

Abstract
Photoactivatable fluorophores are essential tools for studying the dynamic molecular interactions within important biological systems with high spatiotemporal resolution. However, currently developed photoactivatable fluorophores based on conventional dyes have several limitations including reduced photoactivation efficiency, cytotoxicity, large molecular size, and complicated organic synthesis. To overcome these challenges, we herein report a class of photoactivatable fluorescent N-hydroxyoxindoles formed through the intramolecular photocyclization of substituted o-nitrophenyl ethanol (ONPE). These oxindole fluorophores afford excellent photoactivation efficiency with ultra-high fluorescence enhancement (up to 800-fold) and are small in size. Furthermore, the oxindole derivatives show exceptional biocompatibility by generating water as the only photolytic side product. Moreover, structure–activity relationship analysis clearly revealed the strong correlation between the fluorescent properties and the substituent groups, which can serve as a guideline for the further development of ONPE-based fluorescent probes with desired photophysical and biological properties. As a proof-of-concept, we demonstrated the capability of a new substituted ONPE that has an uncaging wavelength of 365–405 nm and an excitation/emission at 515 and 620 nm, for the selective imaging of a cancer cell line (Hela cells) and a human neural stem cell line (hNSCs).
Keywords: cage compounds, dyes/pigments, fluorescence imaging, oxindole, fluorescent probes
Introduction
Photoactivatable fluorophores that exist in stable dark states but can be subsequently photoactivated to a fluorescent state for high photon emission[1] are crucial tools for advanced microscopy imaging such as super-resolution imaging,[2] selective cell/biomolecule imaging,[3] as well as for investigating dynamic biomolecular processes in living cells,[4] because of their exquisite spatiotemporal control over fluorescence by using light.[5] To date, several strategies have been reported for the design of photoactivatable fluorophores. The most popular strategy is to build “caged fluorophores” by direct structural modification of conventional organic fluorophores (e.g., coumarin, resorufin, fluorescein, rhodamine and their derivatives) with photolabile protecting groups (e.g., o-nitrobenzyl alcohol and its various derivatives including 4,5-dimethoxy-2-nitrobenzyl alcohol, 1-(2′-nitrophenyl)ethyl alcohol, and 1-(4′, 5′-dime-thoxy-2′-nitrophenyl) ethyl, and other caging moieties).[6] These systems restore the fluorophore after photoactivation mainly through the removal of photolabile groups, which retain the advantages of the used fluorophores such as high water solubility and strong fluorescent emission. However, such covalent structural modifications usually involve complicated organic synthesis with an unintended loss of biocompatibility. For example, cytotoxicity of the photolysis byproducts of o-nitrobenzyl groups is generally conceived as unfavorable.[7] Furthermore, reduced photoactivation efficiency was occasionally observed in these systems, which may be due to the inner filtering effect of the latent fluorophores. It should be noted that such limitations are also present in photoactivatable fluorophores that built on FRET-based dyes;[8] photo-release of fluorophores from fluorescence quenchers;[9] and photo-switching of the spiropyran-merocyanine-based nanoparticles.[10] To overcome these challenges faced by the aforementioned “caged fluorophores”, researchers have successfully developed photo-activatable fluorophores based on strategies such as the intra-molecular photocyclization of o-hydroxycinnamate to coumarin and the intramolecular “photo click” reaction of tetrazolealkene to pyrazoline, which are more attractive as they prevent the utilization of latent fluorophores and potential toxic caging groups, thus having higher fluorescence turn-on ratio and better biocompatibility.[11] However, to achieve the ultimate goal of spatial-temporal selective imaging in dynamic biological systems, the design of small-sized photoactivatable dyes with further advancements in the speed and resolution of activation, reduced inner filter effect, and equally importantly, simplified synthetic routes, is highly desired.
Herein, we report the development of a new class of photo-activatable fluorophore based on a substituted o-nitrophenyl ethanol (ONPE) structure, capable of rapid and ultra-efficient photoactivation (up to 800-fold fluorescence turn-on ratio) with negligible inner-filter effect and noncytotoxic side products. In contrast with conventional caged dyes, our self-caged fluorophore is smaller in size (single benzene moiety vs. 3–4 benzene moiety-based conventional dyes, such as fluorescein and rhodamine) and therefore involves facile synthesis and minimal steric perturbation in biological systems, which is of particular importance in probing dynamic molecular processes.[5c] Furthermore, we demonstrate a structure–activity relationship between the different substituted ONPEs and their corresponding fluorescent properties, which can serve as a guideline for the design and development of new photoactivatable fluorophores with desired optical properties. Finally, we demonstrate the application of a photoactivatable dye with excitation–emission in the green-red region for spatially controlled imaging of human neural stem cells (hNSCs) and cervical cancer (HeLa) cells.
Results and Discussion
O-Nitrophenyl ethanol and its derivatives have been utilized as photolabile protecting groups for the caging and highly efficient photorelease of biomolecules including amino acids and nucleotides, with a low cytotoxic o-nitrostyrene as the major photolysis product.[12] However, it is surprising to find that a buffered methanolic solution of substituted ONPE 1 (3:1 v/v, methanol/PBS, pH7.4, 10 mM) was nonfluorescent prior to photo irradiation, but exhibited bright-green fluorescence upon brief exposure to UV light (254 nm, 3 mW cm−2, 1 min) (Figure 1 A, inset), indicating a fluorescent product other than o-nitrostyrene, because o-nitrostyrene is nonfluorescent. A closer examination of the absorption spectrum revealed that the photolysis proceeded with a decrease in absorption peak of 1 (254 nm), and a concomitant formation of a new strong absorption peak centered at 324 nm (Figure 1 B). This was accompanied by a remarkable fluorescence enhancement that reached a maximum of about 800-fold enhancement after 20 min of UV exposure. It has been reported that UV photoirradiation of ONPE leads to an intramolecular photocyclization[13] in which the ethanol group was first selectively oxidized to acetic acid by the excited nitro group, which was followed by the cyclization of the intermediate to form N-hydroxyoxindole and water as the side product (Figure 1 A). The photo conversion is highly efficient and was not dependent on the solvent.[13] It was expected that our ONPE derivative 1 would also undergo a similar photocyclization under UV irradiation (λ= 254 nm) with a corresponding substituted oxindole 1a as the photolysis product. To demonstrate this, the photoreaction was monitored by HPLC-MS, which showed that the progressive photolysis of 1 upon photoirradiation resulted in the formation of a major product with a molecular weight of 207.2 g mol−1 assigned to the substituted oxindole compound 1 a (Figure 1 C and the Supporting Information, Figure S1). Further content analysis of the HPLC monitoring showed that the photolysis of 1 and the photogeneration of 1 a have 50 % symmetry (Figure 1 D), indicating that the photoreaction is clean and proceeds without the formation of byproducts. Although the photocyclization of ONPE to N-hydroxyloxindole has been reported as early as 1970s,[13,14] to the best of our knowledge, our system is the first demonstration of an ONPE-based photo-activatable fluorophore.
Figure 1.
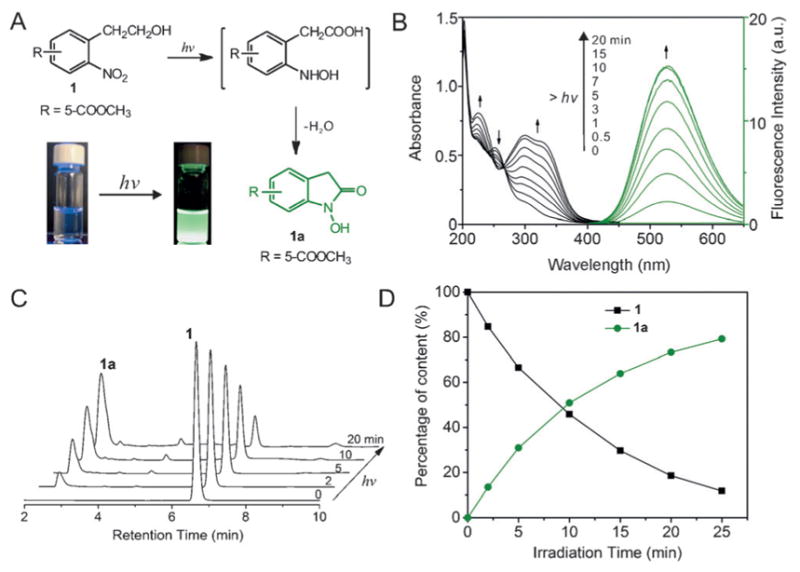
A) Photocyclization of ONPE to N-hydroxyoxindole. Inset shows the photographs of a solution of 1 under UV lamp before and after UV irradiation; B) Absorption and fluorescence spectra depicting the photolysis of 1 in MeOH/PBS (3:1, v/v) under UV light irradiation (254 nm); C) HPLC analysis of the photolytic reaction reveals progressive photolysis of 1 upon irradiation and formation of a major photolytic product (1 a). The signal was recorded with a UV-absorption detector at the wavelength of 299 nm, which is the isosbestic point of the absorption spectrum shown in (B); D) Content analysis of the 1 and 1 a during the photolysis shows 50 % symmetry indicating a clean photolysis reaction without any byproducts. Analysis was done using integrated area of 1 and 1 a on the HPLC chromatogram.
To modulate the fluorescence properties and understand the structure–activity relationship of this photoinduced fluorescent oxindole, we synthesized a series of ONPEs with different substituted groups on the meta- and/or para-position and studied the fluorescence profiles of their respective photoreactions. As summarized in Table 1 (for details, see the Supporting Information, Figures S2–S10), in general, meta-substituted electron-withdrawing (EW) groups such as carbonyl and cyanide groups slightly affect the absorption of ONPEs, in which only minor blueshifts in absorption maxima were observed compared with the spectrum of the non-substituted ONPE. On the other hand, ONPE substituted with meta-electron-donating (ED) group such as the methoxy group enhanced the absorbance and generated a new strong absorption peak at wavelength beyond 300 nm. Such change in absorption spectrum would benefit the photolysis of ONPE through photoirradiation with a long wavelength. In contrast to this, a much stronger blue-shift and redshift induced by EW and ED groups, respectively, was observed in the absorption of para-substituted ONPEs (2 and 10). Furthermore, it is interesting to find that only the ONPEs with strong EW groups showed significant fluorescence enhancement after photoirradiation (compounds 1–6, 21 to 800-fold fluorescence enhancements). In particular, compound 1, which possesses an meta-methyl acetate group, demonstrated a large difference in absorption peaks before and after photo irradiation (254 nm vs. 334 nm), and a large Stokes shift (186 nm) as well as an 800-fold fluorescence enhancement. The separated photoactivation and excitation wavelength is important for highly efficient photolysis and allows for real-time quantification of uncaging. Hydrolysis of the substituted methyl acetate in 1 to acid (6) led to a photolysis product with a weak fluorescence. It should be noted that only a negligible fluorescence enhancement was observed in case of non-substituted ONPE (7) or compounds with meta- or para-substituted strong ED groups (8, 9, and 10). Moreover, photolysis of the derivatives such as the esterification product of ONPE led to generation of non-fluorescent o-nitrostyrene (the Supporting Information, Figure S11). A closer look into the fluorescence properties of the photogenerated oxindoles revealed an extremely large Stock’s shift (≈200 nm) in oxindoles with meta-substituted EW groups, which is larger than that in meta-substituted oxindole with ED groups and para-substituted oxindoles. These results demonstrated a strong structural significance of the meta-substituted strong EW groups and 1′-unmodified ethyl hydroxyl group for the highly sensitive photo-activation of ONPE with strong fluorescent N-hydroxyoxindole product.
Table 1.
Optical properties of o-nitrophenylenthanols with various substituted groups and their corresponding oxindoles.[a]
| |||||||
|---|---|---|---|---|---|---|---|
| Compds | R1 | R2 | λmax[c] [nm] | λmax[d] [nm] | λem[e] [nm] | φ[f] | F/F0[g] |
| 1–1a | COOCH3 | H | 254 | 334 | 520 | 0.36 | 800 |
| 2–2a | H | COOCH3 | 230 | 290 | 401 | 0.15 | 200 |
| 3–3a | COOCH3 | OCH3 | 334 | 321 | 502 | 0.17 | 230 |
| 4–4a | CN | H | 254 | 290 | 514 | 0.047 | 32 |
| 5–5a | CHO | H | 258 | 317 | 585 | 0.13 | 161 |
| 6–6a | COOH | H | 262 | 282 | 485 | 0.027 | 21 |
| 7–7a | H | H | 260 | 281 | 450 | 0.001 | 3 |
| 8–8a | CH2OH | H | 273 | 321 | 420 | 0.001 | 5 |
| 9–9a | OCH3 | H | 312 | 325 | 458 | 0.001 | 5 |
| 10 | H | OCH3 | 345 | 275 | 457 | 0.002 | 6 |
All the spectra were recorded in MeOH/PBS (3:1, v/v) solution with a concentration of 10 μM.
Fluorescence emission was not detected.
Absorption maxima in the 225–450 nm region for compound 1–10.
Absorption maxima in the 250–450 nm region for compound 1a–10 a.
Fluorescence emission in the 400–650 nm region.
Fluorescence quantum yields were measured using fluorescein (1a, 3a, 4a, and 5a) and DAPI (2a and 6a–10a) as standards.
Obtained by comparing the integrated area of emission.
We further evaluated the fluorescence properties of 1 a in detail. As shown in Figure 2 A, oxinidole 1a has a maximum absorption at 324 nm, and a broad fluorescence emission peak centered at 520 nm, with a fluorescence quantum yield of 36 % in MeOH/PBS solution (3:1 v/v, pH7.4). It has an emission lifetime of 2.51 ns (the Supporting Information, Figure S12) and showed a better photostability than the common laboratory used fluorophore 4′,6-diamidino-2-phenylindole (DAPI; the Supporting Information, Figure S13). The fluorescence emission was found to be dependent on the polarity of the solvent used, with maximum emission peak shift from 516 nm to 535 nm observed when the solvent was changed from PBS (pH 7.4, 10 mM) to methanol (the Supporting Information, Figure S14). In addition, the photophysical properties of 1a were found to be pH-dependent. At pH lower than 4, compound 1 a had an absorption peak at 281 nm. However, an increase in pH (>4) resulted in a decrease of the absorption at 281 nm, while generating a new absorption peak at 324 nm with an isosbestic point at 299 nm (Figure 2 B). This pH-induced change in the absorption spectrum indicated the deprotonation of the N-hydroxyl moiety in 1 a as shown in Figure 2 D (upper panel) and revealed a pKa of 6.5. The absorption maxima peaks at 281 nm and 324 nm can be assigned to the protonated and deprotonated form of 1 a, respectively. On the other hand, the fluorescence spectrum of 1 a demonstrated an emission peak at 520 nm over the entire range of pH from 3 to 10 (the Supporting Information, Figure S15). Whereas the emission wavelength of 1 a is pH-independent, the emission intensity is highly dependent on the solution pH as well as excitation wavelengths. As shown in Figure 2C, fluorescence quenching and enhancement following increase in the solution pH were observed using excitation wavelength of 281 nm and 324 nm, respectively. However, the emission intensity remained constant under all pH conditions when using the isosbestic point as excitation wavelength (299 nm). These results strongly suggest a photoinduced proton transfer within the excited 1 a (1 a*), in which 1 a* undergoes rapid deprotonation of the N-hydroxyl group to its fluorescent alkaline form (Figure 2 D, bottom panel). This can be further supported by the computing of the electron density of the HOMO and LUMO for 1 a. As shown in Figure 2 E, the electron density of the HOMO for 1a is distributed evenly over the entire π-conjugated system and is highly rich in the N-hydroxyl moiety. In contrast, a misdistribution of the electron density was observed for the LUMO of 1a, in which it is mainly localized in the benzyl part and is strongly weakened on N-hydroxyl group. Such an electron-deficient oxygen would strengthen the acidity of the N-hydroxyl group, thus representing the photoinduced deprotonation of 1 a. A similar photochemical reaction has been demonstrated in the trisodium salt of 8-hydroxypyrene-1,3,6-trisulfonic acid (HOPSA),[15] a well-known excitation-ratiometric pH sensor, thus indicating the great potential of our 1a as a new photoactivat-able excitation-ratiometric pH sensor.
Figure 2.
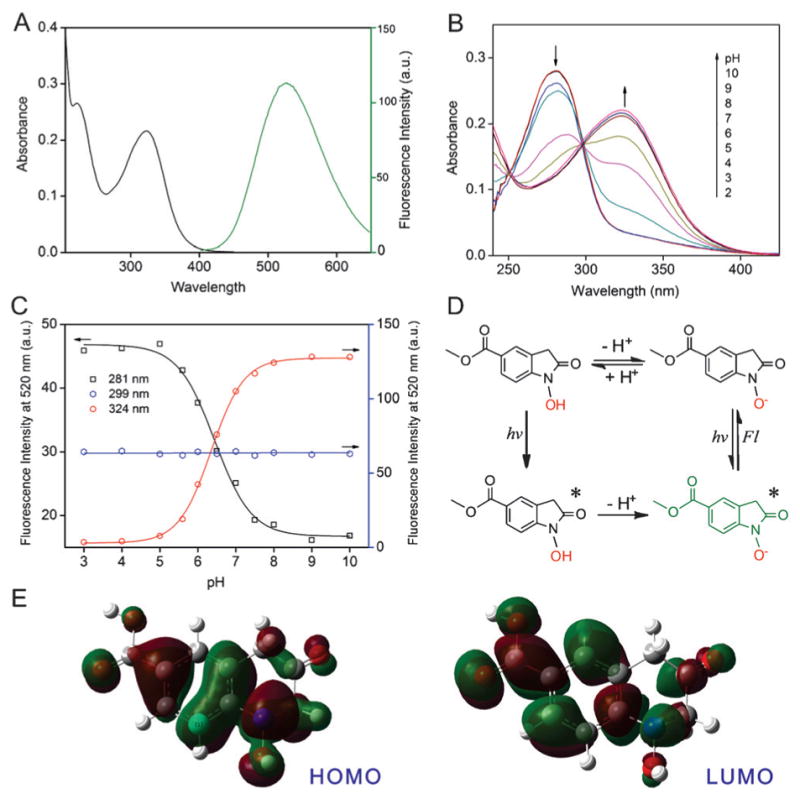
A) Absorption and fluorescence emission spectra of 1 a in MeOH/PBS (3:1, v/v, pH7.4, 10 mM PBS) solution; B) pH-dependent UV/Vis absorption spectrum of 1 a; C) pH-dependent fluorescence emission intensity of 1 a with different excitation wavelengths (detailed emission spectra were shown in Figure S15, the Supporting Information); D) Schematic illustration of the photo-induced deprotonation of the N-hydroxyl group of 1 a at the excited state; E) Electron density of HOMO and LUMO for 1 a computed by density functional theory (DFT).
On the basis of the astonishing fluorescence properties seen in an extremely small single benzyl moiety-containing structure, including the large stokes shift, solvent-polarity dependent emission, as well as the importance of the presence of EW group, it is reasonable to further propose a photoinduced excited keto–enol tautomerization of 1 a in which the enol form acts as the fluorescent molecule, as illustrated in Figure 3. Compared with the structure of the keto form, the enol form of 1 a has an extended conjugation and an electron pull (carbonyl)–push (hydroxyl) structure displaying typical intramolecular charge transfer (ICT) fluorescence and resulting in a fluorescence profile with large Stokes shift and polarity-sensitive emission.[16] Such keto–enol tautomerization for 1 a is proposed to be photoinduced and can only be observed at the excited state of 1a and not at ground state as not seen in 1H NMR spectrum. Our hypothesis was further supported by the DFT simulations (Figure 3), which revealed a 66.6 kJ mol−1 and a −171.6 kJ mol−1 in the change of the free Gibbs energy at the ground and excited states of the aforementioned keto–enol tautomerism (for more details see the Supporting Information, Figure S16). This demonstrated that the keto and the enol form is the thermostable molecular configuration for the ground and excited state of 1 a, respectively. To further verify this hypothesis, we synthesized methyl 3-(tert-butyl)-4-nitro-benzoate as a control compound (C1, the Supporting Information, Figure S17), which can be photolyzed to produce a N-hydroxyoxindole without α-proton (C1 a).[17] Such a molecule would therefore not undergo the keto–enol tautomerization, thus resulting in a smaller π-conjugation system compared with that of 1a. As expected, a fluorescence enhancement (106-fold) in the UV region (350 nm) with a much smaller Stokes shift (62 nm) was observed for the photolysis of C1 (the Supporting Information, Figure S17), indicating an emission from the local excited state of C1 a, in contrast to the ICT fluorescence of 1 a. These results strongly support the hypothesis on the photoinduced keto–enol tautomerization of 1 a and offer a deeper understanding in the fluorescence properties of oxindole. Taken together with the importance of meta-substituted EW groups of benzene and the hydroxyl on ethyl alcohol, a correlation between the molecular structure and the photophysical properties could potentially serve as a guideline for the design of new ONPE-based photoactivatable dyes with desired photophysical properties.
Figure 3.
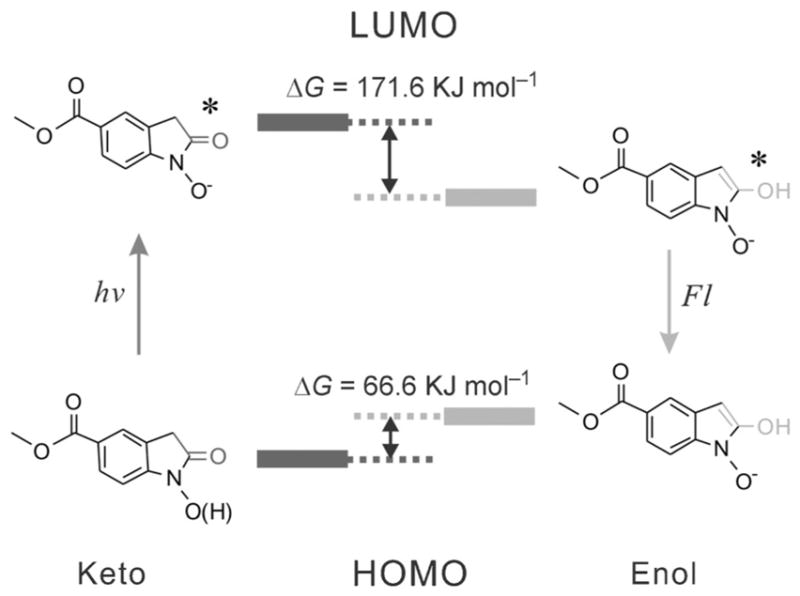
Schematic illustration of the proposed excited-state keto–enol tautomerization of 1 a, whereas only the enol form is the fluorescent moiety. HOMO and LUMO calculations of the keto and enol forms computed by DFT calculations show that enol form of 1 a is more stable at the excited state than the keto form.
As a proof-of-concept, we showed the development of a new ONPE-based photoactivatable fluorescence dye with excitation–emission in the green-red region, which is promising for the application of spatially-controlled cell-selective imaging. As shown in Figure 4A, ONPE was substituted with a strong EW group, 2-dicyanomethylene-3-cyano-4, 5, 5-trimethyl-2, 5-dihydrofuran (TCF)[18] through a conjugated double bond (11). Therefore, the resulting photolysis product (11a) would not only retain the intramolecular electron pull–push property, but also has an extended π-conjugated system, thus endowing the dye with a biocompatible optical window for the photolysis and excitation–emission-based fluorescence imaging. As expected, the new designed 11 showed broad absorption in UV to visible region, with an absorption maximum at 396 nm (Figure 4 B). Upon irradiating a methanol/PBS (3:1 v/v) solution of 11 with 365 nm light, a decrease in the absorption of 11 at 365 nm and an increase in absorption at wavelength longer than 450 nm was observed, indicating the photolysis of 11. Simultaneously, a 30-fold enhancement in fluorescence emission at 620 nm was observed with a 20 min of exposure. The photolysis of 11 was further followed by the HPLC-MS analysis (Figure 4 C and the Supporting Information, Figure S18), demonstrating that the photoreaction is clean and produces 11 a as the major photolysis compound, which had an excitation at wavelength longer than 500 nm and emission at 620 nm, with a fluorescence quantum yield of 2.6 %. Furthermore, in contrast to the widely used photolabile o-nitrobenzyl compounds that show significant photolysis-induced cytotoxicity due to the formation of highly reactive o-nitrosobenzyl products[8a], it was expected that our ONPE-based photoactivatable fluorophores would be highly biocompatible due to the formation of water molecule as the only side product during the photolysis (Figure 1 A). To verify this, we evaluated the cytotoxicity of OPNE 11 and its correspondent oxindole 11a in human neural stem cells (hNSCs) and cervical cancer (HeLa) cells using cell proliferation assays. As shown in Figure 4 D, a negligible effect on cell viability (>80 %) was observed for both 11 and 11a over a wide range of concentrations (0–50 μM). Overall, these results indicated that the new developed photoactivatable fluorophore 11 has great potential for cell-selective-imaging-related bio-applications.
Figure 4.
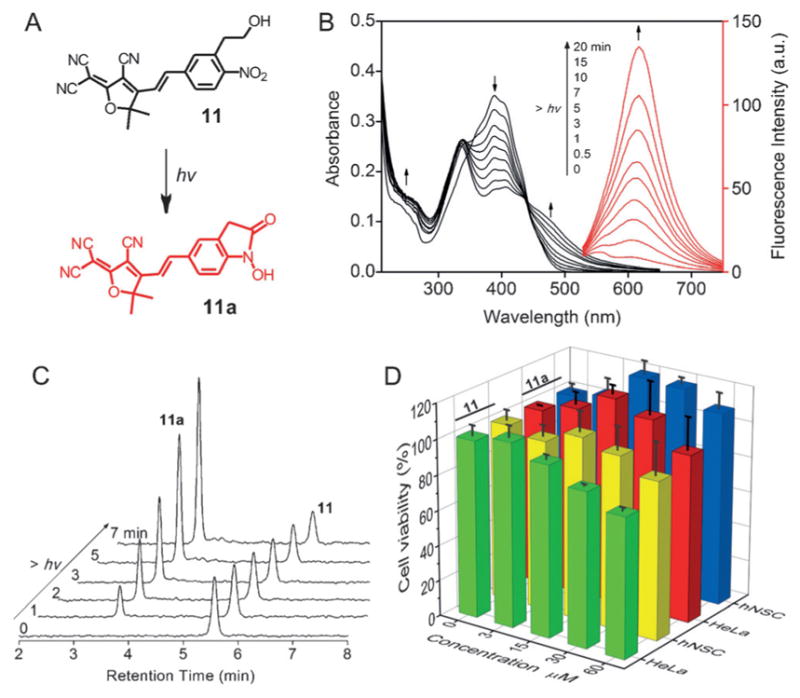
A) Molecular structures of the photoactivatable dye 11 and its corresponding photolysis product 11a; B) UV/Vis absorption and fluorescence emission spectra analysis of the photolysis of 11 in MeOH/PBS (3:1, v/v) under UV light irradiation (photolysis, 365 nm, 3 mW cm−2; excitation, 480 nm); C) HPLC monitoring revealed that the photolysis is clean and produces 11a as the major product; D) Analysis of viability and proliferation of HeLa cells and hNSCs in the presence of increasing concentrations of 11 and 11a.
We further examined the intracellular photoactivatable properties of 11. As shown in Figure 5 A, HeLa cells incubated with 11 (5 μM) showed negligible fluorescence before UV activation (t = 0). However, a dramatic time-dependent fluorescence enhancement was observed once the cells were irradiated with UV light (365 nm) (Figure 5A–G). A 3 min UV exposure led to a 30-fold fluorescence enhancement of the cell without any significant photobleaching (Figure 5E), thus highlighting the robust photostability of 11a. With this information in hand, we finally went on to study the spatially controlled cell-selective imaging using 11 through a photomask-assisted photolysis. As shown in Figure S20 (the Supporting Information), only the cells unprotected by photomask showed remarkable fluorescence after UV exposure. We further demonstrated the enhanced spatially controlled single-cell-selective imaging utilizing a confocal microscope. The broad absorption band of 11 enabled the photoactivation to proceed at 405 nm irradiation. As demonstrated in Figure 5 J, only the selected human neural stem cell (hNSC) irradiated with blue light (405 nm, 15 s) showed bright-red fluorescence, indicating successful rapid fluorescence enhancement of 11 a. In contrast, the region beyond the selected cell demonstrated weak fluorescence enhancement (16 % intensity as compared to the activated region).
Figure 5.

A–H) Time-dependent photoactivation of 11 in HeLa cells. The photoactivation was performed on a Nikon Ti microscope using the DAPI excitation (Nikonen slight C-HGFI lamp, 130 W) with a 20 × objective lens. The fluorescent images were collected using Texas Red channel (excitation 550–580 nm, emission 600–650 nm). Dye concentration is 5 μM, scale bar is 100 μm; I–K) Spatially-controlled photoactivation of 11 in hNSCs, I) DIC, J) fluorescence, and K) merged images of a single hNSC with photoactivated fluorescence. The selected cell was first irradiated by a 405 nm excitation from the confocal microscope (5.95 mW, 15 s) and the fluorescence imaging was recorded with an excitation at 515 nm. Scale bar is 20 μm.
Conclusion
We have developed a new class of photoactivatable fluorescent probes based on substituted o-nitrophenyl ethanol (ONPE) structures. These self-caged fluorophores are small in molecular size, show ultra-high (up to 800-fold) photo-induced fluorescence enhancement with good biocompatibility and hence can be readily adopted for applications such as monitoring dynamic molecular processes in living cells. Furthermore, we have revealed a structure–activity relationship between the substituted ONPE probes and their corresponding emission profiles, which can serve as a guideline for designing fluorescent probes with desired photophysical properties. As a proof-of-concept, an ONPE derivative with a Vis excitation (> 500 nm) and an NIR emission (620 nm) has been developed and its application in the spatially controlled selective labeling of single cancer and stem cells has been demonstrated. Together, these ONPE-based caged fluorophores can evolve into valuable probes for the convenient super-resolution imaging of biological samples and thereby permit spatiotemporally controlled mapping of biomolecular processes.
Experimental Section
Materials and methods
All starting materials were purchased from Sigma-Aldrich and used without further purification. UV/Vis absorption spectra were recorded on a Varian Cary 50 spectrophotometer. Fluorescence spectra were record on a Varian Cary Eclipse fluorescence spectrophotometer. 1H NMR and 13C NMR spectra were acquired on Varian 300, 400 or 500 MHz NMR spectrometer. ESI-MS was collected on Finnigan LCQ™ DUO LC/MS spectrometer. HPLC analysis was performed on Agilent 1100 series HPLC system. Luminescence digital photographs were taken with a Nikon D3000 camera. Cell fluorescence images were acquired on Nikon eclipse Ti inverted epifluorescence microscope with a Nikon Lens Light C-HGFI 130 W lamp. Confocal imaging was done using a Zeiss LSM 710-META confocal microscope. The photolysis of various ONPEs was carried out by exposure of ONPEs solution (methanol/PBS, 3:1 v/v) to a UV-lamp with 254 nm irradiation (3 mWcm−2). The procedures for the synthesis of various OPNEs are provided in the Supporting Information.
Cell culture
Hela cells and human neural stem cells (hNSCs, Millipore) were used for this project. For Hela cells, the cells were cultured in the DMEM-based medium with supplementation of 10 % FBS and 1 % streptomycin-penicillin. The cells were detached from the plate and split after reaching 90 % confluency. For hNSCs, the cells were cultured in a polylysine (10 μg mL−1, 12 h) and laminin (20 μg mL−1, 4 h) pre-coated 24 well plate, with an initial seeding density of 60,000 cells per well. A manufacturer’s protocol (ReNcell VM media, Millipore in the presence of basic fibroblast growth factor with a concentration of 20 ngmL−1, Millipore) was used for the expansion and culturing of hNSCs under 37 °C in a humidified atmosphere of 5 % CO2. The hNScs were detached and splitted after reaching 90 % confluency.
Viability assay
Cell viabilities for the Hela cells and hNSCs at different concentrations (0, 3, 15, 30, 50 μM) of oxindole derivatives were determined after 7 days of culturing using the Presto Blue assay following standard protocols described by the manufacture (Life Technologies). All the conditions were conducted in triplicates and averaged for the final data analysis. The final data is represented as the fluorescent emission at 590 nm under the excitation of 570 nm from the assay, and considering the Hela cells and hNSCs are 100 % viable when no oxindole derivatives are added.
Cell-selective imaging
For the delivery of oxindole derivatives (compound 11) into Hela cells and hNSCs, a low concentration of 11 was first fully dissolved in Giboco Opti-MEM media and ReNcell VM media supplemented with basic fibroblast growth factor, respectively, at a low concentration of 5 μM was used with a transfection time of 4 h. The cell media was then changed to normal growth media. After 4 more hours of culturing, the cells were imaged. For Hela cells, a Nikon eclipse Ti inverted epifluorescence microscope was used for bright field and fluorescent image collection, after the Hela cells were activated under the DAPI channel for 0, 10, 30, 60, and 180 seconds. The images were collected under identical exposure time, acquisition time and have the same contrast and brightness. For hNSCs, a Zeiss LSM 510-META confocal microscope equipped with an Axiovert 200 inverted Scope was used for the subcellular activation of fluorescence for 11, with an activation time of 10 seconds and a laser wavelength of 405 nm. All the cells were imaged under live culturing chambers (5 % CO2, humidified air under 37 °C) and no noticeable change of cell viability was observed throughout the imaging process.
Supplementary Material
Acknowledgments
K.-B. Lee acknowledges financial support from the NIH Director’s Innovator Award [1DP20D006462-01], National Institute of Neurological Disorders and Stroke (NINDS) [1R21NS085569-01], the NSF 9CHE-1429062 and CBET-12365080, the N.J. Commission on Spinal Cord grant [09-3085-SCR-E-0], and ACS Petroleum Research Fund. We are also grateful to Prof. Chuanliu Wu for his kind support for the fluorescence lifetime measurements.
Footnotes
Supporting information for this article is available on the WWW under http://dx.doi.org/10.1002/chem.201600547.
References
- 1.FernQndez-Su Qrez M, Ting AY. Nat Rev Mol Cell Biol. 2008;9:929. doi: 10.1038/nrm2531. [DOI] [PubMed] [Google Scholar]
- 2.a) Hell SW. Science. 2007;316:1153. doi: 10.1126/science.1137395. [DOI] [PubMed] [Google Scholar]; b) Rust MJ, Bates M, Zhuang XW. Nat Methods. 2006;3:793. doi: 10.1038/nmeth929. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Betzig E, Patterson GH, Sougrat R, Lindwasser OW, Olenych S, Bonifacino JS, Davidson MW, Lippincott-Schwartz J, Hess HF. Science. 2006;313:1642. doi: 10.1126/science.1127344. [DOI] [PubMed] [Google Scholar]
- 3.a) Kobayashi T, Urano Y, Kamiya M, Ueno T, Kojima H, Nagano T. J Am Chem Soc. 2007;129:6696. doi: 10.1021/ja070376d. [DOI] [PubMed] [Google Scholar]; b) Li WH, Zheng G. Photochem Photobiol Sci. 2012;11:460. doi: 10.1039/c2pp05342j. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Puliti D, Warther D, Orange C, Specht a, Goeldner M. Bioorg Med Chem. 2011;19:1023. doi: 10.1016/j.bmc.2010.07.011. [DOI] [PubMed] [Google Scholar]
- 4.a) Theriot JA. Nature. 1991;352:126. doi: 10.1038/352126a0. [DOI] [PubMed] [Google Scholar]; b) Cramer L, Mitchison TJ. J Cell Biol. 1993;122:833. doi: 10.1083/jcb.122.4.833. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Paul PH, Garguilo MG, Rakestraw DJ. Anal Chem. 1998;70:2459. doi: 10.1021/ac9709662. [DOI] [PubMed] [Google Scholar]
- 5.a) Kaur G, Costa MW, Nefzger CM, Silva J, Fierro-Gonzalez JC, Polo JM, Bell TDM, Plachta N. Nat Commun. 2013;4:1637. doi: 10.1038/ncomms2657. [DOI] [PubMed] [Google Scholar]; b) Specht A, Bolze F, Omran Z, Nicoud JF, Goeldner M. HFSP J. 2009;3:255. doi: 10.2976/1.3132954. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Shestopalov IA, Pitt CLW, Chen JK. Nat Chem Biol. 2012;8:270. doi: 10.1038/nchembio.772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.a) Mitchison TJ, Sawin KE, Theriot JA, Gee K, Mallavarapu A. Methods Enzymol. 1998;291:63. doi: 10.1016/s0076-6879(98)91007-2. [DOI] [PubMed] [Google Scholar]; b) Lord SJ, Conley NR, Lee HL, Samuel R, Liu N, Twieg RJ, Moerner WE. J Am Chem Soc. 2008;130:9204. doi: 10.1021/ja802883k. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Belov VN, Wurm CA, Boyarskiy VP, Jakobs S, Hell SW. Angew Chem Int Ed. 2010;49:3520. doi: 10.1002/anie.201000150. [DOI] [PubMed] [Google Scholar]; Angew Chem. 2010;122:3598. [Google Scholar]
- 7.a) KlQn P, Solomek T, Bochet CG, Blanc A, Givens R, Rubina M, Popik V, Kostikov A, Wirz J. Chem Rev. 2013;113:119. doi: 10.1021/cr300177k. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Zhao H, Sterner ES, Coughlin EB, Theato P. Macromolecules. 2012;45:1723. [Google Scholar]; c) Bochet CG. J Chem Soc Faraday Trans. 1;2002:125. [Google Scholar]; d) Lai JP, Xu YY, Mu X, Wu XL, Li C, Zheng JS, Wu CL, Chen JB, Zhao YB. Chem Commun. 2011;47:3822. doi: 10.1039/c0cc03361h. [DOI] [PubMed] [Google Scholar]
- 8.a) Zhao YR, Zheng Q, Dakin K, Xu K, Martinez ML, Li WH. J Am Chem Soc. 2004;126:4653. doi: 10.1021/ja036958m. [DOI] [PubMed] [Google Scholar]; b) Zheng GH, Guo YM, Li WH. J Am Chem Soc. 2007;129:10616. doi: 10.1021/ja071427+. [DOI] [PubMed] [Google Scholar]; c) Warther D, Bolze F, Leonard J, Gug S, Specht A, Puliti D, Sun XH, Kessler P, Lutz Y, Vonesch JL. J Am Chem Soc. 2010;132:2585. doi: 10.1021/ja9074562. [DOI] [PubMed] [Google Scholar]
- 9.a) Maurel D, Banala S, Laroche T, Johnsson K. ACS Chem Biol. 2010;5:507. doi: 10.1021/cb1000229. [DOI] [PubMed] [Google Scholar]; b) Ragab SS, Swaminathan S, Baker JD, Raymo FM. Phys Chem Chem Phys. 2013;15:14851. doi: 10.1039/c3cp51580j. [DOI] [PubMed] [Google Scholar]
- 10.a) Zhu MQ, Zhu LY, Han JJ, Wu WW, Hurst JK, Li ADQ. J Am Chem Soc. 2006;128:4303. doi: 10.1021/ja0567642. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Uno SN, Kamiya M, Yoshihara T, Sugawara K, Okabe K, Tarhan MC, Fujita H, Funatsu T, Okada Y, Tobita S. Nat Chem. 2014;6:681. doi: 10.1038/nchem.2002. [DOI] [PubMed] [Google Scholar]
- 11.a) Gagey N, Neveu P, Benbrahim C, Goetz B, Aujard I, Baudin JB, Jullien L. J Am Chem Soc. 2007;129:9986. doi: 10.1021/ja0722022. [DOI] [PubMed] [Google Scholar]; b) Gagey N, Neveu P, Jullien L. Angew Chem Int Ed. 2007;46:2467. doi: 10.1002/anie.200604598. [DOI] [PubMed] [Google Scholar]; Angew Chem. 2007;119:2519. [Google Scholar]; c) Yu ZP, Ho LY, Lin Q. J Am Chem Soc. 2011;133:11912. doi: 10.1021/ja204758c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.a) Hasan A, Stengele KP, Giegrich H, Cornwell P, Isham KR, Sachleben RA, Pfleiderer W, Foote RS. Tetrahedron. 1997;53:4247. [Google Scholar]; b) Specht A, Thomann JS, Alarcon K, Wittayanan W, Ogden D, Furuta T, Kurakawa Y, Goeldner M. ChemBioChem. 2006;7:1690. doi: 10.1002/cbic.200600111. [DOI] [PubMed] [Google Scholar]
- 13.Bakke J. Acta Chem Scand. 1970;24:2650. [Google Scholar]
- 14.Walbert S, Pfleiderer W, Steiner UE. Helv Chim Acta. 2001;84:1601. [Google Scholar]
- 15.Han JY, Burgess K. Chem Rev. 2010;110:2709. doi: 10.1021/cr900249z. [DOI] [PubMed] [Google Scholar]
- 16.Lakowicz JR. Principles of fluorescence spectroscopy. 3. Springer; New York: 2006. [Google Scholar]
- 17.Dopp D. Chem Commun. 1968:1284. [Google Scholar]
- 18.Zhou XF, Su FY, Tian YQ, Youngbull C, Johnson RH, Meldrum DR. J Am Chem Soc. 2011;133:18530. doi: 10.1021/ja207345s. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.