

Abstract
Cardiovascular autonomic dysfunctions including neurogenic orthostatic hypotension, supine hypertension and post-prandial hypotension are relatively common in patients with Parkinson disease. Recent evidence suggest that early autonomic impairment such as cardiac autonomic denervation and even neurogenic orthostatic hypotension occur prior to the appearance of the typical motor deficits associated with the disease. When neurogenic orthostatic hypotension develops, patients with Parkinson disease have increased risk of mortality, falls, and trauma related to falls. Neurogenic orthostatic hypotension reduces quality of life, contributes to cognitive decline and physical deconditioning. The co-existence of supine hypertension complicates the treatment of neurogenic orthostatic hypotension because it involves the use of drugs with opposing effects. Furthermore, treatment of neurogenic orthostatic hypotension is challenging because of few therapeutic options; in the past twenty years, the US Food and Drug Administration approved only two drugs for the treatment of this condition. Small open label or randomized studies using acute doses of different pharmacologic probes suggest benefit of other drugs as well, which could be used in individual patients under close monitoring. This review describes the pathophysiology of neurogenic orthostatic hypotension and supine hypertension in Parkinson disease. We discuss the mode of action and therapeutic efficacy of different pharmacologic agents used in the treatment of patients with cardiovascular autonomic failure.
1. Autonomic dysfunction in Parkinson disease
Parkinson disease (PD) is the second most common neurodegenerative disease after Alzheimer disease with the median annual incidence rates of 14 per 100,000 people in the total population. The incidence of PD is low before the age of 50 years, but increases rapidly from there; people aged 65 years and older have an annual incidence rate of 160 per 100,000 or 11 times more the reported incidence in the total population. There are more males than females with PD, male to female ratio ranges from around 1.3 to 2.0 in the majority of the studies, and higher prevalence is observed mostly among white non-hispanics. [1, 2]
Parkinson disease has been traditionally classified as a movement disorder and the diagnosis is based on the presence of three classic motor features: tremor, rigidity, and bradykinesia (paucity of movement), which are the result of loss dopaminergic cells in the substantia nigra pars compacta in the midbrain. Prior to the appearance of motor symptoms, however, the neurodegenerative process has long begun. There is a period that could last for decades with minimal clinical findings but a silent spread of synuclein- driven neurodegeneration in different areas of the nervous system. [3, 4] Braak et. al [5, 6] showed that the earliest abnormal depositions of α-synuclein occur in the anterior olfactory nucleus, the dorsal motor nucleus of the vagus in the medulla and the enteric plexus in the gut. The clinical correlation is reduced sense of smell (hyposmia) and constipation.
Another prominent non-motor symptom that precedes the development of motor deficits in patients with PD is REM sleep behavior disorder. This parasomnia is defined as enactment of dreams and is due to loss of the normal physiologic paralysis (sleep atonia) during REM sleep. The neuroanatomical lesion is located near the mesencephalic and pontine tegmentum. [7] The evidence that RBD predicts a CNS synucleinopathy is very strong; approximately 80–90% of patients with RBD confirmed by polysomnography developed PD, Lewy body dementia (LBD) or multiple system atrophy (MSA).[8, 9]
In our prospective natural history study of synucleinopathies, patients with RBD, as well as neurogenic orthostatic hypotension (nOH) and hyposmia, phenoconverted to PD and LBD after an average of 9 years of illness.[9] Patients that phenoconverted to MSA also had RBD and OH but their sense of smell was preserved and they had a shorter duration of illness (5 years). Similarly, a retrospective study with review of medical records showed a two-fold increase risk of developing PD in patients with OH. [10]
Orthostatic hypotension, defined as a drop in systolic blood pressure of 20 mm Hg or more or diastolic blood pressure fall of greater than 10 mm Hg within 3 minutes of standing.[11] is relatively common in patients with PD. A recent literature review of 25 cross-sectional studies that included 5,070 PD patients estimated a pooled OH prevalence of 30% (95% CI: 23 to 38%).[10]. OH can be symptomatic or asymptomatic. It is estimated that 16 to 18% [12] of PD patients have symptomatic OH.[13] Symptomatic OH is considered one of the most debilitating conditions among patients with PD. [14]
Antiparkinson medications such as levodopa and dopamine agonists can trigger or worsen OH, but OH can also occur in untreated patients because the disease process affects autonomic reflexes indicating a neurogenic origin (nOH). In a patient with PD and nOH, a simple bedside test to screen for autonomic dysfunction is to determine the increase in heart rate (HR) in response to the decrease in blood pressure on standing. A healthy autonomic reflex arch would induce an increase in sympathetic activity and withdrawal of parasympathetic cardiac modulation in response to volume shift during standing. An increase in HR equal or less than 15 bpm in patients with nOH suggest autonomic impairment.[11]
2. Impact of neurogenic orthostatic hypotension in Parkinson disease
In a recent meta-analysis, the presence of OH has been recognized as a risk factor for overall all-cause mortality and cardiovascular outcomes such as myocardial infarction, congestive heart failure and stroke.[15] The majority of epidemiological studies did not discern whether OH was associated or not with a neurodegenerative diseases such as PD. However, the presence of nOH in patients with PD was associated with higher mortality and a significantly elevated number of clinical events such as bone fractures, head trauma and syncope.[16]
2.1 Neurogenic orthostatic hypotension and falls in Parkinson disease
In the elderly, falls are significant clinical events associated with high mortality. Falls rank among the most common reasons for hospital admissions in patients with PD.[17–19] A systematic review of 22 studies of falls in patients with PD reported that 60.5% of patients enrolled reported at least one fall, and of these patients, 68% experienced recurrent falls.[20] It is debatable whether falls in PD relates to nOH or occur as a result of impaired postural reflexes, it could certainly be that both contribute. A recent study[21] showed that PD patients with nOH have a higher prevalence of medically-attended falls, hospitalizations and clinic visits than PD patients without nOH which results in higher healthcare costs.[21] Additionally, close relationships have been described between the occurrence of falls and increased caregiver burden in patients with PD.[22]
There is a paucity of data on the effect of nOH on quality of life in PD; patients with PD with impaired cardiovascular regulation reported orthostatic dizziness which significantly impact their daily activities.[8] Greater decrease in blood pressure has been observed in patients with PD and depression than in their non-depressed controls. Of note, the degree of the change in blood pressure associates with depression scaling.[23]
2.2 Cognitive Deficit and Neurogenic Orthostatic Hypotension
The connection between nOH and cognitive deficit is a controversial topic. Patients with PD, and nOH had more significant impairment in sustained attention and memory than those with PD without nOH.[24, 25] The pathophysiology of cognitive impairment in patients with nOH and PD may be multifactorial. It is possible that the neurodegenerative process itself may cause cognitive impairment and autonomic dysfunction in parallel by affecting common areas in the brain or substrates for neurotransmitters. On the other hand, decreased cerebral perfusion particularly in the frontal lobe during nOH episodes could also explain the cognitive impairment. Indeed, previous studies have found an association between nOH and poorer global cognitive function.[26, 27] During a head up tilt angle able to induce nOH, patients scored significantly worse in cognitive performance affecting both global cognitive functioning and specific tasks, mainly exploring executive functions. These patients had normal brain structures assessed by brain magnetic resonance imaging and normal Mini Mental State Examination in supine position.[28, 29]
3. Pathophysiology of Autonomic Dysfunction in Parkinson disease
Although PD is classically thought of as a disease of the central nervous system, nOH in these patients is the result of degeneration of peripheral post-ganglionic sympathetic nerve fibers.[30] The most notable evidence of degeneration of postganglionic sympathetic fibers in PD comes from neuroimaging studies of the heart. Utilizing I-MIBG SPECT[31, 32] or 6-[18F] fluorodopamine PET [30] a characteristic loss of uptake by myocardial sympathetic fibers has been convincingly shown in many studies involving hundreds of patients with PD.[33] The neuroimaging studies have been confirmed by immunohistochemistry of post mortem myocardium of PD patients showing loss of tyrosine hydroxylase staining.[34]
The loss of sympathetic innervation is distal-to-proximal and involvement of the autonomic ganglia seems to be a later phenomenon. Cardiac autonomic denervation frequently occurs in the early stages of the disease, sometimes before the development of motor symptoms.[35] Cardiac autonomic denervation does not cause OH and its clinical correlation with specific symptoms is uncertain.
There is a diagnostic value when obtaining cardiac imaging; patients with MSA most frequently have normal cardiac autonomic innervation compared to those with PD or pure autonomic failure (PAF) who have impaired uptake. Review of the literature suggests that values below 1.75[32] for the heart to mediastinum ratio in cardiac MIBG indicates cardiac autonomic denervation. The overall sensitivity to positively identify patients with PD was 89.7%, and the specificity to discriminate them from patients with multiple system atrophy was 94.6%.[36] These tests should be carefully interpreted in patients with a history of type 2 diabetes mellitus or in patients taking serotonin, norepinephrine reuptake receptor inhibitors (SNRIs) such as venlafaxine, desvenlafaxine, bupropion, duloxetine, atomoxetine commonly used for the treatment of depression or attention deficit hyperactivity disorders. Furthermore, use of droxidopa for the treatment of OH will also affect the results.
After assuming the upright position gravitational forces shift about 700 ml of blood volume to the lower part of the body particularly the splanchnic circulation.[37] Normally, the baroreflex compensates for the fall in cardiac preload by causing sympathetic activation and vasoconstriction. In patients with PD the loss of post-ganglionic sympathetic neurons causes inadequate vasoconstriction and patients develop neurogenic orthostatic hypotension (Fig 1).
Figure 1.
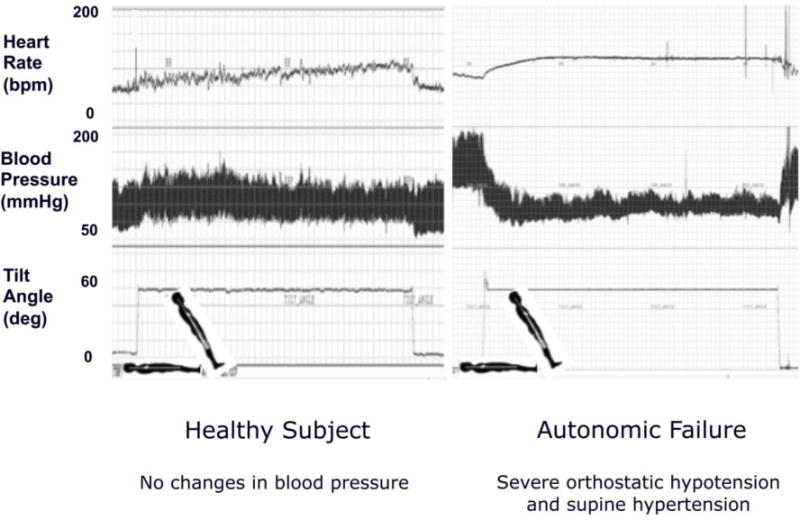
Continuous heart rate (HR) and blood pressure monitoring during 70-degree head up tilt in a normal control (left) and in patient with autonomic failure (right).
4. Pathophysiology of Symptomatic Neurogenic Orthostatic Hypotension
Symptomatic nOH is defined as the presence of nOH and pre-syncopal symptoms such as dizziness, lightheadedness, weakness and blurred vision. The severity of these symptoms correlates with the decrease in blood pressure during head up tilt. The underlying mechanism for the development of symptoms is reduced cerebral blood flow when blood pressure is below the cerebral autoregulatory range (low break point).
In normal circumstances, the autoregulatory range is between ~80–160 mm hg SBP. Within this range, changes in blood pressure only elicit minimal changes in cerebral blood flow. A study in patients with severe nOH reported an expansion of these autoregulatory ranges.[38]. However, when the patients reached their low break point, cerebral autoregulation fails, and the decrease in blood pressure elicits substantial changes in cerebral blood flow. In PD patients, symptomatic nOH was associated with a mean blood pressure below 75 mm Hg. This cut-point had a sensitivity of 97% and a specificity of 98% for detecting symptomatic nOH and appears to be a useful benchmark when deciding whether the benefits of initiating pharmacological treatment of nOH outweighs the risks of exacerbating supine hypertension.[12] Further studies, however, need to be conducted to validate this cut-point particularly in PD patients with OH and supine hypertension.
5. Treatment of Symptomatic neurogenic Orthostatic Hypotension
Neurogenic orthostatic hypotension causes substantial disability. Treatment should be directed to ameliorate symptoms, improving patient’s functional status and reduce the risk of nOH-related falls and syncope. Rather than arbitrary blood pressure goals, treatment should be focused on reducing symptoms.
5.1 Non-pharmacologic strategies
The first line of treatment is removing medications that predispose or exacerbate nOH. In patients with PD, lowering the dose of antiparkinson medication, particularly dopamine agonist should be considered. PD patients with hypertension should reduce or discontinue anti-hypertensive medications particularly direct vasodilators, diuretics, sympatholytic agents and alpha-1 blockers. Patients with benign prostatic hypertrophy should discontinue alpha-1 blockers including selective ones such as tamsulosin (Flomax©).[39] Lastly, hidden sympatholytics such as tizanidine, an alpha-2 adrenergic agonist (like clonidine), commonly prescribed for the treatment of muscle spasms should not be used in patients with PD and nOH.
Second, patients should be educated on physical counter-maneuvers to reduce the gravitational blood pooling in the lower extremities. The impaired postural reflexes in PD make it difficult to perform counter-maneuvers such as squatting or leg crossing, and are not recommended because they increase the risk of falls. We emphasize simple measures such as moving gradually from supine to standing positions and avoiding standing motionless, particularly when exposed to heat.
Devices to reduce venous pooling, such as custom-fitted thigh or waist high compression stockings and abdominal binders are useful when graded pressure of at least 30 to 40 mmHg is applied to the lower body.[40, 41] However, these devices are difficult to put on and may be uncomfortable to wear, limiting patient’s compliance. In our clinical practice, an alternative that is acceptable to some patients is the use of tight “biker shorts”.
PD patients without hypertension should be encouraged to employ approaches to improve central volume. Salt consumption can be increased to 6 to 9 g of sodium chloride per day (with 1 g sodium chloride tablets taken with each meal if needed). Water intake can also be increased up to 2 to 3 liters per day, with rapid water ingestion (16 ounces in 3 to 4 minutes) used as a rescue measure to increase systolic blood pressure and relieve orthostatic symptoms.[42] The pressor effects of water peak at 30 minutes and can last for up to 2 hours, with effects mediated by activation of the sympathetic nervous system.[43]
In PD patients with hypertension, sleeping with the head of the bed tilted up 6 to 9 inches can also prevent volume depletion by reducing nocturnal pressure natriuresis.[44, 45]
Lastly, patients with PD and nOH also develop post-prandial hypotension because their impaired autonomic reflexes fail to compensate for splanchnic blood pooling after meals. Because the magnitude of the decrease in blood pressure depends on the size[46] and the composition of the meal, particularly the amount of carbohydrates; [47, 48] patients should eat small frequent meals and eat the carbohydrate-loaded meals close to bedtime. Combining caffeinated drinks or drinking 16 ounces of water with meals may also help to reduce symptoms associated with post-prandial hypotension.[49] Of note, post-prandial hypotension is one of the earliest abnormal cardiovascular manifestations in patients with PD. Postprandial hypotension may be misdiagnose as a motor off state with worsened rigidity and bradykinesia.[50] Non-pharmacologic approaches are very cost-effective and have an important role in the treatment strategy for patients with nOH.
5.2 Pharmacological Therapy
There are only two FDA approved drugs for the treatment of orthostatic hypotension in the U.S. The α-1 adrenergic agonist, midodrine, and the synthetic norepinephrine precursor, droxidopa. The majority of clinical trials enrolled patients with different types of α-synucleopathies but also patients with non-diabetic autonomic neuropathies (NDAN). Hence, the information discussed in the following section applies to all forms of autonomic failure, NDAN, and PD with nOH.
In this section, we will also discuss data from small clinical studies that tested the effect of different pharmacological agents that are commercially available and in some cases are used as off-label indication for nOH and nOH-related symptoms.
We have divided the pharmacological agents according to their mode of action in the following groups: agents that increase norepinephrine levels; agents that potentiate residual sympathetic activity; direct vasoconstrictors; preferential venoconstrictors; plasma volume expansors; agents that prevent glucose absorption in post-prandial hypotension (Table 1).
Table 1.
Pharmacological Agents Used in the Treatment of Neurogenic Orthostatic Hypotension and Autonomic Failure
| Medications | Mode of action | Doses |
|---|---|---|
| Droxidopa** | Increase norepinephrine levels | 100–600 mg t.i.d. |
| Yohimbine (require compounding) | Stimulate residual SNS activity by antagonizing on α2 receptors | 5.4 mg t.i.d. |
| Pyridostigmine | Enhances ganglionic transmission and SNS activity, acetylcholinesterase inhibitor, peripheral action | 30–60 mg t.i.d. |
| Atomoxetine | Stimulate SNS by blocking norepinephrine reuptake | 10–18 mg b.i.d. |
| Midodrine* | Direct vasoconstrictor, α1 adrenergic agonist | 2.5–10 mg t.i.d. |
| Ergotamine and caffeine (caffergot) avoid coronary artery disease | Direct vasoconstrictor, venoconstrictor | (1 mg ergotamine, 100 caffeine) once a day |
| Octreotide | Splanchnic vasoconstrictor | 12.5–50 ug SQ, once a day |
|
Fludrocortisone avoid heart failure |
Volume expansor, synthetic mineralocorticosteroid | 0.1–0.3 mg/day |
|
Acarbose avoid inflammatory bowel disease |
Reduce post-prandial glucose and decrease insulin release and other vasoactive gut peptides | 50–100 mg, ten minutes before large meal |
Approved for the treatment of orthostatic hypotension (US, Europe, Japan);
Approved for the treatment of neurogenic orthostatic hypotension (US and Japan); t.i.d. three times a day; SQ subcutaneous
These medications should not be prescribed at fixed intervals but should be tailored to the patient’s activities and are best given on a PRN basis. Most of the medication needs to be taken 45 to 60 minutes before upright activities. Evening doses should be avoided because of increased risk of developing supine hypertension.
5.2.1. Agents that increase norepinephrine levels
Physiopathology and targeted pathway
In healthy subjects, circulating levels of plasma NE, the sympathetic neurotransmitter, more than double when standing. This increase in circulating NE levels is the result of sympathetic activation in response to orthostatic stress. Patients with autonomic failure including those with PD with nOH are unable to increase NE levels appropriately while standing. The blunted NE release could be the results of peripheral autonomic denervation or impairment of central autonomic centers that control peripheral sympathetic activation. Drugs that increase NE levels could improve upright blood pressure and symptoms by increasing the availability of NE in the synapse while standing, promoting a vasoconstrictor effect or by acting in autonomic centers in the CNS.
Droxidopa is the only agent in this category; this medication was approved in the US and Japan for the treatment of nOH. Droxidopa is a prodrug that is converted into norepinephrine by dopa decarboxylase, the same enzyme that converts levodopa into dopamine.[51] The approval was based on three randomized, double blind clinical trials (RCT) that showed the efficacy of droxidopa for the relief of symptoms associated with nOH.[52–55]. In study NOH301 and NOH 306, were placebo-controlled and the outcome was symptom improvement. NOH302 was a randomized withdrawal design. In a recent integrated analyses of compiled data from these RCT with a pooled sample of 225 in droxidopa versus 235 in the placebo group. Compare to placebo, droxidopa showed to decrease symptoms of dizziness and lightheadedness scores and measurements of activities of daily living (standing long time, walking a short time, walking a long time) and significantly increase upright blood pressure (11.5±20.5 vs. 4.8±21 for placebo). Droxidopa was well tolerated and among side effects supine hypertension seems to be predominant but with very low prevalence (≤7.9% vs. ≤4.6% for placebo).[56] Of note, these clinical trials assessed efficacy in the ≤2 to 10 week period, and currently a longer RCT is examining long-term efficacy.
Nevertheless, study NOH306 included falls as secondary endpoint in patients with PD and nOH. A total of 197 patients were included in this analysis (92 droxidopa versus 105 placebo). In the droxidopa group, the fall rate was 0.4 falls per patient-week; in the placebo group, the rate was 1.05 falls per patient-week, P=0.014). These results translate into a 77% relative risk reduction. It is important to point out that the statistical significance was found using post-hoc analyses. Further studies powered to assess the effect of droxidopa on falls, as primary endpoint should be conducted to confirm these findings.[57]
5.2.2. Agents that potentiate sympathetic activity
Physiopathology and targeted pathway
Although PD is classically thought of as a CNS disorder, nOH in PD results from degeneration of peripheral sympathetic neurons and impaired ability to release norepinephrine. The degree by which tonic norepinephrine release is impaired varies significantly among patients and depends in part on the severity of the autonomic damage. Patients with PD may have different degrees of residual sympathetic activity. This residual sympathetic activity can be harnessed with pharmacological probes acting centrally or in peripheral autonomic nerves producing an increase in blood pressure; these effects can be used for the treatment of nOH. Even small increases in plasma norepinephrine induced by these agents may lead to exaggerated rises in blood pressure because of autonomic denervation-induced-up-regulation of adrenoreceptors, and inability to buffer hemodynamic changes due to lack of baroreflex capacity.
Yohimbine increases norepinephrine release from sympathetic nerves by augmenting central sympathetic outflow and by interfering with inhibitory modulation of pre-synaptic α-2 adrenoreceptors. As expected from its pharmacological actions, which facilitates sympathetic activation, yohimbine increases plasma norepinephrine in a dose-dependent manner, which translates into an increase in blood pressure and heart rate. In nOH patients, the effect can be achieved at a very low dose (5.4 mg) given their denervation hypesensitivity.[58] Yohimbine is rapidly absorbed after oral administration with a peak plasma concentration occurring at 30 minutes and a half-life of 5 hours. The effect of yohimbine on orthostatic tolerance was first studied by Robertson and collaborators[59] in 12 patients with autonomic failure. Intravenous administration of yohimbine (4–64 ug/kg/iv bolus) increased norepinephrine spillover and blood pressure and reduced the hypotensive response to head up tilt in these patients. In open label studies, oral administration of 5.4 mg of yohimbine increased blood pressure by 50 mm Hg with a peak effect at 75 minutes.[60] More recently, in a single-blind, placebo-controlled, crossover study, the same dose of yohimbine increased standing diastolic blood pressure by 11 mm Hg and reduced pre-syncopal symptoms, particularly lightheadedness in 31 patients with autonomic failure.[61] No long-term studies are available showing sustained effect on blood pressure or symptoms.
Yohimbine appears well tolerated in autonomic failure patients, with no report of adverse events in short term clinical studies. Yohimbine requires compounding; the therapeutic dose is 5.4 mg p.o. up to three times a day. It can be also available as a herbal supplement.
Pyridostigmine inhibits the enzyme acetylcholinesterase and increases the transmission of impulses from cholinergic neurons across the synaptic cleft. After oral administration, pyridostigmine achieved peak plasma concentration at around 1.7 hours later and had a short half-life of 1–2 hours. Because all ganglionic neurotransmission is cholinergic, pyridostigmine increases both sympathetic and parasympathetic nerve function. The appeal of this pharmacologic approach is that it should be silent under conditions of low sympathetic tone such as in supine posture, but will potentiate the sympathetic activation that occurs on standing. Two studies have reported the beneficial effect of pyridostigmine as a treatment for nOH in patients with autonomic failure. The effect of 60 mg of pyridostigmine on standing diastolic blood pressure was first tested in an open label study in 15 patients with autonomic failure [62]. Pyridostigmine significantly increased orthostatic blood pressure and reduced the fall in blood pressure associated with head up tilt. In a subsequent double-blind, randomized 4-way crossover study, pyridostigmine increased upright systolic blood pressure by only 4 mm Hg compared with placebo, the combination with 5 mg of midodrine was slightly more effective.[63] The improvement in orthostatic blood pressure in both studies was associated with a significant improvement in orthostatic symptom without causing supine hypertension. Our study, however, did not yield the same positive results; pyridostigmine did not improve orthostatic tolerance or symptoms, perhaps because our autonomic failure patients were more severely affected than in previous studies.[61] It is possible that the response to pyridostigmine is proportional to the degree of residual sympathetic tone and therefore, this agent could be more effective in patients with some degree of residual sympathetic tone.
Atomoxetine is a selective blocker of the neuronal norepinephrine transporter (NET) commonly used for the treatment of attention deficit hyperactivity disorder in children and adults. After oral administration, the time to achieve peak plasma concentration is within 2 hours and its elimination half-life is 5 hours. [64] Atomoxetine blocks the activity of the norepinephrine transporter, therefore inhibits the reuptake of NE and increases neurotransmitter concentrations in the neuroeffector junction which induces a pressor effect through stimulation of α1-adrenergic receptors in the vascular wall. This effect is more noticeable in patients with normal NE concentrations and with normal peripheral autonomic nerves such as those with MSA. In a proof-of-concept clinical trial,[65] we determined the effect of 18 mg of atomoxetine on seated and standing blood pressure in 21 patients with autonomic failure (10 MSA and 11 PAF). These preliminary data showed that an acute dose of 18 mg of atomoxetine increased seated systolic blood pressure (SBP) by ~50 mm Hg compared with placebo in MSA. In MSA patients, atomoxetine increased plasma NE levels by 26% suggesting a mechanistic link between the neurohumoral response and its hemodynamic effect. No significant blood pressor responses were observed in PAF patients who had very low plasma NE. The sympathetic nervous system is maximally activated upon standing; plasma NE levels doubled on standing in normal subjects but failed to increase in autonomic failure patients. In a second trial, we found that atomoxetine by preventing the clearance of NE in the synapse would induce a greater blood pressure (BP) response upon standing compared with the direct α-1 adrenergic vasoconstrictor, midodrine. In a separate crossover study,[66] we compared the effect of a single dose of 18 mg of atomoxetine with midodrine (5,10 mg) and placebo on standing SBP and clinical symptoms in 69 patients with nOH and autonomic failure. Our results showed that atomoxetine and midodrine increased seated BP at the same magnitude. However, 18 mg of atomoxetine was more effective than midodrine in improving standing SBP (+7.5 mm Hg). Equally important, atomoxetine but not midodrine induced a significant reduction in clinical symptoms (lightheadedness and dizziness) compared with placebo.[66] Both studies strongly suggest that atomoxetine could be a potential treatment for nOH in autonomic failure, particularly in patients with MSA. We are currently conducting the first clinical trial to determine the effect of chronic use of atomoxetine in nOH (Clinicaltrials.gov NCT02784535).
5.2.3. Direct vasoconstrictors
Physiopathology and targeted pathway
Patient with autonomic failure have sympathetic denervation-induced-up-regulation of vascular adrenoreceptors; these patients have hypersensitivity to α-1 adrenergic receptor agonists that promote vasoconstriction and increase blood pressure. The inability to buffer hemodynamic changes due to lack of baroreflex capacity makes this effect more prominent. Of note, patient with autonomic failure, have a 10-fold elevation of blood pressure with acute i.v boluses of the α-1 adrenergic receptor agonist phenylephrine.
Midodrine is and oral α-1 adrenergic receptor agonist. It is both an arterial and venous vasoconstrictor. It has an elimination half-life of 0.5 hours and is undetectable in plasma 2 hours after an oral dose. The agent undergoes enzymatic hydrolysis in the systemic circulation to form the active agent, desglymidodrine. Desglymidodrine is 15 times more potent than midodrine and is primarily responsible for the therapeutic effect. The absolute bioavailability of desglymidodrine is 93% and the elimination half-life is 2–4 hours, the duration of action of midodrine is approximately 4 hours. It is excreted mainly in the urine. The sensitivity to this agent varies among patients and the dose should be titrated from 5 to 10 mg three times a day. The peak pressor effect occurs 1 hour post-ingestion. The efficacy of midodrine in increasing blood pressure has been shown in double-blind placebo controlled trials.[67, 68] There was a significant linear relationship between midodrine dosage and mean systolic blood pressure in a dose-response study. Furthermore, midodrine improved standing systolic blood pressure and symptoms (dizziness/lightheadedness, weakness/fatigue, syncope, low energy level, impaired ability to stand) compared with placebo. The effect of midodrine on standing blood pressure and ability to stand up has been superior compared with ephedrine, but no comparison with other agents was carried out. Midodrine should be administered with caution in patients with hepatic dysfunction and is contraindicated in acute renal failure, urinary retention, pheochromocytoma and thyrotoxicosis.
5.2.4. Preferential venoconstrictors
Physiopathology and targeted pathway
OH in autonomic failure results from the decrease in pre-load during orthostasis. The gravitational blood pooling is secondary to increase in venous capacitance, mostly in splanchnic circulation. In this section, we will also discuss the effect of both arterial and venous vasoconstrictor and an agent that primarily induces splanchnic vasoconstriction. Arguably, drugs that improve venous return may be more effective that arterial vasoconstrictors.
Caffergot (ergotamine and caffeine), the pressor effect is mostly related to the use of ergotamine which is an α-adrenergic agonist and potent venoconstrictor. This combination increases blood pressure and improves symptoms, but oral bioavailability is variable, and there is concern about its long-term use particularly in patients with coronary artery disease. In patients with refractory autonomic failure this could be an alternative treatment when everything else fails. Previous small studies, ranging from 1 to 10 patients, have shown that ergotamine increases standing blood pressure and improves symptoms in patients with nOH when given by inhaled, intramuscular, or oral routes of administration.[69–73] We previously demonstrated that caffergot (1 mg ergotamine and 100 mg caffeine improves standing time and may have a more potent pressor effect than midodrine.[74] The safety or efficacy of long-term use has not been tested.
Octreotide is a somatostatin analog very effective, even when other drugs fail; this is in part due to its ability to constrict the splanchnic circulation where most of the orthostatic blood pooling occurs. A study reported no changes in plasma norepinephrine after octreotide suggesting no effect on NE pool.[75] Octreotide use is limited by the need for parenteral administration, and by gastrointestinal symptoms such as abdominal pain, nausea and vomiting in susceptible patients. The effective dose is 12.5 ug SQ; in patients with refractory nOH, doses of 25 ug SQ may be more appropriate in refractory cases. The effect of octreotide on orthostatic tolerance as measured by standing time 1 hour after the administration was comparable to midodrine and their combined use seems to potentiate this effect. Octreotide may be also effective for the treatment of post-prandial hypotension.[76, 77]
5.2.5 Plasma Volume Expansors
Physiopathology and targeted pathway
Dietary modifications such as high salt diet and drugs that promote sodium or free water re-absorption have been used in an attempt to increase cardiac pre-load in patients with OH. Among these drugs, the most popular is fludrocortisone, a synthetic mineralocorticoid, which has been used for more than 40 years in patients with OH. Previous studies suggest that mineralocorticoids might have volume-independent effects promoting cardiac fibrosis and endothelial dysfunction and there is concern that this drug could induce or worsen heart failure.
Fludrocortisone increases renal sodium reabsorption and expands plasma volume through its mineralocorticoid effect. This effect however is believed to be transient, and after 1–2 weeks of continuous use, plasma volume returns to baseline values.[78] A previous study reported that fludrocortisone potentiates the blood pressure effect of infused norepinephrine by increasing peripheral vascular resistance.[78] The overall effect of this agent in patients with OH is a sustained increase in blood pressure, both in the supine and upright posture.[78] Fludrocortisone is readily absorbed after oral administration. Its elimination half-life is around 2–3 hours. Recommended starting dose is 0.1 mg a day, maximum doses are 0.3–0.5 mg a day. Only two descriptive case series assessed the effects of fludrocortisone for the treatment of OH, one in patients with diabetes mellitus[79] and one in patients with Parkinson disease on Levodopa.[80] Assessment of the efficacy of this medication was based on reporting of pre-syncopal symptoms and improvements in postural hypotension as measured by supine and standing blood pressure during treatment. These studies suggested that fludrocortisone was effective in the treatment of OH based on the improvement of these parameters.[80] [81] Hypokalemia will develop in nearly 50% of patients, and it can appear within the first week of treatment. Hypomagnesemia has also been described in about 5% of patients. This agent should not be use in patients with prevalent heart failure. Our review of the available literature with a pooled sample of 121 patients with OH from four case series showed that 45% reported adverse events, with heart failure being the most frequent, affecting 11%.[80–83]
5.2.6. Agents that prevent glucose absorption for the treatment of postprandial OH
Physiopathology and targeted pathway
More than 90% of patients with OH have post-prandial hypotension (PPH). PPH is defined as a decrease in blood pressure of more than 20 mm Hg within 30 minutes after a meal. Impaired autonomic reflexes and failure to compensate for increased blood pooling in splanchnic circulation cause PPH. Postprandial hypotension occurs independently of postural changes and worsens pre-syncopal symptoms by reducing upright blood pressure after meals. Possible underlying mechanisms include release of vasoactive gut peptides in response to glucose absorption. Insulin is one of these peptides, however is not the solely explanation because this condition is observed in type I diabetes mellitus characterized by dependence on exogenous insulin.[84]
Acarbose inhibits alpha-glucosidase in the brush border of the small intestine, delaying glucose absorption by decreasing the breakdown of complex carbohydrates. These actions also decrease the release of gastrointestinal hormones and slower gastric emptying.[85] We showed that acute administration of 100 mg of acarbose, twenty minutes before a standardized meal reduced the postprandial fall in systolic blood pressure by 17 mmHg compared with placebo in autonomic failure patients. This effect was associated with an improvement in total peripheral resistance but no significant changes in cardiac output.[86] No serious side effects have been reported in these studies, but acarbose was only used acutely. Chronic treatment with acarbose can be associated with an increased rate of flatulence and loose stools as a result of specific drug effects on carbohydrate digestion. To minimize gastrointestinal adverse effects, treatment should be initiated with a low dose and gradually increased until adequate effects are achieved. In patients with autonomic failure suffering from PPH, we start treatment with 50 mg of acarbose once a day, twenty minutes before eating, usually their largest meal. The dose can be titrated up to 100 mg three times a day. That said, however, it is important to emphasize that this medication should be taken, as needed only when patients eat large carbohydrate meals or when they need to be upright after a meal. Acarbose is contraindicated in patients with diabetic ketoacidosis, cirrhosis, inflammatory bowel disease, ulcerative colitis, intestinal obstruction or any chronic intestinal disease that could disrupt digestion or absorption.
6. Pathophysiology of Supine Hypertension
Supine HTN is arbitrarily defined as a systolic blood pressure ≥150 mm Hg and diastolic blood pressure ≥90 mm Hg in the supine position.[87] It is estimated that around 50–60% of patients with OH would also develop supine HTN.[88] This condition is often missed because blood pressure is usually assessed in the seated position. The pathophysiology of supine HTN is multifactorial; medications use to treat OH with a long half-life like the mineralocorticoid, fludrocortisone, may cause supine HTN.[78] Supine HTN is also observed in treatment-naïve patients. It is unclear if these patients have de novo HTN or if they have prevalent HTN worsened by absence of baroreflex buffering that occurred as a result of autonomic failure.
Uncontrolled supine HTN has negative consequences in patients with OH. It limits the use of pressor agents to treat OH and induce pressure natriuresis particularly at night that worsen OH and OH-related symptoms in the morning. Furthermore, supine HTN in patients with OH associates with left ventricular hypertrophy and renal impairment.[89, 90]
In patients with MSA, supine HTN is likely caused by residual sympathetic activity acting on hypersensitive adrenoreceptors unrestrained because of ineffective baroreflex buffering. The ganglionic blocker, trimethaphan that completely inhibit sympathetic and parasympathetic transmission at the level of the autonomic ganglia normalizes blood pressure in MSA patients.[91]
In patients with peripheral autonomic failure (PD and PAF), the cause of supine HTN is unknown. Supine HTN is sustained during ganglionic blocker infusion, which indicates that residual sympathetic activity is not an important contributor. A study showing low intravascular volume and cardiac output, [92] suggest that HTN is probably caused by increased peripheral vascular resistance. Biaggioni and collaborators showed that in patients with autonomic failure renin activity is very low, almost undetectable;[93] and there is an excess of the vasodilator nitric oxide.[94] Hence, neither of these factors can account for the increased vasoconstriction that causes HTN. Recent studies, however, suggest that although aldosterone, is found in normal concentrations, angiotensin II found in excess, and inappropriate mineralocorticoid receptor activation may contributes to supine HTN. [95, 96] Small studies looking at the effect of anti-HTN agents that targeted specific receptors for these peptides showed promising results, although only the acute effects were tested. Further studies that assess the chronic effect of these drugs are warranted.
7. Treatment of Supine Hypertension
We recommend to initiate treatment if the patient has evidence of target organ damage (left ventricular hypertrophy, impaired renal function) or if the patient has persistent systolic blood pressure (SBP) >180 mm Hg and diastolic blood pressure (DBP) > 110 mm Hg. Treatment should be individualized in subjects with a SBP 160–180 mm Hg and DBP 90–110 mm Hg.[11] It is noteworthy that supine blood pressure throughout the day and night varies significantly. Patients with autonomic failure have lower blood pressures in the morning and as the day progresses seated and supine blood pressures increase; the highest blood pressure values are usually observed in the evenings prior to bedtime. Dipping, which is described as the decrease in blood pressure at night occurred in one-third of autonomic failure patients with supine HTN. [97] In these patients, multiple blood pressure measurements throughout the night, which can be achieved with an ambulatory blood pressure monitor, would help to determine if anti-HTN treatment is required.
7.1 Non-pharmacological Strategies
During the daytime, the most effective approach to manage HTN is to avoid the supine position particularly when using compression devices or within hours after taken pressor agents to treat OH. Other conservative measures to treat supine HTN include eating a sweet snack (≈400 Kcal) around bedtime to elicit transient post-prandial hypotension and elevating the head of the bed by 6 to 9 inches using a mattress wedge or an electric bed. A recent study suggests that passive heat application using a commercial heating pad over the abdomen and pelvis had blood pressure-lowering effect in autonomic failure patient with supine HTN. [98]
7.2. Pharmacological Therapy
The ideal pharmacological agent for the treatment of supine HTN should have the following characteristics: its effect should be long enough to control blood pressure during the night, but short enough to disappear by the following morning; it should reduce pressure natriuresis and volume depletion at bedtime to prevent worsening of OH in the morning and should prevent end-organ damage and reduce cardiovascular risk in these patients. Based on these characteristics and understanding of the pathophysiology of supine HTN in patients with nOH, we have tested a selected number of anti-HTN agents that are commercially available. In this section we discuss the results of small clinical trials that measure the acute effect of these agents on supine blood pressure in patients with supine HTN and nOH. We have divided the pharmacological agents according to their mode of action in 4 groups: agents that reduce residual sympathetic tone; direct vasodilators; agents that potentiate nitric oxide function; mineralocorticosteroid receptor antagonist and angiotensin receptor blockers, (Table 2).
Table 2.
Pharmacological Agents Used in the Treatment of Supine Hypertension in Autonomic Failure
| Medications | Mode of action | Doses |
|---|---|---|
| Clonidine | Central sympatholytic agent, reduce residual sympathetic activity. Drug of choice in MSA patients with supine HTN | 0.1 mg evening |
| Nitroglycerin transdermal patch | Nitric oxide donor. Target increased peripheral vascular resistance. Recommended in PAF and PD patients | 0.1–0.2 mg/hr applied at bedtime, remove in the morning |
| Short acting nifedipine | Calcium channel blocker. Target increased peripheral vascular resistance. Recommended in patients with refractory supine HTN. | 10–30 mg bedtime |
| Sildenafil citrate | Potentiate nitric oxide effect | 25 mg bedtime |
| Nebivolol | Third generation beta blocker with vasodilatory properties. Potentiate nitric oxide function | 2.5–5 mg bedtime |
| Eplerenone | Selective mineralocorticoid antagonist. Target inappropriate MR receptor activation in autonomic failure | 50 mg bedtime |
| Losartan | AT1 receptor blocker. Target increased Angiotensin II levels in autonomic failure | 50 mg bedtime |
MR, mineralocorticoid receptor.
7.2.1. Agents that reduce residual sympathetic tone
Physiopathology and targeted pathways
In many patients with autonomic failure, particularly those with MSA, supine hypertension is driven by residual sympathetic tone acting on hypersensitive adrenoreceptors and unrestrained by the impairment of baroreflex buffering. We will discuss the role of selective α2-adrenergic agonists that act centrally to reduce sympathetic outflow. The only selective α2-adrenergic agonist tested in patients with nOH and supine HTN is clonidine.
Clonidine is a central sympatholytic agent use for the treatment of essential hypertension. We tested the effect of 0.1 mg clonidine p.o. versus placebo on supine HTN in 23 patients with autonomic failure. The medication was given at bedtime, the maximum decrease in blood pressure with clonidine compared to placebo was ≈26 mm Hg and occurred 6 hours after medication administration. The decrease in blood pressure with clonidine correlated with the decrease in blood pressure with the ganglionic blocker, trimethaphan, indicating that clonidine blood pressure-lowering effect was due to a reduction of residual sympathetic activity. Clonidine also reduced nocturnal sodium excretion, however we did not find an improvement in morning nOH or orthostatic tolerance, in part because the hypotensive effect carried over into the morning. This agent should be administered in the afternoon to prevent the residual hypotensive effect in the morning. This could be a treatment option for patients with MSA and severe supine HTN.
7.2.2. Direct vasodilators
Physiopathology and targeted pathway
Supine HTN in patients with conditions that causes peripheral autonomic denervation (PAF and PD) results from increased peripheral vascular resistance, which is not driven by residual sympathetic activity. Hence, short-acting vasodilators are central in the treatment of supine HTN. In this drug group, we discuss two different classes: nitric oxide donors and calcium channel blockers.
Nitroglycerin transdermal patches and short acting nifedipine effectively reduce supine hypertension in patients with autonomic failure. Jordan and collaborators[99] compared the effects of 0.05 to 0.2 mg/h transdermal nitroglycerin and 30 mg of oral nifedipine versus placebo in a single blind fashion. The maximal decrease in blood pressure was 36 ± 10 and 37 ±9 mm Hg, respectively, four hours after the drug administration.[99] Even though blood pressure control was achieved, neither of these drugs reduced pressure natriuresis or improved orthostatic tolerance in the morning. Nighttime urinary volume and sodium excretion was similar with nitroglycerin and placebo. On the other hand, nifedipine increased fractional sodium excretion and worsened orthostatic tolerance. Of interest, these agents are not the first line treatment for essential hypertension, because they provoke reflex sympathetic and renin activation, and decreased survival in the elderly.[100]However in patients with autonomic failure these effects are not present, because baseline sympathetic activity is low, they lack baroreflex modulation, and baseline renin activity is very low. Headache is a limiting side effect.
In treatment-naïve patients, we recommend initiating treatment with 0.1 mg/hr nitroglycerin transdermal patch, patients should be instructed to remove the patch before standing in the morning. Nifedipine is a much more potent vasodepressor agent, and the initial treatment should start at doses of 10 mg at bedtime.
7.2.3. Agents that potentiate nitric oxide function
Physiopathology and targeted pathway
Patients with autonomic failure, nOH and supine HTN have increased nitric oxide function.[94] Inhibition of nitric oxide synthase enzyme (that eliminates nitric oxide production) induced a larger increase in blood pressure in autonomic failure as compared to controls. This exaggerated response to nitric oxide-mediated vasodilation can be harnessed for the treatment of supine HTN. We discuss the effect of two agents that potentiate nitric oxide function (i) phosphodiesterase 5 inhibitor, sildenafil citrate and (ii) third-generation beta-blocker with nitric oxide potentiating effect, nebivolol.
Sildenafil citrate is a phosphodiesterase 5 inhibitor approved for the treatment of erectile dysfunction and pulmonary hypertension. It blocks the degradation of cyclic GMP, which potentiate nitric oxide function, a potent vasodilator. We tested the effect of 25 mg sildenafil compared with placebo on eight patients with supine HTN and nOH. The maximal decrease in systolic blood pressure with sildenafil was ~30 mm Hg, and this effect occur approximately 8 hours after administration.[94] In some patients with PAF, sildenafil may induce hypotension lasting for several hours. Sildenafil should not be taken in combination with nitric oxide donors. Unfortunately, sildenafil does not have any effect on sodium excretion in autonomic failure patients. No side effects were observed with acute use, the limiting factor for the use of sildenafil citrate in supine HTN is the cost of the medication. However, generic forms are now available.
Nebivolol is a selective β1-adrenergic receptor blocker and is considered a third-generation β-blocker with unique vasodilatory actions. The vasodilatory effect of nebivolol is nitric oxide-dependent, this medication is thought to increase bioavailability of endothelium-derived nitric oxide. The blood pressure-lowering effect of 5 mg nebivolol, 25 mg sildenafil, 50 mg metoprolol and placebo on supine HTN was tested in 20 patients with supine HTN and nOH in a double-blind crossover fashion.[101] Nebivolol significantly lowered blood pressure (~40 mm Hg) at night in patients who also responded to 25 mg of sildenafil citrate. Similar to sildenafil, nebivolol had a neutral effect on sodium excretion. Of note, despite its potent blood pressure-lowering effect, this medication does not worsen morning nOH. Nebivolol is well-tolerated, we recommend to initiate treatment with nebivolol at doses of 2.5 mg in the evening.
7.2.4. Angiotensin receptor blockers and mineralocorticoid receptor antagonist
Physiopathology and targeted pathway
The renin-angiotensin system contributes to the development of hypertension via actions of angiotensin II (Ang II) acting on AT1 receptors that promote vasoconstriction and aldosterone release. Patients with nOH have very low plasma renin activity, impaired renin responses to postural changes and loss of renin immunoreactive cells in autopsied kidneys. [93] These characteristics explain why angiotensin converting enzyme inhibitors have no blood pressure-lowering effect in patients with OH and supine HTN.[95] Nevertheless, angiotensin II, which is the downstream product of renin and angiotensin I, is elevated in nOH. [95] These findings suggest that renin-independent mechanisms are involved in the synthesis of this peptide. Furthermore aldosterone levels are normal; this hormone is released from the adrenal cortex, via stimulation of Ang II. We discuss two medications that independently target these pathways the mineralocorticoid receptor antagonist, eplerenone, and the AT1 receptor blocker, losartan.
Eplerenone is a selective mineralocorticoid receptor antagonist; the acute blood pressure-lowering effect of 50 mg of eplerenone versus placebo was tested in ten patients with autonomic failure and supine HTN.[96] Eplerenone reached peak plasma concentration ≈1.5 hour after oral intake, the half-life of this medication is 4–6 hours. Eplerenone maximally decreased blood pressure by ≈32 mm Hg, eight hours after administration. The blood pressure effect of eplerenone is not related to its diuretic effect. Overnight sodium excretion was similar during eplerenone versus placebo and this medication does not negatively impact morning nOH. Multiple mechanisms could explain the blood pressure-lowering effect including blockade of aldosterone, cortisol, Angiotensin II or hormone-independent ligands.[96] This study only evaluated the acute effect of eplerenone on supine HTN, further studies are needed to determine if chronic administration of this medication worsen morning nOH.
Losartan is an AT1 receptor blocker (ARB), the effect of this medication on supine HTN was tested in 11 patients with severe autonomic failure. Losartan 50 mg successfully decreased blood pressure by ~32 mm Hg at 6 hours after administration.[95] Furthermore, losartan is the only medication that reduces pressure natriuresis and fluid loss as measured by changes in body weight in autonomic failure patients. Losartan had a neutral effect on morning nOH.[95] Treatment with ARB may have additional beneficial effects beyond the treatment of supine HTN. Previous studies have suggested that this class improves cerebral autoregulation in hypertensive patients with stroke[102] and diabetes.[103] Treatment with ARB may be a good choice for patients with nOH and severe supine HTN.
Conclusion
Cardiovascular autonomic dysfunction is highly prevalent in patients with PD. Recent evidence suggest that autonomic dysfunction including nOH may precede the appearance of motor symptoms. Neurogenic orthostatic hypotension in patients with PD has deleterious effect; it increases the risk of medical-attended falls and hospitalizations. Furthermore, nOH has been associated with depression and cognitive decline in PD patients. Treatment of nOH is challenging because of few therapeutic options with only two FDA-approved drugs (midodrine and droxidopa) in the past twenty years. Small studies suggest that fludrocortisone, octreotide, pyridostigmine, ergotamine/caffeine and yohimbine can be used for the treatment of nOH. A new class of drugs, NET blocker, showed promising results for the treatment of nOH and we are currently conducting the first clinical trial to evaluate the chronic effect of atomoxetine, a selective NET blocker for the treatment of symptomatic nOH.
Cardiovascular co-morbidities such as supine HTN and post-prandial hypotension associates with nOH and complicates its treatment. Acarbose is the only available medication that specifically targets postprandial hypotension. Often, medications with opposing effects such as pressor agents and short acting anti-HTN are used in combination in patients with OH and supine HTN. The majority of the literature that addresses the therapeutic management of this condition relies on small clinical trials that investigate the acute effects of short-acting anti-HTN medications. Among them short acting nifedipine, nitroglycerin transdermal patches, sildenafil, nebivolol, losartan and eplerenone showed blood pressure-lowering effect.
Key Points.
Cardiovascular autonomic dysfunction, particularly neurogenic orthostatic hypotension (nOH), is relatively common in patients with PD and occurs before the development of motor symptoms.
Neurogenic orthostatic hypotension has deleterious effects; it increases the risk of medical-attended falls and hospitalizations and is associated with depression and cognitive decline.
Treatment of nOH is challenging because of few therapeutic options. Only midodrine and droxidopa (approved in the US and Japan) have evidence from randomized clinical trials supporting its use for the treatment of nOH.
Supine hypertension occurs in 50–60% of patients with nOH. Treatment for this condition should be individualized based on the severity of hypertension and target organ damage.
Acknowledgments
C.A.S. was supported by Doris Duke Foundation Career Development Award. C.A.S. and H.K. received grant support from Office of Orphan Products Development. Food and Drug Administration, Grant #FD-R-04778-01-A3
Statement regarding funding for the article/study
The authors did not receive funding for the preparation and writing of the present manuscript.
C.A.S. has received research grant from Doris Duke Foundation. C.A.S. has received speaker honorarium from Lundbeck Pharmaceuticals. C.A.S. and H.K. received consulting honoraria from Lundbeck and Theravance Biopharma. C.A.S is member of the Board for the American Autonomic Society.
Footnotes
Compliance with Ethical Standards
Disclosure of Potential Conflict of Interest
References
- 1.Ascherio A, Schwarzschild MA. The epidemiology of Parkinson’s disease: risk factors and prevention. Lancet Neurol. 2016 Nov;15(12):1257–72. doi: 10.1016/S1474-4422(16)30230-7. [DOI] [PubMed] [Google Scholar]
- 2.Wright Willis A, Evanoff BA, Lian M, Criswell SR, Racette BA. Geographic and ethnic variation in Parkinson disease: a population-based study of US Medicare beneficiaries. Neuroepidemiology. 2010;34(3):143–51. doi: 10.1159/000275491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Fearnley JM, Lees AJ. Ageing and Parkinson’s disease: substantia nigra regional selectivity. Brain. 1991 Oct;114(Pt 5):2283–301. doi: 10.1093/brain/114.5.2283. [DOI] [PubMed] [Google Scholar]
- 4.de la Fuente-Fernandez R, Schulzer M, Kuramoto L, Cragg J, Ramachandiran N, Au WL, et al. Age-specific progression of nigrostriatal dysfunction in Parkinson’s disease. Ann Neurol. 2011 May;69(5):803–10. doi: 10.1002/ana.22284. [DOI] [PubMed] [Google Scholar]
- 5.Braak H, Del Tredici K. Invited Article: Nervous system pathology in sporadic Parkinson disease. Neurology. 2008 May;70(20):13. 1916–25. doi: 10.1212/01.wnl.0000312279.49272.9f. [DOI] [PubMed] [Google Scholar]
- 6.Braak H, Del Tredici K, Rub U, de Vos RA, Jansen Steur EN, Braak E. Staging of brain pathology related to sporadic Parkinson’s disease. Neurobiol Aging. 2003 Mar-Apr;24(2):197–211. doi: 10.1016/s0197-4580(02)00065-9. [DOI] [PubMed] [Google Scholar]
- 7.Boeve BF, Silber MH, Saper CB, Ferman TJ, Dickson DW, Parisi JE, et al. Pathophysiology of REM sleep behaviour disorder and relevance to neurodegenerative disease. Brain. 2007;130(Pt 11):2770–88. doi: 10.1093/brain/awm056. 11/2007. [DOI] [PubMed] [Google Scholar]
- 8.Postuma RB, Gagnon JF, Pelletier A, Montplaisir J. Prodromal autonomic symptoms and signs in Parkinson’s disease and dementia with Lewy bodies. Mov Disord. 2013 May;28(5):597–604. doi: 10.1002/mds.25445. [DOI] [PubMed] [Google Scholar]
- 9.Kaufmann H, Norcliffe-Kaufmann L, Palma JA, Biaggioni I, Low PA, Singer W, et al. Natural history of pure autonomic failure: A United States prospective cohort. Ann Neurol. 2017 Feb;81(2):287–97. doi: 10.1002/ana.24877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Velseboer DC, de Haan RJ, Wieling W, Goldstein DS, de Bie RM. Prevalence of orthostatic hypotension in Parkinson’s disease: a systematic review and meta-analysis. Parkinsonism Relat Disord. 2011 Dec;17(10):724–9. doi: 10.1016/j.parkreldis.2011.04.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gibbons CH, Schmidt P, Biaggioni I, Frazier-Mills C, Freeman R, Isaacson S, et al. The recommendations of a consensus panel for the screening, diagnosis, and treatment of neurogenic orthostatic hypotension and associated supine hypertension. J Neurol. 2017 Jan;:03. doi: 10.1007/s00415-016-8375-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Palma JA, Gomez-Esteban JC, Norcliffe-Kaufmann L, Martinez J, Tijero B, Berganzo K, et al. Orthostatic hypotension in Parkinson disease: how much you fall or how low you go? Mov Disord. 2015 Apr;30(5):15. 639–45. doi: 10.1002/mds.26079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ha AD, Brown CH, York MK, Jankovic J. The prevalence of symptomatic orthostatic hypotension in patients with Parkinson’s disease and atypical parkinsonism. ParkinsonismRelat Disord. 2011;17(8):625–8. doi: 10.1016/j.parkreldis.2011.05.020. 9/2011. [DOI] [PubMed] [Google Scholar]
- 14.Hely MA, Morris JG, Reid WG, Trafficante R. Sydney Multicenter Study of Parkinson’s disease: non-L-dopa-responsive problems dominate at 15 years. Mov Disord. 2005 Feb;20(2):190–9. doi: 10.1002/mds.20324. [DOI] [PubMed] [Google Scholar]
- 15.Xin W, Lin Z, Mi S. Orthostatic hypotension and mortality risk: a meta-analysis of cohort studies. Heart. 2014 Mar;100(5):406–13. doi: 10.1136/heartjnl-2013-304121. [DOI] [PubMed] [Google Scholar]
- 16.Pathak A, Lapeyre-Mestre M, Montastruc JL, Senard JM. Heat-related morbidity in patients with orthostatic hypotension and primary autonomic failure. Mov Disord. 2005 Sep;20(9):1213–9. doi: 10.1002/mds.20571. [DOI] [PubMed] [Google Scholar]
- 17.Stolze H, Klebe S, Zechlin C, Baecker C, Friege L, Deuschl G. Falls in frequent neurological diseases–prevalence, risk factors and aetiology. J Neurol. 2004 Jan;251(1):79–84. doi: 10.1007/s00415-004-0276-8. [DOI] [PubMed] [Google Scholar]
- 18.Woodford H, Walker R. Emergency hospital admissions in idiopathic Parkinson’s disease. Mov Disord. 2005 Sep;20(9):1104–8. doi: 10.1002/mds.20485. [DOI] [PubMed] [Google Scholar]
- 19.Low V, Ben-Shlomo Y, Coward E, Fletcher S, Walker R, Clarke CE. Measuring the burden and mortality of hospitalisation in Parkinson’s disease: A cross-sectional analysis of the English Hospital Episodes Statistics database 2009–2013. Parkinsonism Relat Disord. 2015 May;21(5):449–54. doi: 10.1016/j.parkreldis.2015.01.017. [DOI] [PubMed] [Google Scholar]
- 20.Allen NE, Schwarzel AK, Canning CG. Recurrent falls in Parkinson’s disease: a systematic review. Parkinsons Dis. 2013;2013:906274. doi: 10.1155/2013/906274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Francois C, Biaggioni I, Shibao C, Ogbonnaya A, Shih HC, Farrelly E, et al. Fall-related healthcare use and costs in neurogenic orthostatic hypotension with Parkinson’s disease. J Med Econ. 2017 May;20(5):525–32. doi: 10.1080/13696998.2017.1284668. [DOI] [PubMed] [Google Scholar]
- 22.Schrag A, Hovris A, Morley D, Quinn N, Jahanshahi M. Caregiver-burden in parkinson’s disease is closely associated with psychiatric symptoms, falls, and disability. Parkinsonism Relat Disord. 2006 Jan;12(1):35–41. doi: 10.1016/j.parkreldis.2005.06.011. [DOI] [PubMed] [Google Scholar]
- 23.Magerkurth C, Schnitzer R, Braune S. Symptoms of autonomic failure in Parkinson’s disease: prevalence and impact on daily life. ClinAutonRes. 2005;15(2):76–82. doi: 10.1007/s10286-005-0253-z. 4/2005. [DOI] [PubMed] [Google Scholar]
- 24.Pilleri M, Facchini S, Gasparoli E, Biundo R, Bernardi L, Marchetti M, et al. Cognitive and MRI correlates of orthostatic hypotension in Parkinson’s disease. J Neurol. 2013 Jan;260(1):253–9. doi: 10.1007/s00415-012-6627-y. [DOI] [PubMed] [Google Scholar]
- 25.Allcock LM, Kenny RA, Mosimann UP, Tordoff S, Wesnes KA, Hildreth AJ, et al. Orthostatic hypotension in Parkinson’s disease: association with cognitive decline? Int J Geriatr Psychiatry. 2006 Aug;21(8):778–83. doi: 10.1002/gps.1562. [DOI] [PubMed] [Google Scholar]
- 26.Frewen J, Savva GM, Boyle G, Finucane C, Kenny RA. Cognitive performance in orthostatic hypotension: findings from a nationally representative sample. J Am Geriatr Soc. 2014 Jan;62(1):117–22. doi: 10.1111/jgs.12592. [DOI] [PubMed] [Google Scholar]
- 27.Mehrabian S, Duron E, Labouree F, Rollot F, Bune A, Traykov L, et al. Relationship between orthostatic hypotension and cognitive impairment in the elderly. J Neurol Sci. 2010 Dec 15;299(1–2):45–8. doi: 10.1016/j.jns.2010.08.056. [DOI] [PubMed] [Google Scholar]
- 28.Poda R, Guaraldi P, Solieri L, Calandra-Buonaura G, Marano G, Gallassi R, et al. Standing worsens cognitive functions in patients with neurogenic orthostatic hypotension. Neurol Sci. 2012 Apr;33(2):469–73. doi: 10.1007/s10072-011-0746-6. [DOI] [PubMed] [Google Scholar]
- 29.Centi J, Freeman R, Gibbons CH, Neargarder S, Canova AO, Cronin-Golomb A. Effects of orthostatic hypotension on cognition in Parkinson disease. Neurology. 2017 Jan;88(1):03. 17–24. doi: 10.1212/WNL.0000000000003452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Li ST, Dendi R, Holmes C, Goldstein DS. Progressive loss of cardiac sympathetic innervation in Parkinson’s disease. AnnNeurol. 2002;52(2):220–3. doi: 10.1002/ana.10236. 8/2002. [DOI] [PubMed] [Google Scholar]
- 31.Mitsui J, Saito Y, Momose T, Shimizu J, Arai N, Shibahara J, et al. Pathology of the sympathetic nervous system corresponding to the decreased cardiac uptake in 123I-metaiodobenzylguanidine (MIBG) scintigraphy in a patient with Parkinson disease. JNeurolSci. 2006;243(1–2):101–4. doi: 10.1016/j.jns.2005.11.034. 4/15/2006. [DOI] [PubMed] [Google Scholar]
- 32.Braune S, Reinhardt M, Schnitzer R, Riedel A, Lucking CH. Cardiac uptake of [123I]MIBG separates Parkinson’s disease from multiple system atrophy. Neurology. 1999;53(5):1020–5. doi: 10.1212/wnl.53.5.1020. 9/22/1999. [DOI] [PubMed] [Google Scholar]
- 33.Kaufmann H, Goldstein DS. Autonomic dysfunction in Parkinson disease. Handb Clin Neurol. 2013;117:259–78. doi: 10.1016/B978-0-444-53491-0.00021-3. [DOI] [PubMed] [Google Scholar]
- 34.Orimo S, Uchihara T, Nakamura A, Mori F, Kakita A, Wakabayashi K, et al. Axonal alpha-synuclein aggregates herald centripetal degeneration of cardiac sympathetic nerve in Parkinson’s disease. Brain. 2008 Mar;131(Pt 3):642–50. doi: 10.1093/brain/awm302. [DOI] [PubMed] [Google Scholar]
- 35.Palma JA, Kaufmann H. Autonomic disorders predicting Parkinson’s disease. Parkinsonism Relat Disord. 2014 Jan;20(Suppl 1):S94–8. doi: 10.1016/S1353-8020(13)70024-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Braune S. The role of cardiac metaiodobenzylguanidine uptake in the differential diagnosis of parkinsonian syndromes. Clinical autonomic research: official journal of the Clinical Autonomic Research Society. 2001 Dec;11(6):351–5. doi: 10.1007/BF02292766. [DOI] [PubMed] [Google Scholar]
- 37.Diedrich A, Biaggioni I. Segmental orthostatic fluid shifts. ClinAutonRes. 2004;14(3):146–7. doi: 10.1007/s10286-004-0188-9. 6/2004. [DOI] [PubMed] [Google Scholar]
- 38.Novak V, Novak P, Spies JM, Low PA. Autoregulation of cerebral blood flow in orthostatic hypotension. Stroke; a journal of cerebral circulation. 1998 Jan;29(1):104–11. doi: 10.1161/01.str.29.1.104. [DOI] [PubMed] [Google Scholar]
- 39.Bird ST, Delaney JA, Brophy JM, Etminan M, Skeldon SC, Hartzema AG. Tamsulosin treatment for benign prostatic hyperplasia and risk of severe hypotension in men aged 40–85 years in the United States: risk window analyses using between and within patient methodology. BMJ. 2013 Nov 05;347:f6320. doi: 10.1136/bmj.f6320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Podoleanu C, Maggi R, Brignole M, Croci F, Incze A, Solano A, et al. Lower limb and abdominal compression bandages prevent progressive orthostatic hypotension in elderly persons: a randomized single-blind controlled study. J Am Coll Cardiol. 2006 Oct;48(7):03. 1425–32. doi: 10.1016/j.jacc.2006.06.052. [DOI] [PubMed] [Google Scholar]
- 41.Smit AA, Wieling W, Fujimura J, Denq JC, Opfer-Gehrking TL, Akarriou M, et al. Use of lower abdominal compression to combat orthostatic hypotension in patients with autonomic dysfunction. ClinAutonRes. 2004;14(3):167–75. doi: 10.1007/s10286-004-0187-x. 6/2004. [DOI] [PubMed] [Google Scholar]
- 42.Jordan J, Shannon JR, Grogan E, Biaggioni I, Robertson D. A potent pressor response elicited by drinking water. Lancet. 1999;353(9154):723. doi: 10.1016/S0140-6736(99)99015-3. 2/27/1999. [DOI] [PubMed] [Google Scholar]
- 43.Jordan J. Effect of water drinking on sympathetic nervous activity and blood pressure. Curr Hypertens Rep. 2005 Feb;7(1):17–20. doi: 10.1007/s11906-005-0050-z. [DOI] [PubMed] [Google Scholar]
- 44.Ten Harkel AD, Baisch F, Karenmaker JM. Increased orthostatic blood pressure variability after prolonged head-down tilt. Acta PhysiolScand. 1992;144:89–99. 1992. [PubMed] [Google Scholar]
- 45.van Lieshout JJ, Ten Harkel AD, Wieling W. Fludrocortisone and sleeping in the head-up position limit the postural decrease in cardiac output in autonomic failure. ClinAutonRes. 2000;10(1):35–42. doi: 10.1007/BF02291388. 2/2000. [DOI] [PubMed] [Google Scholar]
- 46.Puvi-Rajasingham S, Mathias CJ. Effect of meal size on post-prandial blood pressure and on postural hypotension in primary autonomic failure. Clinical autonomic research: official journal of the Clinical Autonomic Research Society. 1996;6(2):111–4. doi: 10.1007/BF02291232. 4/1996. [DOI] [PubMed] [Google Scholar]
- 47.Mathias CJ. Postprandial hypotension. Pathophysiological mechanisms and clinical implications in different disorders. Hypertension. 1991;18(5):694–704. doi: 10.1161/01.hyp.18.5.694. 11/1991. [DOI] [PubMed] [Google Scholar]
- 48.Visvanathan R, Chen R, Garcia M, Horowitz M, Chapman I. The effects of drinks made from simple sugars on blood pressure in healthy older people. BrJNutr. 2005;93(5):575–9. doi: 10.1079/bjn20051401. 5/2005. [DOI] [PubMed] [Google Scholar]
- 49.Onrot J, Goldberg MR, Biaggioni I, Hollister AS, Kincaid D, Robertson D. Hemodynamic and humoral effects of caffeine in human autonomic failure. NEnglJMed. 1985;313:549–54. doi: 10.1056/NEJM198508293130905. 1985. [DOI] [PubMed] [Google Scholar]
- 50.Chaudhuri KR, Ellis C, Love-Jones S, Thomaides T, Clift S, Mathias CJ, et al. Postprandial hypotension and parkinsonian state in Parkinson’s disease. Mov Disord. 1997 Nov;12(6):877–84. doi: 10.1002/mds.870120608. [DOI] [PubMed] [Google Scholar]
- 51.Kaufmann H, Norcliffe-Kaufmann L, Palma JA. Droxidopa in neurogenic orthostatic hypotension. Expert Rev Cardiovasc Ther. 2015;13(8):875–91. doi: 10.1586/14779072.2015.1057504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Biaggioni I, Freeman R, Mathias CJ, Low P, Hewitt LA, Kaufmann H, et al. Randomized withdrawal study of patients with symptomatic neurogenic orthostatic hypotension responsive to droxidopa. Hypertension. 2015 Jan;65(1):101–7. doi: 10.1161/HYPERTENSIONAHA.114.04035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Hauser RA, Isaacson S, Lisk JP, Hewitt LA, Rowse G. Droxidopa for the short-term treatment of symptomatic neurogenic orthostatic hypotension in Parkinson’s disease (nOH306B) Mov Disord. 2015 Apr;30(5):15. 646–54. doi: 10.1002/mds.26086. [DOI] [PubMed] [Google Scholar]
- 54.Chatrchyan S, Khachatryan V, Sirunyan AM, Tumasyan A, Adam W, Bergauer T, et al. Measurement of inclusive W and Z boson production cross sections in pp collisions at sqrt[s] = 8TeV. Phys Rev Lett. 2014 May 16;112(19):191802. doi: 10.1103/PhysRevLett.112.191802. [DOI] [PubMed] [Google Scholar]
- 55.Kaufmann H, Freeman R, Biaggioni I, Low P, Pedder S, Hewitt LA, et al. Droxidopa for neurogenic orthostatic hypotension: a randomized, placebo-controlled, phase 3 trial. Neurology. 2014;83(4):328–35. doi: 10.1212/WNL.0000000000000615. 7/22/2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Biaggioni I, Arthur Hewitt L, Rowse GJ, Kaufmann H. Integrated analysis of droxidopa trials for neurogenic orthostatic hypotension. BMC Neurol. 2017 May;17(1):12. 90. doi: 10.1186/s12883-017-0867-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Hauser RA, Heritier S, Rowse GJ, Hewitt LA, Isaacson SH. Droxidopa and Reduced Falls in a Trial of Parkinson Disease Patients With Neurogenic Orthostatic Hypotension. Clin Neuropharmacol. 2016 Sep-Oct;39(5):220–6. doi: 10.1097/WNF.0000000000000168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Goldberg MR, Hollister AS, Robertson D. Influence of yohimbine on blood pressure, autonomic reflexes and plasma catecholamines in humans. Hypertension. 1983;5:772–8. doi: 10.1161/01.hyp.5.5.772. 1983. [DOI] [PubMed] [Google Scholar]
- 59.Onrot J, Goldberg MR, Biaggioni I, Wiley R, Hollister AS, Robertson D. Oral yohimbine in human autonomic failure. Neurology. 1987;37:215–20. doi: 10.1212/wnl.37.2.215. 1987. [DOI] [PubMed] [Google Scholar]
- 60.Jordan J, Shannon JR, Biaggioni I, Norman R, Black BK, Robertson D. Contrasting actions of pressor agents in severe autonomic failure. American Journal of Medicine. 1998;105(2):116–24. doi: 10.1016/s0002-9343(98)00193-4. 8/1998. [DOI] [PubMed] [Google Scholar]
- 61.Shibao C, Okamoto LE, Gamboa A, Yu C, Diedrich A, Raj SR, et al. Comparative efficacy of yohimbine against pyridostigmine for the treatment of orthostatic hypotension in autonomic failure. Hypertension. 2010 Nov;56(5):847–51. doi: 10.1161/HYPERTENSIONAHA.110.154898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Singer W, Opfer-Gehrking TL, McPhee BR, Hilz MJ, Bharucha AE, Low PA. Acetylcholinesterase inhibition: a novel approach in the treatment of neurogenic orthostatic hypotension. JNeurolNeurosurgPsychiatry. 2003;74(9):1294–8. doi: 10.1136/jnnp.74.9.1294. 9/2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Singer W, Sandroni P, Opfer-Gehrking TL, Suarez GA, Klein CM, Hines S, et al. Pyridostigmine treatment trial in neurogenic orthostatic hypotension. Arch Neurol. 2006 Apr;63(4):513–8. doi: 10.1001/archneur.63.4.noc50340. [DOI] [PubMed] [Google Scholar]
- 64.Simpson D, Plosker GL. Atomoxetine: a review of its use in adults with attention deficit hyperactivity disorder. Drugs. 2004;64(2):205–22. doi: 10.2165/00003495-200464020-00005. [DOI] [PubMed] [Google Scholar]
- 65.Shibao C, Raj SR, Gamboa A, Diedrich A, Choi L, Black BK, et al. Norepinephrine transporter blockade with atomoxetine induces hypertension in patients with impaired autonomic function. Hypertension. 2007;50(1):47–53. doi: 10.1161/HYPERTENSIONAHA.107.089961. 7/2007. [DOI] [PubMed] [Google Scholar]
- 66.Ramirez CE, Okamoto LE, Arnold AC, Gamboa A, Diedrich A, Choi L, et al. Efficacy of atomoxetine versus midodrine for the treatment of orthostatic hypotension in autonomic failure. Hypertension. 2014 Dec;64(6):1235–40. doi: 10.1161/HYPERTENSIONAHA.114.04225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Low PA, Gilden JL, Freeman R, Sheng KN, McElligott MA. Efficacy of midodrine vs placebo in neurogenic orthostatic hypotension. A randomized, double-blind multicenter study. Midodrine Study Group. Jama. 1997;277(13):1046–51. 4/2/1997. [PubMed] [Google Scholar]
- 68.Wright RA, Kaufmann HC, Perera R, Opfer-Gehrking TL, McElligott MA, Sheng KN, et al. A double-blind, dose-response study of midodrine in neurogenic orthostatic hypotension. Neurology. 1998;51(1):120–4. doi: 10.1212/wnl.51.1.120. 7/1998. [DOI] [PubMed] [Google Scholar]
- 69.Bevegard S, Castenfors J, Lindblad LE. Haemodynamic effects of dihydroergotamine in patients with postural hypotension. Acta MedScand. 1974;196:473–7. doi: 10.1111/j.0954-6820.1974.tb01044.x. 1974. [DOI] [PubMed] [Google Scholar]
- 70.Benowitz NL, Byrd R, Schambelan M, Rosenberg J, Roizen MF. Dihydroergotamine treatment for orthostatic hypotension from Vacor rodenticide. AnnIntMed. 1980;92:387–8. doi: 10.7326/0003-4819-92-3-387. 1980. [DOI] [PubMed] [Google Scholar]
- 71.Chobanian AV, Tifft CP, Faxon DP, Creager MLA, Sackel H. Treatment of orthostatic hypotension with ergotamine. Circulation. 1983;67:602–9. doi: 10.1161/01.cir.67.3.602. 1983. [DOI] [PubMed] [Google Scholar]
- 72.Hoeldtke RD, Cavanaugh ST, Hughes JD, Polansky M. Treatment of orthostatic hypotension with dihydroergotamine and caffeine. AnnIntMed. 1986;105:168–73. doi: 10.7326/0003-4819-105-2-168. 1986. [DOI] [PubMed] [Google Scholar]
- 73.Biaggioni I, Zygmunt D, Haile V, Robertson D. Pressor effect of inhaled ergotamine in orthostatic hypotension. AmJCardiol. 1990;65:89–92. doi: 10.1016/0002-9149(90)90031-u. 1990. [DOI] [PubMed] [Google Scholar]
- 74.Arnold AC, Ramirez CE, Choi L, Okamoto LE, Gamboa A, Diedrich A, et al. Combination ergotamine and caffeine improves seated blood pressure and presyncopal symptoms in autonomic failure. Front Physiol. 2014;5:270. doi: 10.3389/fphys.2014.00270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Armstrong E, Mathias CJ. The effects of the somatostatin analogue, octreotide, on postural hypotension, before and after food ingestion, in primary autonomic failure. ClinAutonRes. 1991;1(2):135–40. doi: 10.1007/BF01826210. 6/1991. [DOI] [PubMed] [Google Scholar]
- 76.Hoeldtke RD, O’Dorisio TM, Boden G. Treatment of autonomic neuropathy with a somatostatin analog SMS 201-995. Lancet. 1986;2:602–5. doi: 10.1016/s0140-6736(86)92428-1. 1986. [DOI] [PubMed] [Google Scholar]
- 77.Hoeldtke RD, Dworkin GE, Gaspar SR, Israel BC, Boden G. Effect of the somatostatin analogue SMS-201-995 on the adrenergic response to glucose ingestion in patients with post prandial hypotension. AmJMed. 1989;86:673–7. doi: 10.1016/0002-9343(89)90442-7. 1989. [DOI] [PubMed] [Google Scholar]
- 78.Chobanian AV, Volicer L, Tifft CP, Gavras H, Liang CS, Faxon D. Mineralocorticoid-induced hypertension in patients with orthostatic hypotension. NEnglJMed. 1979;301:68–73. doi: 10.1056/NEJM197907123010202. 1979. [DOI] [PubMed] [Google Scholar]
- 79.Campbell IW, Ewing DJ, Clarke BF. 9-a-fluorohydrocortisone in the treatment of postural hypotension in diabetic autonomic neuropathy. Diabetes. 1975;24:381–4. doi: 10.2337/diab.24.4.381. 1975. [DOI] [PubMed] [Google Scholar]
- 80.Hoehn MM. Levodopa-induced postural hypotension. Treatment with fludrocortisone. Archives of Neurology. 1975;32:50–1. doi: 10.1001/archneur.1975.00490430072013. 1975. [DOI] [PubMed] [Google Scholar]
- 81.Campbell IW, Ewing DJ, Clarke BF. Therapeutic experience with fludrocortisone in diabetic postural hypotension. British Medical Journal. 1976;1(6014):872–4. doi: 10.1136/bmj.1.6014.872. 4/10/1976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Hussain RM, McIntosh SJ, Lawson J, Kenny RA. Fludrocortisone in the treatment of hypotensive disorders in the elderly. Heart. 1996;76(6):507–9. doi: 10.1136/hrt.76.6.507. 12/1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Pathak A, Raoul V, Montastruc JL, Senard JM. Adverse drug reactions related to drugs used in orthostatic hypotension: a prospective and systematic pharmacovigilance study in France. EurJClinPharmacol. 2005;61(5–6):471–4. doi: 10.1007/s00228-005-0941-6. 7/2005. [DOI] [PubMed] [Google Scholar]
- 84.Maule S, Tredici M, Dematteis A, Matteoda C, Chiandussi L. Postprandial hypotension treated with acarbose in a patient with type 1 diabetes mellitus. Clin Auton Res. 2004 Dec;14(6):405–7. doi: 10.1007/s10286-004-0220-0. [DOI] [PubMed] [Google Scholar]
- 85.Lyons TJ, McLoughlin JC, Shaw C, Buchanan KD. Effect of acarbose on biochemical responses and clinical symptoms in dumping syndrome. Digestion. 1985;31(2–3):89–96. doi: 10.1159/000199185. 1985. [DOI] [PubMed] [Google Scholar]
- 86.Shibao C, Gamboa A, Diedrich A, Dossett C, Choi L, Farley G, et al. Acarbose, an alpha-glucosidase inhibitor, attenuates postprandial hypotension in autonomic failure. Hypertension. 2007;50(1):54–61. doi: 10.1161/HYPERTENSIONAHA.107.091355. 7/2007. [DOI] [PubMed] [Google Scholar]
- 87.Biaggioni I, Robertson RM. Hypertension in orthostatic hypotension and autonomic dysfunction. CardiolClin. 20(2):291–301. doi: 10.1016/s0733-8651(01)00005-4. 2002. 2002. [DOI] [PubMed] [Google Scholar]
- 88.Arnold AC, Biaggioni I. Management approaches to hypertension in autonomic failure. CurrOpinNephrolHypertens. 2012;21(5):481–5. doi: 10.1097/MNH.0b013e328356c52f. 9/2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Vagaonescu TD, Saadia D, Tuhrim S, Phillips RA, Kaufmann H. Hypertensive cardiovascular damage in patients with primary autonomic failure. Lancet. 2000;355(9205):725–6. doi: 10.1016/S0140-6736(99)05320-9. 2/26/2000. [DOI] [PubMed] [Google Scholar]
- 90.Garland EM, Gamboa A, Okamoto L, Raj SR, Black BK, Davis TL, et al. Renal impairment of pure autonomic failure. Hypertension. 2009 Nov;54(5):1057–61. doi: 10.1161/HYPERTENSIONAHA.109.136853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Shannon JR, Jordan J, Diedrich A, Pohar B, Black BK, Robertson D, et al. Sympathetically mediated hypertension in autonomic failure. Circulation. 2000;101(23):2710–5. doi: 10.1161/01.cir.101.23.2710. 6/13/2000. [DOI] [PubMed] [Google Scholar]
- 92.Ibrahim MM, Tarazi RC, Dustan HP, Bravo EL. Idiopathic orthostatic hypotension: circulatory dynamics in chronic autonomic insufficiency. AmJCardiol. 1974;34:288–94. doi: 10.1016/0002-9149(74)90029-0. 1974. [DOI] [PubMed] [Google Scholar]
- 93.Biaggioni I, Garcia F, Inagami T, Haile V. Hyporeninemic normoaldosteronism in severe autonomic failure. Journal of Clinical Endocrinology & Metabolism. 1993;76:580–6. doi: 10.1210/jcem.76.3.7680352. 1993. [DOI] [PubMed] [Google Scholar]
- 94.Gamboa A, Shibao C, Diedrich A, Paranjape SY, Farley G, Christman B, et al. Excessive nitric oxide function and blood pressure regulation in patients with autonomic failure. Hypertension. 2008 Jun;51(6):1531–6. doi: 10.1161/HYPERTENSIONAHA.107.105171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Arnold AC, Okamoto LE, Gamboa A, Shibao C, Raj SR, Robertson D, et al. Angiotensin II, independent of plasma renin activity, contributes to the hypertension of autonomic failure. Hypertension. 2013 Mar;61(3):701–6. doi: 10.1161/HYPERTENSIONAHA.111.00377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Arnold AC, Okamoto LE, Gamboa A, Black BK, Raj SR, Elijovich F, et al. Mineralocorticoid Receptor Activation Contributes to the Supine Hypertension of Autonomic Failure. Hypertension. 2016 Feb;67(2):424–9. doi: 10.1161/HYPERTENSIONAHA.115.06617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Okamoto LE, Gamboa A, Shibao C, Black BK, Diedrich A, Raj SR, et al. Nocturnal blood pressure dipping in the hypertension of autonomic failure. Hypertension. 2009 Feb;53(2):363–9. doi: 10.1161/HYPERTENSIONAHA.108.124552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Okamoto LE, Celedonio JE, Gamboa A, Shibao CA, Raj SR, Diedrich A, et al. Abstract 022: Blood Pressure-Lowering Effect of Local Passive Heat in Autonomic Failure Patients with Supine Hypertension. Hypertension. 2016;68(Suppl 1):A022–A. [Google Scholar]
- 99.Jordan J, Shannon JR, Pohar B, Paranjape SY, Robertson D, Robertson RM, et al. Contrasting effects of vasodilators on blood pressure and sodium balance in the hypertension of autonomic failure. Journal of the American Society of Nephrology. 1999;10(1):35–42. doi: 10.1681/ASN.V10135. 1/1999. [DOI] [PubMed] [Google Scholar]
- 100.Pahor M, Guralnik JM, Corti MC, Foley DJ, Carbonin P, Havlik RJ. Long-term survival and use of antihypertensive medications in older persons. J Am Geriatr Soc. 1995 Nov;43(11):1191–7. doi: 10.1111/j.1532-5415.1995.tb07393.x. [DOI] [PubMed] [Google Scholar]
- 101.Okamoto LE, Gamboa A, Shibao CA, Arnold AC, Choi L, Black BK, et al. Nebivolol, but not metoprolol, lowers blood pressure in nitric oxide-sensitive human hypertension. Hypertension. 2014 Dec;64(6):1241–7. doi: 10.1161/HYPERTENSIONAHA.114.04116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Moriwaki H, Uno H, Nagakane Y, Hayashida K, Miyashita K, Naritomi H. Losartan, an angiotensin II (AT1) receptor antagonist, preserves cerebral blood flow in hypertensive patients with a history of stroke. J Hum Hypertens. 2004 Oct;18(10):693–9. doi: 10.1038/sj.jhh.1001735. [DOI] [PubMed] [Google Scholar]
- 103.Kario K, Ishikawa J, Hoshide S, Matsui Y, Morinari M, Eguchi K, et al. Diabetic brain damage in hypertension: role of renin-angiotensin system. Hypertension. 2005 May;45(5):887–93. doi: 10.1161/01.HYP.0000163460.07639.3f. [DOI] [PubMed] [Google Scholar]