

Abstract
We report the silylation of primary C–H bonds located β to secondary and tertiary alcohols by exploiting perfluorinated esters as traceless directing groups. The conversion of a secondary or tertiary alcohol to a perfluoroalkyl ester and conversion of the ester to the corresponding silyl acetals by hydrosilylation allows for selective β-C(sp3)–H silylation catalyzed by the combination of [Ir(cod)OMe]2 and Me4Phen (3,4,7,8-tetramethyl-1,10-phenanthroline) to form 6-membered dioxasilinane. Tamao-Fleming oxidation of these dioxasilinane leads to 1,2 diols. The developed sequence was applied to a series of natural products containing hydroxyl groups.
Graphical Abstract
INTRODUCTION
Site-selective functionalization of specific C–H bonds in the presence of an array of C–H bonds and functional groups is transforming the logic by which organic molecules are synthesized and modified.1 Among existing C–H functionalization reactions, the borylation2 and silylation3 of C–H bonds are particularly valuable because the silicon and boron-based products undergo a range of cross-coupling and oxidative functionalization reactions.
The hydroxyl group is the most commonly encountered functional group in natural compounds, making alcohol-directed C–H functionalization reactions particularly important for the selective, direct modification of complex molecules.4 The C–H bonds located α to the hydroxyl group are the most reactive and are cleaved during radical and oxidative processes.5 The C–H bonds δ to an alcohol undergo functionalization by 1,5-hydrogen atom abstraction of highly energetic oxygen-centered radicals.6 The functionalization of alcohols at the γ and β positions is more limited.7
Previously, our group developed Ir-catalyzed silylations of unactivated γ-C(sp3)–H bonds in alcohols to form 1,3 diols (see Scheme 1a).8 In this approach, (hydrido)silyl ethers obtained by the dehydrogenative coupling of the alcohol with diethylsilane undergo functionalization of primary C(sp3)–H bonds at the position γ to the alcohol. Subsequent Tamao-Fleming oxidation of the resulting 5-membered oxasilolanes furnishes the 1,3-diol product.8b However, this approach, without modification, is not applicable to the functionalization of C–H bonds located β to the alcohol because the reaction would require the formation of strained 4-membered ring oxasiletane (see Scheme 1a).
Scheme 1.

Alcohol-Directed C–H Functionalization
Dong reported an alternative strategy that led to the β functionalization of alcohols. Pd-catalyzed β-acetoxylation9 and β-tosylation10 reactions of alcohols occur with an O-alkyl oxime as directing group (see Scheme 1b). Although providing novel selectivity, the multistep installation and reductive removal of this directing group, along with the moderate selectivity for monofunctionalization of the many identical C–H bonds, are drawbacks of this method, particularly when functionalizing complex molecules containing an array of functional groups. Thus, we have sought alternative strategies for the silylation of C–H bonds located β to an alcohol.
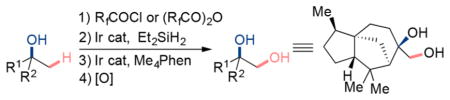
To functionalize the β-C(sp3)–H bonds of alcohols, we envisioned a strategy by which an ester, converted to a silyl acetal, would serve as a readily cleaved directing group. Jeon recently used silyl acetals as directing groups for the ortho-silylation of phenols.11 Our proposed strategy for the functionalization of β-C(sp3)–H bonds is outlined in Scheme 1c. Ester 2 would be accessed by a simple acylation of alcohol 1. Hydrosilylation of ester 2 would generate the silylacetal 3, which could undergo C(sp3)–H functionalization at the β position to form the 6-membered dioxasilinane 4. Subsequent Tamao-Fleming oxidation of the dioxasilinane 4 would provide the corresponding 1,2-diol.
We report the successful implementation of this strategy by which the silylation of primary C–H bonds located β to secondary and tertiary alcohols occurs with perfluorinated esters as traceless directing groups. The hydrosilylation of a perfluoroalkyl ester, subsequent in situ silylation of a C(sp3)–H bond, and subsequent Tamao-Fleming oxidation at the Si–C bond leads to the introduction of a hydroxyl group at the primary carbon located in the position β to an alcohol. This sequence is suitable for the derivatization of C–H bonds located β to both secondary and tertiary alcohols including those in natural products.
RESULTS AND DISCUSSION
To achieve the silylation of C–H bonds β to alcohols by this strategy (Scheme 1c), several challenges needed to be addressed. First, the silylacetal intermediate 3 must be stable during the C(sp3)–H silylation. Although a silylacetal was applied by Jeon for the functionalization of aromatic C–H bonds ortho to a phenol, an acetal that is stable to the conditions for cleavage of an unactivated, aliphatic C–H bond is needed.11 Second, a system that forms a 7-membered metalacyclic intermediate by C(sp3)–H activation is needed.12 Third, the catalyst used for ester reduction must be compatible with the catalyst used for C–H silylation if the process is to be run without isolation of intermediates. By virtue of the ester intermediate and silane reagent, the stability and reactivity of the intermediate silylacetal can be tuned by the acyl group that will be removed in situ during the Tamao-Fleming oxidation.
Effect of the Silylacetal Substituent on the C–H Silylation Reaction
The feasibility of the planned reaction sequence was assessed by conducting reactions with several catalysts and esters derived from 6-methylheptan-2-ol. A full list of results with esters is given in the Supporting Information. A selection of the results is shown in Scheme 1. The choice of ester was important for the success of the envisaged transformation; we investigated formate 2a, acetate 2b, pivalate 2c, and trifluoroacetate 2d. The electronic and steric properties of these esters vary among this series. The hydrosilylation of all four esters with diethylsilane proceeded in the presence of catalytic (1,5-cyclooctadiene)(methoxy)iridium(I) dimer ([Ir-(cod)OMe]2, cod = 1,5-cyclooctadiene) to complete conversion (by 1H NMR spectroscopy) to form the intermediate silylacetals 3a–3d. The crude silylacetals were then used for the C–H bond activation step after evaporation of the solvent and other volatile materials.
The C–H silylation reactions of silylacetals 3a–3d were conducted with catalysts based on Ir and Rh. The reaction of trifluoromethylacetal 3d in the presence of [Ir(cod)OMe]2, 3,4,7,8-tetramethyl-1,10-phenanthroline (Me4Phen) as catalyst and norbornene (nbe) as hydrogen acceptor provided 45% yield (by 1H NMR spectroscopy) of the desired dioxasilinane 4d over two steps. Side-products resulting from the disproportionation of silylacetals 3d accounted for 12% of the total conversion. The same procedure with 6-methylheptan-2-yl formate 2c formed the 6-membered silinane 4a in 24% yield over the two steps. The analogous reactions with methylacetal 3b and tert-butylacetal 3c furnished a complex mixture without the formation of expected dioxasilinane 4b and 4c, respectively.
We also tested rhodium catalysts that had been reported for the silylation of aromatic C–H bonds. The reactions of trifluoromethylacetal 3d with chlorobis(cyclooctene)rhodium- (I) dimer ([Rh(coe)2Cl]2, coe = cyclooctene) and 5,5′-bis[di(3,5-di-tert-butyl-4-methoxyphenyl)phosphino]-4,4′-bi-1,3-benzodioxole (DTBM-SEGPHOS) furnished the dioxasilinane 4d in 28% yield, as determined by 1H NMR spectroscopy. In these rhodium-catalyzed reactions, a side product resulting from the hydrosilylation of norbornene with the trifluoromethylacetal 3d formed in 17% yield.
The observed reactivity of the series of acetals 3a–3d can be rationalized in terms of the stability and steric properties of the directing groups. The stability of silylacetal intermediates 3a–3d is revealed by comparing the hydration constants of formaldehyde, acetaldehyde, pivalaldehyde, and trifluoro-acetaldehyde (see Scheme 2).13 Generally, the presence of electron-withdrawing groups adjacent to the carbonyl group of an aldehyde increase the stability of the hydrate relative to the free aldehyde. Consequently, formaldehyde and trifluoroacetaldehyde exist primarily as a geminal diol in aqueous solution, while pivalaldehyde exists primarily as a free aldehyde in aqueous solution. Consistent with this greater stability of these acetals, trifluoroacetaldehyde silylacetal 3d and formaldehyde silylacetal 3a furnished the desired dioxasilinane 4, but the substrates corresponding to less stable acetals did not. Although the silylacetals of formaldehyde and trifluoro-acetaldehyde are stable (Khydration > 102), the trifluoro-acetaldehyde silylacetal 3b furnished the product of C–H silylation in higher yield than the formaldehyde silylacetal 3c (45% vs 24%, see Scheme 2). This difference in reactivity parallels the greater steric hindrance of the trifluoromethyl group (A-value (–CF3) = 2.1 kcal/mol) of trifluoroacetate than of the hydrogen (A-value (–H) = 0 kcal/mol) of the formate (see Scheme 2).14 The more hindered trifluoromethyl group will impart a greater Thorpe-Ingold effect on the cyclization reaction.15
Scheme 2. Initial Evaluation of Directing Groups for β-C(sp3)–H Silylation.

a[Ir(cod)OMe]2 (1.0 mol %), Et2SiH2 (4.0 equiv), THF (0.5 M), rt or 50°C, 24 h, N2. b[Rh(coe)2Cl]2 (2.0 mol %), DTBM-SEGPHOS (6.0 mol %), nbe (1.5 equiv), THF (0.1 M), 100 °C, 16 h, N2. c[Ir(cod)OMe]2 (2.0 mol %), Me4Phen (6.0 mol %), nbe (1.5 equiv), THF (0.1 M), 100 °C, 16 h, N2.
Because the reaction conducted with the combination of [Ir(cod)OMe]2 and Me4Phen furnished the desired dioxasilinane in higher yield than the reactions conducted with combination of [Rh(coe)2Cl]2 and DTBM-SEGPHOS, we tested a series of reaction conditions for the dehydrogenative silylation of trifluoroacetal 3d catalyzed by [Ir(cod)OMe]2. Evaluation of the ligand, hydrogen acceptor, solvent, concentration, and temperature did not result in an increase in reaction yield (refer to the Supporting Information). Thus, we modified the substituent on the acetal to achieve high yields for the dehydrogenative silylation step.
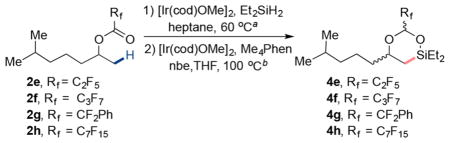
Considering the initially observed effect of the stability and the steric hindrance of silylacetal on the silylation step, we evaluated a series of perfluorinated esters of 6-methylheptan-2-ol with varied steric properties (2e–2h, Table 1). The reduction of perfluoropropanoate 2e with diethylsilane (4.0 equiv) in the presence of [Ir(cod)OMe]2 (2.0 mol %) in heptane at 60 °C led to complete formation of the silylacetal (see entry 2, Table 1). Intramolecular C–H silylation of perfluoroethyl silylacetal 3e in the presence of [Ir(cod)OMe]2 (2.0 mol %), Me4Phen (6.0 mol %), and nbe (1.5 equiv) in THF (0.1 M) at 100 °C formed the desired 6-membered dioxasilinane 4e in 68% yield (by 1H NMR spectroscopy) over two steps.
Table 1.
Evaluation of Fluorinated Directing Group
| |||
|---|---|---|---|
| entry | ester | conversionc | yieldd |
| 1 | 2d, Rf = CF3 | 100% | 45% |
| 2 | 2e, Rf = C2F5 | 100% | 68% |
| 3 | 2f, Rf = C3F7 | 96% | 78% |
| 4 | 2g, Rf = CF2Ph | 98% | 75% |
| 5 | 2h, Rf = C7F15 | 95% | 84%(75%)e |
Conditions for hydrosilylation of ester: [Ir(cod)OMe]2 (1.0 mol %), Et2SiH2 (4.0 equiv), heptane (0.5 M), 60 °C, 24–48 h, N2.
Conditions for β-C(sp3)–H silylation: [Ir(cod)OMe]2 (2.0 mol %), Me4Phen (6.0 mol %), nbe (1.5 equiv), THF (0.1 M), 100 °C, 16 h, N2.
Conversion for the hydrosilylation step determined by 1H NMR spectroscopy.
Overall yield for the two step determined by 1H NMR spectroscopy using CH2Br2 as internal standard.
Isolated yield.
This positive effect of the larger perfluoroalkyl group on the yield led us to test the perfluorobutanoate 2f under the same reaction conditions (see entry 3, Table 1). The hydrosilylation of the ester occurred in 96% conversion, and the overall yield for the formation of dioxasilinane 4f from the perfluorobutanoate 2f was 78%. Similar results were obtained with the 2,2-difluoro-2-phenylacetate 4g (75% yield, entry 4, Table 1); only trace amounts of product resulting from C(sp2)–H bond silylation of the phenyl ring of the acetal were observed. Finally, subjecting the perfluorooctanoate ester 2h to the sequence of hydrosilylation and β-C(sp3)–H silylation yielded the dioxasilinane 4h in 84% yield over the two steps.
After identifying conditions for the hydrosilylation of the esters and dehydrogenative silylation of the C(sp3)–H bonds, we tested if dioxasilinane 4h would undergo Tamao-Fleming oxidation to furnish 1,2-diols. After evaluating a series of conditions for the Tamao-Fleming oxidation, we found that isolated silinane 4h converts to the corresponding 1,2-diol in 72% yield (1H NMR) in the presence of hydrogen peroxide, potassium bicarbonate, and potassium fluoride in methanol at 60°C (Scheme 3). Moreover, we found that these conditions for oxidation can be used without isolating the silinane 4h. The crude silinane 4h formed 6-methylheptane-1,2-diol 5a in 59% isolated yield over the three steps, without isolation of any intermediates.
Scheme 3. Tamao-Fleming Oxidation.

aHydrosilylation of ester: [Ir(cod)OMe]2 (1.0 mol %), Et2SiH2 (4.0 equiv), heptane (0.5 M), 60 °C, 48 h, N2. bC–H silylation step: [Ir(cod)OMe]2 (2.0 mol %), Me4Phen (6.0 mol %), nbe (1.5 equiv), THF (0.1 M), 100 °C, 16 h, N2. cOxidation step: KHCO3 (4.0 equiv), KF (4.0 equiv), 50% aqueous H2O2 (10 equiv), MeOH, 60 °C, 2 h. dDetermined by 1H NMR spectroscopy using CH2Br2 as internal standard. eOverall isolated yield for the steps 1–3.
Scope of β-C–H Silylation Reaction
Studies to evaluate the selectivity and functional-group tolerance of the developed sequence comprising hydrosilylation and C–H silylation are summarized in Scheme 4. Substrates containing both primary and either secondary (4h), tertiary (4i, 4j), or benzylic (4q, 4r) β-C(sp3)–H bonds underwent silylation exclusively at the primary C–H bonds. Moreover, products resulting from potential intermolecular silylation of aromatic C–H bonds were not observed. Esters containing a TBDPS-protected alcohol (4n), an alkyl halide (4p), or a trisubstituted alkene (4s) underwent the sequence comprising hydrosilylation and C–H bond silylation without interference of the auxiliary functional groups. In addition, substrates bearing aryl chloride (4m), bromide (4r), and fluoride (4q) moieties underwent the same sequence to furnish the six-membered ring dioxasilinanes in 73–76% yield. Methylenedioxy protected catechol (4o) and methyl phenol ether (4l) were also compatible with the reaction conditions. Esters, amides, ketones, and nitroarenes were not tolerated; these groups were reduced under the conditions for hydrosilylation. Substrates containing coordinating groups, such as nitriles and thiols, underwent the hydrosilylation step in low conversion.
Scheme 4. Scope of C–H Silylation Reaction.

aHydrosilylation of ester: [Ir(cod)OMe]2 (1.0 mol %), Et2SiH2 (4.0 equiv), heptane (0.5 M), 60 °C, 48 h, N2. bC–H silylation step: [Ir(cod)OMe]2 (2.0 mol %), Me4Phen (6.0 mol %), nbe (1.5 equiv), THF (0.1 M), 100 °C, 16 h, N2. cDetermined by 1H NMR spectroscopy using CH2Br2 as internal standard. dIsolated yield for the 1,2 diols over the three steps, oxidation step: KHCO3 (4.0 equiv), KF (4.0 equiv), 50% aqueous H2O2 (10 equiv), MeOH, 60 °C, 2 h.
The hydrosilylation of perfluorooctanoates of tertiary alcohols under the developed conditions did not furnish the desired silylacetals, presumably due to the high steric hindrance. Thus, we examined a series of esters as directing groups for the hydrosilylation and C–H silylation of tertiary alcohols (refer to the Supporting Information for details). Among the investigated esters, only the hydrosilylation of trifluoroacetate and 2,2-difluoro-2-phenylacetate proceeded to high conversion. The silylacetal derived from a tertiary 2,2-difluoro-2-phenylacetate provided substantial amounts of products resulting from silylation of the C(sp2)–H bond of the phenyl group in the difluorobenzyl substituent, leading to low yield of the C(sp3)–H silylation product. However, the hydrosilylation of the trifluoroacetate of 2-methyl-4-phenylbutan-2-ol 1t and subsequent dehydrogenative silylation occurred to form the dioxasilinane 4t in 72% yield (by 1H NMR spectroscopy) over the two steps (Scheme 3). Because of the instability of the dioxasilinane bearing a trifluoromethyl group, we isolated the 1,2-diol after the Tamao-Fleming oxidation in 52% yield over three steps. Thus, simple modification of the substituent on the directing group can match the steric and electronic requirements to achieve oxidation of the β C–H bond in high yield for both secondary and tertiary alcohols.
Hydroxylation of Natural Products Derivatives
The developed sequence of hydrosilylation, C–H silylation, and oxidation was applicable to the selective hydroxylation of a series of natural products derivatives containing tertiary and secondary alcohols (Scheme 5). The perfluorooctanoate (Rf = nC7F15) substituent was a suitable traceless directing group for reactions of natural products containing secondary 2-methyl alcohols. The developed sequence led to the introduction of a second hydroxyl group in the β position of the alcohols derived from geranylacetone (5aa), dihydro-β-ionone (5ab), dihydro-α-ionone (5ac), and the 27-nor-25-ketocholesterol (5ad). The combination of hydrosilylation and C–H silylation occurred in 69–76% yield (determined by 1H NMR spectroscopy) over the two steps. The resulting dioxasilinane was subjected to Tamao-Fleming oxidation to furnish the final 1,2-diols in 41–54% isolated yield over the three-step sequence.
Scheme 5. Application of C–H Silylation/Oxidation Sequence to Natural Product Derivatives.

aHydrosilylation of ester: [Ir(cod)OMe]2 (1.0 mol %), Et2SiH2 (4.0 equiv), heptane (0.5 M), 60 °C, 48 h, N2. bC–H silylation step: [Ir(cod)OMe]2 (2.0 mol %), Me4Phen (6.0 mol %), nbe (1.5 equiv), THF (0.1 M), 100 °C, 16 h, N2. cOxidation step: KHCO3 (4.0 equiv), KF(4.0 equiv), 50% aqueous H2O2(10 equiv), MeOH, 60 °C, 2 h. dIsolated yield for the 1,2 diols over the three steps. eYield for the dioxasilinane determined by 1H NMR spectroscopy using CH2Br2 as internal standard. fStarting dihydro-α-ionone was purchased as the mixture of cyclohex-2-ene and cyclohex-3-ene derivatives in 4:1 ratio.
Natural products containing tertiary alcohols underwent the sequence when bearing a removable directing group containing a trifluoromethyl substituent. The developed strategy led to hydroxylation of the primary β C–H bonds of (+)-cedrol (5ah), ledol (5ag), (–)-α-bisabolol (5af), and α-terpinol (5ae) to form the coresponding 1,2-diols. The diols were isolated in 21–36% yield over the three-step sequence. Although occurring in modest yield, this sequence led to the formation of readily isolable amounts of the modified products. The initial introduction of the directing group (TFAA, pyridine, DCM) did not require isolation by column chromatography.
The complementarity of our developed method to functionalize alcohols is well illustrated in the case of (+)-cedrol (5ah), a sesquiterpenoid used in the perfume industry extracted from Texas cedarwood oil. Direct chemical or microbial16 oxidation of cedrol has been studied extensively. Generally, the oxidation reactions of cedrol with m-chlorobenzoic acid and ruthenium or iron catalysts led to the introduction of a hydroxyl group at the tertiary C(2) carbon (5ak).17 The oxidation of cedrol with the “Gif IV system” resulted in the introduction of ketone functionality at the C(10) position (5ai).18 Finally, methods based on the generation of oxygen-centered radicals, followed by 1,5-hydrogen atom transfer, led to etherification of the distal methyl group (C(14) position, 5aj).19 None of the developed methods has led to the selective oxidation of primary C–H bonds located β to the alcohol. Thus, the method we report offers new opportunities for selective alcohol oxidation.
CONCLUSION
We have developed an Ir-catalyzed functionalization of C–H bonds located β to hydroxyl functionality by incorporating traceless perfluorinated ester directing groups. Diethyl silane plays a dual role in the overall process, acting as the reductant for the hydrosilylation of the ester and as the reagent for silylation of the C–H bond, in this case to obtain six-membered dioxasilinanes. The silicon-containing intermediates undergo oxidation to 1,2-diols, leading to a net alcohol directed β-C(sp3)–H hydroxylation. The iridium catalysts react preferentially with primary β-C(sp3)–H bonds over secondary, benzylic, or tertiary C–H bonds, leading to a selectivity that complements that of previously developed oxidation methods. Thus, this functionalization of an alkyl C–H bond enriches the currently available toolbox of C–H bond functionalization reactions suitable for diversification of complex structures.
Supplementary Material
Acknowledgments
We thank the NIH (GM 115812) and The Dow Chemical Company for support of this work. A.B. thanks the Swiss National Science Foundation for a postdoctoral fellowship. T.W.B. gratefully acknowledges the National Science Foundation for a predoctoral fellowship.
Footnotes
Notes
The authors declare no competing financial interest.
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/jacs.7b12150.
Experimental details, synthetic procedures, spectral data (PDF)
References
- 1.(a) Jazzar R, Hitce J, Renaudat A, Sofack-Kreutzer J, Baudoin O. Chem - Eur J. 2010;16:2654–2672. doi: 10.1002/chem.200902374. [DOI] [PubMed] [Google Scholar]; (b) Gutekunst WR, Baran PS. Chem Soc Rev. 2011;40:1976–1991. doi: 10.1039/c0cs00182a. [DOI] [PubMed] [Google Scholar]; (c) McMurray L, O’Hara F, Gaunt MJ. Chem Soc Rev. 2011;40:1885–1898. doi: 10.1039/c1cs15013h. [DOI] [PubMed] [Google Scholar]; (d) Brückl T, Baxter RD, Ishihara Y, Baran PS. Acc Chem Res. 2012;45:826–839. doi: 10.1021/ar200194b. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Yamaguchi J, Yamaguchi AD, Itami K. Angew Chem, Int Ed. 2012;51:8960–9009. doi: 10.1002/anie.201201666. [DOI] [PubMed] [Google Scholar]; (f) He J, Wasa M, Chan KSL, Shao Q, Yu JQ. Chem Rev. 2017;117:8754–8786. doi: 10.1021/acs.chemrev.6b00622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.(a) Hartwig JF. Chem Soc Rev. 2011;40:1992–2002. doi: 10.1039/c0cs00156b. [DOI] [PubMed] [Google Scholar]; (b) Mkhalid IAI, Barnard JH, Marder TB, Murphy JM, Hartwig JF. Chem Rev. 2010;110:890–931. doi: 10.1021/cr900206p. [DOI] [PubMed] [Google Scholar]
- 3.(a) Ghavtadze N, Melkonyan FS, Gulevich AV, Huang C, Gevorgyan V. Nat Chem. 2014;6:122. doi: 10.1038/nchem.1841. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Cheng C, Hartwig JF. Chem Rev. 2015;115:8946–8975. doi: 10.1021/cr5006414. [DOI] [PubMed] [Google Scholar]; (c) Kon K, Suzuki H, Takada K, Kohari Y, Namikoshi T, Watanabe S, Murata M. ChemCatCh-em. 2016;8:2202–2205. [Google Scholar]; (d) Fukumoto Y, Hirano M, Chatani N. ACS Catal. 2017;7:3152–3156. [Google Scholar]
- 4.Henkel T, Brunne RM, Müller H, Reichel F. Angew Chem, Int Ed. 1999;38:643–647. doi: 10.1002/(SICI)1521-3773(19990301)38:5<643::AID-ANIE643>3.0.CO;2-G. [DOI] [PubMed] [Google Scholar]
- 5.(a) Zhang SY, Zhang FM, Tu YQ. Chem Soc Rev. 2011;40:1937–1949. doi: 10.1039/c0cs00063a. [DOI] [PubMed] [Google Scholar]; (b) Ketcham JM, Shin I, Montgomery TP, Krische MJ. Angew Chem, Int Ed. 2014;53:9142–9150. doi: 10.1002/anie.201403873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.(a) Barton DHR, Beaton JM, Geller LE, Pechet MM. J Am Chem Soc. 1960;82:2640–2641. [Google Scholar]; (b) Hartung J, Gottwald T, Špehar K. Synthesis. 2002;2002:1469–1498. [Google Scholar]
- 7.(a) Doyle MP, Kalinin AV, Ene DG. J Am Chem Soc. 1996;118:8837–8846. [Google Scholar]; (b) Espino CG, Wehn PM, Chow J, Du Bois J. J Am Chem Soc. 2001;123:6935–6936. [Google Scholar]; (c) Chen K, Richter JM, Baran PS. J Am Chem Soc. 2008;130:7247–7249. doi: 10.1021/ja802491q. [DOI] [PubMed] [Google Scholar]
- 8.(a) Simmons EM, Hartwig JF. J Am Chem Soc. 2010;132:17092–17095. doi: 10.1021/ja1086547. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Simmons EM, Hartwig JF. Nature. 2012;483:70–73. doi: 10.1038/nature10785. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Li B, Driess M, Hartwig JF. J Am Chem Soc. 2014;136:6586–6589. doi: 10.1021/ja5026479. [DOI] [PubMed] [Google Scholar]; (d) Lee T, Wilson TW, Berg R, Ryberg P, Hartwig JF. J Am Chem Soc. 2015;137:6742–6745. doi: 10.1021/jacs.5b03091. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Lee T, Hartwig JF. Angew Chem, Int Ed. 2016;55:8723–8727. doi: 10.1002/anie.201603153. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Su B, Zhou TG, Li XW, Shao XR, Xu PL, Wu WL, Hartwig JF, Shi ZJ. Angew Chem, Int Ed. 2017;56:1092–1096. doi: 10.1002/anie.201609939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ren Z, Mo F, Dong G. J Am Chem Soc. 2012;134:16991–16994. doi: 10.1021/ja3082186. [DOI] [PubMed] [Google Scholar]
- 10.Xu Y, Yan G, Ren Z, Dong G. Nat Chem. 2015;7:829–834. doi: 10.1038/nchem.2326. [DOI] [PubMed] [Google Scholar]
- 11.Hua Y, Asgari P, Avullala T, Jeon J. J Am Chem Soc. 2016;138:7982–7991. doi: 10.1021/jacs.6b04018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.(a) Saget T, Cramer N. Angew Chem, Int Ed. 2012;51:12842–12845. doi: 10.1002/anie.201207959. [DOI] [PubMed] [Google Scholar]; (b) Piou T, Bunescu A, Wang Q, Neuville L, Zhu J. Angew Chem, Int Ed. 2013;52:12385–12389. doi: 10.1002/anie.201306532. [DOI] [PubMed] [Google Scholar]
- 13.Guthrie JP. Can J Chem. 1975;53:898–906. [Google Scholar]
- 14.Hirsch JA. Topics in Stereochemistry. 1967;1:199–222. [Google Scholar]
- 15.Jung ME, Piizzi G. Chem Rev. 2005;105:1735–1766. doi: 10.1021/cr940337h. [DOI] [PubMed] [Google Scholar]
- 16.Fraga BM, Guillermo R, Hanson JR, Truneh A. Phytochemistry. 1996;42:1583–1586. [Google Scholar]
- 17.(a) Tenaglia A, Terranova E, Waegell B. J Org Chem. 1992;57:5523–5528. [Google Scholar]; (b) Gómez L, Canta M, Font D, Prat I, Ribas X, Costas M. J Org Chem. 2013;78:1421–1433. doi: 10.1021/jo302196q. [DOI] [PubMed] [Google Scholar]; (c) Jana S, Ghosh M, Ambule M, Sen Gupta S. Org Lett. 2017;19:746–749. doi: 10.1021/acs.orglett.6b03359. [DOI] [PubMed] [Google Scholar]; (d) Mack JB, Gipson JD, Du Bois J, Sigman MS. J Am Chem Soc. 2017;139:9503–9506. doi: 10.1021/jacs.7b05469. [DOI] [PubMed] [Google Scholar]
- 18.Barton DHR, Beloeil JC, Billion A, Boivin J, Lallemand JY, Lelandais P, Mergui S. Helv Chim Acta. 1987;70:2187–2200. [Google Scholar]
- 19.(a) Brun P, Waegell B. Tetrahedron. 1976;32:1137–1145. [Google Scholar]; (b) Dorta RL, Francisco CG, Freire R, Suárez E. Tetrahedron Lett. 1988;29:5429–5432. [Google Scholar]; (c) Hung K, Condakes ML, Morikawa T, Maimone TJ. J Am Chem Soc. 2016;138:16616–16619. doi: 10.1021/jacs.6b11739. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.