

Abstract
Integrin binding to bioengineered hydrogel scaffolds is essential for tissue regrowth and regeneration, yet not all integrin binding can lead to tissue repair. Here, we show that through engineering hydrogel materials to promote α3/α5β1 integrin binding, we can promote the formation of a space filling and mature vasculature compared to hydrogel materials that promote a αvβ3 integrin binding. In vitro, α3/α5β1 scaffolds promoted endothelial cells to sprout and branch, forming organized extensive networks that eventually reached and anastomosed with neighboring branches. In vivo, α3/α5β1 scaffolds delivering vascular endothelial growth factor (VEGF) promoted non-tortuous blood vessel formation and non-leaky blood vessels by 10-days post stroke. In contrast, materials that promote αvβ3 integrin binding promoted endothelial sprout clumping in vitro and leaky vessels in vivo. This work shows that precisely controlled integrin activation from a biomaterial can be harnessed to direct therapeutic vessel regeneration and reduce VEGF induced vascular permeability in vivo.
The design of therapeutic angiogenic materials to treat cardiovascular diseases, such as deficient blood supply to the heart, limbs, and brain, has primarily been driven by the delivery of angiogenic factors within a scaffold. Recently, the synergy between extracellular matrix (ECM) integrin ligands and angiogenic factor delivery has been demonstrated 1 and proposed as a therapeutic strategy to design next generation agiogneic materials 2. However, specific integrin engagement from engineered materials has not been explored as a vascular morphogenic signal.
Integrins are a family of heterodimeric transmembrane proteins, composed of alpha (α) and beta (β) subunits, that bind to both the extracellular matrix (ECM) and the cellular cytoskeleton, providing both mechanical and biochemical signaling3. Integrins have been associated with processes ranging from cell structure and adhesion to cell differentiation and survival3–5, which are cell behaviors critical to tissue morphogenesis, homeostasis and repair. In endothelial cells, at least seven α and β heterodimers are expressed, including αvβ3, α3β1 and α5β1, which have been implicated in vascular morphogenesis and vessel patterning 6, 7. In particular, β1 and β3 integrins have been implicated in vascular lumen formation6, 8, tight cell-cell junction formation6, 9, 10, and the recruitment of mural cells to the vessels for stabilization11. β1 integrin expression is slowly upregulated in brain vessels over time as vessels mature12 and αvβ3 expression in resting endothelial cells (ECs) is low, while αvβ3 expression in cytokine-activated ECs or tumor ECs is upregulated 13–15. Further, both up-regulation and abolishment of β1 and β3 integrin activation have shown to be related to pathological angiogenesis3, 9, 10, 16, 17. While excessive suprabasal expression of β1 integrin in skin has been shown to induce a psoriasis phenotype16, the knockout of β1 integrin resulted in weakening of endothelial cell junctions and induction of blood leakage in a retinal angiogenesis assay9. Likewise, the upregulation of β3 integrin leads to enhanced endothelial cell permeability18 while the abolition of β3 integrin leads to intrauterine bleeding, defective coronary capillaries, and enhanced tumor angiogenesis (induced by the compensatory VEGF increase after β3 knockout)10, 17, 19.
The incorporation of integrin binding peptides such as RGD derived from natural ECM proteins to biomaterials is a popular approach to promote integrin engagement20–22. Though integrin-binding peptides can support cell attachment, migration, and differentiation, they have severely reduced binding affinity and specificity compared to the same peptide presented within its associated full-length 3D protein structure. In the context of fibronectin (Fn), recombinant fragments of the 9th type III repeat and 10th type III repeat (Fn III9-10) have been expressed and incorporated into matrices to present RGD sequences in their correct 3D structural context to improve binding affinity and modulate growth factor signaling 1. However, without the rest of the full-length protein these protein fragments do not contain the natural switches that modulate integrin engagement (e.g. native fibronectin binds several integrin pairs depending on the level of extension of the protein23, 24) and thus, lack complete specificity. To improve specificity, recombinant fragments of Fn III9-10 have been engineered to promote α3/α5β1 specific binding, enhanced mesenchymal stem cell differentiation toward bone25, and modulation of epithelial to mesenchymal transition26. Here we explore specific integrin engagement as a vascular morphogenic signal within engineered matrices.
Fibronectin fragments with tunable integrin binding
Immediately following injury, fibrin and fibronectin are the major ECM constituents of the provisional pre-vascularized matrix. In our study, recombinant fibronectin fragments of Fn III9-10 were designed to preferentially bind α3/α5β1 or αvβ3 integrin through introduction of a Leu-Pro point mutation at position 1408 or a flexible linker (4XGly) spanning between to the two domains 25, 27, 28. Though both recombinant fragments can bind αvβ3 integrin via the RGD sequence, we and others consistently observe a preference of the Leu-Pro mutant to bind synergy-dependent integrins, like α5β1 integrin in cell – material interactions26, 29. In this manuscript, we refer to the Leu-Pro mutated, or stabilized, fragment as 9*10 and the 4xGly insertion mutated fragment as 9(4G)10. For ease of immobilization onto surfaces and incorporation into natural and synthetic hydrogel biomaterials both fragments were produced with an N-terminal cysteine residue to allow Michael type addition modifications and a factor XIIIa substrate sequence 30 to allow enzymatic conjugation.
Fibronectin fragment coated surfaces were first used to verify the specific integrin activations on human umbilical vein endothelial cells (HUVEC). HUVEC were able to attach and spread on either fragment-modified surface (Fig. S1a-c). As expected, clear αvβ3 integrin staining was observed on Fn9(4G)10 coated surfaces but not Fn9*10 coated surfaces, while clear β1 integrin staining was observed at cell edges on Fn9*10 coated surfaces but not on the Fn9(4G)10 coated ones (Fig. S1a). The actin cytoskeleton for cells cultured on αvβ3 specific Fn9(4G)10 surfaces showed more short and disoriented actin fibers compared with α3/α5β1 specific Fn9*10 surfaces, where actin fibers reached extensive lengths (Fig. S1c), suggesting that differential integrin activation from immobilized fragments impacts EC cytoskeleton arrangement in 2D. Addition of VEGF to ECs plated on αvβ3 specific surfaces significantly increased EC migration compared to cells seeded on blank surfaces or α3/α5β1 specific surfaces, while no differences were observed for EC proliferation or integrin binding (Fig S1. d-f). Together these findings confirm that ECs alter their cellular behavior depending on the integrin binding specificity dominating their attachment to the surface.
Integrin stimulation guides endothelial cell sprouting patterns
We are interested in using VEGF, the master regulator of angiogenesis31, to induce EC sprouting and angiogenesis in vivo. Thus, we looked at the influence of fibronectin mediated cell adhesion on VEGF induced vascular sprouting in a three-dimensional assay. Two types of commercially available fibrinogen were used, one that contained fibronectin (Fib1) and one that is fibronectin depleted (Fib3). EC coated beads were suspended in the fibrin matrices and cultured in the presence of soluble VEGF for 7-days following the protocol of Hughes et al (Fig. S2a)32. At day 7, the cultures were fixed, stained for actin, and quantified for the number of sprouts, number of branching points and total network length per bead. Sprouting and branching points in Fib3 was significantly decreased compared with Fib1, suggesting that the presence of native fibronectin is critical for EC sprouting (Fig. 1a and Fig. S2b). Addition of exogenous fibronectin to Fib3 partially rescued EC sprouting, resulting in statistically increased sprouting, branching points and total network length (Fig. S2b); however, regardless of the amount of exogenous fibronectin added, the level of sprouting in Fib3 was significantly lower than that observed in Fib1 (Fig. 1a,b and Fig. S2b). This indicated that besides the endogenous fibronectin within the Fib1 matrices, other factors removed during the Fib3 preparation may also be important for EC sprouting in fibrin. Among all the tested fibronectin concentrations, it was also observed that 1μM was the lowest concentration that achieved statistically increased sprouting, branching points and total network length (Fig. S2b). Thus, we incorporated equal molar concentration of Fn III9-10 fragments (2μM), in our in vitro studies; each fibronectin protein contains two copies of Fn III9-10.
Figure 1. Endothelial cell sprouting in αvβ3 specific matrices displays clustered branches.
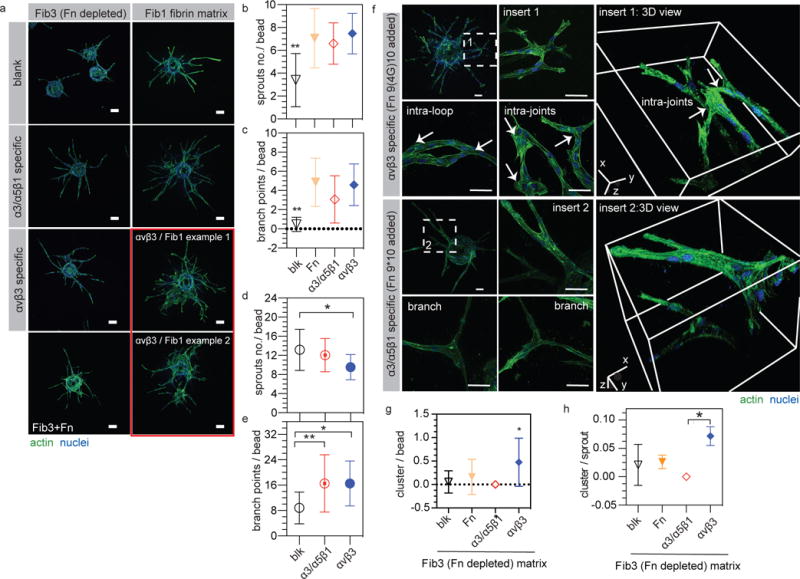
(a) HUVEC sprouting in FXIIIa stabilized fibronectin containing fibrin gel (Fib1) and fibronectin depleted fibribrin gel (Fib3). Scale bar: 100 μm. (b,c) Quantification of sprout number and branch points per bead in Fib3 matrices. (d,e) Quantification of sprout number and branch points per bead in Fib1 matrices. (f) Representative images and 3D views of HUVEC branch structures in both αvβ3 and α3/α5β1 specific Fib3 matrices. Intra-loop and intra-joint branch structures were observed in αvβ3 specific matrices while organized branch structures were observed in α3/α5β1 specific matrices. Scale bar: 50 μm. (g) Quantification of branch clusters per bead and (h) branch cluster number per sprout. Blank = blk = no fibronecitn fragment added, α3/α5β1 specific matrices = gel + 2μM Fn9*10, αvβ3 specific matrices = gel + 2μM Fn9(4G)10, Fn matrices = Fib3 gel + 1μM Fn. For all quantifications n ≥ 15 HUVEC coated beads, from 3 independent gels. Each bead is treated as its own independent sample. Statistical analyses were performed using Prism (GraphPad, San Diego, CA). Brown-Forsythe test was performed to ensure that the variance of the data is similar between the groups that are being statistically compared. Data were analyzed using a one-way analysis of variance (ANOVA) followed by a Tukey post-hoc test and a 95% confidence interval. All plots represent mean ± SD. * and ** indicate P < 0.05 and P < 0.01, respectively. An alternative statistical analysis for this data treating experiments performed on different days as the independent sample can be found on Figure S3.
We next tested the role of α3/α5β1-specific (Fn9*10 fragment added) and αvβ3-specific (Fn9(4G)10 fragment added) matrices on EC sprouting. The number of sprouts, branch points or total network length showed no significant difference among full length fibronectin, α3/α5β1-specific and αvβ3-specific conditions, indicating that integrin engagement is a major factor that affects sprouting (Fig. 1b,c and Fig. S2c). Between α3/α5β1-specific and αvβ3-specific conditions we observed no differences in sprout number, branch points or total network length (Fig. 1b-e, Fig. S2c,d and Fig. S3a-c,e-g); however, we observed sprouting “clusters” in the αvβ3-specific matrices but not on blank, fibronectin, or α3/α5β1-specific matrices (Fig. 1a,f). The clusters are merged vessel branch clumps from the same bead, which can either locate close to the surface of the beads or on the sprouts. They are always presented as chaotic branch bundles, thus the number of branches and sprouts within them are difficult to discern. Using high resolution z-stack confocal imaging we found that the clusters were associated with intra-loop and intra-joint structures both within and between neighboring sprouts, which were not observed in α3/α5β1-specific matrices (Fig. 1f). The quantification of the number of clusters per bead, sprout or branch, showed statistically significant occurrence in αvβ3-specific gels compared with the blank, fibronectin and α3/α5β1-specific Fib1 or α3/α5β1-specific Fib3 matrices (Fig. 1g,h, Fig. S3d,h and Fig. S4a-f). Taken together, these results show that although both Fn fragments enhanced sprouting and branching of ECs, they lead to different vascular patterns in the resulting EC network. In particular, αvβ3-specific matrices promoted the formation of a pathological vascular network33 containing intra-loop and intra-joint features.
The involvement and importance of integrins in EC sprouting was further confirmed through function blocking antibody studies. VEGF induced EC sprouting in α3/α5β1-specific matrices in the presence of function blocking antibodies against either α5 or β1 completely inhibited the sprouting process (Fig. 2a and Fig. S4g,h). In contrast, EC sprouting in αvβ3-specific matrices in the presence of function blocking antibodies against αv did not impact sprouting, while function blocking antibodies against β3 again completely inhibited the sprouting process (Fig. 2a and Fig. S4g,h). These results indicate that α5, β3, and β1 are essential to EC sprouting but αv is not. We next explored whether blocking αv normalized the resulting vasculature by decreasing the number of intra-loop and intra-joint structures. Surprisingly, blocking αv binding decreased the number of branch clusters (Fig. 2b), suggesting that αv binding is responsible for the sprout clusters formed in the αvβ3-specific matrices. These results are in agreement with previous studies showing that activation of αvβ3 results in increased vascular permeability in 2D18 while inhibition of αv reduces vascular permeability34. Interestingly, we have recently demonstrated in a retinal angiogenesis model that tips cells mechanically unfold the integrin binding domain of Fn at their leading edge, resulting in a Fn9(4G)10 and thus avb3 binding character. The conformation of Fn’s integrin binding domain returns to its native, Fn9*10-like conformation during vessel maturation 35
Figure 2. Endothelial cell sprount clustering is partially rescued with αv blocking.
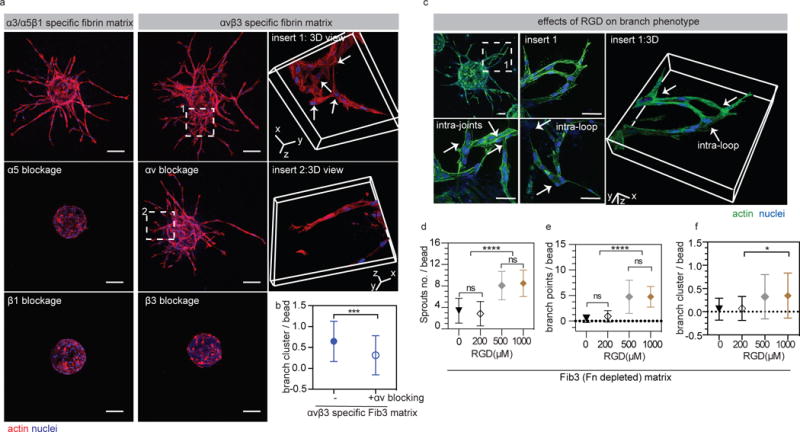
(a) HUVEC sprouting in the presence of integrin αv, α5, β1 or β3 (5μg/ml changed daily) blocking antibodies. Blocking α5, β1 or β3 completely blocks sprouting, while blocking αv does not. α3/α5β1 specific matrices = gel + 2μM Fn9*10, αvβ3 specific matrices = gel + 2μM Fn9(4G)10. Scale bars, 100 μm. (b) comparison of branch cluster occurrence with and without αv blocking (n=51 HUVEC coated beads from three independent gels). (c) Images showing HUVEC sprouts in RGD (1000μM) modified FXIIIa stabilized Fn depleted fibrin matrices. RGD modified matrices promote HUVEC sprouting and branch cluster formation. Scale bar: 50 μm (d,e) Quantification of HUVEC sprouting in RGD modified Fib 3 matrices at different concentrations. (f) Quantification of branch clusters per bead. For quantification n ≥ 15 HUVEC coated beads, from 3 independent gels. Each bead is treated as its own independent sample. Statistical analyses were performed using Prism (GraphPad, San Diego, CA). Brown-Forsythe test was performed to ensure that the variance of the data is similar between the groups that are being statistically compared. Data were analyzed using a one-way analysis of variance (ANOVA) followed by a Tukey post-hoc test and a 95% confidence interval. All plots represent mean ± SD. *,*** and **** indicate P < 0.05, P < 0.001 and P < 0.0001.
Matrices modified with RGD peptides show similar vascular pattern as Fn9(4G)10
Next, we sought to examine vascular patterns utilizing the ubiquitously used synthetic peptide derived from fibronectin, RGD. RGD peptides with α2PI1-8 sequences (H-NQEQVSPLRGDSPG-NH2) were incorporated within fibrin matrices using FXIIIa enzyme. As observed with the fibronectin fragments, the incorporation of RGD within Fib3 matrices resulted in enhanced EC sprouting (Fig. 2c-e and Fig. S4i,j). Similar to αvβ3-specific matrices, RGD modified hydrogels resulted in intra-loop and intra-joint structures (Fig. 2c,f). These results suggest that RGD binding without the presence of the proline-histidine-serine-arginine-asparagine (PHSRN) “synergy” domain on the 9th type III repeat36, 37, result in pathological angiogenesis, which may come from the preferential αvβ3 binding38. It should be noted that the effect of enhanced sprouting and disorganized structures was only observed for a much higher concentration of RGD peptide (> 500μM) compared with the concentration of fibronectin fragments used (2μM).
Upregulation of αvβ3 and alterations in its activation state have been associated with disease states such as cancer39 and fibrosis40 and have been widely used as cancer targeting ligands in drug delivery applications41, yet RGD is the most widely used integrin binding peptide to modify biomaterials. We do not mean to suggest that the use of RGD modified biomaterials for therapeutic angiogenesis is inherently flawed; rather, we believe that the incorporation conditions for RGD peptides such as presentation, concentration, and other neighboring ligands should be studied to ensure that the desired revascularization pattern is obtained. For example, clustering RGD within hydrogels has been shown to upregulate the expression of β1 integrins in MSCs 42 and immobilization of VEGF has leads to β1 recruitment43.
Integrin stimulation guides vascular anastomosis
During the angiogenesis process, sprouts from parental vessels fuse with other sprouts or pre-existing blood vessels for the purposes of supplying blood and oxygen to surrounding tissues44. To test the effect of αvβ3 and α3/α5β1 specific matrices on EC sprout anastomosis, a similar bead assay was performed using stably transfected ECs expressing enhanced green fluorescent protein (EGFP) (Fig. S5a). HDFs were seeded within the fibrin matrix to yield more robust and long-lasting tubes for culture. Normal anastomosis results in the binding of tip cells through a single contact45. Clear single tip-tip contact or paralleled tip interaction were observed in Fib3 and Fib3 + Fn9*10 conditions (Fig. S5b,c). In contrast, Fib3 + Fn9(4G) matrices promoted multiple tip-tip contacts, resulting in independent contact sites and loop structures (Fig. S5b,c). Thus, consistent with our observations in EC sprouting morphogenesis, inter-loop and inter-joint structures are observed in anastomosed sprouts within αvβ3-specific matrices.
αv activation leads to pathological vasculature through VE-cadherin disruption
Next, we investigated possible mechanisms for the observed differences in the vascular patterns generated by α3/α5β1-specific and αvβ3-specific matrices. Vessel sprouts lacking VE-cadherin display irregular anastomosis, characterized by multiple tip-tip contact sites and disturbed junctional connections45, similar to our observations in Fn9(4G)10 matrices. We examined VE-cadherin distribution on ECs cultured in vitro on αvβ3 or α3/α5β1 modified surfaces. Cells seeded on α3/α5β1 specific surfaces showed significantly increased amounts of VE-cadherin signal at cell-cell junctions compared with αvβ3 specific conditions both with and without VEGF (Fig. 3a,b and Fig. S6a). Obvious absence and significantly lowered VE-cadherin signals between adjacent cells were observed on αvβ3 specific surfaces, indicating VE-cadherin disruption (Fig. 3a,b and Fig. S6a). VE-cadherin grey value analysis was performed on randomly-chosen cell-cell junctions from Fig. 3a. α3/α5β1 specific surfaces led to thicker VE-cadherin layers with stronger signal at cell-cell junction when compared with αvβ3 specific surfaces (Fig. 3c,d). EC sprouting in αvβ3 specific matrices was also characterized by reduced VE-cadherin staining on sprout shunts and cell-cell junctions compared with EC sprouting in α3/α5β1 specific matrices (Fig. 3e). To confirm that increased αv integrin binding is responsible for the decrease in VE-cadherin staining, αv integrin binding was disrupted using function-blocking antibodies as previously done. VE-cadherin staining after αv blocking in αvβ3 specific fibrin matrices showed EC cells with increased VE-cadherin staining similar to what was observed in α3/α5β1 specific matrices, strongly suggesting that αv binding is responsible for the reduction in VE-cadherin localized at cell-cell junctions. The effect of αv blocking was observed in both Fib1 and Fib3 matrices (Fig. 3e and Fig. S6b,c). VE-cadherin is an important cell-cell junction protein responsible for shifting endothelial cell response to VEGF 46, but also functions to maintain low permeability of endothelial cell layers18. Partial knockout of VE-cadherin can lead to vascular instability and hemorrhages47. VE-cadherin function can be disrupted by upregulation of αvβ3 integrin, enhancing endothelial cell permeability18. Taken together, these findings support the idea that VE-cadherin-related pathological vasculature was caused by αv activation in αvβ3 specific matrices and demonstrate that αv blockage can be utilized to rescue the pathological effects.
Figure 3. αvβ3 integrin engagement disrupts VE-cadherin in HUEVEs.
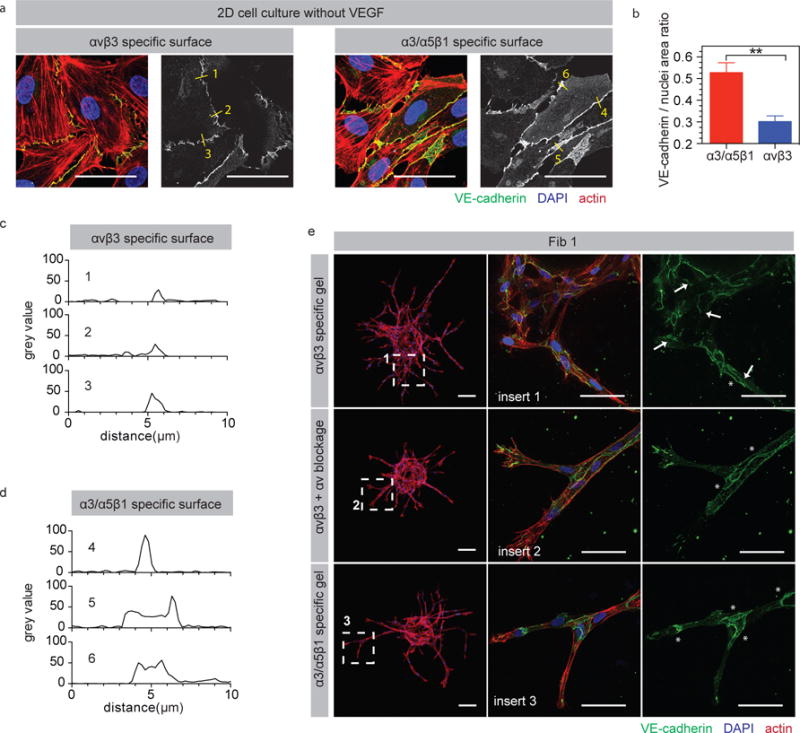
(a) Microscopic analysis of VE-cadherin distribution for HUVECs seeded on surfaces modified with α3/α5β1 or αvβ3 specific surfaces < 24-hours after plating. Scale bar: 50 μm. (b) Quantification of VE-Cadherin to nuclei positive area ratio for HUVECs seeded on surfaces modified with α3/α5β1 or αvβ3 specific surfaces < 24-hours after plating. Quantification was performed from 3 independent cultures performed at three different times. Statistical analyses were performed using Prism (GraphPad, San Diego, CA) using a two-tailed unpaired t-test. Plot represents mean ± SD. ** indicate P < 0.01. (c) Grey value distribution analysis for 3 random cell-cell junctions on α3/α5β1 specific surface from (a). (d) Grey value distribution analysis for 3 random cell-cell junctions on αvβ3 specific surface from (a). (e) Microscopic analysis of VE-cadherin signals for HUVECs seeded in α3/α5β1 specific matrices = gel + 2μM Fn9*10, αvβ3 specific matrices = gel + 2μM Fn9(4G)10, and αvβ3 specific matrices + αv blocking antibody = gel + 2μM Fn9(4G)10 + αv blocking antibody (5μg/ml changed daily). Arrows represent reduced and missing VE-Cadherin signal while stars represent coherent and clear VE-Cadherin signal on sprout shunts and cell-cell junctions. Scale bar: 100 μm (whole bead) and 50 μm (sprouts).
Integrin stimulation from a bioengineered matrix guides vascular patterns in vivo
The fibronectin fragments were tested in a modified matrigel plug assay that uses bioengineered hyaluronic acid (HA) hydrogels instead of matrigel to assess angiogenesis in mice (Fig 4a and Fig. S7a-d). HA hydrogels are formed through crosslinking HA molecules using Michael type addition chemistry between acrylamide groups introduced to the backbone of hyaluronic acid and dithiol crosslinker containing protease degradable peptides48. Fn fragments were also introduced to this protease degradable hydrogel matrix backbone to mediate integrin binding using the same Michael type chemistry through the cysteine in the fragment N-terminus. VEGF was incorporated into the system using a controlled release system based on single protein nanocapsules (nV) previously developed in our laboratory49 (Fig. S7e). Hydrogels containing fibronectin fragments and nV were implanted subcutaneously (Fig. S7c,f). Evaluation of isolectin perfused whole mount sections was performed 14-days after implantation using light sheet (Fig. S8a) and confocal microscopy (Fig. 4). HA hydrogels that do not contain fibronectin fragments (blk) resulted in the least vessel sprouting on the hydrogel surface and vessel infiltration within the hydrogel compared with fragment conditions and was similar to normal skin (Fig. 4b) even with the presence of nV, demonstrating that integrin binding is essential for angiogenesis to occur in vivo (Fig. 4c and Fig. S8b-d). HA hydrogels modified with either fibronectin fragment supported an angiogenic response and similar infiltration (Fig. S8); however, the morphology of the vessels on the gel surfaces was significantly different. The α3/α5β1 specific HA gels (Fn9*10 immobilized) displayed non-tortuous vessels displaying similar features as the normal mouse vasculature (control) while αvβ3 specific gels displayed tortuous and unorganized vessels that appeared to clump with one another (Fig. 4b-d). While vessels on α5β1 specific gel surfaces presented organized and even vessel distribution like the blank gel control, vessels on αvβ3 specific gel surfaces yielded uneven distributions, which originated from regional tortuous and unorganized vessel clumps (Fig. 4e,f). These results show that similar to in vitro sprouting and anastomosis, integrin specific materials can dictate vascular patterning in vivo in a non-diseased model.
Figure 4. Modulation of vascular patterning in skin using integrin specific hydrogels.
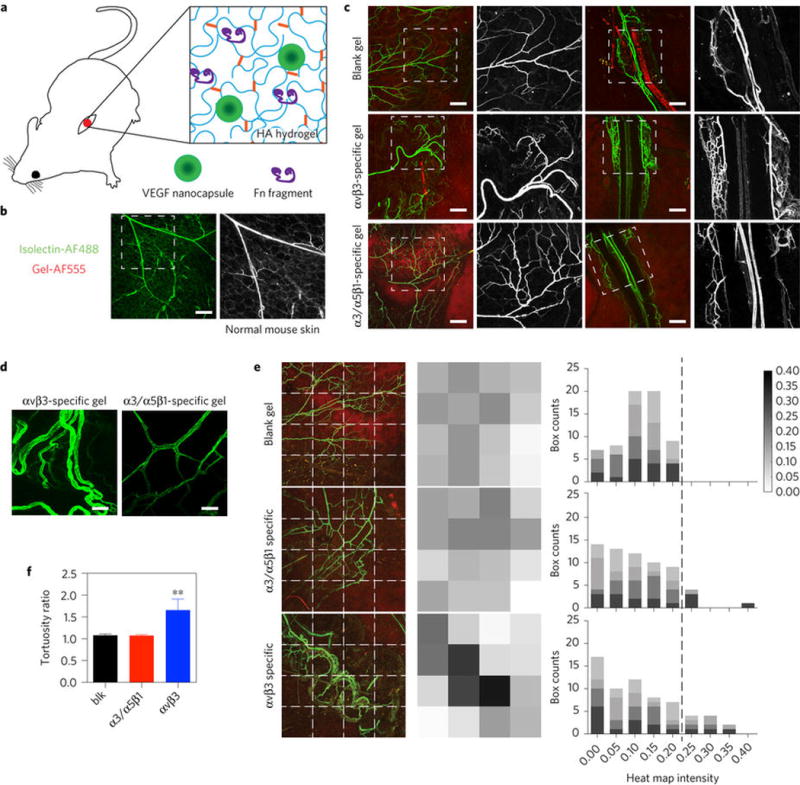
a, A modified Matrigel plug assay using bioengineered hyaluronic acid (HA) hydrogels was utilized to assess angiogenesis in mice. b, Images of vessels on normal mouse skin. Scale bars, 200 μm. c, Images of isolectin-modified vessels at the surface of the implant 2 weeks after implantation. Scale bar, 200 μm. d, Representative ×40 images of vessels on the surface of α3/α5β1-specific and αvβ3-specific HA matrices. Scale bar, 50 μm. e, Heat map analysis of vessel distribution on the surfaces of blank, α3/α5β1-specific and αvβ3-specific matrices (n = 4 individual implants). Darker box indicates higher vessel density in that region while white indicates the absence of vessels. Plot represents the histogram of grey values. Blank gel: HA gel; α3/α5β1-specific matrices: HA gel + 10 μM Fn9*10 + 200 ng VEGF nanocapsules; αvβ3-specific matrices: HA gel + 10 μM Fn9(4G)10 + 200 ng VEGF nanocapsules. f, Vessel tortuosity comparison among blank, α3/α5β1-specific and αvβ3-specific conditions (n = 4 implants). Statistical analyses were performed using Prism (GraphPad). The Brown–Forsythe test was performed to ensure that the variance of the data is similar between the groups that are being statistically compared. Data were analysed using a one-way analysis of variance followed by a Tukey post hoc test and a 95% confidence interval. All plots represent mean s.d., and ** indicates P < 0.01.
α3/α5β1 integrin binding reduces VEGF induced vascular permeability after stroke
We next sought to evaluate the angiogenic response of integrin specific materials on VEGF-induced angiogenesis in a murine model of stroke. VEGF is one of the essential molecules in normal post-stroke angiogenesis 50. However, The delivery of VEGF after stroke, has been complicated by the induction of a disordered and permeable vasculature51, similar to other organs 52–54. Given that fibronectin is upregulated in the provisional matrix after stroke, but is only located in blood vessels in normal brain (Fig. 5a and Fig. S9a), we believe that the injection of an integrin specific hydrogel into the stroke cavity can modulate vascular patterning after stroke. Adult mice were submitted to a cerebral artery occlusion (MCAo). 5-days after stroke, HA hydrogels (containing nV and fragments) were injected directly into the stroke cavity (Fig. 5b). Ten days post-transplantation, animals injected with fibronectin fragment containing hydrogels were perfused with tomato lectin before sacrifice for the purpose of studying perfused vessel morphology while other animals were directly perfused with 4% PFA and sacrificed.
Figure 5. Integrin specific hydrogels modulate vascular patterning and permeability after stroke.
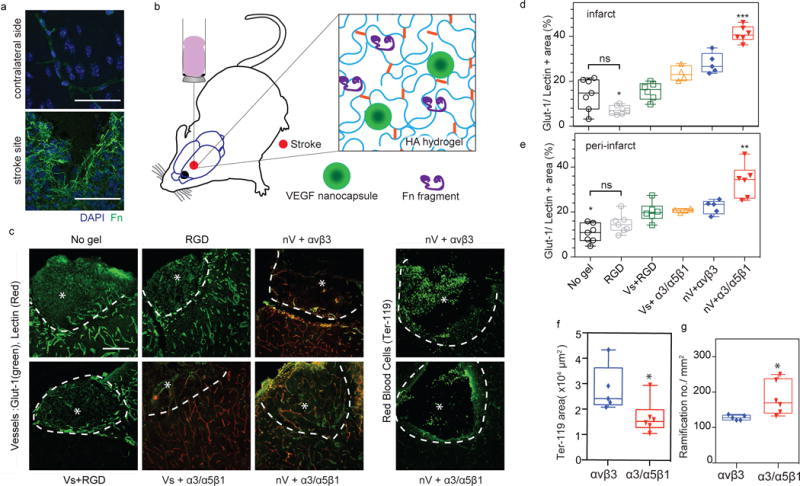
A middle cerebral artery occlusion ischaemic stroke model was utilized to look at the effects of injected integrin-specific HA matrices on stroke repair 10 days post-injection (injection was performed 5 days post-stroke). a, Fibronectin location in normal and stroke brain 15 days post-stroke. Scale bars, 50 μm. b, Schematic illustration of the hydrogel injected into the mouse brain after stroke. VEGF nanocapsules (nV) were designed to slowly release VEGF. c, Fluorescent microscopy showing brain vasculature in both the infarct and peri-infarct (stained for Glut-1 or Glut-1 plus tomato lectin intravascular perfusion) as well as leaked red blood cells (stained for Ter-119). The asterisk indicates the stroke site while the white dashed curve indicates the boundary between the infarct (stroke site) and peri-infarct (the area adjacent to the stroke site). Scale bars, 100 μm. d,e, Quantification of the total vessel area (perfused and not prefused) in the infarct (inside the stroke) and peri-infarct (around the stroke) areas. f, Quantification of Ter-119-positive red blood cell area. g, The morpho-analysis of growing vessels in the peri-infarct area for vessel ramification. All plots represent mean ± s.d. Each dot in the plots represents an individual mouse. Statistical analyses were performed using Prism (GraphPad). Data in d,e were analysed using a one-way analysis of variance followed by a Tukey post hoc test and a 95% confidence interval. Data in f,g were analysed using a two-tailed unpaired test. *, ** and *** indicate P < 0.05, P < 0.01 and P < 0.001; NS, not significant.
Sections were all stained for Glut-1, a glucose transporter expressed on brain endothelial cells and the positively stained vascular area was quantified in both the infarct and peri-infarct areas (Fig. 5c). To compare the vascular bed in all conditions, both Glut-1 stained only and Glut-1 plus tomato lectin stained in tomato lectin-perfused animals were quantified. Glut-1 stained for all vessels while tomato lectin only stained for perfused vessels. Tomato lectin alters Glut-1 staining such that in tomato lectin-perfused animals the combination of both stains reveals the vascular bed the same as Glut-1 alone in tomato lectin-unperfused animals (Fig. S9b). As expected, all the VEGF containing hydrogels showed a greater vascular area percentage than RGD only gels in the infarct and the peri-infarct regions. However, we found that the vascular area was significantly increased in the HA gel + nV + Fn9*10 (α3/α5β1 specific) condition compared with any other group in both regions (Fig. 5d,e), suggesting a strong role of activated α3/α5β1 integrin binding in promoting the angiogenesis process.
In order to evaluate the quality of these newly formed vascular network, both their permeability and structure were studied. For this, Ter-119, a red blood cell marker was used to assess vessel permeability (Fig. 5c). The results showed a significantly reduced positive area for Ter-119 in α3/α5β1 specific HA hydrogels, suggesting a beneficial effect of the activation of α3/α5β1 in reducing VEGF induced vascular permeability (Fig. 5f). In addition, the morpho-analysis of tomato lectin-perfused vessels was performed by quantifying the number of vascular ramifications growing out of a common vascular tree. We found that the number of ramifications per mm2 was again significantly greater in α3/α5β1 specific HA hydrogels (Fig. 5g). These results demonstrate for the first time that hydrogels with specific integrin activation can modulate vascular patterning after stroke and could reduce soluble VEGF induced vascular permeability.
Integrin-specific scaffold for therapeutic angiogenesis
Integrin binding is a fundamental design parameter for engineering matrices for tissue repair and regeneration. The incorporation of integrin binding molecules within engineered matrices ranges from peptides such as RGD55, to protein fragments56, and natural proteins57, with the primary purpose to promote cell spreading and migration within these matrices. However, integrin binding ligands are often overlooked as bioactive cues, capable of dictating morphogenesis and guiding tissue repair. Our work shows that integrin stimulation from engineered matrices is a morphogenic signal that can be harnessed to generate either a normal vasculature or a diseased vasculature depending on the integrin being engaged (Fig. 6).
Figure 6.
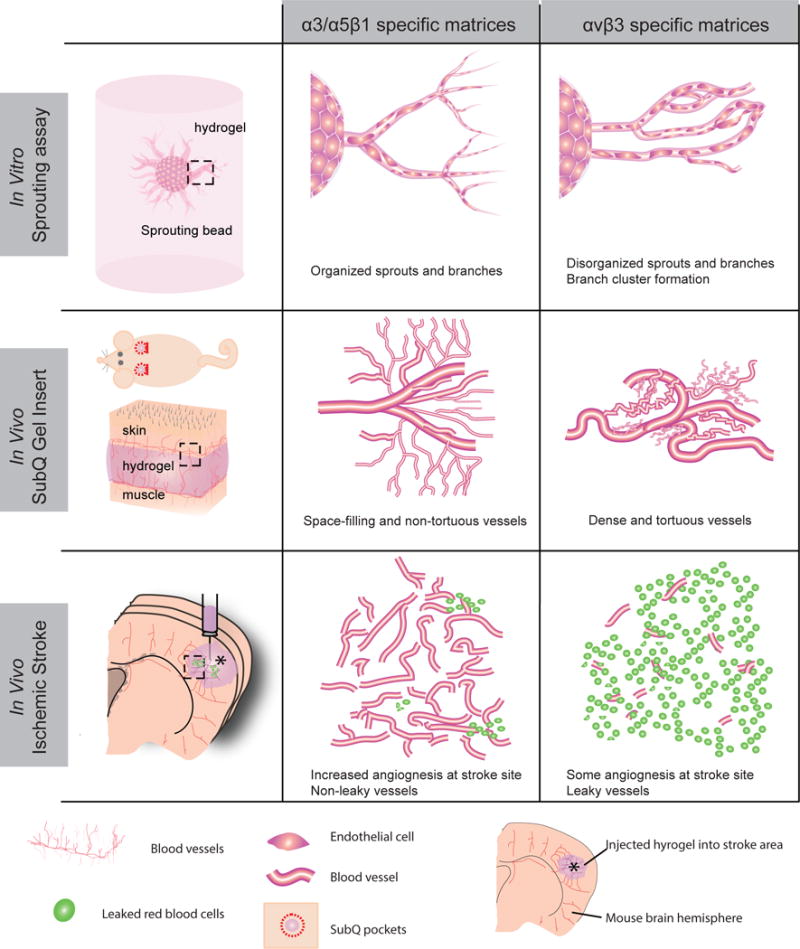
α3/α5β1 and αvβ3 integrin-specific materials regulate vascular patterning in vitro and in vivo
Methods
Recombinant fibronectin fragments
Recombinant fibronectin fragments of the 9th type III repeat (Fn III9) and 10th type III repeat (Fn III10) were designed to preferentially bind α3/α5β1 or αvβ3 integrin respectively. To achieve α3/α5β1 specificity, the thermodynamic stability of Fn III9 was increased through a leucine to proline point mutation at position 1408. This mutation has been previously shown to stabilize the integrin-binding domain of fibronectin, i.e. Fn III9-10, and enhance its binding selectivity to synergy-dependent β1 integrins, including both α5β1 and α3β125, 27, 28.To achieve αvβ3 integrin specificity, four (4) glycine residues were then inserted into the linker region between Fn III9 and Fn III10. The 4xGly insertion both physically separates the synergy (PHSRN) and RGD sites located on Fn III9 and Fn III10, respectively, and introduces torsional flexibility between the two domains, resulting in a complete disruption of α3/α5β1 integrin binding and promoting a αvβ3 integrin preference. Though both recombinant fragments can theoretically bind αvβ3 integrin via the RGD sequence, we and others consistently observe a preference of the stabilized mutant to bind synergy-dependent integrins, like α5β1 integrin in cell material interactions26, 29. In this manuscript, we called the Leu-Pro mutated, or stabilized, fragment 9*10 and the 4xGly insertion mutated fragment 9(4G)10. For ease of immobilization onto surfaces and incorporation into natural and synthetic hydrogel biomaterials both fragments were produced with an N-terminal cysteine residue to allow Michael type addition modifications and a factor XIIIa substrate sequence, consisting of residues 1–8 of the protein alpha2 plasmin inhibitor (α2PI1-8, NQEQVSPL)30 to allow enzymatic conjugation.
The expression system for these recombinant Fn fragments has been optimized from prior publications (Markowski et al.) to create a high throughput production process29. The expression vectors were transformed into MDS™42 LowMut ΔrecA (6262) chemically competent cells and individual clones were picked for successive expansion and increased protein yields. Transformed cells were grown in 2xYT media with Kanamycin (50 μg/mL) and protein production was induced by addition of 1.5mM IPTG. Incubation at 37°C with shaking for approximately 16 hours allows maximal protein production. Cultures were then centrifuged and lysed via sonication and freeze-thaw cycling. Recombinant Fn9-10-tdtomato-10 His were purified on a HisTrap nickel column (AKTSA Start, GE Healthcare) via affinity chromatography. Tdtomato and 10-His tag were removed by utilizing the thrombin cleavage site and bovine thrombin (Sigma-Aldrich) to separate the fragment and the tdtomato fusion protein. A Benzamidine column bound the free bovine thrombin and the His tag allows collection of C-terminal tdtomato in the HisTrap again and the Fn fragments (Fn9*10 or Fn9(4G)10) were released. The purity of Fn9-10 protein fragments was then analyzed by SDS-PAGE as mentioned in previous works25, 26.
Immobilization of Fn9*10 or Fn9(4G)10 on homogenous gold surface
Standard laboratory microscope glass slides were sequentially washed with acetone, isopropyl alcohol and methanol before gold deposition in e-beam evaporator. Deposition parameter: 5nm titanium at 0.3 Å/s deposition rate, followed by 30 nm gold at 0.5 Å/s deposition rate. Gold slides were then functionalized with 1% HS-C11-EG6-NH2(11-Mercaptoundecyl)hexa(ethylene glycol) amine, ProChimia Surfaces) and 99% HS-C11-EG4-OH (11-Mercaptoundecyl)tetra(ethylene glycol), Sigma-Aldrich). A total of 100mg of EMCH (N-[e-Maleimidocaproic acid]hydrazide, Fisher Scientific, PI-22106) was first dissolved in anhydrous DMSO(Dimethyl sulfoxide) to make 50Mm stock. 5 mg/ml Heparin (Alfa Aesar, A16198) solution in 100mM 2-(N-morpholino)ethanesulfonic acid (MES) pH6 buffer was then mixed with EMCH, NHS (N-Hydroxysuccinimide, Sigma-Aldrich) and EDC (1-(3-Dimethylaminopropyl)-3-ethylcarbodiimide hydrochloride, Fisher Scientific) sequentially. Mole ratio of COOH in heparin/EMCH/NHS/EDC=1:1:1:10. The reaction continued for 6 hours with gentle shaking followed by dialysis. The dialyzed samples were then lyophilized and sent for NMR for modification verification. The modified heparin was then conjugated to SAMs (99% EG-OH, 1% EG-NH2) formed on gold slides via EDC/NHS method as previously stated. Either Fn9*10 or Fn9(4G)10 (1000ng/slide, 53.3ng/cm2) was incubated with the modified heparin-coated surfaces overnight at 4 °C, followed by three PBS washes.
ELISA on gold surface
Modified gold surfaces was Argon-dried and then assembled together with PDMS sheet that has two 8mm circular wells followed by 60ul/well 0.1%BSA-PBS as blocking buffer for 1 hour at room temperature. After aspiration, 60ul/well of Anti-Fibronectin primary antibody (1:2000 dilution in blocking buffer, ab299, Abcam) was added for 2 hours at room temperature. After 3 washes using 0.05% Tween-20+PBS (washing buffer), 60ul/well of streptavidin-HRP (1:5000 dilution in blocking buffer, #DY998, R&D Systems) was added for 1hour at room temperature. After 3 washes, 60ul/well of TMB substrate (#7004L, Cell signaling) was added, incubated for 8 min in dark and then transferred to 96-well plates containing 1M H2SO4. The absorbance was measured at 450nm and normalized against absorbance at 550nm.
Primary cells
HUVEC cells (LONZA, CC-2519); HDF cells (Thermo Fisher Scientific, C0135C). Both cell types are primary and not cell lines, which were tested negative for mycoplasma, bacteria, yeast, fungi, HIV-1, hepatitis B and hepatitis C.
Cell Proliferation Assay
Slides immobilized with Fn9*10 or Fn9(4G)10 were washed twice with sterile PBS and then blew dry in cell culture hood for well assembly with 8-wells ibidi sticky-bottom device (ibidi, #80828). A total of 5000 HUVECs in EGM-2 stripped off fibronectin were seeded in each well with 2ng/ml of VEGF and the cell number was assayed after 48 hours using Cyquant assay. This experiment was performed 4 times on different days. Each time, at least 8 wells were seeded and analyzed for each condition. The plot generated is from averages of the biological (different days) replicates. The analysis was performed blindly by a different observer from whom that set up the experiments.
Cell Migration Assay
Slides immobilized with Fn9*10 or Fn9(4G)10 were washed twice with sterile PBS and then blew dry in cell culture hood for well assembly with PDMS sheets containing two 8mm holes. 5000 HUVECs pre-stained with SP-DilC18(3) lipophilic red fluorescence dye (Life Technologies) were seeded in each PDMS well on surface in EGM-2 w/o Fibronectin/VEGF medium and allowed for cell attachment for 3 hours in cell incubator. The slides were then transferred into incubation system of Zeiss LSM 780 confocal for 10× phase time-lapse tracking. In this experiment, single cell tracking was performed and each individual cell was considered a biological replicate. Within the two PDMS wells, two 10× regions (at least 2mm apart) in each well were monitored via time lapse microscopy to have total of 4-time series of 10× images per condition. Thus, images were taken at 4 different locations over time span of 7 hours with 15 min interval. These 10× images time series were then divided into 16 equally sized square sub-regions. For each sub-region time-laps image series, one cell was blindly circled out and its movement was detected by automated Mathlab program. The graphs were directly outputted by the Mathlab program and was composed of 64 individual cells path per condition. n=64 in each condition was used for quantification purpose.
2D Immunofluorescence Staining on Fragments Coated Cell Culture Plates
Glass-bottom 24-well plates were first incubated with 500μl of 2 μM Fn9*10 or Fn9(4G)10 per well overnight. Wells were incubated with 5% BSA-PBS for 1 hour at room temperature before the seeding of HUVECs which were pre-starved in EBM-2 media for 6 hours. A total of 50000 HUVECs in EGM-2 media stripped off fibronectin were seeded in each well without VEGF and fixed after 18 hrs. Cell samples were first fixed in 1% PFA for 20 min, washed twice with PBS for 5 min. Samples were incubated at room temperature for 30min in blocking buffer: PBS+ 5% Normal Goat Serum. Primary antibodies were prepared as follows in blocking buffer: Mouse anti-αvβ3 (clone LM609, Millipore; MAB1976) – 1:100, Mouse anti-α5β1(clone HA5, Millipore; MAB1999) – 1:100. Samples were incubated with primary antibodies overnight at 4°C, followed by Secondary antibodies (1:500), rhodamine phalloidin (1:500) and 2μg/ml DAPI for 1 hour in the dark at room temperature. Imaging was performed using a Nikon C2 confocal.
2D Immunofluorescence Staining on Fragments immobilized Gold Surfaces
Slides immobilized with Fn9*10 or Fn9(4G)10 were washed twice with sterile PBS and then blew dry in cell culture hood for well assembly with 12-well customized white Teflon wells. A total of 5000 HUVECs in EGM-2 stripped off fibronectin were seeded in each well with or without 2ng/ml of VEGF and fixed after 24 hrs. Cell samples were first fixed in 4% PFA for 15 min, washed twice with PBS for 5 min each before incubating with PBS+0.1% Triton for 3 min. After washing the samples again with PBS, samples were incubated at room temperature for 30min in blocking buffer: PBS+ 2% Normal Goat Serum. Primary antibodies were prepared as follows in blocking buffer: Rabbit anti-mouse and human VEGFR-2 (Cell Signaling Technology; #2479L) – 1:200, Mouse anti-human PECAM-1 (R&D; #BBA7) – 1:200, Monoclonal mouse anti-Vinculin antibody (Sigma-Aldrich, #V9131) – 1:400, Mouse anti-αvβ3 antibody (EMD Millipore, MAB1976) – 1:200. Samples were incubated with primary antibodies overnight at 4°C, followed by Secondary antibodies (1:500), Alexa Fluor 488 phalloidin (1:500) and 2μg/ml DAPI for 1 hour in the dark at room temperature. Imaging was performed using a Zeiss confocal and images were analyzed using Image J.
Sprouting Assay with blank, full length Fibronectin, Fn9*10 or Fn9(4G)10 fibrin gels
The sprouting fibrin bead assay was performed as previously described by others57 with some modifications. HUVEC coated beads were resuspended at a concentration of 500 beads/ml in 2 mg/ml fibrinogen (Fib1 or Fib3), 1 U/ml factor XIII and 0.04 U/ml aprotinin at a pH of 7.4 with or without 2 μM Fn9*10, 2 μM Fn9(4G)10, or 0.54 μM full length Fibronectin (Millipore, FC010). A total of 250 ul of this fibrinogen/bead solution was added to 0.16 units of thrombin in one well of glass-bottom 24-well plates. Fibrinogen/bead solution was allowed to clot for 5 min at room temperature and then at 37°C and 5% CO2 for 20 min. EGM-2 w/o fibronectin was added to each well and equilibrated with the fibrin clot for 30 min at 37°C and 5% CO2. Medium was removed from the well and replaced with 1 ml of fresh EGM-2 w/o Fibronectin. A total of 20,000 HDFs were plated on top of the clot and the medium was changed every other day. Bead assays were monitored for 7 days and then fixed in 4% PFA for 20 min. Samples were washed three times in PBS for 5 min each, blocked for 2 hours at room temperature in a blocking buffer of PBS+ 0.05% Tween-20 + 5% Normal Goat Serum. Samples were then stained with Alexa Fluor 488 phalloidin or rhodamine phalloidin (1:500) and 2μg/ml DAPI for 1 hour in the dark at room temperature. Imaging was performed using a Nikon C2 confocal and images were analyzed using Image J. At least three independent gels were evaluated per condition per sprouting experiment. Quantifications of at least 5 beads from each gel were analyzed (only sprouts with a length of one bead diameter were included). Since beads contain a small number of HUVECs that can vary widely in sprouting behavior, and each bead was surrounded by its own microenvironment, we consider each bead as an independent sprouting sample. Additionally, sprouting data were analyzed across experiments performed in different days taking each day as one biological replicate (in supplementary information). The trends from both types of analyses are the same.
Sprouting Assay with RGD Presence
Sprouting assay was performed as previously described in Fib3 fibrin gels with 200, 500 or 1000 μM of α2PI1-8-RGD (H-NQEQVSPLRGDSPG-NH2, GenScript).
Anastomosis Sprouting Assay with Fn9*10 and Fn9(4G)10
The anastomosis assay was performed similarly to the sprouting assay with minor modifications. Briefly, EGFP-HUVEC were used to coat the dextran-coated Cytodex 3 microcarriers. EGFP-HUVEC coated beads (500 beads/ml) were resuspeded in 2 mg/ml Fib3 fibrinogen, 1 U/ml factor XIII and 0.04 U/ml aprotinin, 80,000 cells/ml HDF at a pH of 7.4 with 2 μM of Fn9*10 or Fn9(4G)10. This fibrinogen/bead solution (250 μl) was added to 0.16 units of thrombin in one well of glass-bottom 24-well plates. Fibrinogen/HUVEC bead/HDF cells solution was allowed to clot for 5 min at room temperature and then at 37°C and 5% CO2 for 20 min. EGM-2 w/o Fibronectin was added to each well and equilibrated with the fibrin clot for 30 min at 37°C and 5% CO2. Medium was removed from the well and replaced with 1 ml of fresh EGM-2 w/o Fibronectin and later was changed every other day. Bead assays were monitored for 11 days. Three independent wells were evaluated per condition. Imaging was performed using a Nikon C2 confocal. Tip-tip contact distribution from each condition were analyzed within the 170μm of working distance of 60× objective.
Flow Cytometry Analysis of to test the Efficiency of αv Blocking Functional Antibody P3G8
HUVEC cells were harvested from 25 cm2 cell culture flask using 1ml of 2mg/ml Collagenase (Thermo Fisher Scientific, ICN1951091) for incubation of 30min at 37°C. The reaction was later quenched with 1mM EDTA in PBS for 5min before centrifugation at 200g for 5min. 60,000 cells per well were then added to 96-well round-bottom plate (Corning, 3365) which already contained 100 μl of serial dilution of P3G8 (Developmental Studies Hybridoma Bank) starting at concentration of 1μg/ml. The plate was then incubated in cell incubator for 30min before centrifuged at 1200rpm for 2min. 200 μl of FACS buffer (2% FBS in PBS) per well was then added to resuspend the cells pellet. This washing step was repeated twice before Goat-anti-Mouse AF488 was diluted in 1:4000 and added into the wells. The secondary antibody incubation was done in dark outside of incubator for 20min. Cells were then centrifuged and washed 3 times like described above. The final cell pellets were resuspended in FACS buffer. Analysis was performed using a MACS Quant VYB and the data was analyzed using FLOWJO. Triplicates were done for each condition with at least 9700 events/sample. The data was gated such that the negative control had 0.5% positive events (Fig. S10).
Integrin Blocking Sprouting Assay
Sprouting assays were performed as previously described. For Fn9*10 gels, HUVEC beads were suspended at a concentration of 500 beads/ml in 2 mg/ml fibrinogen (Fib1 or Fib3), 1 U/ml factor XIII, 0.04 U/ml aprotinin and 2 μM (high dosage) or 0.267 μM (low dosage) Fn9*10 at a pH of 7.4 with or without 5μg/ml of β1 integrin blocking antibody (AIIB2, Developmental Studies Hybridoma Bank) or α5 integrin blocking antibody (BIIG2, Developmental Studies Hybridoma Bank). The blocking antibody (5μg/ml) in fresh fibronectin-free EGM-2 medium was replenished every day.
For Fn9(4G)10 gels, HUVEC beads were suspended at a concentration of 500 beads/ml in 2 mg/ml fibrinogen (Fib1 or Fib3), 1 U/ml factor XIII, 0.04 U/ml aprotinin and 2 μM (high dosage) or 0.239 μM (low dosage) Fn9(4G)10 at a pH of 7.4 with or without 5μg/ml of β3 integrin blocking antibody (9H5, Developmental Studies Hybridoma Bank) or αv integrin blocking antibody (P3G8, Developmental Studies Hybridoma Bank). The blocking antibody (5μg/ml) in fresh fibronectin-free EGM-2 medium was replenished every day.
VE-cadherin Staining on 3D Integrin-Blocking Assay
Gel samples were first fixed in 1% PFA for 15 min, blocked for 2 hours at room temperature in a blocking buffer of PBS+ 0.05% Tween-20 + 5% Normal Goat Serum. Samples were then incubated in a primary antibody directed against VE Cadherin (Rabbit, Abcam; ab33168, 1:200) overnight at 4°C, followed by a secondary antibody (1:500), rhodamine phalloidin (1:500) and 2μg/ml DAPI for 1 hour. Imaging was performed using a Nikon C2 confocal and images were analyzed using Image J.
VE-cadherin Staining on 2D Fragments-Coated Surfaces
A total of 500ul of 2 μM Fn9*10 or Fn9(4G)10 in PBS buffer was added into sterile 24 well glass-bottom plates (MatTek Corporation) for 1 hour in cell incubator, followed by 0.1% heat-deactivated sterile BSA-PBS buffer for 1 hour. HUVECs cells pre-starved for 6 hours in EBM-2 were collected, resupspended in EGM-2 medium w/o Fibronectin either with or without VEGF and seeded into fragments and BSA treated wells. Cell density of 50,000 cells per well in 24-well glass-bottom plates was used. After 12 hours, cells were fixed in 1% PFA for 15 min and stained for VE-cadherin. Cell samples were first fixed in 1% PFA for 15 min, washed three times with 1XPBS for 5 min before blocked for 2 hrs at room temperature in blocking buffer: 1XPBS+ 0.1%BSA + 0.1% Tween-20 + 0.3M Glycine+ 10% Normal Goat Serum. Samples were incubated in a primary antibody Rabbit anti-VE Cadherin (Abcam; ab3316, 1:500) overnight at 4°C, followed by a secondary antibodies Donkey anti-Rabbit (1:500), rhodamine phalloidin (1:500) and 2μg/ml DAPI for 1 hour. Imaging was performed using a Nikon C2 confocal and images were analyzed using Image J. At least three independent wells were evaluated per condition on a same-day experiment. The experiment was repeated three times on different days. Similar trend was observed. The data reported in the main text figures represent one of these experiments. Images were taken per well by a blinded observer and analyzed blindly by a different person from that who took the images. Grey value distributions were analyzed using Image J.
Hyaluronic Acid-Acrylate Synthesis
Sodium hyaluronan was modified to contain acrylate functionalities as previously described58.
Vascular Endothelial Growth Factor Nanocapsules Synthesis
Plasmin-degradable nanocapsules of VEGF (nV) wer formed through in situ radical polymerization of acrylate and acrylamide containing monomers and peptide crosslinkers around a protein core as previously described48 (Fig. S7e). Capsules with four different compositions (100:0, 75:25, 50:50, 25:75 L peptide:D peptide) were mixed together. The L to D peptide compositions were, to render the capsules degradable at different rates.
HA Hydrogel Storage Modulus Optimization
HA hydrogel was formed in 0.3M pH 8.2 HEPES buffer, following steps as below.
Tube 1: HA-ADH-Ac in HEPES buffer (ADH modification is 65.62% and Ac modification is 13.33%) was incubated with fibronectin fragments of for 20 min. Tube 2: Poly(ethylene glycol) dithiol (MW 1000, Sigma-Aldrich, #717142) and Alexa Fluor 555 C2 Maleimide (Thermo Fisher Scientific, #A-20346) solutions in HEPES buffer were mixed together at equal moles for 20 min to generate fresh SH-PEG-AF555. Tube 1 was then mixed with Tube 2 mixture for 20 min before nanocapsules of VEGF was added. Di-cysteine modified Matrix Metalloprotease (MMP) (Ac-GCRDGPQGIWGQDRCG-NH2) (GenScript) sensitive crosslinker was added in the end to initiate gelation. Gelation was allowed for 30 min at 37°C. To determine the storage modulus range, gels with different thiol to acrylate (R ratio) were tested. Pre-swelled HA hydrogels (8mm in diameter and 1mm thickness) were placed between 8mm (diameter) rheological discs at normal force of 0.01N using a plate-to-plate rheometer (Anton paar physica mcr 301 Rheometer). The storage modulus was measured under constant 1% amplitude, from 10 to 0.1 rad/s angular frequency and was optimized to be 350Pa. Three independent gels were casted and evaluated per R ratio condition (Fig. S11a).
HA Gel Formula for SubQ Mice Model
HA hydrogel was synthesized as described above. Briefly, HA-ADH-Ac is dissolved into 0.08mg/ml solution in 0.3 M HEPES buffer (pH8.2). The solution is then incubated with 10μM Fn9*10 or Fn9(4G)10 (this amount of fibronectin fragment has been shown to sufficiently promote cell spreading in HA hydrogel in vitro, Fig. S11b) for 20 min. SH-PEG-AF555, nanoVEGF, and MMP crosslinker are added sequentially. R ratio of 0.60 was used for animal experiment. The final composition of the hydrogel is 3.5% HA, MMP crosslinker 3.335 mM, SH-PEG-AF555 10 μM, Fn fragments 10μM, VEGF 200ng/50μL gel, G′ = 350Pa.
SubQ Mice Model
G*Power was first used to compute the required sample size. Using two-tailed t-test on previous SubQ data and high variance assumption for the tortuosity measurement, we found the required sample size to be within the range of 3 to 6. We choose to start with 6 samples (2 gel samples on each animal) per group.
All in vivo studies were conducted in compliance with the NIH Guide for Care and Use of Laboratory Animals and UCLA ARC standards. Gel implantation was performed as previously described59. At day 7, the clips closing the incision were taken off and after 2 weeks, each mouse was injected with 100ul of 1mg/ml of isolectin GS-IB4-AF488 conjugate (ThermoFisher Scientific, #I21411) through the left external jugular vein before and sacrificed by isoflurane overdose. The implant hydrogels (total of 6 blank gels, 6 Fn9*10 gels, 6 Fn9(4G)10 gels) were then collected and fixed in 1% PFA for 16 hours at 4°C. Due to the variance in sample collection process, the membrane layers attached to the implants were intact only for 4 gel implant per condition. Thus, for confocal imaging, only those with intact membranes were analyzed.
SubQ Mice Model Quantification
Samples were first imaged using a Nikon C2 confocal to visualize the superficial vascular network on the surface of the sample. For space filling analysis, confocal images were converted to binary images and analyzed using the Matlab software package. Images were divided into equally sized regions using window sizes of 256 pixels in length for 4×4 heat map images. The fraction of non-white pixels in each region was calculated and utilized to generate an associated heat map indicating the degree of vascular signaling present in that region. Color map scale bars were set in grayscale.
Light sheet microscopy was then used to image the vascular infiltration in the implanted gel. Briefly, fixed hydrogel samples were inserted into a transparent 6mm tube. The tubes were then filled with 0.3% agarose solution in PBS. After the agarose gel solidified, samples were fixed in position and sheet confocal images were taken at 4× magnification for whole-mount samples (3–5μm step size, 6000 images total). After 3D rendering, 100μm-thick samples were sliced out to merge into single-plane maximum intensity projection image (none-overlapping samples, 4 to 6 different slices from each gel sample, 6 individual implant gels per condition).
Ischemic Stroke Model
G*Power was first used to compute the required sample size. Using two-tailed t-test on previous stroke data and high variance assumption for the vessel density measurement, we found the required sample size to be within the range of 3 to 9. We used 7 or 8 animals per group.
Animal procedures were performed in accordance with the US National Institutes of Health Animal Protection Guidelines and the University of California Los Angeles Chancellor’s Animal Research Committee. Focal and permanent cortical stroke was induced by a middle cerebral artery occlusion (MCAo) on young adult C57BL/6 male mice (8–12 weeks) obtained from Jackson Laboratories. The male mice were chosen because females of this age have a complete and cyclic menstruations. The increased systemic inflammation that is associated with their cycle could interfere with the results of the experiments. Briefly, under isoflurane anesthesia (2–2.5% in a 70% N2O/30% O2 mixture), a small craniotomy was performed over the left parietal cortex. One anterior branch of the distal middle cerebral artery was then exposed, electrocoagulated and cut. Body temperature was maintained at 36.9 ± 0.4 °C with a heating pad throughout the operation. In this model, ischemic cellular damage is localized to somatosensory and motor cortex60.
Brain Hydrogel Transplantation
No randomization was used in this experiment. In order to avoid any confusion, all the animals from a cage were injected with the same treatment. Each cage represented one condition, with 2 cages per condition since the n is superior to 4, the max number of animal allowed per cage. Animals were labeled with their condition and a number attributed in the order of surgery. Five days later, HA hydrogel precursor (see Table for composition) was loaded into a 25 μl Hamilton syringe (Hamilton, Reno, NV) connected to a syringe pump. The solution was then injected in liquid form directly into the stroke cavity using a 30-gauge needle at stereotaxic coordinates 0.26 mm anterior/posterior (AP), 3 mm medial/lateral (ML), and 1 mm dorsal/ventral (DV) with an infusion speed of 1 μl/min. The needle was withdrawn from the mouse brain immediately after the injection was complete. The final composition of the hydrogel is 3.5% HA, MMP crosslinker 3.335 mM, Fn fragments 10μM, VEGF 200ng/6μL gel, G′ = 350Pa.
Ten days following the hydrogel transplantation, animals injected with fibronectin fragment (Vs+ Fn9*10, nV+Fn9*10 and nV+Fn9(4G)10) containing hydrogels were perfused with DyLight 594 labeled Lycopersicon Esculentum (Tomato) Lectin (Vector Laboratories, # DL-1177) through the left through external jugular vein and then sacrificed by isoflurane overdose. Other mice conditions (No gel, HA-RGD and Vs+HA-RGD) were perfused with 4% PFA and sacrificed.
Brain tissue processing
Mice brains were harvested and post-fixed in 4% PFA overnight or perfused with PFA before harvesting, then cryoprotected in 30% sucrose in phosphate buffer for 24 hours and frozen. Tangential cortical sections of 30 μm-thick were sliced using a cryostat and directly mounted on gelatin-subbed glass slides. Brain sections were then washed in PBS and permeabilized and blocked in 0.3% Triton and 10% Normal Donkey Serum before being immunohistochemically stained. The primary antibody Rat anti-Ter-119 (R&D Systems, #MAB1125, 1:200), Rabbit anti-Glut-1 (Glucose Transporter1, Abcam, 1:400) or Rabbit anti-Fibronectin (Millipore, AB2040, 1:200) were incubated overnight at +4°C followed by secondary antibodies Donkey anti-rat and rabbit- AF488 (Thermo Fisher Scientific, 1:200) for 1 hour at room temperature. After 3× 10 minute washes in PBS, the slides were dehydrated in ascending ethanol baths, dewaxed in xylene and coverslipped over fluorescent mounting medium (Dako). Once brain sections were stained, a different experimenter attributed a random number to each animal to keep the main experimenter blind. The images were blindly taken and analyzes were performed without identifying the different samples to the different conditions.
Microscopy and Morphoanalysis
Analyses were performed on microscope images of 3 coronal brain levels at +0.80 mm, −0.80 mm and −1.20 mm according to bregma, which consistently contained the cortical infarct area. Each image represents a maximum intensity projection of 10 to 12 Z-stacks, 0.85μm apart, captured at a 20× magnification with a Nikon C2 confocal microscope using the NIS Element software. Each n represents one mouse.
To quantify the vascular bed in the no gel and gel conditions, Glut-1 stained only or Gut-1 plus tomato lectin in tomato lectin-perfused animals were quantified. Tomato lectin alters Glut-1 staining such that in lectin-perfused animals the combination of both stains reveals the vascular bed the same as Glut-1 alone in tomato lectin-unperfused animals (Fig. S9b).
The vascular area
The vascular area (stained by Glut-1 only or by both tomato lectin and Glut-1) in the infarct and peri-infarct areas was quantified in 8 randomly chosen regions of interest (ROI) of 0.3 mm2 in both regions. In each ROI, the positive area was measured using pixel threshold on 8-bit converted images (ImageJ v1.43, Bethesda, Maryland, USA) and expressed as the area fraction of positive signal per ROI. Values were then averaged across all ROI and sections, and expressed as the average positive area per animal.
The evaluation of perfused vascular ramifications
The evaluation of perfused vascular ramifications allows for a quantitative analysis of the vessel architecture, by counting manually the number of branching points on positively tomato lectin perfused vessels of the peri-infarct per mm2.
Statistical Analysis
Statistical analyses were performed using Prism (GraphPad, San Diego, CA). Brown-Forsythe test was performed to test the variance similarity between the groups that are being statistically compared. The variance is similar between the groups that are being statistically compared. Data were analyzed using a one-way analysis of variance (ANOVA) followed by a Tukey post-hoc test and a 95% confidence interval.
For 2D VE-cadherin staining analysis (n=3) and stroke model quantification (minimum n=5), two-tailed unpaired test was used when only two groups were compared. The results are expressed as mean ± SD. Single, double, triple and quadruple asterisks represent p < 0.05, p < 0.01, p < 0.001 and p < 0.0001, respectively. A p value < 0.05 was considered statistically significant.
Supplementary Material
Acknowledgments
The authors would like to thank Prof. Yvonne Chen and Ximin Chen for their help with flow cytometry. The authors would like to acknowledge Developmental Studies Hybridoma Bank (DSHB) for providing antibodies P3G8, AIIB2, BIIG2 and 9H5. This work was supported by National Institutes of Health R01NS079691(TS).
Footnotes
Author contributions
SL contributed with conceptual design, experimental execution, troubleshooting. LRN contributed with conceptual design, experimental execution, and troubleshooting for experiments involving the MCAo stroke model (Fig. 5). HB contributed with conceptual design, troubleshooting and production of the Fn9*10 and Fn9(4G)10 fibronectin fragments. PF contributed with conceptual design and troubleshooting of sheet confocal imaging for the modified matrigel plug assay. YL contributed with the conceptual design and experimental execution of the Matlab code used for cell migration analysis in Fig. S1e,f. EN contributed with experimental execution and troubleshooting for experiments involving VE-Cadherin quantification. RD contributed with the conceptual design and experimental execution of the Matlab code used for space filling analysis in Fig. 4d. STC contributed with the conceptual design, data interpretation for Fig, 5. THB contributed with conceptual design and troubleshooting of the Fn9*10 and Fn9(4G)10 fibronectin fragments. TS contributed with conceptual design, oversaw all experimental design and interpretation. While SL and TS wrote the first draft of the manuscript all authors read and gave comments, especially with regards of their experimental section and analysis.
Competing Financial Interests
Nothing to disclose
Data availability statement
The data generated during and/or analyzed during the current study are available from the corresponding author on reasonable request.
References
- 1.Martino MM, et al. Engineering the growth factor microenvironment with fibronectin domains to promote wound and bone tissue healing. Sci Transl Med. 2011;3:100ra189. doi: 10.1126/scitranslmed.3002614. [DOI] [PubMed] [Google Scholar]
- 2.Briquez PS, Clegg LE, Martino MM, Gabhann FM, Hubbell JA. Design principles for therapeutic angiogenic materials. 2016;1:15006. [Google Scholar]
- 3.Hynes RO. Integrins: bidirectional allosteric signaling machines. Cell. 2002;110:673–687. doi: 10.1016/s0092-8674(02)00971-6. [DOI] [PubMed] [Google Scholar]
- 4.Hynes RO. The Extracellular Matrix: Not Just Pretty Fibrils. Science. 2009;326:1216–1219. doi: 10.1126/science.1176009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Giancotti FG, Ruoslahti E. Integrin signalingScience. 1999;285:1028–1032. doi: 10.1126/science.285.5430.1028. [DOI] [PubMed] [Google Scholar]
- 6.Zovein AC, et al. beta 1 Integrin Establishes Endothelial Cell Polarity and Arteriolar Lumen Formation via a Par3-Dependent Mechanism. Dev Cell. 2010;18:39–51. doi: 10.1016/j.devcel.2009.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rupp PA, Little CD. Integrins in vascular development. Circ Res. 2001;89:566–572. doi: 10.1161/hh1901.097747. [DOI] [PubMed] [Google Scholar]
- 8.Bayless KJ, Salazar R, Davis GE. RGD-dependent vacuolation and lumen formation observed during endothelial cell morphogenesis in three-dimensional fibrin matrices involves the alpha(v)beta(3) and alpha(5)beta(1) integrins. Am J Pathol. 2000;156:1673–1683. doi: 10.1016/s0002-9440(10)65038-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yamamoto H, et al. Integrin beta1 controls VE-cadherin localization and blood vessel stability. Nat Commun. 2015;6:6429. doi: 10.1038/ncomms7429. [DOI] [PubMed] [Google Scholar]
- 10.Hodivala-Dilke KM, et al. Beta3-integrin-deficient mice are a model for Glanzmann thrombasthenia showing placental defects and reduced survival. J Clin Invest. 1999;103:229–238. doi: 10.1172/JCI5487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Abraham S, Kogata N, Fassler R, Adams RH. Integrin beta1 subunit controls mural cell adhesion, spreading, and blood vessel wall stability. Circ Res. 2008;102:562–570. doi: 10.1161/CIRCRESAHA.107.167908. [DOI] [PubMed] [Google Scholar]
- 12.Milner R, Campbell IL. Developmental regulation of beta1 integrins during angiogenesis in the central nervous system. Mol Cell Neurosci. 2002;20:616–626. doi: 10.1006/mcne.2002.1151. [DOI] [PubMed] [Google Scholar]
- 13.Brooks PC, Clark RA, Cheresh DA. Requirement of vascular integrin alpha v beta 3 for angiogenesis. Science. 1994;264:569–571. doi: 10.1126/science.7512751. [DOI] [PubMed] [Google Scholar]
- 14.Tonnesen MG, Feng X, Clark RA. Angiogenesis in wound healing. J Investig Dermatol Symp Proc. 2000;5:40–46. doi: 10.1046/j.1087-0024.2000.00014.x. [DOI] [PubMed] [Google Scholar]
- 15.Friedlander M, et al. Definition of two angiogenic pathways by distinct alpha v integrins. Science. 1995;270:1500–1502. doi: 10.1126/science.270.5241.1500. [DOI] [PubMed] [Google Scholar]
- 16.Carroll JM, Romero MR, Watt FM. Suprabasal integrin expression in the epidermis of transgenic mice results in developmental defects and a phenotype resembling psoriasis. Cell. 1995;83:957–968. doi: 10.1016/0092-8674(95)90211-2. [DOI] [PubMed] [Google Scholar]
- 17.Reynolds LE, et al. Enhanced pathological angiogenesis in mice lacking beta3 integrin or beta3 and beta5 integrins. Nat Med. 2002;8:27–34. doi: 10.1038/nm0102-27. [DOI] [PubMed] [Google Scholar]
- 18.Alghisi GC, Ponsonnet L, Ruegg C. The integrin antagonist cilengitide activates alphaVbeta3, disrupts VE-cadherin localization at cell junctions and enhances permeability in endothelial cells. PloS one. 2009;4:e4449. doi: 10.1371/journal.pone.0004449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Weis SM, et al. Cooperation between VEGF and beta3 integrin during cardiac vascular development. Blood. 2007;109:1962–1970. doi: 10.1182/blood-2005-10-038893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mhanna R, et al. GFOGER-modified MMP-sensitive polyethylene glycol hydrogels induce chondrogenic differentiation of human mesenchymal stem cells. Tissue Eng Part A. 2014;20:1165–1174. doi: 10.1089/ten.tea.2013.0519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Shekaran A, et al. Bone regeneration using an alpha 2 beta 1 integrin-specific hydrogel as a BMP-2 delivery vehicle. Biomaterials. 2014;35:5453–5461. doi: 10.1016/j.biomaterials.2014.03.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lee ST, et al. Engineering integrin signaling for promoting embryonic stem cell self-renewal in a precisely defined niche. Biomaterials. 2010;31:1219–1226. doi: 10.1016/j.biomaterials.2009.10.054. [DOI] [PubMed] [Google Scholar]
- 23.Krammer A, Craig D, Thomas WE, Schulten K, Vogel V. A structural model for force regulated integrin binding to fibronectin’s RGD-synergy site. Matrix Biol. 2002;21:139–147. doi: 10.1016/s0945-053x(01)00197-4. [DOI] [PubMed] [Google Scholar]
- 24.Grant RP, Spitzfaden C, Altroff H, Campbell ID, Mardon HJ. Structural requirements for biological activity of the ninth and tenth FIII domains of human fibronectin. J Biol Chem. 1997;272:6159–6166. doi: 10.1074/jbc.272.10.6159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Martino MM, et al. Controlling integrin specificity and stem cell differentiation in 2D and 3D environments through regulation of fibronectin domain stability. Biomaterials. 2009;30:1089–1097. doi: 10.1016/j.biomaterials.2008.10.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Brown AC, Rowe JA, Barker TH. Guiding epithelial cell phenotypes with engineered integrin-specific recombinant fibronectin fragments. Tissue Eng Part A. 2011;17:139–150. doi: 10.1089/ten.tea.2010.0199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Altroff H, et al. Interdomain tilt angle determines integrin-dependent function of the ninth and tenth FIII domains of human fibronectin. J Biol Chem. 2004;279:55995–56003. doi: 10.1074/jbc.M406976200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Brown AC, Dysart MM, Clarke KC, Stabenfeldt SE, Barker TH. Integrin alpha3beta1 Binding to Fibronectin Is Dependent on the Ninth Type III Repeat. J Biol Chem. 2015;290:25534–25547. doi: 10.1074/jbc.M115.656702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Markowski MC, Brown AC, Barker TH. Directing epithelial to mesenchymal transition through engineered microenvironments displaying orthogonal adhesive and mechanical cues. J Biomed Mater Res A. 2012;100:2119–2127. doi: 10.1002/jbm.a.34068. [DOI] [PubMed] [Google Scholar]
- 30.Schense JC, Hubbell JA. Cross-linking exogenous bifunctional peptides into fibrin gels with factor XIIIa. Bioconjug Chem. 1999;10:75–81. doi: 10.1021/bc9800769. [DOI] [PubMed] [Google Scholar]
- 31.Hoeben A, et al. Vascular endothelial growth factor and angiogenesis. Pharmacol Rev. 2004;56:549–580. doi: 10.1124/pr.56.4.3. [DOI] [PubMed] [Google Scholar]
- 32.Nakatsu MN, Davis J, Hughes CC. Optimized fibrin gel bead assay for the study of angiogenesis. J Vis Exp. 2007;186 doi: 10.3791/186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Carmeliet P, Jain RK. Angiogenesis in cancer and other diseases. Nature. 2000;407:249–257. doi: 10.1038/35025220. [DOI] [PubMed] [Google Scholar]
- 34.Santulli RJ, et al. Studies with an orally bioavailable alpha V integrin antagonist in animal models of ocular vasculopathy: retinal neovascularization in mice and retinal vascular permeability in diabetic rats. J Pharmacol Exp Ther. 2008;324:894–901. doi: 10.1124/jpet.107.131656. [DOI] [PubMed] [Google Scholar]
- 35.Cao L, et al. Detection of an Integrin-Binding Mechanoswitch within Fibronectin during Tissue Formation and Fibrosis. ACS Nano. 2017;11:7110–7117. doi: 10.1021/acsnano.7b02755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Aota S, Nomizu M, Yamada KM. The short amino acid sequence Pro-His-Ser-Arg-Asn in human fibronectin enhances cell-adhesive function. J Biol Chem. 1994;269:24756–24761. [PubMed] [Google Scholar]
- 37.Danen EH, et al. Requirement for the synergy site for cell adhesion to fibronectin depends on the activation state of integrin alpha 5 beta 1. J Biol Chem. 1995;270:21612–21618. doi: 10.1074/jbc.270.37.21612. [DOI] [PubMed] [Google Scholar]
- 38.Mogford JE, Davis GE, Platts SH, Meininger GA. Vascular smooth muscle alpha v beta 3 integrin mediates arteriolar vasodilation in response to RGD peptides. Circ Res. 1996;79:821–826. doi: 10.1161/01.res.79.4.821. [DOI] [PubMed] [Google Scholar]
- 39.Desgrosellier JS, Cheresh DA. Integrins in cancer: biological implications and therapeutic opportunities. Nat Rev Cancer. 2010;10:9–22. doi: 10.1038/nrc2748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Henderson NC, et al. Targeting of alphav integrin identifies a core molecular pathway that regulates fibrosis in several organs. Nat Med. 2013;19:1617–1624. doi: 10.1038/nm.3282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Liu Z, Wang F, Chen X. Integrin alpha(v)beta(3)-Targeted Cancer Therapy. Drug Dev Res. 2008;69:329–339. doi: 10.1002/ddr.20265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lam J, Segura T. The modulation of MSC integrin expression by RGD presentation. Biomaterials. 2013;34:3938–3947. doi: 10.1016/j.biomaterials.2013.01.091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Chen TT, et al. Anchorage of VEGF to the extracellular matrix conveys differential signaling responses to endothelial cells. J Cell Biol. 2010;188:595–609. doi: 10.1083/jcb.200906044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Adams RH, Alitalo K. Molecular regulation of angiogenesis and lymphangiogenesis. Nat Rev Mol Cell Bio. 2007;8:464–478. doi: 10.1038/nrm2183. [DOI] [PubMed] [Google Scholar]
- 45.Lenard A, et al. In vivo analysis reveals a highly stereotypic morphogenetic pathway of vascular anastomosis. Dev Cell. 2013;25:492–506. doi: 10.1016/j.devcel.2013.05.010. [DOI] [PubMed] [Google Scholar]
- 46.Wallez Y, Vilgrain I, Huber P. Angiogenesis: the VE-cadherin switch. Trends in cardiovascular medicine. 2006;16:55–59. doi: 10.1016/j.tcm.2005.11.008. [DOI] [PubMed] [Google Scholar]
- 47.Montero-Balaguer M, et al. Stable vascular connections and remodeling require full expression of VE-cadherin in zebrafish embryos. PloS one. 2009;4:e5772. doi: 10.1371/journal.pone.0005772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lei Y, Segura T. DNA delivery from matrix metalloproteinase degradable poly(ethylene glycol) hydrogels to mouse cloned mesenchymal stem cells. Biomaterials. 2009;30:254–265. doi: 10.1016/j.biomaterials.2008.09.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zhu S, Nih L, Carmichael ST, Lu Y, Segura T. Enzyme-Responsive Delivery of Multiple Proteins with Spatiotemporal Control. Adv Mater. 2015;27:3620–3625. doi: 10.1002/adma.201500417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Andres RH, et al. The CCR2/CCL2 Interaction Mediates the Transendothelial Recruitment of Intravascularly Delivered Neural Stem Cells to the Ischemic Brain. Stroke. 2011 doi: 10.1161/STROKEAHA.110.606368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Arai K, Jin G, Navaratna D, Lo EH. Brain angiogenesis in developmental and pathological processes: neurovascular injury and angiogenic recovery after stroke. Febs J. 2009;276:4644–4652. doi: 10.1111/j.1742-4658.2009.07176.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Giacca M, Zacchigna S. VEGF gene therapy: therapeutic angiogenesis in the clinic and beyond. Gene Ther. 2012;19:622–629. doi: 10.1038/gt.2012.17. [DOI] [PubMed] [Google Scholar]
- 53.Roberts WG, Palade GE. Increased microvascular permeability and endothelial fenestration induced by vascular endothelial growth factor. J Cell Sci. 1995;108(Pt 6):2369–2379. doi: 10.1242/jcs.108.6.2369. [DOI] [PubMed] [Google Scholar]
- 54.Esser S, et al. Vascular endothelial growth factor induces endothelial fenestrations in vitro. J Cell Biol. 1998;140:947–959. doi: 10.1083/jcb.140.4.947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lin HB, Zhao ZC, Garcia-Echeverria C, Rich DH, Cooper SL. Synthesis of a novel polyurethane co-polymer containing covalently attached RGD peptide. Journal of biomaterials science Polymer edition. 1992;3:217–227. doi: 10.1163/156856292x00132. [DOI] [PubMed] [Google Scholar]
- 56.Ghosh K, Ren XD, Shu XZ, Prestwich GD, Clark RA. Fibronectin functional domains coupled to hyaluronan stimulate adult human dermal fibroblast responses critical for wound healing. Tissue Eng. 2006;12:601–613. doi: 10.1089/ten.2006.12.601. [DOI] [PubMed] [Google Scholar]
- 57.Hou S, et al. The repair of brain lesion by implantation of hyaluronic acid hydrogels modified with laminin. J Neurosci Methods. 2005;148:60–70. doi: 10.1016/j.jneumeth.2005.04.016. [DOI] [PubMed] [Google Scholar]
- 58.Nakatsu MN, et al. Angiogenic sprouting and capillary lumen formation modeled by human umbilical vein endothelial cells (HUVEC) in fibrin gels: the role of fibroblasts and Angiopoietin-1. Microvasc Res. 2003;66:102–112. doi: 10.1016/s0026-2862(03)00045-1. [DOI] [PubMed] [Google Scholar]
- 59.Lei Y, Gojgini S, Lam J, Segura T. The spreading, migration and proliferation of mouse mesenchymal stem cells cultured inside hyaluronic acid hydrogels. Biomaterials. 2011;32:39–47. doi: 10.1016/j.biomaterials.2010.08.103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Cam C, Segura T. Chemical sintering generates uniform porous hyaluronic acid hydrogels. Acta biomaterialia. 2014;10:205–213. doi: 10.1016/j.actbio.2013.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Carmichael ST. Rodent models of focal stroke: size, mechanism, and purpose. NeuroRx. 2005;2:396–409. doi: 10.1602/neurorx.2.3.396. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.