

Abstract
Purpose of review
To provide an overview of recent research of how HIV integration relates to productive and latent infection and implications for cure strategies.
Recent findings
How and where HIV integrates provides new insights into how HIV persists on antiretroviral therapy (ART). Clonal expansion of infected cells with the same integration site demonstrates that T cell proliferation is an important factor in HIV persistence, however the driver of proliferation remains unclear. Clones with identical integration sites harbouring defective provirus can accumulate in HIV-infected individuals on ART and defective proviruses can express RNA and produce protein. HIV integration sites differ in clonally expanded and non-expanded cells and in latently and productively infected cells and this influences basal and inducible transcription. There is a growing number of cellular proteins that can alter the pattern of integration to favour latency. Understanding these pathways may identify new interventions to eliminate latently infected cells.
Summary
Using advances in analysing HIV integration sites, T cell proliferation of latently infected cells is thought to play a major role in HIV persistence. Clonal expansion has been demonstrated with both defective and intact viruses. Production of viral RNA and protein from defective viruses may play a role in driving chronic immune activation. The site of integration may determine the likelihood of proliferation and the degree of basal and induced transcription.
Finally, host factors and gene expression at the time of infection may determine the integration site. Together these new insights may lead to novel approaches to elimination of latently infected cells.
Keywords: HIV integration, clonal expansion, HIV latency, HIV reservoir, HIV persistence
Introduction
Integration of HIV into the host genome is a critical step in the HIV life cycle. Following HIV integration, productive or latent infection can be established. While productively infected cells have a short half life, latency is established in cells that are long lived or can undergo proliferation [1, 2]. These latently infected cells persist on ART and are the main barrier to an effective cure once ART is ceased [3-5].
Recently, our understanding of latency has been significantly changed. In productive infection, it was shown many years ago that HIV preferentially integrates into genes [6, 7] and the integration site is heterogenous [8]. In contrast, in CD4+ T-cells from HIV-infected individuals on ART, there is expansion of cells with HIV integration at specific sites or in specific genes [8-10]. The driver for expansion on ART of cells with ia specific integration sites, remains a critical unanswered question and could be due to homeostatic proliferation [1] although this would not explain why one infected cell is favoured over another, antigen-driven expansion or alternatively, a consequence of the site of integration (reviewed in [11]). For example, the site of integration may render a cell less susceptible to apoptosis or increase the likelihood of clonal expansion.
One of the major controversies has been whether proliferation of infected cells with a specific integration site was possible with an intact rather than a defective virus. A defective provirus is defined by the presence of inversions, hypermutation, premature stop-codons, large internal deletions, frameshift mutation, packaging signal mutation or mutations in the major splice donor site [12, 13]. One of the earlier studies of integration sites in CD4+ T-cells from individuals on ART demonstrated that all clonally expanded cells harboured defective provirus [8]. A subsequent study analysing cells from an HIV-infected individual on ART with malignancy, intact virus was sequenced from a specific clone and full length construction of this virus clearly showed the virus was replication competent [14]. Subsequently, other groups sequenced RNA from supernatants collected in viral outgrowth assays (VOA) using CD4+ T-cells from HIV infected individuals on ART and demonstrated that the majority of intact replication competent virus was derived from clonally expanded cells [15-17]. Furthermore, the majority of intact virus was found in effector memory, rather than central memory T-cells [12].
What remains unclear, is the overall contribution of the clonally expanded cells with intact or replication competent virus to the total reservoir, or more importantly to the virus that rebounds post ART cessation. This remains an extremely important question for the field to answer. In addition, if clonally expanded cells contain intact or replication competent virus, how can these cells divide without virus activation and cell death? What allows these cells to survive and proliferate? If clonal expansion is the major factor maintaining latently infected cells on ART, why don’t we see an increase in latently infected cells over time, instead of steady state or a slight decrease over time? [18]. Finally, if clonal expansions are stochastic and there are expansions and then contractions, what factors are regulating this?
The role of replication competent and defective virus in clonal expansion
Defective viruses and their potential function
Defective proviruses rapidly accumulate on prolonged ART with >95% of proviruses being defective and not replication competent [19, 20]. In a recent case study of an HIV-infected individual on ART with an exceptionally high frequency of cells with integrated HIV (31,070 per million CD4+ T cells), which was several logs higher than the frequency of cells with replication competent virus as measured by a modified quantitative viral outgrowth assay (mQVOA) or Tat/rev Induced Limiting Dilution Assay (TILDA), the vast majority of integrated virus was defective [21]. Single-genome sequencing of the viral envelope revealed a unique sequence and integration site sequencing demonstrated that 83% of effector memory cells contained virus integrated in chromosome 14, in the Ribosomal Protein S6 Kinase A5 (RPS6KA5) gene [21] required for stress induced phosphorylation of transcription factors [22].
Insights into the kinetics of clonally expanded latently infected cells on ART can be determined using site directed PCR, rather than sequencing. Using a multiplex droplet digital PCR that quantified proviral DNA with separate primers in the LTR, gag, tat/rev exons 1 and 2, the LTR:Gag ratio increased after a median of 14.4 years on ART consistent with an increase in defective virus [23]. In this study, one integration site, which represented 20% of the total integration sites, was located in the HORMA Domain Containing 2 (HORMAD2) gene, essential for synapsis surveillance during meiosis and known to be defective [23]. This clone was detected after six months on ART and the expansion over time of this clone, suggests a survival advantage or some other form of selection [23]. One explanation could be that cells that contain replication competent virus, or express viral proteins are preferentially lost over time, potentially due to immune recognition and therefore virus with large internal deletions will increase over time [24].
Although defective viruses are not able to replicate upon cessation of ART, defective provirus may still be transcriptionally active and produce viral proteins [24-26]. In another case study of an HIV-infected individual on ART, direct amplicon sequencing of the genomic DNA revealed a dominant clone with a 2.4kb deletion leaving Gag, Pol and Nef intact [26]. This partially deleted provirus could still produce Gag and Nef proteins as detected by western blot and flow cytometry [26]. Persistent antigen presentation could potentially drive chronic immune activation and CD8+ T-cell exhaustion, as shown recently [24]. Together these data demonstrate that defective proviruses accumulate during ART, and although not replication competent, these defective viruses can still produce HIV proteins, and potentially contribute to adverse outcomes (figure 1). Therefore, we might not be able to completely ignore defective viruses that persist on ART.
Figure 1.
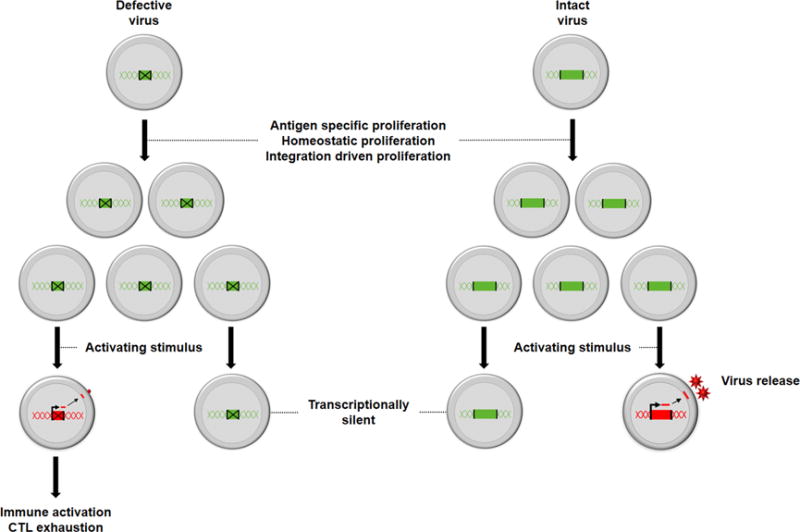
The different fates of cells that are latently infected with defective and intact virus. Latently infected cells with intact or defective virus can undergo proliferation. Following activation, defective virus (green with cross) is unable to produce replication competent virus. but can produce cell associated HIV RNA and protein leading to immune activation and cytotoxic lymphocyte (CTL) exhaustion. Intact virus (green) can produce and release virions leading to a persistent pool of replication competent viruses that can rebound once ART is stopped. In the absence of an activation stimulus, both intact and defective virus can persist in a latent form.
Intact viruses and clonal expansion
Over the last 12 months, multiple groups have now clearly demonstrated that expanded clones of infected cells on ART, can also contain intact virus, which is replication competent [14-17]. In one individual with a squamous cell carcinoma, a single infected clone with intact replication competent virus accounted for 4% of the total number of infected cells [9, 14]. Using single cell RNA sequencing, they showed that a fraction of these clones could express HIV RNA whereas the majority >95% remained latently infected [27]. Interestingly, this clones of intact and defective infected cells had a similar level of expression of cell associated HIV RNA [27].
Another way to determinethe relationship of HIV RNA expression to integration sites or clonal expansion, is to assess individuals with drug resistant virus [28]. In a recent longitudinal study of individuals on ART infected with drug resistant and wild type virus, 4.5% of cells infected with wild type virus expressed cell associated HIV RNA at low levels, whereas almost 20% of cells infected with drug resistant provirus expressed high levels of cell associated HIV RNA, potentially representing recent infection of these cells [28]. This data also shows that some clonally expanded cells with replication competent viruses can produce virus while other infected cells remain latent (figure 1). Additionally, the study demonstrated that the chromatin environment surrounding the same integration site in clonally expanded cells can differ which will lead to different outcomes for the infected cell.
Using Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analysis [29] we recently demonstrated using CD4+ T-cells from HIV-infected individuals on ART, that clonally expanded cells with identical integration sites were in pathways associated with evasion of apoptosis and cell proliferation [30]. Further studies are still needed to show a direct link between the site of HIV integration and an effect on proliferation or cell survival and thereby likelihood of clonal expansion.
Models for studying HIV integration sites and impact on transcription
The chromatin environment surrounding an HIV integration site critically determines how much HIV RNA is expressed. For example, an increase in histone acetylation or a reduction in methylation will result in an increase in HIV transcription [31, 32]. Modification of the chromatin environment with latency reversing agents (LRA) is one approach being investigated to eliminate latency (reviewed in [33-35]. Therefore, response of a latently infected cells to an LRA may be directly related to the site of HIV integration.
Using an adapted dual fluorescent reporter virus (with Green fluorescent protein (conformation-sensitive, csGFP) expression following productive infection and monomeric Kusabira-Orange (mKO) fluorescent protein following integration (GKO)), both latent and productive infection can be quantified in vitro [36]. In some cells latently infected cells, virus could be activated to express GFP with a stimulus such as anti CD3/anti CD28 or an LRA [33, 37, 38] (inducible latency) while in other cells, there was HIV integration, but csGFP expression was not observed (non-inducible latency) [39]. In this model, non-induced latent viruses were found more commonly in intergenic regions and further away from activation enhancers and active transcribed regions [40]. Interestingly, non-induced viruses compared to productive infection were more frequently located in lamin-associated domains (LADs) [40]. Furthermore, HIV can be integrated in the same gene, referred to as recurrent integrated genes (RIG) and these are usually outside LADs [7].
In response to the histone deacetylase inhibitor (HDACi) vorinostat, an LRA with low potency, only 5% of cells produced the productive expression marker, consistent with insufficient HIV RNA transcription and/or HIV protein production, as previously described [33, 41, 42]. It is possible that the absence of Nef in the reporter viruses may be relevant and lead to reduced virus expression. Following the removal of Nef, using Clustered Regularly Short Palindromic Repeats/CRISPR-associated system 9 (CRISPR/Cas9) in a T cell line [43] infected with envelope deleted fluorescent reporter HIV, there was a reduction in fluorescent reporter protein after activation with LRAs [44].
Another approach used CRISPR/Cas9 to introduce an HIV-derived reporter cassette in a known integration site. In this study, the investigators introduced HIV in a T cell line, at two sites in the BACH2 gene at intron 5, a well described RIG [9]. Integration at these sites led to the production of the reporter protein after LRA administration [45]. The utilization of CRISPR/Cas9 to knock in an HIV reporter construct at a particular position in the human genome opens the possibility of investigating the functional impact of a clonally expanded specific integration site.
Assessment of integration sites using human tissue and animal models
Studying HIV integration in latently infected cells in vitro, using cell lines or primary CD4+ T cells, does not necessarily recapitulate what happens in vivo, where there are multiple other factors driving HIV persistence including proliferation and the location of infected cells in tissue [30, 46, 47]. In matched blood and rectal tissue samples from two HIV infected individuals on ART, we observed that clonal expansion of CD4+ T cells as measured by HIV integration sites was lower in rectal tissues compared to blood [30]. Further work is needed in matched blood and tissue samples from HIV infected individuals on ART, to determine whether the driver for clonal expansion is site specific.
Similar to studies in HIV-infected individuals on ART [8-10, 30], in SIV infected rhesus macaques (RM) on suppressive ART, SIV was integrated in highly transcribed genes and gene rich regions with evidence of clonal expansion [48]. In contrast to human studies where access to tissue is limited, here multiple compartments were examined including blood, lymph node and spleen. Similar levels of clonal expansion of cells with identical integration sites were found in two or more different sites [48]. The similarity of HIV integration patterns in RM and human studies is an important finding and will hopefully allow for a far more extensive understanding of factors driving clonal expansion in tissues, including the central nervous system.
Host factor control of HIV integration
It is now apparent that interactions of the virus with specific host proteins will influence the site of HIV integration. Specifically, microtubules, the nuclear pore proteins and host proteins such as Lens Epithelium-Derived Growth Factor (LEDGF) can determine where HIV integrates which in turn can determines the transcriptional activity of the integrated provirus.
HIV entry into the nucleus
After HIV binds to the CD4 receptor and chemokine co-receptor on the target cell, the viral capsid is released into the cytoplasm. The HIV capsid traffics to the nuclear pore through interaction with microtubules [49-51]. Dharan et al. demonstrated that a microtubular adaptor protein bicaudual D homolog 2 (BICD2) binds the HIV capsid [52]. CRISPR/Cas9 knock-out of BICD2 showed that movement of the viral core to the nucleus was disrupted and postulated that this interaction would be a novel therapeutic target to inhibit integration [52] (figure 2).
Figure 2.
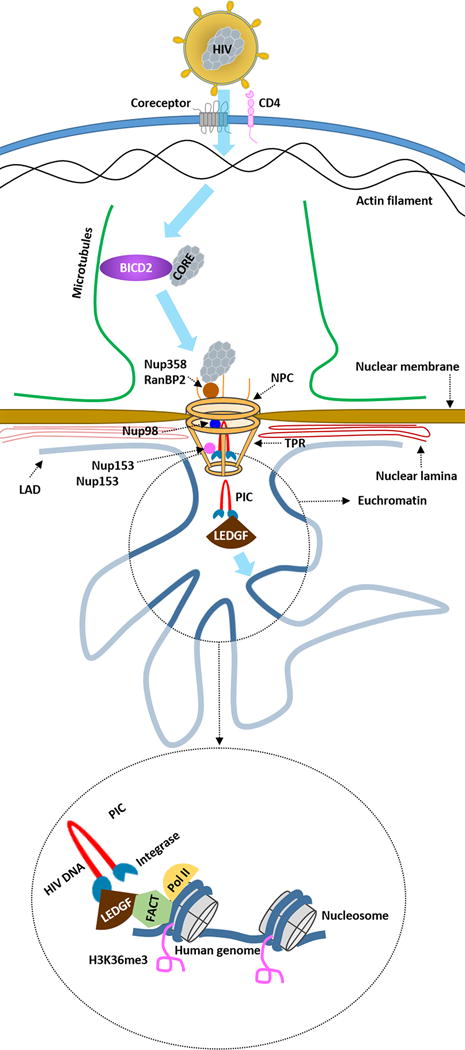
Schematic overview of host factors that have a direct impact on the site of HIV integration and basal and inducible transcription of the integrated provirus. BICD2 = bicaudual D homolog 2; NPC = Nuclear Pore Complex; Nup = Nucleoporin; RanBP2= RAN Binding Protein 2; LAD = lamin-associated domains; TPR= Translocated Promoter Region; PIC = Preintegration complex; LEDGF = Lens Epithelium-Derived Growth Factor; FACT = Facilitates Chromatin Transcription; Pol II = RNA polymerase II; H3K36me3 = trimethylated histone H3 at lysine 36.
If normal trafficking of HIV to the nucleus occurs, the viral capsid docks with the nuclear pore complex through the nuclear pore protein Nup358/RanBP2. Subsequently, Nup153 and Nup98 interact with the viral pre-integration complex (PIC) and transport the HIV genome across an intact nuclear membrane [53-55]. Nup153 is thought to interact with the PIC and anchors itself to Translocated Promoter Region (Tpr), a nuclear basket protein [56]. One important function of Tpr is exclusion of heterochromatin near the nuclear pore and it is thought that Tpr recruits highly transcribed genes to the immediate proximity of the nuclear pore [57]. Knock down of Tpr by short hairpin RNA (shRNA) in a T cell line demonstrated that HIV integration occurred more distal from H3K36me3 chromatin marks, which are associated with actively transcribed genes [58]. Additionally knockdown of Tpr led to HIV integration in genes involved in in cell cycle, DNA repair and transcription [59] (figure 2).
Host factors guiding HIV integration to active transcriptional sites
Beyond the nuclear pore proteins there are other host proteins that are critically important for HIV integration, one of the most important being, LEDGF/p75 [60]. LEDGF/p75 interacts with the PIC by binding HIV integrase at one end and host chromatin at the other end, to tether the PIC to the target host genome [61]. LEDGF/p75 targets vesicular stomatitis virus protein G (VSV-g) pseudotyped HIV, in HEK293T cells, to intron-dense, highly spliced genes that are transcribed by RNA polymerase II [62], which has been proven to be a co-factor in HIV integration [63, 64].
The Facilitates chromatin transcription (FACT) complex was recently shown to bind to LEDGF/p75 [65]. FACT concentrates in the vicinity of HIV integration sites and RNA polymerase II in vitro [64] (figure 2). It is thought that FACT remodels the chromatin structure generating partially dissociated nucleosomes leading to chromatin structures that favour both RNA transcription and HIV integration [64].
Inhibition of the interaction between LEDGF/p75 and HIV integrase with compounds called LEDGINS strongly inhibits infectivity of HIV in vitro [66]. However, LEDGINs also changed the patterns of HIV integration to sites more distal from epigenetic markers associated with transcription [67, 68]. Interestingly, HIV infection in the presence of LEDGIN reduced the response of the infected cell to LRAs [68]. In summary, it is now clear that multiple host proteins can guide HIV integration to a specific site. In turn, these sites may enhance or inhibit HIV transcription therefore having a significant effect on latency or the response to an LRA. Given that latency is established within days of infection, it remains unclear whether targeting these early events will translate into an effective intervention.
Conclusions and implications
In conclusion, over the past 12 months, our understanding of the features of HIV integration in latently infected cells has advanced significantly. First, it is now clear that expanded clones of infected cells with identical integration sites can harbour both intact and defective viruses. Second, the location of integration influences basal and inducible transcription which has significant implications for activity of LRAs. Finally, multiple host proteins determine the site of integration and could be potential new targets to inhibit the establishment of latency.
Clonal expansion of infected cells that harbour intact replication competent are a major challenge for strategies aimed at eliminating latency [27, 40]. It will be critical to understand the drivers for expansion in order to develop a targeted intervention. [9, 10, 30, 45]. One potential approach is to inhibit proliferation of infected clones could be the inhibition of Janus Kinase I (JAK-I) which was shown to inhibit seeding of the viral reservoir and blocked proliferation of infected cells resulting in a reduction of infected cells in vitro [69]. Whether this will translate to in vivo efficacy remains to be determined. New interventions could also be developed to inhibit protein expression from clonally expanded cells infected with a defective virus [24-26, 70] as a potential strategy to reduce chronic immune activation which is detrimental for individuals living with HIV [71]. Inhibition of tat induced transcription could inhibit production of replication competent and defective viral RNA thereby reducing HIV antigen presentation [72]. Finally, a detailed understanding of all the events that lead to HIV integration could identify new targets for interventions, which may be important if there is ongoing infection of cells on ART [73, 74].
In summary, we have learnt that far more mechanistic studies are needed to understand HIV integration, HIV transcription and clonal expansion of infected cells in individuals on ART. Insights from T cell biologists who understand the fundamental drivers for T cell memory and homeostasis may prove to be helpful. Furthermore, the availability and application of new bioinformatic tools, single cell sequencing, CRISP/Cas9 libraries, dual reporter viruses and robust animal models will likely accelerate our understanding of HIV persistence of ART and lead to new interventions.
Key bullet points.
Defective provirus accumulates during prolonged ART.
Defective provirus can produce HIV RNA, viral protein and contributes to chronic immune activation.
HIV transcription levels differs in clonally expanded cells with the same integration sites.
Integration sites in SIV infected rhesus macaques demonstrate a similar pattern as integration site patterns in humans.
Inhibition of LEDGF/p75, HIV pre-integration complex interaction results in an HIV integration patterns resembling latent HIV integration site patterns.
Acknowledgments
We would like to thank Dr Ulrike Lange and Dr Francesca Di Nunzio for providing information for this review.
Financial support and sponsorship
This work was supported by a program grant from the Australian NHMRC; the National Institute of Allergy and Infectious Disease (NIAID), US National Institutes of Health (NIH) (Delaney AIDS Research Enterprise, DARE; UM1AI126611) and the American Foundation for AIDS Research.
Footnotes
Conflicts of interest
S.R.L.’s institution has received funding from Merck, Viiv, Gilead and Tetralogic for investigator initiated research and from Merck, Viiv and Gilead for educational activities. The remaining authors have no conflicts of interest.
REFERENCES AND RECOMMENDED READING
Papers of particular interest, published within the annual period of review, have been highlighted as: ■ of special interest ■■ of outstanding interest
- 1.Chomont N, El-Far M, Ancuta P, et al. HIV reservoir size and persistence are driven by T cell survival and homeostatic proliferation. Nature medicine. 2009;15(8):893–900. doi: 10.1038/nm.1972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chun T-W, Stuyver L, Mizell SB, et al. Presence of an inducible HIV-1 latent reservoir during highly active antiretroviral therapy. Proceedings of the National Academy of Sciences. 1997;94(24):13193–7. doi: 10.1073/pnas.94.24.13193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chun T-W, Davey RT, Jr, Engel D, et al. AIDS: re-emergence of HIV after stopping therapy. Nature. 1999;401(6756):874. doi: 10.1038/44755. [DOI] [PubMed] [Google Scholar]
- 4.Chun TW, Stuyver L, Mizell SB, et al. Presence of an inducible HIV-1 latent reservoir during highly active antiretroviral therapy. Proc Natl Acad Sci U S A. 1997;94(24):13193–7. doi: 10.1073/pnas.94.24.13193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Finzi D, Hermankova M, Pierson T, et al. Identification of a reservoir for HIV-1 in patients on highly active antiretroviral therapy. Science. 1997;278(5341):1295–300. doi: 10.1126/science.278.5341.1295. [DOI] [PubMed] [Google Scholar]
- 6.Schröder AR, Shinn P, Chen H, et al. HIV-1 integration in the human genome favors active genes and local hotspots. Cell. 2002;110(4):521–9. doi: 10.1016/s0092-8674(02)00864-4. [DOI] [PubMed] [Google Scholar]
- 7.Marini B, Kertesz-Farkas A, Ali H, et al. Nuclear architecture dictates HIV-1 integration site selection. Nature. 2015;521(7551):227–31. doi: 10.1038/nature14226. [DOI] [PubMed] [Google Scholar]
- 8.Cohn LB, Silva IT, Oliveira TY, et al. HIV-1 integration landscape during latent and active infection. Cell. 2015;160(3):420–32. doi: 10.1016/j.cell.2015.01.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Maldarelli F, Wu X, Su L, et al. Specific HIV integration sites are linked to clonal expansion and persistence of infected cells. Science. 2014;345(6193):179–83. doi: 10.1126/science.1254194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wagner TA, McLaughlin S, Garg K, et al. Proliferation of cells with HIV integrated into cancer genes contributes to persistent infection. Science. 2014;345(6196):570–3. doi: 10.1126/science.1256304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kwon KJ, Siliciano RF. HIV persistence: clonal expansion of cells in the latent reservoir. The Journal of Clinical Investigation. 2017;127(7) doi: 10.1172/JCI95329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hiener B, Horsburgh BA, Eden J-S, et al. Identification of Genetically Intact HIV-1 Proviruses in Specific CD4+ T Cells from Effectively Treated Participants. Cell Reports. 2017;21(3):813–22. doi: 10.1016/j.celrep.2017.09.081. ■■ This study demonstrated that clonally expanded cells can harbour intact replication competent HIV. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bruner KM, Murray AJ, Pollack RA, et al. Defective proviruses rapidly accumulate during acute HIV-1 infection. Nature medicine. 2016;22(9):1043–9. doi: 10.1038/nm.4156. ■ This study demonstrated that defective provirus are a large part of the viral reservoir. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Simonetti FR, Sobolewski MD, Fyne E, et al. Clonally expanded CD4+ T cells can produce infectious HIV-1 in vivo. Proceedings of the National Academy of Sciences. 2016;113(7):1883–8. doi: 10.1073/pnas.1522675113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bui JK, Sobolewski MD, Keele BF, et al. Proviruses with identical sequences comprise a large fraction of the replication-competent HIV reservoir. PLoS pathogens. 2017;13(3):e1006283. doi: 10.1371/journal.ppat.1006283. ■■ This study demonstrated over 50% of virus obtained from VAO were expanded clones. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hosmane NN, Kwon KJ, Bruner KM, et al. Proliferation of latently infected CD4+ T cells carrying replication-competent HIV-1: Potential role in latent reservoir dynamics. Journal of Experimental Medicine. 2017 doi: 10.1084/jem.20170193. jem.20170193. ■ This study also demonstrated proliferation of cells infected with intact virus and that intact virus often required more than one round of stimulation to be expressed. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Freund NT, Wang H, Scharf L, et al. Coexistence of potent HIV-1 broadly neutralizing antibodies and antibody-sensitive viruses in a viremic controller. Science translational medicine. 2017;9(373):eaal2144. doi: 10.1126/scitranslmed.aal2144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Siliciano JD, Kajdas J, Finzi D, et al. Long-term follow-up studies confirm the stability of the latent reservoir for HIV-1 in resting CD4+ T cells. Nature medicine. 2003;9(6):727–8. doi: 10.1038/nm880. [DOI] [PubMed] [Google Scholar]
- 19.Bruner K, Pollack R, Murray A, et al. Rapid accumulation of defective proviruses complicates HIV-1 reservoir measurements. Nat Med. 2016 [Google Scholar]
- 20.Ho Y-C, Shan L, Hosmane NN, et al. Replication-competent noninduced proviruses in the latent reservoir increase barrier to HIV-1 cure. Cell. 2013;155(3):540–51. doi: 10.1016/j.cell.2013.09.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Fromentin R, et al. In vivo massive expansion of a T-cell clone carrying a defective HIV genome: implication for the measurement of the HIV reservoir. 9th IAS Conference on HIV Science (IAS 2017); Paris, France. 2017. p. MOPEA0067. [Google Scholar]
- 22.Deak M, Clifton AD, Lucocq JM, Alessi DR. Mitogen-and stress-activated protein kinase-1 (MSK1) is directly activated by MAPK and SAPK2/p38, and may mediate activation of CREB. The EMBO journal. 1998;17(15):4426–41. doi: 10.1093/emboj/17.15.4426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Anderson E, Hill S, Bell J, et al., editors. JOURNAL OF THE INTERNATIONAL AIDS SOCIETY. INT AIDS SOCIETY AVENUE DE FRANCE 23; GENEVA, 1202, SWITZERLAND: 2017. Accumulation and persistence of deleted HIV proviruses following prolonged ART. [Google Scholar]
- 24.Pollack RA, Jones RB, Pertea M, et al. Defective HIV-1 Proviruses Are Expressed and Can Be Recognized by Cytotoxic T Lymphocytes, which Shape the Proviral Landscape. Cell Host & Microbe. 2017;21(4):494–506.e4. doi: 10.1016/j.chom.2017.03.008. ■■ This study demonstrated that defective proviruses can still produce protein and be recognized by CTLs through MHC-I thereby selecting for proviruses with large internal deletions. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Imamichi H, Dewar RL, Adelsberger JW, et al. Defective HIV-1 proviruses produce novel protein-coding RNA species in HIV-infected patients on combination antiretroviral therapy. Proceedings of the National Academy of Sciences. 2016;113(31):8783–8. doi: 10.1073/pnas.1609057113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Imamichi H, Smith M, Pau A, et al., editors. JOURNAL OF THE INTERNATIONAL AIDS SOCIETY. INT AIDS SOCIETY AVENUE DE FRANCE 23; GENEVA, 1202, SWITZERLAND: 2017. Evidence of production of HIV-1 proteins from “defective” HIV-1 proviruses in vivo: Implication for persistent immune activation and HIV-1 pathogenesis. [Google Scholar]
- 27.Kearney MF, et al. Characterizing HIV Expression in vivo. Conference on Retroviruses and Oppertunistic Infections; Seattle, Washington. 2017. ■ This study demonstrated that clonally expanded infected cells do not uniformly express HIV RNA and that there was no difference in HIV RNA expression from defective or intact HIV. [Google Scholar]
- 28.Musick A, Spindler J, Sobolewski M, et al., editors. JOURNAL OF THE INTERNATIONAL AIDS SOCIETY. INT AIDS SOCIETY AVENUE DE FRANCE 23; GENEVA, 1202, SWITZERLAND: 2017. A higher fraction of drug-resistant proviruses express unspliced HIV RNA during ART compared to the archival wild-type proviruses that comprise the HIV-1 reservoir. [Google Scholar]
- 29.Wang J, Duncan D, Shi Z, Zhang B. WEB-based gene set analysis toolkit (WebGestalt): update 2013. Nucleic acids research. 2013;41(W1):W77–W83. doi: 10.1093/nar/gkt439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Symons J, Chopra A, Leary S, et al., editors. JOURNAL OF THE INTERNATIONAL AIDS SOCIETY. INT AIDS SOCIETY AVENUE DE FRANCE 23; GENEVA, 1202, SWITZERLAND: 2017. HIV integration sites in CD4 T cells from virally suppressed individuals show clonal expansion but no preferential location in oncogenes. [Google Scholar]
- 31.Van Lint C, Emiliani S, Ott M, Verdin E. Transcriptional activation and chromatin remodeling of the HIV-1 promoter in response to histone acetylation. The EMBO journal. 1996;15(5):1112. [PMC free article] [PubMed] [Google Scholar]
- 32.Bednarik DP, Cook J, Pitha P. Inactivation of the HIV LTR by DNA CpG methylation: evidence for a role in latency. The EMBO journal. 1990;9(4):1157. doi: 10.1002/j.1460-2075.1990.tb08222.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rasmussen TA, Lewin SR. Shocking HIV out of hiding: where are we with clinical trials of latency reversing agents? Current Opinion in HIV and AIDS. 2016;11(4):394–401. doi: 10.1097/COH.0000000000000279. [DOI] [PubMed] [Google Scholar]
- 34.Delagrèverie HM, Delaugerre C, Lewin SR, et al., editors. Open forum infectious diseases. Oxford University Press; 2016. Ongoing clinical trials of human immunodeficiency virus latency-reversing and immunomodulatory agents. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Rasmussen TA, Anderson JL, Wightman F, Lewin SR. Cancer therapies in HIV cure research. Current Opinion in HIV and AIDS. 2017;12(1):96–104. doi: 10.1097/COH.0000000000000328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chavez L, Calvanese V, Verdin E. HIV latency is established directly and early in both resting and activated primary CD4 T cells. PLoS pathogens. 2015;11(6):e1004955. doi: 10.1371/journal.ppat.1004955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Elliott JH, Wightman F, Solomon A, et al. Activation of HIV transcription with short-course vorinostat in HIV-infected patients on suppressive antiretroviral therapy. PLoS pathogens. 2014;10(11):e1004473. doi: 10.1371/journal.ppat.1004473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Rasmussen TA, Tolstrup M, Brinkmann CR, et al. Panobinostat, a histone deacetylase inhibitor, for latent-virus reactivation in HIV-infected patients on suppressive antiretroviral therapy: a phase 1/2, single group, clinical trial. The Lancet HIV. 2014;1(1):e13–e21. doi: 10.1016/S2352-3018(14)70014-1. [DOI] [PubMed] [Google Scholar]
- 39.Procopio FA, Fromentin R, Kulpa DA, et al. A novel assay to measure the magnitude of the inducible viral reservoir in HIV-infected individuals. EBioMedicine. 2015;2(8):874–83. doi: 10.1016/j.ebiom.2015.06.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Battivelli E, et al. HIV-1 integration sites reveal heterogeneity in the latent population and explain the limited induction of latent proviruses. 9th IAS Conference on HIV Science (IAS 2017); Paris, France. 2017. p. MOPEA0045. ■■ This study demonstrated that there is a difference in HIV integration site patterns between productive, inducible latent and non inducible latent HIV infection. [Google Scholar]
- 41.Wu G, Swanson M, Talla A, et al. HDAC inhibition induces HIV-1 protein and enables immune-based clearance following latency reversal. JCI insight. 2017;2(16) doi: 10.1172/jci.insight.92901. ■ This study demonstrated that the LRA vorinostat and panobinostat were are able to induce HIV protein production allowing for recognition and elimination by envelope-specific antibodies. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Archin NM, Kirchherr JL, Sung JA, et al. Interval dosing with the HDAC inhibitor vorinostat effectively reverses HIV latency. The Journal of Clinical Investigation. 2017;127(8):3126–35. doi: 10.1172/JCI92684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Foley GE, Lazarus H, Farber S, et al. Continuous culture of human lymphoblasts from peripheral blood of a child with acute leukemia. Cancer. 1965;18(4):522–9. doi: 10.1002/1097-0142(196504)18:4<522::aid-cncr2820180418>3.0.co;2-j. [DOI] [PubMed] [Google Scholar]
- 44.Kuang XT, et al. HIV-1 Nef is required for efficient latency reversal in a T cell line. 9th IAS Conference on HIV Science (IAS 2017); Paris, France. 2017. p. WEPEA0223. [Google Scholar]
- 45.Ulrike LC, et al. Generation of HIV latency reporter cell lines by targeted genome engineering to explore effects of proviral integration site choice. 9th IAS Conference on HIV Science (IAS 2017); Paris, France. 2017. p. MOPEA0038. [Google Scholar]
- 46.Khoury G, Fromentin R, Solomon A, et al. Human Immunodeficiency Virus Persistence and T-Cell Activation in Blood, Rectal, and Lymph Node Tissue in Human Immunodeficiency Virus–Infected Individuals Receiving Suppressive Antiretroviral Therapy. The Journal of infectious diseases. 2017;215(6):911–9. doi: 10.1093/infdis/jix039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Symons J, Chopra A, Malatinkova E, et al. HIV integration sites in latently infected cell lines: evidence of ongoing replication. Retrovirology. 2017;14(1):2. doi: 10.1186/s12977-016-0325-2. ■ This study identified a new high throughput robotic based strategy to characterise HIV integration sites and applied the method to define integration sites in latently infected cell lines. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lifson J, et al. Expanded clones in SIV-infected macaques on antiretroviral therapy recapitulate key features of expanded clones in HIV infection: implications for evaluation of “HIV Cure” interventions. 9th IAS Conference on HIV Science (IAS 2017); Paris, France. 2017. p. MOLBPEA04. [Google Scholar]
- 49.McDonald D, Vodicka MA, Lucero G, et al. Visualization of the intracellular behavior of HIV in living cells. The Journal of cell biology. 2002;159(3):441–52. doi: 10.1083/jcb.200203150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Arhel N, Genovesio A, Kim K-A, et al. Quantitative four-dimensional tracking of cytoplasmic and nuclear HIV-1 complexes. Nature methods. 2006;3(10):817. doi: 10.1038/nmeth928. [DOI] [PubMed] [Google Scholar]
- 51.Lukic Z, Dharan A, Fricke T, et al. HIV-1 uncoating is facilitated by dynein and kinesin 1. Journal of virology. 2014;88(23):13613–25. doi: 10.1128/JVI.02219-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Dharan A, Opp S, Diaz-Griffero F, Campbell E, editors. JOURNAL OF THE INTERNATIONAL AIDS SOCIETY. INT AIDS SOCIETY AVENUE DE FRANCE 23; GENEVA, 1202, SWITZERLAND: 2017. Bicaudal D2 facilitates the cytoplasmic trafficking and nuclear import of HIV-1 genomes during infection. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Di Nunzio F, Danckaert A, Fricke T, et al. Human nucleoporins promote HIV-1 docking at the nuclear pore, nuclear import and integration. PloS one. 2012;7(9):e46037. doi: 10.1371/journal.pone.0046037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Dharan A, Talley S, Tripathi A, et al. KIF5B and Nup358 cooperatively mediate the nuclear import of HIV-1 during infection. PLoS pathogens. 2016;12(6):e1005700. doi: 10.1371/journal.ppat.1005700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.König R, Zhou Y, Elleder D, et al. Global analysis of host-pathogen interactions that regulate early-stage HIV-1 replication. Cell. 2008;135(1):49–60. doi: 10.1016/j.cell.2008.07.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Fontoura BM, Dales S, Blobel G, Zhong H. The nucleoporin Nup98 associates with the intranuclear filamentous protein network of TPR. Proceedings of the National Academy of Sciences. 2001;98(6):3208–13. doi: 10.1073/pnas.061014698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Krull S, Dörries J, Boysen B, et al. Protein Tpr is required for establishing nuclear pore-associated zones of heterochromatin exclusion. The EMBO journal. 2010;29(10):1659–73. doi: 10.1038/emboj.2010.54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Lelek M, Casartelli N, Pellin D, et al. Chromatin organization at the nuclear pore favours HIV replication. Nature communications. 2015;6 doi: 10.1038/ncomms7483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Di Nunzio F, et al. The nuclear pore orchestrates HIV-1 nuclear import and sculpts the chromatin landscape near integration sites. 9th IAS Conference on HIV Science (IAS 2017); Paris, France. 2017. p. MOPEA0042. [Google Scholar]
- 60.Shun M-C, Raghavendra NK, Vandegraaff N, et al. LEDGF/p75 functions downstream from preintegration complex formation to effect gene-specific HIV-1 integration. Genes & development. 2007;21(14):1767–78. doi: 10.1101/gad.1565107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Cherepanov P, Maertens G, Proost P, et al. HIV-1 integrase forms stable tetramers and associates with LEDGF/p75 protein in human cells. Journal of Biological Chemistry. 2003;278(1):372–81. doi: 10.1074/jbc.M209278200. [DOI] [PubMed] [Google Scholar]
- 62.Singh PK, Plumb MR, Ferris AL, et al. LEDGF/p75 interacts with mRNA splicing factors and targets HIV-1 integration to highly spliced genes. Genes & development. 2015;29(21):2287–97. doi: 10.1101/gad.267609.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Parissi VN. Regulation of retroviral integration by RNA polymerase II associated factors and chromatin structure. 9th IAS Conference on HIV Science (IAS 2017); Paris, France. 2017. p. MOPEA0029. [Google Scholar]
- 64.Matysiak J, Lesbats P, Mauro E, et al. Modulation of chromatin structure by the FACT histone chaperone complex regulates HIV-1 integration. Retrovirology. 2017;14(1):39. doi: 10.1186/s12977-017-0363-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Belotserkovskaya R, Reinberg D. Facts about FACT and transcript elongation through chromatin. Current opinion in genetics & development. 2004;14(2):139–46. doi: 10.1016/j.gde.2004.02.004. [DOI] [PubMed] [Google Scholar]
- 66.Christ F, Shaw S, Demeulemeester J, et al. Small-molecule inhibitors of the LEDGF/p75 binding site of integrase block HIV replication and modulate integrase multimerization. Antimicrobial agents and chemotherapy. 2012;56(8):4365–74. doi: 10.1128/AAC.00717-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Gijsbers R, Ronen K, Vets S, et al. LEDGF hybrids efficiently retarget lentiviral integration into heterochromatin. Molecular Therapy. 2010;18(3):552–60. doi: 10.1038/mt.2010.36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Bruggemans A, et al. Ledgins hamper the establishment of a reactivation competent HIV reservoir. 9th IAS Conference on HIV Science (IAS 2017); Paris, France. 2017. p. MOLBPEA13. [Google Scholar]
- 69.Gavegnano C, et al. Jak Inhibitors Employ Novel Mechanisms to Block Reservoir Seeding and HIV Persistence. 9th IAS Conference on HIV Science (IAS 2017); Paris, France. 2017. [Google Scholar]
- 70.Wiegand A, Spindler J, Hong FF, et al. Single-cell analysis of HIV-1 transcriptional activity reveals expression of proviruses in expanded clones during ART. Proc Natl Acad Sci U S A. 2017;114(18):E3659–E68. doi: 10.1073/pnas.1617961114. ■ A new method that can detect RNA production on a single cells level showed that on average 7% of proviruses express RNA on ART and a similar proportion of cells with either intact or defective virus express RNA. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Paiardini M, Müller-Trutwin M. HIV-associated chronic immune activation. Immunological reviews. 2013;254(1):78–101. doi: 10.1111/imr.12079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Mousseau G, Kessing CF, Fromentin R, et al. The Tat inhibitor didehydro-cortistatin A prevents HIV-1 reactivation from latency. MBio. 2015;6(4):e00465–15. doi: 10.1128/mBio.00465-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Henrich T, Hatano H, Hill A, et al., editors. JOURNAL OF THE INTERNATIONAL AIDS SOCIETY. INT AIDS SOCIETY AVENUE DE FRANCE 23; GENEVA, 1202, SWITZERLAND: 2017. Prolonged HIV-1 remission and viral rebound in an individual treated during hyperacute infection. [Google Scholar]
- 74.Whitney JB, Hill AL, Sanisetty S, et al. Rapid seeding of the viral reservoir prior to SIV viremia in rhesus monkeys. Nature. 2014;512(7512):74. doi: 10.1038/nature13594. [DOI] [PMC free article] [PubMed] [Google Scholar]