

Abstract
Nucleoside triphosphates play a central role in biology, but efforts to study these roles have proven difficult because the levels of triphosphates are tightly regulated in a cell and because individual triphosphates can be difficult to label or modify. In addition, many synthetic biology efforts are focused on the development of unnatural nucleoside triphosphates that perform specific functions in the cellular environment. In general, both of these efforts would be facilitated by a general means to directly introduce desired triphosphates into cells. Previously, we demonstrated that recombinant expression of a nucleoside triphosphate transporter from Phaeodactylum tricornutum (PtNTT2) in Escherichia coli functions to import triphosphates that are added to the media. Here, to explore the generality and utility of this approach, we report a structure activity-relationship study of PtNTT2. Using a conventional competitive uptake inhibition assay, we characterize the effects of nucleobase, sugar, and triphosphate modification, and then develop an LC-MS/MS assay to directly measure the effects of the modifications on import. Lastly, we use the transporter to import radiolabeled or 2′-fluoro-modified triphosphates and quantify their incorporation into DNA and RNA. The results demonstrate the general utility of the PtNTT2-mediated import of natural or modified nucleoside triphosphates for different molecular or synthetic biology applications.
Graphical abstract
INTRODUCTION
Nucleoside triphosphates play a central role in virtually all aspects of biology. In addition to their fundamental roles as substrates for the synthesis of DNA and RNA and as the currency of cellular energy, nucleoside triphosphates can regulate transcription, translation, and inter- or intracellular signaling, and they are also required for glycogen, lipid, and cofactor synthesis. Unfortunately, characterizing the roles of nucleoside triphosphates in the cell is complicated by our inability to selectively label and/or experimentally control specific triphosphate concentrations. In addition, many recent efforts in synthetic biology have focused on the use of sugar- or nucleobase-modified triphosphates, for example, for the creation of novel biopolymers1,2 or the expansion of the genetic alphabet,3–5 and ultimately all of these efforts require the availability of unnatural nucleoside triphosphates within the cell.
One possible route to the introduction of natural or unnatural triphosphates into cells is the passive diffusion or facilitated uptake of the corresponding nucleosides across the cell membrane(s),6–9 followed by their phosphorylation via the kinases of the nucleoside salvage pathway.10 Indeed, this strategy is employed by many nucleoside prodrugs, such as azidothymidine (AZT) and gemcitabine, which are active as triphosphates. However, this approach is unlikely to be general, because many organisms, including fungi and bacteria, disproportionately or exclusively rely on de novo synthesis, and have lost some or all of the salvage kinases.11–14 Furthermore, even if salvage kinases are present, their recognition of unnatural analogs may not be sufficient.15 For example, gemcitabine is inactive against many Gram-negative bacteria due to poor conversion of the free nucleoside to the monophosphate, while AZT is inactive against many Gram-positive bacteria, at least in part, due to poor conversion of the monophosphate to the diphosphate.16 In principle, these challenges may be overcome via heterologous expression of kinases with lower specificity.17–21 However, the addition of exogenous kinase activity may perturb the balance of natural triphosphates within the cell, which is known to be toxic.22–24 Regardless, whether with native or engineered pathways, relying on the activation of free nucleosides to produce the desired triphosphates is less than optimal, because it requires three steps of activation, and activation must compete with nucleoside degradation, because both eukaryotes and prokaryotes utilize nucleosides as sources of carbon and nitrogen.25–27 This is likely to increase the challenge of achieving controlled intracellular concentrations of the triphosphates, which is likely to be problematic with many applications.
As part of an effort to create semi-synthetic organisms that by virtue of an unnatural base pair store24,28 and retrieve29 increased genetic information, we have reported that the constituent unnatural nucleoside triphosphates, which bear nucleobases with little homology to their natural counterparts, are imported directly into Escherichia coli through a heterologously expressed nucleoside triphosphate transporter (NTT) from Phaeodactylum tricornutum (PtNTT2).30 In its native algae, PtNTT2 is thought to mediate the counter exchange of nucleoside triphosphates across the outer plastidial membrane from the cytoplasm to the stroma, where they are required for DNA and RNA synthesis. When expressed in E. coli, PtNTT2 is inserted into the cytoplasmic membrane, where it transports nucleoside triphosphates into the cytoplasm after they first diffuse from the media into the periplasm, presumably through porins. While PtNTT2 is selective for triphosphates over other nucleotides, its ability to transport all eight deoxy- and ribonucleoside triphosphates endows it with the broadest substrate scope of any known NTT, although the different natural substrates are transported with somewhat different efficiencies. Further, our observation that PtNTT2 may be used to import triphosphates with wholly unnatural nucleobases indicates that the substrate scope of PtNTT2 may extend far beyond the natural nucleotides. This suggests that PtNTT2 may be a useful tool for a variety of molecular and synthetic biology applications requiring the availability of unnatural nucleoside triphosphates within a cell.
The standard assay used to characterize triphosphate uptake with PtNTT2, as well as other NTTs, is based on the use of radiolabeled substrates.30 However, radiolabeled versions of many triphosphates of interest are not commercially available and are challenging to synthesize. To increase the range of substrates that may be examined, inhibition of the uptake of a commercially available radiolabeled substrate has also been used.30 However, this assay by definition only characterizes the ability of the triphosphate of interest to competitively bind the NTT and does not actually characterize its uptake. Here, as part of an effort to develop NTTs as general tools for molecular and synthetic biology, we report a structure activity-relationship (SAR) study of PtNTT2 using the standard inhibition assay, to characterize transporter binding, and we develop a more general LC-MS/MS assay to characterize uptake directly, including the uptake of interesting unnatural triphosphates whose characterization is not possible with traditional assays. The data constitute the first SAR study of NTT activity and clarify the scope of analogs that might be made available in vivo for molecular and synthetic biology applications. Finally, we demonstrate the utility of PtNTT2 for the characterization of DNA replication and transcription in E. coli with both natural and modified nucleotides.
RESULTS AND DISCUSSION
As with many membrane proteins, expression of native PtNTT2 is somewhat toxic in E. coli. However, we recently reported that removal of the protein’s N-terminal residues 1–65, which are normally removed in its native host, resulting in the variant PtNTT2(66-575), largely eliminates toxicity while preserving at least some level of activity.28 This allowed us to construct E. coli YZ2,28 which has PtNTT2(66-575) integrated into the chromosome under the control of the constitutive lacUV5 promoter. Chromosomal expression of PtNTT2(66-575) reduces variation in transporter expression, and presumably in uptake, compared to plasmid-based expression, due to reduced fluctuation in gene copy number. However, it is possible that deletion of the N-terminus alters activity. Thus, we first characterized the ability of the truncated transporter to import [α32P]ATP, using the standard radioactivity assay. Control experiments employing the BL21(DE3) CmR strain, which bears a chloramphenicol resistance marker at the locus that would otherwise carry the transporter, showed no detectable uptake, while YZ2 showed robust uptake. To characterize activity we determined normalized initial velocities of triphosphate uptake (expressed in the units fmol•cell−1•hr−1), and then plotted them against the concentration of [α32P]ATP added. Fitting the resulting curve to the Michaelis-Menten equation yielded the apparent KM and Vmax values, 186 ± 21.1 μM and 1.74 ± 0.091 × 10−2 fmol•cell−1•hr−1, respectively. Although we cannot compare Vmax as the absolute amount of transporter produced in strain YZ2 is unknown, the KM is in good agreement with that reported previously for the intact transporter,30 indicating that the truncation does not significantly alter activity.
Analysis of inhibition
To elucidate the SARs governing nucleoside triphosphate binding, we first employed the standard assay based on the inhibition of [α32P]-ATP uptake.30 Briefly, YZ2 cells were grown to an OD600 of ~0.3 in 2×YT media, after which [α32P]-ATP (50 μM) and a 10-fold excess of the triphosphate of interest was added. The cells were incubated for 10 min, during which time degradation due to extracellular phosphatases is insignificant,24 and then transferred by vacuum to filter paper, which was washed to remove excess triphosphate and subjected to phosphorimaging to determine [α32P]-ATP uptake. We initially focused on characterizing the SARs associated with the nucleobase and specifically on CTP with C5-modifications (Fig. 1), because CTP is the most efficiently imported natural substrate,30 and because such modifications are important in nature.31–33 Both 5-iodo-2′-cytidine triphosphate (5-ICTP) and 5-iodo-2′-deoxycytidine triphosphate (5-IdCTP) potently inhibited [α32P]-ATP uptake (96.6 ± 1.3 % and 73.6 ± 1.8 % inhibition, respectively). In fact, both of these analogs inhibited uptake more than or as much as their respective natural counterparts (70.5 ± 4.9% and 76.3 ± 2.8%, respectively). 5-hydroxymethyl cytidine (5-hmCTP) and 5-allyl amine cytidine (5-aaCTP) also potently inhibited uptake, but slightly less efficiently than their iodinated counterpart (76.3 ± 2.9% and 64.7 ± 9.3% inhibition, respectively). Based on this competitive inhibition data, it appears that the transporter is relatively tolerant of substitution at the C5-position of pyrimidine, with iodo substitution actually increasing inhibition.
Figure 1.
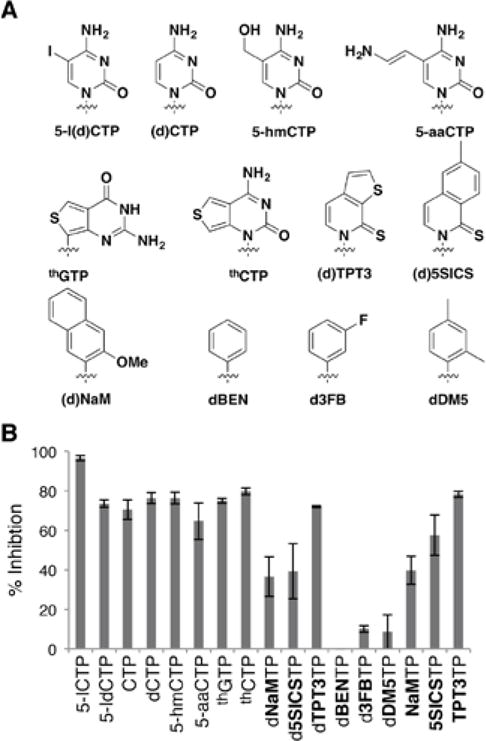
(A) Structure of nucleobase modified nucleoside triphosphates used for the inhibition assay; sugar and phosphates omitted for clarity. (B) Percent inhibition of uptake of [α32P]ATP (50 μM) by the analogs indicated (500 μM each). Data shown are the average and s.e.m. of 3 independent trials.
To explore more dramatic nucleobase modifications, we examined inhibition with thieno-CTP (thCTP) and thieno-GTP (thGTP) (Fig. 1). Interestingly, both thGTP and thCTP analogs mediated relatively potent inhibition of [α32P]-ATP uptake (75 ± 1.2% and 79.8 ± 1.8%, respectively). We next explored the effect of even more drastic nucleobase modifications with the unnatural triphosphates dNaMTP, d5SICSTP, and dTPT3TP, which were used to expand the genetic alphabet and code of E. coli,24,28,29 as well as several precursors explored during their development34–38 (Fig. 1). The monocyclic derivatives dBENTP, d3FBTP, and dDM5TP showed little to no inhibition (≤10%), while dNaMTP and d5SICSTP showed moderate inhibition (36.6 ± 10.0%, 39.3 ± 14.0%) and dTPT3TP showed significant inhibition (72.1 ± 0.5%). Overall, these differences suggest that competitive binding is favored by large aromatic surface area, and possibly the nature of the substituent ortho to the glycosidic linkage, for example, its ability to participate in hydrogen bond (H-bond) formation, as is known to be important for polymerase recognition.39–41 Finally, we characterized the ribonucleoside triphosphate variants, NaMTP, 5SICSTP, and TPT3TP,29,42 and found that 5SICSTP appeared to inhibit uptake slightly more than its deoxy counterpart (57.6 ± 10.2%), but that NaMTP and TPT3TP did not (39.8 ± 7.1% and 78.3 ± 1.5%, respectively), suggesting that the role of the 2′-OH group may be context dependent.
To further characterize the SARs associated with the sugar moiety, we explored the contribution of each hydroxyl group within the adenosine triphosphate scaffold (Fig. 2). As reported previously, ATP is a more potent inhibitor of PtNTT2 than dATP (57.2 ± 7.1% vs. 43.5 ± 6.6% inhibition). While ddATP showed only modest inhibition (32.2 ± 4.4%), 3′-dATP (cordyceptin) inhibits [α32P]ATP more potently than either natural analog (71.3 ± 4.8% inhibition). This demonstrates that both OH groups contribute to competitive inhibition, but their contributions are not additive, with the 3′-OH actually reducing binding when a 2′-OH is already present.
Figure 2.
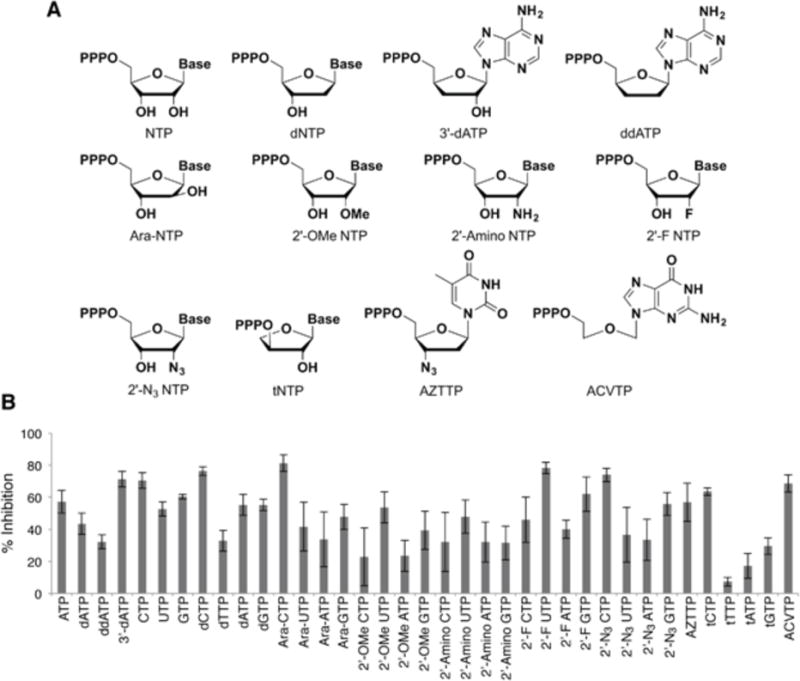
(A) Structure of sugar modified nucleoside triphosphate analogs used for the inhibition assay. Base, one of the natural nucleobases; PPPO, triphosphate. (B) Percent inhibition of uptake of [α32P]ATP (50 μM) by the analogs indicated (500 μM each). Data shown are the average and s.e.m. of 3 independent trials.
To further explore the role of the 2′-substituent, we characterized analogs with altered stereochemistry or with unnatural substituents (Fig. 2). We first characterized all four arabinose (Ara) triphosphates. In the case of the Ara-CTP, the altered stereochemistry increased inhibition (81.3 ± 5.2% versus 70.5 ± 4.9%), while for Ara-UTP, Ara-ATP, and Ara-GTP, it decreased inhibition (41.7 ± 15.2% versus 52.7 ± 4.4%, 33.8 ± 17.2% versus 57.2 ± 7.1%, and 47.8 ± 7.9% versus 60.4 ± 1.4%, respectively). In general, the 2′-methoxy and 2′-amino modified analogues are less potent inhibitors than their natural counterparts, with a reduction in inhibition of at least 20% in all cases relative to the parent ribonucleotide. In contrast, the 2′-fluoro (F) and 2′-azido (N3) modifications reduced binding only with the adenosine analogue (40.1 ± 5.6% and 33.4 ± 13.0% inhibition, respectively), and with 2′-F analog, this reduction was only to the level observed with dATP.
To explore the effects of 3′-modification, we synthesized the triphosphate of azidothymidine (AZTTP, Supporting Information), and found that the 3′-N3 substituent increased inhibition relative to dTTP (56.9 ± 12.0% vs. 32.9 ± 6.5%). This is in contrast to the effects at the 2′-position discussed above, where the azide substituent did not significantly alter competitive inhibition relative to dTTP.
To explore more dramatic changes to the sugar, we examined the threose nucleic acid (TNA) analogs43,44 of the natural triphosphates (tNTPs; Fig. 2), which have generated significant synthetic biology interest.45,46 Despite their significantly different topology, tGTP, tATP, and tTTP still competitively inhibit uptake, although significantly less so than their natural counterparts, while uptake inhibition by tCTP and CTP was virtually identical. Finally, we synthesized the triphosphate of the GTP analog acyclovir (ACVTP, Fig. 2; Supporting Information), which lacks C2′ and C3′ atoms altogether, and surprisingly, found that it inhibits ATP uptake (68.6 ± 5.5%) better than either dGTP (55.3 ± 3.6%) or GTP (60.4 ± 1.4%). This data further emphasizes the complex role of the sugar moiety.
Previous work by Haferkamp and coworkers demonstrated that PtNTT2 does not import ADP or AMP,30 however its tolerance of triphosphate modification has not been explored. Thus to examine the SARs associated with the triphosphate moiety, we characterized a variety of derivatives with intact, but modified triphosphates (Fig. 3). First, we explored the ability of the α-phosphorothioate of each natural triphosphate, as well as the γ-phosphorothioate of ATP, which replaces a non-bridging oxygen atom at the α- or γ-phosphate with a sulfur atom (in each case a racemic mixture of R and S enantiomers was employed), to inhibit ATP uptake. Inhibition observed with each of these analogs was similar to that observed for the respective unmodified parent nucleotide. To explore the role of a bridging oxygen, we synthesized several analogs of AZT triphosphate in which the β,γ-bridging oxygen was replaced with a methylene (β,γ-CH2-AZT), a dichloromethylene (β,γ-CCl2-AZT), or a difluoromethylene (β,γ-CF2-AZT; Supporting Information). Such modifications have been used as biochemical probes,47–50 as well as non-hydrolyzable triphosphate analogs.51 In contrast to modification of a non-bridging oxygen, β,γ-CH2-AZT and β,γ-CF2-AZT showed a reduction in the inhibition of [α32P]ATP uptake relative to AZTTP (31.0 ± 20.0% and 32.6 ± 18.0%, respectively), while β,γ-CCl2-AZT showed only a slight reduction in inhibition.(50.5 ± 14.3%) Thus, though apparently context dependent, the β,γ oxygen appears to contribute more to transporter binding than the α phosphate.
Figure 3.
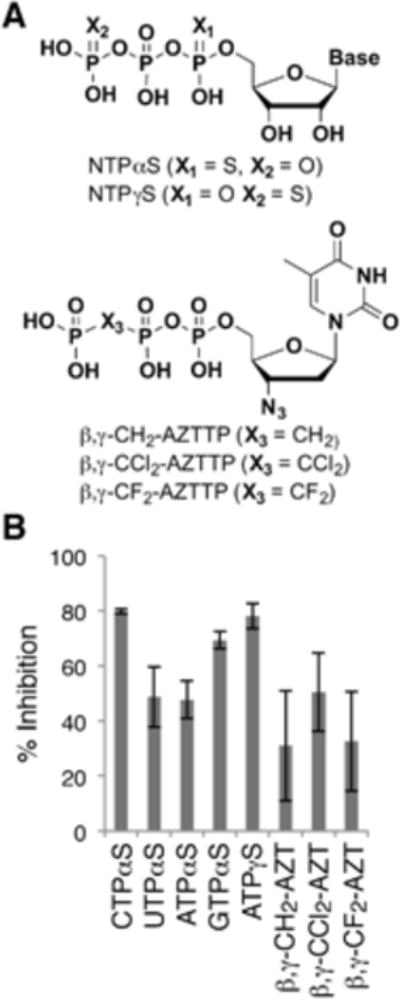
(A) Structure of triphosphate-modified nucleoside triphosphates used for the inhibition assay. Base, one of the natural nucleobases. (B) Percent inhibition of uptake of [α32P]ATP (50 μM) by the analogs indicated (500 μM each). Data shown are the average and s.e.m. of 3 independent trials.
Direct analysis of uptake
The traditional assay to directly characterize triphosphate uptake relies on radiolabeled substrates and is straightforward to employ. Similarly, the analysis of intrinsically fluorescent analogs is also straightforward. To use these assays to characterize PtNTT2 SARs, we explored the uptake of thCTP and thGTP, which are intrinsically fluorescent, and 5-IdCTP radiolabeled with 125I (Supporting Information). We found that the thCTP and thGTP are imported more efficiently than 5-IdCTP, consistent with PtNTT2 efficiently importing analogs bearing natural-like nucleobases with increased aromatic surface area, and suggesting that the potent inhibition mediated by 5-ICTP results from non-productive binding.
Many triphosphates of interest are neither intrinsically fluorescent nor readily amenable to radiolabeling. Thus, we explored the development of a quantitative LC-MS/MS assay that would permit the general characterization of modified nucleoside triphosphate uptake. Briefly, exponentially growing YZ2 cells were treated with varying concentrations of nucleoside triphosphate and incubated for 1 h at 37 °C. Cells were then pelleted and washed before extracting intracellular nucleotides with acidic acetonitrile.52 Nucleotides were then fully degraded to their corresponding nucleosides through the addition of shrimp alkaline phosphatase, and then quantified by LC-MS/MS. To correct for any passive uptake of the free nucleoside produced by extracellular degradation of the triphosphate, we subtracted the signal from a no transporter BL21(DE3) CmR control strain for each concentration of added triphosphate. The concentrations of nucleoside detected thus represents the amount of triphosphate imported, and includes all factors that may affect import, such as extracellular degradation and diffusion into the periplasm. Initial apparent velocities were determined using an external calibration curve constructed using free nucleosides and then plotted against the concentration of triphosphate added to the media. The resulting curves were fit to the Michaelis-Menten equation to determine apparent KM (μM) and apparent Vmax (fmol•cell−1•hr−1) values (Table 1), with Vmax/KM being taken as a measure of uptake efficiency.
Table 1.
Rates of uptake for various nucleoside triphosphates by PtNTT2 as determined by LC-MS/MS.a
| Triphosphate | Vmax (fmol•cell−1•hr−1) | KM (μM) | Vmax / KM |
|---|---|---|---|
| ATPb | (1.74 ± 0.091) × 10−2 | 186.0 ± 21.1 | (9.34 ± 0.32) × 10−5 |
| dATP | (1.24 ± 0.34) × 10−3 | 365.8 ± 47.9 | (3.38 ± 0.54) × 10−6 |
| 3′-dATP | (1.04 ± 0.29) × 10−3 | 162.7 ± 58.8 | (6.41 ± 0.31) × 10−6 |
| ddATP | (2.3 ± 0.81) × 10−3 | 766.6 ± 374.7 | (3.00 ± 0.74) × 10−6 |
| 2′-F ATP | (7.75 ± 2.3) × 10−5 | 478.7 ± 262.4 | (1.64 ± 0.28) × 10−7 |
| tATP | –c | –c | (1.21 ± 0.17) × 10−7 |
| c3dATP | (1.75 ± 0.21) × 10−4 | 388.2 ± 74.6 | (4.81 ± 0.96) × 10−7 |
| c7dATP | –c | –c | (8.75 ± 1.3) × 10−7 |
| dNaM | –c | –c | (1.94 ± 0.15) × 10−8 |
| dTPT3 | (7.50 ± 0.73) × 10−5 | 116.9 ± 11.4 | (6.68 ± 1.38) × 10−7 |
Data shown are the average and s.e.m. of 3 independent trials.
Assayed by radiolabeling.
not determined.
To validate the assay, we first characterized the uptake of dATP using [α32P]dATP and the standard radioactivity-based assay (Supporting Information). Uptake of dATP was found to proceed with a KM of 171 ± 78.4 μM and a Vmax of 1.88 ± 0.49 × 10−3 fmol•cell−1•hr−1. This KM is in good agreement with that reported previously (270.6 μM).30 With the LC-MS/MS assay, we found the uptake of dATP to proceed with a KM of 366 ± 48 μM and a Vmax of 1.24 ± 0.34 × 10−3 fmol•cell−1•hr−1, which we deemed to be in sufficient agreement with the results of the conventional assay. Using the validated LC-MS/MS assay, we proceeded to characterize the uptake of 3′-dATP, which we found to proceed with a Vmax that is virtually identical to that of dATP (1.04 ± 0.29 × 10−3 fmol•cell−1•hr−1) and a KM that is 2-fold reduced (163 ± 58.8 μM). We next characterized the uptake of ddATP, which we found to proceed with a 2-fold increase in both Vmax and KM (2.3 ± 0.81 × 10−3 fmol•cell−1•hr−1 and 767 ± 375 μM, respectively), relative to dATP. Comparison of ATP, dATP, 3′-dATP, and ddATP reveals that while both hydroxyl groups contribute similarly to turnover, the 2′-OH contributes more to productive binding, with the 3′-OH contributing only if the 2′-OH is absent. The data also suggests that the uniquely potent inhibition of transporter function mediated by 3′-dATP (see above) results from non-productive binding, and that the 3′-OH plays an important role by reducing it. Regardless, both ddATP and 3′-dATP are imported with second order rate constants that are similar to or even greater than that for the import of dATP.
To further explore the role of the 2′-substituent, we examined the uptake of the 2′-F analog of adenosine triphosphate. Import of 2′-F ATP proceeded with a KM that is similar to that observed for dATP (478 ± 262 μM), but with a Vmax that is 16-fold reduced (7.75 ± 2.3 × 10−5 fmol•cell−1•hr−1). Thus, while the addition of a 2′-OH to dATP (i.e. ATP) slightly increases binding (2-fold) and significantly increases turnover (14-fold), a fluoro substituent at the same position has little effect on binding but a significant and opposite effect on turnover. The deleterious effect on turnover is unlikely to result from an alteration of sugar conformation (both substituents favor the C3′-endo conformation of the sugar ring53) and thus must result from differences in size, electronegativity, and perhaps most likely, H-bonding potential. Regardless, 2′-F ATP is imported with a second order rate constant that is only ~20-fold reduced relative to dATP.
As a final examination of sugar modifications, we explored the uptake of the TNA analog tATP, which we found was too inefficient to determine KM and a Vmax independently. However, we were able to determine the second order rate constant, which was found to be 1.21 ± 0.17 × 10−7 mL•cell−1•hr−1, which is only ~30-fold reduced relative to dATP.
To explore the effects of nucleobase modifications, we characterized the uptake of c3dATP and c7dATP. The 3-deaza analog was imported with a KM of 388 ± 75 μM and a Vmax of 1.75 ± 0.21 × 10−4 fmol•cell−1•hr−1, while the 7-deaza analog was imported with a KM too high to determine (>1 mM), but with a Vmax/KM of 8.75 ± 1.3 × 10−7 mL•cell−1•hr−1. Thus, the elimination of N7 results in an at least 3-fold reduction in binding with little effect on turnover, while elimination of N3 has little effect on binding, but reduces turnover 7-fold. This suggests that the increased competitive inhibition observed with the nucleotides with bicyclic unnatural nucleobase analogs, relative to monocyclic analogs (see above), likely results from increased aromatic surface area. Regardless, these analogs are imported with an efficiency that is only 4- to 7-fold reduced relative to dATP.
Finally, we demonstrated previously that the PtNTT2-mediated import of dNaMTP, d5SICSTP, and dTPT3TP is sufficient to enable replication of DNA containing the corresponding unnatural base pair.24,28,29 In addition, the inhibition data presented above demonstrates that PtNTT2 effectively binds these unnatural triphosphates, as well as others with predominantly hydrophobic nucleobases that are very different from natural nucleobases. Using the LC-MS/MS assay, we observed a KM for dNaMTP that was too high to determine independently of Vmax (KM >1 mM), but the Vmax/KM was found to be 1.94 ± 0.15 × 10-8 mL•cell-1•hr-1. In contrast, dTPT3TP was found to be imported more efficiently, with a KM of 117 ±11.4 μM and a Vmax of 7.50 ± 0.73 × 10-5 fmol•cell-1•hr-1. The more efficient import of dTPT3TP may result from the altered size, electrostatics, or H-bonding potential of its nucleobase.
Use of PtNTT2 to study replication and transcription in vivo
The specificity of DNA and RNA polymerases is ubiquitously studied in vitro with model polymerases that are amenable to recombinant expression. However, this does not include the main replicative polymerases, which thus have not been extensively characterized. Moreover, it is not clear whether behavior in vitro translates to the natural in vivo environment. Thus, to explore replication in vivo, studies typically rely on the uptake of modified nucleotides and their subsequent three-step activation to the corresponding triphosphates. To explore the more controlled delivery of triphosphates, E. coli YZ2 harboring a pUC19 plasmid was grown to an OD600 of ~0.3 in the presence of an individual [α32P]-labeled dNTP or NTP, and the bulk RNA or plasmid DNA was then recovered and analyzed by PAGE and phosphorimaging to quantify the extent of radiolabeling.
Analysis of bulk RNA revealed that the addition of each [α32P]-labeled NTP resulted in efficient RNA labeling (Fig. 4). Interestingly, we also observed significant labeling of plasmid DNA with each NTP. The addition of [α32P]-labeled dNTPs resulted in low but detectable levels of RNA labeling, and as expected, the import of [α32P]-dGTP, [α32P]-dATP, and [α32P]-dTTP resulted in the efficient labeling of plasmid DNA (Fig. 4). Interestingly, through comparison of the extent of labeling, it was clear that with the exception of [α32P]-GTP, the addition of each [α32P]-dNTP resulted in the less efficient labeling of DNA than the corresponding NTP. One possible explanation for this cross-compartment labeling is greater import and/or degradation and subsequent phosphate exchange with the NTP substrates.
Figure 4.
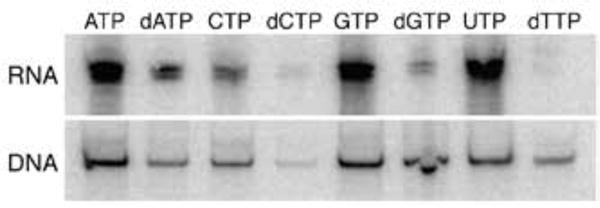
RNA and DNA radiolabeled with [α32P]-labeled analogs of each natural nucleotide.
The results with the addition of [α32P]-dCTP were strikingly different than with the other [α32P]-labeled dNTPs. In this case we observed significant toxicity (Fig. S5), and even after normalization for the amount of DNA analyzed, we observed significantly less radioactivity in the recovered DNA (Fig. 4). Previous studies have demonstrated PtNTT2 imports dCTP more efficiently than the other dNTPs30), indicating that these observations are not related to low levels up uptake. To more directly explore the fate of the imported dCTP, we examined the incorporation of dC into RNA using an adaptation of the developed LC-MS/MS assay. Briefly, E. coli YZ2 was grown in 2×YT media, and at an OD600 of 0.3, 250 μM of dCTP was added. After 90 min of growth, cellular RNA was extracted and degraded using a mixture of benzonase, phosphodiesterase I, and calf intestinal phosphatase, a cocktail which has been shown to non-specifically degrade even modified nucleic acids to free nucleosides.54,55 The degraded nucleosides were then analyzed by LC-MS/MS to determine the level of dC incorporation relative to C (Table 2). Interestingly, we found that 3% of the C had been replaced with dC. This clearly indicates that at the elevated concentrations of dCTP afforded by PtNTT2, dCTP is used by E. coli RNA polymerase only ~30-fold less efficiently than the corresponding natural substrate, and also demonstrates that at least some of the cross-compartment labeling observed with the radioactive substrates results from actual nucleotide incorporation, as opposed to phosphate exchange. This makes the above described inefficient labeling of DNA with [α32P]-dCTP all the more surprising, and it suggests that the labeling of RNA may be more efficient than the labeling of DNA, possibly due to differences in the polymerases and/or their accessibility.
Table 2.
Percent labeling of RNA and DNA from nucleoside triphosphates imported by PtNTT2 relative to the corresponding natural nucleotide.a
| Triphosphate | RNA labeling (%) | DNA labeling (%) |
|---|---|---|
| 2′-F UTP | <0.3 | -b |
| 2′-F GTP | 1.58 ± 0.16 | -b |
| 2′-F ATP | 0.78 ± 0.19 | -b |
| 2′-F CTP | 9.11 ± 0.98 | 2.77 ± 0.16 |
| dCTP | 3.03 ± 0.29 | -b |
Data shown are the average and s.e.m. of 3 independent trials.
not determined.
Based on the growing interest in the replacement of the natural nucleotides of DNA or RNA in an organism with modified analogs,54–58 we explored the import and fate of 2′-F modified triphosphates. We first examined the import and incorporation of each unnatural 2′-F triphosphate into RNA, using the same assay described above, but with the addition of 250 μM of 2′-F ATP, 2′-F GTP, 2′-F UTP, or 2′-F CTP to the media. While the addition of the 2′-F ATP, GTP, or UTP analogs has little effect on growth, the addition of the 2′-F CTP analog resulted in a reduced rate of growth (Fig. S5). Upon analysis, we were unable to detect the incorporation of 2′-F U, but 2′-F A and 2′-F G were detected at 0.8% and 1.6% of the level of their natural counterparts, respectively, and remarkably, 2′-F C was detected at greater than 9%. (Table 2) As with its radiolabeled counterpart, the toxicity and greater incorporation of the analog likely results from increased import by PtNTT2. Indeed, cell growth and analog incorporation levels similar to those observed with the other modified NTPs were observed with the addition of less 2′-F CTP to the media (Supporting Information). This data demonstrates that the 2′-F modified substrates, at their imported concentrations, are incorporated into RNA only ~10 to 100-fold less efficiently than the natural NTP substrates.
The data described above suggest that providing modified substrates for DNA synthesis may be more challenging than for RNA synthesis. Thus, to determine whether a 2′-F substrate could be incorporated into DNA, we grew E. coli YZ2 harboring a pUC19 plasmid in media containing 250 μM 2′-F CTP. Proceeding as above, we found that 2.77 ± 0.16% of the dC residues of pUC19 were replaced with 2′-F C (Table 2). Thus, along with our previously reported results with dNaMTP, d5SICSTP, and dTPT3TP,24,28,29 this data demonstrates that PtNTT2 imported substrates may access an active replication fork and participate in the synthesis of significant amounts of modified DNA, although apparently less efficiently than with RNA.
CONCLUSIONS
The ability to directly import nucleoside triphosphates into E. coli allows for the controlled introduction of labeled or modified nucleotides for molecular or synthetic biological applications. However, many potential applications depend on the import of modified analogs, and thus on the substrate scope of PtNTT2. The competitive binding SAR data presented here demonstrates that susbstituents at the 5-position are generally well tolerated. The data also reveals that increased nucleobase surface area can be tolerated, as can at least in some cases, dramatic alterations. At the sugar, both 2′- and 3′-OH groups contribute to competitive binding, but in a non-additive manner, while other substituents, or even other sugar topologies, can have variable effects, depending on the type of modification and the nucleotide scaffold. Finally, the β,γ-bridging oxygen appears to be generally more important for binding than either the α- or γ-nonbridging oxygen, potentially providing an important mechanism to control the specificity of nucleoside triphosphates relative to di- and monophosphates.
While convenient to measure, uptake inhibition is at best an approximate proxy for import, and the direct SARs generated using the developed LC-MS/MS assay in fact reveal that the uptake inhibition assay is complicated by significant non-productive binding. This direct SAR data is to our knowledge the first reported for a transporter, and it reveals that while the H-bond acceptor imine moieties of a nucleobase can facilitate uptake, either by increased binding or turnover, at least in the cases examined they are not required for efficient import. It also revealed that while both ribotyl hydroxyl groups facilitate uptake, the 3′-OH only contributes in the absence of a 2′-OH, and again, that neither are actually required for efficient uptake. While we were unable to characterize the role of the triphosphate moieties with the LC-MS/MS assay, as the analysis currently relies on their removal, the data clearly indicate that the majority of PtNTT2 substrate recognition is focused on the this region, which is consistent with the uptake inhibition SAR data.
Our initial study of the utility of the NTT not only demonstrated that the imported triphosphates can be used by the cell, but also revealed that the efficiency with which NTPs label RNA relative to DNA is much higher than the efficiency with which dNTPs label DNA relative to RNA. While this may in part be due to phosphate exchange, the surprisingly efficient incorporation of dC into RNA with the import of dCTP demonstrates a significant level of polymerase-mediated nucleotide incorporation. It is thus possible that the increased efficiency with which RNA is labeled may be due to decreased substrate specificity of RNA polymerase, which is consistent with the lower fidelity of transcription versus replication. However, another more provocative explanation is that while the total cellular concentration of NTPs may be available for transcription, only a more difficult to access subpopulation of the total dNTP pool may be available for replication. Indeed, DNA replication is highly compartmentalized, and supramolecular complexes of proteins involved in dNTP synthesis and replication have been identified and are thought to produce dNTPs from their ribonucletide precursors, and then channel them directly to the replication fork.59,60 Nonetheless, the data demonstrates that both RNA and DNA are labeled by 2′-F CTP, suggesting that PtNTT2 may be used to study the synthesis of DNA or RNA.
Perhaps most importantly, the data clearly demonstrate that modified nucleoside triphosphates can be made available within E. coli. In fact, of all the analogs examined, which include analogs with a wide range of nucleobase and sugar modifications, the efficiency of dNaMTP import is the lowest: however, we have already shown that the import of dNaMTP is sufficient to support the replication of an unnatural base pair on a high copy plasmid. Thus the data suggest that all of the analogs tested, and likely many others, are likely to be imported into E. coli by PtNTT2 with an efficiency that is sufficient for molecular and synthetic biology applications.
Supplementary Material
Acknowledgments
Financial support was provided by the National Institutes of Health (GM118178 to F.E.R.) and the National Science Foundation Graduate Research Fellowship (DGE-1346837 to A.W.F. and M.P.L.)
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website.
Methods, Supporting Figures and Tables (PDF)
Author Contributions
All authors have given approval to the final version of the manuscript.
References
- 1.Chen T, Hongdilokkul N, Liu Z, Thirunavukarasu D, Romesberg FE. Curr Opin Chem Biol. 2016;34:80–87. doi: 10.1016/j.cbpa.2016.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Houlihan G, Arangundy-Franklin S, Holliger P. Acc Chem Res. 2017;50:1079–1087. doi: 10.1021/acs.accounts.7b00056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Malyshev DA, Romesberg FE. Angew Chem Int Ed. 2015;54:11930–11944. doi: 10.1002/anie.201502890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hirao I, Kimoto M, Yamashige R. Acc Chem Res. 2012;45:2055–2065. doi: 10.1021/ar200257x. [DOI] [PubMed] [Google Scholar]
- 5.Benner SA, Karalkar NB, Hoshika S, Laos R, Shaw RW, Matsuura M, Fajardo D, Moussatche P. Cold Spring Harb Perspect Biol. 2016;8:a023770. doi: 10.1101/cshperspect.a023770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Craig JE, Zhang Y, Gallagher MP. Mol Microbiol. 1994;11:1159–1168. doi: 10.1111/j.1365-2958.1994.tb00392.x. [DOI] [PubMed] [Google Scholar]
- 7.Norholm MH, Dandanell G. J Bacteriol. 2001;183:4900–4904. doi: 10.1128/JB.183.16.4900-4904.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Westh Hansen SE, Jensen N, Munch-Petersen A. Eur J Biochem. 1987;168:385–391. doi: 10.1111/j.1432-1033.1987.tb13431.x. [DOI] [PubMed] [Google Scholar]
- 9.Ye J, van den Berg B. EMBO J. 2004;23:3187–3195. doi: 10.1038/sj.emboj.7600330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Henderson JF, Paterson ARP. Nucleotide Metabolism: An Introduction. Academic Press; New York, NY: 1973. [Google Scholar]
- 11.Sandrini MP, Piskur J. Trends Biochem Sci. 2005;30:225–228. doi: 10.1016/j.tibs.2005.03.003. [DOI] [PubMed] [Google Scholar]
- 12.Konrad A, Yarunova E, Tinta T, Piskur J, Liberles DA. Gene. 2012;492:117–120. doi: 10.1016/j.gene.2011.10.039. [DOI] [PubMed] [Google Scholar]
- 13.Sivakumar S, Porter-Goff M, Patel PK, Benoit K, Rhind N. Methods. 2004;33:213–219. doi: 10.1016/j.ymeth.2003.11.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Vernis L, Piskur J, Diffley JF. Nucleic Acids Res. 2003;31:e120. doi: 10.1093/nar/gng121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yan H, Tsai MD. Adv Enzymol Relat Areas Mol Biol. 1999;73:103–143. doi: 10.1002/9780470123195.ch4. [DOI] [PubMed] [Google Scholar]
- 16.Sandrini MP, Clausen AR, On SL, Aarestrup FM, Munch-Petersen B, Piskur J. J Antimicrob Chemother. 2007;60:510–520. doi: 10.1093/jac/dkm240. [DOI] [PubMed] [Google Scholar]
- 17.Wu Y, Fa M, Tae EL, Schultz PG, Romesberg FE. J Am Chem Soc. 2002;124:14626–14630. doi: 10.1021/ja028050m. [DOI] [PubMed] [Google Scholar]
- 18.Matsuura MF, Winiger CB, Shaw RW, Kim MJ, Kim MS, Daugherty AB, Chen F, Moussatche P, Moses JD, Lutz S, Benner SA. ACS Synth Biol. 2017;6:388–394. doi: 10.1021/acssynbio.6b00228. [DOI] [PubMed] [Google Scholar]
- 19.Black ME, Newcomb TG, Wilson HM, Loeb LA. Proc Natl Acad Sci USA. 1996;93:3525–3529. doi: 10.1073/pnas.93.8.3525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Johansson M, van Rompay AR, Degreve B, Balzarini J, Karlsson A. J Biol Chem. 1999;274:23814–23819. doi: 10.1074/jbc.274.34.23814. [DOI] [PubMed] [Google Scholar]
- 21.Munch-Petersen A, Jensen N. Eur J Biochem. 1990;190:547–551. doi: 10.1111/j.1432-1033.1990.tb15608.x. [DOI] [PubMed] [Google Scholar]
- 22.Yoshioka A, Tanaka S, Hiraoka O, Koyama Y, Hirota Y, Ayusawa D, Seno T, Garrett C, Wataya Y. J Biol Chem. 1987;262:8235–8241. [PubMed] [Google Scholar]
- 23.Chou IN, Zeiger J, Rapaport E. Proc Natl Acad Sci USA. 1984;81:2401–2405. doi: 10.1073/pnas.81.8.2401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Malyshev DA, Dhami K, Lavergne T, Chen T, Dai N, Foster JM, Correa IR, Jr, Romesberg FE. Nature. 2014;509:385–388. doi: 10.1038/nature13314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hantke K. FEBS Lett. 1976;70:109–112. doi: 10.1016/0014-5793(76)80737-5. [DOI] [PubMed] [Google Scholar]
- 26.McKeown M, Kahn M, Hanawalt P. J Bacteriol. 1976;126:814–822. doi: 10.1128/jb.126.2.814-822.1976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Acimovic Y, Coe IR. Mol Biol Evol. 2002;19:2199–2210. doi: 10.1093/oxfordjournals.molbev.a004044. [DOI] [PubMed] [Google Scholar]
- 28.Zhang Y, Lamb B, Feldman AW, Zhou AX, Lavergne T, Li L, Romesberg FE. Proc Natl Acad Sci USA. 2017;114:1317–1322. doi: 10.1073/pnas.1616443114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhang Y, Ptacin JL, Fischer EC, Aerni HR, Caffaro CE, San Jose K, Feldman AW, Turner CR, Romesberg FE. Nature. 2017;551:644–647. doi: 10.1038/nature24659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ast M, Gruber A, Schmitz-Esser S, Neuhaus HE, Kroth PG, Horn M, Haferkamp I. Proc Natl Acad Sci USA. 2009;106:3621–3626. doi: 10.1073/pnas.0808862106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Nabel CS, Manning SA, Kohli RM. ACS Chem Biol. 2012;7:20–30. doi: 10.1021/cb2002895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sanchez-Romero MA, Cota I, Casadesus J. Curr Opin Microbiol. 2015;25:9–16. doi: 10.1016/j.mib.2015.03.004. [DOI] [PubMed] [Google Scholar]
- 33.Munzel M, Globisch D, Carell T. Angew Chem Int Ed. 2011;50:6460–6468. doi: 10.1002/anie.201101547. [DOI] [PubMed] [Google Scholar]
- 34.Henry AA, Olsen AG, Matsuda S, Yu C, Geierstanger BH, Romesberg FE. J Am Chem Soc. 2004;126:6923–6931. doi: 10.1021/ja049961u. [DOI] [PubMed] [Google Scholar]
- 35.Leconte AM, Hwang GT, Matsuda S, Capek P, Hari Y, Romesberg FE. J Am Chem Soc. 2008;130:2336–2343. doi: 10.1021/ja078223d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Li L, Degardin M, Lavergne T, Malyshev DA, Dhami K, Ordoukhanian P, Romesberg FE. J Am Chem Soc. 2014;136:826–829. doi: 10.1021/ja408814g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Seo YJ, Hwang GT, Ordoukhanian P, Romesberg FE. J Am Chem Soc. 2009;131:3246–3252. doi: 10.1021/ja807853m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Matsuda S, Henry AA, Romesberg FE. J Am Chem Soc. 2006;128:6369–6375. doi: 10.1021/ja057575m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Meyer AS, Blandino M, Spratt TE. J Biol Chem. 2004;279:33043–33046. doi: 10.1074/jbc.C400232200. [DOI] [PubMed] [Google Scholar]
- 40.Morales JC, Kool ET. J Am Chem Soc. 1999;121:2323–2324. doi: 10.1021/ja983502+. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Spratt TE. Biochemistry. 2001;40:2647–2652. doi: 10.1021/bi002641c. [DOI] [PubMed] [Google Scholar]
- 42.Seo YJ, Matsuda S, Romesberg FE. J Am Chem Soc. 2009;131:5046–5047. doi: 10.1021/ja9006996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sau SP, Fahmi NE, Liao JY, Bala S, Chaput JC. J Org Chem. 2016;81:2302–2307. doi: 10.1021/acs.joc.5b02768. [DOI] [PubMed] [Google Scholar]
- 44.Sau SP, Chaput JC. Org Lett. 2017;19:4379–4382. doi: 10.1021/acs.orglett.7b02099. [DOI] [PubMed] [Google Scholar]
- 45.Chaput JC, Yu H, Zhang S. Chem Biol. 2012;19:1360–1371. doi: 10.1016/j.chembiol.2012.10.011. [DOI] [PubMed] [Google Scholar]
- 46.Pinheiro VB, Taylor AI, Cozens C, Abramov M, Renders M, Zhang S, Chaput JC, Wengel J, Peak-Chew SY, McLaughlin SH, Herdewijn P, Holliger P. Science. 2012;336:341–344. doi: 10.1126/science.1217622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sucato CA, Upton TG, Kashemirov BA, Batra VK, Martinek V, Xiang Y, Beard WA, Pedersen LC, Wilson SH, McKenna CE, Florian J, Warshel A, Goodman MF. Biochemistry. 2007;46:461–471. doi: 10.1021/bi061517b. [DOI] [PubMed] [Google Scholar]
- 48.Sucato CA, Upton TG, Kashemirov BA, Osuna J, Oertell K, Beard WA, Wilson SH, Florian J, Warshel A, McKenna CE, Goodman MF. Biochemistry. 2008;47:870–879. doi: 10.1021/bi7014162. [DOI] [PubMed] [Google Scholar]
- 49.Batra VK, Pedersen LC, Beard WA, Wilson SH, Kashemirov BA, Upton TG, Goodman MF, McKenna CE. J Am Chem Soc. 2010;132:7617–7625. doi: 10.1021/ja909370k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Oertell K, Chamberlain BT, Wu Y, Ferri E, Kashemirov BA, Beard WA, Wilson SH, McKenna CE, Goodman MF. Biochemistry. 2014;53:1842–1848. doi: 10.1021/bi500101z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Feldman AW, Dien VT, Romesberg FE. J Am Chem Soc. 2017;139:2464–2467. doi: 10.1021/jacs.6b12731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Rabinowitz JD, Kimball E. Anal Chem. 2007;79:6167–6173. doi: 10.1021/ac070470c. [DOI] [PubMed] [Google Scholar]
- 53.Manoharan M, Akinc A, Pandey RK, Qin J, Hadwiger P, John M, Mills K, Charisse K, Maier MA, Nechev L, Greene EM, Pallan PS, Rozners E, Rajeev KG, Egli M. Angew Chem Int Ed. 2011;50:2284–2288. doi: 10.1002/anie.201006519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Mehta AP, Li H, Reed SA, Supekova L, Javahishvili T, Schultz PG. J Am Chem Soc. 2016;138:7272–7275. doi: 10.1021/jacs.6b03904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Mehta AP, Li H, Reed SA, Supekova L, Javahishvili T, Schultz PG. J Am Chem Soc. 2016;138:14230–14233. doi: 10.1021/jacs.6b09661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Nainar S, Beasley S, Fazio M, Kubota M, Dai N, Correa IR, Jr, Spitale RC. Chembiochem. 2016;17:2149–2152. doi: 10.1002/cbic.201600300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Pezo V, Liu FW, Abramov M, Froeyen M, Herdewijn P, Marliere P. Angew Chem Int Ed. 2013;52:8139–8143. doi: 10.1002/anie.201303288. [DOI] [PubMed] [Google Scholar]
- 58.Marliere P, Patrouix J, Doring V, Herdewijn P, Tricot S, Cruveiller S, Bouzon M, Mutzel R. Angew Chem Int Ed. 2011;50:7109–7114. doi: 10.1002/anie.201100535. [DOI] [PubMed] [Google Scholar]
- 59.Murthy S, Reddy GP. J Cell Physiol. 2006;209:711–717. doi: 10.1002/jcp.20842. [DOI] [PubMed] [Google Scholar]
- 60.Mathews CK. In: Genetic Consequences of Nucleotide Pool Imbalance. de Serres FJ, editor. Springer; Boston, MA: 1985. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.