

Abstract
We profiled three novel T. gondii inhibitors identified from an antimalarial phenotypic high throughput screen (HTS) campaign: styryl 4-oxo-1,3-benzoxazin-4-one KG3, tetrahydrobenzo[b]pyran KG7, and benzoquinone hydrazone KG8. These compounds inhibit T. gondii in vitro with IC50 values ranging from 0.3 to 2 μM, comparable to that of 0.25 to 1.5 μM for the control drug pyrimethamine. KG3 had no measurable cytotoxicity against five mammalian cell lines, whereas KG7 and KG8 inhibited the growth of 2 of 5 cell lines with KG8 being the least selective for T. gondii. None of the compounds were mutagenic in an Ames assay. Experimental gLogD7.4 and calculated PSA values for the three compounds were well within the ranges predicted to be favorable for good ADME, even though each compound had relatively low aqueous solubility. All three compounds were metabolically unstable, especially KG3 and KG7. Multiple IP doses of 5 mg/kg KG7 and KG8 increased survival in a T. gondii mouse model. Despite their liabilities, we suggest that these compounds are useful starting points for chemical prospecting, scaffold-hopping, and optimization.
Keywords: Toxoplasma gondii, drug discovery, lead compounds, anti-parasitics, Plasmodium falciparum
Graphical abstract
1. Introduction
The protozoan parasite Toxoplasma gondii (T. gondii) is the most common parasitic infection in the world with a 30-50% prevalence in the human population (1). The initial infection frequently occurs upon ingestion of contaminated food and water or exposure to environmentally persistent oocysts shed by infected members of the Felidae family (2-3). Upon the initial exposure to T. gondii, acute toxoplasmosis can occur subclinically (approximately 25%), or can be manifested as self-resolving flu-like symptoms (2, 4). The parasite will often persist in a slow-growing cyst stage of infection under continual host immune surveillance. When the host immune system is weakened, the latent infection (chronic toxoplasmosis) can re-activate and cause severe tissue damage such as toxoplasmic encephalitis (5). Special populations such as HIV and AIDS patients require life-long prophylactic treatment to control parasitic latent infections, but such treatment does not lead to clearance of the parasite.
Pyrimethamine remains the most effective treatment and is commonly used in combination with sulfonamides (6). Pyrimethamine administration can lead to dose-dependent bone marrow toxicity in the treated host, which may be partially controlled by co-administration of folinic acid supplements (7). In addition, patients can be allergic to the adjunctive medication sulfadiazine (8). While acute toxoplasmosis is treatable with these drugs, the latent infection remains recalcitrant to therapy of any length (9). For these reasons, there exists a need to identify better drugs for treatment of toxoplasmosis and ultimately clearance of infection (9-10).
In 2010 Guigemende et al. performed a cell-based high-throughput screen (HTS) to identify novel antimalarial chemotypes. This work identified several compounds that show promise against both Plasmodium falciparum (P. falciparum) and T. gondii. As these two apicomplexan parasites display close evolutionary relationships, they may share similar drug sensitivities (11). However, this screen suggested that T. gondii may be more chemoresistant: while 561 P. falciparum hits emerged from a chemical library of over 300,000 compounds, only 23 had measurable activity against T. gondii (12).
From this filtered group of compounds with activity against both parasites (12), three were selected for further characterization with T. gondii: styryl 4-oxo-1,3-benzoxazin-4-one SJ000033445 (KG3), tetrahydrobenzo[b]pyran SJ000144280 (KG7), and benzoquinone hydrazone SJ000296485 (KG8) (Figure 1). Each of these chemical structures appeared to be singleton scaffolds, making their characterization useful for further work in generating derivatives to optimize activity. In addition to verifying in vitro activity against multiple strains of T. gondii, the mammalian cell cytotoxicity profile was expanded, in vitro ADME properties were profiled, and mutagenicity was assessed. Lastly, the compounds were examined in vivo for their capacity to increase survivorship following an acute lethal challenge with T. gondii tachyzoites. Our evaluation of these compounds demonstrates statistically significant but incomplete in vivo survivorship following acute parasite infection, likely hampered by metabolic instability.
Figure 1. Chemical structures.
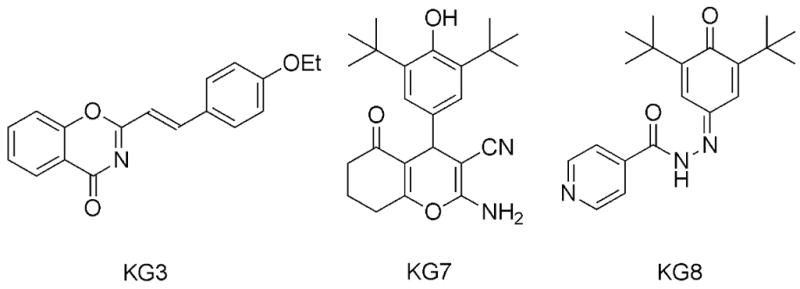
Styryl 4-oxo-1,3-benzoxazin-4-one (KG3), Tetrahydrobenzo[b]pyran (KG7), and Benzoquinone hydrazone (KG8).
2. Materials and Methods
Compounds
Compounds were obtained from ChemDiv (San Diego, California).
Cell Maintenance
Human foreskin fibroblasts (HFF) and murine macrophages were obtained from American Type Culture Collection (ATCC). All cell lines and parasite strains were maintained in D10 media which consisted of DMEM media (Lonza) supplemented with 10% heat inactivated Hyclone bovine serum (GE Healthcare Life Sciences), HyClone 2 mM L-glutamine (GE Healthcare Life Sciences), 100 μg/mL penicillin and streptomycin (Corning), 20% Medium 199 (Corning) and gentamicin sulfate (Corning) at 37°C with 5% CO2. Type I strain of T. gondii constitutively expressing red fluorescent dimerized Tomato (“RH-dTom”) and a type II strain, PRU expressing the same fluorophore (“PRU-dTom”) were used in assays.
Cell Toxicity Assay
Bone marrow derived murine macrophages were allowed to grow until confluent in 96 well plates. Once confluent, an increasing concentration of compound (0 to 100 μM) was added and incubated for 24 h. Alamar blue (10 mM) was then added to each well and incubated for 4 h. A BioTek Synergy HT plate reader was then used to determine fluorescence.
IC50 Assay
HFF cells were cultured in 96 well plates at 20,000 cells per well and allowed to grow until confluent. Then 2,000 tachyzoites were then added to each well and incubated for 12 h allowing for invasion of host cells. Media was then replaced and compounds were added at increasing concentration from 0 to 100 μM in duplicate. All compounds were dissolved in DMSO; the concentration of DMSO did not exceed 1% in all assays. A fluorescent reading was then taken with a BioTek Snergy HT plate reader at day 5 post-infection.
Host Cell and Extracellular Parasite Pre-treatment Assay
HFF cells were cultured in 96 well plates at 20,000 cells per well and allowed to grow until confluent. Once confluent, 10 μM of each compound was added to the wells. After 24 h, media was aspirated and cells were washed three times with D10 media. Cells were then infected with either 2,000 RH-dTom or PRU-dTom tachyzoites and fluorescently quantified 5 day post-infection. Assays were performed in triplicate. To evaluate extracellular parasite responses to compound exposure, RH-dTom tachyzoites were isolated from culture and resuspended at 1×106 tachyzoites/mL in D10 media. Tachyzoites were treated with 10 μM of compound and incubated at 37 °C for 4 h. After treatment, HFF cells were then infected with treated tachyzoites at 20,000 tachyzoites/mL and tachyzoite growth was fluorescently quantified 5 day post-infection.
Physicochemical Parameters and ADME Characteristics
pKa, PSA, and cLogP/cLogD values were assessed by the ChemAxon chemistry cartridge via JChem for Excel Software (version 16.4.11). To determine kinetic aq. solubility, compounds were dissolved into DMSO and then spiked into either pH 6.5 phosphate buffer or 0.01 M HCl (pH 2.0) with the final concentration being DMSO 1%. After 30 min at room temperature, samples were analyzed using nephelometry to determine a solubility range (13). Partition coefficient values were estimated at pH 7.4 by correlation of their chromatographic retention properties against those of standard compounds with known partition coefficient values by gradient HPLC (14). Chromatography-based protein binding values (cPPB) were determined by correlation of compound chromatographic retention properties using a human albumin column against those of a standard series of compounds with known protein binding values (15). To evaluate metabolic stability, compounds (1 μM) were incubated with liver microsomes from humans and mice at 37°C at a protein concentration of 0.4 mg/mL. Metabolic reactions were initiated by the addition of an NADPH-regenerating system. The reaction was quenched at varying time points (2, 30, and 60 min) and compound concentrations determined by LCMS.
Bacterial Reverse Mutation Assay
A modified form of the Ames assay (Environmental Bio-Detection Products Inc.) with Salmonella typhimurium (TA100 strain) was used in to specifically detect point mutagenicity. Compounds were tested at concentrations of 3x the averaged T. gondii IC50 values in sets of 48 replicates. A count of revertant colonies was performed and compared to the natural revertant control with the unpaired Students t-test to assess statistical significance (p-value <0.05).
In vivo T. gondii Studies
Swiss Webster CFW mice (Charles River, Wilmington, MA) were infected by IP injection of 20,000 T. gondii RH-dTom tachyzoites. At 24 h post-infection, test compounds dissolved in DMSO and then diluted with water to their respective concentration. All solutions were subsequently treated with hydrochloric acid or sodium hydroxide until dissolved, and the volume of DMSO administered was below the previously established toxic dose (17, 18). Compounds were administered in twice daily IP doses for 7 consecutive days (KG3, n=3, KG7 and KG8, n=2). Doses of all three compounds were selected empirically by determining the dose at which drug exposure caused murine toxicity (in all cases, murine hypoactivity) and dividing by 2. Pyrimethamine was included as a positive control (n=3) and untreated infected mice as the negative control (n=8). Mice were monitored for toxicity or illness throughout the study. Mouse survival was quantified through a Kaplan-Meier survival curve and a Student’s t-test was used to determine significance. All in vivo studies were institutionally approved and carried out under IACUC #12-062-10.
3. Results
In vitro Activity Against T. gondii
KG3 had an IC50 value of 0.27 μM against the RH-dTom (type I strain); KG7 and KG8, with respective IC50 values of 1.3 and 2.3 μM, were only slightly less potent (Table 1). Against the PRU-dTom (type II) strain of T. gondii, KG3 was still the most effective with an IC50 of 1.4 μM while KG7 and KG8 had respective IC50 values of 2.1 and 2.5 μM (Table 1), all with standard deviations less than 10%.
Table 1.
Toxoplasma gondii growth inhibition.
| Identifier | T. gondii RH-dTom IC50 (μM)a | T. gondii PRU-dTom IC50 (μM) |
|---|---|---|
| KG3 | 0.27 | 1.4 (s.d. = 0.04) |
| KG7 | 1.3 | 2.1 (s.d. = 0.17) |
| KG8 | 2.3 | 2.5 (s.d. = 0.12) |
| Pyrimethamine | 1.5 | 0.79 (s.d. = 0.07) |
Host Cell Cytotoxicity
Human foreskin fibroblast (HFF), kidney (HEK293), hepatocytes (HC04), B lymphocyte (RAJI), (13) and the murine-derived macrophage (NR-9456) cell lines were used to determine cytotoxic effects. At the concentrations tested, KG3 did not inhibit the growth of any of the five cell lines, whereas KG7 and KG8 inhibited the growth of 2/5 cell lines. Of the three compounds, KG8 was the most cytotoxic with a mean in vitro therapeutic index of 15 (Table 2).
Table 2.
Cytotoxicity.
| Identifier | HFF IC50a (μM) | HEK293 IC50a (μM) | HC04 IC50a (μM) | RAJI IC50a (μM) | NR-9456 IC50 (μM) | Mean Therapeutic Index (TI) | Ames Assay (TA100) |
|---|---|---|---|---|---|---|---|
| KG3 | >40 | >40 | >40 | >40 | >100 | >61 | Negative |
| KG7 | >36 | >36 | 3.0 | 3.0 | >100 | 23 | Negative |
| KG8 | >28 | 0.97 | >28 | 0.68 | >100 | 15 | Negative |
| Pyrimethamine | >100 | >100 | >100 | >100 | >100 | >114 | Not tested |
IC50 data for these cell lines was included from [Guigemende 2010], where the compounds were initially screened in human cell lines: human foreskin flibroblast (HFF), kidney (HEK293), hepatocytes (HC04), and B lymphocyte (RAJI). This host cytoxicity profile was expanded to include an additional model organism and cell type for a broader inquiry into cytotoxicity: murine-derived macrophage (NR-9456). In vitro therapeutic index (TI) was derived by dividing the mammalian cell IC50 by the averaged T. gondii IC50 (Table 1) and averaging these across all cell types, serving as a general marker of mammalian cell safety and frames the therapeutic window.
Pre-Exposure Assays
To further understand possible mode(s) of action, host cells were exposed to 10 μM of compounds prior to infection. Under these conditions, KG3 decreased T. gondii growth by 21% (Figure 2), whereas KG7 and KG8 had no effect. To assess direct compound effects on T. gondii, recently released extracellular tachyzoites were exposed to 10 μM of compound for 4 h. Compared to untreated tachyzoites, KG3 and KG7 decreased T. gondii growth rate by 32 and 16%, respectively. In contrast, KG8 did not inhibit the growth of pre-treated tachyzoites (Figure 2).
Figure 2. Host cell pre-treatment and extracellular tachyzoite exposure results in reduced growth.
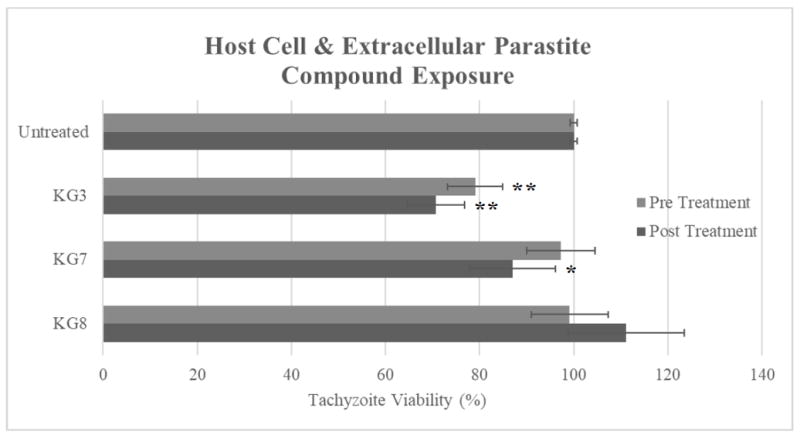
HFF cells were pretreated with the indicated compound at 10 μM for 24 h (pre-treatment). Cells were then washed with media and infected with 2,000 RH-dTom tachyzoites. Isolated RH-dTom tachyzoites were also treated with respective compound at 10 μM for 4 h (post-treatment) and were then allowed to infect HFF cells in a 96 well plate at 2,000 tachyzoites/mL. Fluorescent plate readings were taken at day 4 post-infection for both experiments. p value <0.05(*); p value <0.001(**)
Bacterial Reverse Mutation Assay
Data from an Ames assay for DNA mutagenesis revealed that none of the compounds showed DNA point mutations in Salmonella when compared to that of the natural revertant control (Table 2). However, KG7 appeared to inhibit S. typhimurium growth.
Physicochemical and in vitro ADME
Experimental gLogD7.4 values of 2.8 for KG3 and 3.9 for both KG7 and KG8 indicate that these compounds are not unduly hydrophobic, although they had relatively low aq. solubilities ranging from 1.1 to 6.3 μg/mL at pH 6.5 (Table 3). In this respect, all three compounds are unionized at physiological pH. The calculated polar surface area (PSA) values ranging from 48 to 96 A2 for KG3, KG7, and KG8 indicate that the polarity of these compounds will not be a rate-limiting factor for membrane permeability (16). Plasma protein binding was relatively high for all three compounds, especially for KG8 with a cPPB value of >99.5%. Metabolic stability was tested in both human and mouse liver microsomes. As the predicted intrinsic clearance (CLint) and hepatic extraction ratio (EH) values indicate, KG3 and KG7 were rapidly metabolized in both human and mouse microsomes, whereas KG8 was somewhat more stable to metabolism.
Table 3.
Physiochemical parameters, solubility, and plasma protein binding of KG3, KG7, and KG8.
| Compound ID | cPPBa (% bound) | PSA (Å2)b | gLogD (pH 7.4) | Sol6.5c (μg/mL) |
|---|---|---|---|---|
| KG3 | 99 | 47.9 | 2.8 | 1.2 - 2.2 |
| KG7 | 93 | 96.3 | 3.9 | 3.1 - 6.3 |
| KG8 | >99.5 | 71.4 | 3.9 | 3.1 - 6.3 |
Plasma protein binding estimated using a chromatographic method.
Calculated through ChemAxon chemistry cartridge with JChem from Excel.
Kinetic solubility after 30 min at room temperature.
In vivo Efficacy
For preliminary in vivo efficacy experiments, mice were infected with a lethal dose of T. gondii strain RH tachyzoites and treated with twice daily IP doses of KG3 (20 mg/kg), KG7 (5 mg/kg), and KG8 (5 mg/kg); the chosen compound doses were proportional to their in vitro IC50 values. In this experiment, KG3 spared 33% of mice, and KG7 and KG8 spared 50%; for comparison, treatment of 21 mg/kg pyrimethamine in this same model led to 100% sparing. (Figure 3). The low metabolic stability of these compounds could be responsible for their low in vivo efficacy.
Figure 3. In vivo Survival Following Compound Treatment.
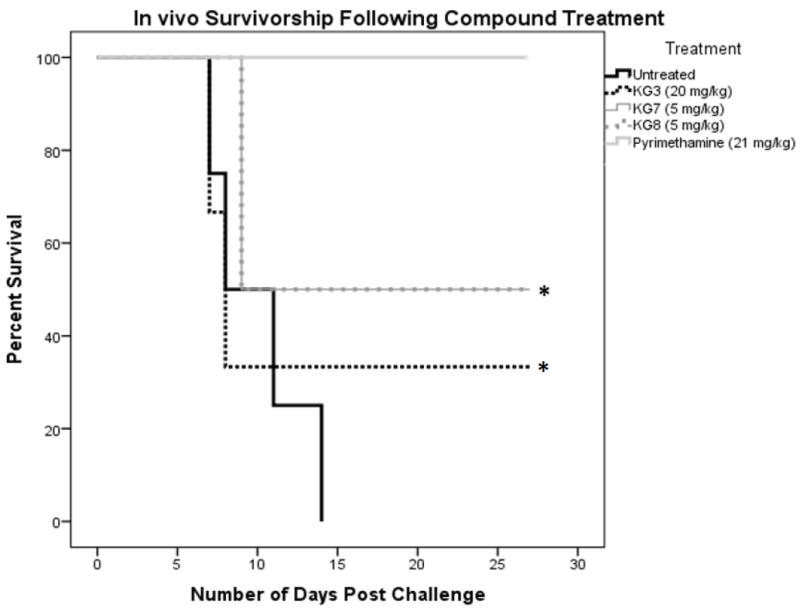
CFW mice were treated with compounds at respective amounts twice per day for 10 d. Survival was quantified over 30 d. Survival was plotted as a Kaplan-Meier curve with a calculated p value <0.05 (*)
4. Discussion
We characterized styryl 4-oxo-1,3-benzoxazin-4-one (KG3), tetrahydrobenzo[b]pyran (KG7), and benzoquinone hydrazone (KG8), three new T. gondii inhibitors that were identified from data generated in a phenotypic HTS campaign to discover novel antimalarial chemotypes. These compounds inhibit T. gondii in vitro with potencies comparable to that for the control drug pyrimethamine. Of the three compounds evaluated, KG3 was the most potent when parasites were exposed to compound within the host cell. When host cells were exposed to compounds prior to infection and subsequently washed, only KG3 had any subsequent effect on parasite growth. This result may be due to the relative potency of KG3 compared to the KG7 and KG8. However, when extracellular tachyzoites were exposed to compound, both KG3 and KG7 decreased subsequent intracellular parasite growth. This suggests that KG8 may be effective only during the intracellular replication phase of the parasite, or possibly by affecting parasite egress (17). In addition, treatment of infected mice with KG7 and KG8 significantly increased survival. Notably, KG3 had no measurable cytotoxicity against five mammalian cell lines whereas KG7 and KG8 inhibited the growth of 2 out of 5 cell lines. None of the compounds were mutagenic in an Ames assay.
Experimental gLogD7.4 and calculated PSA values for the three compounds were well within the ranges predicted to be favorable for good ADME, even though each compound had relatively low aq. solubility. Plasma protein binding was relatively high for all three compounds, and all three compounds were metabolically unstable. The metabolic instability of KG7 and KG8 is likely due in part to their twin tert-butyl groups (19-20). The benzoquinone substructure of KG8 could be another site of metabolic (and chemical) reactivity.
To contemplate potential optimization of KG3, KG7, or KG8, it is useful to consider their chemical and biological pedigree. KG3 and evaluated analogs (21) have been little investigated for their biological properties. For example, KG3 was inactive in the one assay listed in PubChem and the corresponding quinazolinones were screened in only five assays and appear to inhibit tubulin polymerization. With its scarcity of sp3 carbon atoms (22), KG3 clearly has some structural liabilities. Similar to KG3, almost no biological data has been published for KG7 and its analogs (23). For example, data from PubChem revealed that KG7 was active against several protozoan parasites with an in vitro selectivity index of approximately 10. KG8 and closely related analogues inhibit cyclooxgenase-2 and 5-lipoxygenase (24) and potentiate nerve growth factor (25). Interestingly, the closely related compound poloxin is an inhibitor of polo-like kinase 1(26), and kinases are thought to be promising T. gondii drug targets (27). KG8 is also a hydrazone derivative of the antibiotic isoniazid; however, isoniazid showed no efficacy against Toxoplasma gondii in vitro (data not shown).
Determining mode of action would further be useful in understanding how these compounds interact directly with the parasite and if any novel molecular targets have been identified. To aid in target identification, the results from the extracellular tachyzoite exposure experiment suggest that KG8 most likely affects the parasite following invasion of the host cell while KG3 and KG7 may affect the parasite extracellularly. Describing the mode of action more specifically would particularly be useful for drug optimization for target binding efficiency and specificity reducing the need for further non-directed chemical analogue synthesis. Furthermore, these three compounds have yet to be examined for blood brain permeability, or more importantly, their ability to reduce T. gondii cyst burden in animal models.
In summary, while each of these three compounds has liabilities to overcome, they do constitute interesting starting points for chemical prospecting, scaffold-hopping, and optimization to pursue novel leads with efficacy for the treatment of toxoplasmosis and malaria.
Table 4.
Metabolic stability of KG3, KG7, and KG8.
| Compound ID | Microsome species | T1/2 (min) | CLint, in vitro (μL/min/mg protein) | Predicted EH |
|---|---|---|---|---|
| KG3 | Human | 9 | 129 | 0.88 |
| Mouse | <2 | >886 | N/A | |
| KG7 | Human | 2 | 746 | 0.97 |
| Mouse | <2 | >886 | >0.94 | |
| KG8 | Human | 47 | 37 | 0.6 |
| Mouse | 10 | 180 | N/A |
Metabolic stability was assessed in both human and mouse microsomes. The half-life (T1/2), intrinsic clearance in vitro (CLint), and the predicted hepatic extraction ratios (EH) were determined.
Highlights.
Novel chemotypes with in vitro efficacy for T. gondii and P. falciparum.
Stability experiments suggest sub-optimal metabolic stability.
Chemotypes increase in vivo survivorship following acute T. gondii challenge.
Efficacious early-leads for development as anti-toxoplasma chemotherapies.
Acknowledgments
Funding: We thank the U.S. National Institutes of Health [P20GM103427 and AI116723], Nebraska Research Initiative, and the GRACA and FUSE programs of the University of Nebraska at Omaha for support of this work. None of the mentioned agencies participated in study design, data collection, analysis, or the decision for publication.
Footnotes
Conflicts of interest: none
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Flegr J, Prandota J, Sovičková M, Israili ZH. Toxoplasmosis–a global threat. Correlation of latent toxoplasmosis with specific disease burden in a set of 88 countries. PloS one. 2014;9:e90203. doi: 10.1371/journal.pone.0090203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Jones JL, Dargelas V, Roberts J, Press C, Remington JS, Montoya JG. Risk factors for Toxoplasma gondii infection in the United States. Clinical Infectious Diseases. 2009;49:878–884. doi: 10.1086/605433. [DOI] [PubMed] [Google Scholar]
- 3.Grothen DC, Zach SJ, Davis PH. Detection of Intestinal Pathogens in River, Shore, and Drinking Water in Lima, Peru. J Genomics. 2017;5:4–11. doi: 10.7150/jgen.18378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Weiss LM, Dubey JP. Toxoplasmosis: A history of clinical observations. Int J Parasitol. 2009;39:895–901. doi: 10.1016/j.ijpara.2009.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kamerkar S, Davis PH. Toxoplasma on the brain: understanding host-pathogen interactions in chronic CNS infection. J Parasitol Res. 2012;2012:589295. doi: 10.1155/2012/589295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fung HB, Kirschenbaum HL. Treatment regimens for patients with toxoplasmic encephalitis. Clin Ther. 1996;18:1037–56. doi: 10.1016/s0149-2918(96)80059-2. [DOI] [PubMed] [Google Scholar]
- 7.Montoya JG, Boothroyd JC, Kovacs JA. Toxoplasma gondii. In: Bennett JE, Dolin R, Blaser MJ, editors. Mandell, Douglas, and Bennett’s Principles and Practice of Infectious Diseases. 8. Elsevier Saunders; Philadelphia, PA: 2015. [Google Scholar]
- 8.Cohn JA, McMeeking A, Cohen W, Jacobs J, Holzman RS. Evaluation of the policy of empiric treatment of suspected Toxoplasma encephalitis in patients with the acquired immunodeficiency syndrome. Am J Med. 1989;86:521–527. doi: 10.1016/0002-9343(89)90378-1. [DOI] [PubMed] [Google Scholar]
- 9.Neville AJ, Zach SJ, Wang X, Larson JJ, Judge AK, Davis LA, Vennerstrom JL, Davis PH. Clinically Available Medicines Demonstrating Anti-Toxoplasma Activity. Antimicrob Agents Chemother. 2015;59:7161–9. doi: 10.1128/AAC.02009-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.McFarland MM, Zach SJ, Wang X, Potluri LP, Neville AJ, Vennerstrom JL, Davis PH. A Review of Experimental Compounds Demonstrating Anti-Toxoplasma Activity. Antimicrobial Agents and Chemotherapy. 2016 doi: 10.1128/AAC.01176-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Imming P, Sinning C, Meyer A. Drugs, their targets and the nature and number of drug targets. Nat Rev Drug Discov. 2006;5:821–34. doi: 10.1038/nrd2132. [DOI] [PubMed] [Google Scholar]
- 12.Guiguemde WA, Shelat AA, Bouck D, Duffy S, Crowther GJ, Davis PH, Smithson DC, Connelly M, Clark J, Zhu F, Jiménez-Díaz MB, Martinez MS, Wilson EB, Tripathi AK, Gut J, Sharlow ER, Bathurst I, El Mazouni F, Fowble JW, Forquer I, McGinley PL, Castro S, Angulo-Barturen I, Ferrer S, Rosenthal PJ, Derisi JL, Sullivan DJ, Lazo JS, Roos DS, Riscoe MK, Phillips MA, Rathod PK, Van Voorhis WC, Avery VM, Guy RK. Chemical genetics of Plasmodium falciparum. Nature. 2010;465:311–315. doi: 10.1038/nature09099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Beven K. On the future of distributed modelling in hydrology. Hydrological Processes. 2000;14:3183–3184. [Google Scholar]
- 14.Lombardo F, Shalaeva MY, Tupper KA, Gao F. ElogD(oct): a tool for lipophilicity determination in drug discovery. 2. Basic and neutral compounds. J Med Chem. 19:2490–7. doi: 10.1021/jm0100990. [DOI] [PubMed] [Google Scholar]
- 15.Valko K, Nunhuck S, Bevan C, Abraham MH, Reynolds DP. Fast gradient HPLC method to determine compounds binding to human serum albumin. Relationships with octanol/water and immobilized artificial membrane lipophilicity. J Pharm Sci. 2003;92:2236–48. doi: 10.1002/jps.10494. [DOI] [PubMed] [Google Scholar]
- 16.Palm K, Stenberg P, Luthman K, Artursson P. Polar molecular surface properties predict the intestinal absorption of drugs in humans. Pharm Res. 1997;14:568–571. doi: 10.1023/a:1012188625088. [DOI] [PubMed] [Google Scholar]
- 17.Chandramohanadas R, Davis PH, Beling DP, Harbut MB, Darling C, Velmourougane G, Lee MY, Greer PA, Roos DS, Greenbaum DC. Apicomplexan parasites co-opt host calpains to facilitate their escape from infected cells. Science. 2009;324:794–7. doi: 10.1126/science.1171085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Caujolle FM, Caujolle DH, Cros SB, Calvet MM. Limits of toxic and teratogenic tolerance of dimethyl sulfoxide. Ann N Y Acad Sci. 1967 Mar 15;141(1):110–26. doi: 10.1111/j.1749-6632.1967.tb34871.x. [DOI] [PubMed] [Google Scholar]
- 19.Jacob SW, Rosenbaum EE. The toxicology of dimethyl sulfoxide (DMSO) Headache. 1966 Oct;6(3):127–36. doi: 10.1111/j.1526-4610.1966.hed0603127.x. [DOI] [PubMed] [Google Scholar]
- 20.Barnes-Seeman D, Jain M, Bell L, Ferreira S, Cohen S, Chen X-H, Amin J, Snodgrass B, Hatsis P. Metabolically stable tert-butyl replacement. ACS Med Chem Lett. 2013;4:514–516. doi: 10.1021/ml400045j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Westphal MV, Wolfstädter BT, Plancher J-M, Gatfield J, Carreira EM. Evaluation of tert-butyl Isosteres: Case studies of physicochemical and pharmacokinetic properties, efficacies, and activities. ChemMedChem. 2015;10:461–469. doi: 10.1002/cmdc.201402502. [DOI] [PubMed] [Google Scholar]
- 22.Suvorova EY, Vikrishchu NI, Popov LD, Starikova ZA, Vikrishchuk AD, Zhdanov YA. Reactions of 4-oxo-1,3-benzoxazinium perchlorates with guanidines. Russ J Org Chem. 2007;43:1553–1558. [Google Scholar]
- 23.Lovering F, Bikker J, Humblet C. Escape from flatland: increasing saturation as an approach to improving clinical success. J Med Chem. 2009;52:6752–6756. doi: 10.1021/jm901241e. [DOI] [PubMed] [Google Scholar]
- 24.Abdel-Latif FF, Masahaly MM, El-Gawish EH. Synthesis of heterocycles through reactions of nucleophiles with acrylonitriles. Part 15. Synthesis of some new functionalized benzo[b]pyrans and indeno[1,2-b]pyrans of potential biological activity. J Chem Res S. 1995;5:178–179. [Google Scholar]
- 25.Misra S, Ghatak S, Patil N, Dandawate P, Ambike V, Adsule S, Unni D, Venkateswara Swamy K, Padhye S. Novel dual cyclooxygenase and lipoxygenase inhibitors targeting hyaluronan-CD44v6 pathway and inducing cytotoxicity in colon cancer cells. Bioorg Med Chem. 2013;21:2551–2559. doi: 10.1016/j.bmc.2013.02.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Eguchi T, Kanai S, Kakinuma K, Okazaki T, Mizoue K. Synthesis of NG-061 and its analogs, and their biological evaluation as an enhancer of nerve growth factor. Chem Pharm Bull. 2000;48:1470–1473. doi: 10.1248/cpb.48.1470. [DOI] [PubMed] [Google Scholar]
- 27.Liao C, Park JE, Park Bang JK, Nicklaus MC, Lee KS. Probing binding modes of small molecule inhibitors to the polo-box domain of human polo-like kinase 1. ACS Med Chem Lett. 2010;1:110–114. doi: 10.1021/ml100020e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Meneceur P, Bouldouyre MA, Aubert D, Villena I, Menotti J, Sauvage V, Garin JF, Derouin F. In vitro susceptibility of various genotypic strains of Toxoplasma gondii to pyrimethamine, sulfadiazine, and atovaquone. Antimicrob Agents Chemother. 2008;52:1269–77. doi: 10.1128/AAC.01203-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Peixoto L, Chen F, Harb OS, Davis PH, Beiting DP, Brownback CS, Ouloguem D, Roos DS. Integrative genomic approaches highlight a family of parasite-specific kinases that regulate host responses. Cell Host Microbe. 2010;8:208–18. doi: 10.1016/j.chom.2010.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]