and delivered to HeLa cells, which exhibited a decrease in cellular viability (due to DOX) and green fluorescence from calcein. As such, this new soft nanoparticle-based GO hybrid system exhibited the ability to codeliver molecules with opposite properties (e.g., DOX is cationic and hydrophobic while calcein is negatively charged and hydrophilic) using a single platform.
Last, while the delivery of small molecule drugs has been extensively explored using graphene–nanoparticle composites, there have been limited studies demonstrating their use for gene delivery. PEGylated, polyethylenimine (PEI)-grafted graphene/Au composites (PPGA) have recently been used for the delivery of small interfering RNA (siRNA) into human promyelocytic leukemia cells (HL-60).227 The polymer coatings served specific functions, wherein the cationic PEI was used to bind electrostatically with negative charged siRNA while the PEG promoted high water dispersibility and excellent blood compatibility. The PPGA composites showed biocompatibility for in vitro cultures when used at concentrations below 20 μg mL−1, as well as knockdown of Bcl-2 protein expression at a PPGA/siRNA mass ratio of 100:4. Moreover, because these composites also contained AuNPs, the photothermal effect of PPGA was demonstrated under NIR laser irradiation. Finally, Wang et al. developed a strategy to combine chemotherapy and gene therapy using a single chitosan magnetic–graphene (CMG) nanocomposite platform.228 The CMG nanocomposites were synthesized by the in situ growth of IONPs on the rGO surface, followed by the covalent binding of chitosan. Thereafter, DOX was physically adsorbed onto the CMG composites (via interaction with GO) and a green fluorescent protein (GFP) reporter DNA plasmid (via interaction with positively charged chitosan). These DOX-CMG-GFP complexes (30 μg of DOX and 25 μg of GFP-pDNA per mouse) were administered intravenously to LLC1 tumor bearing mice, whereby the presence of DOX (red) and GFP (green) expression was observed in frozen mouse sections (Figure 35) after 24 and 48 h. Biodistribution studies further showed significant accumulation in the tumor site, with lower accumulation in the liver and spleen. Moreover, the MNPs in the CMG composites allowed for MR imaging.
Figure 35.
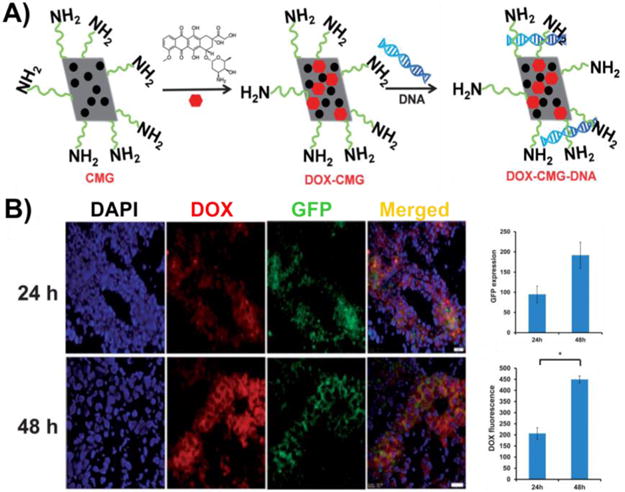
Multifunctional chitosan magnetic-graphene nanoparticles for tumor-targeted codelivery of drugs, genes, and MRI contrast agents. (A) Schematic showing the generation of DOX-loaded chitosan functionalized magnetic graphene complexed with DNA plasmids encoding GFP (DOX-CMG-GFP-DNA) complexes. (B) GFP expression and DOX fluorescence in frozen sections from mice injected with DOX-CMG-GFP-DNA (30 μg of DOX and 25 μg of GFP-DNA), examined using a fluorescent microscope at 400× magnification. Expression was quantified and normalized to control background. Reprinted with permission from ref 228. Copyright 2013 Royal Society of Chemistry.
4.4. Multifunctional and Multimodal Approaches
Graphene–nanoparticle hybrid composites are robust and versatile platforms that can be used in numerous distinct biological applications as described above, including imaging, PTT, and drug/gene delivery. In particular, the ability to exploit the intrinsic properties of these hybrids to achieve a combined, multifunctional therapy has drawn immense attention. As an example, combining chemotherapeutic drugs and other molecules with hyperthermia has been a common approach to enhance therapeutic efficacy.229 Dai and co-workers initially demonstrated such an integrated methodology using ultrasmall iron–cobalt (FeCo) MNPs (~4 nm) coated with a single- or few-layer graphitic carbon shell (GC).230 The rationale for this platform was that the highly magnetic FeCo core (r2 = 185 mM−1 s−1) would provide a high saturation magnetization for MRI contrast, while the GC shell would enable DOX loading (via π–π stacking) as well as absorbance of NIR light for photothermal effects. The FeCo/GC nanocrystals showed a pH-dependent release, wherein the release of DOX from FeCo/GC was observed to be a slow, continuous process that was about 3 times greater at pH 5.5 than pH 7.4 after 8 days. Moreover, the FeCo/GC had a slower DOX release profile than single-walled CNTs (~1 nm diameter). This was possibly caused by the stronger interaction between DOX and the graphitic sidewall of the hybrids as well as the more planar surface of the GC used in this system, which resulted in higher drug loading and slower release. Thermal imaging of the FeCo/GC solution further showed a temperature increase of ~35 °C following only 10 min of NIR light exposure (808 nm, 0.3 W cm−2). In vitro studies with MCF-7 breast cancer cells, which employed PTT with DOX-loaded nanocrystals, more than doubled the toxicity of the FeCo/GC-Dox, causing more cell death than DOX in its free form with or without NIR irradiation. Similarly, Ma et al. demonstrated an approach for a combination therapy using GO–magnetic IONP hybrids for DOX delivery, PTT, and in vivo MR imaging in 4T1 tumor-bearing mice.231 Last, the codelivery of multiple anticancer drugs, camptothecin (CPT) and methotrexate (MTX), using IONP–GO hybrids in combination with NIR-induced PTT was recently demonstrated in vivo to achieve a tumor inhibitory rate of 73.9% in S-180 sarcoma-bearing Balb/c mice.232
Utilizing nanoparticles with varying compositions to construct graphene–hybrid nanostructures can permit enhanced effects upon photoirradiation. For instance, high photothermal energy conversion efficiencies have been reported for hybrids composed of gold nanostructures with GO or rGO.209,233 To this end, NGO-enwrapped AuNRs were observed to not only increase the solution temperature to above 60 °C within 3 min of NIR light exposure (808 nm, 4 W cm−2) but also had enhanced biocompatibility over CTAB-coated AuNRs alone.234 At a higher power intensity of 6 W cm−2, the temperature of the solution (0.2 mg mL−1) increased to 75 °C within 2 min. The further loading of DOX and targeted delivery to hepatoma Huh-7 cells led to 82% cell death after NIR irradiation, which was about 1.5-fold and 4-fold more efficient than separate treatments of chemotherapy and PTT, respectively. However, by exploiting the unique luminescence properties of UCNPs, Zhang and co-workers reported a theranostic platform that afforded NIR-based imaging and both PTT and photodynamic therapy (PDT).235 While PTT uses light to induce local heating, PDT employs light to activate a photosensitive drug to facilitate the generation of reactive oxygen species. After covalently grafting the UCNPs with NGO, the nanocomposites were loaded with the photosensitizer phthalocyanine (ZnPc). These UCNP-NGO/ZnPc hybrids allowed for (1) imaging of cells and whole-body animals with upconversion luminescence, (2) conversion of 808 nm laser energy (2 W cm−2) into thermal energy for PTT (heating to 58 °C within 3 min), and (3) generation of cytotoxic singlet oxygen from ZnPc under 630 nm light exposure for PDT. Importantly, the authors found that a synergistic effect could be achieved due to the combination of noninvasive PDT and PTT, thereby improving therapeutic efficiency and decreasing the dose-limiting toxicity and tissue damage from overheating (50% and 75% cell viability for PDT and PTT alone, respectively, versus 15% when combined using UCNP-NGO/ZnPc hybrid). Such integrated platforms allow for the integration of diagnostics and therapies in a single nanocomposite.
Graphene-based MSNPs have also served as a reliable nanocomposite for multifunctional therapies. Wang et al. developed a mesoporous silica-coated graphene nanosheet for the targeted treatment of glioma cells.236 Mesoporous silica generated on GO sheets was highly loaded with DOX (1.27 μg of DOX per μg of composite), attributed to encapsulation within the mesopores as well as adsorption on the GO surface. As for the photothermal effects, irradiation with NIR light (808 nm, 6 W cm−2) caused the temperature of the nanocomposite solution (50 μg mL−1) to exceed 50 °C within 2 min. The surface was further modified with a targeting peptide against IL-13 protein, which targets the receptor chain 2 of interleukin 13 that is overexpressed in malignant tumors including glioma. In vitro incubation with U251 glioma cells showed that combining hybrid composite-based DOX delivery with NIR irradiation mediated the highest rate of cell death (combination index = 0.504 [<1]) as compared to a single treatment of the composite with DOX only (chemotherapy alone) or the composite with NIR irradiation only (PTT alone). In a more recent study, Wang et al. modified the above-described system to further incorporate MNPs (Fe3O4) for magnetic targeting and MRI.237 The targeting peptide-modified magnetic graphene-based mesoporous silica (MGMSPI) showed enhanced targeting with a magnet in glioma-bearing mice, and NIR-treated, DOX-loaded MGMSPI showed the maximal decrease in cell viability when administered to in vitro U251 cultures.
Finally, while MSNPs assembled on GO enhance drug loading due to the high surface area to volume ratio, further coating with amorphous carbon produced “nanocookie-like structures” that enabled on-demand, NIR-responsive chemotherapy and hyperthermia (Figure 36A–C).238 The loading capacity of a model hydrophobic drug, CPT, was found to be 0.878 mmol g−1, which was much higher than that for rGO (0.433 mmol g−1) and porous silica (0.336 mmol g−1). This is likely due to the affinity between CPT and the carbonaceous surface, along with the presence of the massive surface area from the porous SiNP support (~720 m2 g−1). Interestingly, irradiation with NIR light (808 nm, 2 W cm−2) not only induced local heating (due to GO), but also facilitated burst-like drug release. This feature proved to be critical to enhance combined therapy, in which in vitro studies with MDA-MB-231 breast cancer cells revealed ~90% cell death with a total nanocookie-CPT concentration of 0.878 μM and 5 min of NIR irradiation. Further studies with nude mice bearing MDA-MB-231 tumor cells showed that the size of the tumor decreased in the nanocookie + NIR and nanocookie-CPT + NIR treated mice in the first 14 days, while it increased in other treatments (Figure 36D). In addition, after 14 days, the tumor size of nanocookie + NIR treated mice began to increase again, but the nanocookie-CPT + NIR treated mice showed a continual decrease in tumor size and the tumors eventually disappeared after 30 days (Figure 36E). Overall, this type of recent work provides strong evidence that combining small molecule drugs, hyperthermia, and imaging in a single multifunctional graphene–nanoparticle composite can enhance cancer therapeutics.
Figure 36.
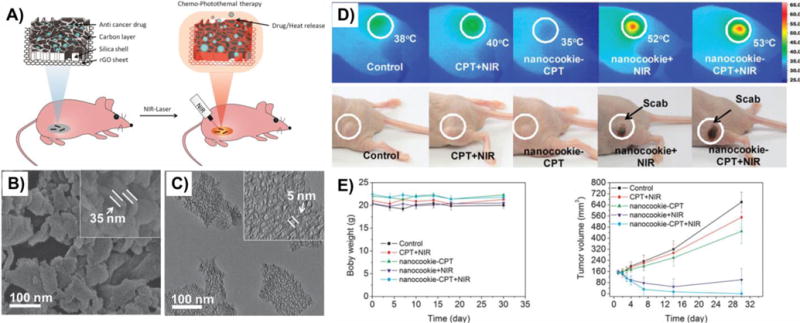
rGO/carbon/mesoporous silica nanocookies for NIR-triggered photochemothermal therapy. (A) Schematic illustration of chemo-photothermal therapy using reduced graphene oxide/carbon/mesoporous silica nanocookies, under NIR light-control. (B) FE-SEM and (C) low magnification TEM images of nanocookies. Inset: High magnification images of a nanocookie. (D) Infrared thermal images of a MDA-MB-231 tumor-bearing nude mouse treated with PBS+NIR (control), CPT+NIR, nanocookie-CPT (no NIR), nanocookie+NIR, nanocookie-CPT+NIR treatment. Color bar on right shows temperature in °C. Pictures at day 4 after NIR irradiation (808 nm, 0.75 W cm−2, 5 min, 1 min interval for every min treatment) show the tumor turned into a scab at the site injected with nanocookie+NIR and nanocookie-CPT+NIR. White circles represent tumor sites. (E) Changes in body weight and tumor volumes following treatment. Reprinted with permission from ref 238. Copyright 2014 Wiley.
4.5. Stem Cell and Tissue Engineering Approaches
Tissue engineering is a growing field of study that combines living cells and biocompatible materials to create constructs that can potentially repair or replace the function of living tissues and organs.239 In regard to living cells, stem cells are an attractive cell source for tissue engineering applications because they exhibit the ability to self-renew and differentiate into specific cell lineages. However, guiding stem cell differentiation toward a desired cell type tends to require precise control over the biochemical and physical microenvironmental cues. In this regard, engineering cellular microenvironments to promote stem cell attachment, growth, viability, and differentiation has been a major area of focus. As such, graphene has proven to be a promising candidate for stem cell engineering, exhibiting excellent biocompatibility and tunable functionalities for cultures of embryonic stem cells (ESCs), induced pluripotent stem cells (iPSCs), mesenchymal stem cells (MSCs), and neural stem cells (NSCs).10a,240 Moreover, recent work has further explored ways to arrange graphene-based nanomaterials into three-dimensional architectures, with the goal of designing tissue-like transplantable scaffolds.241 With the increasing interest in adding greater functionality to cellular scaffolds, a handful of studies have investigated the potential of graphene–nanoparticle hybrids for stem cell and tissue engineering with great success.
Graphene-coated surfaces have been found to be conducive to protein attachment, potentially due the hydrophobic and hydrophilic patches that are found on the surface of proteins.242 As mentioned previously, an early study conducted by Deng et al. demonstrated the incorporation of proteins on the GO surface to efficiently assemble nanoparticles of different size, shape composition, and surface properties.102 Using BSA as a model protein, BSA-coated GO were used to assemble presynthesized metallic nanoparticles (Au, Pt, Pd, Ag) and/or nonmetallic nanoparticles (latex). The work concluded that multiple interactions mediated via specific chemical groups led to the adsorption of protein (e.g., cysteine, lysine, histidine residues) and that the GO surface could serve as a “universal adhesive” to facilitate the attachment of nanomaterials and potentially cells.
The nanoscale topography of hybrid surfaces can also present a unique extracellular microenvironment to control stem cell behavior (e.g., cell shape, adhesion, proliferation, and differentiation).243 For this purpose, nanotopographical substrates composed of rGO and positively charged polysaccharide chitosan have been fabricated with the hypothesis that the unique, biocompatible nanotopography could provide an effective environment for the differentiation of human MSCs.241b The mixture of rGO and chitosan at varying ratios (0–5% w/w) produced nanoparticle-like composites, which, upon spin-coating onto bare glass substrates, introduced surface roughness that ranged from 0.9 to 7.7 nm. While it was observed that the presence of higher concentrations of graphene on the substrates decreased the proliferation rate of human MSCs, concentrations of less than 0.1 mg mL−1 had minimal cytotoxicity. On the other hand, cellular adhesion was found to be greatest on the 5% rGO–chitosan substrates and provided a suitable environment for proliferation. These nanocomposite substrates were further observed to enhance bone differentiation, both with and without osteogenic-induction media, as well as neurogenesis under neurogenic conditions. In this way, the unique nanoscale topographical cues of the graphene–chitosan nanocomposites are likely to play a crucial role for human MSC differentiation, although the exact mechanism is not yet understood. Along these lines, in a recent study from our group, Solanki et al. generated arrays of graphene–nanoparticle hybrid structures to guide the differentiation and alignment of human NSCs.244 Positively charged SiNPs (300 nm) were assembled on glass substrates using a centrifugation process, after which negatively charged GO was deposited (Figure 37A). These hybrid films were further coated with the extracellular matrix protein laminin to facilitate adhesion and growth of human NSCs. After 2 weeks of culture, the graphene–nanoparticle hybrid nanostructures were observed to promote a higher efficiency of differentiation into the neuronal cell lineage. Interestingly, cellular extensions were seen to spread in a unidirectional manner after 5 days of culture. This axonal alignment was observed to be maximal on the hybrid films, in which the SiNP–GO substrates showed the smallest variation in the angle of orientation of axons as compared to control substrates (Figure 37B). This behavior was further observed on hybrid films generated on flexible and biocompatible polymeric substrates, underscoring the potential of using the graphene–nanoparticle hybrids for guided neural tissue engineering. However, the authors note that further study is required to fully understand the underlying mechanisms governing the observed neuronal differentiation and axonal alignment.
Figure 37.
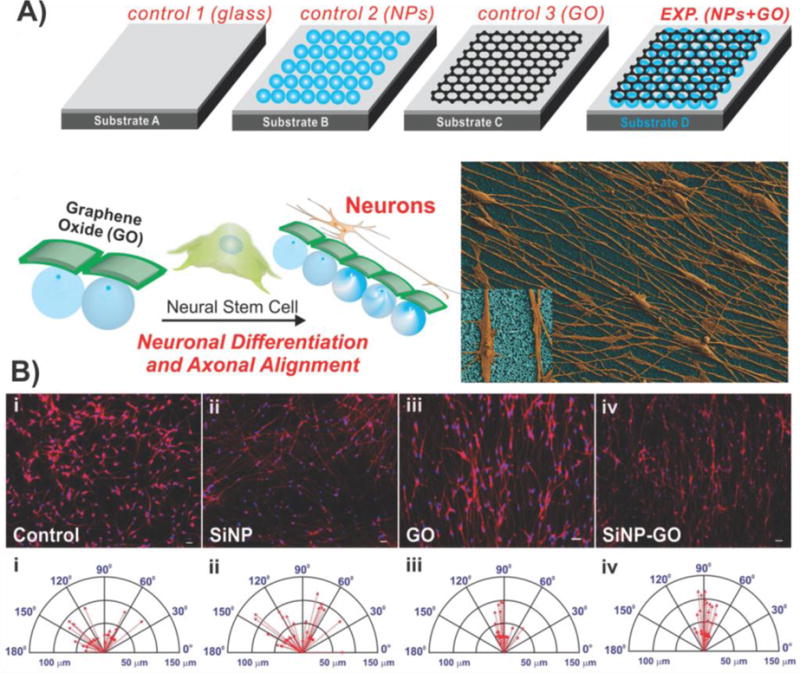
Graphene–nanoparticle hybrids to guide neural stem cell differentiation and axonal alignment. (A) Schematic diagram depicting the varying conditions and influence of nanoparticle (NP) monolayers coated with graphene oxide (GO) on the alignment of axons extending from human NSCs, and the differentiation into neurons. The differentiated human NSCs (orange) and the NPs-coated with GO (blue) in the SEM image show enhanced neuronal differentiation and axonal alignment. (B) Differentiated human NSCs, immunostained with TuJ1 (red), show no alignment of axons on glass (control) and SiNP films, whereas the axons are significantly aligned on GO and SiNP-GO films. Scale bar: 10 um. Compass plots show a large variation in the angle of orientation of axons on glass (±42°) and SiNPs (±46.11°) and minimal variation on GO (±17.8°) and SiNP-GO (±9.16°). Plots also show that axons extending on SiNP and SiNP-GO are longer than those extending on glass and GO. Reprinted with permission from ref 244. Copyright 2013 Wiley.
Finally, an essential step in conducting stem cell studies is the ability to monitor the differentiation process and identify the stage of differentiation, which can give insight into the development of effective stem cell-based therapies. Many of the conventional techniques used to distinguish undifferentiated versus differentiated stem cells, such as immunostaining, fluorescence-activated cell sorting (FACS), and Western blotting, tend to be laborious, time-consuming, and destructive. A graphene–nanoparticle platform based on SERS detection was recently shown to be an effective and highly sensitive tool that could distinguish undifferentiated and differentiated stem cells, while maintaining cell viability.245 In particular, GO-encapsulated AuNP films were generated on ITO surfaces, followed by use for the culture of mouse NSCs. On the basis of several reports, undifferentiated stem cells have been found to contain molecules bearing a high number of C=C bonds (high degree of saturation) as compared to differentiated cells.246 Given that such bonds have strong affinity to GO, the GO–Au nanoparticle films were effectively used to measure the SERS signal and distinguish such chemical heterogeneity between C=C bonds (1656 cm−1) and C–H bonds (1470 cm−1). By measuring differences in the SERS signal, the differentiation state of the NSCs was detected and validated via immunostaining. These results were further verified by detecting the electrochemical signals with CV in a microgap configuration containing GO–AuNP films. As such, the use of GO–nanoparticle composites provided unique multifunctionalities wherein the GO–AuNP films could act as a biocompatible substrate for cell differentiation as well as SERS and electrochemical detection to provide a noninvasive and effective method for the in situ monitoring of stem cell differentiation.
4.6. Biocompatibility of Graphene–Nanoparticle Hybrid Materials
Despite the recent rise in popularity of graphene–nanoparticle hybrid materials, for these materials to reach their full potential for bioapplications, especially those involving cellular interactions, they must first prove to be biocompatible both in vitro and in vivo. A limited number of studies have focused on the biocompatibility of graphene–nanoparticle hybrids; however, in this section, we will review what has been reported to date. In particular, we will begin by highlighting what is currently known about the biocompatibility of graphene materials in vitro and in vivo, where significantly more work has been performed. Following this, we will review the present understanding of the biocompatibility of graphene–nanoparticle hybrid materials with a primary focus on in vitro studies utilizing mammalian cell models and preliminary in vivo studies, which have predominantly concentrated on the use of mouse models of human cancers.
4.6.1. Biocompatibility of Graphene Materials
A significant number of studies have investigated the biological effects of graphene and its derivatives.247 In particular, the biocompatibility of graphene materials has been found to depend significantly on their physicochemical properties (e.g., hydrophilicity, physical dimensions), concentration, time of exposure, and route of administration. In vitro, hydrophilic forms (e.g., GO) and small sizes of graphene are better able to penetrate the cell membrane, where they can translocate into organelles in the cytoplasm (e.g., lysosomes, mitochondrion, endoplasm, and nucleus),248 and are generally less toxic than hydrophobic forms (e.g., graphene and rGO) and larger sizes of graphene (Figure 38). For example, Wang et al. reported that the delivery of GO (up to 10 μg mL−1) only slightly decreases the viability of human fibroblast cells (less than 20% even when exposed for 4 days) (Figure 38A).248 However, as would be expected, increasing the exposure time and concentration of graphene led to further decreases in cell viability, where it was reported that GO concentrations above 50 μg mL−1 exhibited obvious cytotoxicity even after only 1 day of exposure (>20%).248 On the other hand, pristine graphene, which is highly hydrophobic, can also result in significant toxicity, wherein exposure of Vero cells to 10 μg mL−1 of pristine graphene for 24 h resulted in a 10–15% decrease in cell viability (as compared to <5% decrease for GO from Wang et al.’s study).249
Figure 38.
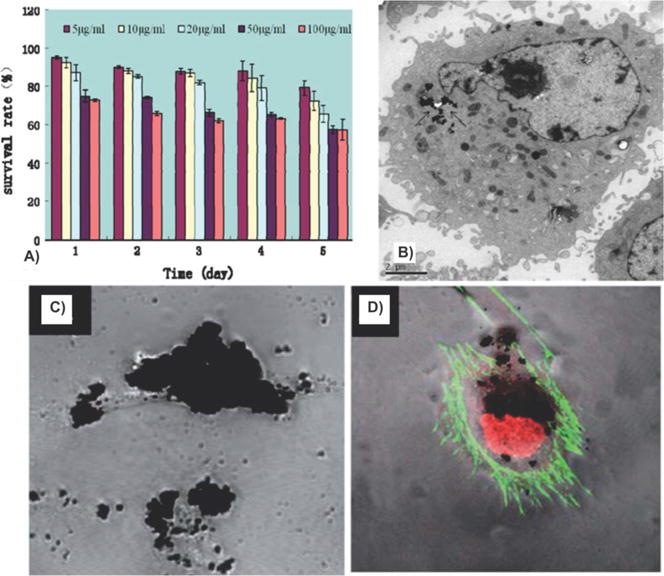
Biocompatibility of graphene materials. Effects of GO on human fibroblast cells. (A) The survival rate at different concentrations of GO and at different periods of exposure. (B) TEM picture showing the location of GO inside human fibroblast cells as indicated by the black arrows. Reprinted with permission from ref 248. Copyright 2011 Springer. (C) Differential interference contrast image showing the accumulation of pristine graphene on the plasma membrane of Vero cells. (D) Fluorescence confocal microscopy of cytoskeletal F-actin arrangement of cells treated with pristine graphene. Reprinted with permission from ref 249. Copyright 2011 Royal Society of Chemistry.
In terms of their ability to be uptaken by cells, possible explanations for the dependence of cell uptake on hydrophobicity include the fact that hydrophobic forms of graphene are hindered by both repulsive interactions as well as agglomeration in physiological medium. However, despite not being able to enter the cell, graphene can accumulate on the cell membrane resulting in membrane deformation, destabilization of the cytoskeleton, and intracellular stress, which may lead to apoptosis (Figure 38C and D). Fortunately, there are numerous methods available to improve the biocompatibility of hydrophobic derivatives of graphene including functionalization and/or coating the graphene with functional groups or polymers. For instance, Sasidharan and colleagues found that by making pristine graphene more hydrophilic via carboxyl functionalization, it was possible to significantly enhance its biocompatibility.249 In particular, carboxyl functionalization was achieved by treatment of graphene with mild acid. As a consequence, 24 h exposure of Vero cells to up to 300 μg mL−1 of this hydrophilic functionalized graphene did not significantly affect viability (<5% decrease). Finally, despite the biocompatibility shown by hydrophilic forms of graphene, it should be mentioned that reports have claimed that all forms of graphene, regardless of their hydrophobicity, have the ability to generate reactive oxygen species in mammalian cells, which may lead to apoptosis and should be taken into consideration when using graphene materials.250
Graphene materials, especially GO, have also been shown to strongly promote cell adhesion. In particular, numerous studies have utilized graphene and its derivatives as substrates for the culture of various cell types including stem cells (e.g., iPSCs, NSCs). In the case of NSCs, Park et al. offered the first demonstration that laminin-coated graphene could enhance and increase the differentiation of human NSCs into neurons rather than glial cells, which occurred in the absence of added biochemical motifs.240c Of particular importance, the authors found that culturing NSCs on graphene did not negatively affect their proliferation or their differentiation ability. Similarly, a number of studies have utilized rGO substrates to culture and differentiate human NSCs into neurons.251 As with Park et al.’s study, it was found that rGO did not affect proliferation or differentiation capability and, in fact, accelerated the differentiation of NSCs into neurons.251 Besides NSCs, Chen and co-workers demonstrated that both graphene and GO could support mouse iPSC culture and allowed for spontaneous differentiation. Specifically, in comparison to glass substrates, iPSCs grown on graphene were found to exhibit similar degrees of cell adhesion and proliferation, while iPSCs cultured on GO adhered and proliferated at a faster rate. Interestingly, graphene was found to favorably maintain the iPSCs in an undifferentiated state. On the contrary, GO expedited the differentiation process resulting in ectodermal and mesodermal lineages, wherein it was reported that GO augmented endodermal differentiation while graphene tended to suppress iPSC differentiation toward this lineage.240a
Besides hydrophobicity, the size of the sheets also plays a major role in their biocompatibility. To determine the relationship between size and biocompatibility, Liao and coworkers fabricated graphene as well as different sized GO sheets via sonication (from 342 to 765 nm) and tested their biological effect on red blood cells (RBCs) and human fibroblasts.250a It was found that all of the GO and graphene sheets exhibited dose-dependent hemolytic activity on RBCs. In the case of the GO samples, the size played a critical role in the extent of hemolysis, wherein smaller GO sheets exhibited higher hemolytic activity than larger GO sheets. As compared to the GO sheets, which are better dispersed due to their high surface oxygen content, graphene tended to aggregate and showed lower hemolytic activity. Interestingly, covering the GO sheets with chitosan eliminated their hemolytic activity, further demonstrating that coating and functionalization of graphene materials plays a significant role in their biocompatibility. Finally, the authors confirmed that GO was more biocompatible than graphene when incubated with human fibroblasts but did not study the effect of size on biocompatibility in these cells.
In vivo, the majority of studies have reported that there are no significant side effects (e.g., no occurrences of animal death) when graphene materials are administered into mouse models; however, some studies have reported accumulation as well as inflammation and, in rare cases, fibrosis. In addition to physicochemical properties, concentration, and exposure time, which were mentioned previously for in vitro biocompatibility, the administration route of graphene materials and the characteristics of the animals used also significantly affect the biocompatibility of graphene. Most studies report no occurrence of adult animal’s death, even for graphene material doses up to 10 mg kg−1.44a,252 For instance, Wang et al. reported that the injection of 4–10 mg kg−1 of GO showed no signs of toxicity.248 However, one-half of the mice treated with >15 mg kg−1 died within 7 days of injection. Histopathology revealed that major airways in the lung were blocked by GO conglomeration leading to suffocation in 15% of the GO-administered mice. Further investigation determined that when administered intravenously, GO also generally accumulated in the reticuloendothelial system (RES) such as the liver and spleen, which had been confirmed in other studies as well.44a,248,253 In mice that did survive high doses of GO administration, the mice appeared weak and lost 10% of their body weight within the first week following administration, but symptoms disappeared after 1 week as evidenced by normal eating and weight. This could be explained by fecal elimination of accumulated graphene materials via bile as well as renal elimination when the graphene materials had a sufficiently small size.248,253 Finally, in terms of the rate of clearance of graphene materials based on size, small size graphene sheets (single sheets with diameter of 10 nm or less) were found to have a t1/2 (elimination half-life) close to 0.4 h, while the t1/2 values for larger sheets were observed to be greater than 5 h.253,254 It should also be noted that the functionalization and/or coating of graphene materials makes a significant difference on their in vivo biocompatibility. For example, Yang and colleagues determined that even 20 mg kg−1 of PEGylated graphene did not cause appreciable toxicity in mice during a period of three months as evidenced by blood biochemistry, hematological analysis, and histological examinations.253
Overall, graphene materials appear to be biocompatible depending on their physicochemical properties (e.g., more hydrophilic), concentration, time of exposure, and route of administration. Moreover, functionalization and polymer coatings (e.g., chitosan, PEGylation) can enhance biocompatibility. However, graphene materials are nondegradable materials with great potential for cellular internalization. As such, more detailed characterization and investigation of the long-term adverse effects of graphene materials remains to be fully elucidated before they can be readily applied in the clinic.
4.6.2. Biocompatibility of Graphene–Nanoparticle Hybrid Materials
For graphene–nanoparticle hybrid materials, there are currently limited biocompatibility studies available; however, preliminary results have fallen in line with the biocompatibility seen with graphene materials and, as such, are promising. Currently, the majority of the studies reporting biocompatibility for graphene–nanoparticle hybrids have primarily focused on graphene–MNP hybrids. Studies using this type of graphene–nanoparticle hybrid have indicated that these hybrids are biocompatible up to iron concentrations as high as 80 μg mL−1.194,197 In particular, Chen et al. determined that HeLa cells could be incubated with GO-Fe3O4 MNP composites with different iron concentrations of 10, 20, 40, and 80 μg mL−1 while maintaining low cytotoxicities (100%, 96%, 92%, and 91% cell viability, respectively).194 This was confirmed by Chen and co-workers wherein drug delivery and optical imaging were demonstrated using DOX-GO-PEG-β-FeOOH nanocomposites.195 Moreover, fluorescence images that were taken of HeLa cells incubated with DOX-GO-PEG-β-FeOOH for 24 h indicated that the DOX loaded on GO-PEG-β-FeOOH primarily remained in the cytoplasm, where the DOX was released and subsequently found to accumulate in the cell nuclei after 36 h of incubation. Finally, Yang and colleagues reported that their rGO-IONP-PEG nanocomposites were biocompatible both in vitro and in vivo and that they could be used for image-guided PTT.207 In particular, their preliminary in vivo results suggested that PEGylated graphene-based nanocomposites (up to 200 uL of 20 mg mL−1 solution injected intravenously) showed no obvious toxicity in mice (mice bearing 4T1 tumors in their right shoulders) with most accumulation occurring in the RES (e.g., liver and spleen) and the tumor (Figure 39A and B), when followed for at least 2 weeks.
Figure 39.
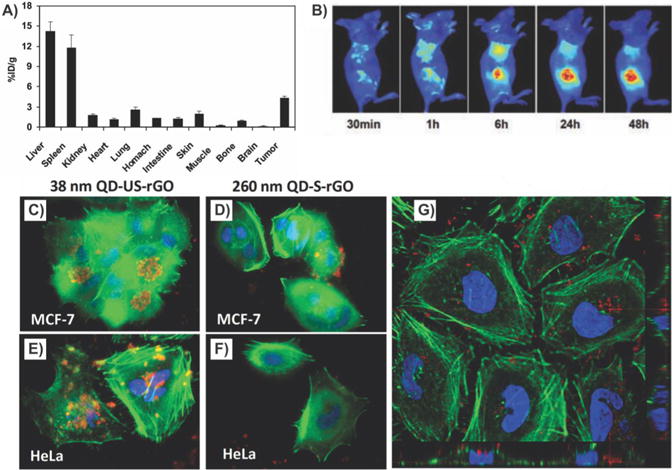
Biocompatibility of graphene–nanoparticle hybid materials. (A) Biodistribution of 125I-RGO-IONP-PEG in 4T1 tumor-bearing mice. High uptake was observed in the RES (e.g., liver and spleen) as well as in the tumor. (B) Fluorescence imaging using Cy5 labeled RGO-IONP-PEG showing biodistribution. Reprinted with permission from ref 207. Copyright 2012 Wiley. (C–F) Cellular uptake of FA-QD-S-rGO in MCF-7 cells (C) and HeLa cells (E) showing QD fluorescence (red-orange) in many regions, and that of FA-QD-S-rGO in MCF-7 cells (D) and HeLa cells (F), showing QDs mostly at the cell surface. (G) Confocal images of multiple cross sections (bottom and right), exhibiting various locations of the QD-rGOs within the MCF-7 cells. Reprinted with permission from ref 212. Copyright 2012 Wiley.
Graphene nanoparticle hybrid materials composed of noble metal (e.g., Ag and Au) nanoparticles and graphene and its derivatives have also proven to be biocompatible. For instance, for GO–AgNP hybrids, a cell viability of 95% was obtained following exposure of A549 (adenocarcinoma human alveolar basal epithelial) cells to 1 mg mL−1 of the GO–AgNPs for 24 h.254 Moreover, due to the properties of graphene–noble metal nanoparticle hybrids, they offer the ability to act as a SERS agent. As such, a number of studies have utilized these hybrids to study the specific cell uptake mechanism of graphene–nanoparticle hybrids as well as to determine their cellular distribution. From these studies, it was determined that GO–noble metal nanoparticle hybrids are generally uptaken via clathrin-mediated, energy-dependent endocytosis and are then distributed in the cytoplasm.179,198,255 In particular, Huang and colleagues used GO–AuNPs to study the mechanisms of cellular uptake of graphene–hybrid materials in Ca Ski cells via SERS.255 In this work, the authors reported that, in addition to distribution throughout the cytoplasm, the graphene–nanoparticle hybrids could not cross the nuclear membrane. Moreover, cell uptake was detectable (via SERS) starting 4 h after incubation with GO–AuNPs, reached a maximum after 6 h, and weakened thereafter, until the signal was barely above noise level (by 12 h of incubation). This suggested that the AuNPs, which were covalently conjugated via EDC/NHS coupling, may have been released from the GO sheets in the cellular environment. In vivo, GO-IONP-Au-PEG hybrids were intratumorally injected at a concentration of 50 ug mL−1 (40 uL) into subcutaneous 4T1 tumor bearing BALB/c mice. While cytotoxicity was not studied in particular, the authors reported no side effects and a similar growth speed for tumors in hybrid-injected and control mice.211
Reports on MSNP–GO hybrid materials also suggest that these graphene–nanoparticle hybrids are biocompatible for both in vitro and in vivo cell studies. In vitro, MSNP–GO hybrids did not exhibit considerable cytotoxicity at low to moderate concentrations (1–100 μg mL−1). For example, He and colleagues confirmed the biocompatibility of FA-targeted MSNP–GO nanoparticles.224 In this case, the mesoporous silica shell was loaded with DOX and whose release was controlled by photoinduced pH activation. In the absence of activation, the MSNP–GO hybrids were biocompatible up to 100 μg mL−1 with HeLa and L02 cell viability being maintained above 80% after 24 h of incubation. Moreover, the authors found that the FA-targeted hybrids exhibited selective cell internalization via receptor-mediated endocytosis. Similarly, Sreejith et al. reported that MSNP–GO hybrids loaded with squaraine dyes were also biocompatible but at a lower dose (incubation of 1 μg mL−1 in HeLa cells for 24 h resulted in a >80% viability).202 In vivo, nude mice have been shown to be able to withstand up to 5 mg kg−1 of MSNP–GO hybrids without any reported toxicities. In this case, Chen et al. demonstrated that their nanocookies (5 mg kg−1 via intravenous injection), as described in a previous section, could be used to effectively release CPT and induce PTT therapy, thereby eradicating subcutaneous MDA-MB-231 breast tumors in nude mice.238 In particular, the tumors were found to have disappeared 14 days post treatment and did not reappear again even after 2 months, demonstrating the prolonged biocompatibility of their platform. Moreover, no significant toxicities (e.g., weight loss, change in behavior) were reported. Last, in support of these findings, Wang and colleagues determined that their targeting peptide-modified MGMSPI, which were loaded with DOX, were also well-tolerated following intraperitonial injection in nude mice bearing intracranial U251 brain tumors at doses up to 6 mg mL−1 (150 uL).237
Finally, although less has been reported on the biocompatibility of other types of graphene–nanoparticle hybrids, GO–UCNP and GO–QD hybrids have been demonstrated to be biocompatible. In the case of GO–UCNPs, HeLa cells and KB (human nasopharyngeal epidermal cancer) cells incubated with NGO–UCNP concentrations as high as 320 mg mL−1 for 24 h resulted in cell viabilities higher than 90%.236 In vivo injection was also performed, where it was found that subcutaneous injection of 200 μg mL−1 of NGO–UCNPs into white Kunming mice caused no significant toxicities and could be used for NIR-mediated imaging and PTT/PDT cancer therapy. On the other hand, GO–QDs have also been found to be biocompatible but at a lower concentration than for GO–UCNPs.204,212 Sheng and co-workers found that AIZS-GO-PEG hybrid nanoparticles were biocompatible in NIH/3T3 cells (mouse fibroblasts) when incubated for 24 h at concentrations up to 100 mg mL−1.204 Similarly, Hu et al. found that the viability of HeLa cells remained high (92%) after exposure to 20 mg L−1 of rGO–QD nanocomposites (CdSe/ZnS QDs) for 12–48 h.212 In addition, uptake of the QD–rGO composites into the cytoplasm of the HeLa cells could be clearly visualized, wherein it was found that US-rGO (38 nm) localized throughout the cytoplasm while S-rGO (260 nm) was mainly found on the cell surface (Figure 39C–G). Importantly, the authors reported that the QD–rGO composites appear to be localized as discrete dots within the HeLa cells following incubation, suggesting a lack of QD leaching from the nanocomposites.
Overall, although in depth and long-term in vivo studies still remain to be performed, preliminary results suggest that graphene–nanoparticle hybrids composed of GO tend to be highly biocompatible, which may be by virtue of their highly hydrophilic nature as well as the coatings used (e.g., PEGylation). It should be noted that the majority of the studies performed for graphene–nanoparticle hybrid materials have used cancer cell lines. While the results are promising, these cell lines tend to be more resistant to apoptotic cell death, and, as a result, this must be taken into consideration when evaluating the reported findings. Looking forward, to accelerate the translation of graphene–nanoparticle hybrids to the clinic, there is a need for more detailed studies looking at the biocompatibility of different graphene–nanoparticle composites with a variety of properties including the size of the nanoparticles, size of the graphene sheets, concentration, surface charge and/or coating, and long-term stability and in vivo fate.
5. CONCLUDING REMARKS AND FUTURE PERSPECTIVES
Graphene is a fascinating material that has garnered ever-increasing interest from the scientific community over the past decade. Because of its exceptional properties, graphene is already being used in a wide variety of applications including electronics, energy, sensing, and even bioapplications such as biosensors, drug delivery, stem cell differentiation, and cellular imaging. However, in the last several years, the incessant search for new avenues that would benefit from graphene has led to the advent of graphene–nanoparticle hybrid structures, which combine the unique and advantageous properties of nanomaterials/nanotechnology with those of graphene to produce advantageous and often synergistic effects. In this Review, we have sought to provide insight into the synthesis and characterization of graphene–nanoparticle hybrid materials as well as their implementation to a host of bioapplications that fall into two general categories: (1) biosensing and (2) cellular applications such as imaging, PTT, multifunctional drug delivery, and the control of stem cell differentiation.
In terms of the synthesis of graphene–nanoparticle hybrids, numerous avenues are currently available for the preparation of hybrids composed of graphene-based nanomaterials and various nanoparticles. These include in situ (e.g., reduction, hydrothermal, and electrochemical) and ex situ (e.g., covalent, electrostatic, and π–π stacking) methods. However, the optimal method depends greatly on the requirements of the desired application. For example, in biosensing applications, wherein the electrical properties of graphene in the graphene–nanoparticle hybrids should be maximized, noncovalent ex situ methods such as electrostatic interactions and π–π stacking are most desirable as the use of presynthesized nanoparticles eliminates any possible incompatibilities between nanoparticle synthesis and assembly as well as any alterations to the structure of graphene. On the other hand, in situ methods require less time and fewer steps to complete and can be used in applications where a narrower size distribution as well as precise control over the size, shape, and density of the nanoparticles that decorate the graphene sheets is unnecessary. For instance, most cellular applications such as stem cell differentiation, drug delivery, or cellular imaging do not require maximizing the electrical properties of the hybrids and, as such, have used in situ methods. It should be noted that ex situ methods have also been used for cellular application, especially for stem cell differentiation as well as applications using graphene-encapsulated nanoparticles.
As for the bioapplications of graphene–nanoparticle hybrids, these structures have mainly been applied to biosensing and biomedicine. In the case of biosensing, we covered three main types: (1) electronic, (2) electrochemical, and (3) optical. These biosensors, which each have their own advantages and disadvantages, have all benefited from the application of graphene–nanoparticle hybrids with achievable sensitivities and selectivities that are comparable to or better than gold standards including other carbon-based nanomaterials. In particular, electronic sensors wherein graphene–nanoparticle hybrids are applied to FET-based devices can currently achieve the highest sensitivity with a LOD in the attomolar range. In addition, the use of hybrids provides enhanced surface area for analyte binding while amplifying the achievable signal and electrical conductivity. Hybrid sensors that utilize optical mechanisms also have great promise as they can potentially allow for the detection of single molecules (e.g., SERS) as well as the quantification of molecules in living cells in a nondestructive manner (e.g., FRET and SERS). Similarly, although hybrid electrochemical sensors lag behind in terms of their achievable sensitivity, they allow for direct conversion of a biological recognition event to an electrical signal at a low cost and complexity. Moreover, hybrid structures can help overcome the poor utilization coefficient of aggregated nanoparticles as well as improve the electron transfer that occurs between the analyte and the electrode. Finally, graphene–nanoparticle hybrid structures offer the ability to develop novel sensing mechanisms. For example, our group has recently developed a label-free polypeptide-based biosensor for enzyme detection using a graphene–AuNP composite (Figure 40).110 In this platform, graphene was first deposited between Au electrodes. A layer of functional polypeptide linker followed by AuNPs was then assembled on the graphene layer. In this way, enzymatic degradation of the functional polypeptide linker by the enzyme of interest resulted in release of the AuNPs and a measurable shift in electrical hysteresis. Using this method, we achieved the sensitive (LOD 1 μM) and selective detection of Carboxypeptidase B, a predictor for severe acute pancreatitis. In terms of its mechanism, the AuNPs assembled on the graphene surface using a functional polypeptide linker have the ability to store charge resulting in a measurable hysteresis upon their release from the device surface.
Figure 40.
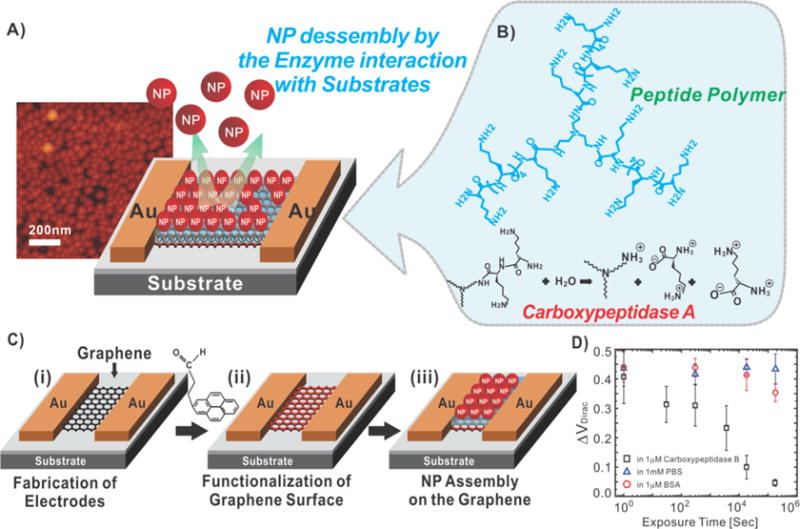
Hysteresis-based enzyme detection using a graphene–nanoparticle hybrid sensor. (A) Graphene–nanoparticle hybrid devices for enzyme sensing. (B) Chemical structure of the functional polypeptide linker molecule. (C) Fabrication process of the hybrid biosensor. Fabrication of the graphene channel between the Au electrodes (i). Functionalization of the graphene surface with hydrophilic molecules (ii). Assembly of the functional peptide linker molecules and AuNPs on the polypeptide layer (iii). (D) Change in VDirac under various periods of exposure to a 1 μM solution of carboxypeptidase B in PBS, 1 mM PBS solution, and 1 mM solution of BSA in PBS. Reprinted with permission from ref 110. Copyright 2012 Wiley.
For graphene–nanoparticle hybrid application to biomedicine, we covered five topics of interest: (1) bioimaging, (2) PTTs, (3) drug/gene delivery, (4) combined multifunctional therapies, and (5) stem cell/tissue engineering. In this case, graphene–nanoparticle hybrid materials show immense potential due to their intrinsic unique physical, chemical, magnetic, electrical, plasmonic, and mechanical properties. Specifically, similar to the case with biosensors, the application of graphene–nanoparticle hybrids has seen great success with different combinations being used to attain different multi-functionalities of interest. Therefore, graphene–nanoparticle hybrids have led to the enhancement of image contrast, cancer therapies, and stem cell differentiation.
While the future prospects of graphene–nanoparticle hybrids are very bright, our ability to synthesize graphene and its derivatives with controllable sizes, shapes, and defects at a low-cost and high-yield manner as well as our ability to control the size, composition, morphology, and crystallinity of the various incorporated nanoparticles remain critical bottlenecks to the advancement of graphene–nanoparticle hybrids. Moreover, while significant progress has been made to achieve precise control over the density and specific arrangement of nanoparticles that are assembled on graphene and its derivatives, there is still significant room for improvement. As such, there is a need to gain a deeper understanding of the underlying mechanism of graphene–nanoparticle formation to better control their synthesis, assembly, and adhesion. This is especially true for biosensing and imaging applications wherein the nanostructure and charge transfer behavior are particularly important. This will also allow for better control over stem cell differentiation, the distribution and targeting of hybrids in the body, as well as lower the limits of achievable sensitivity and selectivity for biosensing. Finally, significant investigation will be required to assess the toxicity and biodistribution of such hybrid material in vivo as well as the targeting of nanocomposites to improve the efficacy of treatments and reduce off-site toxicity. In addition to biodistribution and long-term safety, understanding the cellular interaction and the precise mechanism of action guiding changes in cellular behavior is equally important for regenerative medicine and tissue engineering.
In conclusion, we have highlighted an up-and-coming field wherein graphene–nanoparticle hybrid structures can bring synergistic advantages to a wide variety of bioapplications. While promising, the field of graphene–nanoparticle hybrids remains in its infancy, and a number of challenging issues must be addressed before its maximum potential can be achieved. Moreover, we imagine that the maturation of this technology will result in the use of graphene–nanoparticle hybrids in a much broader range of bioapplications, while the high-quality and large-scale fabrication of graphene–nanoparticle hybrids will minimize costs and lead to its commercialization. We hope that this Review has inspired interest from various disciplines that will benefit from the development of graphene–nanoparticle hybrids for bioapplications.
Acknowledgments
K.-B.L. acknowledges financial support from the NIH Director’s Innovator Award [1DP20D006462-01], National Institute of Neurological Disorders and Stroke (NINNDS) [1R21NS085569-01], the NSF 9CHE-1429062 and CBET-12365080, the N.J. Commission on Spinal Cord grant [CSCR13ERG005], the Busch Biomedical Grant Program, the Collaborative Research Travel Grant (CRTG) from the Burroughs Wellcome Fund, American Cryostem Corp., and the Rutgers Faculty Research Grant Program. P.T.Y. would also like to acknowledge the NIH Biotechnology Training Grant. Finally, S.S. acknowledges NSF DGE 0801620, Integrative Graduate Education and Research Traineeship (IGERT) on the Integrated Science and Engineering of Stem Cells.
ABBREVIATIONS
- 2D
two-dimensional
- 5-FU
fluorouracil
- AA
ascorbic acid
- AFM
atomic force microscopy
- AFP
alpha fetoprotein
- AgNP
silver nanoparticle
- AIZS
zinc-doped AgInS
- aM
attomolar
- APTES
(3-aminopropyl)triethoxysilane
- AuNP
gold nanoparticle
- AuNR
gold nanorod
- AuNS
gold nanoshell
- BSA
bovine serum albumin
- CEA
carcinoembryonic antigen
- CMG
chitosan magnetic–graphene nanocomposite
- CNT
carbon nanotube
- conA
concanavalin A
- CPT
camptothecin
- CT
computed tomography
- CV
cyclic voltammetry
- CVD
chemical vapor deposition
- DA
dopamine
- DOPC
dioleoyl-sn-glycero-3-phosphocholine
- DOX
doxorubicin
- EDC
1-ethyl-3-(3-(dimethylamino)propyl)carbodiimide
- EGFR
epidermal growth factor receptor
- EP
electrophoretic preconcentration
- ERGO
electrochemically reduced graphene oxide
- ESCs
embryonic stem cells
- FA
folic acid
- FACS
fluorescence-activated cell sorting
- FB1
fumonisin B1
- FET
field-effect transistors
- fM
femtomolar
- FR
folate receptor
- FRET
fluorescence resonance energy transfer
- GC
graphitic carbon shell
- GCE
glassy carbon electrode
- GFP
green fluorescent protein
- GNC
gold nanocluster
- GO
graphene oxide
- GSGHs
graphene–mesoporous silica–AuNP hybrids
- hCG
human chorionic gonadotrophin
- HER2
human epidermal growth factor receptor 2
- HRP
horseradish peroxidase
- HUVEC
human umbilical vein endothelial cells
- ID
injected dose
- IGAuNPs
intracellularly grown gold nanoparticles
- IgG
immunoglobulin G
- IgM
immunoglobulin M
- IONP
iron oxide nanoparticle
- iPSCs
induced pluripotent stem cells
- ITO
indium tin oxide
- LOD
limit of detection
- LSPR
localized surface plasmon resonance
- MB
molecular beacon
- MGMSPI
magnetic graphene-based meosporous silica
- MGPNs
multilayered graphene petal nanosheets
- MNP
magnetic nanoparticle
- MRI
magnetic resonance imaging
- MSCs
mesenchymal stem cells
- MSNP
mesoporous silica nanoparticle
- MTX
methotrexate
- MUA
11-mercaptoundecanoic acid
- NHS
N-hydroxysuccinimide
- NIR
near-infrared
- NMR
nuclear magnetic resonance
- NSCs
neural stem cells
- NSET
nanometal surface energy transfer
- OTA
ochratoxin A
- PAG
photoacid generator
- PBA
1-pyrenebutyric acid
- PBS
phosphate-buffered saline
- PCR
polymerase chain reaction
- PDDA
poly(diallyldimethylammonium chloride)
- PDT
photodynamic therapy
- PEGylated
polyethyelene glycol-coated
- PEI
polyethylenimine
- pM
picomolar
- PPGA
PEGylated, polyethylenimine-grafted graphene/Au composites
- PTBO
poly(toluidine blue O)
- PtNP
platinum nanoparticle
- PTT
photothermal therapy
- PVP
polyvinylpyrrolidone
- QDs
quantum dots
- r2
relaxivity rate
- RBCs
red blood cells
- RES
reticuloendothelial system
- rGO
reduced graphene oxide
- S-rGO
small reduced graphene oxide
- SALDI-MS
surface-assisted laser desorption/ionization mass spectrometry
- SCE
saturated calomel electrode
- SERS
surface-enhanced Raman spectroscopy
- SiNPs
silica nanoparticles
- siRNA
small interfering RNA
- ssDNA
single-stranded DNA
- T1
spin–lattice longitudinal relaxation time
- t1/2
elimination half-life
- T2
spin–spin transverse relaxation time
- Trf
transferrin
- TRGO
thermally reduced graphene oxide
- UA
uric acid
- UCNPs
upconversion nanoparticles
- US-rGO
ultrasmall reduced graphene oxide
- UV
ultraviolet
Biographies

Perry To-tien Yin received his B.S. in Biomedical Engineering from Columbia University in 2010 and is currently pursuing his Ph.D. in Biomedical Engineering at Rutgers University, where he plans to graduate in 2015. His doctoral research, under the supervision of Prof. Ki-Bum Lee, focuses on the application of multifunctional nanoparticles for the detection and treatment of breast, ovarian, and brain cancer with particular emphasis on the application of magnetic nanoparticles for magnetic hyperthermia-based treatments. He has published more than 10 peer-reviewed articles in journals that include Advanced Materials, Angewandte Chemie International Edition, ACS Nano, and Small.

Shreyas Shah received his B.S. in Biomedical Engineering from Rutgers, The State University of New Jersey in 2010 and is currently pursuing his Ph.D. in Chemistry & Chemical Biology at Rutgers University, where he plans to graduate in 2015. Under the supervision of Prof. Ki-Bum Lee, his doctoral research focuses on designing nanomaterial-based 2D/3D scaffolds with temporal and spatial control of microenvironmental cues to selectively guide stem cell behavior. He is particularly interested in applying these unique biomaterials for applications in neural regeneration and the treatment of central nervous system diseases and injuries. His work has been published in high impact peer-reviewed journals, including Advanced Materials, Small, Scientific Reports, and Journal of the American Chemical Society.

Manish Chhowalla is a Professor and the Associate Chair of the Materials Science and Engineering Department at Rutgers University. He was also the Director of Nanotechnology for Clean Energy NSF IGERT Program (2009–2014) and the Donald H. Jacobs Chair in Applied Physics (2009–2011). From June 2009 to July 2010 he was a Professor in the Department of Materials at the Imperial College London. He has won the NSF CAREER Award for young scientists as well as the Sigma Xi Outstanding Young Investigator for the Mid-Atlantic Region. Before Rutgers, he was a Royal Academy of Engineering Postdoctoral Research Fellow at the University of Cambridge, which is also where he completed his Ph.D. in Electrical Engineering. Prior to his Ph.D., he worked for Multi-Arc Inc. (now Ion Bond) where he developed one of the first applications of “amorphous diamond” thin films. He is interested in understanding the role of disorder in determining material properties as well as phase engineering in low dimensional materials.

Ki-Bum Lee is an Associate Professor of Chemistry and Chemical Biology at Rutgers University, where he has been a faculty member since 2008. He received his Ph.D. in Chemistry from Northwestern University (with Chad. A. Mirkin, 2004) and completed his postdoctoral training at The Scripps Research Institute (with Peter G. Schultz, 2007). The primary research interest of Prof. Lee’s group is to develop and integrate nanotechnologies and chemical functional genomics to modulate signaling pathways in cells (e.g., stem cells and cancer cells) toward specific cell lineages or behaviors. In recognition of his outstanding scientific achievement at Rutgers, Dr. Lee has received several awards including the NIH Director’s New Innovator Awards (2009), Board of Trustees Research Award for Scholarly Excellence (2013), Johnson and Johnson Proof-of-Concept Award (2011), Faculty Research Grant Award (2012), and New Jersey Spinal Cord Research Award (2009). He is the first author, coauthor, and corresponding author of approximately 48 articles published in high-profile journals including Science, Cell Stem Cell, Journal of the American Chemical Society, Angewandte Chemie International Edition, Nano Letters, ACS Nano, Advanced Materials, Biomaterials, Scientific Reports, Lab Chip, Small, Physical Chemistry Chemical Physics, Nanomedicine, and Cancer Research, which are highly cited (>5000).
Footnotes
Notes
The authors declare no competing financial interest.
References
- 1.(a) Castro Neto AH, Guinea F, Peres NMR, Novoselov KS, Geim AK. Rev Mod Phys. 2009;81:109. [Google Scholar]; (b) Geim AK, Novoselov KS. Nat Mater. 2007;6:183. doi: 10.1038/nmat1849. [DOI] [PubMed] [Google Scholar]; (c) Novoselov KS, Fal’ko VI, Colombo L, Gellert PR, Schwab MG, Kim K. Nature. 2012;490:192. doi: 10.1038/nature11458. [DOI] [PubMed] [Google Scholar]
- 2.Stoller MD, Park SJ, Zhu YW, An JH, Ruoff RS. Nano Lett. 2008;8:3498. doi: 10.1021/nl802558y. [DOI] [PubMed] [Google Scholar]
- 3.Lee C, Wei XD, Kysar JW, Hone J. Science. 2008;321:385. doi: 10.1126/science.1157996. [DOI] [PubMed] [Google Scholar]
- 4.Balandin AA, Ghosh S, Bao W, Calizo I, Teweldebrhan D, Miao F, Lau CN. Nano Lett. 2008;8:902. doi: 10.1021/nl0731872. [DOI] [PubMed] [Google Scholar]
- 5.(a) Bolotin KI, Sikes KJ, Jiang Z, Klima M, Fudenberg G, Hone J, Kim P, Stormer HL. Solid State Commun. 2008;146:351. [Google Scholar]; (b) Gomez-Navarro C, Weitz RT, Bittner AM, Scolari M, Mews A, Burghard M, Kern K. Nano Lett. 2007;7:3499. doi: 10.1021/nl072090c. [DOI] [PubMed] [Google Scholar]
- 6.Bonaccorso F, Sun Z, Hasan T, Ferrari AC. Nat Photonics. 2010;4:611. [Google Scholar]
- 7.Chung C, Kim YK, Shin D, Ryoo SR, Hong BH, Min DH. Acc Chem Res. 2013;46:2211. doi: 10.1021/ar300159f. [DOI] [PubMed] [Google Scholar]
- 8.Wang XM, Zhang WH. New Carbon Mater. 2013;28:321. [Google Scholar]
- 9.(a) Shao YY, Wang J, Wu H, Liu J, Aksay IA, Lin YH. Electroanalysis. 2010;22:1027. [Google Scholar]; (b) Pumera M. Mater Today. 2011;14:308. [Google Scholar]
- 10.(a) Lee WC, Lim CHYX, Shi H, Tang LAL, Wang Y, Lim CT, Loh KP. ACS Nano. 2011;5:7334. doi: 10.1021/nn202190c. [DOI] [PubMed] [Google Scholar]; (b) Sun XM, Liu Z, Welsher K, Robinson JT, Goodwin A, Zaric S, Dai HJ. Nano Res. 2008;1:203. doi: 10.1007/s12274-008-8021-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.(a) Shah B, Yin PT, Ghoshal S, Lee KB. Angew Chem, Int Ed. 2013;52:6190. doi: 10.1002/anie.201302245. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Bai S, Shen X. RSC Adv. 2012;2:64. [Google Scholar]; (c) Wu ZS, Zhou GM, Yin LC, Ren W, Li F, Cheng HM. Nano Energy. 2012;1:107. [Google Scholar]
- 12.Tjoa V, Jun W, Dravid V, Mhaisalkar S, Mathews N. J Mater Chem. 2011;21:15593. [Google Scholar]
- 13.Artiles MS, Rout CS, Fisher TS. Adv Drug Delivery Rev. 2011;63:1352. doi: 10.1016/j.addr.2011.07.005. [DOI] [PubMed] [Google Scholar]
- 14.Yang K, Feng LZ, Shi XZ, Liu Z. Chem Soc Rev. 2013;42:530. doi: 10.1039/c2cs35342c. [DOI] [PubMed] [Google Scholar]
- 15.(a) Choi W, Lahiri I, Seelaboyina R, Kang YS. Crit Rev Solid State. 2010;35:52. [Google Scholar]; (b) Georgakilas V, Otyepka M, Bourlinos AB, Chandra V, Kim N, Kemp KC, Hobza P, Zboril R, Kim KS. Chem Rev. 2012;112:6156. doi: 10.1021/cr3000412. [DOI] [PubMed] [Google Scholar]
- 16.Xu WG, Xiao JQ, Chen YF, Chen YB, Ling X, Zhang J. Adv Mater. 2013;25:928. doi: 10.1002/adma.201204355. [DOI] [PubMed] [Google Scholar]
- 17.Emtsev KV, Bostwick A, Horn K, Jobst J, Kellogg GL, Ley L, McChesney JL, Ohta T, Reshanov SA, Rohrl J, Rotenberg E, Schmid AK, Waldmann D, Weber HB, Seyller T. Nat Mater. 2009;8:203. doi: 10.1038/nmat2382. [DOI] [PubMed] [Google Scholar]
- 18.(a) Zhang YB, Small JP, Pontius WV, Kim P. Appl Phys Lett. 2005;86:073104. [Google Scholar]; (b) Novoselov KS, Geim AK, Morozov SV, Jiang D, Zhang Y, Dubonos SV, Grigorieva IV, Firsov AA. Science. 2004;306:666. doi: 10.1126/science.1102896. [DOI] [PubMed] [Google Scholar]; (c) Hernandez Y, Nicolosi V, Lotya M, Blighe FM, Sun ZY, De S, McGovern IT, Holland B, Byrne M, Gun’ko YK, Boland JJ, Niraj P, Duesberg G, Krishnamurthy S, Goodhue R, Hutchison J, Scardaci V, Ferrari AC, Coleman JN. Nat Nanotechnol. 2008;3:563. doi: 10.1038/nnano.2008.215. [DOI] [PubMed] [Google Scholar]
- 19.(a) Pei SF, Zhao JP, Du JH, Ren WC, Cheng HM. Carbon. 2010;48:4466. [Google Scholar]; (b) Chua CK, Pumera M. Chem Soc Rev. 2014;43:291. doi: 10.1039/c3cs60303b. [DOI] [PubMed] [Google Scholar]
- 20.(a) Eda G, Chhowalla M. Adv Mater. 2010;22:2392. doi: 10.1002/adma.200903689. [DOI] [PubMed] [Google Scholar]; (b) Chen D, Feng H, Li J. Chem Rev. 2012;112:6027. doi: 10.1021/cr300115g. [DOI] [PubMed] [Google Scholar]
- 21.Zhu YW, Murali S, Cai WW, Li XS, Suk JW, Potts JR, Ruoff RS. Adv Mater. 2010;22:3906. doi: 10.1002/adma.201001068. [DOI] [PubMed] [Google Scholar]
- 22.Dreyer DR, Park S, Bielawski CW, Ruoff RS. Chem Soc Rev. 2010;39:228. doi: 10.1039/b917103g. [DOI] [PubMed] [Google Scholar]
- 23.Park S, Ruoff RS. Nat Nanotechnol. 2009;4:217. doi: 10.1038/nnano.2009.58. [DOI] [PubMed] [Google Scholar]
- 24.Compton OC, Nguyen ST. Small. 2010;6:711. doi: 10.1002/smll.200901934. [DOI] [PubMed] [Google Scholar]
- 25.Pei SF, Cheng HM. Carbon. 2012;50:3210. [Google Scholar]
- 26.Zhou M, Wang YL, Zhai YM, Zhai JF, Ren W, Wang FA, Dong SJ. Chem—Eur J. 2009;15:6116. doi: 10.1002/chem.200900596. [DOI] [PubMed] [Google Scholar]
- 27.(a) Schniepp HC, Li JL, McAllister MJ, Sai H, Herrera-Alonso M, Adamson DH, Prud’homme RK, Car R, Saville DA, Aksay IA. J Phys Chem B. 2006;110:8535. doi: 10.1021/jp060936f. [DOI] [PubMed] [Google Scholar]; (b) Kuila T, Mishra AK, Khanra P, Kim NH, Lee JH. Nanoscale. 2013;5:52. doi: 10.1039/c2nr32703a. [DOI] [PubMed] [Google Scholar]
- 28.Kong BS, Geng JX, Jung HT. Chem Commun. 2009;16:2174. doi: 10.1039/b821920f. [DOI] [PubMed] [Google Scholar]
- 29.(a) Muszynski R, Seger B, Kamat PV. J Phys Chem C. 2008;112:5263. [Google Scholar]; (b) Goncalves G, Marques PAAP, Granadeiro CM, Nogueira HIS, Singh MK, Gracio J. Chem Mater. 2009;21:4796. [Google Scholar]; (c) Hong WJ, Bai H, Xu YX, Yao ZY, Gu ZZ, Shi GQ. J Phys Chem C. 2010;114:1822. [Google Scholar]
- 30.Tien HW, Huang YL, Yang SY, Wang JY, Ma CCM. Carbon. 2011;49:1550. [Google Scholar]
- 31.(a) Qiu JD, Wang GC, Liang RP, Xia XH, Yu HW. J Phys Chem C. 2011;115:15639. [Google Scholar]; (b) Li YJ, Gao W, Ci LJ, Wang CM, Ajayan PM. Carbon. 2010;48:1124. [Google Scholar]
- 32.(a) Lin J, Raji ARO, Nan KW, Peng ZW, Yan Z, Samuel ELG, Natelson D, Tour JM. Adv Funct Mater. 2014;24:2044. [Google Scholar]; (b) Yang X, Chen CL, Li JX, Zhao GX, Ren XM, Wang XK. RSC Adv. 2012;2:8821. [Google Scholar]
- 33.Liang YY, Wang HL, Casalongue HS, Chen Z, Dai HJ. Nano Res. 2010;3:701. [Google Scholar]
- 34.(a) Kim YJ, Lee JH, Yi GC. Appl Phys Lett. 2009;95:73701. doi: 10.1063/1.3206910. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Son DI, Kwon BW, Park DH, Seo WS, Yi Y, Angadi B, Lee CL, Choi WK. Nat Nanotechnol. 2012;7:465. doi: 10.1038/nnano.2012.71. [DOI] [PubMed] [Google Scholar]
- 35.(a) Lin J, Peng Z, Xiang C, Ruan G, Yan Z, Natelson D, Tour JM. ACS Nano. 2013;7:6001. doi: 10.1021/nn4016899. [DOI] [PubMed] [Google Scholar]; (b) Su DW, Ahn HJ, Wang GX. Chem Commun. 2013;49:3131. doi: 10.1039/c3cc40448j. [DOI] [PubMed] [Google Scholar]
- 36.(a) Deng S, Tjoa V, Fan HM, Tan HR, Sayle DC, Olivo M, Mhaisalkar S, Wei J, Sow CH. J Am Chem Soc. 2012;134:4905. doi: 10.1021/ja211683m. [DOI] [PubMed] [Google Scholar]; (b) Li BJ, Cao HQ, Yin G, Lu YX, Yin JF. J Mater Chem. 2011;21:10645. [Google Scholar]
- 37.(a) Peng LL, Peng X, Liu BR, Wu CZ, Xie Y, Yu GH. Nano Lett. 2013;13:2151. doi: 10.1021/nl400600x. [DOI] [PubMed] [Google Scholar]; (b) He Y, Chen W, Li X, Zhang Z, Fu J, Zhao C, Xie E. ACS Nano. 2013;7:174. doi: 10.1021/nn304833s. [DOI] [PubMed] [Google Scholar]
- 38.(a) Zhou GM, Wang DW, Yin LC, Li N, Li F, Cheng HM. ACS Nano. 2012;6:3214. doi: 10.1021/nn300098m. [DOI] [PubMed] [Google Scholar]; (b) Yang HB, Guai GH, Guo CX, Song QL, Jiang SP, Wang YL, Zhang W, Li CM. J Phys Chem C. 2011;115:12209. [Google Scholar]
- 39.(a) Xu C, Wang X, Zhu JW. J Phys Chem C. 2008;112:19841. [Google Scholar]; (b) Chen Y, Li Y, Sun D, Tian DB, Zhang JR, Zhu JJ. J Mater Chem. 2011;21:7604. [Google Scholar]
- 40.Gao W, Alemany LB, Ci LJ, Ajayan PM. Nat Chem. 2009;1:403. doi: 10.1038/nchem.281. [DOI] [PubMed] [Google Scholar]
- 41.Zhuo QQ, Ma YY, Gao J, Zhang PP, Xia YJ, Tian YM, Sun XX, Zhong J, Sun XH. Inorg Chem. 2013;52:3141. doi: 10.1021/ic302608g. [DOI] [PubMed] [Google Scholar]
- 42.Jain PK, Huang XH, El-Sayed IH, El-Sayed MA. Acc Chem Res. 2008;41:1578. doi: 10.1021/ar7002804. [DOI] [PubMed] [Google Scholar]
- 43.(a) Eustis S, El-Sayed MA. Chem Soc Rev. 2006;35:209. doi: 10.1039/b514191e. [DOI] [PubMed] [Google Scholar]; (b) Liao HW, Nehl CL, Hafner JH. Nanomedicine (London, UK) 2006;1:201. doi: 10.2217/17435889.1.2.201. [DOI] [PubMed] [Google Scholar]
- 44.(a) Duch MC, Budinger GRS, Liang YT, Soberanes S, Urich D, Chiarella SE, Campochiaro LA, Gonzalez A, Chandel NS, Hersam MC, Mutlu GM. Nano Lett. 2011;11:5201. doi: 10.1021/nl202515a. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Huang J, Zhang LM, Chen BA, Ji N, Chen FH, Zhang Y, Zhang ZJ. Nanoscale. 2010;2:2733. doi: 10.1039/c0nr00473a. [DOI] [PubMed] [Google Scholar]
- 45.Zhang H, Hines D, Akins DL. Dalton Trans. 2014;43:2670. doi: 10.1039/c3dt52573b. [DOI] [PubMed] [Google Scholar]
- 46.(a) Lim EJ, Choi SM, Seo MH, Kim Y, Lee S, Kim WB. Electrochem Commun. 2013;28:100. [Google Scholar]; (b) Wang Y, Li SS, Yeh YC, Yu CC, Chen HL, Li FC, Chang YM, Chen CW. Nanoscale. 2013;5:1687. doi: 10.1039/c2nr33220e. [DOI] [PubMed] [Google Scholar]
- 47.(a) Subrahmanyam KS, Manna AK, Pati SK, Rao CNR. Chem Phys Lett. 2010;497:70. [Google Scholar]; (b) Li YM, Tang LH, Li JH. Electrochem Commun. 2009;11:846. [Google Scholar]; (c) Seger B, Kamat PV. J Phys Chem C. 2009;113:7990. [Google Scholar]
- 48.(a) Zhang S, Shao YY, Liao HG, Liu J, Aksay IA, Yin GP, Lin YH. Chem Mater. 2011;23:1079. [Google Scholar]; (b) Yang P, Jin SY, Xu QZ, Yu SH. Small. 2013;9:199. doi: 10.1002/smll.201201008. [DOI] [PubMed] [Google Scholar]; (c) Wang R, Wu Z, Chen C, Qin Z, Zhu H, Wang G, Wang H, Wu C, Dong W, Fan W, Wang J. Chem Commun (Cambridge, U K) 2013;49:8250. doi: 10.1039/c3cc43948h. [DOI] [PubMed] [Google Scholar]; (d) Gao LN, Yue WB, Tao SS, Fan LZ. Langmuir. 2013;29:957. doi: 10.1021/la303663x. [DOI] [PubMed] [Google Scholar]
- 49.Guo SJ, Dong SJ, Wang EK. ACS Nano. 2010;4:547. doi: 10.1021/nn9014483. [DOI] [PubMed] [Google Scholar]
- 50.(a) Guo SJ, Dong SJ, Wang EK. J Phys Chem C. 2009;113:5485. [Google Scholar]; (b) Guo SJ, Wang L, Dong SJ, Wang EK. J Phys Chem C. 2008;112:13510. [Google Scholar]
- 51.Jasuja K, Linn J, Melton S, Berry V. J Phys Chem Lett. 2010;1:1853. [Google Scholar]
- 52.Han YJ, Luo ZM, Yuwen LH, Tian J, Zhu XR, Wang LH. Appl Surf Sci. 2013;266:188. [Google Scholar]
- 53.Kundu P, Nethravathi C, Deshpande PA, Rajamathi M, Madras G, Ravishankar N. Chem Mater. 2011;23:2772. [Google Scholar]
- 54.Chen SQ, Wang Y. J Mater Chem. 2010;20:9735. [Google Scholar]
- 55.Tsuji M, Hashimoto M, Nishizawa Y, Tsuji T. Mater Lett. 2004;58:2326. [Google Scholar]
- 56.Zhou Y, Bao QL, Tang LAL, Zhong YL, Loh KP. Chem Mater. 2009;21:2950. [Google Scholar]
- 57.(a) Zou WB, Zhu JW, Sun YX, Wang X. Mater Chem Phys. 2011;125:617. [Google Scholar]; (b) Wu JL, Shen XP, Jiang L, Wang K, Chen KM. Appl Surf Sci. 2010;256:2826. [Google Scholar]; (c) Saravanakumar B, Mohan R, Kim SJ. Mater Res Bull. 2013;48:878. [Google Scholar]
- 58.(a) Shen JF, Shi M, Yan B, Ma HW, Li N, Ye MX. Nano Res. 2011;4:795. [Google Scholar]; (b) Shen JF, Yan B, Shi M, Ma HW, Li N, Ye MX. J Mater Chem. 2011;21:3415. [Google Scholar]; (c) Perera SD, Mariano RG, Vu K, Nour N, Seitz O, Chabal Y, Balkus KJ. ACS Catal. 2012;2:949. [Google Scholar]
- 59.(a) Zhou KF, Zhu YH, Yang XL, Li CZ. Electroanalysis. 2011;23:862. [Google Scholar]; (b) Castrillon M, Mayoral A, Urtizberea A, Marquina C, Irusta S, Meier JG, Santamaria J. Nanotechnology. 2013;24:505702. doi: 10.1088/0957-4484/24/50/505702. [DOI] [PubMed] [Google Scholar]
- 60.(a) Huang XD, Zhou XF, Zhou L, Qian K, Wang YH, Liu ZP, Yu CZ. ChemPhysChem. 2011;12:278. doi: 10.1002/cphc.201000376. [DOI] [PubMed] [Google Scholar]; (b) Park SK, Yu SH, Pinna N, Woo S, Jang B, Chung YH, Cho YH, Sung YE, Piao Y. J Mater Chem. 2012;22:2520. [Google Scholar]
- 61.(a) Wu ZS, Wang DW, Ren W, Zhao J, Zhou G, Li F, Cheng HM. Adv Funct Mater. 2010;20:3595. [Google Scholar]; (b) Wu ZS, Ren WC, Wen L, Gao LB, Zhao JP, Chen ZP, Zhou GM, Li F, Cheng HM. ACS Nano. 2010;4:3187. doi: 10.1021/nn100740x. [DOI] [PubMed] [Google Scholar]
- 62.Ren LL, Huang S, Fan W, Liu TX. Appl Surf Sci. 2011;258:1132. [Google Scholar]
- 63.Yan SC, Shi Y, Zhao B, Lu T, Hu D, Xu X, Wu JS, Chen JS. J Alloys Compd. 2013;570:65. [Google Scholar]
- 64.Xue LP, Shen CF, Zheng MB, Lu HL, Li NW, Ji GB, Pan LJ, Cao JM. Mater Lett. 2011;65:198. [Google Scholar]
- 65.Su YJ, Lu XN, Xie MM, Geng HJ, Wei H, Yang Z, Zhang YF. Nanoscale. 2013;5:8889. doi: 10.1039/c3nr02992a. [DOI] [PubMed] [Google Scholar]
- 66.(a) Chang K, Chen WX. J Mater Chem. 2011;21:17175. [Google Scholar]; (b) Huang GC, Chen T, Chen WX, Wang Z, Chang K, Ma L, Huang FH, Chen DY, Lee JY. Small. 2013;9:3693. doi: 10.1002/smll.201300415. [DOI] [PubMed] [Google Scholar]
- 67.Yuan CZ, Hou LR, Yang L, Fan CG, Li DK, Li JM, Shen LF, Zhang F, Zhang XG. Mater Lett. 2011;65:374. [Google Scholar]
- 68.Lu ZS, Guo CX, Yang HB, Qiao Y, Guo J, Li CM. J Colloid Interface Sci. 2011;353:588. doi: 10.1016/j.jcis.2010.10.007. [DOI] [PubMed] [Google Scholar]
- 69.Medintz IL, Uyeda HT, Goldman ER, Mattoussi H. Nat Mater. 2005;4:435. doi: 10.1038/nmat1390. [DOI] [PubMed] [Google Scholar]
- 70.Resch-Genger U, Grabolle M, Cavaliere-Jaricot S, Nitschke R, Nann T. Nat Methods. 2008;5:763. doi: 10.1038/nmeth.1248. [DOI] [PubMed] [Google Scholar]
- 71.(a) Jasieniak J, Smith L, van Embden J, Mulvaney P, Califano M. J Phys Chem C. 2009;113:19468. [Google Scholar]; (b) Yu WW, Qu LH, Guo WZ, Peng XG. Chem Mater. 2003;15:2854. [Google Scholar]
- 72.(a) Alivisatos P. Nat Biotechnol. 2004;22:47. doi: 10.1038/nbt927. [DOI] [PubMed] [Google Scholar]; (b) Niemeyer CM. Angew Chem, Int Ed. 2001;40:4128. doi: 10.1002/1521-3773(20011119)40:22<4128::AID-ANIE4128>3.0.CO;2-S. [DOI] [PubMed] [Google Scholar]; (c) Kim JY, Voznyy O, Zhitomirsky D, Sargent EH. Adv Mater. 2013;25:4986. doi: 10.1002/adma.201301947. [DOI] [PubMed] [Google Scholar]
- 73.Zhang N, Zhang YH, Pan XY, Fu XZ, Liu SQ, Xu YJ. J Phys Chem C. 2011;115:23501. [Google Scholar]
- 74.(a) Shen JF, Shi M, Yan B, Ma HW, Li N, Ye MX. J Mater Chem. 2011;21:7795. [Google Scholar]; (b) Zhou XZ, Huang X, Qi XY, Wu SX, Xue C, Boey FYC, Yan QY, Chen P, Zhang H. J Phys Chem C. 2009;113:10842. [Google Scholar]
- 75.Zhang H, Chen S, Quan X, Yu HT, Zhao HM. J Mater Chem. 2011;21:12986. [Google Scholar]
- 76.Lee SH, Kakati N, Jee SH, Maiti J, Yoon YS. Mater Lett. 2011;65:3281. [Google Scholar]
- 77.(a) Bijani S, Martinez L, Gabas M, Dalchiele EA, Ramos-Barrado JR. J Phys Chem C. 2009;113:19482. [Google Scholar]; (b) Therese GHA, Kamath PV. Chem Mater. 2000;12:1195. [Google Scholar]
- 78.(a) Ding L, Liu YP, Zhai JP, Bond AM, Zhang J. Electroanalysis. 2014;26:121. [Google Scholar]; (b) Hu YJ, Jin JA, Wu P, Zhang H, Cai CX. Electrochim Acta. 2010;56:491. [Google Scholar]
- 79.Golsheikh AM, Huang NM, Lim HN, Zakaria R, Yin CY. Carbon. 2013;62:405. [Google Scholar]
- 80.(a) Zhou YG, Chen JJ, Wang FB, Sheng ZH, Xia XH. Chem Commun. 2010;46:5951. doi: 10.1039/c0cc00394h. [DOI] [PubMed] [Google Scholar]; (b) Claussen JC, Kumar A, Jaroch DB, Khawaja MH, Hibbard AB, Porterfield DM, Fisher TS. Adv Funct Mater. 2012;22:3399. [Google Scholar]
- 81.Wu SX, Yin ZY, He QY, Lu G, Yan QY, Zhang H. J Phys Chem C. 2011;115:15973. [Google Scholar]
- 82.Han KH, Tao M. Sol Energy Mater Sol Cells. 2009;93:153. [Google Scholar]
- 83.Wu SX, Yin ZY, He QY, Lu G, Zhou XZ, Zhang H. J Mater Chem. 2011;21:3467. [Google Scholar]
- 84.Wu SX, Yin ZY, He QY, Huang XA, Zhou XZ, Zhang H. J Phys Chem C. 2010;114:11816. [Google Scholar]
- 85.(a) He FA, Fan JT, Song F, Zhang LM, Chan HLW. Nanoscale. 2011;3:1182. doi: 10.1039/c0nr00672f. [DOI] [PubMed] [Google Scholar]; (b) Yang X, Xu MS, Qiu WM, Chen XQ, Deng M, Zhang JL, Iwai H, Watanabe E, Chen HZ. J Mater Chem. 2011;21:8096. [Google Scholar]
- 86.He FA, Fan JT, Ma D, Zhang LM, Leung C, Chan HL. Carbon. 2010;48:3139. [Google Scholar]
- 87.Pham TA, Choi BC, Jeong YT. Nanotechnology. 2010;21:465603. doi: 10.1088/0957-4484/21/46/465603. [DOI] [PubMed] [Google Scholar]
- 88.Herron N, Wang Y, Eckert H. J Am Chem Soc. 1990;112:1322. [Google Scholar]
- 89.Ismaili H, Geng D, Sun AX, Kantzas TT, Workentin MS. Langmuir. 2011;27:13261. doi: 10.1021/la202815g. [DOI] [PubMed] [Google Scholar]
- 90.Bjork J, Hanke F, Palma CA, Samori P, Cecchini M, Persson M. J Phys Chem Lett. 2010;1:3407. [Google Scholar]
- 91.Wang W, Guo SR, Penchev M, Zhong JB, Lin J, Bao DD, Vullev V, Ozkan M, Ozkan CS. J Nanosci Nanotechnol. 2012;12:6913. doi: 10.1166/jnn.2012.6507. [DOI] [PubMed] [Google Scholar]
- 92.Zhang XR, Li SG, Jin X, Zhang SS. Chem Commun. 2011;47:4929. doi: 10.1039/c1cc10830a. [DOI] [PubMed] [Google Scholar]
- 93.Bourlinos AB, Georgakilas V, Zboril R, Steriotis TA, Stubos AK. Small. 2009;5:1841. doi: 10.1002/smll.200900242. [DOI] [PubMed] [Google Scholar]
- 94.(a) Lin Y, Zhang K, Chen WF, Liu YD, Geng ZG, Zeng J, Pan N, Yan LF, Wang XP, Hou JG. ACS Nano. 2010;4:3033. doi: 10.1021/nn100134j. [DOI] [PubMed] [Google Scholar]; (b) Geng XM, Niu L, Xing ZY, Song RS, Liu GT, Sun MT, Cheng GS, Zhong HJ, Liu ZH, Zhang ZJ, Sun LF, Xu HX, Lu L, Liu LW. Adv Mater. 2010;22:638. doi: 10.1002/adma.200902871. [DOI] [PubMed] [Google Scholar]
- 95.Patil AJ, Vickery JL, Scott TB, Mann S. Adv Mater. 2009;21:3159. [Google Scholar]
- 96.Liu J, Li Y, Li Y, Li J, Deng Z. J Mater Chem. 2010;20:900. [Google Scholar]
- 97.Wang Y, Zhen SJ, Zhang Y, Li YF, Huang CZ. J Phys Chem C. 2011;115:12815. [Google Scholar]
- 98.Zhu GX, Liu YJ, Xu Z, Jiang TA, Zhang C, Li X, Qi G. ChemPhysChem. 2010;11:2432. doi: 10.1002/cphc.201000227. [DOI] [PubMed] [Google Scholar]
- 99.Zhai DY, Li BH, Du HD, Gao GY, Gan L, He YB, Yang QH, Kang FY. Carbon. 2012;50:5034. [Google Scholar]
- 100.Lu GH, Mao S, Park S, Ruoff RS, Chen JH. Nano Res. 2009;2:192. [Google Scholar]
- 101.(a) Jiang T, Kuila T, Kim NH, Ku BC, Lee JH. Compos Sci Technol. 2013;79:115. [Google Scholar]; (b) Myung S, Solanki A, Kim C, Park J, Kim KS, Lee KB. Adv Mater. 2011;23:2221. doi: 10.1002/adma.201100014. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Howarter JA, Youngblood JP. Langmuir. 2006;22:11142. doi: 10.1021/la061240g. [DOI] [PubMed] [Google Scholar]
- 102.Liu JB, Fu SH, Yuan B, Li YL, Deng ZX. J Am Chem Soc. 2010;132:7279. doi: 10.1021/ja100938r. [DOI] [PubMed] [Google Scholar]
- 103.Li JH, Kuang DZ, Feng YL, Zhang FX, Xu ZF, Liu MQ, Wang DP. Biosens Bioelectron. 2013;42:198. doi: 10.1016/j.bios.2012.10.029. [DOI] [PubMed] [Google Scholar]
- 104.Yang SB, Feng XL, Ivanovici S, Mullen K. Angew Chem, Int Ed. 2010;49:8408. doi: 10.1002/anie.201003485. [DOI] [PubMed] [Google Scholar]
- 105.Lee JK, Smith KB, Hayner CM, Kung HH. Chem Commun. 2010;46:2025. doi: 10.1039/b919738a. [DOI] [PubMed] [Google Scholar]
- 106.Zhang J, Zhu ZP, Tang YP, Feng XL. J Mater Chem A. 2013;1:3752. [Google Scholar]
- 107.Ji G, Ding B, Sha Z, Wu JS, Ma Y, Lee JY. Nanoscale. 2013;5:5965. doi: 10.1039/c3nr00085k. [DOI] [PubMed] [Google Scholar]
- 108.Lin SX, Shen CM, Lu DB, Wang CM, Gao HJ. Carbon. 2013;53:112. [Google Scholar]
- 109.Kawasaki H, Nakai K, Arakawa R, Athanassiou EK, Grass RN, Stark WJ. Anal Chem. 2012;84:9268. doi: 10.1021/ac302004g. [DOI] [PubMed] [Google Scholar]
- 110.Myung S, Yin PT, Kim C, Park J, Solanki A, Reyes PI, Lu YC, Kim KS, Lee KB. Adv Mater. 2012;24:6081. doi: 10.1002/adma.201202961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Zhou M, Cai TW, Pu F, Chen H, Wang Z, Zhang HY, Guan SY. ACS Appl Mater Interfaces. 2013;5:3449. doi: 10.1021/am400521n. [DOI] [PubMed] [Google Scholar]
- 112.Luo JY, Zhao X, Wu JS, Jang HD, Kung HH, Huang JX. J Phys Chem Lett. 2012;3:1824. doi: 10.1021/jz3006892. [DOI] [PubMed] [Google Scholar]
- 113.Ronkainen NJ, Halsall HB, Heineman WR. Chem Soc Rev. 2010;39:1747. doi: 10.1039/b714449k. [DOI] [PubMed] [Google Scholar]
- 114.Kuila T, Bose S, Khanra P, Mishra AK, Kim NH, Lee JH. Biosens Bioelectron. 2011;26:4637. doi: 10.1016/j.bios.2011.05.039. [DOI] [PubMed] [Google Scholar]
- 115.(a) Pingarron JM, Yanez-Sedeno P, Gonzalez-Cortes A. Electrochim Acta. 2008;53:5848. [Google Scholar]; (b) Haun JB, Yoon TJ, Lee H, Weissleder R. WIREs Nanomed Nanobiotechnol. 2010;2:291. doi: 10.1002/wnan.84. [DOI] [PubMed] [Google Scholar]
- 116.Liu S, Guo XF. NPG Asia Mater. 2012;4:1. [Google Scholar]
- 117.Lee CS, Kim SK, Kim M. Sensors. 2009;9:7111. doi: 10.3390/s90907111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Allen BL, Kichambare PD, Star A. Adv Mater. 2007;19:1439. [Google Scholar]
- 119.Chen KI, Li BR, Chen YT. Nano Today. 2011;6:131. [Google Scholar]
- 120.(a) Lee K, Nair PR, Scott A, Alam MA, Janes DB. J Appl Phys. 2009;105:102046. doi: 10.1063/1.3116630. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Makowski MS, Ivanisevic A. Small. 2011;7:1863. doi: 10.1002/smll.201100211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.(a) Yang WR, Ratinac KR, Ringer SP, Thordarson P, Gooding JJ, Braet F. Angew Chem, Int Ed. 2010;49:2114. doi: 10.1002/anie.200903463. [DOI] [PubMed] [Google Scholar]; (b) Ohno Y, Maehashi K, Yamashiro Y, Matsumoto K. Nano Lett. 2009;9:3318. doi: 10.1021/nl901596m. [DOI] [PubMed] [Google Scholar]; (c) Kim DJ, Sohn IY, Jung JH, Yoon OJ, Lee NE, Park JS. Biosens Bioelectron. 2013;41:621. doi: 10.1016/j.bios.2012.09.040. [DOI] [PubMed] [Google Scholar]
- 122.Zhang YJ, Geng MK, Zhang H, He Y, Peng C, Huang Q, Fan CH. Chin Sci Bull. 2012;57:3086. [Google Scholar]
- 123.Dinh DA, Hui KS, Hui KN, Cho YR, Zhou W, Hong XT, Chun HH. Appl Surf Sci. 2014;298:62. [Google Scholar]
- 124.Mao S, Lu GH, Yu KH, Bo Z, Chen JH. Adv Mater. 2010;22:3521. doi: 10.1002/adma.201000520. [DOI] [PubMed] [Google Scholar]
- 125.Abe M, Murata K, Ataka T, Ifuku Y, Matsumoto K. J Nanosci Nanotechnol. 2009;9:1947. doi: 10.1166/jnn.2009.424. [DOI] [PubMed] [Google Scholar]
- 126.Mao S, Yu KH, Lu GH, Chen JH. Nano Res. 2011;4:921. [Google Scholar]
- 127.(a) Kwon OS, Lee SH, Park SJ, An JH, Song HS, Kim T, Oh JH, Bae J, Yoon H, Park TH, Jang J. Adv Mater. 2013;25:4177. doi: 10.1002/adma.201301523. [DOI] [PubMed] [Google Scholar]; (b) Kim DJ, Park HC, Sohn IY, Jung JH, Yoon OJ, Park JS, Yoon MY, Lee NE. Small. 2013;9:3352. doi: 10.1002/smll.201203245. [DOI] [PubMed] [Google Scholar]
- 128.Melisko ME, Glantz M, Rugo HS. Nat Clin Pract Oncol. 2009;6:25. doi: 10.1038/ncponc1243. [DOI] [PubMed] [Google Scholar]
- 129.Wang J. Nucleic Acids Res. 2000;28:3011. doi: 10.1093/nar/28.16.3011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Yin ZY, He QY, Huang X, Zhang J, Wu SX, Chen P, Lu G, Chen P, Zhang QC, Yan QY, Zhang H. Nanoscale. 2012;4:293. doi: 10.1039/c1nr11149c. [DOI] [PubMed] [Google Scholar]
- 131.Chikkaveeraiah BV, Bhirde AA, Morgan NY, Eden HS, Chen X. ACS Nano. 2012;6:6546. doi: 10.1021/nn3023969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Ambrosi A, Pumera M. Chem—Eur J. 2010;16:10946. doi: 10.1002/chem.201001584. [DOI] [PubMed] [Google Scholar]
- 133.Alwarappan S, Erdem A, Liu C, Li CZ. J Phys Chem C. 2009;113:8853. [Google Scholar]
- 134.Luo XL, Morrin A, Killard AJ, Smyth MR. Electroanalysis. 2006;18:319. [Google Scholar]
- 135.Xiao Y, Patolsky F, Katz E, Hainfeld JF, Willner I. Science. 2003;299:1877. doi: 10.1126/science.1080664. [DOI] [PubMed] [Google Scholar]
- 136.Park S, Boo H, Chung TD. Anal Chim Acta. 2006;556:46. doi: 10.1016/j.aca.2005.05.080. [DOI] [PubMed] [Google Scholar]
- 137.Shan CS, Yang HF, Han DX, Zhang QX, Ivaska A, Niu L. Biosens Bioelectron. 2010;25:1070. doi: 10.1016/j.bios.2009.09.024. [DOI] [PubMed] [Google Scholar]
- 138.Liu Y, Wang MK, Zhao F, Xu ZA, Dong SJ. Biosens Bioelectron. 2005;21:984. doi: 10.1016/j.bios.2005.03.003. [DOI] [PubMed] [Google Scholar]
- 139.Shan CS, Yang HF, Song JF, Han DX, Ivaska A, Niu L. Anal Chem. 2009;81:2378. doi: 10.1021/ac802193c. [DOI] [PubMed] [Google Scholar]
- 140.Yang J, Deng SY, Lei JP, Ju HX, Gunasekaran S. Biosens Bioelectron. 2011;29:159. doi: 10.1016/j.bios.2011.08.011. [DOI] [PubMed] [Google Scholar]
- 141.(a) Zhang J, Sasaki K, Sutter E, Adzic RR. Science. 2007;315:220. doi: 10.1126/science.1134569. [DOI] [PubMed] [Google Scholar]; (b) Zhang S, Shao YY, Yin GP, Lin YH. Angew Chem, Int Ed. 2010;49:2211. doi: 10.1002/anie.200906987. [DOI] [PubMed] [Google Scholar]
- 142.Zhang J, Lei JP, Pan R, Leng CA, Hu Z, Ju HX. Chem Commun. 2011;47:668. doi: 10.1039/c0cc04198j. [DOI] [PubMed] [Google Scholar]
- 143.Qi T, Liao J, Li Y, Peng J, Li W, Chu B, Li H, Wei Y, Qian Z. Biosens Bioelectron. 2014;61:245. doi: 10.1016/j.bios.2014.05.021. [DOI] [PubMed] [Google Scholar]
- 144.Lu J, Liu S, Ge S, Yan M, Yu J, Hu X. Biosens Bioelectron. 2012;33:29. doi: 10.1016/j.bios.2011.11.054. [DOI] [PubMed] [Google Scholar]
- 145.Lin L, Liu Y, Tang L, Li J. Analyst. 2011;136:4732. doi: 10.1039/c1an15610a. [DOI] [PubMed] [Google Scholar]
- 146.Du Y, Guo SJ, Dong SJ, Wang EK. Biomaterials. 2011;32:8584. doi: 10.1016/j.biomaterials.2011.07.091. [DOI] [PubMed] [Google Scholar]
- 147.Grieshaber D, MacKenzie R, Voros J, Reimhult E. Sensors. 2008;8:1400. doi: 10.3390/s80314000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Sun CL, Lee HH, Yang JM, Wu CC. Biosens Bioelectron. 2011;26:3450. doi: 10.1016/j.bios.2011.01.023. [DOI] [PubMed] [Google Scholar]
- 149.Luo J, Jiang SS, Zhang HY, Jiang JQ, Liu XY. Anal Chim Acta. 2012;709:47. doi: 10.1016/j.aca.2011.10.025. [DOI] [PubMed] [Google Scholar]
- 150.Chen QW, Zhang LY, Chen G. Anal Chem. 2012;84:171. doi: 10.1021/ac2022772. [DOI] [PubMed] [Google Scholar]
- 151.Qu WD, Zhang LY, Chen G. Biosens Bioelectron. 2013;42:430. doi: 10.1016/j.bios.2012.11.011. [DOI] [PubMed] [Google Scholar]
- 152.Chang HC, Wang XM, Shiu KK, Zhu YL, Wang JL, Li QW, Chen BA, Jiang H. Biosens Bioelectron. 2013;41:789. doi: 10.1016/j.bios.2012.10.001. [DOI] [PubMed] [Google Scholar]
- 153.Peng G, Tisch U, Adams O, Hakim M, Shehada N, Broza YY, Billan S, Abdah-Bortnyak R, Kuten A, Haick H. Nat Nanotechnol. 2009;4:669. doi: 10.1038/nnano.2009.235. [DOI] [PubMed] [Google Scholar]
- 154.(a) Nair RR, Blake P, Grigorenko AN, Novoselov KS, Booth TJ, Stauber T, Peres NMR, Geim AK. Science. 2008;320:1308. doi: 10.1126/science.1156965. [DOI] [PubMed] [Google Scholar]; (b) Johari P, Shenoy VB. ACS Nano. 2011;5:7640. doi: 10.1021/nn202732t. [DOI] [PubMed] [Google Scholar]
- 155.(a) Shang JZ, Ma L, Li JW, Ai W, Yu T, Gurzadyan GG. Sci Rep. 2012;2:792. doi: 10.1038/srep00792. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Thomas HR, Valles C, Young RJ, Kinloch IA, Wilson NR, Rourke JP. J Mater Chem C. 2013;1:338. [Google Scholar]
- 156.(a) Matte HSSR, Subrahmanyam KS, Rao KV, George SJ, Rao CNR. Chem Phys Lett. 2011;506:260. [Google Scholar]; (b) Dong HF, Gao WC, Yan F, Ji HX, Ju HX. Anal Chem. 2010;82:5511. doi: 10.1021/ac100852z. [DOI] [PubMed] [Google Scholar]; (c) Wu SJ, Duan N, Ma XY, Xia Y, Wang HG, Wang ZP, Zhang Q. Anal Chem. 2012;84:6263. doi: 10.1021/ac301534w. [DOI] [PubMed] [Google Scholar]
- 157.Malard LM, Pimenta MA, Dresselhaus G, Dresselhaus MS. Phys Rep. 2009;473:51. [Google Scholar]
- 158.Roy R, Hohng S, Ha T. Nat Methods. 2008;5:507. doi: 10.1038/nmeth.1208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.(a) Jares-Erijman EA, Jovin TM. Nat Biotechnol. 2003;21:1387. doi: 10.1038/nbt896. [DOI] [PubMed] [Google Scholar]; (b) Nagai T, Yamada S, Tominaga T, Ichikawa M, Miyawaki A. Proc Natl Acad Sci USA. 2004;101:10554. doi: 10.1073/pnas.0400417101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Swathi RS, Sebastian KL. J Chem Phys. 2009;130:086101. doi: 10.1063/1.3077292. [DOI] [PubMed] [Google Scholar]
- 161.Cheng L, Wang C, Liu Z. Nanoscale. 2013;5:23. doi: 10.1039/c2nr32311g. [DOI] [PubMed] [Google Scholar]
- 162.Zhang CL, Yuan YX, Zhang SM, Wang YH, Liu ZH. Angew Chem, Int Ed. 2011;50:6851. doi: 10.1002/anie.201100769. [DOI] [PubMed] [Google Scholar]
- 163.Li M, Zhou XJ, Guo SW, Wu NQ. Biosens Bioelectron. 2013;43:69. doi: 10.1016/j.bios.2012.11.039. [DOI] [PubMed] [Google Scholar]
- 164.(a) Wang ZH, Qin YX, Wang C, Sun LJ, Lu XL, Lu XQ. Appl Surf Sci. 2012;258:2017. [Google Scholar]; (b) Zhang M, Yin BC, Tan WH, Ye BC. Biosens Bioelectron. 2011;26:3260. doi: 10.1016/j.bios.2010.12.037. [DOI] [PubMed] [Google Scholar]; (c) Li T, Wang E, Dong S. Anal Chem. 2010;82:1515. doi: 10.1021/ac902638v. [DOI] [PubMed] [Google Scholar]
- 165.Tao Y, Lin YH, Huang ZZ, Ren JS, Qu XG. Analyst. 2012;137:2588. doi: 10.1039/c2an35373c. [DOI] [PubMed] [Google Scholar]
- 166.Fu XL, Lou TT, Chen ZP, Lin M, Feng WW, Chen LX. ACS Appl Mater Interfaces. 2012;4:1080. doi: 10.1021/am201711j. [DOI] [PubMed] [Google Scholar]
- 167.Ahijado-Guzman R, Gomez-Puertas P, Alvarez-Puebla RA, Rivas G, Liz-Marzan LM. ACS Nano. 2012;6:7514. doi: 10.1021/nn302825u. [DOI] [PubMed] [Google Scholar]
- 168.(a) Graham D, Thompson DG, Smith WE, Faulds K. Nat Nanotechnol. 2008;3:548. doi: 10.1038/nnano.2008.189. [DOI] [PubMed] [Google Scholar]; (b) Le Ru EC, Blackie E, Meyer M, Etchegoin PG. J Phys Chem C. 2007;111:13794. [Google Scholar]
- 169.Das G, Chirumamilla M, Toma A, Gopalakrishnan A, Zaccaria RP, Alabastri A, Leoncini M, Di Fabrizio E. Sci Rep. 2013;3:1792. doi: 10.1038/srep01792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Hutter E, Fendler JH. Adv Mater. 2004;16:1685. [Google Scholar]
- 171.Rycenga M, Camargo PHC, Li WY, Moran CH, Xia YN. J Phys Chem Lett. 2010;1:696. doi: 10.1021/jz900286a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Xu WG, Mao NN, Zhang J. Small. 2013;9:1206. doi: 10.1002/smll.201203097. [DOI] [PubMed] [Google Scholar]
- 173.Otto A. J Raman Spectrosc. 2005;36:497. [Google Scholar]
- 174.He SJ, Liu KK, Su S, Yan J, Mao XH, Wang DF, He Y, Li LJ, Song SP, Fan CH. Anal Chem. 2012;84:4622. doi: 10.1021/ac300577d. [DOI] [PubMed] [Google Scholar]
- 175.Ren W, Fang YX, Wang EK. ACS Nano. 2011;5:6425. doi: 10.1021/nn201606r. [DOI] [PubMed] [Google Scholar]
- 176.Xie YF, Li Y, Niu L, Wang HY, Qian H, Yao WR. Talanta. 2012;100:32. doi: 10.1016/j.talanta.2012.07.080. [DOI] [PubMed] [Google Scholar]
- 177.Zhang YW, Liu S, Wang L, Qin XY, Tian JQ, Lu WB, Chang GH, Sun XP. RSC Adv. 2012;2:538. [Google Scholar]
- 178.Li YT, Qu LL, Li DW, Song QX, Fathi F, Long YT. Biosens Bioelectron. 2013;43:94. doi: 10.1016/j.bios.2012.12.005. [DOI] [PubMed] [Google Scholar]
- 179.Liu ZM, Hu CF, Li SX, Zhang W, Guo ZY. Anal Chem. 2012;84:10338. doi: 10.1021/ac3023907. [DOI] [PubMed] [Google Scholar]
- 180.Sun YG, Xia YN. Science. 2002;298:2176. doi: 10.1126/science.1077229. [DOI] [PubMed] [Google Scholar]
- 181.Liu ZM, Guo ZY, Zhong HQ, Qin XC, Wan MM, Yang BW. Phys Chem Chem Phys. 2013;15:2961. doi: 10.1039/c2cp43715e. [DOI] [PubMed] [Google Scholar]
- 182.Zhang Z, Xu F, Yang W, Guo M, Wang X, Zhang B, Tang J. Chem Commun (Cambridge, U K) 2011;47:6440. doi: 10.1039/c1cc11125f. [DOI] [PubMed] [Google Scholar]
- 183.Jiang W, KimBetty YS, Rutka JT, ChanWarren CW. Nat Nanotechnol. 2008;3:145. doi: 10.1038/nnano.2008.30. [DOI] [PubMed] [Google Scholar]
- 184.(a) Hoffmann F, Cornelius M, Morell J, Froeba M. Angew Chem, Int Ed. 2006;45:3216. doi: 10.1002/anie.200503075. [DOI] [PubMed] [Google Scholar]; (b) Wang F, Liu X. Chem Soc Rev. 2009;38:976. doi: 10.1039/b809132n. [DOI] [PubMed] [Google Scholar]; (c) Murray CB, Norris DJ, Bawendi MG. J Am Chem Soc. 1993;115:8706. [Google Scholar]; (d) Soppimath KS, Aminabhavi TM, Kulkarni AR, Rudzinski WE. J Controlled Release. 2001;70:1. doi: 10.1016/s0168-3659(00)00339-4. [DOI] [PubMed] [Google Scholar]; (e) Laurent S, Forge D, Port M, Roch A, Robic C, Elst LV, Muller RN. Chem Rev. 2008;108:2064. doi: 10.1021/cr068445e. [DOI] [PubMed] [Google Scholar]
- 185.(a) Solanki A, Shah S, Yin PT, Lee KB. Sci Rep. 2013;3:1553. doi: 10.1038/srep01553. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Chatteriee DK, Rufalhah AJ, Zhang Y. Biomaterials. 2008;29:937. doi: 10.1016/j.biomaterials.2007.10.051. [DOI] [PubMed] [Google Scholar]; (c) Michalet X, Pinaud FF, Bentolila LA, Tsay JM, Doose S, Li JJ, Sundaresan G, Wu AM, Gambhir SS, Weiss S. Science. 2005;307:538. doi: 10.1126/science.1104274. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Pankhurst QA, Connolly J, Jones SK, Dobson J. J Phys D: Appl Phys. 2003;36:R167. [Google Scholar]; (e) Sperling RA, Rivera Gil P, Zhang F, Zanella M, Parak WJ. Chem Soc Rev. 2008;37:1896. doi: 10.1039/b712170a. [DOI] [PubMed] [Google Scholar]; (f) Agasti SS, Rana S, Park MH, Kim CK, You CC, Rotello VM. Adv Drug Delivery Rev. 2010;62:316. doi: 10.1016/j.addr.2009.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Goenka S, Sant V, Sant S. J Controlled Release. 2014;173:75. doi: 10.1016/j.jconrel.2013.10.017. [DOI] [PubMed] [Google Scholar]
- 187.Kim JH, Park K, Nam HY, Lee S, Kim K, Kwon IC. Prog Polym Sci. 2007;32:1031. [Google Scholar]
- 188.Sharma P, Brown S, Walter G, Santra S, Moudgil B. Adv Colloid Interface Sci. 2006;123–126:471. doi: 10.1016/j.cis.2006.05.026. [DOI] [PubMed] [Google Scholar]
- 189.Sun C, Lee JSH, Zhang M. Adv Drug Delivery Rev. 2008;60:1252. doi: 10.1016/j.addr.2008.03.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Ai H, Flask C, Weinberg B, Shuai XT, Pagel MD, Farrell D, Duerk J, Gao J. Adv Mater. 2005;17:1949. [Google Scholar]
- 191.(a) Raynal I, Prigent P, Peyramaure S, Najid A, Rebuzzi C, Corot C. Invest Radiol. 2004;39:56. doi: 10.1097/01.rli.0000101027.57021.28. [DOI] [PubMed] [Google Scholar]; (b) Rogers WJ, Basu P. Atherosclerosis. 2005;178:67. doi: 10.1016/j.atherosclerosis.2004.08.017. [DOI] [PubMed] [Google Scholar]
- 192.(a) Szabo T, Bakandritsos A, Tzitzios V, Devlin E, Petridis D, Dekany I. J Phys Chem B. 2008;112:14461. doi: 10.1021/jp802879a. [DOI] [PubMed] [Google Scholar]; (b) Singh VK, Patra MK, Manoth M, Gowd GS, Vadera SR, Kumar N. New Carbon Mater. 2009;24:147. [Google Scholar]
- 193.Cong HP, He JJ, Lu Y, Yu SH. Small. 2010;6:169. doi: 10.1002/smll.200901360. [DOI] [PubMed] [Google Scholar]
- 194.Chen W, Yi P, Zhang Y, Zhang L, Deng Z, Zhang Z. ACS Appl Mater Interfaces. 2011;3:4085. doi: 10.1021/am2009647. [DOI] [PubMed] [Google Scholar]
- 195.Chen ML, Shen LM, Chen S, Wang H, Chen XW, Wang JH. J Mater Chem B. 2013;1:2582. doi: 10.1039/c3tb20234h. [DOI] [PubMed] [Google Scholar]
- 196.Zeng LY, Ren WZ, Zheng JJ, Wu AG, Cui P. Appl Surf Sci. 2012;258:2570. [Google Scholar]
- 197.Liu Q, Wei L, Wang J, Peng F, Luo D, Cui R, Niu Y, Qin X, Liu Y, Sun H, Yang J, Li Y. Nanoscale. 2012;4:7084. doi: 10.1039/c2nr32525j. [DOI] [PubMed] [Google Scholar]
- 198.Huang J, Zong C, Shen H, Lui M, Chen B, Ren B, Zhang Z. Small. 2012;8:2577. doi: 10.1002/smll.201102743. [DOI] [PubMed] [Google Scholar]
- 199.Zwicke GL, Mansoori GA, Jeffery CJ. Nano Rev. 2012;3:18496. doi: 10.3402/nano.v3i0.18496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Weitman SD, Lark RH, Coney LR, Fort DW, Frasca V, Zurawski VR, Kamen BA. Cancer Res. 1992;52:3396. [PubMed] [Google Scholar]
- 201.Ntziachristos V. Annu Rev Biomed Eng. 2006;8:1. doi: 10.1146/annurev.bioeng.8.061505.095831. [DOI] [PubMed] [Google Scholar]
- 202.Sreejith S, Ma X, Zhao Y. J Am Chem Soc. 2012;134:17346. doi: 10.1021/ja305352d. [DOI] [PubMed] [Google Scholar]
- 203.Subramaniam P, Lee SJ, Shah S, Patel S, Starovoytov V, Lee KB. Adv Mater. 2012;24:4014. doi: 10.1002/adma.201201019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Sheng Y, Tang X, Peng E, Xue J. J Mater Chem B. 2013;1:512. doi: 10.1039/c2tb00123c. [DOI] [PubMed] [Google Scholar]
- 205.(a) Zhou F, Xing D, Ou Z, Wu B, Resasco DE, Chen WR. J Biomed Opt. 2009;14:021009. doi: 10.1117/1.3078803. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Johannsen M, Gneveckow U, Eckelt L, Feussner A, Waldofner N, Scholz R, Deger S, Wust P, Loening SA, Jordan A. Int J Hyperthermia. 2005;21:637. doi: 10.1080/02656730500158360. [DOI] [PubMed] [Google Scholar]
- 206.Jaque D, Martinez Maestro L, del Rosal B, Haro-Gonzalez P, Benayas A, Plaza JL, Martin Rodriguez E, Garcia Sole J. Nanoscale. 2014;6:9494. doi: 10.1039/c4nr00708e. [DOI] [PubMed] [Google Scholar]
- 207.Yang K, Hu L, Ma X, Ye S, Cheng L, Shi X, Li C, Li Y, Liu Z. Adv Mater. 2012;24:1868. doi: 10.1002/adma.201104964. [DOI] [PubMed] [Google Scholar]
- 208.Yang K, Feng L, Hong H, Cai W, Liu Z. Nat Protoc. 2013;8:2392. doi: 10.1038/nprot.2013.146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Lim DK, Barhoumi A, Wylie RG, Reznor G, Langer RS, Kohane DS. Nano Lett. 2013;13:4075. doi: 10.1021/nl4014315. [DOI] [PubMed] [Google Scholar]
- 210.(a) Huschka R, Zuloaga J, Knight MW, Brown LV, Nordlander P, Halas NJ. J Am Chem Soc. 2011;133:12247. doi: 10.1021/ja204578e. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Echtermeyer TJ, Britnell L, Jasnos PK, Lombardo A, Gorbachev RV, Grigorenko AN, Geim AK, Ferrari AC, Novoselov KS. Nat Commun. 2011;2:458. doi: 10.1038/ncomms1464. [DOI] [PubMed] [Google Scholar]
- 211.Shi X, Gong H, Li Y, Wang C, Cheng L, Liu Z. Biomaterials. 2013;34:4786. doi: 10.1016/j.biomaterials.2013.03.023. [DOI] [PubMed] [Google Scholar]
- 212.Hu SH, Chen YW, Hung WT, Chen IW, Chen SY. Adv Mater. 2012;24:1748. doi: 10.1002/adma.201104070. [DOI] [PubMed] [Google Scholar]
- 213.(a) Panyam J, Labhasetwar V. Adv Drug Delivery Rev. 2003;55:329. doi: 10.1016/s0169-409x(02)00228-4. [DOI] [PubMed] [Google Scholar]; (b) Shah S, Solanki A, Sasmal PK, Lee KB. J Am Chem Soc. 2013;135:15682. doi: 10.1021/ja4071738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Farokhzad OC, Langer R. ACS Nano. 2009;3:16. doi: 10.1021/nn900002m. [DOI] [PubMed] [Google Scholar]
- 215.Yang X, Zhang X, Ma Y, Huang Y, Wang Y, Chen Y. J Mater Chem. 2009;19:2710. [Google Scholar]
- 216.Balcioglu M, Rana M, Yigit MV. J Mater Chem B. 2013;1:6187. doi: 10.1039/c3tb20992j. [DOI] [PubMed] [Google Scholar]
- 217.Wang C, Li J, Amatore C, Chen Y, Jiang H, Wang XM. Angew Chem, Int Ed. 2011;50:11644. doi: 10.1002/anie.201105573. [DOI] [PubMed] [Google Scholar]
- 218.Ma X, Qu QY, Zhao Y, Luo Z, Zhao Y, Ng KW, Zhao YL. J Mater Chem B. 2013;1:6495. doi: 10.1039/c3tb21385d. [DOI] [PubMed] [Google Scholar]
- 219.Fahmy TM, Fong PM, Goyal A, Saltzman WM. Mater Today. 2005;8:18. [Google Scholar]
- 220.Fan X, Jiao G, Gao L, Jin P, Li X. J Mater Chem B. 2013;1:2658. doi: 10.1039/c3tb00493g. [DOI] [PubMed] [Google Scholar]
- 221.Chen ML, He YJ, Chen XW, Wang JH. Bioconjugate Chem. 2013;24:387. doi: 10.1021/bc3004809. [DOI] [PubMed] [Google Scholar]
- 222.Hogemann-Savellano D, Bos E, Blondet C, Sato F, Abe T, Josephson L, Weissleder R, Gaudet J, Sgroi D, Peters PJ, Basilion JP. Neoplasia. 2003;5:495. doi: 10.1016/s1476-5586(03)80034-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223.Shah S, Sasmal PK, Lee KB. J Mater Chem B. 2014;2:7685. doi: 10.1039/C4TB01436G. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224.He D, He X, Wang K, Zou Z, Yang X, Li X. Langmuir. 2014;30:7182. doi: 10.1021/la501075c. [DOI] [PubMed] [Google Scholar]
- 225.Shirai M, Tsunooka M. Prog Polym Sci. 1996;21:1. [Google Scholar]
- 226.Wang F, Liu B, Ip AC, Liu J. Adv Mater. 2013;25:4087. doi: 10.1002/adma.201301183. [DOI] [PubMed] [Google Scholar]
- 227.Cheng FF, Chen W, Hu LH, Chen G, Miao HT, Li C, Zhu JJ. J Mater Chem B. 2013;1:4956. doi: 10.1039/c3tb20656d. [DOI] [PubMed] [Google Scholar]
- 228.Wang C, Ravi S, Garapati US, Das M, Howell M, Mallela J, Alwarapan S, Mohapatra SS, Mohapatra S. J Mater Chem B. 2013;1:4396. doi: 10.1039/C3TB20452A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229.(a) Yin PT, Shah BP, Lee KB. Small. 2014;10:4106. doi: 10.1002/smll.201400963. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Shah BP, Pasquale N, De G, Tan T, Ma J, Lee KB. ACS Nano. 2014;8:9379. doi: 10.1021/nn503431x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230.Sherlock SP, Tabakman SM, Xie L, Dai H. ACS Nano. 2011;5:1505. doi: 10.1021/nn103415x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231.Ma X, Tao H, Yang K, Feng L, Cheng L, Shi X, Li Y, Guo L, Liu Z. Nano Res. 2012;5:199. [Google Scholar]
- 232.Shen JM, Gao FY, Guan LP, Su W, Yang YJ, Li QR, Jin ZC. RSC Adv. 2014;4:18473. [Google Scholar]
- 233.Zedan AF, Moussa S, Terner J, Atkinson G, El-Shall MS. ACS Nano. 2013;7:627. doi: 10.1021/nn304775h. [DOI] [PubMed] [Google Scholar]
- 234.Xu C, Yang D, Mei L, Li Q, Zhu H, Wang T. ACS Appl Mater Interfaces. 2013;5:12911. doi: 10.1021/am404714w. [DOI] [PubMed] [Google Scholar]
- 235.Wang Y, Wang H, Liu D, Song S, Wang X, Zhang H. Biomaterials. 2013;34:7715. doi: 10.1016/j.biomaterials.2013.06.045. [DOI] [PubMed] [Google Scholar]
- 236.Wang Y, Wang K, Zhao J, Liu X, Bu J, Yan X, Huang R. J Am Chem Soc. 2013;135:4799. doi: 10.1021/ja312221g. [DOI] [PubMed] [Google Scholar]
- 237.Wang Y, Huang R, Liang G, Zhang Z, Zhang P, Yu S, Kong J. Small. 2014;10:109. doi: 10.1002/smll.201301297. [DOI] [PubMed] [Google Scholar]
- 238.Chen YW, Chen PJ, Hu SH, Chen IW, Chen SY. Adv Funct Mater. 2014;24:451. [Google Scholar]
- 239.Berthiaume F, Maguire TJ, Yarmush ML. Annu Rev Chem Biomol Eng. 2011;2:403. doi: 10.1146/annurev-chembioeng-061010-114257. [DOI] [PubMed] [Google Scholar]
- 240.(a) Chen GY, Pang DWP, Hwang SM, Tuan HY, Hu YC. Biomaterials. 2012;33:418. doi: 10.1016/j.biomaterials.2011.09.071. [DOI] [PubMed] [Google Scholar]; (b) Nayak TR, Andersen H, Makam VS, Khaw C, Bae S, Xu X, Ee PLR, Ahn JH, Hong BH, Pastorin G, Özyilmaz B. ACS Nano. 2011;5:4670. doi: 10.1021/nn200500h. [DOI] [PubMed] [Google Scholar]; (c) Park SY, Park J, Sim SH, Sung MG, Kim KS, Hong BH, Hong S. Adv Mater. 2011;23:H263. doi: 10.1002/adma.201101503. [DOI] [PubMed] [Google Scholar]; (d) Sebaa M, Nguyen TY, Paul RK, Mulchandani A, Liu H. Mater Lett. 2013;92:122. [Google Scholar]
- 241.(a) Shah S, Yin PT, Uehara TM, Chueng STD, Yang L, Lee KB. Adv Mater. 2014;26:3673. doi: 10.1002/adma.201400523. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Kim J, Kim YR, Kim Y, Lim KT, Seonwoo H, Park S, Cho SP, Hong BH, Choung PH, Chung TD, Choung YH, Chung JH. J Mater Chem B. 2013;1:933. doi: 10.1039/c2tb00274d. [DOI] [PubMed] [Google Scholar]
- 242.Hu W, Peng C, Lv M, Li X, Zhang Y, Chen N, Fan C, Huang Q. ACS Nano. 2011;5:3693. doi: 10.1021/nn200021j. [DOI] [PubMed] [Google Scholar]
- 243.(a) Watt FM, Huck WTS. Nat Rev Mol Cell Biol. 2013;14:467. doi: 10.1038/nrm3620. [DOI] [PubMed] [Google Scholar]; (b) Solanki A, Shah S, Memoli KA, Park SY, Hong S, Lee KB. Small. 2010;6:2509. doi: 10.1002/smll.201001341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 244.Solanki A, Chueng STD, Yin PT, Kappera R, Chhowalla M, Lee KB. Adv Mater. 2013;25:5477. doi: 10.1002/adma.201302219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 245.Kim TH, Lee KB, Choi JW. Biomaterials. 2013;34:8660. doi: 10.1016/j.biomaterials.2013.07.101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 246.(a) Ramm Sander P, Hau P, Koch S, Schutze K, Bogdahn U, Kalbitzer HR, Aigner L. Trends Biotechnol. 2013;31:204. doi: 10.1016/j.tibtech.2013.01.008. [DOI] [PubMed] [Google Scholar]; (b) Yanes O, Clark J, Wong DM, Patti GJ, Sánchez-Ruiz A, Benton HP, Trauger SA, Desponts C, Ding S, Siuzdak G. Nat Chem Biol. 2010;6:411. doi: 10.1038/nchembio.364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 247.Pinto AM, Goncalves IC, Magalhaes FD. Colloids Surf, B. 2013;111:188. doi: 10.1016/j.colsurfb.2013.05.022. [DOI] [PubMed] [Google Scholar]
- 248.Wang K, Ruan J, Song H, Zhang JL, Wo Y, Guo SW, Cui DX. Nanoscale Res Lett. 2011;6:8. doi: 10.1007/s11671-010-9751-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 249.Sasidharan A, Panchakarla LS, Chandran P, Menon D, Nair S, Rao CNR, Koyakutty M. Nanoscale. 2011;3:2461. doi: 10.1039/c1nr10172b. [DOI] [PubMed] [Google Scholar]
- 250.(a) Liao KH, Lin YS, Macosko CW, Haynes CL. ACS Appl Mater Interfaces. 2011;3:2607. doi: 10.1021/am200428v. [DOI] [PubMed] [Google Scholar]; (b) Gollavelli G, Ling YC. Biomaterials. 2012;33:2532. doi: 10.1016/j.biomaterials.2011.12.010. [DOI] [PubMed] [Google Scholar]
- 251.(a) Akhavan O, Ghaderi E, Abouei E, Hatamie S, Ghasemi E. Carbon. 2014;66:395. [Google Scholar]; (b) Akhavan O, Ghaderi E. J Mater Chem B. 2013;1:6291. doi: 10.1039/c3tb21085e. [DOI] [PubMed] [Google Scholar]
- 252.Li B, Yang JZ, Huang Q, Zhang Y, Peng C, Zhang YJ, He Y, Shi JY, Li WX, Hu J, Fan CH. NPG Asia Mater. 2013;5:e44. [Google Scholar]
- 253.Yang K, Wan JM, Zhang SA, Zhang YJ, Lee ST, Liu ZA. ACS Nano. 2011;5:516. doi: 10.1021/nn1024303. [DOI] [PubMed] [Google Scholar]
- 254.Zhang XY, Yin JL, Peng C, Hu WQ, Zhu ZY, Li WX, Fan CH, Huang Q. Carbon. 2011;49:986. [Google Scholar]
- 255.Huang J, Zong C, Shen H, Liu M, Chen BA, Ren B, Zhang ZJ. Small. 2012;8:2577. doi: 10.1002/smll.201102743. [DOI] [PubMed] [Google Scholar]