

Abstract
Endogenous dynorphin signaling via the kappa-opioid receptor (KOR) in the nucleus accumbens (NAcc) powerfully mediates negative affective states and stress reactivity. Excitatory inputs from the hippocampus and amygdala play a fundamental role in shaping the activity of both NAcc D1 and D2 MSNs, which encode positive and negative motivational valences, respectively. However, a circuit-based mechanism by which KOR modulation of excitation-inhibition balance modifies D1 and D2 MSN activity is lacking. Here, we provide a comprehensive synaptic framework wherein presynaptic KOR inhibition decreases excitatory drive of D1 MSN activity by the amygdala, but not hippocampus. Conversely, presynaptic inhibition by KORs of inhibitory synapses on D2 MSNs enhances integration of excitatory drive by the amygdala and hippocampus. In conclusion, we describe a circuit-based mechanism showing differential gating of afferent control of D1 and D2 MSN activity by KORs in a pathway specific manner.
Introduction
Dynorphin (Dyn), an endogenous opioid receptor peptide, and its receptor, the kappa-opioid receptor (KOR), are powerful effectors of stress-induced alterations in reward processing and dysphoric states (Chavkin et al., 1982; Van’t Veer and Carlezon, 2013). Dyn and KORs are highly expressed in the NAcc (Gerfen et al., 1990; Meng et al., 1993; Meshul and McGinty, 2000; Svingos et al., 1999). Activation of NAcc KORs, pharmacologically or by endogenous Dyn, produces aversion and depressive-like states (Bruchas et al., 2010; Muschamp et al., 2011; Tejeda et al., 2012; Wee and Koob, 2010). Increased Dyn expression in ventral and dorsal striatum of addicted and suicide individuals (Hurd and Herkenham, 1993; Hurd et al., 1997), and in animal models of addiction and depression (Bruchas et al., 2010; Carlezon et al., 1998; Pliakas et al., 2001; Schlosburg et al., 2013; Shirayama et al., 2004), has led to the hypothesis that the NAcc Dyn/KOR system underlies negative affective states and heightened stress reactivity in various psychiatric disorders. However, the mechanism by which KORs shape NAcc activity to modify affective and stress-related behaviors has yet to be identified.
The NAcc is a hub of a distributed motivational circuit of the brain. NAcc medium-sized spiny neurons (MSNs) integrate excitatory inputs, including basolateral amygdala (BLA) and ventral hippocampus (VH) afferents, with local inhibition to dictate spiking activity (Britt et al., 2012; O’Donnell and Grace, 1995). NAcc MSNs are segregated into dopamine (DA) D1 receptor-containing MSNs (D1 MSNs) that express Dyn and D2 receptor-containing MSNs (D2 MSNs) (Gerfen et al., 1990; Kreitzer, 2009). D1 MSN activity is a critical mediator of reward learning, whereas optogenetic activation of D2 MSNs attenuates reward and is aversive (Calipari et al., 2016; Ferguson et al., 2011; Kravitz et al., 2012; Lobo et al., 2010). As MSNs do not intrinsically generate action potentials, MSN activity is driven by excitatory synapses (O’Donnell and Grace, 1995). Optogenetic activation of VH and BLA afferents to the NAcc is rewarding and these pathways mediate natural and drug reward-related behaviors (Britt et al., 2012; Pascoli et al., 2014; Stuber et al., 2011). VH and BLA pathways to the NAcc have also been implicated in mediating negative behavioral outcomes after chronic stress (Bagot et al., 2015). In neocortical regions, excitation-inhibition balance is critical for fine-tuned network activity (Isaacson and Scanziani, 2011). How excitatory synapses interact with inhibition to gate NAcc D1 and D2 MSN output is not well understood. KORs activation inhibits release of both glutamate and GABA (Hjelmstad and Fields 2003). However, an understanding of how KORs shape synaptic transmission from specific glutamatergic afferents and local circuit GABAergic synapses is lacking. Moreover, it is not known whether KORs differentially regulate D1 and D2 MSNs. Elucidating the role of KORs in regulating limbic glutamatergic afferents and GABAergic microcircuitry within the NAcc will provide a comprehensive physiological framework wherein the NAcc KOR system can control mood and emotion. To this end, we utilized a combination of whole-cell slice electrophysiology, optogenetics, anatomy, and transgenic mice to delineate the cellular basis for KOR regulation of NAcc afferents and microcircuits, and ultimately NAcc D1 and D2 MSN output.
Results
Glutamatergic Pathway-Specific Regulation by KORs
In the NAcc, KOR activation inhibits glutamatergic release (Hjelmstad and Fields, 2003; Mu et al., 2011). In agreement with previous reports, bath application of the KOR agonist, U69,593 (U69) produces a long-term depression (LTD) of electrically-evoked EPSCs (eEPSCs) in NAcc MSNs in the medial shell (Figure S1). However, KOR mRNA is expressed in various cortical and limbic excitatory regions innervating the NAcc (Meng et al., 1993). It is unclear whether KOR modulates VH and BLA afferents in a pathway-specific manner. To determine whether KORs negatively modulate BLA and VH inputs, we injected AAV-CaMKII-ChR2-eYFP into the BLA or VH of wildtype (WT) mice and conducted whole-cell slice electrophysiology recordings from MSNs in the NAcc medial shell (Figure 1A). In agreement with previous findings, VH-evoked optical EPSCs (VH oEPSCs) were larger than BLA oEPSCs, and were mediated by glutamatergic transmission (Figure S2) (Britt et al., 2012). U69 inhibited BLA oEPSCs, but not VH oEPSCs (Figure 1B), suggesting that KORs confer pathway-specific effects. To determine whether KOR inhibition occurred via a pre-synaptic site of action, we examined the effects of U69 on paired-pulse ratios (PPRs) of BLA and VH oEPSCs (Figure S3A). However, it is unclear whether KOR regulation of BLA synapses occurs presynaptically, as there was no change in the PPR of VH or BLA oEPSCs. To confirm that lack of a U69 effect on VH oEPSCs was not due to a general lack of presynaptic inhibition in this pathway, we bath applied the GABA-B receptor agonist baclofen and observed a reduction of VH oEPSCs (Figure 1C). Baclofen inhibition of VH oEPSCs was associated with an increase in the PPR (Figure S3B), suggesting a pre-synaptic site of GABA-B receptor action on VH synapses. Inhibition of BLA oEPSCs was prevented by pre-treatment with the KOR antagonist nor-BNI (Figure 1D), however inhibition was not reversed by nor-BNI application after U69 (Figure 1E). These results suggest that once KORs are activated, downstream signaling maintains KOR-dependent inhibition of BLA synapses.
Figure 1. Pathway Specific KOR Modulation of Glutamatergic Afferents to NAcc.
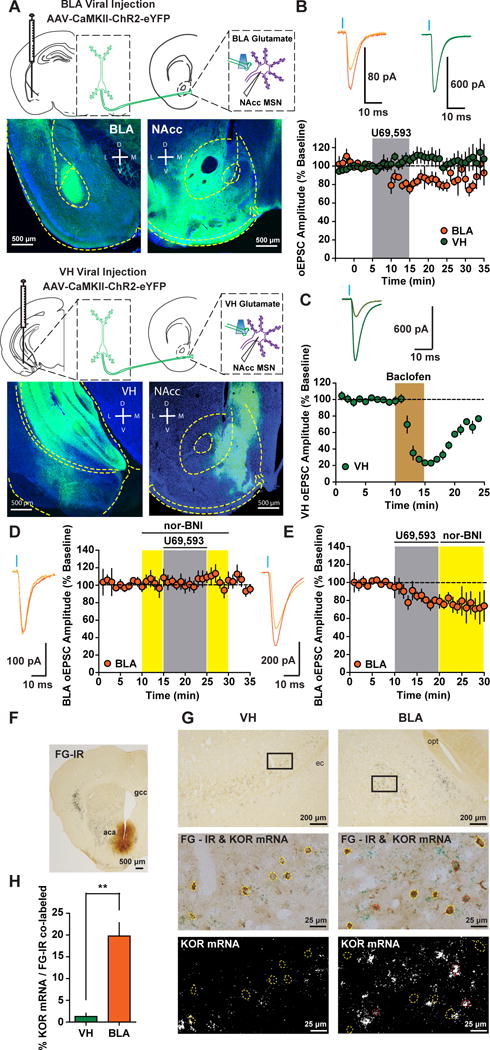
(A) AAV-CaMKII-ChR2-eYFP expression in the BLA and VH and expression of ChR2-eYFP-containing terminals in the NAcc. (B) Time course of the effect of KOR activation with U69 (1 μM) on the amplitude (expressed as a percentage of baseline) of optically-evoked EPSCs (oEPSCs) in medial shell NAcc MSNs from animals expressing ChR2-eYFP in the BLA (n=8) or VH (n=7; Two-way ANOVA Afferent x Time Interaction; F(19, 247)=3.34; p<0.0001). Representative oEPSCs recorded in NAcc MSNs from BLA (orange traces) and VH (green traces) during baseline (dark traces) and after the KOR agonist U69 (light traces). (C) Time course and representative traces of baclofen-mediated inhibition of VH EPSCs (n=4). (D) Time course and representative traces of nor-BNI pretreatment on U69 on BLA oEPSCs (n=8). (E) Time course representative traces of nor-BNI treatment after U69 on BLA oEPSCs (n=5). (F) Low magnification of the fluorogold (FG) injection site in the NAcc. aca, anterior commissure; gcc, genu of the corpus callosum. (G) Top, low magnification of FG-immunoreactive (IR) cells in the BLA and VH. Squares delimit areas shown for high magnification images in middle and bottom panels. Middle, FG-IR neurons in the BLA and VH (yellow and red outline) and FG labeled cells expressing KOR mRNA (red outline). Bottom, dark field illumination of KOR mRNA in BLA and VH. opt, optic tract; ec, external capsule. (H) Percentage cells expressing KOR-mRNA and fluorogold in the BLA and VH (t(4)=5.854; p=0.004; n=3). Data in this figure, and all figures hereafter, are expressed as mean ± SEM.
To determine whether differential KOR modulation of VH and BLA afferents was due to differential KOR expression, we injected the retrograde tracer fluorogold (FG) in the NAcc, and performed KOR mRNA in situ hybrization in the BLA and VH (Figure 1F,G). The number of cells projecting to the NAcc from the VH exceeded the number of cells from the BLA (Figure S2) (Britt et al., 2012). FG-positive cells in the BLA also contained KOR mRNA, whereas in the VH, FG-positive cells did not express KOR mRNA (Figure 1G,H). Collectively, these results suggest that KORs inhibit glutamatergic synapses onto NAcc MSNs in a pathway-specific manner, and inhibition is dictated by differential KOR expression (Figure 8A).
Figure 8. Model of Pathway- and Cell Type-Specific KOR Modulation of D1 and D2 MSN Activity.
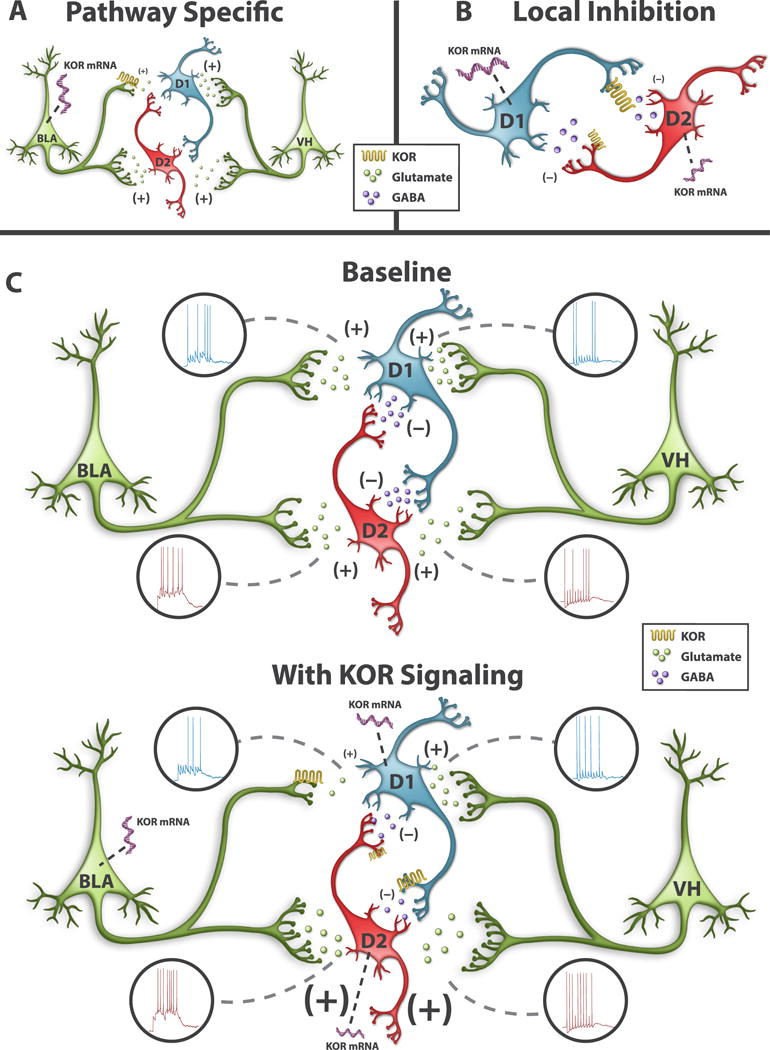
(A) Pathway-specific KOR modulation of excitatory synapses in the NAcc. KORs inhibit BLA, but not VH, afferents into NAcc MSNs. NAcc-projecting BLA neurons, express KOR mRNA, whereas KOR mRNA expression is absent in NAcc-projecting VH neurons. KORs only inhibit BLA inputs to D1 MSNs. (B) KOR modulation of MSN collaterals. KORs inhibit D1 MSN collaterals more strongly than D2 MSN collaterals. KOR mRNA expression is higher in D1 MSNs than D2 MSNs. (C) Afferent control of D1 and D2 MSNs by the VH and BLA during basal conditions (top) and after increased KOR signaling (bottom). The strength of excitation-spiking coupling between afferent and post-synaptic neurons is depicted at individual afferent synapses by the number of action potentials, the size of the presynaptic terminal, and the size of the glutamate or GABA. KOR inhibition of BLA synapses in D1 MSNs decreases BLA-driven spiking in D1 MSNs, whereas VH afferent drive of D1 MSNs is unchanged. In D2 MSNs, KOR disinhibits VH- and BLA-driven spiking, by inhibiting D1 MSN collaterals.
KOR Inhibition of BLA Afferents is Mediated by Presynaptic KORs
We next determined whether KORs on BLA terminals mediate inhibition of BLA oEPSCs, we injected a cocktail of AAV-Syn-Cre-GFP and AAV-EF1α-DIO-ChR2-eYFP (McDevitt et al., 2014) into the BLA of WT and KORloxP/loxP mice (Van’t Veer et al., 2013)(Figure 2A). Utilizing this strategy, ChR2-expressing cells should lack KOR expression in KORloxP/loxP mice, while KOR expression remains intact in ChR2-expressing cells in WT mice. We confirmed that KOR expression was reduced in ChR2-expressing cells by utilizing in situ hybridization of KOR and eYFP mRNA. The percentage of BLA eYFP-positive cells that were also KOR mRNA-positive was significantly decreased in KORloxP/loxP mice relative to WTs (Figure 2B). If KOR inhibition is mediated by KORs on BLA terminals, then BLA oEPSCs in NAcc MSNs from KORloxP/loxP mice should be insensitive to U69. Bath application of U69 inhibited BLA-oEPSCs in NAcc MSNs of WT mice, an effect that was absent in KORloxP/loxP mice (Figure 2C). This provides unequivocal evidence that KOR inhibition of BLA synapses onto MSNs is mediated by KOR on BLA terminals (Figure 8A), functional evidence of our knockdown approach, and confirms specificity of U69 at the KOR.
Figure 2. KORs on BLA Terminals Mediate KOR Inhibition of BLA Synapses and Modulate Basal Synaptic Efficacy.
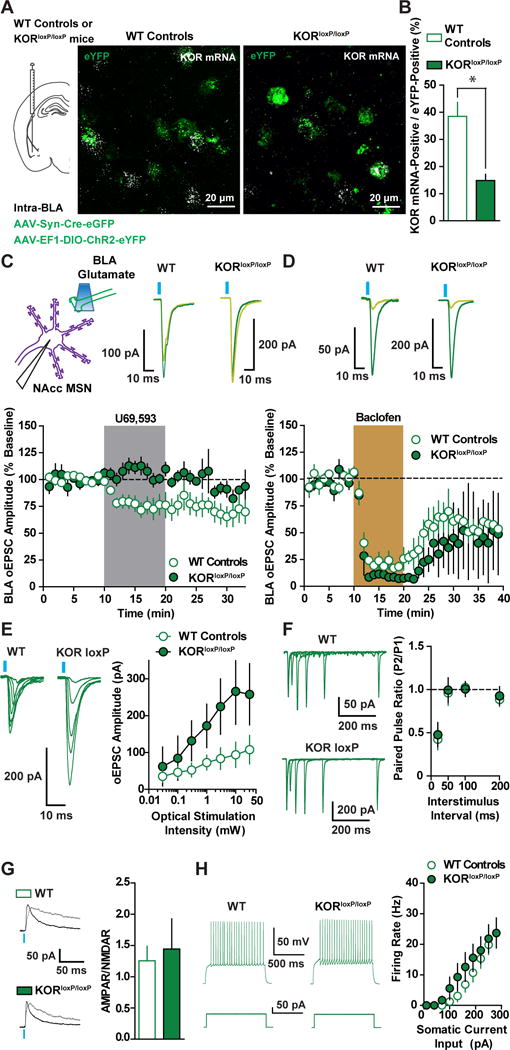
(A) Representative image of ChR2-YFP and eYFP- and KOR-mRNA (puncta) in the BLA of a WT control and a KORloxP/loxP mouse after injection of AAV-EF1α-DIO-ChR2-eYFP and AAV-Syn-Cre-eGFP into BLA. (B) The percentage of eYFP-positive cells with KOR mRNA was decreased in KORloxP/loxP mice (n=3) relative to WT controls (n=2) (t(3)=4.881; p=0.016), as was the proportion of cells expressing KOR mRNA in eYFP cells in WT (49/125; 38.5%) and KORloxP/loxP (25/173; 14.9%) mice (Fishers Exact Test; p=0.0164). (C) Representative traces and time course of U69 (1 μM) effects on BLA oEPSCs in WT controls (n=7) and KORloxP/loxP mice (n=6; ANOVA Genotype x Time Interaction; F(19, 190)=4.76; p<0.0001). (D) Representative traces and time course showing GABAB receptor inhibition of BLA oEPSCs with baclofen (5 μM) in WT (n=5) and KORloxP/loxP mice (n=3; ANOVA Genotype x Time Interaction; F(19, 114)=0.7; p=0.81). (E) Representative traces and mean BLA oEPSC amplitude in response to graded optical stimulation intensities in KORloxP/loxP (n=5) and WT mice (n=6; ANOVA Genotype x Time Interaction; F(6, 54)=3.62; p=0.004). (F) Mean PPR and representative traces of BLA oEPSCs in MSNs of KORloxP/loxP (n=7) and WT mice (n=9; ANOVA Genotype x Time Interaction; F(4, 56)=0.1; p=0.98). (G) BLA KOR genetic ablation does not alter BLA to NAcc MSN synaptic strength (t(7)=0.32; p=0.76). Representative traces of AMPAR and NMDAR EPSCs at +40 mV from WT control (n=4) and KORloxP/loxP mice (n=5). (H) Intrinsic excitability of NAcc MSNs from WT (n=12) and KORloxP/loxP mice (n=7) does not differ (ANOVA Genotype x Time Interaction; F(12, 204)=1.13; p=0.34).
To test the possibility that genetic ablation of KOR in BLA neurons renders BLA synapses insensitive to presynaptic inhibition of glutamate release, we determined whether GABA-B-mediated inhibition was still intact in BLA synapses in KORloxP/loxP mice. Baclofen produced a similar depression of BLA oEPSCs in MSNs from WT control mice and KORloxP/loxP mice (Figure 2D), suggesting presynaptic inhibition is still present after BLA KOR knockdown.
We next examined whether KORs on BLA terminals regulate basal synaptic transmission in the BLA to NAcc pathway of KORloxP/loxP mice expressing AAV-Syn-Cre-GFP and AAV-EF1α-DIO-ChR2-eYFP in the BLA. We first determined the input-output relationship between optical stimulation intensity and BLA-evoked oEPSC amplitude in MSNs between KORloxP/loxP and WT mice. KORloxP/loxP mice displayed increased BLA oEPSC amplitudes relative to WT controls at higher stimulation intensities (Figure 2E), suggesting KORs inhibit the recruitment of BLA synapses onto MSNs. PPRs of BLA-oEPSCs across multiple pulse intervals were not significantly different between WT controls and KORloxP/loxP mice (Figure 2F). Thus, KOR inhibition of synaptic efficacy is not mediated by a change in probability of release. The increased input-output relationship in KORloxP/loxP mice could be a consequence of increased postsynaptic strength. Optically-evoked BLA AMPAR to NMDAR ratios (AMPAR/NMDAR), an index of postsynaptic strength, were similar in WT and KORloxP/loxP mice (Figure 2G), suggesting that the increased input/output curve in KORloxP/loxP mice is mediated by enhanced recruitment of BLA afferents. Synaptic-driven homeostatic plasticity of intrinsic excitability, wherein long-term changes in synaptic activity modifies intrinsic excitability, has been reported in the NAcc (Ishikawa et al., 2009). We failed to detect a significant change in intrinsic excitability in MSNs from WT controls and KORloxP/loxP mice, suggesting that lack of KOR inhibition does not change NAcc MSN intrinsic excitability (Figure 2H). Collectively, these results suggest that KORs negatively modulate synaptic efficacy of BLA synapses in the NAcc via a presynaptic site of action (Figure 8A).
KOR Regulation of MSN-MSN Collateral GABAergic Transmission
KORs inhibit GABAergic synapses onto MSNs via a presynaptic site of action (Hjelmstad and Fields, 2003). One fundamental question is whether KORs would inhibit lateral inhibition from D1 and D2 MSNs to other MSNs. In the dorsal and ventral striatum, Dyn mRNA expression is restricted to D1 mRNA-expressing MSNs (Gerfen et al., 1990), while adenosine2A (A2A) receptor expression is restricted to D2 MSNs (Kreitzer 2009). We utilized ProDyn-IRES-Cre (PDyn-iCre) and A2A-Cre mice to examine the role of KORs in regulating local D1 and D2 MSN output, respectively, within the NAcc. We first validated that PDyn-iCre mice (Al-Hasani et al., 2015; Krashes et al., 2014) provide selective genetic access to D1 MSNs in the NAcc by injecting AAV-DIO-eYFP into the NAcc and immunostaining for substance P, a peptide expressed in D1-MSNs (Figure 3A). In PDyn-iCre mice injected with intra-NAcc AAV-DIO-eYFP, 86.3% of eYFP-positive cells were also substance P positive (Figure 3B; 776 eYFP/Substance P double positive out of 899 total eYFP positive cells). Moreover, with intra-NAcc AAV-DIO-ChR2-eYFP injection, ChR2-positive cells in A2A-Cre mice, which express Cre in D2 MSNs, had enhanced intrinsic excitability relative to ChR2-positive cells from PDyn-iCre mice (Figure 3B), consistent with reports demonstrating decreased excitability in D1 MSNs relative to D2 MSNs (Grueter et al., 2010; Kreitzer and Malenka, 2007). Thus, the PDyn-iCre mouse is a useful tool to gain genetic access to D1 MSNs in the NAcc (Al-Hasani et al., 2015).
Figure 3. KOR Modulation of GABAergic Transmission between MSNs.
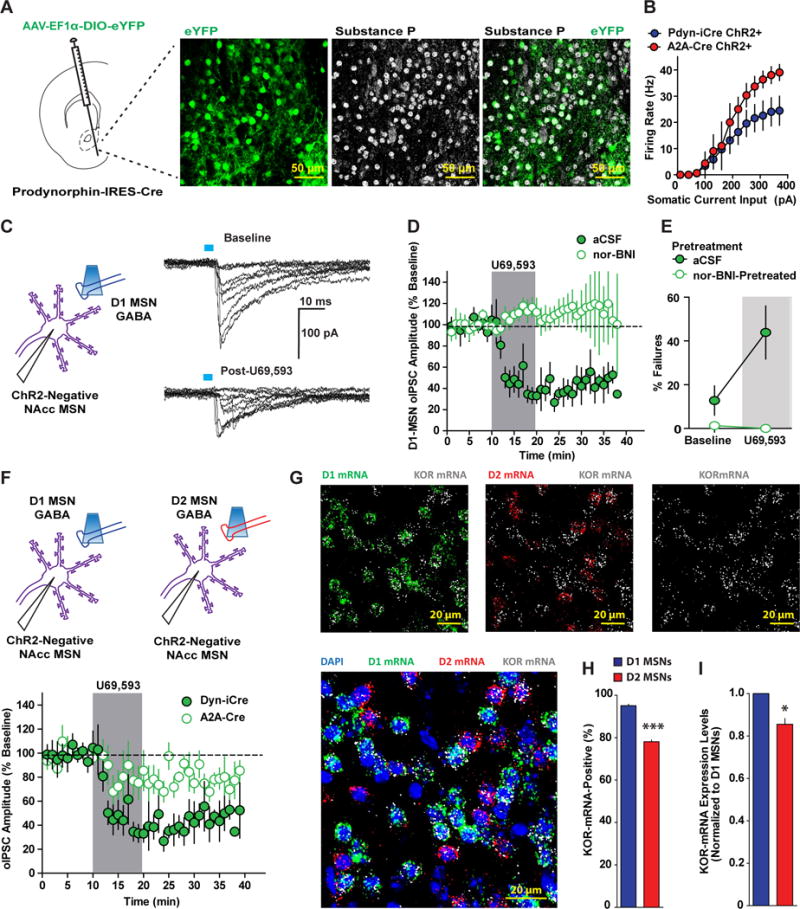
(A) Experimental schematic depicting AAV-EF1α-DIO-eYFP injection into the NAcc of PDyn-IRES-Cre (PDyn-iCre) mice. Colocalization of eYFP-positive and substance P-positive NAcc MSNs in PDyn-iCre mice (n=4). (B) Mean firing rate evoked by depolarizing current pulses in ChR2-positive MSNs from PDyn-iCre (n=7) and A2A-Cre (n=3) mice injected with AAV-EF1α-DIO-ChR2-eYFP into the NAcc. (C) ChR2-negative MSNs were recorded and GABA-A IPSCs from D1-MSNs (D1-MSN oIPSCs) were evoked in PDyn-iCre mice expressing ChR2-eYFP in NAcc. Representative traces of D1-MSN oIPSCs in ChR2-negative MSNs at baseline (top) and after U69 (bottom) recorded with a KCl-based internal solution. (D) U69 (1 μM) inhibition of D1 MSN oIPSCs recorded in ChR2-negative MSNs from PDyn-iCre mice expressing AAV-EF1α-DIO-ChR2-eYFP in the NAcc (filled green circles; n=6). Pretreatment with nor-BNI (100 nM) blocked U69 inhibition of D1 MSN oIPSCs (open green circles; n=5; Two-way ANOVA Treatment x Time Interaction; F(19, 171)=5.84; p<0.0001). (E) Percentage of D1-MSN oIPSC failures at baseline and after U69 (shaded region) in cells recorded in regular aCSF (filled green circles) or in aCSF-containing nor-BNI (Two-way ANOVA Treatment x Time Interaction; F(1, 9)=15.25; p=0.0036). (F) Time course of U69 (1 μM) inhibition of D1 MSN (filled green circles; n=6) and D2 MSN (open green circles; n=5) oIPSCs recorded in ChR2-negative MSNs from PDyn-iCre and A2A-Cre mice, respectively (Two-way ANOVA Genotype x Time Interaction; F(19,209)=1.72; p=0.0342). (G) Representative image of RNAscope in situ hybridization of DAPI (blue), D1 mRNA (green), D2 mRNA (red), and KOR mRNA (white) expression. Top left, D1 and KOR mRNA expression. Top middle, D2 and KOR mRNA expression. Top right, KOR mRNA expression. Bottom left, combined D1, D2, and KOR mRNA expression. (H) Percentage of D1 and D2 mRNA-positive MSNs that co-express KOR mRNA (n=4; t(7)=21.13; p<0.0001). (I) Mean relative expression levels of KOR mRNA (mean integrated density/area) relative to D1 MSNs in KOR mRNA-positive D1 and D2 MSNs (t(7)=5.284; p=0.0011).
We determined whether D1-MSN collaterals were inhibited by KORs by injecting PDyn-iCre mice with AAV- DIO-ChR2-eYFP into NAcc and recording in regions with dense ChR2-eYFP expression. We recorded from ChR2-negative neurons with optically-evoked Dyn-MSN GABA-A oIPSCs (D1-MSNs oIPSC; Figure 3C). Under these conditions, recorded neurons are likely D2 MSNs or the remaining fraction of uninfected D1 MSNs. As collaterals from MSNs to other MSNs elicit small inhibitory currents (Tepper et al., 2004; Wilson, 2007), we utilized a KCl-based internal solution to detect GABA-A currents as large inward currents at a -70 mV holding potential. Consistent with a low probability of release and connection between MSNs (Planert et al., 2010; Tunstall et al., 2002), Dyn-MSN optical stimulation elicits GABA-A currents with occasional failures (Figure 3C). U69 inhibited optically-evoked D1-MSN oIPSCs (Figure 3D) and increased the number of D1-MSN oIPSC failures (Figure 3E), indicative of a reduction in pre-synaptic probability of release and/or number of release sites. D1-MSN oIPSC inhibition and increase in failure by U69 were blocked by nor-BNI pre-treatment. Interestingly, basal D1-MSN oIPSC failures in nor-BNI-treated cells was significantly smaller than of untreated cells (Figure 3E), suggesting KORs modulate basal D1 MSN lateral inhibition.
To determine whether KORs modulate D2 MSN collaterals, we injected AAV- DIO-ChR2-eYFP into NAcc of A2A-Cre mice and recorded from ChR2-negative neurons while evoking D2-MSN oIPSCs (Figure 3F). Under these conditions, recordings are biased towards D1 MSNs and non-infected D2 MSNs. D2 MSN oIPSCs were larger in amplitude (Figure S4A) and exhibited less failures than D1 MSN collaterals (Figure S4B), consistent with a higher probability of synaptic connections between D2 MSNs and neighboring than D1 MSNs (Planert et al., 2010). U69 inhibited A2A-MSN oIPSCs, albeit this effect was not as pronounced as that seen on D1-MSN oIPSCs (Figure 3F). Thus, KORs have a significantly stronger control over D1 MSN lateral inhibition than D2 MSN lateral inhibition.
A possibility for greater KOR inhibition of D1 MSN collaterals than D2 MSN collaterals is differential expression of KORs between D1 and D2 MSNs. To this end, using triple-labeling in situ hybridization to detect D1, D2, and KOR mRNA, we determined whether KOR is differentially expressed in D1 and D2 MSNs (Figure 3G). The proportion of D1 MSNs expressing KOR mRNA was significantly higher than D2 MSNs (Figure 3H). Furthermore, the relative expression level of KOR mRNA within KOR-positive D2 MSNs cells was 85% of KOR-positive D1 MSNs (Figure 3I). NAcc GABAergic interneurons are also a source of GABA for MSNs, which may be regulated by KORs. To determine whether KORs were expressed in GABAergic interneurons, we utilized in-situ hybridization to determine if somatostatin mRNA- and parvalbumin mRNA-positive interneurons express KOR mRNA (Figure S5). KOR mRNA expression in somatostatin- and parvalbumin-positive interneurons was sparse and weak relative to MSNs (Figure S5A, B), suggesting KORs are strategically-poised to regulate MSN collaterals. Thus, KORs modulate MSN collaterals, and modulation of D1 MSN output to other MSNs is stronger than D2 MSN output, presumably due to enhanced KOR mRNA expression in D1 MSNs (Figure 8B).
Differential KOR Modulation of Glutamatergic Synapses onto D1- and D2-MSNs
It is unclear if KORs differentially inhibit glutamatergic synapses onto D1 and D2 MSNs. As our previous experiments were agnostic to MSN identity, we utilized D1-tdtomato mice expressing tdtomato under control of the D1 receptor promoter to determine whether presynaptic KOR regulation differed between D1 and D2 MSNs. D1 MSNs were identified by tdtomato fluorescence, while D2 MSNs were identified by lack of td-tomato fluorescence and electrophysiological properties indicative of MSNs. D1 and D2 MSNs can be distinguished with this strategy, as D2 MSNs (tdtomato negative) had increased excitability relative to D1 MSNs (td-tomato positive; Figure S6), consistent with other reports (Grueter et al., 2010; Kreitzer and Malenka, 2007). We determined whether KOR modulation of eEPSCs would differ between D1 and D2 MSNs (Figure 4A). U69 inhibited eEPSCs in D1 MSNs, while inhibition was not consistently observed in D2-MSNs (Figure 4B). We determined if pre-synaptic KOR modulation of glutamatergic synapses differed between D1 and D2 MSNs by examining the effects of U69 on miniature EPSCs (mEPSCs; Figure 4C). U69 decreased mEPSC frequency, but not amplitude, in D1 MSNs, an effect not observed in vehicle-treated MSNs and U69-treated D2 MSNs (Figure 4D). Thus, KORs preferentially inhibit glutamate release onto D1 MSNs.
Figure 4. KORs Differentially Regulate Glutamatergic Synaptic Transmission in D1 and D2 MSNs.
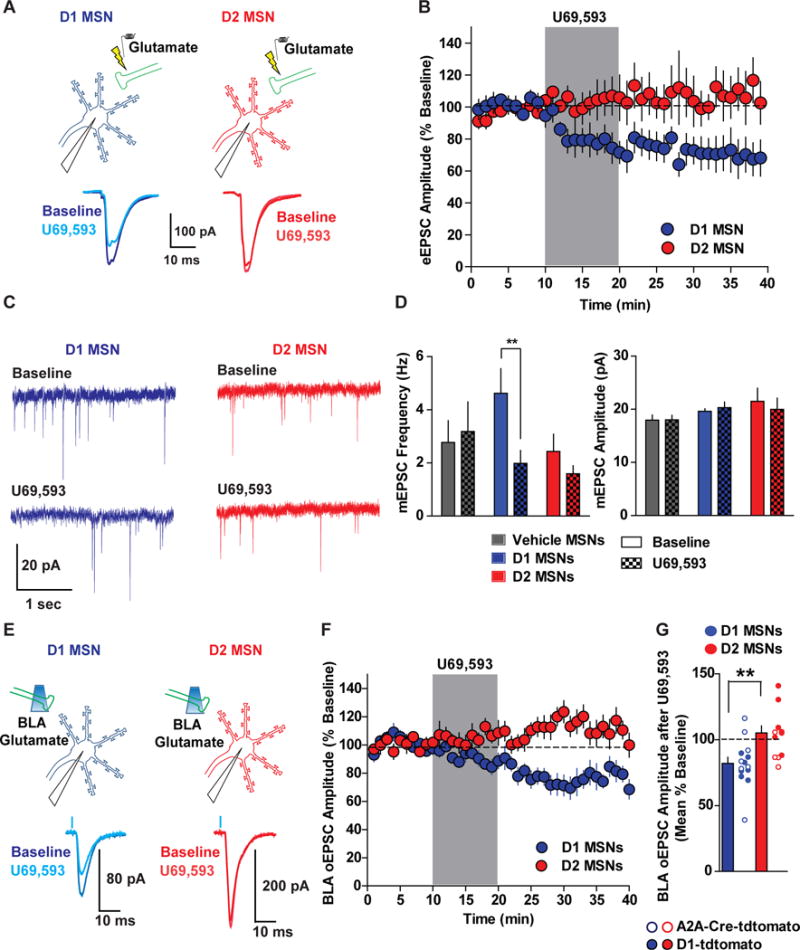
(A) D1 MSNs (tdtomato positive) and D2 MSNs (tdtomato negative) MSNs were recorded from D1-tdtomato mice and eEPSCs were evoked by electrical stimulation. Representative traces of eEPSCs in D1 and D2 MSNs at baseline and after U69. (B) U69 (1 μM) reliably inhibited eEPSCs in D1 (blue circles; n=6), but not in D2 MSNs (red circles; n=10; Two-way ANOVA Cell Type x Time Interaction; F(19,266)=2.79; p=0.0001). (C) Representative traces of mEPSCs recorded from D1 (blue traces) and D2 MSNs (red traces) before (top) and after application of U69 (bottom). (D) mEPSC frequency (Hz) and amplitude (pA) during baseline (solid bars) and after U69 (patterned bars) in vehicle-treated MSNs (grey bars; n=8) and U69-treated D1 (blue bars; n=9) and D2 MSNs (red bars; n=8). U69 decreased mEPSC frequency in D1 MSNs, but had no significant effect in D2 MSNs (Two-way ANOVA Cell Type x Time Interaction; F(2,22)=10.46; p=0.0006). U69 had no effect on mEPSC amplitude (mEPSC Frequency; Two-way ANOVA Cell Type x Time Interaction; F(2,22)=1.95; p=0.17). (E) D1 and D2 MSNs were recorded in D1-tdtomato and A2A-Cre/floxed-tdtomato mice and U69 effects on BLA oEPSCs were determined. (F) KOR inhibition of BLA oEPSCs in D1 MSNs (n=13), but not in D2 MSNs (n=12; Two-way ANOVA Cell Type x Time Interaction; F(19,437)=2.43; p=0.0007). (G) Mean percent of baseline oEPSC in pooled D1 and D2 MSNs from both genotypes (t(24)=3.22; p=0.0037), which did not differ between D1 and D2 MSNs identified in D1-tdtomato and A2A-Cre/floxed-tdtomato mice (Two-way ANOVA Main effect of Cell-Type (D1 vs D2); F(1, 22)=9.70; p=0.0051; No Cell-type x Genotype interaction or main effect of Genotype; p>0.05).
We subsequently determined whether KOR modulation of BLA afferents is cell-type specific by injecting D1-tdtomato mice with intra-BLA AAV-CaMKII-ChR2-eYFP and recording BLA oEPSCs in D1 and D2 MSNs (Figure 4E). KOR activation inhibited BLA oEPSCs in D1 MSNs, but not D2 MSNs (Figure 4F). As D1-tdtomato mice have been reported to display abnormal electrophysiological properties (Bagetta et al., 2011), we injected AAV-CaMKII-ChR2-eYFP into the BLA of A2A-Cre mice crossed with tdtomato reporter mice. Here, D2 MSNs were identified by presence of tdtomato and D1 MSNs were lacking tdtomato. KOR activation inhibited BLA oEPSCs in D1 MSNs, but not D2 MSNs, in A2A-Cre-td-tomato mice (Figure 4G). Thus, differential regulation of excitatory afferents onto D1 and D2 MSNs is not due to altered synaptic plasticity due to transgene expression in D1-tdtomato and A2A-Cre-tdtomato mice. Collectively, these results suggest that the KORs are poised to regulate excitatory drive of D1-MSNs more reliably than excitatory synapses in D2-MSNs (Figure 8A).
NAcc D1 MSNs project to the ventral tegmental area (VTA) and substantia nigra (SNc) in the midbrain and the lateral hypothalamus (Bocklisch et al., 2013; O’Connor et al., 2015), but it is unclear whether presynaptic KOR regulation of glutamatergic synapses differs between D1 MSN efferents. We determined whether presynaptic KOR regulation of glutamate release onto D1 MSNs projecting to the VTA/SNc (herein referred to as midbrain) or the LH would differ. By injecting red retrobeads into the midbrain and green retrobeads into the LH (Figure 5A) we first identified whether midbrain- and LH-projecting NAcc MSNs were sending projections to only one target and/or collateralizing to both LH and midbrain. Midbrain- and LH-projecting NAcc MSNs were spatially segregated and largely non-overlapping, with the majority of midbrain-projecting MSNs localized to lateral NAcc shell and core, and LH-projecting MSNs localized to medial and lateral shell and core (Figure 5A). To conservatively ascertain levels of overlap, we analyzed regions where midbrain- and LH-projecting cells were both present. 22.4% (202/901) of red retrobead-positive cells (midbrain-projecting) also contained green retrobeads (LH-projecting). Similarly, 24.3% (202/832) of green retrobead-containing MSNs (LH-projecting) also contained red retrobeads (midbrain-projecting). Thus, midbrain- and LH-projecting MSNs are largely non-overlapping populations, with subsets of cells projecting to both (Figure 5A). To confirm that midbrain and LH-projecting MSNs were primarily D1 MSNs, we injected green retrobeads into the LH or midbrain of D1-tdtomato mice and observed that 95.6% (439/458) midbrain- and 94.1% (386/410) LH-projecting MSNs were tdtomato-positive D1 MSNs (Figure 5B). Thus, NAcc outputs to the midbrain and LH primarily consist of largely, non-overlapping D1 MSNs.
Figure 5. Pre-synaptic KOR Regulation of Glutamatergic Synapses Depends on NAcc Sub-Region but not efferent.
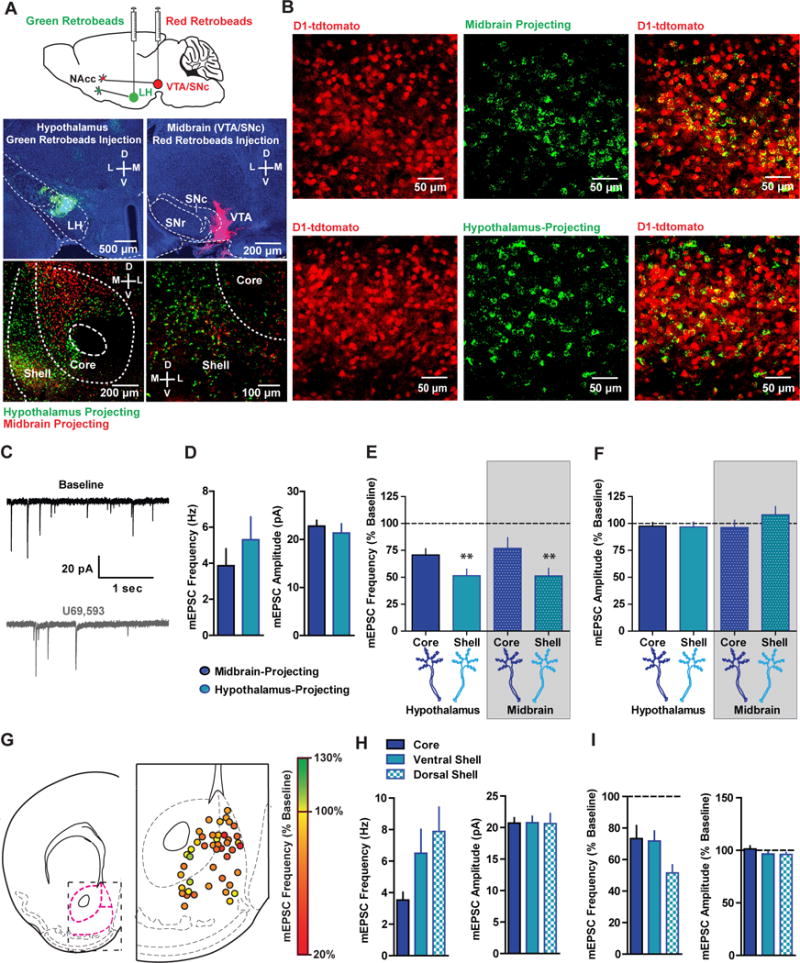
(A) Top, WT mice were injected with unilateral fluorescent Green XI and Red XI retrobeads into the LH and ipsilateral midbrain, respectively. Bottom, red and green retrobead labeling in the NAcc at low (left) and high (right) magnification. (B) Representative image of green fluorescent retrobead labeling in the NAcc of D1-tdtomato mice (n=3) injected with green retrobeads in the LH or midbrain demonstrating that 95.9% (439/458) of midbrain-projecting and 94.15% (386/410) of LH-projecting MSNs are D1 MSNs. C) Representative traces of mEPSCs during baseline (black) and after U69 (grey) from a midbrain-projecting MSN in the NAcc shell. (D) Basal mEPSC frequency and amplitude in LH- (n=14) and midbrain-projecting (n=12) D1 MSNs (Frequency, t=0.89(24); p=0.38; Amplitude, t=0.67(24); p=0.52). (E) Mean normalized mEPSC frequency (expressed as a percentage of baseline) and (F) amplitude after U69 in D1 MSNs in the core and shell projecting to the LH and midbrain (mEPSC frequency; Two-way ANOVA; Sub-region main effect; F(1,21)=9.18; p=0.006; ** p<0.01 pooled shell vs pooled core). (G) Map of presynaptic KOR inhibition of mEPSC frequency (expressed as a percentage of baseline) in D1 MSNs from D1-tdtomato mice (n=43). (H) Basal mEPSC frequency and amplitude in D1 MSNs recorded in the NAcc core (n=15), ventral shell (n=16), and dorsomedial shell (n=17; ANOVA; F(2,45)=3.104; p=0.05; * p<0.05 pooled shell vs core). (I) mEPSC frequency and amplitude (expressed as a percentage of baseline) in D1 MSNs recorded in the NAcc core (n=13), ventral shell (n=16), and dorsomedial shell after U69 (n=17; ANOVA; F(2,43)=3.749; p=0.04; * p<0.05).
To examine whether KOR regulation of presynaptic glutamate release differed between LH- and midbrain-projecting MSNs, WT mice were injected with retrobeads into the midbrain or LH. Six to seven days after injection, retrobead-positive MSNs in NAcc core and shell were patched, and the effects of U69 application on mEPSC frequency was determined (Figure 5C). Basal mEPSC frequency and amplitude did not differ between LH- and midbrain-projecting cells (Figure 5D). Similar to results obtained in D1-tdtomato mice, U69 decreased mEPSC frequency (Figure 5E), but not amplitude (Figure 5F), demonstrating KORs presynaptically inhibit glutamate release onto D1 MSNs utilizing a non-genetic approach to identify D1 MSNs. Specifically, U69 inhibited mEPSC frequency in a similar manner in LH- and midbrain-projecting cells, independent of whether they were localized in core or shell. However, U69 decreased mEPSC frequency more robustly in shell D1 MSNs than core D1 MSNs, regardless of whether they projected to the LH or the midbrain. Thus, KORs inhibit presynaptic glutamate release onto D1 MSNs projecting to both the LH and midbrain, and this effect is significantly more robust in the shell than core. We further studied sub-regional differences in KOR regulation of glutamate release onto D1 MSNs by determining the effects of U69 on mEPSCs in a large population of D1 MSNs across the NAcc in D1-tdtomato mice (Fig 5G). Basal mEPSC frequency was significantly smaller in NAcc core vs shell subregions (Fig 5H), without significant differences in mEPSC amplitude (Fig 5H). KOR inhibition of mEPSC frequency in D1 MSNs was observed throughout the NAcc, however this effect was significantly stronger in mediodorsal NAcc shell than the ventral shell and core (Fig 5 G,I). Together, these results reveal that presynaptic KOR modulation of glutamate release onto D1 MSNs does not differ between LH- and midbrain-projecting D1 MSNs, and, though it is more robust in the dorsomedial NAcc shell, it is present throughout the NAcc.
KORs Differentially Regulate Excitation/Inhibition Balance in D1- and D2-MSNs
KORs inhibit GABAergic output from D1 and D2 MSN collaterals, however, it is currently not known whether KORs differentially inhibit GABAergic inputs onto D1 and D2 MSNs. We patched D1 and D2 MSNs from D1-tdtomato mice and examined the effects of U69 on mIPSCs to determine KOR regulation of GABA synapses (Figure 6A). mIPSC frequency and amplitude did not differ between D1 and D2 MSNs (Figure S7A). U69 significantly decreased mIPSC frequency in D2 MSNs without affecting amplitude, an effect not observed in vehicle-treated MSNs and U69-treated D1 MSNs (Figure 6B, C). U69 also decreased electrically-evoked IPSC amplitude in both D1 and D2 MSNs, however, this effect was larger in D2 MSNs than D1 MSNs (Figure S7B). Collectively, these results suggest that KORs preferentially inhibit GABAergic synapses onto D2 MSNs relative to D1 MSNs, via a presynaptic site of action.
Figure 6. KOR Activation Alters Excitation/Inhibition Balance in a Cell Type-Specific Manner.
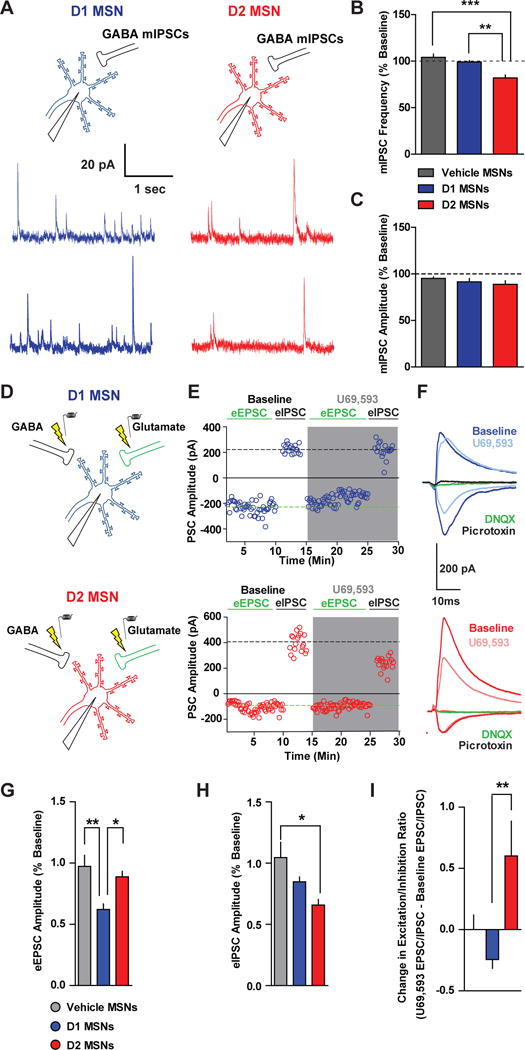
(A) D1 MSNs or D2 MSNs were patched from D1-tdtomato mice and mIPSCs were recorded at 0 mV. Representative traces of mIPSCs during baseline and after U69 (1 μM) in D1 MSNs (blue traces) and in D2 MSNs (red traces). (B) Mean mIPSC frequency (expressed as a percentage of basal frequency) after vehicle (grey; n=9) or U69 in D1 MSNs (blue; n=7) and in D2 MSNs (red; n=8; One-way ANOVA; F(2,23)=15.3; p<0.0001; ***, ** p < 0.05). (C) Mean mIPSC amplitude after vehicle or U69 in D1 MSNs and in D2 MSNs (One-way ANOVA; F(2,23)=1.172; p=0.33). (D) D1 MSNs and D2 MSNs were patched and eEPSCs were recorded at the reversal potential of GABA IPSCs, while eIPSCs were recorded at the reversal potential of glutamatergic EPSCs. (E) Representative postsynaptic currents (PSCs) D1 (top) and D2 (bottom) MSNs during baseline at -55 mV (eEPSCs) and 0 mV (eIPSCs) at baseline, and during U69 application (grey shaded region). (F) Traces from representative D1 and D2 MSNs. eEPSCs recorded at -55 mV were abolished by application of DNQX (10 μM), while eIPSCs were completely abolished by application of picrotoxin (100 μM). (G) Relative eEPSC amplitudes (expressed as a percentage of baseline) after vehicle (n=9) or U69 (1 μM) in D1 MSNs (n=8) and D2 MSNs (n=8; One-way ANOVA; F(2,24)=7.39; p=0.0035; **, * p < 0.05). (H) Relative eIPSC amplitudes (expressed as a percentage of baseline) after vehicle or U69 (One-way ANOVA; F(2,24)=5.29; p=0.013; **, p < 0.05). (I) Change in excitation-inhibition ratio after vehicle or U69 (eEPSC/eIPSCU69 - eEPSC/eIPSCBaseline) in D1 MSNs and D2 MSNs (One-way ANOVA; F(2,24)=5.76; p=0.0097; **, p < 0.05).
If KORs are exerting more control of glutamatergic synapses onto D1 MSNs than D2 MSNs, and more net inhibitory control over GABAergic synapses in D2 MSNs than D1 MSNs, then KORs may serve as a gate to dynamically switch excitation-inhibition balance in D1 and D2 MSNs. To determine whether KORs were differentially regulating excitation-inhibition balance in D1 and D2 MSNs, we recorded biophysically-isolated basal AMPAR eEPSCs at -70 or -55 mV and basal GABA-A eIPSCs at 0 mV (Figure 6D,E). We then applied U69 and subsequently recorded AMPAR eEPSCs at -70 or -55 mV and baseline GABA-A eIPSCs at 0 mV. eEPSCs and eIPSCs are blocked by DNQX and picrotoxin (PTX), respectively (Figure 6F). U69 decreased the AMPAR eEPSC amplitude relative to baseline in D1 MSNs, but not D2 MSNs (Figure 6E, G). Moreover, a significant decrease in GABA-eIPSCs was only observed in D2 MSNs (Figure 6E, H). Thus, U69 decreased the excitation-inhibition ratio in D1 MSNs, and increased it in D2 MSNs (Figure 6I). As D1 and D2 MSN excitation-inihibition ratio is similar between D1 and D2 MSNs (Figure S7C), this would imply that KOR activation may shift the balance of excitation and inhibition in D1 and D2 MSNs by decreasing excitatory drive of D1 MSNs, and favoring disinhibition of D2 MSNs.
NAcc KOR Regulation of Pathway-Specific Excitation-Inhibition Balance Decreases Excitatory Drive of D1 MSNs and Disinhibits D2 MSN Firing
To directly delineate whether presynaptic KOR inhibition of glutamate and GABA onto D1 and D2 MSNs, respectively, would differentially impact synaptically-driven spiking in D1 and D2 MSNs, we examined the effects of U69 on electrical stimulation-evoked spiking (Figure 7A). A ten pulse, 20 Hz electrical train was utilized to evoke spikes in D1 and D2 MSNs. Evoked spikes were mediated by glutamatergic transmission and inhibited by GABA-A receptors, as PTX bath application enhanced MSN spiking, while DNQX/AP-5 blocked spiking (Figure S8A). In D1 MSNs, U69 decreased the number of evoked spikes per train relative to baseline (Figure 7B, C). In D2 MSNs, an increase in synaptically-driven spiking was observed after U69 application (Figure 7B, C). To determine whether KOR activation altered the pattern of evoked firing, we analyzed the interspike interval (ISI) and the latency to first spike. There was no difference in the ISI after vehicle or U69 in D1 or D2 MSNs (Figure S8B, C). In D2 MSNs, U69 decreased the latency to first spike, an effect absent in D1 MSNs and controls. Thus, KORs decrease excitatory drive of D1 MSNs without changing the pattern of activity, while KOR-mediated disinhibition in D2 MSNs allows for spiking to occur earlier.
Figure 7. KOR Activation Preferentially Decreases Excitatory Drive of D1 MSNs and Disinhibits D2 MSNs.
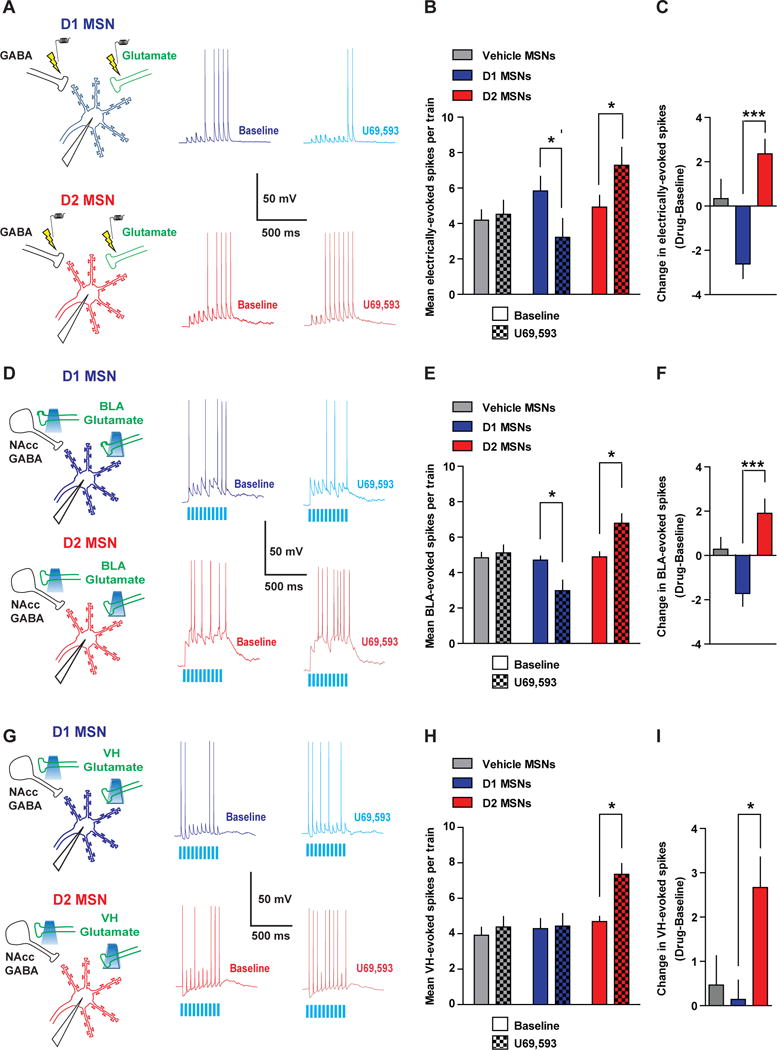
(A) D1 or D2 MSNs were patched and spiking was driven by extracellular stimulation using a bipolar stimulating electrode. Representative traces of synaptically-driven spiking in D1 and D2 MSN during baseline and after U69. (B) Mean electrically-evoked spikes during baseline (solid) and after vehicle or U69 (checkered) in MSNs treated with vehicle (grey, n=10) or D1 (blue, n=8) and D2 (red, n=7) MSNs treated with U69 (Two-way ANOVA Cell Type x Drug Interaction; F(2,22)=9.58; p=0.001). (C) Mean change in evoked spiking in vehicle MSNs, and U69-treated D1 and D2 MSNs (One-way ANOVA; F(2,24)=9.585; p=0.001; *** p < 0.05). (D) Spiking was evoked in D1 and D2 MSNs by optogenetically activating BLA glutamatergic terminals, which recruits feed-forward/collateral inhibition. Representative traces of optogenetic BLA stimulation-evoked spiking in D1 and D2 MSNs during baseline and after U69 in D1 and D2 MSNs. (E) Mean BLA-evoked spikes during baseline and after vehicle or U69 in MSNs treated with vehicle (n=10) or D1 (n=10) and D2 MSNs (n=7) treated with U69 (Two-way ANOVA Cell Type x Drug Interaction; F(2,24)=9.25; p=0.0011). (F) The mean change in BLA-evoked spiking in vehicle MSNs, and U69-treated D1 and D2 MSNs (One-way ANOVA; F(2,26)=9.25; p=0.0011; *** p < 0.05). (G) Representative traces of optogenetic VH stimulation-evoked spiking in D1 and D2 MSNs during baseline and after U69. (H) Mean VH-evoked spikes during baseline and after vehicle or U69 in MSNs treated with vehicle (n=7) or D1 (n=7) and D2 MSNs (n=7) treated with U69 (Two-way ANOVA Cell Type x Drug Interaction; F(2,18)=5.14; p=0.017). (I) Mean change in VH-evoked spiking in vehicle MSNs, and U69-treated D1 and D2 MSNs (One-way ANOVA; F(2,20)=5.14; p=0.0017; * p < 0.05).
We next determined whether KOR activation would inhibit spiking evoked by optogenetic activation of BLA afferents in D1 and D2 MSNs (Figure 7D). Similar to that observed with electrical stimulation, BLA-evoked spiking was enhanced by PTX, suggesting that feedforward inhibition was recruited to inhibit BLA-evoked spiking, and BLA-evoked spiking was completely abolished by DNQX/AP-5 (Figure S8D). Interestingly, PTX increased D2 MSN firing rate more robustly than D1 MSNs, suggesting that BLA-evoked spiking in D2 MSNs is under stronger GABAergic control. U69 decreased BLA-evoked spiking in D1 MSNs, while it increased spiking in D2 MSNs (Figure 7E, F). In D2 MSNs, U69 decreased both the latency to first spike and ISI, and this was not observed in D1 MSNs or vehicle MSNs (Figure S8E,F). Thus, KORs decrease BLA excitatory drive of D1 MSNs, and disinhibit D2 MSNs by allowing them to spike earlier and at an increased firing rate (Figure 8C).
As VH afferents are not directly modulated by KOR, KOR modulation of excitation-inhibition balance is predicted to result in a different pattern of D1 and D2 MSN output than that observed for electrically- and BLA-evoked spiking. To this end, we determined the effects of U69 on optogenetic VH-evoked spiking in D1 and D2 MSNs (Figure 7G). PTX significantly increased VH-evoked spiking in a similar manner in both D1 and D2 MSNs (Figure S8G). U69, however, did not modify VH-evoked firing in D1 MSNs (Figure 7H, I), consistent with a lack of effect of U69 on VH-evoked oEPSCs. This result also suggests that KOR modulation of electrically- and BLA-evoked D1 MSN spiking is not due to a change in intrinsic excitability. KOR activation with U69 enhanced VH-evoked spiking in D2 MSNs. Moreover, firing pattern, as assessed by the ISI and latency to first spike, was unchanged in vehicle-treated MSNs and U69-treated D1 MSNs (Figure S8H, I). In D2 MSNs, U69 decreased the ISI, suggesting that KORs allow VH afferents to drive spiking at a higher rate (Figure S8H). Thus, the KOR system acts as a pathway-specific filter to gate BLA, but not VH, control of D1 MSNs, while facilitating integration of both VH and BLA excitatory drive of D2 MSNs (Figure 8C).
Discussion
Here we describe a novel synaptic framework wherein pathway- and cell type-specific KOR modulation of excitatory and inhibitory balance shapes information flow into and out of the NAcc (Figure 8C). Activation of NAcc KORs inhibits glutamatergic synaptic transmission from the BLA, but not VH, via a presynaptic site of action. Accordingly, KOR mRNA is expressed in NAcc-projecting BLA neurons, but not those originating in the VH. Within local microcircuits, KORs inhibit GABAergic collaterals from D1 MSNs more robustly than D2 MSN collaterals via enhanced KOR expression in D1 MSNs. Glutamatergic synapses in D1 MSNs are more sensitive than those on D2 MSNs to KOR inhibition. This effect is independent on the output (hypothalamus vs midbrain) of D1 MSNs, but is significantly more robust in NAcc shell than core D1 MSNs. Conversely, KOR modulation of GABAergic synapses was significantly stronger onto D2 MSNs than D1 MSNs, as KORs decreased excitation/inhibition balance in D1 MSNs, while increasing it in D2 MSNs. The net result dichotomous KOR modulation of excitation and inhibition in D1 and D2 MSNs is decreased BLA, but not VH, synaptic drive of D1 MSNs and disinhibition of BLA and VH excitatory drive of D2 MSNs. Thus, KORs play a role in NAcc information processing by inhibiting limbic excitatory drive of D1 MSNs in a pathway-specific manner and amplifying D2 MSN output via disinhibition in a pathway-independent manner (Figure 8C).
Here we demonstrate that the BLA is a KOR-sensitive glutamatergic afferent. Moreover, we did not detect an effect of U69 on VH glutamatergic synapses. This is in agreement with our anatomical findings that KOR mRNA is expressed in NAcc-projecting BLA neurons, but not in NAcc-projecting VH neurons. As BLA terminal expression is “patchy” in the dorsomedial NAcc shell and KOR inhibition of glutamate release is strongest in this region, it is possible that there is at least one more KOR-sensitive input to the shell. BLA afferents in the prefrontal cortex and bed nucleus of the stria terminalis are inhibited by KORs (Crowley et al., 2016; Tejeda et al., 2015), suggesting that presynaptic modulation of BLA output is a shared principle among different BLA efferents. Interestingly, baclofen inhibited both VH- and BLA-evoked oEPSCs suggesting that NAcc GABA-B receptors may not confer the same pathway specificity as KORs. Thus, the NAcc may have signals that broadly decrease excitatory drive, presynaptically, across various pathways (i.e. GABA-B) and modulatory signals that confer pathway-specific modulation (i.e. KORs). Indeed, mu-opioid receptors have been shown to produce pathway-specific effects in the dorsal striatum (Atwood et al., 2014). This is consistent with our hypothesis that endogenous opioid receptor systems contribute to information processing by virtue of pathway-specific regulation of synaptic transmission. KOR inhibition of BLA-evoked oEPSCs was observed in D1 MSNs, however, KOR activation failed to modify BLA-evoked oEPSCs in D2 MSNs. A possibility is that distinct BLA neurons project to D1 and D2 MSNs, and KORs are preferentially expressed in the former, not the latter. Another intriguing scenario is that BLA neurons that express KOR target both D1 and D2 MSNs, but functional KOR is preferentially targeted to BLA synapses on D1 MSNs, rather than D2 MSNs. Thus, KORs may act as cell-type specific filters on BLA afferents by selectively depressing BLA synapses onto D1 MSNs, but largely leaving glutamatergic synaptic transmission onto D2 MSNs intact (Figure 8A,C).
D1 MSNs and D2 MSNs are synaptically connected to both D1 and D2 MSNs (Planert et al., 2010). KORs inhibit D1 MSN collaterals more strongly than D2 MSN collaterals. In our experiments examining KOR regulation of D1 and D2 MSN collaterals onto ChR2-negative cells, it is likely that sampling was biased towards D2 and D1 MSNs, respectively. It is tempting to speculate that D1 MSN collaterals to D2 MSNs are more strongly inhibited than D2 MSNs to D1 MSNs. This is consistent with more reliable effects of U69 on GABAergic transmission onto D2, than D1, MSNs. KOR regulation of D1 MSN GABA output may be restricted to NAcc collaterals, but not projections, as U69 does not modify GABA synapses from NAcc to VTA DA neurons (Matsui et al., 2014). Connections between MSNs elicit weak IPSPs/IPSCs recorded at the soma, largely due to distribution of MSN collaterals on distal dendrites of MSNs (Koos et al., 2004; Tunstall et al., 2002). In pyramidal neurons, GABA synapses at distal dendrites inhibit synaptic integration (Lovett-Barron et al., 2012; Palmer et al., 2012). Likewise, MSN collaterals are hypothesized to play a role in sculpting synaptic integration by shunting incoming excitatory signals, while interneuronal inhibition regulates spike output (Tepper et al., 2004). KOR mRNA was sparsely and weakly expressed in somatostatin- and parvalbumin-expressing NAcc interneurons relative to D1 and D2 MSNs, suggesting KORs are preferentially expressed in NAcc MSNs and are poised to fine tune dendritic inhibition by MSNs. By depressing MSN collateral dendritic inhibition, KORs may increase the window of synaptic integration of excitatory potentials and increase the probability of action potential generation. Consistent with this notion, electrical and optogenetic excitation-driven spiking was facilitated by U69 in D2 MSNs. Thus, KORs also indirectly control information flow from afferents by regulating local inhibitory connections between MSNs (Figure 8B,C).
NAcc KORs may alter encoding properties of D1 and D2 MSNs via presynaptic control of glutamate and GABA. KORs may serve as a mechanism by which D1 MSN-mediated Dyn release, in response to strong glutamate and DA activation of D1 MSNs (Atwood et al., 2014; Gerfen et al., 1990; Moratalla et al., 1996; Wang et al., 1994), can decrease excitation-inhibition balance and, ultimately, excitation-spiking coupling in a pathway-specific manner in D1 MSNs. Conversely, presynaptic inhibition of GABA release on D2 MSNs increases excitation-inhibition balance, consequentially facilitating excitatory drive of D2 MSNs. This effect is of interest as disinhibition of spiking by delta-opioid systems has been recently reported in D1 MSNs in dorsal striatal striosomes (Banghart et al., 2015). Since Dyn signaling is mobilized in an activity-dependent manner, then Dyn/KOR signaling could be a mechanism by which activity states of D1 MSNs influence ongoing activity of both D1 and D2 MSNs. Balanced excitation and inhibition is a critical mediator of oscillatory activity in cortical networks, and dynamic shifts in this balance is a candidate mechanism to regulate synchronized spiking behavior in neuronal ensembles (Isaacson and Scanziani, 2011; Sohal et al., 2009). Control of excitation-inhibition balance by KORs is a mechanism by which both NAcc D1 and D2 MSNs neuronal ensembles can alter synaptic integration and adapt spike output in the face of strong DAergic and glutamatergic activity in a pathway-specific manner.
By modulating various neurotransmitter systems, KORs may precisely gate how D1 and D2 MSNs encode behaviors. KORs expressed within the NAcc inhibit DA release (Chefer et al., 2013; Ehrich et al., 2015), which is predicted to have similar network effects to those produced by KOR regulation of excitation-inhibition balance. DA D1 receptor signaling is necessary for long-term potentiation (LTP) of glutamatergic inputs, including the BLA (Floresco et al., 2001; Shen et al., 2008), while D2 receptor stimulation promotes LTD of glutamatergic synapses on D2 MSNs (Kreitzer and Malenka, 2007; Shen et al., 2008). Thus, KOR-mediated inhibition of DA release is predicted to dampen DA-induced synaptic strengthening in D1 MSNs and decrease DA-dependent LTD of excitatory synapses onto D2 MSNs. The net effect of KOR inhibition of glutamate, GABA, and DA would be to decrease excitation-spiking coupling from KOR-expressing afferents onto D1 MSNs and promote D2 MSN activity via removal of GABA inhibition and LTD of excitatory synapses. This strengthens our hypothesis that the KOR system is a modulatory system that alters D1 and D2 MSN network dynamics (Figure 8C).
NAcc KOR signaling is a modulator of various motivationally-charged behaviors. NAcc KOR signaling produces aversion, depressive-like behavior, increased food intake, and mediates drug-seeking behavior driven by negative reinforcement (Bruchas et al., 2010; Crowley and Kash, 2015; Muschamp et al., 2011; Tejeda et al., 2012). Modulation of diverse behaviors may be due, at least in part, by the virtue that D1 MSN efferents terminate in various brain regions participating in different behaviors, including the VTA and LH. As subsets of NAcc D1 MSNs project to the LH and inhibit feeding behavior (O’Connor et al., 2015), KOR signaling may inhibit excitatory synapses in LH-projecting D1 MSNs and disinhibit LH neurons controlling feeding. NAcc KORs may limit reward-seeking behavior, which relies on concomitant BLA to NAcc, and BLA-driven, DA transmission (Ambroggi et al., 2008; Jones et al., 2010; Stuber et al., 2011) by inhibiting both BLA and DA output. In agreement with the notion that KORs regulate diverse behaviors depending on the information being encoded by NAcc subpopulation, optogenetic stimulation of dorsomedial or ventromedial NAcc shell Dyn MSNs produced KOR-dependent reward or aversion, respectively (Al-Hasani et al., 2015). Whereas NAcc KOR signaling has consistently been reported to induce negative affect, a conditioned place preference is formed when dorsomedial NAcc shell KOR agonist microinjections are paired with a neutral environment (Castro and Berridge, 2014). As NAcc KORs inhibit neurotransmission throughout the NAcc, differences in KOR-mediated behavioral effects may be dictated by differential afferent inputs to different NAcc sub-regions and/or differential connectivity of NAcc MSNs with downstream targets. DA D3-receptor containing D1 MSNs are also concentrated in dorsomedial shell (Gangarossa et al., 2013), and these receptors oppose D1 receptor signaling. KOR inhibition of DA release may produce differential effects on D1/D3 MSNs in dorsomedial shell. Thus, NAcc Dyn/KOR may participate in a wide-range of behaviors by virtue of inhibition of neurotransmission, which alters heterogeneous microcircuits within NAcc.
In conclusion, we provide data in support of a comprehensive model wherein KORs regulate NAcc network dynamics by modulating excitatory and inhibitory balance in a pathway- and cell type-specific manner (Figure 8). As an integral part of the basal ganglia, under physiological conditions, the Dyn/KOR system may be a homeostatic mechanism to dampen excessive activity after motivationally-charged stimuli or stress. Based on our model, dysregulation of Dyn/KOR signaling may be involved in the pathogenesis of maladaptive behaviors triggered by stress. Altered KOR regulation of information flow from limbic systems to D1 and D2 MSNs, as well as local circuit inhibition, may result in aberrant NAcc network activity and contribute to negative affective states. Thus, understanding how Dyn/KOR activity in the NAcc regulates neurotransmission under normal and pathological conditions may provide novel therapeutic avenues to restore aberrant NAcc activity and negative affective symptoms in psychiatric conditions.
Experimental Procedures
Please refer to supplemental experimental procedures for a detailed description of each of the procedures employed in this paper. Male and female WT C57/BL6J, D1-tdtomato, A2A-Cre, A19 ROSA26-tdtomato reporter, ProDyn-IRES-Cre (Al-Hasani et al., 2015; Krashes et al., 2014), and KORloxP/loxP mice (Van’t Veer et al., 2013) were used at 2–3 months of age. Whole-cell electrophysiology was conducted as previously described (Britt et al., 2012). Recordings were restricted to the dorsomedial and dorsal portion of the ventromedial shell, unless where otherwise specified. Optogenetic EPSCs (0.1 Hz), IPSCs (0.1 Hz), and synaptically driven spikes (10 pulses at 20 Hz; 0.033 Hz) were evoked using 1 ms 473 nm laser pulses in mice expressing AAV-CaMKII-ChR2-eYFP in the BLA or VH for 5–8 weeks, or AAV-EF1α-DIO-ChR2-eYFP in the NAcc for 4–5 weeks. KOR knockdown in KORloxP/loxP was achieved with an intra-BLA injection of AAV-Syn-Cre-eGFP and AAV1-EF1α-DIO-ChR2-eYFP. mEPSCs and mIPSCs were collected in the presence of TTX and PTX, or TTX and DNQX/AP-5, at -70 and 0 mV, respectively. Retrograde tracing experiments and recordings from retrobead-containing D1 MSNs were conducted 6–7 days after retrobead injection.
For retrograde tracing/KOR in situ hybridization experiments, fluorogold (FG) was ionotophoretically delivered into the NAcc. Sections were subsequently processed for FG immunolabeling and radioactive in situ hybridization labeling KOR mRNA, as previously described (Yamaguchi et al., 2011). RNAscope ISH was conducted as previously described (Tejeda et al., 2013). Probes used were as follows: Drd1-C1, Mm-Drd2-C2, Mm-OPRK-C3, SST-C1, PV-C2, and eYFP-C1. Confocal images were acquired using a confocal microscope (Olympus).
Supplementary Material
Acknowledgments
We thank members of the Bonci laboratory, Drs. Alex Hoffman, Carl Lupica, and Roy Wise for helpful discussions. We thank Drs. Yeka Aponte and Zheng-Xiong Xi for sharing equipment. We thank Dr. Marisela Morales for reagents, resources, and guidance on fluorogold/radioactive in situ hybridization experiments. We thank Drs. Carlos Mejias-Aponte, Bing Liu, Hui-Ling Wang for assistance with fluorogold/radioactive in situ hybridization experiments. We thanks Drs. Robert Steiner and Sasha Kauffman for generously providing KOR cDNA. We thank Drs. Bruce Cohen, Anita Bechtholt, and Ashlee Van’t Veer for their roles in developing KOR-floxed mice This work was funded by the NIDA Intramural Research Program, and a grant from NIDDK to B.B.L.
Footnotes
Author contributions: HAT and AB designed research. HAT, JW, AK, MP, and VK conducted experiments. BL, MK, and WC provided transgenic mice. AB and HAT wrote the manuscript. All authors edited and approved final manuscript.
References
- Al-Hasani R, McCall JG, Shin G, Gomez AM, Schmitz GP, Bernardi JM, Pyo CO, Park SI, Marcinkiewcz CM, Crowley NA, et al. Distinct Subpopulations of Nucleus Accumbens Dynorphin Neurons Drive Aversion and Reward. Neuron. 2015;87:1063–1077. doi: 10.1016/j.neuron.2015.08.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ambroggi F, Ishikawa A, Fields HL, Nicola SM. Basolateral amygdala neurons facilitate reward-seeking behavior by exciting nucleus accumbens neurons. Neuron. 2008;59:648–661. doi: 10.1016/j.neuron.2008.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atwood BK, Kupferschmidt DA, Lovinger DM. Opioids induce dissociable forms of long-term depression of excitatory inputs to the dorsal striatum. Nat Neurosci. 2014;17:540–548. doi: 10.1038/nn.3652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bagetta V, Picconi B, Marinucci S, Sgobio C, Pendolino V, Ghiglieri V, Fusco FR, Giampa C, Calabresi P. Dopamine-dependent long-term depression is expressed in striatal spiny neurons of both direct and indirect pathways: implications for Parkinson’s disease. J Neurosci. 2011;31:12513–12522. doi: 10.1523/JNEUROSCI.2236-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bagot RC, Parise EM, Pena CJ, Zhang HX, Maze I, Chaudhury D, Persaud B, Cachope R, Bolanos-Guzman CA, Cheer JF, et al. Ventral hippocampal afferents to the nucleus accumbens regulate susceptibility to depression. Nat Commun. 2015;6:7062. doi: 10.1038/ncomms8062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banghart MR, Neufeld SQ, Wong NC, Sabatini BL. Enkephalin Disinhibits Mu Opioid Receptor-Rich Striatal Patches via Delta Opioid Receptors. Neuron. 2015;88:1227–1239. doi: 10.1016/j.neuron.2015.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bocklisch C, Pascoli V, Wong JC, House DR, Yvon C, de Roo M, Tan KR, Luscher C. Cocaine disinhibits dopamine neurons by potentiation of GABA transmission in the ventral tegmental area. Science. 2013;341:1521–1525. doi: 10.1126/science.1237059. [DOI] [PubMed] [Google Scholar]
- Britt JP, Benaliouad F, McDevitt RA, Stuber GD, Wise RA, Bonci A. Synaptic and behavioral profile of multiple glutamatergic inputs to the nucleus accumbens. Neuron. 2012;76:790–803. doi: 10.1016/j.neuron.2012.09.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruchas MR, Land BB, Chavkin C. The dynorphin/kappa opioid system as a modulator of stress-induced and pro-addictive behaviors. Brain Res. 2010;1314:44–55. doi: 10.1016/j.brainres.2009.08.062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calipari ES, Bagot RC, Purushothaman I, Davidson TJ, Yorgason JT, Pena CJ, Walker DM, Pirpinias ST, Guise KG, Ramakrishnan C, et al. In vivo imaging identifies temporal signature of D1 and D2 medium spiny neurons in cocaine reward. Proc Natl Acad Sci U S A. 2016;113:2726–2731. doi: 10.1073/pnas.1521238113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlezon WA, Jr, Thome J, Olson VG, Lane-Ladd SB, Brodkin ES, Hiroi N, Duman RS, Neve RL, Nestler EJ. Regulation of cocaine reward by CREB. Science. 1998;282:2272–2275. doi: 10.1126/science.282.5397.2272. [DOI] [PubMed] [Google Scholar]
- Castro DC, Berridge KC. Opioid hedonic hotspot in nucleus accumbens shell: mu, delta, and kappa maps for enhancement of sweetness “liking” and “wanting”. J Neurosci. 2014;34:4239–4250. doi: 10.1523/JNEUROSCI.4458-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chavkin C, James IF, Goldstein A. Dynorphin is a specific endogenous ligand of the kappa opioid receptor. Science. 1982;215:413–415. doi: 10.1126/science.6120570. [DOI] [PubMed] [Google Scholar]
- Chefer VI, Backman CM, Gigante ED, Shippenberg TS. Kappa opioid receptors on dopaminergic neurons are necessary for kappa-mediated place aversion. Neuropsychopharmacology. 2013;38:2623–2631. doi: 10.1038/npp.2013.171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crowley NA, Bloodgood DW, Hardaway JA, Kendra AM, McCall JG, Al-Hasani R, McCall NM, Yu W, Schools ZL, Krashes MJ, et al. Dynorphin Controls the Gain of an Amygdalar Anxiety Circuit. Cell Rep. 2016;14:2774–2783. doi: 10.1016/j.celrep.2016.02.069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crowley NA, Kash TL. Kappa opioid receptor signaling in the brain: Circuitry and implications for treatment. Prog Neuropsychopharmacol Biol Psychiatry. 2015;62:51–60. doi: 10.1016/j.pnpbp.2015.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehrich JM, Messinger DI, Knakal CR, Kuhar JR, Schattauer SS, Bruchas MR, Zweifel LS, Kieffer BL, Phillips PE, Chavkin C. Kappa Opioid Receptor-Induced Aversion Requires p38 MAPK Activation in VTA Dopamine Neurons. J Neurosci. 2015;35:12917–12931. doi: 10.1523/JNEUROSCI.2444-15.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferguson SM, Eskenazi D, Ishikawa M, Wanat MJ, Phillips PE, Dong Y, Roth BL, Neumaier JF. Transient neuronal inhibition reveals opposing roles of indirect and direct pathways in sensitization. Nat Neurosci. 2011;14:22–24. doi: 10.1038/nn.2703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Floresco SB, Blaha CD, Yang CR, Phillips AG. Dopamine D1 and NMDA receptors mediate potentiation of basolateral amygdala-evoked firing of nucleus accumbens neurons. J Neurosci. 2001;21:6370–6376. doi: 10.1523/JNEUROSCI.21-16-06370.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gangarossa G, Espallergues J, de Kerchove d’Exaerde A, El Mestikawy S, Gerfen CR, Herve D, Girault JA, Valjent E. Distribution and compartmental organization of GABAergic medium-sized spiny neurons in the mouse nucleus accumbens. Front Neural Circuits. 2013;7:22. doi: 10.3389/fncir.2013.00022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerfen CR, Engber TM, Mahan LC, Susel Z, Chase TN, Monsma FJ, Jr, Sibley DR. D1 and D2 dopamine receptor-regulated gene expression of striatonigral and striatopallidal neurons. Science. 1990;250:1429–1432. doi: 10.1126/science.2147780. [DOI] [PubMed] [Google Scholar]
- Grueter BA, Brasnjo G, Malenka RC. Postsynaptic TRPV1 triggers cell type-specific long-term depression in the nucleus accumbens. Nat Neurosci. 2010;13:1519–1525. doi: 10.1038/nn.2685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hjelmstad GO, Fields HL. Kappa opioid receptor activation in the nucleus accumbens inhibits glutamate and GABA release through different mechanisms. J Neurophysiol. 2003;89:2389–2395. doi: 10.1152/jn.01115.2002. [DOI] [PubMed] [Google Scholar]
- Hurd YL, Herkenham M. Molecular alterations in the neostriatum of human cocaine addicts. Synapse. 1993;13:357–369. doi: 10.1002/syn.890130408. [DOI] [PubMed] [Google Scholar]
- Hurd YL, Herman MM, Hyde TM, Bigelow LB, Weinberger DR, Kleinman JE. Prodynorphin mRNA expression is increased in the patch vs matrix compartment of the caudate nucleus in suicide subjects. Mol Psychiatry. 1997;2:495–500. doi: 10.1038/sj.mp.4000319. [DOI] [PubMed] [Google Scholar]
- Isaacson JS, Scanziani M. How inhibition shapes cortical activity. Neuron. 2011;72:231–243. doi: 10.1016/j.neuron.2011.09.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishikawa M, Mu P, Moyer JT, Wolf JA, Quock RM, Davies NM, Hu XT, Schluter OM, Dong Y. Homeostatic synapse-driven membrane plasticity in nucleus accumbens neurons. J Neurosci. 2009;29:5820–5831. doi: 10.1523/JNEUROSCI.5703-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones JL, Day JJ, Aragona BJ, Wheeler RA, Wightman RM, Carelli RM. Basolateral amygdala modulates terminal dopamine release in the nucleus accumbens and conditioned responding. Biol Psychiatry. 2010;67:737–744. doi: 10.1016/j.biopsych.2009.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koos T, Tepper JM, Wilson CJ. Comparison of IPSCs evoked by spiny and fast-spiking neurons in the neostriatum. J Neurosci. 2004;24:7916–7922. doi: 10.1523/JNEUROSCI.2163-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krashes MJ, Shah BP, Madara JC, Olson DP, Strochlic DE, Garfield AS, Vong L, Pei H, Watabe-Uchida M, Uchida N, et al. An excitatory paraventricular nucleus to AgRP neuron circuit that drives hunger. Nature. 2014;507:238–242. doi: 10.1038/nature12956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kravitz AV, Tye LD, Kreitzer AC. Distinct roles for direct and indirect pathway striatal neurons in reinforcement. Nat Neurosci. 2012;15:816–818. doi: 10.1038/nn.3100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kreitzer AC. Physiology and pharmacology of striatal neurons. Annu Rev Neurosci. 2009;32:127–147. doi: 10.1146/annurev.neuro.051508.135422. [DOI] [PubMed] [Google Scholar]
- Kreitzer AC, Malenka RC. Endocannabinoid-mediated rescue of striatal LTD and motor deficits in Parkinson’s disease models. Nature. 2007;445:643–647. doi: 10.1038/nature05506. [DOI] [PubMed] [Google Scholar]
- Lobo MK, Covington HE, 3rd, Chaudhury D, Friedman AK, Sun H, Damez-Werno D, Dietz DM, Zaman S, Koo JW, Kennedy PJ, et al. Cell type-specific loss of BDNF signaling mimics optogenetic control of cocaine reward. Science. 2010;330:385–390. doi: 10.1126/science.1188472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lovett-Barron M, Turi GF, Kaifosh P, Lee PH, Bolze F, Sun XH, Nicoud JF, Zemelman BV, Sternson SM, Losonczy A. Regulation of neuronal input transformations by tunable dendritic inhibition. Nat Neurosci. 2012;15:423–430. S421–423. doi: 10.1038/nn.3024. [DOI] [PubMed] [Google Scholar]
- Matsui A, Jarvie BC, Robinson BG, Hentges ST, Williams JT. Separate GABA afferents to dopamine neurons mediate acute action of opioids, development of tolerance, and expression of withdrawal. Neuron. 2014;82:1346–1356. doi: 10.1016/j.neuron.2014.04.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDevitt RA, Tiran-Cappello A, Shen H, Balderas I, Britt JP, Marino RA, Chung SL, Richie CT, Harvey BK, Bonci A. Serotonergic versus nonserotonergic dorsal raphe projection neurons: differential participation in reward circuitry. Cell Rep. 2014;8:1857–1869. doi: 10.1016/j.celrep.2014.08.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meng F, Xie GX, Thompson RC, Mansour A, Goldstein A, Watson SJ, Akil H. Cloning and pharmacological characterization of a rat kappa opioid receptor. Proc Natl Acad Sci U S A. 1993;90:9954–9958. doi: 10.1073/pnas.90.21.9954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meshul CK, McGinty JF. Kappa opioid receptor immunoreactivity in the nucleus accumbens and caudate-putamen is primarily associated with synaptic vesicles in axons. Neuroscience. 2000;96:91–99. doi: 10.1016/s0306-4522(99)90481-5. [DOI] [PubMed] [Google Scholar]
- Moratalla R, Xu M, Tonegawa S, Graybiel AM. Cellular responses to psychomotor stimulant and neuroleptic drugs are abnormal in mice lacking the D1 dopamine receptor. Proc Natl Acad Sci U S A. 1996;93:14928–14933. doi: 10.1073/pnas.93.25.14928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mu P, Neumann PA, Panksepp J, Schluter OM, Dong Y. Exposure to cocaine alters dynorphin-mediated regulation of excitatory synaptic transmission in nucleus accumbens neurons. Biol Psychiatry. 2011;69:228–235. doi: 10.1016/j.biopsych.2010.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muschamp JW, Van’t Veer A, Parsegian A, Gallo MS, Chen M, Neve RL, Meloni EG, Carlezon WA., Jr Activation of CREB in the nucleus accumbens shell produces anhedonia and resistance to extinction of fear in rats. J Neurosci. 2011;31:3095–3103. doi: 10.1523/JNEUROSCI.5973-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Connor EC, Kremer Y, Lefort S, Harada M, Pascoli V, Rohner C, Luscher C. Accumbal D1R Neurons Projecting to Lateral Hypothalamus Authorize Feeding. Neuron. 2015;88:553–564. doi: 10.1016/j.neuron.2015.09.038. [DOI] [PubMed] [Google Scholar]
- O’Donnell P, Grace AA. Synaptic interactions among excitatory afferents to nucleus accumbens neurons: hippocampal gating of prefrontal cortical input. J Neurosci. 1995;15:3622–3639. doi: 10.1523/JNEUROSCI.15-05-03622.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palmer LM, Schulz JM, Murphy SC, Ledergerber D, Murayama M, Larkum ME. The cellular basis of GABA(B)-mediated interhemispheric inhibition. Science. 2012;335:989–993. doi: 10.1126/science.1217276. [DOI] [PubMed] [Google Scholar]
- Pascoli V, Terrier J, Espallergues J, Valjent E, O’Connor EC, Luscher C. Contrasting forms of cocaine-evoked plasticity control components of relapse. Nature. 2014;509:459–464. doi: 10.1038/nature13257. [DOI] [PubMed] [Google Scholar]
- Planert H, Szydlowski SN, Hjorth JJ, Grillner S, Silberberg G. Dynamics of synaptic transmission between fast-spiking interneurons and striatal projection neurons of the direct and indirect pathways. J Neurosci. 2010;30:3499–3507. doi: 10.1523/JNEUROSCI.5139-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pliakas AM, Carlson RR, Neve RL, Konradi C, Nestler EJ, Carlezon WA., Jr Altered responsiveness to cocaine and increased immobility in the forced swim test associated with elevated cAMP response element-binding protein expression in nucleus accumbens. J Neurosci. 2001;21:7397–7403. doi: 10.1523/JNEUROSCI.21-18-07397.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlosburg JE, Whitfield TW, Jr, Park PE, Crawford EF, George O, Vendruscolo LF, Koob GF. Long-term antagonism of kappa opioid receptors prevents escalation of and increased motivation for heroin intake. J Neurosci. 2013;33:19384–19392. doi: 10.1523/JNEUROSCI.1979-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen W, Flajolet M, Greengard P, Surmeier DJ. Dichotomous dopaminergic control of striatal synaptic plasticity. Science. 2008;321:848–851. doi: 10.1126/science.1160575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shirayama Y, Ishida H, Iwata M, Hazama GI, Kawahara R, Duman RS. Stress increases dynorphin immunoreactivity in limbic brain regions and dynorphin antagonism produces antidepressant-like effects. J Neurochem. 2004;90:1258–1268. doi: 10.1111/j.1471-4159.2004.02589.x. [DOI] [PubMed] [Google Scholar]
- Sohal VS, Zhang F, Yizhar O, Deisseroth K. Parvalbumin neurons and gamma rhythms enhance cortical circuit performance. Nature. 2009;459:698–702. doi: 10.1038/nature07991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stuber GD, Sparta DR, Stamatakis AM, van Leeuwen WA, Hardjoprajitno JE, Cho S, Tye KM, Kempadoo KA, Zhang F, Deisseroth K, Bonci A. Excitatory transmission from the amygdala to nucleus accumbens facilitates reward seeking. Nature. 2011;475:377–380. doi: 10.1038/nature10194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Svingos AL, Colago EE, Pickel VM. Cellular sites for dynorphin activation of kappa-opioid receptors in the rat nucleus accumbens shell. J Neurosci. 1999;19:1804–1813. doi: 10.1523/JNEUROSCI.19-05-01804.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tejeda HA, Counotte DS, Oh E, Ramamoorthy S, Schultz-Kuszak KN, Backman CM, Chefer V, O’Donnell P, Shippenberg TS. Prefrontal Cortical Kappa-Opioid Receptor Modulation of Local Neurotransmission and Conditioned Place Aversion. Neuropsychopharmacology. 2013 doi: 10.1038/npp.2013.76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tejeda HA, Hanks AN, Scott L, Mejias-Aponte C, Hughes ZA, O’Donnell P. Prefrontal Cortical Kappa Opioid Receptors Attenuate Responses to Amygdala Inputs. Neuropsychopharmacology. 2015;40:2856–2864. doi: 10.1038/npp.2015.138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tejeda HA, Shippenberg TS, Henriksson R. The dynorphin/kappa-opioid receptor system and its role in psychiatric disorders. Cell Mol Life Sci. 2012;69:857–896. doi: 10.1007/s00018-011-0844-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tepper JM, Koos T, Wilson CJ. GABAergic microcircuits in the neostriatum. Trends Neurosci. 2004;27:662–669. doi: 10.1016/j.tins.2004.08.007. [DOI] [PubMed] [Google Scholar]
- Tunstall MJ, Oorschot DE, Kean A, Wickens JR. Inhibitory interactions between spiny projection neurons in the rat striatum. J Neurophysiol. 2002;88:1263–1269. doi: 10.1152/jn.2002.88.3.1263. [DOI] [PubMed] [Google Scholar]
- Van’t Veer A, Bechtholt AJ, Onvani S, Potter D, Wang Y, Liu-Chen LY, Schutz G, Chartoff EH, Rudolph U, Cohen BM, Carlezon WA., Jr Ablation of kappa-opioid receptors from brain dopamine neurons has anxiolytic-like effects and enhances cocaine-induced plasticity. Neuropsychopharmacology. 2013;38:1585–1597. doi: 10.1038/npp.2013.58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van’t Veer A, Carlezon WA., Jr Role of kappa-opioid receptors in stress and anxiety-related behavior. Psychopharmacology (Berl) 2013;229:435–452. doi: 10.1007/s00213-013-3195-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang JQ, Daunais JB, McGinty JF. NMDA receptors mediate amphetamine-induced upregulation of zif/268 and preprodynorphin mRNA expression in rat striatum. Synapse. 1994;18:343–353. doi: 10.1002/syn.890180410. [DOI] [PubMed] [Google Scholar]
- Wee S, Koob GF. The role of the dynorphin-kappa opioid system in the reinforcing effects of drugs of abuse. Psychopharmacology (Berl) 2010;210:121–135. doi: 10.1007/s00213-010-1825-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson CJ. GABAergic inhibition in the neostriatum. Prog Brain Res. 2007;160:91–110. doi: 10.1016/S0079-6123(06)60006-X. [DOI] [PubMed] [Google Scholar]
- Yamaguchi T, Wang HL, Li X, Ng TH, Morales M. Mesocorticolimbic glutamatergic pathway. J Neurosci. 2011;31:8476–8490. doi: 10.1523/JNEUROSCI.1598-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.