

Abstract
Patients with Pompe disease have realized significant medical benefits due to enzyme replacement therapy (ERT) infusions with alglucosidase alfa. However, regular infusions are time-consuming. Utilizing recommended infusion rates, infusion duration is 3 hours 45 minutes for a patient receiving the standard dose of 20 mg/kg, not including additional time needed for preparation of ERT, assessment of vital signs, intravenous access, and post-infusion Recent studies have demonstrated increased effectiveness of higher dose of ERT (40 mg/kg) infantile-onset Pompe disease (IOPD), which increases the infusion duration to 6 hours 36 minutes. Increased infusion durations compound the psychosocial burden on patients and families and potentially further disrupt family activities and obligations. We developed a stepwise infusion rate escalation protocol to administer higher dose ERT safely while infusion duration, which has been implemented in 15 patients to date. Reported here in detail five patients with IOPD on 40 mg/kg/weekly ERT in whom infusion duration was decreased with individualized, stepwise rate escalation. All patients tolerated rate escalations above the recommended rates without experiencing any infusion associated reactions and experienced a reduction in infusion duration by 1 hour and 24 minutes with a corresponding increase in reported satisfaction. Our experience with ERT rate escalation is presented.
Keywords: Pompe disease, glycogen storage disease type II, neuromuscular disease, enzyme replacement therapy, infusion rate escalation, infusion duration, ERT
INTRODUCTION
Pompe disease is an autosomal recessive lysosomal storage disorder (LSD) caused by deficiency of the enzyme acid alpha-glucosidase (GAA) [1]. GAA degrades lysosomal glycogen and its deficiency leads to accumulation of glycogen in multiple tissues, particularly in skeletal, smooth, and cardiac muscle [2]. Classic infantile-onset Pompe disease (IOPD) is the most severe phenotype with onset of symptoms within the first few days to weeks of life. Patients with classic IOPD present with hypotonia, hypertrophic cardiomyopathy, myopathic facies, enlarged tongue, and without treatment, rarely survive beyond two years of age [2].
Enzyme replacement therapy (ERT) with alglucosidase alfa has led to significant improvement in clinical outcomes in patients with Pompe disease [3–6]. However, response to ERT is variable and dependent on a number of factors including phenotype, age at ERT initiation, extent of preexisting pathology, cross-reactive immunologic material (CRIM) status, and rhGAA IgG antibody titers [7–10]. While the benefits of ERT are considerable, patients often have residual physical impairments including muscle weakness, hearing loss, risk for arrhythmias, hypernasal speech, and dysphagia with risk for aspiration, ptosis, and osteopenia [11].
One strategy to minimize residual physical impairments is to administer ERT at a higher dose and frequency than 20 mg/kg every other week (EOW) as recommended in the Lumizyme® package insert [12–15]. Prior clinical trials and published case reports have demonstrated that patients with IOPD can safely tolerate up to 40 mg/kg (referred to as higher dose ERT) every week [12–14, 16]. A recent study of CRIM-positive IOPD patients receiving higher dose ERT demonstrated respiratory and motor improvements compared with patients treated with 20 mg/kg/EOW [14]. Consequently, ERT at doses greater than 20 mg/kg is increasingly utilized in clinical practice [12, 14, 16].
As the dose of ERT increases, so does the total infusion duration. However, little is known about the effect of longer infusions on the psychosocial wellbeing of patients and their families. Increased infusion duration may compound the psychosocial burden and potentially further disrupt family activities and obligations. A study examining the impact of ERT on patients with LSDs found that while families were generally positive in regards to the health benefits of ERT, time spent getting infusions led to a feeling of “missing out” [17].
For the standard dose of 20 mg/kg, the recommended infusion rates are 1 mg/kg/h (30 minutes), 3 mg/kg/h (30 minutes), 5 mg/kg/h (30 minutes), and 7 mg/kg/h (remainder of the infusion) [15]. Therefore, the infusion duration for 20 mg/kg is 3 hours 45 minutes and 40 mg/kg is 6 hours 36 minutes. This can be a burdensome weekly time commitment for patients and their families. To administer higher dose ERT while reducing infusion duration, safe infusion rate escalation is needed. We developed an infusion rate escalation protocol with the goal of increasing the maximum infusion rate higher than the recommended 7 mg/kg/h and to safely reduce infusion duration. This infusion rate escalation protocol has been implemented successfully in 15 patients with either IOPD or late-onset Pompe disease (LOPD). Details on five patients with IOPD who were receiving higher dose ERT and who completed the infusion rate escalation protocol at Duke University are presented, along with guidelines for safely implementing rate escalation. In two of the five patients, the maximum infusion rate administered was increased to 11 mg/kg/h and the duration of initial rates (1, 3, 5, 7, 9, and 10 mg/kg/h) was shortened further reducing infusion time to 4 hours 45 minutes (data not presented), whereas the other three patients have been infused at a maximum rate of 10 mg/kg/h.
METHODS
Patients were selected for infusion rate escalation based on the following criteria; 1) a confirmed diagnosis of Pompe disease as described previously [2, 18], 2) treatment with higher dose ERT for at least six months, 3) low rhGAA IgG antibody titers (defined as antibody titers of <12,800) [19], and 4) no history of severe IARs. Stepwise infusion rate escalation was performed clinically, not as a research protocol, at Duke University. Clinical data including demographics, GAA variants, infusion history, rhGAA IgG antibody titers (determined by Genzyme), and history of IARs were extracted from patient electronic health records. Written informed consent was obtained for collection and analysis of clinical data under Duke Institutional Review Board (IRB)-approved protocol 00010830 (A long-term follow up study of Pompe disease) and/or protocol 00001562 (Determination of CRIM status in Pompe disease).
Infusion rate escalation
Prior to infusion rate escalation, patients were receiving higher dose ERT at the recommended infusion rates of 1, 3, 5, and 7 mg/kg/h per package insert [15] (Figure 1). The target for escalated ERT infusion rate was a maximum of 10 mg/kg/h, achieved safely and systematically via addition of rates of 9 and 10 mg/kg/h (Figure 1). Patients were transitioned to the target rate in a variable number of phases. Each phase consisted of either adding or removing rates above 7 mg/kg/h in increments of 0.5 mg/kg/h. Rate adjustments were determined by the treating physician (PSK) based on patient circumstances. Each phase continued for a minimum of four infusions and patients were monitored for IARs during each ERT infusion. Patients proceeded to the next phase once tolerance to the increased infusion rate was established. A conservative, stepwise infusion rate escalation example is outlined in Table 1. The exact infusion rate protocol detailed in Table 1 was utilized for Patient 1, all other patients received customized infusion rate escalation, which either combined or further divided the presented phases.
Figure 1.
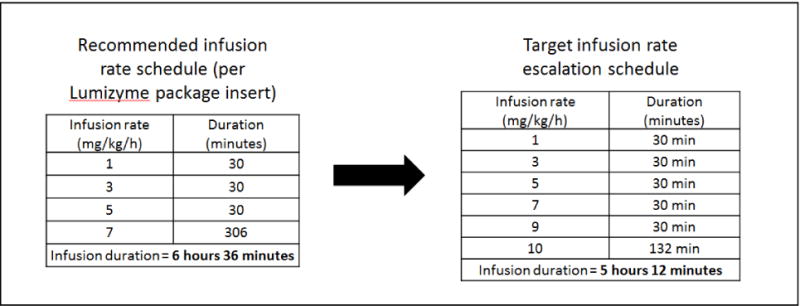
Recommended and escalated infusion rates for patients with Pompe disease receiving ERT with alglucosidase alfa at dose of 40 mg/kg.
Table 1.
Stepwise infusion rate escalation for patients with Pompe disease receiving ERT with alglucosidase alfa.
| Infusion rates (mg/kg/h) administered at specific phase* | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 3 | 5 | 7 | 7.5 | 8 | 8.5 | 9 | 9.5 | 10 | |
| Recommended rate | X | X | X | X | ||||||
| Phase 1 | X | X | X | X | X | X | ||||
| Phase 2 | X | X | X | X | X | X | X | X | ||
| Phase 3 | X | X | X | X | X | X | X | |||
| Phase 4 | X | X | X | X | X | X | ||||
| Phase 5 | X | X | X | X | X | |||||
| Phase 6 | X | X | X | X | X | X | ||||
| Phase 7 | X | X | X | X | X | X | X | |||
| Target | X | X | X | X | X | X | ||||
Infusion rates should be administered for 30 minutes each with the exception of the maximum infusion rate at each phase, which should be administered for the remainder of the infusion.
Psychosocial assessment
A parent or legal guardian of each patient who completed the infusion rate escalation protocol at Duke University was surveyed on the psychosocial impact of infusions on their child and family. Parents/guardians were asked to rate their satisfaction with the amount of time it took for an infusion before and after rate escalation, using a 7-point Likert scale from 1-very dissatisfied to 7-very satisfied. Parents/guardians were also asked to complete the following three qualitative questions; “What do you think about the amount of time needed for infusions?”, “How do you think shortened infusion lengths could impact children and families?”, and “What has been the impact of shortened infusion lengths on your child/family?”
RESULTS
Fifteen patients met the inclusion criteria for infusion rate escalation. Ten patients completed rate escalation at other institutions with guidance from PSK. Five IOPD patients who completed the infusion rate escalation protocol at Duke University are presented. Patient demographics, GAA variant data, age at diagnosis, age at ERT initiation, rhGAA IgG antibody titers prior to rate escalation, and history of IARs are shown in Table 2.
Table 2.
Patient demographics and infusion history
| Patient ID/Gen der | Race | GAA Variant 1 | GAA Variant 2 | Age at diagnosis (months) | Age at ERT initiation (months) | Age at start of rate escalation (months) | rhGA A IgG antibodytiter prior to rate escalation protocol | IARs |
|---|---|---|---|---|---|---|---|---|
| 1/F | Caucasian | c.655G>A | c.655G>A | 6.5 | 7.0 | 106 | 400 | None |
| 2/M | African American | c.-32-13T>G | c.1447G>A | 15.0 | 16.0 | 122 | 800 | None |
| 3/M | Caucasian | c.525delT | c.1642G>T, c.1880C>T | 0.0 | 0.5 | 122 | Negative | None |
| 4/M | Caucasian | c.1933G>A | c.1933G>A | 2.7 | 2.9 | 139 | 400 | Hives |
| 5/M | Hispanic | c.2297A>C | c.2297A>C | 6.8 | 7.3 | 159 | 200 | None |
M, male; F, female; rhGAA, recombinant human acid alpha-glucosidase; IARs, infusion associated reactions
Demographics and ERT dosing history
All patients initiated ERT at the standard dose of 20 mg/kg/EOW. The dose and frequency of ERT gradually increased to 40 mg/kg/weekly because of clinical plateau or decline observed with increasing levels in creatinine kinase (CK), aspartate transaminase (AST), alanine transaminase (ALT), and urinary glucose tetrasaccharide (Glc4), not presented here. All five patients were receiving infusions at home.
Patient 1
Patient 1 is a 10-year-old CRIM-positive, Caucasian female. She was diagnosed with IOPD at age 6.5 months and started receiving ERT with alglucosidase alfa at 20 mg/kg/EOW at 7 months of age. Her dose of ERT increased gradually to 40 mg/kg/weekly at the age of 7 years, 9 months. Rate escalation was initiated at the age of 8 years, 10 months, after 13 months of higher dose ERT.
Patient 2
Patient 2 is an 11-year-old CRIM-positive, African-American male. He was diagnosed with IOPD at age 1 year, 3 months and started ERT with alglucosidase alfa at 20 mg/kg/EOW at the age of 1 year, 4 months. His ERT dose increased gradually to 40 mg/kg/EOW at the age of 9 years, 4 months and 40 mg/kg/weekly at the age of 9 years, 6 months. Rate escalation was initiated at the age of 10 years, 2 months after 10 months of higher dose ERT.
Patient 3
Patient 3 is an 11-year-old CRIM-positive, Caucasian male diagnosed with IOPD at birth, due to a positive family history. He started ERT with alglucosidase alfa at 20 mg/kg/EOW at age 15 days. His ERT dose was increased to 40 mg/kg/EOW at the age of 8 years, 7 months and to 40 mg/kg/weekly at 10 years of age. Rate escalation was initiated at the age of 10 years, 2 months after 19 months of higher dose ERT.
Patient 4
Patient 4 is a 12-year-old CRIM-positive, Caucasian male. He was diagnosed with IOPD at the age of 2.7 months and started ERT at age of 2.9 months with alglucosidase alfa at 20 mg/kg/EOW. His ERT dose increased gradually to 40 mg/kg/weekly at the age of 10 years, 2 months. Rate escalation was initiated at the age of 11 years, 7 months after 17 months of higher dose ERT.
Patient 5
Patient 5 is a 14-year-old CRIM-positive, Hispanic male. He was diagnosed with IOPD at age 6.8 months and started ERT at the age of 7.3 months with alglucosidase alfa at 20 mg/kg/EOW. His ERT dose was increased to 40 mg/kg/EOW at the age of 10 years, 5 months and increased to 40 mg/kg/weekly at 12 years, 2 months. Rate escalation was initiated at the age of 13 years, 3 months after 34 months of higher dose ERT.
Safety monitoring
None of the reported patients had a history of IARs except Patient 4, who began having hives at the age of 6 years without identified triggers. Hives appeared intermittently since that time, and were controlled with the use of cetirizine and diphenhydramine as needed. All patients maintained low rhGAA IgG antibody titers throughout the course of infusion rate escalation.
Infusion rate escalation
Patients received individualized rate escalation as determined by the treating physician based on individual patient circumstances and experiences with rate escalation in prior patients. Phases 1 through 7 of the rate escalation protocol (Table 1) were completed as shown in Patient 1. Phases 1 and 2 were combined in Patients 2, 3, and 5. Phases 6 and 7 were combined in Patient 2 and phases 5, 6, and 7 were combined in Patient 3. Because of Patient 4’s history of hives, dose escalation was conservative with no more than 0.5 mg/kg/h increased at any one time. For example, phase 1 was split into two phases with addition of 7.5 mg/kg/h for weeks 1 to 4 and addition of 8 mg/kg/h for weeks 5 to 10.
All patients have successfully transitioned to the maximum infusion rate of 10 mg/kg/h. At the time of this report, Patients 1, 2, 3, 4, and 5 have been receiving 40 mg/kg/weekly at 10 mg/kg/h for 83 weeks, 86 weeks, 41 weeks, 28 weeks, and 22 weeks respectively. Patient 1 and Patient 3 are now successfully receiving ERT at a maximum infusion rate of 11 mg/kg/h, allowing further reduction in infusion time. Infusion time has been reduced from 6 hours 36 minutes to 5 hours 12 minutes for all, except Patient 1 and Patient 3 where infusion time is now 4 hours 45 minutes.
Psychosocial Results
A parent or guardian of all five patients completed the questionnaire evaluating the psychosocial impact of infusion duration on their child and family. All parents/guardians were generally satisfied with infusion duration before and after rate escalation. Mean satisfaction with infusion duration increased from 5.4/7 prior to infusion rate escalation to 6.4/7 after infusion rate escalation. Two parents/guardians reported increased satisfaction with infusion duration after infusion rate escalation. Reported satisfaction did not change for three parents/guardians. However, two of the parents/guardians without increased satisfaction reported maximum satisfaction (very satisfied) before initiation of infusion rate escalation, leaving no room for improvement.
Parents/guardians reported prioritizing their child’s health and safety of the treatment over infusion duration. One mother wrote, “I want the time to go as fast as possible without thinking that there could be issues. If it needs to be slower for whatever reasons, or better outcomes, then I am fine with it. However, if the outcome is the same…the faster the better!” Parents/guardians reported that shorter infusion durations allow their child time for other activities, reduce boredom and result in less absence from school. Two of the parents/guardians reported that infusion duration, although shorter, did not significantly change after rate escalation. One mother wrote, “It hasn’t changed much! It would be great if infusions were changed drastically by hours.”
DISCUSSION
Enzyme replacement therapy with alglucosidase alfa for IOPD has been available for over a decade, effectively transforming a formerly fatal disease into a treatable, though not yet curable, disorder. While ERT has revolutionized treatment for patients with IOPD and significantly improved clinical outcomes, quality of life remains reduced due to residual physical impairments. Immune modulation and earlier initiation of treatment through newborn screening initiatives have mitigated some of these challenges. An additional approach to maximize therapeutic benefit is to increase the frequency and/or dose of infusions beyond the standard dosing regimen of 20 mg/kg/EOW. Initial reports indicate that patients with IOPD receiving ERT at a dose of 40 mg/kg/weekly experience respiratory and motor improvements without increased risks of IARs [14]. Higher dose ERT increases infusion duration to 6 hours 36 minutes from 3 hours 45 minutes when utilizing the recommended infusion rates. However, the time spent by a patient at an infusion center is longer because of the additional time needed for preparation of the infusion, assessment of vital signs, premedication, intravenous/port access, and post-infusion patient monitoring. Patients with IOPD also receive multidisciplinary care requiring time commitments to various subspecialties in addition to time needed for weekly or EOW ERT infusions. All five patients reported here are either home schooled or attending school, similar to other long-term survivors with IOPD. Parents/guardians were generally satisfied with infusion duration but reported a desire to further minimize infusion duration without compromising treatment efficacy or safety. Shortened infusion duration may translate to more time available for studies and extracurricular activities, and reduced boredom, especially in children as they get older.
This report presents the effectiveness, safety, and psychosocial impact of escalated infusion rates in five patients with IOPD receiving higher dose ERT. All patients tolerated rate escalations above the standard dosing protocol and experienced a reduction in infusion duration of 1 hour 24 minutes with corresponding increase in mean reported satisfaction. In addition, frequency of IARs did not increase after the implementation of rate escalation. Infusion rate escalation can be safely achieved in home infusion settings as many of our patients were receiving home infusion. Successful infusion rate escalation utilizing the protocol described here has also been completed for 10 additional patients receiving infusions at other institutions.
While rate escalation can reduce infusion duration, it should be performed with care as these are increased infusion rates beyond what is recommended in the package insert [15]. Our recommendations for implementing rate escalation are: 1) prior to initiation of rate escalation, patients should complete ERT utilizing recommended infusion rates without significant IARs for at least six months [15], 2) patients should have low rhGAA IgG antibody titers (defined as antibody titers of <12,800) prior to initiating rate escalation, due to the greater likelihood of IARs in patients with high rhGAA antibody titers [4], 3) rate escalations should be performed gradually, in a stepwise fashion as exemplified in Table 1; and 4) patients should be monitored closely and only transitioned to the next phase of rate escalation once patient tolerance has been established to the current infusion rate. These recommendations should be individualized according to patient circumstances and clinical judgement.
Additional efforts into further decreasing infusion duration without compromising safety are warranted. One possibility may be to decrease the 30 minute duration of recommended infusion rates (1, 3, 5, and 7 mg/kg/h) and/or go beyond the maximum rate reported here of 10 mg/kg/h. This has already been completed in one patient to our knowledge, an 18-month-old Caucasian female with the “late-onset” GAA variant who is being treated with ERT at 20 mg/kg/EOW at another institution. For this patient, the infusion rates (1, 3, 5, 7, and 9 mg/kg/h) are utilized for 15 minutes each and the remaining ERT dose is completed at 10 mg/kg/h. Infusion duration has been reduced from 3 hours 45 minutes to 2 hours 50 minutes. In Patient 1 and Patient 3, infusion rate of 11 mg/kg/h has been added along with shortened initial infusion rates (1, 3, 5, 7, 9, and 10 mg/kg/h) to further reduce the infusion duration. For Patient 1 and Patient 3, total infusion duration reduced to 4 hours 45 minutes.
Patient circumstances may add additional challenges to rate escalation. For example, patients undergoing home infusions are constrained by the processing of orders and delivery of new pumps with updated rates, likely slowing transition to the target infusion rate. Ultimately, a careful stepwise method of ERT rate escalation can reduce infusion durations significantly in patients with Pompe disease. The addition of infusion rates 9 and 10 mg/kg/h reduces infusion duration by 1 hour 24 minutes for patients receiving ERT at 40 mg/kg. This approach, while exemplified in patients with Pompe disease receiving ERT at 40 mg/kg, can potentially reduce infusion duration for patients with Pompe disease receiving ERT at any dose and perhaps in patients with other diagnoses treatable with ERT. Although we do not have experience with CRIM-negative IOPD, we believe infusion rate escalation can be safely implemented in patients with CRIM-negative IOPD if the above recommendations are followed carefully.
Synopsis.
A careful stepwise method of enzyme replacement therapy (ERT) rate escalation can safely reduce infusion duration in patients with Pompe disease.
Acknowledgments
The authors thank the patients who participated in this study and their families. We thank Jennifer Coker, BSN, RN, CPN for her help with data acquisition. We thank Dr. Joan Keutzer, PhD for her critical review of the manuscript.
Funding:
This research was supported by a grant from Genzyme Corporation, a Sanofi Company (Cambridge, MA), and in part by the Lysosomal Disease Network, a part of National Institutes of Health Rare Diseases Clinical Research Network (RDCRN). The Lysosomal Disease Network (U54NS065768) is a part of the Rare Diseases Clinical Research Network (RDCRN), an initiative of the Office of Rare Diseases Research (ORDR), and NCATS. This consortium is funded through a collaboration between NCATS, NINDS, and NIDDK. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Disclosures:
PSK has received research/grant support and honoraria from Genzyme Corporation and Amicus Therapeutics. PSK is a member of the Pompe and Gaucher Disease Registry Advisory Board for Genzyme Corporation.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Authors’ Contributions
AKD, CKW, ZBK, SMD, and PSK participated in the design of the study. CKW, SMD, and PSK were involved in the clinical care of the patients at Duke University. Acquisition, analysis and interpretation of data were performed by AKD and CKW. All authors helped to draft the manuscript, and approved the final manuscript.
Conflict of interest
AKD, CKW, HLC, ZBK, and SMD have no financial or proprietary interest in the materials presented herein.
Ethics approval and consent to participate
Written informed consent was obtained from a parent or guardian for all individuals as part of Duke Institutional Review Board approved Pompe long-term follow-up study (Pro00010830) and/or Determination of CRIM status in Pompe disease (Pro00001562).
References
- 1.Hirschhorn R, Reuser AJJ. Glycogen storage disease type II: acid a-glucosidase (acid maltase) deficiency. In: Valle D, Scriver CR, editors. Scriver’s OMMBID the online metabolic & molecular bases of inherited disease. McGraw-Hill; New York: 2009. [Google Scholar]
- 2.Kishnani PS, et al. A retrospective, multinational, multicenter study on the natural history of infantile-onset Pompe disease. J Pediatr. 2006;148(5):671–676. doi: 10.1016/j.jpeds.2005.11.033. [DOI] [PubMed] [Google Scholar]
- 3.Case LE, Kishnani PS. Physical therapy management of Pompe disease. Genet Med. 2006;8(5):318–27. doi: 10.1097/01.gim.0000217789.14470.c5. [DOI] [PubMed] [Google Scholar]
- 4.Kishnani PS, et al. Early treatment with alglucosidase alpha prolongs long-term survival of infants with Pompe disease. Pediatr Res. 2009;66(3):329–35. doi: 10.1203/PDR.0b013e3181b24e94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Nicolino M, et al. Clinical outcomes after long-term treatment with alglucosidase alfa in infants and children with advanced Pompe disease. Genet Med. 2009;11(3):210–9. doi: 10.1097/GIM.0b013e31819d0996. [DOI] [PubMed] [Google Scholar]
- 6.van der Ploeg AT, et al. A randomized study of alglucosidase alfa in late-onset Pompe’s disease. N Engl J Med. 2010;362(15):1396–406. doi: 10.1056/NEJMoa0909859. [DOI] [PubMed] [Google Scholar]
- 7.De Filippi P, et al. Genotype-phenotype correlation in Pompe disease, a step forward. Orphanet J Rare Dis. 2014;9:102. doi: 10.1186/s13023-014-0102-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yanovitch TL, et al. Clinical and histologic ocular findings in pompe disease. J Pediatr Ophthalmol Strabismus. 2010;47(1):34–40. doi: 10.3928/01913913-20100106-08. [DOI] [PubMed] [Google Scholar]
- 9.Banugaria SG, et al. The impact of antibodies on clinical outcomes in diseases treated with therapeutic protein: lessons learned from infantile Pompe disease. Genet Med. 2011;13(8):729–36. doi: 10.1097/GIM.0b013e3182174703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Raben N, et al. Deconstructing Pompe disease by analyzing single muscle fibers: to see a world in a grain of sand. Autophagy. 2007;3(6):546–52. doi: 10.4161/auto.4591. [DOI] [PubMed] [Google Scholar]
- 11.Prater SN, et al. The emerging phenotype of long-term survivors with infantile Pompe disease. Genet Med. 2012;14(9):800–10. doi: 10.1038/gim.2012.44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yanovitch TL, et al. Improvement of bilateral ptosis on higher dose enzyme replacement therapy in Pompe disease. J Neuroophthalmol. 2010;30(2):165–6. doi: 10.1097/WNO.0b013e3181ce162a. [DOI] [PubMed] [Google Scholar]
- 13.Kishnani PS, et al. Recombinant human acid [alpha]-glucosidase: major clinical benefits in infantile-onset Pompe disease. Neurology. 2007;68(2):99–109. doi: 10.1212/01.wnl.0000251268.41188.04. [DOI] [PubMed] [Google Scholar]
- 14.van Gelder CM, et al. Effects of a higher dose of alglucosidase alfa on ventilator-free survival and motor outcome in classic infantile Pompe disease: an open-label single-center study. J Inherit Metab Dis. 2016;393:383–90. doi: 10.1007/s10545-015-9912-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lumizyme R [Package Insert] Genzyme Corporation; Cambridge, MA: Oct, 2016. [Google Scholar]
- 16.Prater SN, et al. Consideration of increased dosing of alglucosidase alfa to achieve improved clinical outcomes in infantile Pompe disease. Molecular Genetics and Metabolism. 114(2):96. [Google Scholar]
- 17.Freedman R, et al. Receiving enzyme replacement therapy for a lysosomal storage disorder: a preliminary exploration of the experiences of young patients and their families. J Genet Couns. 2013;224:517–32. doi: 10.1007/s10897-013-9579-1. [DOI] [PubMed] [Google Scholar]
- 18.van den Hout HM, et al. The natural course of infantile Pompe’s disease: 20 original cases compared with 133 cases from the literature. Pediatrics. 2003;112(2):332–40. doi: 10.1542/peds.112.2.332. [DOI] [PubMed] [Google Scholar]
- 19.Berrier KL, et al. CRIM-negative infantile Pompe disease: characterization of immune responses in patients treated with ERT monotherapy. Genet Med. 2015;17(11):912–8. doi: 10.1038/gim.2015.6. [DOI] [PMC free article] [PubMed] [Google Scholar]