

Abstract
Deficiency of β-Glucocerebrosidase (GBA) activity causes Gaucher Disease (GD). GD can be diagnosed by measuring GBA activity [1] Beutler and Kuhl 1990. In this study, we assayed dried blood spots from a cohort (n=528) enriched for GBA mutation carriers (n=78) and GD patients (n=18) using both the tandem mass spectrometry (MS/MS) and fluorescence assays and their respective synthetic substrates. The MS/MS assay differentiated normal controls, which included GBA mutation carriers, from GD patients with no overlap. The fluorescence assay did not always differentiate normal controls including GBA mutation carriers from GD patients and false positives were observed. The MS/MS assay improved specificity compared to the fluorescence assay.
Keywords: Dried blood spots (DBS), Gaucher Disease (GD), Newborn Screening (NBS), β-Glucocerebrosidase (GBA), Glucocerebroside, Lysosomal Storage Disorder (LSD)
1. Introduction
Gaucher disease (GD; OMIM #230800, 230900, 231000) is an autosomal recessive lysosomal storage disorder (LSD) characterized by β-Glucocerebrosidase deficiency (GBA; EC 3.2.1.45; Acid β-D-Glucosidase; Glucosylceramidase) and the accumulation of its substrate Glucocerebroside (GL-1; Glucosylceramide) in lysosomes [2, 3]. The prevalence of GD ranges from ~1/1,000 live births in Ashkenazi Jewish populations to ~1:50,000 in the general population [4–6]. The progressive nature of GD and the availability of enzyme replacement and substrate reduction therapies create a need for early and accurate diagnosis of patients with GD [7–13]. However, the rarity and clinical heterogeneity of GD leads to diagnostic challenges [3, 14–17]. This study compared two assays used for screening GBA activity in DBS, the MS/MS assay and the fluorescence assay. The MS/MS assay was developed by Li et al and optimized by Zhang et al. [18, 19] and the fluorescence assay was originally developed by Chamoles et al. and optimized by Olivova et al. [20, 21].
Each assay employs a different synthetic substrate instead of the native GBA substrate Cn-Glucocerebroside (n=16–24) [22]. The MS/MS assay uses C12-Glucocerebroside, a synthetic analog of the native substrate with a shorter fatty acyl chain. The substrate is cleaved by GBA extracted from the DBS sample to produce a C12-ceramide product. The product is quantified against a C14-ceramide internal standard by MS/MS [18, 19]. The fluorescence assay utilizes the synthetic substrate 4-methylumbelliferyl β-D-glucopyranoside (4-MUG). GBA hydrolyses 4-MUG to release a fluorescent 4-methylumbelliferon (4-MU) product, which is quantified by fluorometer [20, 21]. The synthetic substrates are outlined in Figure 1.
Figure 1. Synthetic substrates schema.
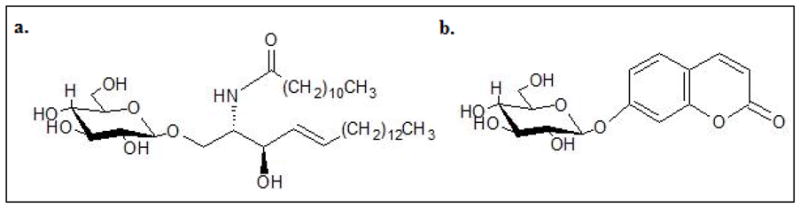
a. C12-Glucocerebroside used in the MS/MS assay b. 4-methylumbelliferyl β-D-glucopyranoside used in the fluorescence assay.
2. Materials and Methods
2.1 Sample collection
Specimens were collected with written informed consent at Columbia University and at Sanofi Genzyme (Cambridge, MA) according to procedure established at Sanofi Genzyme [23]. The blood was drawn into 10 mL BD Vacutainer Blood Collection Tubes with K2EDTA (Franklin Lakes, NJ) and 75 μL aliquots were spotted onto Whatman 903 filter paper (GE Healthcare, Piscataway, NJ). Blood spots were dried for a minimum of 4 h at room temperature. Dried sample cards were stored in sealed plastic bags at −20 °C with a desiccant and a humidity indicator. Cards were shipped at room temperature and stored at −80 °C until analysis.
2.2 Cohort Description and Clinical Status
The full cohort (n=528) contained control DBS (n=510) and GD DBS (n=18). Of these, 515 samples were collected at Columbia University in the “Spot” study, which tested the association between GBA mutations, GBA activity and Parkinson’s disease [24]. The 515 participants signed an informed consent to both enzyme activity analysis at Sanofi Genzyme and genotyping. The remaining 13 DBS samples came from Sanofi Genzyme collection of GD patients who consented to GBA enzyme activity analysis but not genotyping. After genotyping, the control group was further subdivided by GBA genotype into normal DBS (n=432) and carriers (n=78). The control group was defined as unaffected with respect to GD, but some individuals had Parkinson’s disease as described in Alcalay et al. [24].
2.3 Genetic Testing
Samples from the 515 individuals with proper consent were genotyped in two ways. DNA was extracted using a standard salting-out method and all participants were genotyped for eight GBA mutations (p.N409S, p.L483P, p.L29Afs*18, ivs2+1G>A, p.D448G, p.V433L and p.R535H, and the p.[L483P; A495P; V499V] recombinant allele; legacy nomenclature: N370s, L444P, 84GG, ivs2+1G>A, D409G, V394L, R535H and the RecNciI recombinant allele, respectively) and two GBA variants (p.E365K and p.T408M, legacy nomenclature: E326K and T369M, respectively) as previously described [25, 26]. In addition, the GBA locus was fully sequenced in a second round of genotyping as described in Alcalay et al. [24]. The samples were blinded throughout GBA activity analysis.
2.4 GBA Enzyme Activity by Fluorescence Assay
GBA activity was measured as previously described [20] Briefly, one 3.2 mm-diameter punch from each DBS sample was extracted in 200 ;L 0.2 M citrate phosphate buffer, pH 5.2 containing 1% Triton® X-100 (Sigma, St. Louis, MO) and 1% sodium taurodeoxycholate (≥97% purity, Sigma, St. Louis MO) in a 96-well plate. Substrate solution (12.5 mM 4-MUG, Sigma, St. Louis, MO) was prepared either with or without inhibitor (0.5 mM Conduritol B Epoxide, Toronto Research Chemicals). Inhibited or uninhibited substrate solution was mixed 2:1 with DBS extract and incubated for 20 h at 37 °C. To stop the reactions, 100 μL of 0.5 M EDTA (pH 11.5) was added to each well. An eight point 4-MU standard curve (0 – 0.67 μM) was prepared on each plate in duplicate. The plate was read in a fluorometer at 355 nm excitation and 460 nm emission wavelengths. GBA activity was determined by subtracting the background activity measured in the inhibited reaction from that in the uninhibited reaction. Disease cut-off (1.71 μmol/L/h) was established as described in methods section 2.6. The limit of blank (LOB=0.16 μmol/L/h) and limit of detection (LOD=0.41 μmol/L/h) for the fluorescence assay were established previously [20].
2.5 GBA Enzyme Activity by MS/MS Assay
GBA activity was measured as part of an established MS/MS multiplex assay as previously described [18]. The GBA extract from one 3.2 mm punch per DBS sample was combined with C12-glucocerebroside substrate and C14-ceramide internal standard mixtures (The Center for Disease Control and Prevention, Atlanta, Georgia). Sealed 96-well plates were incubated on an orbital shaker at 37°C for 20 h and cleaned up according to the published protocol. Dried sample plates were stored at −20 °C. Prior to MS/MS analysis, plates were thawed and reconstituted with 200 μl of mobile phase (80:20 acetonitrile in water containing 0.2% formic acid). All analytes were monitored on an API 4000 triple quadrupole mass spectrometer (ABSciex, Framingham, Massachusetts, USA) by Multiple Reaction Monitoring (MRM). The enzyme activity of each sample was calculated from the ion abundance ratio of product to internal standard as measured by the mass spectrometer. Background activity of a blank filter paper was subtracted from the DBS activity. Activity was expressed as micromoles of product per liter of whole blood per hour (μmol/l/h). Two QC samples with previously established activity levels for each enzyme and disease positive samples were included in each plate as sample controls. The LOB=0.32 μmol/L/h and LOD=0.5 μmol/L/h for the MS/MS assay were established previously.
2.6 Statistical Analysis and Disease Cut-off
All statistical analysis was performed using the SAS v.9.4 software (SAS Enterprise, Cary, NC). A cut-off point distinguishing the control (normal + carrier) group and the GD group was derived by fitting a normal distribution to the control group (Fcontrol) and the GD group (Fdisease) using a direct estimation of each mean and standard deviation. The cut-off point χcut-off was then defined as the value with equal probability to occur in both, the control and the disease distribution:
where fcontrol and fdisease denoted the probability density function for Fcontrol and Fdisease. Any value χ ≤ χcut-off was more likely to occur in the disease group distribution and any value χ > χcut-off was more likely to occur in the control group.
The GD cut-off for the fluorosecence assay was established as 1.71 μmol/L/h. The GD cut-off for the MS/MS assay was established as 1.49 μmol/L/h.
3. Results and Discussion
The study cohort (n=528) contained control DBS samples (n=510) and GD samples (n=18). The full GBA gene was sequenced in consented samples (n=515) as described in methods. After genotyping, the control group was further subdivided into normal DBS (n=432) and GBA carriers (n=78). The carrier group included any heterozygous GBA sequence variation including non-coding mutations regardless of disease relevance. Table 1 outlines the cohort and genotyping results.
Table 1. Cohort description and genotyping results.
DBS samples were divided by type (control and GD) and further subdivided by genotype (normal, carrier and Gaucher). As noted the genotyping was not performed for 13 of the GD patients from the Sanofi Genzyme collection. Variations in GBA sequence are described in legacy and HGVS nomenclature [27].
| Sample | Mutation or variant nomenclature | Count (n) | |||
|---|---|---|---|---|---|
| Type | GBA Genotype | Legacy (name) | HGVS (protein) | HGVS (nucleotide) | |
| Control n=510 | Normal n=432 | none detected in GBA sequence | 432 | ||
| Carrier n=78 | N370S | p.N409S | c.1226A>G | 35 | |
| L444P | p.L483P | c.1448T>C | 4 | ||
| R496H | p.R535H | c.1604G>A | 4 | ||
| n/a | n/a | c.( −203)A>G1 | 4 | ||
| 84GG | p.L29Afs*18 | c.84dupG | 3 | ||
| ivs2+1G>A | r.(spl?) | c.115+1G>A | 2 | ||
| K-27R | p.K13R | c.38A>G | 2 | ||
| E326K | p.E365K | c.1093G>A | 8 | ||
| T369M | p.T408M | c.1223C>T | 7 | ||
| G202R | p.G241R | c.721G>A | 1 | ||
| V294M | p.V333M | c.997G>A | 1 | ||
| N392S | p.N431S | c.1292A>G | 1 | ||
| T410M | p.T449M | c.1346C>T | 1 | ||
| A456P | p.A495P | c.1483G>C | 1 | ||
| F-37V | p.F3V | c.7T>G | 1 | ||
| Q-8H | p.Q32H | c.96G>C | 1 | ||
| L461P | p.L500P | c.1499T>C | 1 | ||
| S110A | p.S149A | c.445T>G | 1 | ||
| Disease n=18 | Gaucher n=18 | N370S/N370S | p.N409S/p.N409S | c.1226A>G/c.1226A>G | 4 |
| N370S/L444P+A456P | p.N409S/p.[L483P; A495P] | c.1226A>G/c.1448T>C; c.1483G>C2 | 1 | ||
| no genotype available | 13 | ||||
GBA activity was measured in all samples by both the MS/MS and the fluorescence assays. Activity results from the two assays are correlated in Figure 2. The mean activity of the normal and carrier DBS was significantly different (p<0.0001) Table 2, but neither assay could differentiate the carrier and normal DBS populations as expected. The correlation coefficient of the two assays R = 0.8073.
Figure 2. Fluorescence and mass spectrometry assay correlation.

The GBA activity levels in control DBS (normal DBS plus GBA carriers) and GD DBS samples as measured by the MS/MS and the fluorescence (4-MU) assays. The red dotted lines indicates LOD for each assay as described in methods. R=0.8073.
Table 2.
GBA activity distribution, mean and standard deviation in μmol/L/h as measured by the MS/MS and fluorescence assays. LODMS/MS = 0.5 (μmol/L/h).
| Full Cohort n=528 | GBA Activity (μmol/L/h) | ||||
|---|---|---|---|---|---|
| MS/MS Assay | Fluorescence Assay | ||||
| Range | Mean (SD) | Range | Mean (SD) | ||
| Control n=510 | Normal n=432 | 5.07 – 27.63 | 12.11 (3.27) | 1.55 – 9.20 | 3.90 (1.07) |
| Carrier n=78 | 3.32 – 16.37 | 7.93 (2.65) | 0.74 – 5.04 | 2.71 (0.85) | |
| Disease n=18 | Gaucher n=18 | < LOD – 1.25 | 0.40 (0.30) | 0.42 – 1.66 | 0.88 (0.35) |
The mean GBA activity measured in normal controls, GBA carriers and GD DBS by the MS/MS and the fluorescence assays is listed in Table 2. Seven of the GD samples had activity levels below the LOD of the MS/MS assay (0.5 μmol/L/h), while all GD samples tested above the LOD of the 4-MU GBA assay (0.41 μmol/L/h). As expected, the GBA activity of all controls (normal DBS plus carriers) was significantly different from GD DBS activity in both assays (p<0.0001).
The GBA activity distribution span a greater range in the MS/MS assay compared to the fluorescence assay. The GBA activity measured by the MS/MS assay showed a clear separation between GD and normal control samples including GBA mutation carriers as shown in Figure 3. In contrast, the activity distribution measured by the fluorescence assay was continuous between the groups and false positives were observed. Specifically, eight GBA mutation carriers and two normal controls were tested below the GD cut-off of 1.71 μmol/L/h in the fluorescence assay. The GBA activity measured in carriers of specific mutations is reported in Supplementary Tables 1 and 2.
Figure 3. Frequency distribution of GBA enzyme activities in each assay.

a. GBA activity distribution in the MS/MS assay. b. GBA activity distribution in the fluorescence assay.
Both assays showed 100% sensitivity as they correctly detected all 18 GD samples as true positives. The MS/MS assay also showed 100% specificity, while the fluorescence assay specificity was slightly lower at 98.1%. The enzyme activity of GBA carriers clustered at the lower end of the normal activity distribution as observed previously [19, 21]. For GBA carriers in the MS/MS assay the 95% confidence interval (CI) = 2.7 – 13.1 compared to 5.7 – 18.5 for normal controls. For GBA carriers in the fluorescence assay the 95% CI = 1.0 – 4.4 compared to 1.8 – 6.0 for normal controls.
Assay characteristics such as reaction pH, time and temperature are similar in both assays. The difference in the specificity of the two assays may result from the structural differences of their respective synthetic substrates. The MS/MS assay uses a close structural analogue of the natural substrate. The ceramide moiety of the C12-Glucocerebroside substrate differs from the natural substrate only in the length of the fatty acid chain [19, 22]. In addition, the β-linked glucose head group known to bind the active site of GBA is flexible in the C12-Glucocerebroside and rigid in the 4-MUG substrate. This may affect the substrate enzyme configuration during GBA hydrolysis.
The difference between the fluorescence assay and MS/MS assay may be important to newborn screening and diagnostic laboratories [30–32]. False positives observed in GD screening with the fluorescence assay have been reported previously [33, 34]. False-positive test results can cause anxiety to patients and increase costs to the healthcare system. Therefore, borderline activity results in the fluorescence assay should be followed by genotyping.
4. Conclusion
This study demonstrated that the MS/MS GBA activity assay has higher specificity than the fluorescence GBA assay. Since other assay characteristics were similar in both assays, the structural features of the substrates might contribute to the observed differences. Both assays are suitable for GBA activity screening, but borderline results in the fluorescence assay should be followed by genotyping.
Supplementary Material
Acknowledgments
Funding for this study provided by the Parkinson’s Foundation, the National Institutes of Health (K02NS080915 and UL1 TR000040, formerly the National Center for Research Resources, Grant Number UL1 RR024156), and the Brookdale Foundation. The authors would like to thank Karina Sakanaka BS and Paul Greene MD for patient referral and recruitment, and Ms. Judy Hull for facilitating the research collaboration.
References
- 1.Beutler E, Kuhl B. In: Beta-glucosidase. 4. Williams WJ, et al., editors. McGraw-Hill Information Services Co., Health Professions Division; 1990. [Google Scholar]
- 2.Brady RO, et al. Demonstration of a deficiency of glucocerebroside-cleaving enzyme in Gaucher’s disease. J Clin Invest. 1966;45(7):1112–5. doi: 10.1172/JCI105417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Grabowski GA, et al. Gaucher Disease: Phenotypic and Genetic Variation. In: Beaudet AL, et al., editors. The Online Metabolic and Molecular Bases of Inherited Disease. The McGraw-Hill Companies, Inc; New York, NY: 2014. [Google Scholar]
- 4.Meikle PJ, et al. Prevalence of lysosomal storage disorders. Jama. 1999;281(3):249–54. doi: 10.1001/jama.281.3.249. [DOI] [PubMed] [Google Scholar]
- 5.Weinreb NJ, et al. Prevalence of type 1 Gaucher disease in the United States. Arch Intern Med. 2008;168(3):326–7. doi: 10.1001/archinternmed.2007.128. author reply 327–8. [DOI] [PubMed] [Google Scholar]
- 6.Grabowski GA, Petsko GA, Kolodny EH. Gaucher Disease. In: Beaudet AL, et al., editors. The Online Metabolic and Molecular Bases of Inherited Disease. The McGraw-Hill Companies, Inc; New York, NY: 2014. [Google Scholar]
- 7.Cox TM, et al. Eliglustat maintains long-term clinical stability in patients with Gaucher disease type 1 stabilized on enzyme therapy. Blood. 2017;129(17):2375–2383. doi: 10.1182/blood-2016-12-758409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cox TM, et al. Eliglustat compared with imiglucerase in patients with Gaucher’s disease type 1 stabilised on enzyme replacement therapy: a phase 3, randomised, open-label, non-inferiority trial. Lancet. 2015;385(9985):2355–62. doi: 10.1016/S0140-6736(14)61841-9. [DOI] [PubMed] [Google Scholar]
- 9.Lukina E, et al. Eliglustat, an investigational oral therapy for Gaucher disease type 1: Phase 2 trial results after 4years of treatment. Blood Cells Mol Dis. 2014 doi: 10.1016/j.bcmd.2014.04.002. [DOI] [PubMed] [Google Scholar]
- 10.Mistry PK, et al. Osteopenia in Gaucher disease develops early in life: response to imiglucerase enzyme therapy in children, adolescents and adults. Blood Cells Mol Dis. 2011;46(1):66–72. doi: 10.1016/j.bcmd.2010.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pleat R, et al. Stability is maintained in adults with Gaucher disease type 1 switched from velaglucerase alfa to eliglustat or imiglucerase: A sub-analysis of the eliglustat ENCORE trial. Mol Genet Metab Rep. 2016;9:25–28. doi: 10.1016/j.ymgmr.2016.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sims KB, et al. Improvement of bone disease by imiglucerase (Cerezyme) therapy in patients with skeletal manifestations of type 1 Gaucher disease: results of a 48-month longitudinal cohort study. Clin Genet. 2008;73(5):430–40. doi: 10.1111/j.1399-0004.2008.00978.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Weinreb NJ, et al. Long-term clinical outcomes in type 1 Gaucher disease following 10 years of imiglucerase treatment. J Inherit Metab Dis. 2013;36(3):543–53. doi: 10.1007/s10545-012-9528-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hruska KS, et al. Gaucher disease: mutation and polymorphism spectrum in the glucocerebrosidase gene (GBA) Hum Mutat. 2008;29(5):567–83. doi: 10.1002/humu.20676. [DOI] [PubMed] [Google Scholar]
- 15.Sidransky E. Gaucher disease: complexity in a “simple” disorder. Mol Genet Metab. 2004;83(1–2):6–15. doi: 10.1016/j.ymgme.2004.08.015. [DOI] [PubMed] [Google Scholar]
- 16.Mistry PK, et al. A reappraisal of Gaucher disease-diagnosis and disease management algorithms. Am J Hematol. 2011;86(1):110–5. doi: 10.1002/ajh.21888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cassinerio E, Graziadei G, Poggiali E. Gaucher disease: a diagnostic challenge for internists. Eur J Intern Med. 2014;25(2):117–24. doi: 10.1016/j.ejim.2013.09.006. [DOI] [PubMed] [Google Scholar]
- 18.Zhang XK, et al. Multiplex lysosomal enzyme activity assay on dried blood spots using tandem mass spectrometry. Methods Mol Biol. 2010;603:339–50. doi: 10.1007/978-1-60761-459-3_32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Li Y, et al. Direct multiplex assay of lysosomal enzymes in dried blood spots for newborn screening. Clin Chem. 2004;50(10):1785–96. doi: 10.1373/clinchem.2004.035907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Olivova P, et al. An improved high-throughput dried blood spot screening method for Gaucher disease. Clin Chim Acta. 2008;398(1–2):163–4. doi: 10.1016/j.cca.2008.08.024. [DOI] [PubMed] [Google Scholar]
- 21.Chamoles NA, et al. Gaucher and Niemann-Pick diseases--enzymatic diagnosis in dried blood spots on filter paper: retrospective diagnoses in newborn-screening cards. Clin Chim Acta. 2002;317(1–2):191–7. doi: 10.1016/s0009-8981(01)00798-7. [DOI] [PubMed] [Google Scholar]
- 22.Ishibashi Y, Kohyama-Koganeya A, Hirabayashi Y. New insights on glucosylated lipids: metabolism and functions. Biochim Biophys Acta. 2013;1831(9):1475–85. doi: 10.1016/j.bbalip.2013.06.001. [DOI] [PubMed] [Google Scholar]
- 23.Elbin CS, et al. The effect of preparation, storage and shipping of dried blood spots on the activity of five lysosomal enzymes. Clin Chim Acta. 2011;412(13–14):1207–12. doi: 10.1016/j.cca.2011.03.012. [DOI] [PubMed] [Google Scholar]
- 24.Alcalay RN, et al. Glucocerebrosidase activity in Parkinson’s disease with and without GBA mutations. Brain. 2015;138(Pt 9):2648–58. doi: 10.1093/brain/awv179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Alcalay RN, et al. Comparison of Parkinson risk in Ashkenazi Jewish patients with Gaucher disease and GBA heterozygotes. JAMA Neurol. 2014;71(6):752–7. doi: 10.1001/jamaneurol.2014.313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Alcalay RN, et al. Parkinson disease phenotype in Ashkenazi Jews with and without LRRK2 G2019S mutations. Mov Disord. 2013;28(14):1966–71. doi: 10.1002/mds.25647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.den Dunnen JT, et al. HGVS Recommendations for the Description of Sequence Variants: 2016 Update. Hum Mutat. 2016;37(6):564–9. doi: 10.1002/humu.22981. [DOI] [PubMed] [Google Scholar]
- 28.Alfonso P, et al. Characterization of the c.(−203)A>G variant in the glucocerebrosidase gene and its association with phenotype in Gaucher disease. Clin Chim Acta. 2011;412(3–4):365–9. doi: 10.1016/j.cca.2010.11.013. [DOI] [PubMed] [Google Scholar]
- 29.Cormand B, et al. A new gene-pseudogene fusion allele due to a recombination in intron 2 of the glucocerebrosidase gene causes Gaucher disease. Blood Cells Mol Dis. 2000;26(5):409–16. doi: 10.1006/bcmd.2000.0317. [DOI] [PubMed] [Google Scholar]
- 30.Liao HC, et al. Mass Spectrometry but Not Fluorimetry Distinguishes Affected and Pseudodeficiency Patients in Newborn Screening for Pompe Disease. Clin Chem. 2017;63(7):1271–1277. doi: 10.1373/clinchem.2016.269027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Millington DS, Bali DM. Misinformation regarding tandem mass spectrometric vs fluorometric assays to screen newborns for LSDs. Molecular Genetics and Metabolism Reports. 2017;11(Supplement C):72–73. doi: 10.1016/j.ymgmr.2017.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gelb MH, et al. Comparison of tandem mass spectrometry to fluorimetry for newborn screening of LSDs. Mol Genet Metab Rep. 2017;12:80–81. doi: 10.1016/j.ymgmr.2017.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Stroppiano M, et al. Validity of beta-d-glucosidase activity measured in dried blood samples for detection of potential Gaucher disease patients. Clin Biochem. 2014;47(13–14):1293–6. doi: 10.1016/j.clinbiochem.2014.06.005. [DOI] [PubMed] [Google Scholar]
- 34.Liao HC, et al. Detecting multiple lysosomal storage diseases by tandem mass spectrometry - A national newborn screening program in Taiwan. Clin Chim Acta. 2014;431c:80–86. doi: 10.1016/j.cca.2014.01.030. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.