

Abstract
Properties of di(triethylene glycol monomethyl ether) squarate relevant to conjugation of carbohydrates to proteins have been reinvestigated and compared with those of dimethyl squarate. It is concluded that the commercially available, crystalline dimethyl squarate remains the most convenient and efficient reagent for conjugation of amine-containing carbohydrates to proteins by a two-step or one-pot conjugation protocol.
Keywords: Glycoconjugates, squarate, squaric acid chemistry, hydrolytic stability
Graphical Abstract
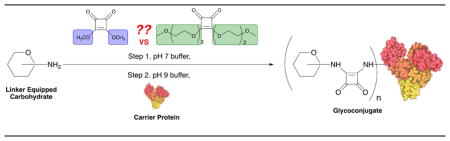
The method for coupling two amines through the squaric acid residue was introduced in Germany by Tietze’s group in 1991 [1]. The conjugation takes place in two steps (Scheme 1). First, at neutral conditions, the first amine reacts with a squaric acid diester to form a monoamide monoester. The latter can react at basic conditions, for example pH 9 [1]. with the same or a different amine to form a diamide. The same laboratory soon recognized [2] that when one of the amines is a functionalized carbohydrate and the other one is a protein important tools in the life sciences, the neoglycoconjugates, could be obtained. The potential of the method was initially not duly recognized, and it took quite a while before this method was rediscovered [3] and then became widely used. Currently, the squaric acid chemistry-based conjugation is considered one of the most powerful methods for making glycoconjugates [4–6]. Many experimental vaccines and tools for the life sciences have been prepared in this way. Although other squaric acid diesters have been used [7, 8], dimethyl (e.g. Ref. [9–12]) and diethyl (e.g Ref. [13–22]) squarates have been the most popular reagents in this regard.
Scheme 1.

Conjugation of amines by squaric acid diester chemistry
We concluded [11] previously that among squarate diesters currently in use, the commercially available dimethyl squarate (1, Fig. 1) was the most convenient reagent for making glycoconjugates. Here, the term ‘squarate reagent’ represents squaric acid diesters and the term ‘conjugation reagent’ is used to describe monoamide monoesters formed from a diester and an amine.
Fig. 1.
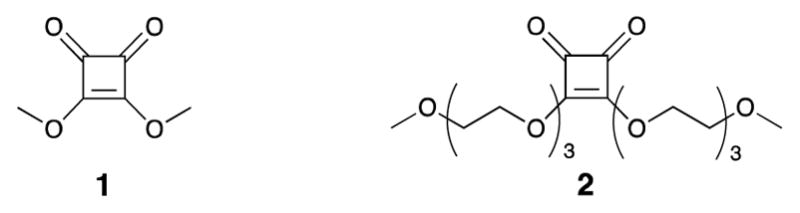
Structures of squaric acid dialkyl esters 1 and 2
When treated with squarate reagents, amines readily yield conjugation reagents which are, unfortunately, prone to saponification at the conditions of conjugation (pH ≥ 9). Thus, conjugation requires use of variable excess of the monoester reagent. When conjugating small oligosaccharides, the rate of the conjugation is relatively high, and the reaction usually takes only a few hours to complete. Therefore, large excess of the conjugation reagent is not required. However, when larger oligosaccharides or polysaccharides are being converted, because of the slower reaction rate, considerable excess of the conjugation reagent is necessary, which eventually ends up as expensive waste.
Wurm et al. [23] have introduced a novel squaric acid-based water-soluble squarate reagent, squaric acid di(tri(ethylene glycol) monomethyl ether)ester (2, Fig. 1). They describe 2 as less prone to hydrolysis than 1 and used the former in a one-pot ligand–protein conjugation in aqueous reaction medium. While one-pot conjugation in water was reported before [10], we were intrigued by the claimed outstanding hydrolytic stability of conjugation reagents made from 2. Having a more stable conjugation reagent would be beneficial in connection with our squaric acid chemistry-driven development of glycoconjugate vaccines from synthetic [12, 24] and particularly bacterial [22, 25, 26] carbohydrates.
Careful examination of the just cited communication [23] revealed that the studies with 2, including hydrolysis kinetics, were performed with crude, unpurified material. This prompted us to firstly, prepare reagent 2 in pure state (c.f. Fig. 2 for comparison of the relevant part of the 1H NMR spectra of 2 [23] with the reagent prepared as described here) and characterize it, secondly, to verify the hydrolytic stability of conjugation reagent prepared from 2 at the conditions of conjugation and, thirdly, compare the utility of the conjugation reagents made from 1 and 2 for making neoglycoconjugates.
Fig. 2.

Comparison of the 1H NMR spectra of 2. A Material reported.[23] B Material described here.
Accordingly, compound 2 was prepared from squaric acid (3) and alcohol 4 as described [23] (Scheme 2) and obtained in the analytically pure state, after chromatography. It is worth mentioning that due to its high boiling point, compound 4 used in excess during the preparation could not be completely removed without chromatography. That also afforded a small amount of 5, which originated from the impurity in the commercial reagent used. Thus, chromatography, which is in this case not a trivial task and was not included in the previous protocol[23], is necessary in order to obtain compound 2 in the pure state. Alcoholic solvents should be avoided during chromatography, to prevent formation of transesterification products. When MeOH was used for purification, variable amount of by-product 6 was isolated. Eventually, MeCN–CH2Cl2 was used for the purification of 2.
Scheme 2.

Synthesis of squarate reagent 2.
A comparison of literature data [23, 27] shows that the hydrolytic stability of squarates 1 and 2 is comparable. About 90% of 1 was found to hydrolyze to the corresponding monoacid after 16 h in 0.5 M pD 7 buffer [27], while it was reported [23] that over 70% of 2 hydrolyzed after ~ 13 h when kept at pD 7 (NMR). However, because both 1 and 2 are relatively cheap commodities, the stability of conjugation reagents at the conditions of conjugation is much more important than that of dialkyl squarate reagents. Therefore, we next evaluated the hydrolytic stability of conjugation reagents prepared from 1 and 2. Wurm et al. reported [23] that conjugation reagent 7 (Fig. 3) made from compound 2 has an estimated half-life in 0.01 M pD 9.5 buffer of 11–12 days. They concluded, from the known [11] half-life data for 8 (2–3 days) that the squarate monoester amides based on squaric acid di(tri(ethylene glycol) monomethyl ether)ester 2 are more stable than those based on 1, and are, therefore, more useful conjugation reagents. However, the comparison presented and the conclusions they made [23] are unsound because the hydrolytic stabilities of the two families of conjugation reagents were determined at different conditions [11, 23]. When Hou et al. tested the stability of 8 (Fig. 3) [11] the reaction medium was a 0.5 M pH 9.0 buffer, where the pH remained relatively stable during the course of the hydrolysis. However, Wurm et al. performed the stability experiments in 0.01 M pH 9.5 buffer. Considering the amount of 7 (43 μmol) and the amount of buffer used (0.7 mL), the capacity of the buffer must have gradually become insufficient as the hydrolysis of 7 progressed. As a result, the pD must have fallen well below 9.5 (this was later proved experimentally, see below). Also, it is not clear why they determined the stability of 7 in 0.01 M buffer (pH 9.5) when they performed the conjugation with the same reagent [23] in 0.1 M buffer (pH 9.1).
Fig. 3.

Structures of conjugation reagents 7 and 8, and byproduct 6.
In order to objectively compare the hydrolytic stability of the two conjugation reagents discussed, we determined stability of conjugation reagents 8 and 10 at pH 9.5 in 0.01 M buffer [23] and also at the conjugation conditions we normally use (0.5 M pH 9.0 buffer). Compound 10 was obtained by derivatization of amine 9 [11] with squarate reagent 2 (Scheme 3). The reaction was uneventful and compound 10 was obtained in 70% yield. In parallel experiments, reagents 8 and 10 were treated under the two different conditions just mentioned (Fig. 4). The progress of the hydrolyses was judged (1H NMR) by monitoring the decrease of the intensity of the squarate ester peaks at δ ~ 4.3 (the signal for the vinylogous methyl squarate group in the squarate derivative 8 appears as two singlets [28]) and at 3.4 ppm (singlet for the methyl group at position 19′ in 10).
Scheme 3.

Preparation of conjugation reagent 10 and its conjugation with BSA.
Fig. 4.

Hydrolytic stability comparison for compound 8 and 10.
As can be seen in Fig. 4, the stability of compounds 8 and 10 is virtually identical; both compounds hydrolyze at a much faster rate in 0.5 M pH 9.0 buffer than in 0.01 M pH 9.5 buffer (see also Experimental). Consequently, considering the hydrolytic stability, there is no advantage to using 10 over 8.
Next, we compared the rate of conjugation to BSA using the two reagents 8 and 10 (Fig. 5). Conjugation with both 8 and 10 is much faster in 0.5 M pH 9.0 buffer than in 0.01 M pH 9.5 buffer [23]. Because both reagents hydrolyze relatively quickly at higher pH, the pH in the mixture conducted in 0.01 M buffer dropped considerably, due to the weak buffer capacity (from 9.5 to 8.6 after 9.5 h and to 8.0 after 48 h). When stronger, 0.5 M pH 9.0 buffer was used, the pH remained almost unchanged (pH 8.8 after 48 h). In parallel conjugations with reagents 8 and 10 in 0.5 M pH 9.0 buffer using ligand–BSA molar ratio = 15, both reactions produced conjugate products within 24 h, with virtually all ligand attached to the protein. For comparison, when conjugations were performed in 0.01 M pH 9.5 buffer with the same initial ligand-BSA ratio, the loading ≤ 10 was reached after 120 h (Fig. 5). Reactions in both buffers were slightly faster involving methyl squarate 8 than those using 10.
Fig. 5.

Conjugation of 8 and 10 to BSA under different conditions.
Lastly, we confirmed the previous showing [10] of the utility of methyl squarate conjugation reagents in one-pot conjugation. In this context, we feel compelled to mention that the emphasis on the novelty of one-pot conjugation and possible benefit of using reagent 2 was unfounded [23]. Without giving due credit to those who accomplished the same many years before using a different water soluble dialkyl squarate [10], Wurm et al. [23] present di(tri(ethylene glycol) monomethyl ether)ester (2) as a unique reagent because “it is a water-soluble squaric acid diester that enables one-pot, two-step peptide–protein conjugation in purely aqueous reaction media”. They carried out the first step of the one-pot conjugation at a squarate–amine molar ratio of 1:1. Because the squarate reagent 2 used was impure and it partially hydrolyzed during conjugation reagent formation, some of the amine present must have remained unchanged. It is reasonable to suggest that in the next, conjugation step at basic conditions, some of that unchanged amine reacted with the squarate conjugation reagent that had been formed, to form a dimer, as we recently showed [27]. When an expensive amine is used in the reaction, this dimer-forming side reaction could be source of waste of precious material. Therefore, for our one-pot two-step conjugation using 1 as squarate reagent, we judiciously altered Wurm et al. [23] protocol.
In brief, amine 9 was treated with 2 equivalents of 1 for 24 h. Excess of 1 ensured virtually complete conversion of 9 and the extended reaction time, during which all unchanged 1 must have hydrolysed [27], ensured absence of the latter in the conjugation step. To carry out the second step, BSA was added into the existing reaction mixture to make the 8:BSA ratio 15:1, and the pH was adjusted to 9.0. After 20 h, the loading 14.5 was achieved, as determined by SELDI-TOF MS. Thus, squarate reagent 1 could be used for one-pot two-step conjugation with excellent yield and efficiency (for details, see the Experimental Section).
To summarize, we have synthesized squarate reagent 2 in the analytically pure state. The hydrolytic stability of the two conjugation reagents, 8 and 10, derived from amine 9 and squarate reagents 1 and 2, respectively, was compared under two conditions. The rate of hydrolysis of conjugation reagents 8 and 10 was found to be very similar. A comparison of the rate of conjugation of 8 and 10 to BSA showed that reaction with 8 is slightly but insignificantly higher, when compared to that with 10. We have also confirmed the previously shown [10] ability of methyl squarate reagent 1 to effect one-pot, two-step conjugation.
In view of the above, we conclude that for conjugating amine-functionalized carbohydrates there is no advantage to using the more labor-intensive, oily squarate reagent 2 over the commercially available, crystalline 1. Thus, to date, squarate reagent 1 can still be considered the most convenient reagent for squaric acid conjugations.
1. Experimental section
1.1. General Methods
All reactions were monitored by thin-layer chromatography (TLC) on silica gel 60 coated glass slides. Column chromatography was performed by elution from prepacked (Varian, Inc.) columns of silica gel with the Isolera Flash Chromatograph (Biotage). Nuclear magnetic resonance (NMR) spectra were measured at 600 MHz for 1H and 150 MHz for 13C with Bruker Avance spectrometers. Solvent peaks were used as internal reference relative to TMS for 1H and 13C chemical shifts (ppm). Assignments of NMR signals were made by homonuclear and heteronuclear two-dimensional correlation spectroscopy, run with the software supplied with the spectrometers. When reporting assignments of NMR signals, nuclei associated with the spacer are denoted with a prime; sugar residues are serially numbered, beginning with the one bearing the aglycon, and are identified by a Roman numeral superscript in listings of signal assignments. Unless stated otherwise, solutions were concentrated (rotary evaporator) at 40°C/2 kPa. Combustion analyses were performed by Atlantic Microlab, Inc., Norcross, GA. The pH values were measured by Mettler Toledo FiveEasy Plus pH meter. The electrode was a MI-410 micro pH electrode from Microelectrodes, Inc. (Bedford, NH U.S.A.). Buffers used were as follows. A: 0.05 M pH 7.0 buffer: BuffAR pH 7.0 reference solution (0.05 M, Mallincrodt, Cat. No. 0031-04); B: 0.5 M borate buffer pH 9.0, made in house (1L) from boric acid (30.9 g), KCl (26.1 g), and KOH (8.42 g), and final adjustment to pH 9.0 by addition of solid KOH; C: 0.01 M pH 9.5 buffer: two stock solutions were made first (stock solution a: 0.1 M sodium hydroxide; stock solution b: 0.05 M sodium tetrahorate). To make buffer C, stock solution a 21 μL was mixed with 79 μL of stock solution b and diluted with 900 μL of water. For hydrolysis experiments, the buffer was made with D2O instead of water).] Triethyleneglycol monomethyl ether (≥97%) was purchased from Sigma-Aldrich.
1.2. 3,4-Di(2-(2-(2-methoxyethoxy)ethoxy)ethoxy)-3-cyclobutene-1,2-dione (2)
3,4-Dihydroxy-3-cyclobutene-1,2-dione (4.6 g, 40.3 mmol, 3) and H2SO4 (0.13 mL, 2.4 mmol) were added with stirring to a 250 mL round-bottom flask containing 60 mL of benzene and triethylene glycol monomethyl ether (15.0 mL, 93.7 mmol, 4). The suspension was vigorously stirred under refluxed in a Soxhlet apparatus for 4 days (~9 hours/day). Benzene was added several times to ensure that the water generated would be efficiently removed. During the reaction time, the colour of the reaction mixture changed from colourless to light-, and later dark-brown. Acid 3 slowly dissolved, and eventually a clear, coloured solution formed. TLC (after neutralization of a sample with solid NaHCO3, 20:1 chloroform-MeOH) showed presence of a small amount of starting material, in addition to a major faster moving product. After concentration, most of triethylene glycol monomethyl ether used in excess at the onset of the reaction was removed at 90°C (~13 Pa).
The crude product (light brown syrup, 20.0 g) was chromatographed (5:1 → 7:3 CH2Cl2-CH3CN), to give first a minor by-product 5 (146 mg, 1%). 1H NMR (600 MHz, CDCl3): 4.81 (m, 4H, H-1, H-1′), 3.83 (m, 2H, H-2), 3.71 (m, 2H, H-2′), 3.67 (m, 2H, H-3), 3.63 (m, 2H, H-4), 3.62 (m, 2H, H-5), 3.53 (m, 2H, H-6), 3.39 (s, 3H, H-3′), 3.37 (s, 3H, H-7); 13C NMR (150 MHz, CDCl3): 189.1, 189.0, 184.0, 183.9, 73.0 (C-1), 72.9 (C-1′), 71.9 (C-6), 70.8 (C-3), 70.7 (C-2′), 70.6 (C-4), 70.6 (C-5), 69.5 (C-2), 59.1 (C-3′), 59.0 (C-7); TOF-HRMS m/z: [M+H]+ calcd for C14H23O8: 319.1393, Found 319.1393.
Eluted next was the title compound 2 (colorless syrup, 14.1 g, 86%). 1H NMR (600 MHz, CDCl3): 4.80 (m, 4H, H-1), 3.82 (m, 4H, H-2), 3.67 (m, 4H, H-3), 3.63 (m, 4H, H-4), 3.61 (m, 4H, H-5), 3.53 (m, 4H, H-6), 3.36 (s, 3H, H-7); 13C NMR (150 MHz, CDCl3): 189.1, 184.0, 73.0 (C-1), 71.9 (C-6), 70.8 (C-3), 70.6 (2C, C-4, C-5), 69.5 (C-2), 59.0 (C-7); TOF-HRMS m/z: [M+H]+ calcd for C18H31O10: 407.1917, Found 407.1922; Anal. Calcd for C18H30O10: C, 53.19; H, 7.44. Found: C, 53.18; H, 7.46.
When MeOH was used during chromatography (not recommended), by-product 6 was also isolated. 1H NMR (600 MHz, CDCl3): 4.79 (m, 2H, H-1), 4.35 (s, 3H, H-1′), 3.81 (m, 2H, H-2), 3.65 (m, 2H, H-3), 3.61 (m, 4H, H-4, H-5), 3.51 (m, 2H, H-6), 3.35 (s, 3H, H-7); 13C NMR (150 MHz, CDCl3): 189.2, 189.1, 184.2, 184.2, 73.0 (C-1), 71.9 (C-6), 70.8 (C-3), 70.6 (2C, C-4, C-5), 69.5 (C-2), 60.9 (C-1′), 59.0 (C-7). TOF-HRMS m/z: [M+H]+ calcd for C12H19O7: 275.1131, Found 275.1129
1.3. Reaction of 3,4-Di(2-(2-(2-methoxyethoxy)ethoxy)ethoxy)-3-cyclobutene-1,2-dione (2) with (2-Aminoethylamido)carbonylpentyl β-D-galactopyranosyl-(1→4)-β-D-glucopyranoside (9) to give the corresponding squarate derivative 10
Amine 9 (200 mg, 0.4 mmol) was dissolved in 10 mL of buffer A. Compound 2 (195 mg, 0.48 mmol) was added and the reaction mixture was kept at room temperature for 4 h, when TLC (CH2Cl2/MeOH 4:1) confirmed absence of the starting material. The mixture was freeze-dried and the white fluffy solid obtained was chromatographed (CH2Cl2/MeOH 4:1), to give compound 10 as a white, amorphous solid (208 mg, 70%). Crystallization from common solvents failed. 1H NMR (600 MHz, D2O): 4.86 (m, 2H, H-13′), 4.47 (d, J = 7.5 Hz, 1H, H-1I), 4.45 (d, J = 7.8 Hz, 1H, H-1II), 3.97 (dd, J = 1.7, 12.3 Hz, H-6Ia), 3.91 (m, 3H, H-14′, H-4II), 3.88 (m, 1H, H-1′a), 3.80 (m, 1H, H-6Ib), 3.79–3.70 (m, 6H, H-6II, H-15′, H-5II, H-8′a), 3.70–3.65 (m, 5H, H-16′, H-17′, H-3II), 3.65–3.62 (m, 3H, H-1′b, H-4I, H-3I) 3.62–3.57 (m, 4H, H-8′b, H-18′, H-5I), 3.54 (dd, J = 7.8, 9.6 Hz, H-2II), 3.42 (m, 2H, H-7′), 3.36 (s, 3H, H-19′), 3.30 (t, J = 7.5 Hz, 1H, H-2I), 2.25–2.20 (m, 2H, H-5′), 1.63–1.58 (m, 2H, H-2′), 1.58–1.52 (m, 2H, H-4′), 1.35–1.30 (m, 2H, H-3′); 13C NMR (150 MHz, D2O): 188.6 and 182.9 (squarate-C), 176.9 (d, C-6′), 176.4 (d, C-10′), 173.2 (d, C-9′), 102.7 (C-1II), 101.8 (C-1I), 78.2 (C-4I), 75.1 (C-5II), 74.5 (C-5I), 74.2 (C-3I), 72.6 (C-2I), 72.3 (d, C-13′), 72.3 (C-3II), 70.8 (C-18′), 70.7 (C-2II), 70.0 (C-1′), 69.6 (d, C-15′), 69.3 (d, C-16′), 69.2 (d, C-17′), 69.0 (d, C-14′), 68.3 (C-4II), 60.8 (C-6II), 59.9 (C-6I), 57.8 (C-19′), 43.8 (d, C-8′), 39.2 (d, C-7′), 35.5 (C-5′), 28.2 (d, C-2′), 24.9 (C-4′), 24.4 (C-3′); TOF-HRMS m/z: [M+H]+ calcd for C31H53N2O18: 741.3293, Found 741.3292.
1.4. Hydrolysis of 8 or 10 in buffer B
Compound 8 or 10 (0.0024 mmol) was dissolved in 600 μL of 0.5 M pD 9.0 buffer to make a 4 mM solution and filtered into an NMR tube (for NMR measurements, in this experiment, the buffer was prepared with D2O). Spectra were measured periodically (Fig. 4).
1.5. Hydrolysis of 8 or 10 in buffer C
Compound 8 or 10 (0.0367 mmol) was dissolved in 600 μL of 0.01 M pD 9.5 buffer to make a 61 mM solution and filtered into NMR tube (for NMR measurements, in this experiment, the buffer was prepared with D2O). Spectra were measured periodically (Fig. 4).
1.6. Conjugation of 8 to BSA in buffer B
Compound 8 (2.0 mg, 0.0033 mmol) and BSA (14.5 mg, 0.000218 mmol) were dissolved in buffer B (820 μL). The clear solution formed was stirred at room temperature and the progress of the reaction was monitored by SELDI-TOF MS. After 48 h, pH of the mixture was 8.92, and 14.4 loading was reached (average MW of conjugate 75,031 Da, as indicated by SELDI-TOF MS (see Supporting Information, Fig. S15). The mixture was diluted with 10 mM aq. (NH4)2CO3 and passed, at 4°C, through a 30 KDa cut-off ultrafiltration membrane, using a Millipore Amicon Ultra 30 KDa ultrafiltration device (for speed/rpm, time and volume of the concentrate, manufacturer’s suggestions were followed). The material that had passed through the membrane was discarded. The retentate was ultrafiltered/washed 7 × with 10 mM aq. (NH4)2CO3, to ensure that all low-molecular mass material had been removed from the conjugate. The retentate was transferred into a storage vial and lyophilized, yielding the conjugate as a white, fluffy solid (14.6 mg, 89%).
1.7. Conjugation of 8 to BSA in buffer C
Compound 8 (2.0 mg, 0.0033 mmol) and BSA (14.5 mg, 0.000218 mmol) were dissolved in buffer C (820 μL). The clear solution formed was stirred at room temperature and the progress of the reaction was monitored by SELDI-TOF MS. After 3 h and 48 h, pH lowered to 8.62 and 8.01 respectively. After 120 h, loading 10.6 was reached (average MW of conjugate 72,580 Da, as indicated by SELDI-TOF MS, see Supporting Information, Fig. S16). Work-up, as described above, yielded 14.9 mg of white fluffy conjugate (94%).
1.8. Conjugation of 10 to BSA in buffer B
Compound 10 (2.0 mg, 0.0027 mmol) and BSA (12.0 mg, 0.000181 mmol) were dissolved in buffer B (675 μL). The clear solution formed was stirred at room temperature and the progress of the reaction was monitored by SELDI-TOF MS. After 48 h, pH was 8.89 and loading 14.6 was reached (average MW of the conjugate, 75,170 Da, as indicated by SELDI-TOF MS, see Supporting Information, Fig. S175). Work-up, as described above yielded 12.7 mg of white fluffy conjugate (93%).
1.9. Conjugation of 10 to BSA in buffer C
Compound 10 (2.0 mg, 0.0027 mmol) and BSA (12.0 mg, 0.000181 mmol) were dissolved in buffer C (675 μL). The clear solution formed was stirred at room temperature and the progress of the reaction was monitored by SELDI-TOF MS. After 3 h and 48 h, pH lowered to 8.45 and 8.04, respectively. After 120 h, loading 8.6 was reached (average MW of conjugate 71,410 Da, as indicated by SELDI-TOF MS, see Supporting Information, Fig. S18). Work-up, as described above, and yielded 11.6 mg of white fluffy conjugate (90%).
1.10. One pot conjugation of 9 and BSA mediated by squarate reagent 1
Compound 9 [11] (10 mg, 0.02 mmol) was added into a reaction vessel containing 1 mL of 0.5 M pH 7.0 phosphate buffer. Squarate reagent 1 (5.7 mg, 0.04 mmol) was added into the clear solution formed and the reaction was kept at room temperature for 24 h (the time needed [27] to completely hydrolyze excess of 1). A solution of BSA (88.8 mg, 0.00133 mmol in 4 mL of Buffer B) was added into the mixture and the pH was adjusted to 9.0 by addition of solid KOH. The progress of the reaction was monitored by SELDI-TOF-MS. After 20 h, loading 14.5 was reached (average MW of conjugate 74,819 Da, as indicated by SELDI-TOF MS). The mixture was worked-up as described above, to afford 90.2 mg of conjugate as a white fluffy solid (91%, conjugation efficiency 97%).
Supplementary Material
Highlights.
The title squaric acid ester was prepared in the analytically pure state.
Stability of conjugation reagents made from two squarate diesters were compared.
An optimized, one-pot protocol for conjugation of carbohydrates to BSA is proposed.
Acknowledgments
This research was supported by the Intramural Research Program of the National Institutes of Health (NIH) and National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Tietze LF, Arlt M, Beller M, Glüsenkamp KH, Jähde E, Rajewsky MF. Chem Ber. 1991;124:1215–1221. [Google Scholar]
- 2.Tietze LF, Schröter C, Gabius S, Brinck U, Goerlach-Graw A, Gabius HJ. Bioconj Chem. 1991;2:148–153. doi: 10.1021/bc00009a003. [DOI] [PubMed] [Google Scholar]
- 3.Kamath VP, Diedrich P, Hindsgaul O. Glycoconj J. 1996;13:315–319. doi: 10.1007/BF00731506. [DOI] [PubMed] [Google Scholar]
- 4.Izumi M, Okumura S, Yuasa H, Hashimoto H. J Carbohydr Chem. 2003;22:317–329. [Google Scholar]
- 5.Wurm FR, Klok HA. Chem Soc Rev. 2013;42:8220–8236. doi: 10.1039/c3cs60153f. [DOI] [PubMed] [Google Scholar]
- 6.Bundle DR. In: Vaccine Design: Innovative Approaches and Novel Strategies. Bagnoli F, Rappuoli R, editors. Caister Academic Press; 2011. pp. 69–107. [Google Scholar]
- 7.Bergh A, Bhattacharyya S, Nilsson UJ. Carbohydr Res. 2002;337:947–949. doi: 10.1016/s0008-6215(02)00080-0. [DOI] [PubMed] [Google Scholar]
- 8.Bergh A, Magnusson BG, Ohlsson J, Wellmar U, Nilsson UJ. Glycoconj J. 2001;18:615–621. doi: 10.1023/a:1020639603070. [DOI] [PubMed] [Google Scholar]
- 9.Tevyashova A, Sztaricskai F, Batta G, Herczegh P, Jeney A. Bioorg Med Chem Lett. 2004;14:4783–4789. doi: 10.1016/j.bmcl.2004.06.072. [DOI] [PubMed] [Google Scholar]
- 10.Pozsgay V, Dubois E, Pannell L. J Org Chem. 1997;62:2832–2846. doi: 10.1021/jo962300y. [DOI] [PubMed] [Google Scholar]
- 11.Hou Sj, Saksena R, Kováč P. Carbohydr Res. 2008;343:196–210. doi: 10.1016/j.carres.2007.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bongat AFG, Saksena R, Adamo R, Fujimoto YSZ, Peterson DC, Fukase K, Vann WF, Kováč P. Glycoconjugate J. 2010;27:69–77. doi: 10.1007/s10719-009-9259-4. [DOI] [PubMed] [Google Scholar]
- 13.Auzanneau FI, Pinto M. Bioorg Med Chem. 1996;4:2003–2010. doi: 10.1016/s0968-0896(96)00183-6. [DOI] [PubMed] [Google Scholar]
- 14.Ellis LA, Mc Vay CS, Probert MA, Zhang Z, Bundle DR, Appleton JA. Glycobiology. 1997;7:383–390. doi: 10.1093/glycob/7.3.383. [DOI] [PubMed] [Google Scholar]
- 15.Zhang J, Yergey A, Kowalak J, Kováč P. Carbohydr Res. 1998;313:15–20. doi: 10.1016/s0008-6215(98)00261-4. [DOI] [PubMed] [Google Scholar]
- 16.Zhang J, Kováč P. Carbohydr Res. 1999;321:157–167. doi: 10.1016/s0008-6215(99)00185-8. [DOI] [PubMed] [Google Scholar]
- 17.Blixt O, Norberg T. Carbohydr Res. 1999;319:80–91. doi: 10.1016/s0008-6215(99)00135-4. [DOI] [PubMed] [Google Scholar]
- 18.Vermeer HJ, Halkes KM, van Kuik JA, Kamerling JP, Vliegenthart JFG. Perkin. 2000;1:2249–2263. [Google Scholar]
- 19.Chernyak A, Oscarson S, Turek D. Carbohydr Res. 2000;329:309–316. doi: 10.1016/s0008-6215(00)00189-0. [DOI] [PubMed] [Google Scholar]
- 20.Saksena R, Ma X, Kováč P. Carbohydr Res. 2003;338:2591–2603. doi: 10.1016/s0008-6215(03)00273-8. [DOI] [PubMed] [Google Scholar]
- 21.Saksena R, Adamo R, Kováč P. Bioorg Med Chem. 2007;15:4283–4310. doi: 10.1016/j.bmc.2007.03.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Xu P, Alam MM, Kalsy A, Charles RC, Calderwood SB, Qadri F, Ryan ET, Kováč P. Bioconj Chem. 2011;21:2179–2185. doi: 10.1021/bc2001984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wurm F, Steinbach T, Klok HA. Chem Commun. 2013;49:7815–7817. doi: 10.1039/c3cc44039g. [DOI] [PubMed] [Google Scholar]
- 24.Chernyak A, Kondo S, Wade TK, Meeks MD, Alzari PM, Fournier JM, Taylor RK, Kováč P, Wade WF. J Infect Dis. 2002;185:950–962. doi: 10.1086/339583. [DOI] [PubMed] [Google Scholar]
- 25.Rollenhagen JE, Kalsy A, Saksena R, Qadri F, Caderwood SB, Kováč P, Wade WF, Ryan ET. Am J Trop Med Hyg Suppl. 2006;75:84–85. [Google Scholar]
- 26.Tarique AA, Kalsy A, Arifuzzaman M, Rollins SM, Charles RC, Leung DT, Harris JB, LaRocque RC, Sheikh A, Bhuiyan MS, Saksena R, Clements JD, Calderwood SB, Qadri F, Kováč P, Ryan ET. Clin Vaccine Immunol. 2012;19:594–602. doi: 10.1128/CVI.05689-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Xu P, Kelly M, Vann WF, Qadri F, Ryan ET, Kováč P. ChemBioChem. 2017;18:799–815. doi: 10.1002/cbic.201600699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nitz M, Bundle DR. J Org Chem. 2001;66:8411–8423. doi: 10.1021/jo010570x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.