

Abstract
Current techniques for the alteration of gene expression in the liver have a number of limitations, including the lack of stable somatic gene transfer and the technical challenges of germline transgenesis. Rapid and stable genetic engineering of the liver would allow systematic, in vivo testing of contributions by many genes to disease. After fumaryl acetoacetate hydrolase (Fah) gene transfer to hepatocytes, selective repopulation of the liver occurs in FAH-deficient mice. This genetic correction is readily mediated with transposons. Using this approach, we show that genes with biological utility can be linked to a selectable Fah transposon cassette. First, net conversion of Fah−/− liver tissue to transgenic tissue, and its outgrowth, was monitored by bioluminescence in vivo from a luciferase gene linked to the FAH gene. Second, coexpressed short hairpin RNAs (shRNAs) stably reduced target gene expression, indicating the potential for loss-of-function assays. Third, a mutant allele of human α1-antitrypsin (hAAT) was linked to Fah and resulted in protein inclusions within hepatocytes, which are the histopathological hallmark of hAAT deficiency disorder. Finally, oncogenes linked to Fah resulted in transformation of transduced hepatocytes.
Conclusion
Coexpression with FAH is an effective technique for lifelong expression of transgenes in adult hepatocytes with applicability to a wide variety of genetic studies in the liver.
Microarray and cancer genome sequencing projects have produced an emergent catalog of genes implicated in liver regeneration, metabolism, cancer, and inherited diseases.1,2 In turn, there is increasing need for in vivo assessment of these genes in a meaningful context. One method for assessing cancer genes is to express putative oncogenes in primordial liver cells, transplant these cells into recipient mice, and measure tumor outgrowth.3 An easier method, in which genes could be directly and stably expressed or suppressed in most hepatocytes in vivo, would facilitate the validation and testing of genes thought to affect liver biology.
Current techniques for gene expression in the liver have limitations. Germline transgenesis establishes stable gene expression, but its expense constrains scale-up or the ready study of genetic interplay. For example, genetic modifiers are thought to affect α1-antitrypsin disease severity in humans,4 yet transgenic mouse models are on a syngeneic background.5-7 A method to selectively and easily express transgenes in hepatocytes would be useful for the study of this and other liver diseases.
Integration and expression of multiple genes is possible using transposon plasmids. The Sleeping Beauty (SB) transposon enables long-term gene expression on gene transfer to the mouse liver.8,9 Delivery is accomplished by a rapid, large-volume injection via the tail vein called “hydrodynamic injection.”10,11 The major shortcoming of this system is that stable integration and expression are restricted to fewer than 1%-2% of hepatocytes after a single delivery.9 One notable exception is with Fah gene delivery to the hepatocytes of FAH-deficient mice, where the initial integration rate may be the same as in wild-type animals but selective repopulation leads to stable Fah gene expression in more than 50% of hepatocytes.12-14 Our results demonstrate that genes encoding luciferase, a short hairpin RNA (shRNA) that targets luciferase, a mutant allele of human α1-antitrypsin (hAAT), the NRAS oncogene, and a p53 shRNA, were all stably expressed in the liver on fumaryl acetoacetate hydrolase (FAH)-mediated repopulation. Thus, we present a facile method for somatic, lifelong manipulation of multiple genes in the mouse liver.
Materials and Methods
Plasmid Construction
A single amino acid substitution, K33A, shown previously to increase transpositional activity by 400% in vitro,15 was incorporated into plasmid pPGK-SB11.16 The resultant PGK-SB-K33A transgene was inserted into pKT2/FAHIL17 outside of the transposon on the 3′ end to generate pKT2/FAHIL//SB.
IRES and luciferase sequences of pKT2/FAHIL were removed to generate pKT2/CaFAH. A pGL3-Promoter vector fragment (Promega Corp.) containing SV40 promoter-luciferase was inserted into pKT2/CaFAH to generate pKT2/Luc-FAH. The U6 promoter-luciferase shRNA fragment of plasmid pSHAG-Ff118 was placed in pKT2/Luc-FAH to generate plasmid pKT2/siLucFAH. Site-directed mutagenesis of the luciferase sequence of pKT2/siLucFAH was performed by 3-step polymerase chain reaction (PCR) mutagenesis to generate pKT2/si*Luc*FAH.
Luciferase complementary DNA (cDNA) from pGL3-Promoter vector was replaced with Fah cDNA to generate pSV40-FAH. The size-minimized Caggs promoter (miniCaggs) from pKT2/mCAG (A. Wilber, unpublished) was excised and combined with an SB-containing fragment of pKT2/FAHIL//SB to make pKT2/CA//SB. The SV40promoter-FAH transgene was inserted into pKT2/CA/SB to make pKT2/FAH-CA//SB. This plasmid contains an EcoRI site in front of the miniCaggs promoter that can be used to insert any cDNA of interest for coexpression with FAH.
The cDNA for hAAT was removed from plasmid pT/hAAT.PGK-SB8 and was inserted into pT2/Caggs19 to generate pT2/Caggs-hAAT. The Z allele of hAAT was made by three-step PCR mutagenesis and was cloned into pT2/Caggs to generate pT2/Caggs-Z.
Complementary DNA encoding wild-type and Z hAAT were inserted into pKT2/FAH-Ca-SB to make pKT2/FAH-hAAT//SB and pKT2/FAH-Z//SB.
The bidirectional promoter from plasmid pKT2/GFP-PGK-CLP-Luc (A. Wilber, unpublished) contains PGK and cytomegalovirus enhancer/EF1α promoters in juxtaposition. The promoter was removed and assembled with FAHIL and NRAS cDNA sequences to generate pKT2/FAHIL-PGK-EF1α-NRAS. The vector MLMS-p53.1224 contains an shRNA that targets mouse p53 and a green fluorescent protein (GFP) transgene.20 The PvuII fragment containing the shRNA and GFP was transferred to an SB transposon plasmid to generate pT2/sh.p53-GFP.
Cell Culture
HT1080 human intestinal carcinoma cells and HeLa human cervical carcinoma cells were maintained on Dulbecco’s modified Eagle’s medium supplemented with 10% fetal bovine serum and 1× penicillin/streptomycin. Transfections were performed using FuGene 6 reagent (Roche Diagnostics) according to the manufacturer’s directions. Luciferase assays were performed using Luciferase Assay System (Promega) according to the manufacturer’s directions.
Mouse Experiments
All animal work was conducted under an institutionally approved animal welfare protocol.
Fah-deficient mice21 were maintained on 7.5 μg/mL 2-(2-nitro-4-trifluoromethylbenzoyl)-1,3-cyclohexanedione (NTBC)22 until injection with the Fah transposon plasmids. Mice between 18 and 25 weeks of age were injected with plasmid DNA by hydrodynamics as described.16 The next day, NTBC was removed from the drinking water. The amount of DNA injected for luciferase expression was 25 μg pKT2/FAHIL//SB; for shRNA expression, 20 μg pKT2/siLucFAH or pKT2/si*Luc*FAH plus 10 μg pPGK-SB11; for hAAT expression, 25 μg pKT2/FAH-hAAT//SB or pKT2/FAH-Z//SB plus 1 μg pKT2/Caggs-Luc23; and for oncogene expression, 1 group of mice was injected with 20 μg pKT2/FAHIL-PGK-EF1α-Nras plus 10 μg pPGK-SB11 and a second group received these plus a third plasmid, 20 μg pT2/sh.p53-GFP. In vivo luciferase imaging was performed at selected time points as described.16 Hepatocytes were isolated for transplantation by collagenase perfusion as described.24
Immunofluorescence
Liver tissue was fixed with 10% formalin and paraffinized. Three-micron sections were hydrated and antigen retrieval was performed by 5 minutes of microwaving in 10 mM sodium citrate, pH 6.0 + 0.1% triton X-100. Slides were washed with phosphate-buffered saline (PBS) plus 0.05% triton X-100 and 0.05% Tween 20, then blocked for 1 hour at room temperature with wash solution plus 10% fetal lamb serum and 2% bovine serum albumin. The slides were then incubated for 1 hour at room temperature with chicken anti-mouse FAH (generated by Dr. Xin Wang) and rabbit anti-hAAT (Dako) at a 1:500 dilution in the block solution. The slides were immersed 3 times in wash solution, and a 1:500 dilution of Alexa Fluor 555 donkey antirabbit immunoglobulin G (IgG) (Invitrogen) and Alexa Fluor 488 goat anti-chicken IgG (Invitrogen) in block solution was added for 1 hour at room temperature. Slides were then washed 3 times, with 1 μg/mL 4′, 6-diuamidino-2-phenylindole included in the second wash, and mounted with Fluoromount-G (Southern Biotech).
Immunohistochemistry
Immunohistochemical staining for FAH was conducted as described.17 NRAS staining was performed with monoclonal mouse-anti-NRAS antibody (Santa Cruz Biotechnology) and Ki67 staining with monoclonal mouse anti-Ki67 antibody (Novocastra). These antibodies were applied to tissue sections using the Mouse on Mouse immunodetection kit (Vector Laboratories) according to the manufacturer’s instructions.
Periodic Acid-Schiff-D Staining
Diastase digestion of tissue sections was performed at 37°C for 15 minutes with 6.7 g/L Amylase Type VI-B (Sigma) in distilled water. The slides were then stained with periodic acid and Schiff’s reagent, counterstained with hematoxylin, dehydrated, and mounted.
Microscopic Imaging
Immunofluorescent images were captured at 100 × magnification using a Zeiss Axioplan 2 (Zeiss Inc.) microscope and Zeiss Axiocam MRm or Canon PowerShot G6 cameras. Multiple optical sections were acquired in each fluorescent channel, and the sections were processed using Axio Vision software (Zeiss Inc.). Adjacent sections (fluorescent/brightfield) were aligned optically using visible tissue landmarks.
Western Blotting
Liver protein was extracted in 1% Triton X-100, 0.5% deoxycholic acid, 10 mM ethylenediamine tetraacetic acid, and 2 mM phenylmethane sulfonylfluoride by grinding and sonication. The lysate was cleared by centrifugation, and 50 μg total protein was boiled for 5 minutes, separated on an acrylamide gel, and transferred onto a nitrocellulose membrane. Blots were blocked with blotto (5% nonfat milk powder in PBS plus 0.1% Tween 20) for 1 hour at room temperature or overnight at 4°C. Primary antibodies were added in blotto solution for 1 hour at room temperature. Blots were washed three times with PBS plus 0.1% Tween 20 before secondary antibody was added then washed again and immersed for 1 minute in chemiluminescent substrate (Pierce). Radiographic paper was then exposed to the blots. For detection of hAAT, rabbit anti-human α1-antitrypsin (Dako) was used at a dilution of 1:5000 in combination with goat anti-rabbit IgG, horseradish peroxidase (HRP) (Sigma) at 1:10,000, or goat anti-hAAT (1:3000, Diasorin) was used with donkey anti-goat IgG, HRP (1:6000; Promega). Albumin was detected with rabbit anti-mouse albumin (1:10,000; Accurate Chemical and Scientific Corp.) in combination with goat anti-rabbit IgG, HRP (1:10,000; Sigma). FAH was detected with rabbit anti-mouse FAH (1:3000; Dr. Xin Wang) in combination with goat anti-rabbit IgG, HRP (1:10,000; Sigma). Luciferase was detected with affinity-purified rabbit anti-firefly luciferase (1:200; Cortex Biochem.). Purified hAAT (Sigma) was used as a standard.
Reverse Transcription PCR
Total RNA was isolated using the TRIzol Reagent (Invitrogen) according to the method provided by the manufacturer. One microgram total RNA was reversed transcribed with random hexamer primers using the Transcriptor First Strand cDNA Synthesis Kit (Roche) according to the manufacturer’s instructions. Subsequent PCR analysis was carried out with 1 μL synthesized cDNA template and various primer sets using standard conditions for nonsaturated PCR.
Results
Rapid and Quantifiable Establishment of Luciferase Expression in Mouse Liver
FAH enzyme deficiency causes neonatal lethality in mice from hepatic toxicity.21 The drug NTBC blocks neonatal lethality in Fah−/−mice by inhibiting 4-hydrozyphenylpyruvate diogenease, an enzyme upstream of FAH in the tyrosine catabolic pathway, enabling colony propagation of Fah−/− mice.22 Correction of FAH deficiency using transfer of the Fah gene results in therapeutic liver repopulation by the FAH gene–corrected Fah−/− hepatocytes, such that the mice no longer require NTBC.12–14
To stably coexpress the reporter luciferase with FAH in repopulating liver cells, the construct pKT2/FAHIL//SB was injected into two Fah−/− mice. NTBC was removed to select for FAH-expressing hepatocytes, and at 1 month postinjection their weights surpassed pretreatment level, indicating therapeutic correction of the metabolic defect (Supplementary Fig. 1A). Immunohistochemistry of liver tissue using an antibody against FAH revealed nodules of liver cells with positive staining surrounded by unstained cells (Fig. 1B). Western analysis of liver lysates indicated that FAH and luciferase were both expressed in the pKT2/FAHIL//SB-repopulated mouse (Fig. 1C).
Fig. 1.
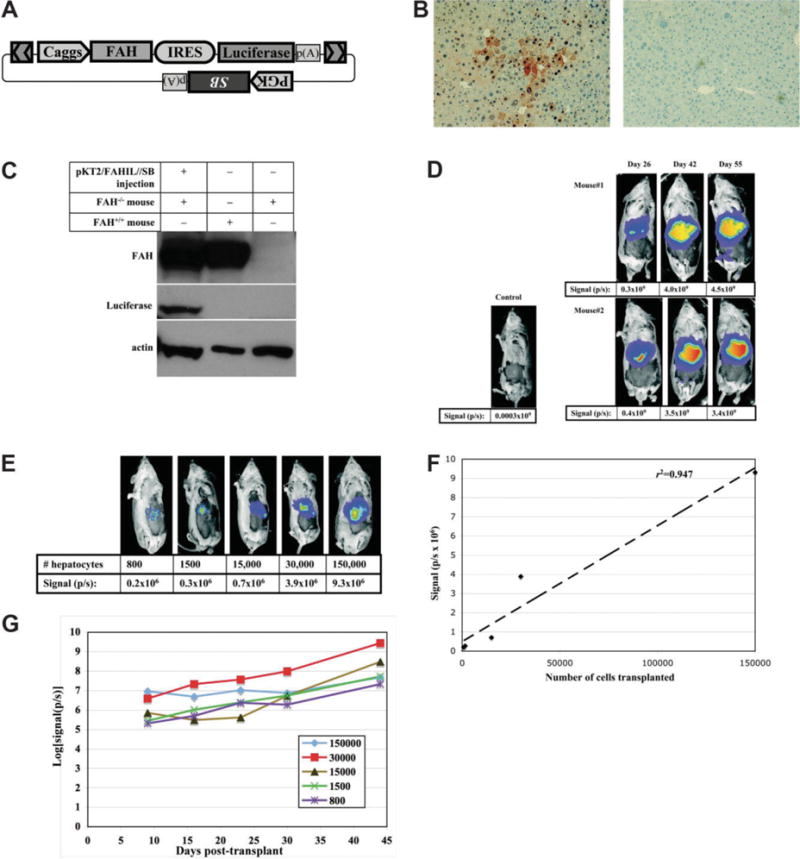
Stable coexpression of luciferase with FAH in the livers of somatically modified and repopulated Fah−/− mice. (A) Schematic of plasmid construct pKT2/FAHIL//SB that contains a transposon with the Caggs promoter driving FAH and luciferase expression, with the coding sequences linked by an internal ribosome entry site (IRES) from encephalomyocarditis virus. In addition, a PGK promoter-driven Sleeping Beauty (SB) transposase transgene is outside of the transposon in the same plasmid. The use of “cis”-type transposon/transposase vectors has been previously shown to mediate effective gene transfer in mouse liver.8 (B) Liver tissue was examined for FAH expression 42 days after repopulation with pKT2/FAHIL-SB (left) or in untreated FAH−/− mice (right). The absolute frequency of nodules, reflecting the initial transduction of hepatocytes, was 1.72 ± 1.48 × 10−4 (~1/5800) for one mouse and 2.32 ± 1.86 × 10−4 (~1/4300) for the other. The overall percentage of tissue expressing FAH was 50% ± 13% and 53% ± 11%, respectively. This is similar to the frequency reported previously for FAH−/− mice using a “trans”-type Sleeping Beauty transposon system.14 (C) Western blot analysis for transgene expression in liver extracts. The first column is an extract from an Fah−/− mouse repopulated using pKT/FAHIL//SB; the second column is wild-type mouse liver extract; and the third column is extract from an untreated, Fah−/− mouse. The blot was probed separately with FAH, luciferase, and actin antibodies. (D, E) Luciferase expression was assayed in vivo after injection with the luciferase substrate luciferin and imaging with a CCD camera. Red indicates regions of highest expression. (D) Fah−/− mice treated with pKT2/FAHIL//SB were evaluated for luciferase expression as the liver repopulated with FAH-transformed liver cells. The numbers below the mice represent the luciferase signal captured as photons emitted per second (p/s). (E) Luciferase expression in Fah−/− recipients, 9 days after transplantation of pKT2/FAHIL//SB-transduced hepatocytes. (F) The luciferase signal (y-axis) is plotted against the number of cells transplanted (x-axis). The best-fit line is shown. (G) Luciferase signals are plotted over time for five Fah−/− recipient mice transplanted with hepatocytes from a pKT2/FAHIL//SB-repopulated mouse. The difference in outgrowth among the recipients reflects a difference in the number of cell doublings (3-10 total) needed to reach normal liver functional capacity.
In vivo bioluminescent imaging was used to track the luciferase expression over time in live animals. The expression was highest in the area of the liver, as seen in previously using hydrodynamic injection16,25 (Fig. 1D). An image taken at postinjection day 26 likely represented an early point in repopulation, as the mice were on NTBC treatment for tyrosinemia-induced weight loss. Luciferase activities measured at 42 and 55 days postinjection were approximately 10-fold higher than the first measurement, indicating that the expression expanded with time. Expansion of transgene expression generally is not seen with standard gene transfer techniques; instead, diminution of gene expression is the norm.16 Therefore, this result demonstrates the ability to link transgene expression with liver repopulation. Sequencing of insertion sites revealed that the transposons integrated into TA dinucleotides, which are characteristic of SB transposase-mediated events (Supplementary Table 1).
To test the stability of expression in dividing hepatocytes, we performed the following transplantation experiment. The hepatocytes from one of the Fah−/− mice repopulated with pKT2/FAHIL//SB were isolated at 55 days postinjection, and suspensions of 800, 1500, 15,000, 30,000, and 150,000 hepatocytes were injected into the spleens of new Fah−/− recipient mice. Nine days later the mice were imaged in vivo for luciferase expression (Fig. 1E). The amount of light emitted was found to have a statistically significant, linear correlation with the number of transplanted hepatocytes (r2 = 0.95; P = 0.005), indicating that luciferase expression can be used as a marker for the relative number of injected hepatocytes (Fig. 1F). NTBC was removed from transplant recipients, and luciferase expression increased by an average of more than 100-fold during 5 weeks of repopulation with the transplanted cells (Fig. 1G).
Transposon-Based Engineering for Gene Knockdown in the Liver
As a proof-of-principle that RNA interference can stably reduce target-gene expression after gene transfer to the mouse liver, we coexpressed, with FAH, a previously reported shRNA that targets the reporter luciferase.18 We constructed a transposon vector, pKT2/siLucFAH, with 3 separate transcriptional units: (1) shRNA; (2) luciferase; and (3) FAH. A control construct was made, pKT2/si*Luc*FAH, with 11 silent mutations in the shRNA-targeted region of luciferase, such that binding is unlikely (Fig. 2A). The energy required for hybridization of the shRNA to the luciferase RNA is increased by more than 26 kcal/mol with the substitutions.26 The plasmids were transiently transfected into tissue culture cells, and luciferase assays indicated a sequence-specific reduction in luciferase expression when the shRNA is combined with the original luciferase sequence (Fig. 2B).
Fig. 2.
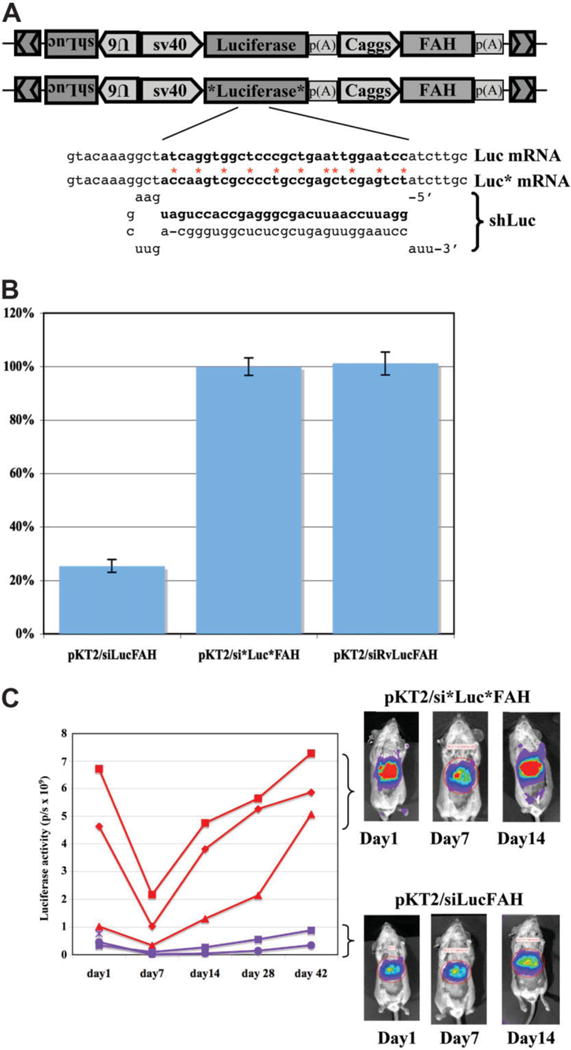
Stable, sequence-specific reduction of target gene expression by shRNA in FAH-repopulated mice. (A) The vector pKT2/siLucFAH is designed to express a luciferase shRNA via the U6 promoter, luciferase by SV40 promoter, and FAH by Caggs. The control vector, pKT2/si*Luc*FAH, is the same as pKT2/siLucFAH except for 11 silent mutations introduced in the shRNA-targeting region. Free energy for RNA:RNA hybridization was calculated by mfold.28 The silent changes increase the energy needed for hybridization by more than 26 kcal/mol. (B) A graph of relative luciferase expression (average ± SEM) 2 days after transfection of HT1080 human intestinal carcinoma cells with the indicated plasmids. pKT2/siRvLuc-FAH is designed to express a control shRNA with the nucleotide sequence of the Luc shRNA reversed, such that there is no complementarity with luciferase RNA. (C) A graph of bioluminescence readings in Fah−/− mice after coinjection pPGK-SB11 plus either pKT2/siLucFAH or pKT2/si*Luc*FAH. Representative images of the mice are shown on the right. Red areas represent highest luciferase expression, yellow second highest, blue weakest. For both experimental groups there was a relative drop in expression at day 7 after injection, which likely reflects loss of unintegrated plasmids as the animals experience tyrosinemia. With pKT2/siLucFAH there was a maintained drop of approximately 90% in luciferase expression through postinjection day 42 (P = 0.004).
Each of the shRNA expression vectors was mixed with Sleeping Beauty transposase plasmid and coinjected into Fah−/− mice. Bioluminescence was measured in vivo at regular intervals, and the mice that received the codon-modified luciferase construct, pKT2/si*Luc*FAH, had 10-fold more robust luciferase expression than the native luciferase construct, pKT2/siLucFAH (Fig. 2C). Overall, the results indicate that shRNA suppressed the target luciferase throughout repopulation.
Modeling of a Gain-of-Function Disease, Human α1-Antitrypsin
We elected to study α1-antitrypsin deficiency, the most common genetic disease of the liver.27 Whereas wild-type hAAT is normally secreted by hepatocytes, the protein expressed by the Z allele (E342K) is abnormally folded and retained in the endoplasmic reticulum, resulting in characteristic protein inclusions within hepatocytes.28 We constructed transposon vectors that contain either wild-type or Z alleles of hAAT together with Fah, with a transposase gene outside of the transposon (Fig. 3A). The wild-type construct (pKT2/FAH-hAAT//SB) and the Z construct (pKT2/FAH-Z//SB) were introduced into FAH-deficient mice by hydrodynamic injection, and NTBC was withdrawn. With each injection mixture, a luciferase expression plasmid (pT2/Caggs-Luc) was included to monitor gene transfer. The luciferase expression levels of the mice determined at 1 day and 1 month postinjection were not different, indicating that gene transfer was the same for both groups (Fig. 3B). The average animal weight of both treatment groups normalized to pretreatment level by 3 weeks postinjection (Supplementary Fig. 1B).
Fig. 3.
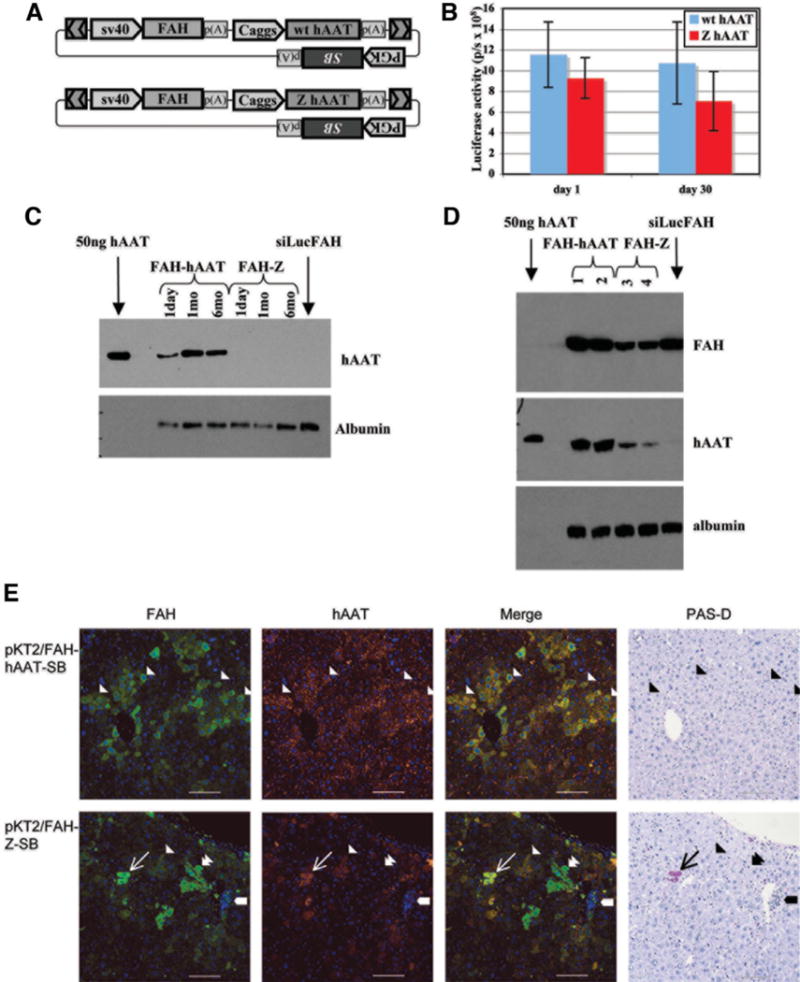
Modeling of a dominant protein folding disorder by somatic transgenesis. Repopulation of FAH-deficient mice with wild-type and Z-mutant hAAT coexpression vectors. (A) The vector pKT2/FAH-hAAT//SB contains a transposon with an Fah gene controlled by the SV40 promoter and an hAAT gene with the Caggs promoter, and has an SB transgene outside of the transposon. The vector pKT2/FAH-Z//SB is the same as pKT2/FAH-hAAT except for a glutamine-to-lysine substitution at amino acid 342 of the hAAT gene, rendering the Z mutant allele. (B) Mice were coinjected with plasmid pT2/Caggs-Luc to monitor gene transfer. The average luciferase activity (± SEM) is graphed for the five mice in each group 1 and 30 days after injection of the plasmids. (C, D) Expression of both wt and Z alleles of hAAT in somatically transgenic livers of Fah−/− mice was assessed at the protein level in sera (C) and liver extracts (D). (C) Western analysis of FAH and hAAT expression in serum samples of mice at 1 day, 1 month, and 6 months after injection of plasmids. One microliter of a 1:50 dilution of serum was loaded into wells. Lane 1 contains 50 ng purified hAAT protein. Serum from an Fah−/− mouse repopulated with pKT2/siLucFAH was included as a control. (D) Western analysis of liver tissue lysates from repopulated mice. Two mice are analyzed for each treatment. (E) Histological analysis of liver tissue after treatment of Fah−/− mice with pKT2/FAH-hAAT//SB. The first column displays FAH immunofluorescence (green), the second column is hAAT expression (red), the third column is a merged image of immunofluorescence, and the fourth column is a PAS-D stain of an adjacent tissue section. The top row shows sections of liver tissue 10 months after treatment of an Fah−/− mouse with pKT2/FAH-hAAT//SB, the bottom row is 10 months after treatment with pKT2/FAH-Z//SB. Single arrowheads illustrate the margins of repopulation nodules. The narrow arrows mark a PAS-D-positive inclusion with correspondent FAH and hAAT immunofluorescence. The thick arrows demarcate an area of inflammation with smaller PAS-D-positive inclusions. Double arrowheads illustrate FAH-positive, hAAT-negative cells. Scale bar represents 100 μm. Nuclei are stained blue using 4′,6-diuamidino-2-phenylindole or hematoxylin.
Six months after plasmid injection, hepatocytes were collected from one mouse in each treatment group for transplantation. For each of the two treatments, 4000 or 400,000 hepatocytes were transplanted into two female and two male FAH-deficient recipient mice. NTBC was withdrawn to select for the transplanted hepatocytes, and the weights of recipient mice were measured (Supplementary Fig 1C). In each group, the male recipients of 4000 hepatocytes died, presumably from the stress of tyrosinemia. The remaining three mice per group exhibited a similar course of weight loss and gain after treatment. Based on the stability of the mouse weight after treatment, there was no apparent difference between Z mutant or wild-type hAAT coexpression in the ability of mice to repopulate. Insertion sites recovered by inverse PCR from DNA-injected mice are consistent with SB transposase-mediated integration (Supplementary Table 1).
Western analysis of the serum of treated mice was performed to determine whether the linked genes are secreted. Wild-type hAAT was detected at 1, 30, and 180 days after injection, at a concentration slightly above the normal range for humans with wild-type hAAT phenotype (0.85-2.15 mg/mL)27 (Fig. 3C).The Z protein was not detectable in the serum. Westerns were carried out on liver extracts to examine transgene expression within the hepatocytes, and both hAAT and FAH proteins were detected with pKT2/FAH-Z//SB and pKT2/FAH-hAAT//SB (Fig. 3D). The results indicate that wild-type hAAT could be secreted, but the Z protein was retained in the liver.
The histopathological hallmark of α1-antitrypsin deficiency in liver is the formation of protein inclusions within hepatocytes that are visible after periodic acid-Schiff staining with diastase pretreatment (PAS-D staining).27 A histological analysis was conducted on livers 10 months after repopulation with pKT2/FAH-Z//SB and pKT2/FAH-hAAT//SB (Fig. 3E). The FAH staining was cytoplasmic for both, and positive cells were in clusters, suggesting clonal outgrowth. FAH staining was noted in approximately 30% to 50% of hepatocytes, a range consistent with previous results with nonviral gene therapy in the FAH model.13,14,17 In mice repopulated with pKT2/FAH-hAAT//SB, the staining with hAAT antibody was fine and granular and was stronger in the area that coincided with FAH expression. Staining of wild-type hAAT was also visible throughout the section in areas around the cells (the interstitium), which likely corresponds to secreted hAAT. In mice repopulated with pKT2/FAH-hAAT//SB, the PAS-D stain showed normal-appearing liver tissue without PAS-D–positive intracytoplasmic inclusions (Fig. 3E).
In liver tissue repopulated using pKT2/FAH-Z//SB, the Z protein was confined within cells and always corresponded to FAH staining (Fig. 3E). Three unique features were found only in mice 10 months after treatment with pKT2/FAH-Z//SB: (1) clusters of cells that stained for FAH but not hAAT, suggesting that there was outgrowth of cells with attenuated expression of the mutant protein (Fig. 3E, double arrowheads); (2) inflammation corresponding to scant Z protein expression and PAS-D–positive material, suggesting an immune reaction to some of the cells with transgenes (Fig. 3E, thick arrows); and (3) large hAAT-positive, FAH-positive, PAS-D–positive globules (Fig. 3E, narrow arrows). Liver tissue samples from Z-homozygous human patients were stained as a control, and displayed PAS-D–positive globules as described in the literature (data not shown).
Expression of Oncogenes Using Somatic Transgenesis Results in Hepatic Tumors
Hepatocellular carcinoma (HCC) results from genetic changes that activate oncogenes or cause loss of tumor suppressors.29 HCC is known to arise in FAH−/− mice that are discontinuously treated with NTBC,22 but no HCC is detected at higher doses of NTBC at 13 months.30 HCC is also detected in two-thirds of mice 1 year after repopulation using retrovirally delivered FAH transgenes.30 The HCC has its origins in FAH-negative, nontransduced cells30 and may result from accumulation of the tyrosine metabolite fumarylacetoacetate, a known mutagen, within these cells.31
To test whether HCC could be induced by expressing oncogenes, a vector was generated that coexpresses FAH, luciferase, and a constitutively activated NRAS oncogene.32 This was administered to two FAH-deficient mice together with a transposase construct. Two other mice received an additional transposon plasmid that expresses an shRNA that targets p53 to promote cancer.20 On withdrawal of NTBC, the mice underwent liver repopulation, as evidenced by stable weight gain (Supplementary Fig. 1D) and increasing luciferase expression (Fig. 4B). At day 43 after injection, liver tissue from mice in both groups was removed by partial hepatectomy and sectioned, and clusters of cells with large nuclei and bizarre mitotic figures were present (data not shown). By 70 days after injection, the mouse with p53 shRNA that had undergone partial hepatectomy developed a massively enlarged abdomen that colocalized with expansive luciferase in vivo expression (Fig. 4B). The second mouse in this group developed an enlarged abdomen by day 112. On necropsy, both mice had massive, expansile, multifocally cystic liver tumors that extended from the diaphragm to the posterior abdomen, filling most of the abdomen (Fig. 4C). In contrast, the mice repopulated with the NRAS construct, but without p53 shRNA, had discrete, nodular tumors in a liver that was otherwise normal in appearance. Transposon insertion sites were recovered and they were consistent with SB-mediated integration (Supplementary Table 1).
Fig. 4.
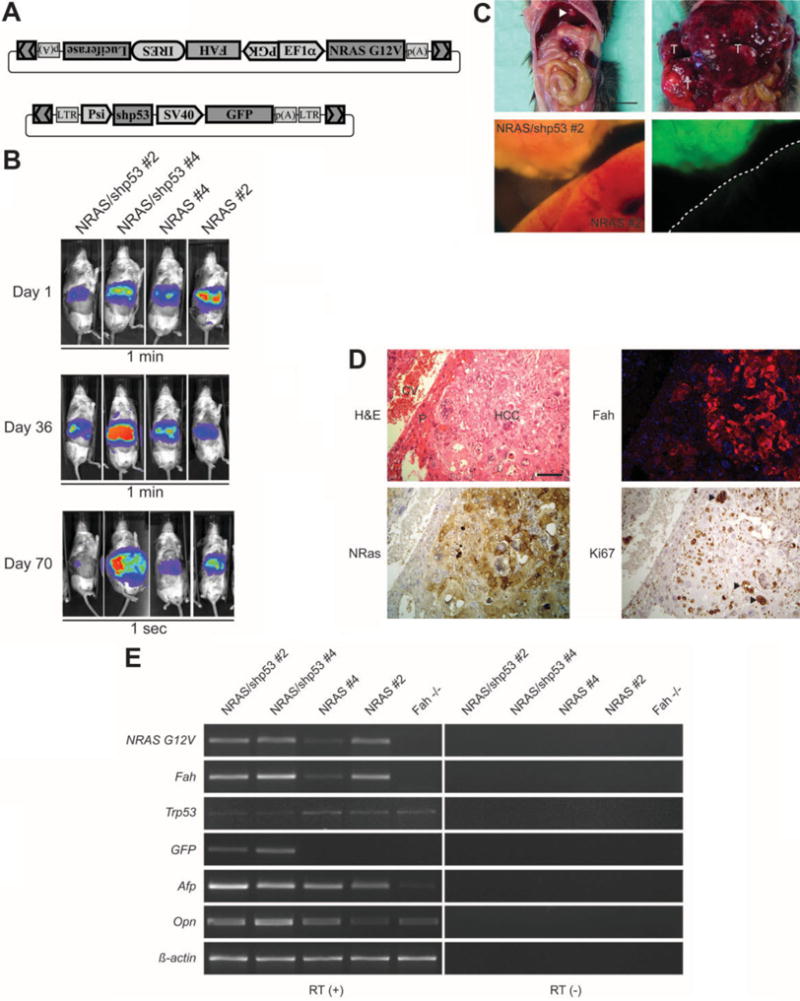
Fulminant hepatocellular carcinomas develop in Fah−/− mice on coexpression of N-ras and shRNA targeting p53. (A) The vector pKT2/FAHIL-NRAS contains a transposon with a bidirectional promoter driving the expression of FAH-IRES-Luciferase and N-ras. The vector pT2/sh.p53-GFP has a transposon with expression of an shRNA that targets p53 as well as a GFP transgene. (B) Luciferase activity was imaged in vivo at the indicated time points after injection of pPGK-SB11 plus pKT2/FAHIL-NRAS and pT2/sh.p53-GFP (left) or pPGK-SB11 plus pKT2/FAHIL-NRAS (right). The images captured at days 1 and 36 postinjection represent a 1-minute exposure, whereas the image at day 70 represents a 1-second exposure, reflecting an amplification in luciferase expression from day 1 to day 70. Note that the expansive abdomen of the second mouse at day 70 is widely expressing luciferase. (C) At postinjection day 112, mice repopulated with pKT2/FAHIL-NRAS alone (top left) or with addition of pT2/sh.p53-GFP (top right) were killed and their abdominal cavity was opened, revealing solitary nodular tumors in the former (arrowhead) and an expansive, cystic liver tumor in the latter (T). The liver tissue was removed and examined with a fluorescent microscope, an analysis that revealed that tumor tissue formation is linked to GFP expression from the construct pT2/sh.p53-GFP. The scale bar represents 0.8 cm. (D) Histopathological analysis of tumor tissue from an Fah−/− mouse injected with pPGK-SB11 plus pKT2/FAHIL-NRAS and pT2/sh.p53-GFP. Hematoxylin-eosin staining (top left) shows that 80% of the hepatic section is composed of cords of large, pleomorphic, anaplastic tumor cells indicative of hepatocellular carcinoma (HCC); a small rim of compressed parenchymal hepatocytes (P) remains adjacent to central vein (CV). Adjacent sections indicate that the tumor tissue expresses FAH and N-ras, and the nuclear proliferation marker Ki-67 is intensely expressed by atypical cells (arrowheads). The scale bar represents 100 μm. (E) Semiquantitative reverse transcription PCR analysis of liver tissue from mice 70 days after injection with pKT2/FAHIL-NRAS and pT2/sh.p53-GFP (first 2 columns) shows expression of the delivered transgenes N-ras, FAH, and GFP, as well as a reduction in p53 levels. With pKT2/FAHIL-NRAS only (third and fourth columns), there is expression of N-ras and FAH, but GFP is not detected and p53 levels are normal. Untreated Fah−/− mice (fifth column) do not express the transgenes. The tumor markers alpha-fetoprotein and osteopontin are elevated with oncogene coexpression. The same reactions were run using samples not treated with reverse transcriptase enzyme as a control.
The p53 shRNA construct has alinked GFP transgene, and the tumor tissue from mice injected with both transposons was found to be strongly GFP positive, demonstrating the presence of the DNA construct encoding the p53 shRNA (Fig. 4C). Microscopic examination of the liver showed that 80% of the tissue consisted of cords of large, pleomorphic, anaplastic hepatocytes arranged in trabeculae and sheets forming nodules multifocally disrupted by variably sized cystic structures (glandular pattern) indicative of hepatocellular carcinoma (Fig. 4D). Small portions of compressed nonneoplastic liver parenchyma remained at the periphery of the tumor. Immuno-histological analysis showed that the tumor cells are positive for FAH, N-ras, and Ki67, a marker of cell proliferation, providing further evidence that tumor formation is linked to the transgene (Fig. 4D).
Reverse transcription PCR was conducted to further assess gene expression in the repopulated liver tissue (Fig. 4E). The analysis confirmed expression of transgenes that were codelivered, including FAH, N-ras, and GFP. Furthermore, p53 expression was reduced in the liver tissue of the mice receiving the shRNA. Two markers of liver cancer were examined by reverse transcription PCR; alpha-fetoprotein33 and osteopontin34 messenger RNA levels were increased in the mice receiving NRAS plus p53 shRNA oncogenes, and alpha-fetoprotein was increased in mice receiving NRAS alone. Taken together, the data indicate a causal relationship between FAH-mediated expansion of cells harboring an oncogene and tumor formation.
Discussion
Many approaches are used for the genetic manipulation of mammalian cells and tissues. Viral and nonviral vectors are capable of mediating gene transfer and expression in somatic tissues. Currently, however, only a relatively low proportion of genetically modified cells can be achieved after somatic modification of mammalian tissues, and most studies have focused on biological systems in which a limited frequency of gene transfer is sufficient to achieve an informative outcome. Germline transgenesis achieves genetic modification of all tissues in the test animal, but evaluation of tissue-specific effects demands a high degree of tissue specificity in regulating expression of the gene of interest.
We have devised a system that allows for the specific genetic modification of a high proportion of cells in the liver after somatic gene transfer. Our results demonstrate that transgenes linked to Fah can be stably expressed in mouse liver after repopulation of the FAH-deficient mouse liver by the initially transfected cells. Genes encoding luciferase, shRNAs, a human dominant-negative gene, and oncogenes were all expressed after proliferation of hepatocytes that were selected by the expression of the codelivered Fah gene. Our method of expression facilitates the stable genetic manipulation of the adult liver.
Luciferase coexpression enabled the tracking, in vivo, of liver cells as they repopulate. The correlation between number of cells transplanted and luciferase signal suggests that luciferase signal can be used as a surrogate marker of the number of cells with established FAH expression. Indeed, we have successfully applied FAH and luciferase coexpression constructs to measure the efficacy of two novel gene transfer methodologies in correcting the FAH mouse.12,17
Recent data suggest that shRNAs expressed in liver can saturate the endogenous RNA interference pathway and cause liver damage and fatality from inability of cells to produce microRNAs.35 Results presented here indicate that an shRNA targeting luciferase was expressed longterm throughout liver repopulation. Furthermore, both experimental and codon-modified constructs used the same shRNA sequences, which rules out the possibility that the observed 90% reduction in expression of the target gene was due to toxicity of the shRNA. Therefore, this result indicates that an shRNA can potently reduce target gene expression in vivo in regenerating and quiescent liver cells without causing lethality.
Liver repopulation of FAH-deficient mice occurred with coexpression of wild-type and Z alleles of hAAT. The lack of difference in the kinetics of repopulation may be a reflection that the disease progression in hAAT is a slow process; only a subset of humans homozygous for the Z allele develop liver disease by their fourth decade.27 Mice transgenic for the Z allele develop pathological inclusions of the mutant protein, but they also develop sheets of hAAT-negative tissue, suggesting selective outgrowth of this tissue.7 In transgenic mice, hepatocytes that contain protein inclusions have a seven-fold decreased proliferation compared with inclusion-devoid hepatocytes.36 In our model, FAH-mediated repopulation occurred with either Z or wild-type coexpression, indicating that any counterselection due to Z expression was not as strong as the positive selection of due to the FAH. Our FAH-mediated expression model does demonstrate, however, full manifestation of the pathological development of hepatocytes inclusions and also demonstrates FAH-positive cells that do not express the Z allele. Previous work indicates unknown genetic modifiers to liver disease progression.4 The FAH coexpression method would be ideal for examining candidate disease-modifying loci by knockdown or expression, for modulation of the disease severity or latency.
There is heightened interest in cancer genome projects that can identify somatic mutations in tumor cells; the result is a list of hundreds of putative oncogenes and tumor suppressors that will have to be characterized for function and significance in biologically relevant models.2 Here, we somatically express an oncogene and target a tumor suppressor gene to cause HCC. Although Fah−/− hepatocytes have an intrinsic propensity to develop HCC from accumulation of fumarylacetoacetate in FAH-de-void cells,30 the HCC observed in our study is linked to FAH expression, has a pronounced onset, and has distinct morphology. Thus, our method of gene expression presents a tool for screening of putative oncogenes for aggressiveness and for cancer phenotype in vivo.
Supplementary Material
Acknowledgments
We thank Perry B. Hackett for manuscript editing.
Supported by the Arnold and Mabel Beckman Foundation (K.J.W., S.C.E., and RS.M), theAlpha1 foundation (K.J.W., C.J.S., and S.C.E.),National Cancer Institute grants R01CA113636 (V.W.K. and D.A.L.) and P30CA077598 (I.M.), University of Minnesota Startup funding (X.W.), the Chinese National 863 Project grant 2006AA02Z474 (Z.H. and X.W.), N.I.H grant DK074561 (Y.C. and X.W.), the Medical Scientist Training Program (K.J.W.), the National Institute of General Medical Sciences (A.W.), and the University of Minnesota Doctoral Dissertation Fellowship (A.W.).
Abbreviations
- cDNA
complementary DNA
- FAH
fumaryl acetoacetate hydrolase
- GFP
green fluorescing protein
- hAAT
human 1-antitrypsin
- HCC
hepatocellular carcinoma
- IgG
immunoglobulin G
- HRP
horseradish peroxidase
- NTBC
2-(2-nitro-4-trifluoromethylbenzoyl)-1,3-cyclohexanedione
- PAS-D
periodic acid-Schiff’s staining with diastase pretreatment
- PBS
phosphate-buffered saline
- PCR
polymerase chain reaction
- SB
Sleeping Beauty
- shRNA
short hairpin RNA
Footnotes
Potential conflict of interest: Nothing to report.
Supplementary material for this article can be found on the HEPATOLOGY Web site (http://interscience.wiley.com/jpages/0270-9139/suppmat/index.html).
References
- 1.Barton MC, Stivers DN. Microarray analysis of hepatic-regulated gene expression: specific applications and nonspecific problems. HEPATOLOGY. 2002;35:727–729. doi: 10.1053/jhep.2002.32145. [DOI] [PubMed] [Google Scholar]
- 2.Haber DA, Settleman J. Cancer: drivers and passengers. Nature. 2007;446:145–146. doi: 10.1038/446145a. [DOI] [PubMed] [Google Scholar]
- 3.Zender L, Spector MS, Xue W, Flemming P, Cordon-Cardo C, Silke J, et al. Identification and validation of oncogenes in liver cancer using an integrative oncogenomic approach. Cell. 2006;125:1253–1267. doi: 10.1016/j.cell.2006.05.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wu Y, Whitman I, Molmenti E, Moore K, Hippenmeyer P, Perlmutter DH. A lag in intracellular degradation of mutant alpha 1-antitrypsin correlates with the liver disease phenotype in homozygous PiZZ alpha 1-antitrypsin deficiency. Proc Natl Acad Sci U S A. 1994;91:9014–9018. doi: 10.1073/pnas.91.19.9014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dycaico MJ, Felts K, Nichols SW, Geller SA, Sorge JA. Neonatal growth delay in alpha-1-antitrypsin disease. Influence of genetic background. Mol Biol Med. 1989;6:137–141. [PubMed] [Google Scholar]
- 6.Dycaico MJ, Grant SG, Felts K, Nichols WS, Geller SA, Hager JH, et al. Neonatal hepatitis induced by alpha 1-antitrypsin: a transgenic mouse model. Science. 1988;242:1409–1412. doi: 10.1126/science.3264419. [DOI] [PubMed] [Google Scholar]
- 7.Geller SA, Nichols WS, Dycaico MJ, Felts KA, Sorge JA. Histopathology of alpha 1-antitrypsin liver disease in a transgenic mouse model. HEPATOLOGY. 1990;12:40–47. doi: 10.1002/hep.1840120108. [DOI] [PubMed] [Google Scholar]
- 8.Mikkelsen JG, Yant SR, Meuse L, Huang Z, Xu H, Kay MA. Helper-independent sleeping beauty transposon-transposase vectors for efficient nonviral gene delivery and persistent gene expression in vivo. Mol Ther. 2003;8:654–665. doi: 10.1016/s1525-0016(03)00216-8. [DOI] [PubMed] [Google Scholar]
- 9.Yant SR, Meuse L, Chiu W, Ivics Z, Izsvak Z, Kay MA. Somatic integration and long-term transgene expression in normal and haemophilic mice using a DNA transposon system. Nat Genet. 2000;25:35–41. doi: 10.1038/75568. [DOI] [PubMed] [Google Scholar]
- 10.Liu Y, Thor A, Shtivelman E, Cao Y, Tu G, Heath TD, et al. Systemic gene delivery expands the repertoire of effective antiangiogenic agents. J Biol Chem. 1999;274:13338–13344. doi: 10.1074/jbc.274.19.13338. [DOI] [PubMed] [Google Scholar]
- 11.Zhang G, Budker V, Wolff JA. High levels of foreign gene expression in hepatocytes after tail vein injections of naked plasmid DNA. Hum Gene Ther. 1999;10:1735–1737. doi: 10.1089/10430349950017734. [DOI] [PubMed] [Google Scholar]
- 12.Balciunas D, Wangensteen KJ, Wilber A, Bell J, Geurts A, Sivasubbu S, et al. Harnessing a high cargo-capacity transposon for genetic applications in vertebrates. PLoS Genet. 2006;2:e169. doi: 10.1371/journal.pgen.0020169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Held PK, Olivares EC, Aguilar CP, Finegold M, Calos MP, Grompe M. In vivo correction of murine hereditary tyrosinemia type I by phiC31 integrase-mediated gene delivery. Mol Ther. 2005;11:399–408. doi: 10.1016/j.ymthe.2004.11.001. [DOI] [PubMed] [Google Scholar]
- 14.Montini E, Held PK, Noll M, Morcinek N, Al-Dhalimy M, Finegold M, et al. In vivo correction of murine tyrosinemia type I by DNA-mediated transposition. Mol Ther. 2002;6:759–769. doi: 10.1006/mthe.2002.0812. [DOI] [PubMed] [Google Scholar]
- 15.Yant SR, Park J, Huang Y, Mikkelsen JG, Kay MA. Mutational analysis of the N-terminal DNA-binding domain of sleeping beauty transposase: critical residues for DNA binding and hyperactivity in mammalian cells. Mol Cell Biol. 2004;24:9239–9247. doi: 10.1128/MCB.24.20.9239-9247.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wilber A, Frandsen JL, Wangensteen KJ, Ekker SC, Wang X, McIvor RS. Dynamic gene expression after systemic delivery of plasmid DNA as determined by in vivo bioluminescence imaging. Hum Gene Ther. 2005;16:1325–1332. doi: 10.1089/hum.2005.16.1325. [DOI] [PubMed] [Google Scholar]
- 17.Wilber A, Wangensteen KJ, Chen Y, Zhuo L, Frandsen JL, Bell JB, et al. Messenger RNA as a source of transposase for sleeping beauty transposon-mediated correction of hereditary tyrosinemia type I. Mol Ther. 2007;15:1280–1287. doi: 10.1038/sj.mt.6300160. [DOI] [PubMed] [Google Scholar]
- 18.Paddison PJ, Caudy AA, Bernstein E, Hannon GJ, Conklin DS. Short hairpin RNAs (shRNAs) induce sequence-specific silencing in mammalian cells. Genes Dev. 2002;16:948–958. doi: 10.1101/gad.981002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ohlfest JR, Lobitz PD, Perkinson SG, Largaespada DA. Integration and long-term expression in xenografted human glioblastoma cells using a plasmid-based transposon system. Mol Ther. 2004;10:260–268. doi: 10.1016/j.ymthe.2004.05.005. [DOI] [PubMed] [Google Scholar]
- 20.Dickins RA, Hemann MT, Zilfou JT, Simpson DR, Ibarra I, Hannon GJ, et al. Probing tumor phenotypes using stable and regulated synthetic microRNA precursors. Nat Genet. 2005;37:1289–1295. doi: 10.1038/ng1651. [DOI] [PubMed] [Google Scholar]
- 21.Grompe M, al-Dhalimy M, Finegold M, Ou CN, Burlingame T, Kennaway NG, et al. Loss of fumarylacetoacetate hydrolase is responsible for the neonatal hepatic dysfunction phenotype of lethal albino mice. Genes Dev. 1993;7:2298–2307. doi: 10.1101/gad.7.12a.2298. [DOI] [PubMed] [Google Scholar]
- 22.Grompe M, Lindstedt S, al-Dhalimy M, Kennaway NG, Papaconstantinou J, Torres-Ramos CA, et al. Pharmacological correction of neonatal lethal hepatic dysfunction in a murine model of hereditary tyrosinaemia type I. Nat Genet. 1995;10:453–460. doi: 10.1038/ng0895-453. [DOI] [PubMed] [Google Scholar]
- 23.Ohlfest JR, Frandsen JL, Fritz S, Lobitz PD, Perkinson SG, Clark KJ, et al. Phenotypic correction and long-term expression of factor VIII in hemophilic mice by immunotolerization and nonviral gene transfer using the Sleeping Beauty transposon system. Blood. 2005;105:2691–2698. doi: 10.1182/blood-2004-09-3496. [DOI] [PubMed] [Google Scholar]
- 24.Overturf K, Al-Dhalimy M, Tanguay R, Brantly M, Ou CN, Finegold M, et al. Hepatocytes corrected by gene therapy are selected in vivo in a murine model of hereditary tyrosinaemia type I. Nat Genet. 1996;12:266–273. doi: 10.1038/ng0396-266. [DOI] [PubMed] [Google Scholar]
- 25.McCaffrey AP, Meuse L, Pham TT, Conklin DS, Hannon GJ, Kay MA. RNA interference in adult mice. Nature. 2002;418:38–39. doi: 10.1038/418038a. [DOI] [PubMed] [Google Scholar]
- 26.Zuker M. Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res. 2003;31:3406–3415. doi: 10.1093/nar/gkg595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Perlmutter DH. Alpha-1-antitrypsin deficiency: diagnosis and treatment. Clin Liver Dis. 2004;8:839–859. viii–ix. doi: 10.1016/j.cld.2004.06.001. [DOI] [PubMed] [Google Scholar]
- 28.Carrell RW, Lomas DA. Alpha1-antitrypsin deficiency: a model for conformational diseases. N Engl J Med. 2002;346:45–53. doi: 10.1056/NEJMra010772. [DOI] [PubMed] [Google Scholar]
- 29.Moradpour D, Blum HE. Pathogenesis of hepatocellular carcinoma. Eur J Gastroenterol Hepatol. 2005;17:477–483. doi: 10.1097/00042737-200505000-00002. [DOI] [PubMed] [Google Scholar]
- 30.Grompe M, Overturf K, al-Dhalimy M, Finegold M. Therapeutic trials in the murine model of hereditary tyrosinaemia type I: a progress report. J Inherit Metab Dis. 1998;21:518–531. doi: 10.1023/a:1005462804271. [DOI] [PubMed] [Google Scholar]
- 31.Jorquera R, Tanguay RM. The mutagenicity of the tyrosine metabolite, fumarylacetoacetate, is enhanced by glutathione depletion. Biochem Biophys Res Commun. 1997;232:42–48. doi: 10.1006/bbrc.1997.6220. [DOI] [PubMed] [Google Scholar]
- 32.Billadeau D, Liu P, Jelinek D, Shah N, LeBien TW, Van Ness B. Activating mutations in the N- and K-ras oncogenes differentially affect the growth properties of the IL-6-dependent myeloma cell line ANBL6. Cancer Res. 1997;57:2268–2275. [PubMed] [Google Scholar]
- 33.Peng SY, Lai PL, Chu JS, Lee PH, Tsung PT, Chen DS, et al. Expression and hypomethylation of alpha-fetoprotein gene in unicentric and multicentric human hepatocellular carcinomas. HEPATOLOGY. 1993;17:35–41. [PubMed] [Google Scholar]
- 34.Gotoh M, Sakamoto M, Kanetaka K, Chuuma M, Hirohashi S. Overexpression of osteopontin in hepatocellular carcinoma. Pathol Int. 2002;52:19–24. doi: 10.1046/j.1440-1827.2002.01316.x. [DOI] [PubMed] [Google Scholar]
- 35.Grimm D, Streetz KL, Jopling CL, Storm TA, Pandey K, Davis CR, et al. Fatality in mice due to oversaturation of cellular microRNA/short hairpin RNA pathways. Nature. 2006;441:537–541. doi: 10.1038/nature04791. [DOI] [PubMed] [Google Scholar]
- 36.Rudnick DA, Liao Y, An JK, Muglia LJ, Perlmutter DH, Teckman JH. Analyses of hepatocellular proliferation in a mouse model of alpha-1-antitrypsin deficiency. HEPATOLOGY. 2004;39:1048–1055. doi: 10.1002/hep.20118. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.