

Abstract
Despite advanced understanding of signaling mediated by local and systemic factors, the role of epigenetic factors in the regulation of bone regeneration remains vague. The DNA methyltransferases (Dnmts) Dnmt3a and Dnmt3b have tissue specific expression patterns and create unique methylation signatures to regulate gene expression. Using a stabilized murine tibia fracture model we find that Dnmt3b is induced early in fracture healing, peaks at 10 days post fracture (dpf), and declines to nearly undetectable levels by 28 dpf. Dnmt3b expression was cell-specific and stage-specific. High levels were observed in chondrogenic lineage cells within the fracture callus. To determine the role of Dnmt3b in fracture healing, Agc1CreERT2;Dnmt3bf/f (Dnmt3bAgc1ER) mice were generated to delete Dnmt3b in chondrogenic cells. Dnmt3bAgc1ER fracture displayed chondrogenesis and chondrocyte maturation defect, and a delay in the later events of angiogenesis, ossification, and bone remodeling. Biomechanical studies demonstrated markedly reduced strength in Dnmt3bAgc1ER fractures and confirmed the delay in repair. The angiogenic response was reduced in both vessel number and volume at 10 and 14 dpf in Dnmt3bAgc1ER mice. Immunohistochemistry showed decreased CD31 expression, consistent with the reduced angiogenesis. Finally, in vitro angiogenesis assays with human umbilical vein endothelial cells (HUVECs) revealed that loss of Dnmt3b in chondrocytes significantly reduced tube formation and endothelial migration. To identify specific angiogenic factors involved in the decreased callus vascularization, a protein array was performed using conditioned media isolated from control and Dnmt3b loss-of-function chondrocytes. Several angiogenic factors, including CXCL12 and osteopontin (OPN) were reduced in chondrocytes following loss of Dnmt3b. DNA methylation analysis further identified hypomethylation in Cxcl12 promoter region. Importantly, the defects in tube formation and cell migration could be rescued by administration of CXCL12 and/or OPN. Altogether, our findings establish that Dnmt3b positively regulates chondrocyte maturation process, and its genetic ablation leads to delayed angiogenesis and fracture repair.
Keywords: DNMT3B, FRACTURE, CHONDROCYTE, ANGIOGENESIS
Introduction
Bone fracture repair occurs through a complex and integrated series of events that lead to regeneration of bone with restoration of mechanical strength and tissue morphometry in the absence of scar formation.(1) Long bones heal primarily via endochondral ossification whereby bone formation occurs through a cartilage intermediate stage, which recapitulates a series of spatiotemporal events during long bone development.(2,3) Following injury, the repair cascade begins with hematoma formation and an inflammatory response, which mediate the recruitment of mesenchymal cells to the fracture site. During endochondral healing, the mesenchymal cells, upon receiving differentiation cues, undergo chondrogenesis to form a cartilaginous soft callus primarily in the more hypoxic environment lacking blood vessels.(4,5) As the cartilage mineralizes it is invaded by blood vessels that mediate an influx of osteoprogenitor cells that produce a central hard callus on top of mineralized cartilage matrixes and an influx of chondroclasts that resorb the calcified cartilage. As a result, the bony callus matures and undergoes remodeling over time, ultimately allowing bone to regain its original architecture and function. However, adequate vascularization is crucial for successful bone repair and lack or inhibition of angiogenesis has been reported as one of the main causes of delayed union or non-union during fracture healing.(6–11) In this regard, diverse local and systemic factors delivered by this neovascularization system control cell fate and differentiation throughout the process of fracture repair. Although cell signaling mediated by local and systemic factors has been extensively investigated, there is limited understanding of the role of epigenetic changes in the regulation of bone regeneration.
DNA methylation, primarily occurring at CpG dinucleotides, is a major mode of epigenetic regulation in the mammalian genome with multiple functions that are essential for normal embryonic development.(12) Of the three catalytic DNA methyltransferase enzymes, Dnmt1, is primarily involved in methylating hemimethylated DNA, leading to the view that Dnmt1 is “maintenance” methyltransferase.(13,14) Conversely, Dnmt3a and Dnmt3b act as de novo DNA methyltransferase modifying unmethylated DNA. Dnmt3a-null mice appear to be grossly normal at birth; however, they do acquire developmental defects postnatally and die shortly after birth.(15) In contrast, Dnmt3b-deletion is embryonically lethal in mice.(15) Ablation of both Dnmt3a and Dnmt3b in murine embryonic stem cells (ESCs) results in progressive loss of differentiation potential with extended passage, although the capacity of self-renewal is largely maintained.(16) Therefore, the two de novo DNA methyltransferases are important for the establishment of DNA patterns during early development. The role of DNA methylation in adult stem cells and lineage development has recently begun to emerge. In this regard, hematopoietic stem cells (HSCs) with single or double deficiency of Dnmt3a and Dnmt3b display enhanced self-renewal and impaired differentiation.(17,18) Thus, de novo Dnmt3 enzymes are thought to be essential to silence pluripotency-associated regulatory elements, allowing differentiation to proceed. Given the comprehensive understanding on how DNA methylation functions during early development and in hematopoietic system, it is of particular interest to investigate whether these principles may extend to other lineages, including chondrocytes.
Recently, using genetically modified murine models, we have identified Dnmt3b as a critical epigenetic factor in the regulation of articular chondrocyte differentiation and in the maintenance of articular cartilage homeostasis.(19) Nonetheless, the relative role of epigenetic factors, the target cells and tissues involved in bone regeneration process, remain to be elucidated. Here, we report that Dnmt3b is induced early during fracture repair with high expression levels observed in chondroprogenitors and chondrocytes within murine fracture callus. Furthermore, ablation of Dnmt3b in chondrocytes during fracture repair leads to a suppression of chondrocyte hypertrophic maturation with persistence of cartilage matrix, delayed development of angiogenesis, as well as perturbation of the later healing phases including endochondral ossification and bone remodeling. Altogether, our findings establish for the first time that epigenetic modulation mediated by Dnmt3b is crucial in the regulation of chondrocyte hypertrophic differentiation during endochondral bone repair process.
Materials and Methods
Experimental animals
All animal studies were done in accordance with approval of the Committees on Animal Resources in University of Rochester Committee and Washington University in St. Louis. Wild-type C57BL/6J male mice and Ai9 Cre reporter mice(20) were purchased from the Jackson Laboratory (Bar Harbor, ME, USA). Dnmt3bf/f(15) mice, originally generated by Dr. En Li, were obtained from the Mutant Mouse Regional Resource Center, Davis, CA (Cat# 29887) R26mTmGf/f,(21) and Agc1CreERT2(22) mice were a generous gift from the laboratory of Dr. Benoit de Crombrugghe (Department of Genetics, University of Texas MD Anderson Cancer Center, Houston, TX). Agc1CreERT2;Dnmt3bf/f (Dnmt3bAgc1ER), Agc1CreERT2;Ai9f/f (Ai9Agc1ER) mice and Crenegative littermate controls were viable and produced in Mendelian ratios. All mice were housed in a room using Microisolator Technology (Seaford, DE) kept at 21°C to 23°C. They had free access to food (LabDiet 5010) and water (Hydropac) at all times.
Tibia fracture model
Open midshaft tibial fractures were created unilaterally in 8-week-old to 10-week-old mice as described.(23) Prior to surgery, mice were anesthetized with 2.5% avertin (15 μL/g body weight). After the mice were anesthetized, a 6-mm-long incision was made in the skin on the anterior side of the tibia. A sterile 26G needle was inserted into the tibial marrow cavity from the proximal end, temporarily withdrawn to facilitate transection of the tibia with a scalpel at midshaft, and then reinserted. The incision was closed with 5-0 nylon sutures. Following surgery, mice were kept in cages after recovery from anesthesia, allowing free unrestricted weight bearing, and buprenorphine was delivered subcutaneously (0.1 mg/kg body weight) to manage pain every 6 to 12 hours, beginning at the time of sedation, for up to 3 days following fractures. Tamoxifen (10 mg/kg body weight) was administrated daily via intraperitoneal injection to Ai9Agc1ER or Dnmt3bAgc1ER mice and their littermate controls from day 1 to day 7 post fracture in order to trace cell lineages targeted by Agc1CreERT2 transgene or to remove Dnmt3b alleles, respectively. Healing of the fractured tibias was confirmed immediately after surgery and monitored during fracture repair using digital radiographs, which were obtained at 0, 7, 10, 14, 21, and 28 days post fracture (dpf) under anesthesia using a Faxitron Cabinet X-Ray System (n ≥ 10 per group per time point).
Histological analyses of fracture calluses
The fractured tibias were collected at 7, 10, 14, 21, and 28 dpf for detailed histological analyses. Excess muscle and soft tissue were excised. Five specimens in each group at each time point were fixed in 10% neutral buffered formalin. These specimens were decalcified for 14 days in 14% EDTA (pH 7.2), processed and embedded in paraffin, and sectioned at a thickness of 3 μm. Sections were stained using Alcian blue/hematoxylin/orange-g (ABH/OG) and tartrate-resistant acid phosphatase (TRAP) in order to analyze the cartilage composition and osteoclast formation in the fracture callus tissues. Cartilage area, bone area, mesenchyme area, and osteoclast surface per bone surface (Oc. S/BS) were quantified on ABH/OG, TRAP-stained sections using the Visiopharm Integrator System. Immunohistochemical (IHC) stainings for Dnmt3b (Abcam, Cambridge, MA, USA; ab16049), CD31 (BD Biosciences, San Jose, CA, USA; 550274), SOX9 (Santa Cruz Biotechnology, Santa Cruz, CA, USA; sc20095), COL1A1 (Abcam; ab21286), COL2A1 (Thermo Fisher Scientific, Waltham, MA, USA; MS235-P) and COL10A1 (Quartet; 1-CO097-05) were performed on paraffin sections following the antigen retrieval and colorimetric development methodologies. Citrate buffer with high-temperature and high-pressure antigen retrieval was used for Dnmt3b, CD31, and Sox9. Proteinase K digestion was used for COL1A1 and pepsin digestion for COL2A1 and COL10A1. Tissues prepared for frozen sections were fixed in 4% paraformaldehyde (PFA) for 2 hours at 4°C, decalcified with 14% EDTA at 4°C for 10 days, infiltrated with gradient sucrose for 3 days, embedded with Tissue-TEK OCT medium, and sectioned at a thickness of 10 μm with a Leica CM1850 cryotome. Immunofluorescent staining of Sox9 was performed on frozen section for fracture tissue. GFP and tdTomato fluoresce from frozen sections was examined by Zeiss upright fluoresce microscope.
Micro–computed tomography assessment of the mineralized callus, callus vascularity, and biomechanical torsion testing
After careful dissection and removal of the intramedullary pin, repaired tibias from days 10, 14, 21, and 28 (10 mice per group per time point) were imaged using a micro–computed tomography (μCT) system (VivaCT 40; Scanco Medical AG, Brüttisellen, Switzerland), with an integration time of 300 ms, a current of 145 mA, and an energy setting of 55 kV. The threshold was chosen using 2D evaluation of several slices in the transverse anatomic plane so that mineralized callus was identified but surrounding soft tissue was excluded. Quantification for the volumes of the bony calluses was determined as previously described using the Scanco analysis software.(24) 3D images were generated using a constant threshold of 275 per-mille for metaphyseal trabecular bone of the contralateral unfractured tibias.
Vascular networks surrounding the fracture calluses were examined using μCT analysis combined with perfusion of a lead chromate–based contrast agent as described.(25) Briefly, the mice were anesthetized and thoracotomy was performed to insert a 20G angiocatheter into the left ventricle. The circulatory system was flushed with 0.9% saline and heparin (100 IU/mL), followed by 10% neutral buffered formalin. Microfil MV-122 (Flow Tech) contrast media was perfused via the same route. Following perfusion, tibias were harvested, intramedullary pin removed, and scanned using μCT system at a 10.5-μm isotropic high-resolution to image bone and vasculature. Following the initial μCT scan, the specimens were then decalcified for 2 weeks in 14% EDTA. After decalcification, the specimens were subjected to the second μCT scan to image only vasculature within the calluses. By registering the 2D slices before and after decalcification, contour lines were drawn to define a volume of interest (VOI) that only included the vasculature in or immediately adjacent to the fracture callus itself. The reconstructed scan images were globally thresholded based on intensity values to render 3D images of the vasculature in new bone callus, excluding the vessels in the surrounding tissues. 3D morphometric analyses, based on direct distance transform methods, were subsequently performed on the 3D images using algorithms that are commonly used to model trabecular bone morphology. This facilitated quantification and analysis of vascular network morphology, including vessel volume and number.
After the μCT imaging of the fracture calluses harvested at 28 dpf, the specimens were moistened with PBS and frozen at −20°C until being thawed for biomechanical testing as described.(26) Briefly, specimens were potted in poly (methyl methacrylate) (PMMA) bone cement (DePuy Orthopaedics) in square aluminum tube holders and allowed to rehydrate in PBS at room temperature for 1 to 2 hours. Specimens were tested in torsion using an EnduraTec TestBench system at 1 degree/s until failure. The torque data was plotted against the rotational deformation to determine the maximum torque, torsional rigidity, and energy to maximum.
Primary chondrocyte cultures and preparation of conditioned culture media
Murine costal chondrocytes were isolated from ribcages of 2-day-old Dnmt3bf/f and initially cultured as described,(27) with modifications. Upon roughly 70% to 80% confluence, cells were transduced with Adeno virus expressing GFP (Ad-GFP) or Adeno virus expressing Cre (Ad-Cre) adenoviruses at a multiplicity of 100 in the presence of polybrene (10 μg/mL) in high-glucose Dulbecco’s modified Eagle’s medium (DMEM; Gibco, Grand Island, NY, USA) supplemented with 2% fetal bovine serum (FBS; Sigma, St. Louis, MO, USA) for 48 hours. Media conditioned during this 48-hour period were then collected and utilized as WT and Dnmt3b loss-of-function (LOF) chondrocyte conditioned media in angiogenic functional assays and protein array analysis. Following viral infection, cells were cultured in complete media containing 50 μg/mL ascorbic acid and 10 mM β-glycerophosphate and allowed to mature for up to 10 days, as indicated. Real-time RT-PCR, alkaline phosphatase (AP), Alizarin red staining, cell proliferation, and cell death detection were performed at indicated time points. Primary murine chondrocytes were also isolated from 2-day-old Dnmt3b transgenic(19) mouse pups, following with Lenti-rtTA or Lenti-GFP transfection for 48 hours and 10 days of culture in DMEM culture medium. Gene expression, AP staining, and Alizarin red staining were performed to examine the chondrocyte maturation process.
Methylation qPCR
Methylation qPCR for Cxcl12 was performed according to the manufacturer’s instruction (Qiagen, Valencia, CA, USA; #335452). Genomic DNA isolated from Dnmt3b LOF and control chondrocytes was enzymatically digested by methylation sensitive or methylation dependent enzymes separately. The enzyme-digested genomic DNA was then used for qPCR and the primer for Cxcl12 methylation (CpG Island 109537) was ordered from Qiagen (#EPMM109537).
Human umbilical vein endothelial cells tube formation, migration assays, and proliferation and apoptosis tests
The tube formation assays were performed as described,(28) with modifications. Briefly, human umbilical vein endothelial cells (HUVECs) were serum-starved for 4 to 6 hours just prior to the assay, and then plated at a density of 4 × 104 cells/well with conditioned media in 96-well tissue culture plates precoated with growth factor-reduced Matrigel (Corning, Inc., Corning, NY, USA). After overnight of culture, five random fields of wells were digitally photographed, and the numbers and length of tubes were measured by Image J software (NIH, Bethesda, MD, USA; https://imagej.nih.gov/ij/). In the HUVEC migration assay, HUVECs were seeded in the upper chamber of the 24-well Millicell inserts (8 μm pore; Corning) at a density of 4 × 104 cells per well in serum-free medium. Conditioned media were added in the bottom chamber. The numbers of migrated HUVECs were stained by crystal violet and counted after overnight of incubation.
BrdU immunostaining analyses were performed by adding 10 μM of BrdU to HUVEC cultures. After 2 hours of incubation, BrdU labeling was detected using the Roche Cell Proliferation ELISA kit (BrdU, colorimetric) (Roche Diagnostics, Mannheim, Germany; 11647229001) per the manufacturer’s instructions. Analyses of apoptotic HUVECs were performed using the Cell Death Detection ELISA kit (Roche; 11774425001) according to the manufacturer’s instructions.
Proteins
The recombinant mouse CXCL12/SDF-1α (460-SD-010) and recombinant mouse osteopontin/OPN (441-OP-050) were both obtained from R&D Systems (Minneapolis, MN, USA), which were used at the final concentration of 100 ng/mL and 500 ng/mL in HUVEC tube formation and cell migration assays, respectively.
Protein array analysis
Proteome Profiler Mouse Angiogenesis Array Kit (R&D Systems; ARY015) was used for angiogenesis-associated factors detection according to the manufacturer’s instructions. Briefly, conditioned media were first mixed with the detection antibody cocktail at room temperature for 1 hour prior to being added to the array membrane. The membrane was then incubated overnight at 4°C with gentle shaking. Following washing, horseradish peroxidase–conjugated streptavidin was added to the membrane, allowing a 30-min incubation at room temperature with gentle shaking. A chemiluminescence imaging system was used to detect and quantify the array signals.
RNA isolation, cDNA synthesis, and quantitative gene expression analyses
In mice administered tibia fractures, at 0, 5, 7, 10, 14, 21, and 28 dpf, tibias were removed and dissected free from muscle and soft tissue, and intramedullary pins were removed. Four millimeters of fracture calluses were excised, flushed of marrow, and flash frozen in liquid nitrogen for mRNA extraction. Frozen fracture calluses were homogenized using the TissueLyser II system, and total RNA was extracted using a Qiagen RNeasy kit (Qiagen; 74104). Total RNA was then reverse transcribed into cDNA using the iScript cDNA synthesis kit (Bio-Rad Laboratories, Hercules, CA, USA) and cDNAs were amplified in a total volume of 20 μL by 40 cycles of denaturation at 95°C for 15 s and annealing at 60°C for 1 min. Primer sequences are available in Table 1.
Table 1.
Primer Sequences for qPCR
| Genes | Sequences |
|---|---|
| Col2a1 | 5′-GCA GAG ATG GAG AAC CTG GTA-3′ 5′-AGC CTT CTC GTC ATA CCCT-3′ |
| Col10a1 | 5′-ATG CCT TGT TCT CCT CTT ACT G-3′ 5′-TGC TGA ACG GTA CCA AAC G-3′ |
| Col1a1 | 5′-GCA TGG CCA AGA AGA CAT CC-3′ 5′-CCT CGG GTT TCC ACG TCT C-3′ |
| Agc1 | 5′-CGT GTT TCC AAG GAA AAG GA-3′ 5′-TGT GCT GAT CAA AGT CCA G-3′ |
| Runx2 | 5′-CGT CCA CTG TCA CTT TAA TAG CTC-3′ 5′-GTA GCC AGG TTC AAC GAT CTG-3′ |
| Mmp13 | 5′-AGA CTG GTA ATG GCA TCA AGG-3′ 5′-GCC ATT TCA TGC TTC CTG ATG-3′ |
| Dnmt1 | 5′-CCT GCC AGG GCT TCA GTG GC-3′ 5′-CAG GCA GCG CAG TGT GAG CT-3′ |
| Dnmt3a | 5′-GCC GAA TTG TGT CTT GGT GGA TGA CA-3′ 5′-CCT GGT GGA ATG CAC TGC AGA AGG A-3′ |
| Dnmt3b | 5-AGC TGC TGA AGC TGT GGA A-3′ 5′-GGT TCG AGT GGC CGA GAT-3′ |
| Sox9 | 5′-CGG CTC CAG CAA GAA CAA G-3′ 5′-TGC GCC CAC ACC ATG A-3′ |
| Bsp | 5′-CAG AAG TGG ATG AAA ACG AG-3′ 5′-CGG TGG CGA GGT GGT CCC AT-3′ |
| Oc | 5′-CTT GAA GAC CGC CTA CAA AC-3′ 5′-GCT GCT GTG ACA TCC ATA C-3′ |
| Actin | 5′-AGA TGT GGA TCA GCA AGC AG-3′ 5′-GCG CAA GTT AGG TTT TGT CA-3′ |
Western blot analysis
Proteins were extracted from 4 mm of fractured calluses of Dnmt3bAgc1ER and littermate control mice at 7 dpf with radioimmunoprecipitation assay buffer (RIPA; Thermo Scientific; 89900) supplemented with proteinase inhibitors and phosphatase inhibitors (Thermo Scientific; 78440) after bone marrow cells were flushed away. Proteins were fractioned in an SDS-polyacrylamide gel and transferred to a polyvinylidene difluoride membrane, and detected with the Dnmt3b antibody (Abcam; ab16049).
Statistics
All results are presented as the mean ±SD. Statistical analyses were performed by two-tailed Student’s t test and two-way analysis of variance (ANOVA) followed by Dunnett’s test as appropriate; a p value <0.05 was considered statistically significant.
Results
Dnmt3b is dominantly expressed in chondrogenic lineage cells within fracture callus
Endochondral bone repair is a precisely spatiotemporal regulated multistage process, including inflammation (days 0 to 5 [D0 to D5] in mice), chondrogenesis (D5–D10), osteogenesis (D14–D21), and bone remodeling (D14–D28).(29,30) In order to examine the temporal expression pattern of DNA methyltransferases during the fracture repair process, real-time qPCR was firstly performed on RNA extracted from the fracture callus of C57BL/6J wild-type (WT) mice at 5, 7, 10, 14, 21, and 28 dpf. Dnmt3b, one of the most critical de novo DNA methyltransferases, was upregulated at the initiation stage of fracture repair, peaking at 10 dpf, and declining thereafter to a nearly undetectable level by 28 dpf, whereas the expression of Dnmt1 and Dnmt3a remained unchanged over time (Fig. 1A), indicating a more important role of Dnmt3b in fracture repair. Interestingly, Dnmt3b displayed a restricted spatial expression pattern within the fracture callus. IHC for Dnmt3b on fracture callus sections from WT mice at multiple time points revealed that Dnmt3b is highly expressed in Sox9 + chondroprogenitors and chondrocytes, whereas the expression of Dnmt3b was nondetectable in osteoblasts, which was also confirmed by RNA sequencing (RNA-Seq) from primary calvarial osteoblast (data not shown) (Fig. B). These findings are consistent with the temporal pattern wherein significant upregulation and downregulation of Dnmt3b were seen during chondrogenesis (D2–D10) and osteogenesis (D14–D28) phases, respectively.(29,30) These results indicate a potential role of Dnmt3b in the regulation of chondrogenesis stage during fracture repair.
Fig. 1.
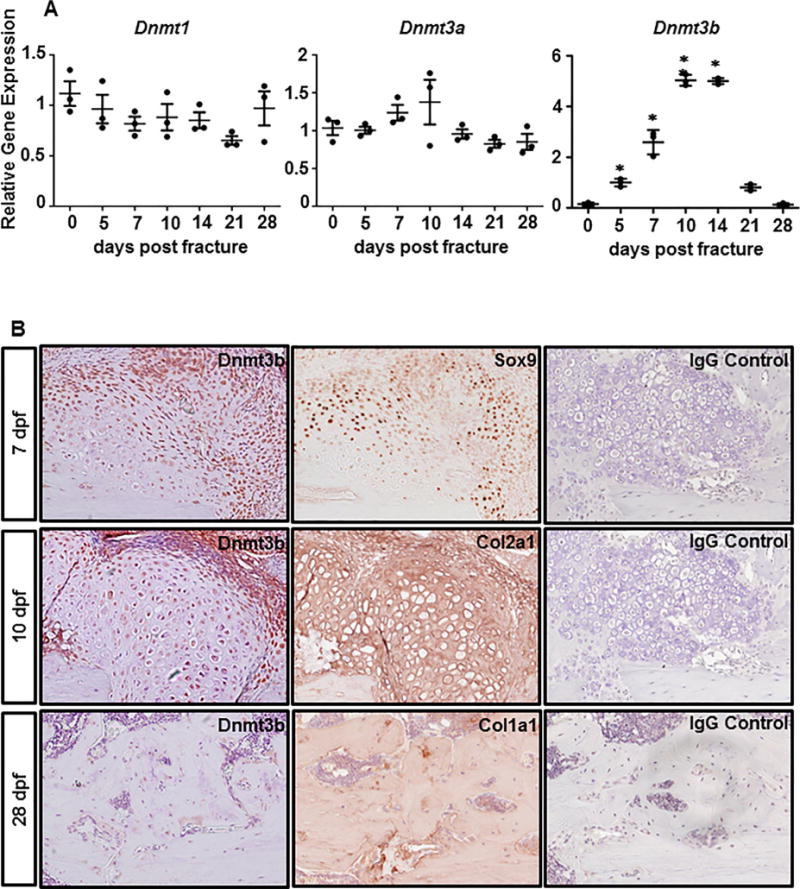
Temporal and spatial expression pattern of Dnmt3b during fracture repair. (A) Real-time qPCR analyses were performed to determine the relative expression of Dnmt1, Dnmt3a, and Dnmt3b in fracture calluses isolated from 2-month-old WT mice at the indicated times during fracture repair. The mRNA abundances were normalized to that of the gene coding β-actin and then were normalized to the 0 dpf. Data are means ±SD of at least three independent experiments.*p < 0.05 compared with 0 dpf by two-tailed Student’s t test. (B) Immunohistochemical analysis of Dnmt3b, Sox9, Col2a1, and Col1a1 expression in the fracture callus sections from 2-month-old WT mice at indicated times during fracture repair. IgG was used as negative controls. Images are representative of at least three independent experiments. WT = wild-type.
Dnmt3b positively regulates chondrocyte hypertrophic maturation during fracture repair
To determine whether Dnmt3b in chondrogenic cells is required during postnatal bone repair, we generated an LOF mouse model in which floxed alleles for Dnmt3b were conditionally deleted specifically within chondrogenic lineage cells using the tamoxifen-inducible Agc1CreERT2 transgene (Supplemental Fig. 1A). We first examined the cell lineages that are targeted by Agc1CreERT2 and contribute to fracture callus during fracture repair by analyzing tibia fractures on Agc1CreERT2;Ai9f/f (Ai9Agc1ER) mice at 5 dpf. Surprisingly, Agc1CreERT2 transgene could be activated no later than 5 dpf with our tamoxifen administration regime and more than 50% of Agc1-expression cells are Sox9+ chondroprogenitors(31,32) within the fracture callus (Supplemental Fig. 1A, B). To assess the recombination efficiency following tamoxifen-induced Cre-mediated gene deletion, we isolated fracture calluses largely in the endochondral bone ossification regions and extracted proteins from both Agc1Cre ERT2;Dnmt3bf/f (Dnmt3bAgc1ER) mutant mice and Cre-negative littermates (Dnmt3bf/f) at 10 dpf. Western blot analyses showed that Dnmt3b was reduced by approximately 50%, which was not unexpected due to the heterogeneous cell populations involved in the healing process (Supplemental Fig. 1C). IHC analyses from WT and Dnmt3bAgc1ER fracture calluses at 10 dpf during the endochondral phase further confirmed an efficient removal of Dnmt3b specifically from chondrocytes (Supplemental Fig. 1D). Both lineage tracing and protein expression results suggest that Agc1Cre ERT2-mediated recombination of Dnmt3b floxed alleles in chondrogenic lineage cells within fracture callus is sufficient.
Next, to determine whether Dnmt3b in chondrocytes is directly required for bone repair, we performed midshaft stabilized tibia fractures on Dnmt3bAgc1ER mutant mice and Cre-negative littermates (Control) at 2 months of age. Following fractures, tamoxifen was administrated using the same strategy aforementioned to target Dnmt3b deletion specifically in chondrocytes. Histological assessments of fracture repair were performed using ABH/OG staining of fracture callus sections at 7, 10, and 14 dpf (Fig. 2A). By 7 dpf, Cre-negative fracture callus was characterized by the formation of a cartilaginous island in the central hypoxic regions at the junction of fracture, and intramembranous ossification directly adjacent to the periosteum both proximally and distally to the fracture site, whereas literally no features of fracture repair were observed in Dnmt3bAgc1ER mutants at this stage. Such differences remained remarkable at 10 dpf. Cre-negative fractures revealed more robust cartilage and primary bone formation with cartilaginous callus peaking at this time; however, the magnitude of reparative response was markedly reduced in the mutant fractures, although fracture callus was apparent. Of note, cartilaginous callus in the control fractures was largely resorbed and replaced by primary bone formation at 14 dpf. In contrast, a central area of cartilage persisted at the junction of fractures of mutant mice, although bone formation was clearly evident at this time. Consistent with histological observations, quantitative histomorphometry (Fig. 2B) performed on ABH/OG-stained fracture callus sections indicated, on one hand, that cartilaginous callus formation begins within a week following fracture and peaks at 10 dpf followed by rapid resolution in both groups. On the other hand, despite similar temporal pattern of chondrogenesis between control and Dnmt3bAgc1ER mutant fractures, the latter exhibited decreased cartilage area within the callus at 10 dpf when peak cartilage formation occurred. Additionally, cartilage maturation was essentially delayed in Dnmt3bAgc1ER fractures because the entire removal of cartilage matrix appeared 1 week later in the mutant (28 dpf) than in the control fractures (21 dpf) (data not shown). These findings are consistent with the molecular changes (Supplemental Fig. 2) associated with the hypertrophic marker, COL10A1, because its expression was markedly less in the Dnmt3bAgc1ER fracture sections at 10 dpf compared with controls, which exhibited the most abundant levels of this matrix protein. By 14 dpf when COL10A1 was minimally visualized in control fractures, its expression persisted pericellularly surrounding the hypertrophic chondrocytes within the mutant fracture callus.
Fig. 2.
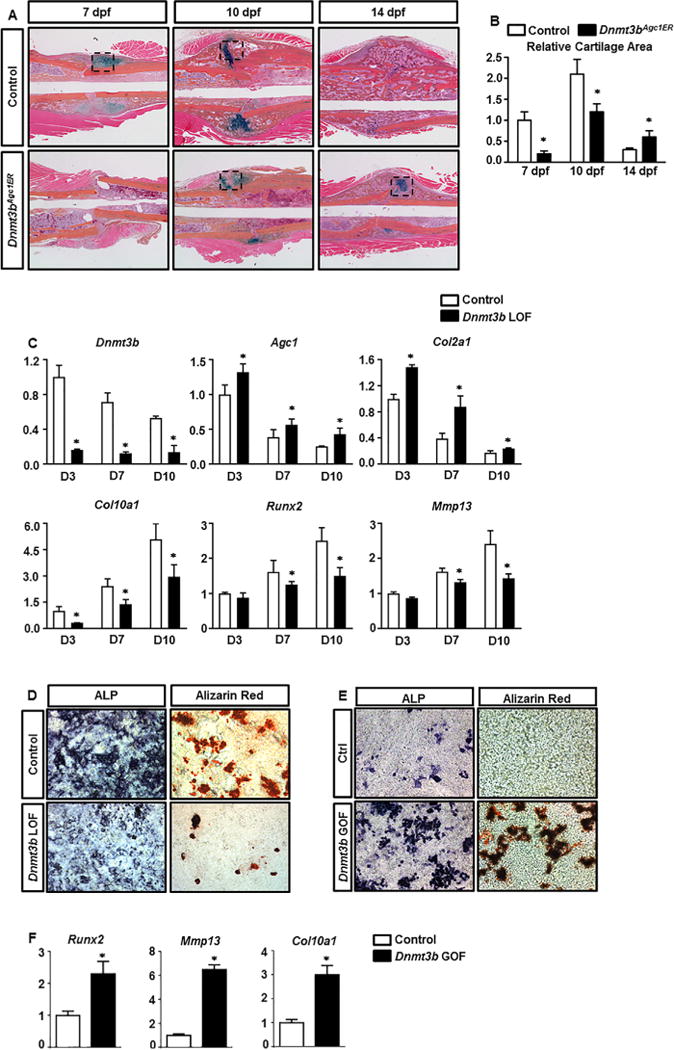
Loss of Dnmt3b in chondrocytes delays cartilage formation, maturation, and removal during fracture repair. (A) ABH/OG staining of fracture callus sections of control and Dnmt3bAgc1ER mice at the indicated times. Data are representative of at least five independent experiments (Black box represents cartilage tissue.). (B) Histomorphometric analyses of cartilage area were performed on fracture callus sections of control and Dnmt3bAgc1ER mice at the indicated times. All results were normalized to the controls at 7 dpf, which were set at 1. Data are means ±SD of at least five independent experiments. *p < 0.05 compared with control by two-way ANOVA followed by Dunnett’s test. (C) P2 Dnmt3bf/f costal chondrocytes were transduced with Ad-GFP or Ad-Cre adenoviruses for 48 hours, and then allowed to mature for up to 10 days. Following 48 hours of viral transductions, RNA was collected from chondrocyte cultures at D3, D7, and D10. Relative abundances of the indicated mRNAs were determined by real-time qPCR analyses. All mRNA abundances were normalized to that of β-actin and then were normalized to the controls. Data are means ±SD of at least three independent experiments. *p < 0.05 compared with control by two-tailed Student’s t test. (D) ALP and Alizarin red staining of control and Dnmt3b LOF chondrocyte cultures at day 5 and day 10 following viral transduction, respectively. (E) ALP and Alizarin red staining of control and Dnmt3b GOF chondrocyte cultures at day 10 following viral transduction. (F) Relative abundances of chondrocyte maturation genes were determined by real-time qPCR analyses from Dnmt3b GOF cells, including Runx2, Mmp13, and Col10a1. Data are representative of at least three independent experiments. ANOVA = analysis of variance; ALP = alkaline phosphatase.
In order to understand the role of Dnmt3b specifically in the chondrocyte lineage, we isolated primary chondrocytes from Dnmt3bf/f pups and treated with either control adenovirus expressing (Ad-GFP) or adenovirus expressing Cre recombinase (Ad-Cre) to delete Dnmt3b. Over the course of a 10-day maturation assay, the control chondrocytes displayed progressive upregulation of genes associated with chondrocyte maturation, including Col10a1, Runx2 and Mmp13, as well as gradual decline of early chondrocyte markers, such as Agc1 and Col2a1, reflecting a normal chondrocyte maturation process. By contrast, without affecting cell proliferation and viability, ablation of Dnmt3b in chondrocyte led to a dramatic increase in early chondrocyte differentiation maker gene expression and a subsequent decrease in later chondrocyte maturation maker gene expression relative to the control chondrocytes throughout the entire maturation process (Fig. 2C; Supplemental Fig. 3), indicating that Dnmt3b positively regulates chondrocyte maturation. This is fully in agreement with the reduced levels of alkaline phosphatase (ALP) and Alizarin red staining in the Dnmt3b LOF cultures at 5 days and 10 days postinfection, respectively (Fig. 2D). We also isolated the primary sternal chondrocytes from Dnmt3b transgenic mice and transfected with Lenti-GFP or Lenti-rtTA for 48 hours to overexpress Dnmt3b in chondrocytes. After 10 days culture with spontaneous maturation, ALP activity and mineralized matrix production were increased in Dnmt3b gain-of-function (GOF) chondrocytes as indicated by an increase in ALP and Alizarin red staining, respectively (Fig. 2E). Gene expression of chondrocyte maturation markers Col10a1, Runx2, and Mmp13 were also increased in Dnmt3b GOF sternal chondrocytes (Fig. 2F). These data, together with the in vivo histological findings (Fig. 2A, B), suggest that Dnmt3b is essential for chondrocyte terminal maturation during fracture repair process.
Elimination of Dnmt3b in chondrocytes results in delayed vascularization during fracture repair
Angiogenesis is an essential step that facilitates successful progression of bone regeneration during fracture repair. The severe impairment of chondrogenesis in the Dnmt3bAgc1ER mutant fractures led us to investigate whether endochondral-mediated angiogenesis of the injury site was affected. To this end, quantitative μCT analyses were used (as described in Materials and Methods) to evaluate vascularization within the fracture callus at 10 and 14 dpf. Representative angiograms of the control fracture calluses demonstrated robust revascularization within the fracture site (Fig. 3A1, A2). In contrast, Dnmt3bAgc1ER mice exhibited a marked reduction of vascularization (Fig. 3A3, A4). Specifically, quantification of vascularity showed significant decrease in both the vessel volume and the number of large and small blood vessels within the fracture callus of Dnmt3bAgc1ER mice (Fig. 3B1–B3). To further confirm these findings, we performed IHC for the endothelial marker, CD31 (Fig. 3A5, A6). During the phase of chondrocyte hypotrophy and cartilage resolution 10 days after injury, specific and abundant expression of CD31 was observed in endothelial cells of presumably regenerating vessels invaded areas of new bone formation, which was flanked by avascular cartilaginous tissue, indicating normal endochondral bone healing in Cre-negative mice. By contrast, staining for CD31 was barely detectable in regions of cartilaginous or bony tissues of fracture callus sections of Dnmt3bAgc1ER mutants.
Fig. 3.
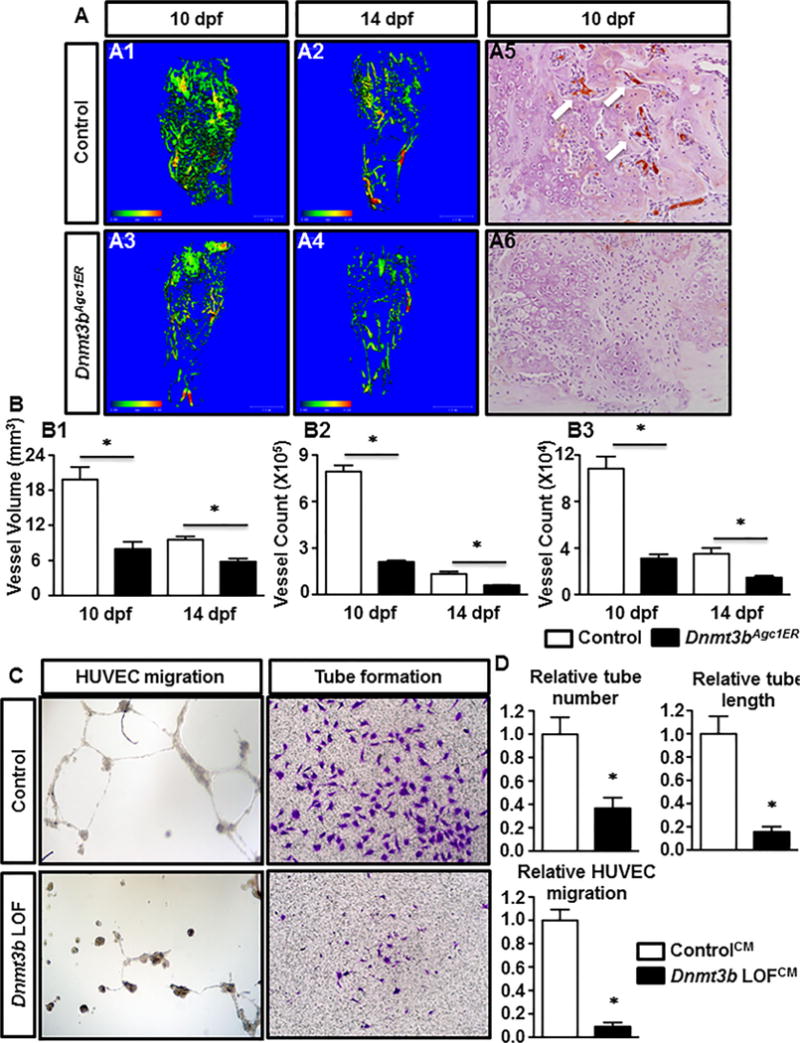
Removal of Dnmt3b in chondrocytes impairs endochondral-mediated vascularization of fracture callus and suppresses the angiogenic capacity of cultured chondrocytes. (A) μCT assessment of newly formed vessels within fracture callus at 10 and 14 dpf from control and Dnmt3bAgc1ER mice (A1–A4). Immunohistochemical analyses of PECAM/CD31 on fracture callus sections from WT and Dnmt3bAgc1ER mice at 10 dpf (A5, A6). Data are representative of at least five independent experiments. (B) Quantification of vascular network morphology, including vessel volume (B1), and vessel counts of large (Ø 0.0105 to 0.0210 mm) (B2) and small (Ø 0.0105 to 0.0210 mm) (B3) vessels. Data are means ±SD of at least five independent experiments. *p < 0.05 compared with control two-way ANOVA followed by Dunnett’s test. (C) HUVECs were seeded onto Matrigel in CM collected from Ad-GFP (ControlCM) or Ad-Cre (Dnmt3b LOFCM) adenoviruses infected primary chondrocytes. After overnight of culture, capillary-like structures were detected by bright field microscope. In the cell migration assay, HUVECs were seeded in the upper chamber of the transwell plates in serum-free medium with ControlCM or Dnmt3b LOFCM added in the bottom chamber. The migrated cells were stained with of crystal violet stain. Data are representative of three independent experiments. (D) Quantifications of HUVEC tube number, tube length, and migrated cell number, which normalized to the ControlCM. Data are expressed as means ±SD of three independent experiments. *p < 0.05 compared with ControlCM by two-tailed Student’s t test. HUVEC = human umbilical vein endothelial cell; CM = conditioned media.
In order to validate our in vivo vascularity findings in Dnmt3bAgc1ER mutant mice, we isolated primary chondrocytes from Dnmt3bf/f pups and treated with either Ad-GFP (Control chondrocyte culture) or Ad-Cre (Dnmt3b LOF chondrocyte culture) adenovirus for 48 hours. Following viral infection, the conditioned media (CM) was collected from Control or Dnmt3b LOF chondrocyte culture (referred to as ControlCM or Dnmt3b LOFCM hereafter) and subsequently used for in vitro angiogenesis assays with HUVECs. HUVEC tube formation with Dnmt3b LOFCM revealed a marked reduction of endothelial tube formation wherein both number and length of tubes were significantly decreased when compared to those treated with ControlCM (Fig. 3C, D). Furthermore, significant differences were also observed in the study of endothelial cell migration. Specifically, ControlCM exhibited a significant effect on HUVEC migration whereas a pronounced lack of cell migration was observed in the presence of Dnmt3b LOFCM (Fig. 3C, D). HUVEC proliferation and apoptosis when cultured with CM were also assessed to determine if any dysregulation in these processes could be part of the underlying cause for the suppressed angiogenic capacity. In fact, ControlCM or Dnmt3b LOFCM displayed no apparent difference in proliferative and apoptotic changes (Supplemental Fig. 4A, B), suggesting reduced endothelial tube formation and migration are unlikely attributed to altered cell growth and death. To further clarify the differential angiogenic capacity produced by conditioned culture media from Control and Dnmt3b LOF chondrocytes, a protein antibody array was employed to detect any alterations in angiogenic factors within the CM from Dnmt3b LOF chondrocytes and we uncovered that the expression of eight angiogenesis-related factors was downregulated in Dnmt3b LOF chondrocytes, of which CXCL12 and Osteopontin (OPN) were the most reduced factors in Dnmt3b LOFCM (Supplemental Fig. 5). Histogram profiles for selected angiogenic factors generated by quantifying the mean spot pixel densities from the array membrane reflected the downregulation of CXCL12 and OPN by 60% and 69%, respectively (Fig. 4A). More importantly, qPCR results further showed that Cxcl12 expression was gradually increased accompanying with chondrocyte maturation, whereas its expression was significantly reduced in Dnmt3b LOF chondrocytes, indicating downregulation of angiogenic factors during delayed chondrocyte maturation upon loss of Dnmt3b (Fig. 4B). In addition, hypomethylation alteration in Cxcl12 gene promoter was observed in Dnmt3b LOF chondrocytes (Fig. 4C). In accordance with these findings, we observed markedly reduced levels of CXCL12 and OPN in hypertrophic chondrocytes within the fracture callus in Dnmt3bAgc1ER mice relative to the Crenegative controls at 10 dpf. This reduction was particularly evident at the interface of avascular cartilaginous callus and vascularized bony callus, consistent with the decreased endochondral-mediated angiogenesis (Fig. 4D1–D6). Next, to determine whether downregulation of CXCL12 and OPN upon loss of Dnmt3b in chondrocytes was directly responsible for the impaired vascularization, exogenous CXCL12 and/or OPN were added to CM and used for the angiogenesis assays (Fig. 4E1–E16). The administration of either CXCL12 or OPN, or both, substantially increased the capacity of tube formation and cell migration of Dnmt3b LOFCM. Notably, addition of both proteins achieved the optimal effect, as reflected by quantitative analyses (Fig. 4F). Together, these results show that Dnmt3b deficiency in chondrocytes results in hypertrophic maturation delay and thus leads to vasculogenic response delay to injury likely via downregulation of certain key angiogenic factors, primarily CXCL12 and OPN.
Fig. 4.
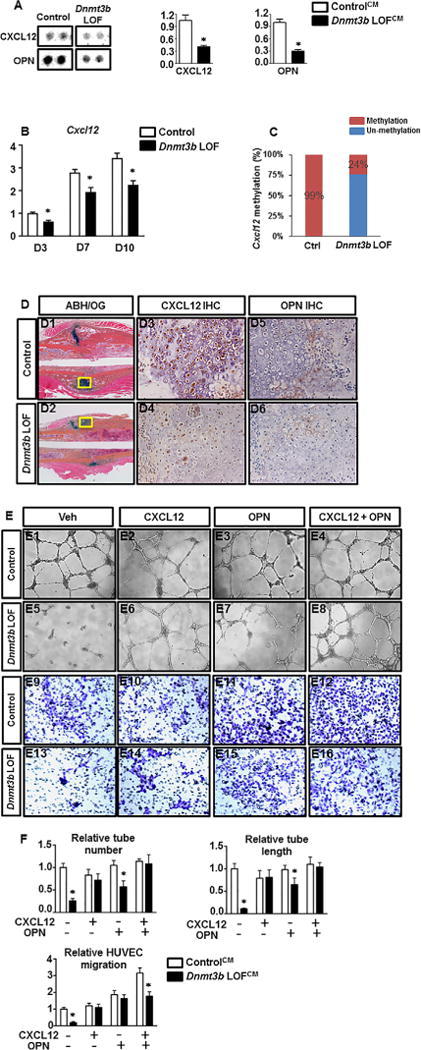
Impaired vascularization in Dnmt3bAgc1ER fractures is likely due to the decreased production of proangiogenic factors by chondrocytes. (A) Images and quantitative results of angiogenesis proteasome array. The ControlCM and Dnmt3b LOFCM were used for the parallel determination of the relative levels of angiogenesis-related factors. Images are representative of four independent experiments. Quantitative results are expressed as means ±SD of three independent experiments. *p < 0.05 compared with ControlCM by two-tailed Student’s t test. (B) P2 Dnmt3bf/f costal chondrocytes were transduced with Ad-GFP or Ad-Cre adenoviruses for 48 hours, and then allowed to mature for up to 10 days. Following 48 hours of viral transductions, RNA was collected from chondrocyte cultures at D3, D7, and D10. Cxcl12 and Opn mRNA level was determined by real-time qPCR analyses. Data are means ±SD of at least three independent experiments. *p < 0.05 compared with control by two-tailed Student’s t test. (C) Methylation pattern in Cxcl12 promoter region was determined by methylation qPCR between Ctrl and Dnmt3b LOF chondrocytes. (D) ABH/OG staining and immunohistochemical analyses for CXCL12 and OPN on fracture callus sections from control and Dnmt3bAgc1ER mice at the indicated times. A centralized cartilaginous region of fracture callus (yellow boxes in D1 and D2) is shown at high magnification in D3–D6. Data are representative of at least five independent experiments. (E) HUVEC tube formation and cell migration assays were performed with HUVECs in control or Dnmt3b conditioned media in the absence or presence of CXCL12 and/or OPN. Bright field images of tube formation are shown in E1–E8. E9–E16 indicate crystal violet staining of migrated cells. All results are representative of three independent experiments. (F) Quantifications of HUVEC tube number, tube length, and migrated cell number. Data are expressed as means ±SD of three independent experiments. *p < 0.05 compared with ControlCM by two-way ANOVA followed by Dunnett’s test.
Ablation of Dnmt3b in chondrocytes results in delayed cortical bridging and bone formation, and altered bony callus bone remodeling
Because we observed defects in cartilage maturation and vascularization within the fracture callus upon loss of Dnmt3b in chondrocytes, we next determined whether the later phases of fracture repair were also affected. μCT analyses (Fig. 5A1–A6) on mineralized calluses of control fractures showed a complete bridging of bony calluses by 14 dpf, followed by a nearly complete remodeling at 28 dpf. Reconstruction of μCT data revealed that mineralized callus volume in control fractures gradually decreased from 14 dpf, indicating progressive callus remodeling (Fig. 5B). By contrast, a thin radiolucent line partially remained in Dnmt3bAgc1ER fracture by 14 dpf. Bone formation appeared to be more pronounced in Dnmt3bAgc1ER mutant fractures at 21 dpf relative to the early stages of fracture pair. Quantitatively, Dnmt3bAgc1ER mutant fractures displayed significantly lower amount of bony callus volume at 14 dpf, though displaying greater mineralized callus at later time points as compared to control fractures (Fig. 5B). The findings that Dnmt3bAgc1ER mutant and control fractures differed in temporal course suggest that loss of Dnmt3b in chondrocytes during fracture repair leads to delay in both bone formation and bone remodeling.
Fig. 5.
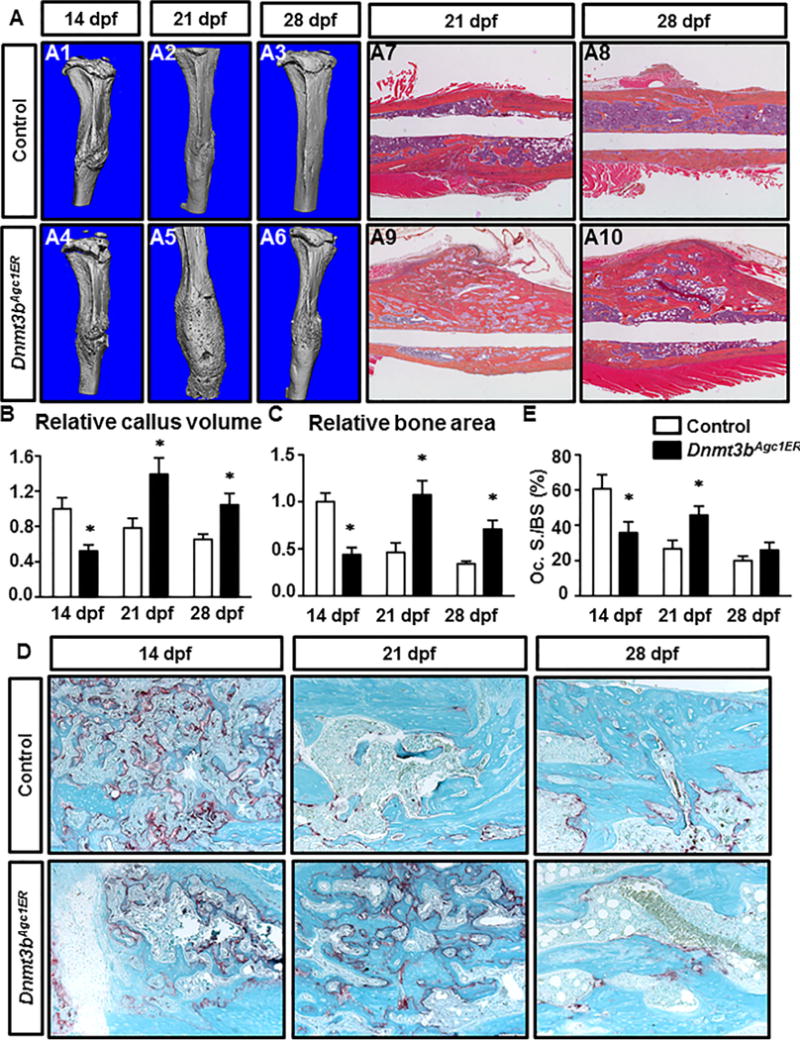
Loss of Dnmt3b in chondrocytes delays bone formation and alters bony callus remodeling during fracture repair. (A) μCT assessment of mineralized bone within fracture callus at the indicated times from control and Dnmt3bAgc1ER mice (A1–A6). ABH/OG staining of fracture callus sections of control and Dnmt3bAgc1ER mice at 21 dpf and 28 dpf (A7–A10). Data are representative of at least five experiments. (B) Quantification of bony callus volume from at the indicated times from control and Dnmt3bAgc1ER mice. All results were normalized to the controls at 14 dpf (Fig. 2), which were set at 1. (C) Histomorphometric analyses of bone area were performed on fracture callus sections of control and Dnmt3bAgc1ER mice at the indicated times. All results were normalized to the controls at 14 dpf (Fig. 2), which were set at 1. (D) TRAP staining of ABH/OG staining of fracture callus sections of control and Dnmt3bAgc1ER mice at the indicated times. Data are representative of at least five experiments. (E) Histomorphometric analyses of the ratio of Oc.S/BS were performed on fracture callus sections of control and Dnmt3bAgc1ER mice at the indicated times. Data are expressed as means ±SD of at least five independent experiments. *p < 0.05 compared with control by two-way ANOVA followed by Dunnett’s test. Oc.S/BS = osteoclast surface/bone surface.
The μCT findings were complemented by histological assessments of fracture repair using ABH/OG staining of fracture calluses at the same time points (Fig. 5A7–A10). By 21 dpf and beyond, fracture repair in the control mice was nearly completed, as illustrated by the restoration of the original lamellar structure of the cortical bone. Alternatively at 21 dpf, Dnmt3bAgc1ER mutants exhibited abundant bony callus with dissipated unmineralized cartilage residuals. By 28 dpf, massive woven bone could still be visualized surrounding the mutant fracture site. Consistent with histological observations, quantitative histomorphometry (Fig. 5C) revealed that bone area progressively increased until 14 dpf and 21 dpf in the control and mutant fractures (data of 10 dpf not shown), respectively, suggesting Dnmt3bAgc1ER mutants experienced delays in both bone formation and subsequent bone remodeling. However, no significant difference in the amount of peak bone formation was detected between the control and mutant fractures. More importantly, the expression of osteoblast genes, including Col1a1, Bsp, and Oc, were similarly expressed in control and Dnmt3b LOF callus at both 14 and 21 dpf (Supplemental Fig. 6A). Altogether, these data suggest that the capabilities of osteogenic differentiation from mesenchymal progenitor/stem cells and osteoblast-mediated bone formation were largely unaffected. Additionally, TRAP staining for osteoclasts was also assessed to determine if osteoclastogenesis was dysregulated in Dnmt3bAgc1ER fractures. The total number of TRAP-positive cells not only peaked one week later in the mutant fractures (Fig. 5D), but the bone remodeling activity, as indicated by the percentage of bone surface covered by osteoclasts (Oc.S/BS), was also lower in the Dnmt3bAgc1ER fractures at 21 dpf when comparing to that of Cre-negative controls at 28 dpf (Fig. 5E). However, the Opg/Rankl ratio was virtually no difference between control and Dnmt3b LOF fracture callus (Supplemental Fig. 6B), suggesting altered bone remodeling observed in Dnmt3bAgc1ER fractures is unlikely due to the deficiency of osteoclast formation and function.
To examine the biomechanical properties of the newly formed bone in these mice, biomechanical torsion testing, which is considered as the definitive measure of fracture healing, was performed on fractured tibias harvested from Dnmt3bAgc1ER and control mice at 28 dpf (Fig. 6). Dnmt3bAgc1ER mutant fractures, though displaying greater volume of bony callus at this point, exhibited significantly lower maximum torque and torsional rigidity (reduced by nearly 41% and 50%, respectively) relative to the control fractures. Furthermore, Dnmt3bAgc1ER mutant fractures required less energy to fail during torsion testing (57% decrease in energy to maximum) when compared to the control group. Collectively, these data confirm that loss of Dnmt3b in chondrocytes markedly lowers biomechanical parameters of the newly formed bone during fracture repair.
Fig. 6.
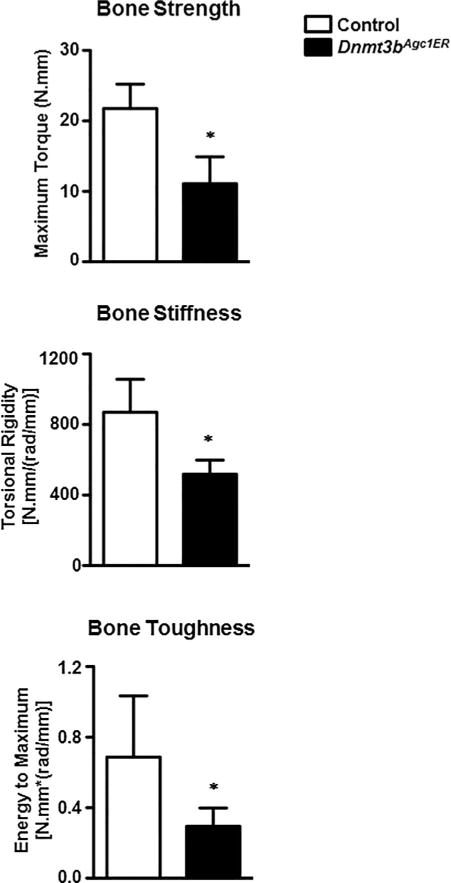
Dnmt3bAgc1ER fractures have remarkably inferior biomechanical properties. Biomechanical torsion testing of control and Dnmt3bAgc1ER fractures at 28 dpf. All biomechanical parameters, including the maximum torque, torsional rigidity, and energy to maximum, which represent the bone strength, bone stiffness, and bone toughness, are expressed as means ±SD of at least five independent experiments. *p < 0.05 compared with control by two-tailed Student’s t test.
Discussion
In this study, we provide the first conclusive genetic evidence that Dnmt3b positively regulates chondrogenesis and chondrocyte maturation during endochondral bone repair process. Using in vivo and in vitro approaches we show that loss of chondrocyte-specific Dnmt3b decreases chondrocyte hypertrophic maturation, resulting in the persistency of unmineralized cartilage within fracture callus during skeletal repair. This study also implicates Dnmt3b in chondrocyte-specific regulation of vascularization of the callus during the early to midstage of healing. In this regard, in vitro data indicate that delayed angiogenesis within fracture callus is likely due to decreased production of key angiogenesis factors accompanying with chondrocyte maturation delay. Furthermore, our results suggest that prolonged bone formation and altered bone remodeling observed throughout the fracture healing process are potentially secondary defects to decreased cartilage maturation and delayed vascularization within fracture callus.
Chondrocyte hypertrophic differentiation, an essential process during endochondral bone regeneration, predominantly recapitulates the cartilage development in the embryonic stage wherein the involvement of transcriptional factors, growth factors, and changes in gene expression have been well studied. However, to date, few reports have analyzed the role of epigenetic regulators within the chondrogenic lineage. Recent studies have been mainly restricted to assessing DNA methylation levels in gene promoters related to chondrocyte phenotypes and chondrogenesis utilizing diverse in vitro models.(33–35) However, the identity and specificity of DNA-methylating enzymes involved in chondrogenic differentiation in vivo remains unknown. The present study demonstrates that Dnmt3b is induced at the initiation stage of fracture repair with abundant expression in chondrogenic lineage cells within murine fracture callus. More importantly, chondrocyte-specific loss of Dnmt3b led to delayed cartilaginous callus formation as well as its maturation with persistence of unmineralized cartilage during fracture repair. Consistently, in vitro data further confirmed that ablation of Dnmt3b in primary sternal chondrocytes inhibited the terminal maturation of chondrocytes, whereas overexpression of Dnmt3b enhanced chondrocyte maturation. Together, these in vivo and in vitro results suggest that Dnmt3b physiologically promotes chondrogenic hypertrophic differentiation and terminal maturation in the context of endochondral bone repair. Despite current limited understanding, we do observe that alterations in chondrogenic differentiation upon Dnmt3b LOF in chondrocytes are broadly reminiscent of differentiation defects associated with Dnmt3b-null ESCs and adult HSCs.(16–18) Moreover, because postnatal bone repair largely recapitulates various aspects of skeletal development, it is reasonable to speculate that the role of Dnmt3b in regulating chondrogenic progression in the context of skeletal repair may be a hallmark of cartilage development. Therefore, it is worth it to further elucidate the role of Dnmt3b in mesenchymal stem cells and progenitors, especially during the chondrogenesis process, which may provide better understanding of the correlation between delayed chondrogenesis and delayed chondrocyte maturation and angiogenesis in the context of Dnmt3b LOF in fracture repair. In addition, clinical observations revealed alarming clinical complications associated with fracture healing, including prolonged healing and nonunion, in elderly patients, smoking, or diabetic patients,(36–38) highlighting the potential deleterious role of chronic systemic inflammation in fracture repair in these patients. However, the underlying mechanism is largely unknown. Other in vitro studies have documented recently that DNA methylation(39) and demethylation(40) enzymes, including Dnmts and Tets, were downregulated in synovial fibroblasts or chondrocytes under IL-1β treatment, indicating DNA methylation is a primary target under inflammatory conditions. Therefore, it will be interesting to elucidate if downregulation of Dnmt3b in chondrocytes induced by inflammatory signals, at least partially, lead to delayed fracture repair under the disease conditions.
Fracture healing is a precisely regulated multistage process during which chondrocyte maturation is a key event coupling the consequential vascularities as well as bony callus formation and remodeling. Adequate blood supply is essential for delivery of growth factors and cytokines to the fracture site leading to the successful bone repair.(41) Indeed, the lack or inhibition of angiogenesis has been reported as a major cause of delayed union or non-union during fracture healing.(9–11,42) During endochondral bone repair, although the initial cartilaginous callus remains avascular, the chondrocytes within the callus become potent stimulators of angiogenesis and vascular invasion as they mature to hypertrophy through secreting a plethora of proangiogenic(43–47) factors that exert chemotactic and proliferative effects on endothelial cells. The coupling of angiogenesis with chondrocyte hypertrophy during endochondral ossification is supported by the findings in which inhibition of chondrocyte differentiation leads to delayed or even absent vascular invasion.(44,48–50) In this study, our data show that chondrocyte-specific removal of Dnmt3b during fracture repair resulted in marked reduction of angiogenesis in the early to mid phases, as reflected by the significant less vessel volume and numbers and diminished expression of CD31 within the callus in the mutant fractures as compared with WT counterparts. In agreement with these in vivo observations, our in vitro results provided further evidence that chondrocytes highly expressed the angiogenic factors accompanying with chondrocyte maturation, while it was inhibited upon loss of Dnmt3b in chondrocytes, thereby leading to decreased endothelial tube formation and migration as well as angiogenesis delay seen in vitro and in vivo, respectively. Through utilizing an angiogenesis array, of all the 53 angiogenesis-related proteins, the levels of two angiogenic factors, CXCL12 and OPN, were distinctly decreased in the matrix of Dnmt3b LOF chondrocytes. Furthermore, the defects in endothelial tube formation could be rescued by exogenous administration of OPN and CXCL12 in vitro, indicating that such angiogenesis suppression was potentially attributed to the decreased release of these two angiogenic factors from Dnmt3b LOF chondrocytes. The critical role for CXCL12, a well-known chemokine expressed in prehypertrophic and hypertrophic chondrocytes,(51,52) in many aspects of bone regeneration have now been established. In addition to regulating the recruitment of stem and progenitor cells,(53–55) Fujio and colleagues(56) have recently shown that SDF-1/CXCL12 stimulated callus formation within the bone fragments through inducing the recruitment of bone marrow endothelial and endothelial progenitor cells, thus improving vascularization. OPN, as previously shown by in situ studies, is expressed in the fracture callus by hypertrophic chondrocytes in the cartilage to bone transitional region, as well as in osteoprogenitors, osteoblasts, osteocytes, and osteoclasts in the hard callus.(57,58) More relevantly, the biological role of OPN during fracture healing has been implicated by a study demonstrating that OPN deficiency alters the function of multiple cell types, resulting in impaired neovascularization, altered mineralization and late remodeling, and reduced biomechanical properties during fracture healing.(59)
Regarding the bone formation, the reduction of the early bony callus volume and area as assessed by the μCT and histomorphometric analyses did not persist, but rather recovered by 21 dpf in Dnmt3b mutant fractures. It is also worth noting that, unlike the cartilaginous callus, both the peak bony callus volume and area were slightly larger in the mutant fractures, suggesting osteoblast mediated bone formation was not adversely altered. Given the close association of angiogenesis and osteogenesis,(60,61) it is not surprising to have the bone formation delay in Dnmt3b mutant mice although Agc1CreERT2; Dnmt3bf/f solely targets deletion of Dnmt3b in chondrocytes but not osteoblasts. Such difference may also be readily explained by the fact that the acute ablation of Dnmt3b beginning at chondrogenesis phase is resolved as chondrocytes terminally differentiate and eventual apoptosis occurs, although angiogenesis was delayed, which is likely due to reduced release of some key angiogenic factors from the supernatant. By contrast, the decreased bone-resorbing activity of osteoclast, as reflected by the lower value of Oc.S/BS in Dnmt3b mutant fractures that occurred 1 week later than in control fractures, was unexpected. Whether this event reflects Dnmt3b-mediated cell-nonautonomous regulation of osteoclastogenesis or an indirect paracrine modulation of osteoclastogenesis mediated by chondrocytes needs to be further determined. Nevertheless, the altered bone remodeling was highlighted by the inferior biomechanical outcomes of the mutant fractures. In addition, given the fact that osteoclasts and endothelial cells are frequently in close contact,(61) and based on the evidence that osteoclast stimulates angiogenesis in vivo,(62) it is very likely that osteoclastogenesis and angiogenesis are mutually regulated during bone regeneration and that delayed angiogenesis observed in the early to mid stages contributes to the defect in osteoclast formation and bone remodeling. This speculation was further reinforced by the observation with regard to the Opg/Rankl ratio in fracture callus, which was virtually no difference between the control and Dnmt3b mutant mice, indicating that the alterations in osteoclast activity or the delayed bone remodeling could possibly be due to the delay in angiogenesis.
Although the underlying mechanisms remain to be explored, this study provides the first genetic evidence clarifying how Dnmt3b controls chondrogenic differentiation and maturation in the context of fracture repair. We also looked into the correlation between delayed chondrocyte maturation and affected angiogenesis in Dnmt3b LOF fractures and further showed the methylation alteration in the angiogenic factor CXCL12 as a proof of concept of epigenetic regulation. In fact, recent studies demonstrate that the role of DNA methylation is complex in regulation of gene expression. In particular, it has been demonstrated that gene transcription is regulated by the combination of DNA methylation pattern across the promoter, enhancer, and gene body.(63,64) Consistent with this concept, Dr. Liang’s group (Yang and colleagues(65))observed that demethylation of the gene body led to downregulation of the gene, although the promoter was unmethylated. Additionally, Weber and colleagues(66) demonstrated that there are two distinct promoters in the genome, the strong CpG island promoter and weak CpG island promoter, which can differently regulate gene expression through DNA methylation; eg, the gene can be suppressed although strong CpG island promoter is unmethylated. Furthermore, Dr. Sachan’s group (Pamnani and colleagues(67))reported that even within the same promoter, the single CpG island methylation (six in total) cannot correlate with Oct4 gene expression during testicular development. Because we only detect the methylation in a single CpG island in Cxcl12 gene, our data cannot provide comprehensive insight into the effect of DNA methylation on Cxcl12 gene expression and cannot directly link a specific CpG site in the Cxcl12 with the gene expression profile. Thus, in order to better understand the epigenetic mechanism underlying genomewide regulation by Dnmt3b both in the context of skeletal development and bone regeneration, further evaluation of the DNA methylation pattern by using more advanced and comprehensive methodologies, eg, bisulfite genomic methylation sequencing, instead of single CpG methylation detection appears to be required. And critical downstream targets should be selected based on the comprehensive whole genome methylation analyses for the further mechanistic study. Taken together, this body of work shows that epigenetic regulation influences many aspects of skeletal repair and highlights the critical role in regulating chondrogenic differentiation implicating Dnmt3b as potential participant in the pathogenesis of impaired fracture repair and other chondrogenesis-associated disorders.
Supplementary Material
Acknowledgments
This work was supported by grants from the National Institutes of Health/National Institute of Arthritis and Musculoskeletal and Skin Diseases (R01 AR054326 and AR049192 to YA, and R01 AR069605 to RJO). We thank Center for Musculoskeletal Research Histology Core members Sarah Mack, Kathy Maltby, and Ashish Thomas for their technical assistance.
Footnotes
Additional Supporting Information may be found in the online version of this article
Disclosures
All authors state that they have no conflicts of interest.
Authors’ roles: Study design: CW, YA, RJO, and JS. Study conduct: CW, RJO, and JS. Data collection: CW and JS. Data analysis: CW, YA, RJO, and JS. Data interpretation: CW, YA, RJO, and JS. Drafting manuscript: CW, YA, RJO, and JS.
References
- 1.Ai-Aql ZS, Alagl AS, Graves DT, Gerstenfeld LC, Einhorn TA. Molecular mechanisms controlling bone formation during fracture healing and distraction osteogenesis. J Dent Res. 2008;87(2):107–18. doi: 10.1177/154405910808700215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Vortkamp A, Pathi S, Peretti GM, Caruso EM, Zaleske DJ, Tabin CJ. Recapitulation of signals regulating embryonic bone formation during postnatal growth and in fracture repair. Mech Dev. 1998;71(1–2):65–76. doi: 10.1016/s0925-4773(97)00203-7. [DOI] [PubMed] [Google Scholar]
- 3.Ferguson C, Alpern E, Miclau T, Helms JA. Does adult fracture repair recapitulate embryonic skeletal formation? Mech Dev. 1999;87(1–2):57–66. doi: 10.1016/s0925-4773(99)00142-2. [DOI] [PubMed] [Google Scholar]
- 4.Taylor DK, Meganck JA, Terkhorn S, et al. Thrombospondin-2 influences the proportion of cartilage and bone during fracture healing. J Bone Miner Res. 2009;24(6):1043–54. doi: 10.1359/jbmr.090101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Burke DP, Kelly DJ. Substrate stiffness and oxygen as regulators of stem cell differentiation during skeletal tissue regeneration: a mechanobiological model. PLoS One. 2012;7(7):e40737. doi: 10.1371/journal.pone.0040737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Brandi ML, Collin-Osdoby P. Vascular biology and the skeleton. J Bone Miner Res. 2006;21(2):183–92. doi: 10.1359/JBMR.050917. [DOI] [PubMed] [Google Scholar]
- 7.Chim SM, Tickner J, Chow ST, et al. Angiogenic factors in bone local environment. Cytokine Growth Factor Rev. 2013;24(3):297–310. doi: 10.1016/j.cytogfr.2013.03.008. [DOI] [PubMed] [Google Scholar]
- 8.Santos MI, Reis RL. Vascularization in bone tissue engineering: physiology, current strategies, major hurdles and future challenges. Macromol Biosci. 2010;10(1):12–27. doi: 10.1002/mabi.200900107. [DOI] [PubMed] [Google Scholar]
- 9.Hausman MR, Schaffler MB, Majeska RJ. Prevention of fracture healing in rats by an inhibitor of angiogenesis. Bone. 2001;29(6):560–4. doi: 10.1016/s8756-3282(01)00608-1. [DOI] [PubMed] [Google Scholar]
- 10.Fang TD, Salim A, Xia W, et al. Angiogenesis is required for successful bone induction during distraction osteogenesis. J Bone Miner Res. 2005;20(7):1114–24. doi: 10.1359/JBMR.050301. [DOI] [PubMed] [Google Scholar]
- 11.Holstein JH, Klein M, Garcia P, et al. Rapamycin affects early fracture healing in mice. Br J Pharmacol. 2008;154(5):1055–62. doi: 10.1038/bjp.2008.167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jaenisch R, Bird A. Epigenetic regulation of gene expression: how the genome integrates intrinsic and environmental signals. Nat Genet. 2003;33(Suppl):245–54. doi: 10.1038/ng1089. [DOI] [PubMed] [Google Scholar]
- 13.Lei H, Oh SP, Okano M, et al. De novo DNA cytosine methyltransferase activities in mouse embryonic stem cells. Development. 1996;122(10):3195–205. doi: 10.1242/dev.122.10.3195. [DOI] [PubMed] [Google Scholar]
- 14.Li E, Bestor TH, Jaenisch R. Targeted mutation of the DNA methyltransferase gene results in embryonic lethality. Cell. 1992;69(6):915–26. doi: 10.1016/0092-8674(92)90611-f. [DOI] [PubMed] [Google Scholar]
- 15.Okano M, Bell DW, Haber DA, Li E. DNA methyltransferases Dnmt3a and Dnmt3b are essential for de novo methylation and mammalian development. Cell. 1999;99(3):247–57. doi: 10.1016/s0092-8674(00)81656-6. [DOI] [PubMed] [Google Scholar]
- 16.Chen T, Ueda Y, Dodge JE, Wang Z, Li E. Establishment and maintenance of genomic methylation patterns in mouse embryonic stem cells by Dnmt3a and Dnmt3b. Mol Cell Biol. 2003;23(16):5594–605. doi: 10.1128/MCB.23.16.5594-5605.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Challen GA, Sun D, Jeong M, et al. Dnmt3a is essential for hematopoietic stem cell differentiation. Nat Genet. 2011;44(1):23–31. doi: 10.1038/ng.1009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Challen GA, Sun D, Mayle A, et al. Dnmt3a and Dnmt3b have overlapping and distinct functions in hematopoietic stem cells. Cell Stem Cell. 2014;15(3):350–64. doi: 10.1016/j.stem.2014.06.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Shen J, Wang C, Li D, et al. DNA methyltransferase 3b regulates articular cartilage homeostasis by altering metabolism. JCI Insight. 2017;2(12):e93612. doi: 10.1172/jci.insight.93612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Madisen L, Zwingman TA, Sunkin SM, et al. A robust and high-throughput Cre reporting and characterization system for the whole mouse brain. Nat Neurosci. 2010;13(1):133–40. doi: 10.1038/nn.2467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Muzumdar MD, Tasic B, Miyamichi K, Li L, Luo L. A global double-fluorescent Cre reporter mouse. Genesis. 2007;45(9):593–605. doi: 10.1002/dvg.20335. [DOI] [PubMed] [Google Scholar]
- 22.Henry SP, Jang CW, Deng JM, Zhang Z, Behringer RR, de Crombrugghe B. Generation of aggrecan-CreERT2 knockin mice for inducible Cre activity in adult cartilage. Genesis. 2009;47(12):805–14. doi: 10.1002/dvg.20564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wang C, Shen J, Yukata K, et al. Transient gamma-secretase inhibition accelerates and enhances fracture repair likely via Notch signaling modulation. Bone. 2015;73:77–89. doi: 10.1016/j.bone.2014.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Reynolds DG, Shaikh S, Papuga MO, et al. muCT-based measurement of cortical bone graft-to-host union. J Bone Miner Res. 2009;24(5):899–907. doi: 10.1359/JBMR.081232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhang X, Xie C, Lin AS, et al. Periosteal progenitor cell fate in segmental cortical bone graft transplantations: implications for functional tissue engineering. J Bone Miner Res. 2005;20(12):2124–37. doi: 10.1359/JBMR.050806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Xie C, Reynolds D, Awad H, et al. Structural bone allograft combined with genetically engineered mesenchymal stem cells as a novel platform for bone tissue engineering. Tissue Eng. 2007;13(3):435–45. doi: 10.1089/ten.2006.0182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gunnell LM, Jonason JH, Loiselle AE, et al. TAK1 regulates cartilage and joint development via the MAPK and BMP signaling pathways. J Bone Miner Res. 2010;25(8):1784–97. doi: 10.1002/jbmr.79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Arnaoutova I, Kleinman HK. In vitro angiogenesis: endothelial cell tube formation on gelled basement membrane extract. Nat Protoc. 2010;5(4):628–35. doi: 10.1038/nprot.2010.6. [DOI] [PubMed] [Google Scholar]
- 29.Pivonka P, Dunstan CR. Role of mathematical modeling in bone fracture healing. Bonekey Rep. 2012;1:221. doi: 10.1038/bonekey.2012.221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Einhorn TA, Gerstenfeld LC. Fracture healing: mechanisms and interventions. Nat Rev Rheumatol. 2015;11(1):45–54. doi: 10.1038/nrrheum.2014.164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ono N, Ono W, Nagasawa T, Kronenberg HM. A subset of chondrogenic cells provides early mesenchymal progenitors in growing bones. Nat Cell Biol. 2014;16(12):1157–67. doi: 10.1038/ncb3067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Akiyama H, Kim JE, Nakashima K, et al. Osteo-chondroprogenitor cells are derived from Sox9 expressing precursors. Proc Natl Acad Sci U S A. 2005;102(41):14665–70. doi: 10.1073/pnas.0504750102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ezura Y, Sekiya I, Koga H, Muneta T, Noda M. Methylation status of CpG islands in the promoter regions of signature genes during chondrogenesis of human synovium-derived mesenchymal stem cells. Arthritis Rheum. 2009;60(5):1416–26. doi: 10.1002/art.24472. [DOI] [PubMed] [Google Scholar]
- 34.Zimmermann P, Boeuf S, Dickhut A, Boehmer S, Olek S, Richter W. Correlation of COL10A1 induction during chondrogenesis of mesenchymal stem cells with demethylation of two CpG sites in the COL10A1 promoter. Arthritis Rheum. 2008;58(9):2743–53. doi: 10.1002/art.23736. [DOI] [PubMed] [Google Scholar]
- 35.Herlofsen SR, Bryne JC, Hoiby T, et al. Genome-wide map of quantified epigenetic changes during in vitro chondrogenic differentiation of primary human mesenchymal stem cells. BMC Genomics. 2013;14:105. doi: 10.1186/1471-2164-14-105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Liuni FM, Rugiero C, Feola M, et al. Impaired healing of fragility fractures in type 2 diabetes: clinical and radiographic assessments and serum cytokine levels. Aging Clin Exp Res. 2015;27(Suppl 1):S37–44. doi: 10.1007/s40520-015-0422-4. [DOI] [PubMed] [Google Scholar]
- 37.Peterson BE, Jiwanlal A, Della Rocca GJ, Crist BD. Orthopedic trauma and aging: it isn’t just about mortality. Geriatr Orthop Surg Rehabil. 2015;6(1):33–6. doi: 10.1177/2151458514565663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Heyes GJ, Tucker A, Marley D, Foster A. Predictors for 1-year mortality following hip fracture: a retrospective review of 465 consecutive patients. Eur J Trauma Emerg Surg. 2017;43(1):113–9. doi: 10.1007/s00068-015-0556-2. [DOI] [PubMed] [Google Scholar]
- 39.Nakano K, Boyle DL, Firestein GS. Regulation of DNA methylation in rheumatoid arthritis synoviocytes. J Immunol. 2013;190(3):1297–303. doi: 10.4049/jimmunol.1202572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Haseeb A, Makki MS, Haqqi TM. Modulation of ten-eleven translocation 1 (TET1), Isocitrate Dehydrogenase (IDH) expression, alpha-Ketoglutarate (alpha-KG), and DNA hydroxymethylation levels by interleukin-1beta in primary human chondrocytes. J Biol Chem. 2014;289(10):6877–85. doi: 10.1074/jbc.M113.512269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hankenson KD, Dishowitz M, Gray C, Schenker M. Angiogenesis in bone regeneration. Injury. 2011;42(6):556–61. doi: 10.1016/j.injury.2011.03.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Stegen S, van Gastel N, Carmeliet G. Bringing new life to damaged bone: the importance of angiogenesis in bone repair and regeneration. Bone. 2015;70:19–27. doi: 10.1016/j.bone.2014.09.017. [DOI] [PubMed] [Google Scholar]
- 43.Colnot CI, Helms JA. A molecular analysis of matrix remodeling and angiogenesis during long bone development. Mech Dev. 2001;100(2):245–50. doi: 10.1016/s0925-4773(00)00532-3. [DOI] [PubMed] [Google Scholar]
- 44.Gerber HP, Vu TH, Ryan AM, Kowalski J, Werb Z, Ferrara N. VEGF couples hypertrophic cartilage remodeling, ossification and angiogenesis during endochondral bone formation. Nat Med. 1999;5(6):623–8. doi: 10.1038/9467. [DOI] [PubMed] [Google Scholar]
- 45.Maes C, Coenegrachts L, Stockmans I, et al. Placental growth factor mediates mesenchymal cell development, cartilage turnover, and bone remodeling during fracture repair. J Clin Invest. 2006;116(5):1230–42. doi: 10.1172/JCI26772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Andrew JG, Hoyland JA, Freemont AJ, Marsh DR. Platelet-derived growth factor expression in normally healing human fractures. Bone. 1995;16(4):455–60. doi: 10.1016/8756-3282(95)90191-4. [DOI] [PubMed] [Google Scholar]
- 47.Vu TH, Shipley JM, Bergers G, et al. MMP-9/gelatinase B is a key regulator of growth plate angiogenesis and apoptosis of hypertrophic chondrocytes. Cell. 1998;93(3):411–22. doi: 10.1016/s0092-8674(00)81169-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Karaplis AC, Luz A, Glowacki J, et al. Lethal skeletal dysplasia from targeted disruption of the parathyroid hormone-related peptide gene. Genes Dev. 1994;8(3):277–89. doi: 10.1101/gad.8.3.277. [DOI] [PubMed] [Google Scholar]
- 49.Schipani E, Lanske B, Hunzelman J, et al. Targeted expression of constitutively active receptors for parathyroid hormone and parathyroid hormone-related peptide delays endochondral bone formation and rescues mice that lack parathyroid hormone-related peptide. Proc Natl Acad Sci U S A. 1997;94(25):13689–94. doi: 10.1073/pnas.94.25.13689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Komori T, Yagi H, Nomura S, et al. Targeted disruption of Cbfa1 results in a complete lack of bone formation owing to maturational arrest of osteoblasts. Cell. 1997;89(5):755–64. doi: 10.1016/s0092-8674(00)80258-5. [DOI] [PubMed] [Google Scholar]
- 51.Toupadakis CA, Wong A, Genetos DC, et al. Long-term administration of AMD3100, an antagonist of SDF-1/CXCR4 signaling, alters fracture repair. J Orthop Res. 2012;30(11):1853–9. doi: 10.1002/jor.22145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Murata K, Kitaori T, Oishi S, et al. Stromal cell-derived factor 1 regulates the actin organization of chondrocytes and chondrocyte hypertrophy. PLoS One. 2012;7(5):e37163. doi: 10.1371/journal.pone.0037163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kitaori T, Ito H, Schwarz EM, et al. Stromal cell-derived factor 1/CXCR4 signaling is critical for the recruitment of mesenchymal stem cells to the fracture site during skeletal repair in a mouse model. Arthritis Rheum. 2009;60(3):813–23. doi: 10.1002/art.24330. [DOI] [PubMed] [Google Scholar]
- 54.Otsuru S, Tamai K, Yamazaki T, Yoshikawa H, Kaneda Y. Circulating bone marrow-derived osteoblast progenitor cells are recruited to the bone-forming site by the CXCR4/stromal cell-derived factor-1 pathway. Stem Cells. 2008;26(1):223–34. doi: 10.1634/stemcells.2007-0515. [DOI] [PubMed] [Google Scholar]
- 55.Granero-Molto F, Weis JA, Miga MI, et al. Regenerative effects of transplanted mesenchymal stem cells in fracture healing. Stem Cells. 2009;27(8):1887–98. doi: 10.1002/stem.103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Fujio M, Yamamoto A, Ando Y, et al. Stromal cell-derived factor-1 enhances distraction osteogenesis-mediated skeletal tissue regeneration through the recruitment of endothelial precursors. Bone. 2011;49(4):693–700. doi: 10.1016/j.bone.2011.06.024. [DOI] [PubMed] [Google Scholar]
- 57.Yamazaki M, Nakajima F, Ogasawara A, Moriya H, Majeska RJ, Einhorn TA. Spatial and temporal distribution of CD44 and osteopontin in fracture callus. J Bone Joint Surg Br. 1999;81(3):508–15. doi: 10.1302/0301-620x.81b3.9398. [DOI] [PubMed] [Google Scholar]
- 58.Hirakawa K, Hirota S, Ikeda T, et al. Localization of the mRNA for bone matrix proteins during fracture healing as determined by in situ hybridization. J Bone Miner Res. 1994;9(10):1551–7. doi: 10.1002/jbmr.5650091007. [DOI] [PubMed] [Google Scholar]
- 59.Duvall CL, Taylor WR, Weiss D, Wojtowicz AM, Guldberg RE. Impaired angiogenesis, early callus formation, and late stage remodeling in fracture healing of osteopontin-deficient mice. J Bone Miner Res. 2007;22(2):286–97. doi: 10.1359/jbmr.061103. [DOI] [PubMed] [Google Scholar]
- 60.Kusumbe AP, Ramasamy SK, Adams RH. Coupling of angiogenesis and osteogenesis by a specific vessel subtype in bone. Nature. 2014;507(7492):323–8. doi: 10.1038/nature13145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Deckers MM, Van Beek ER, Van Der Pluijm FG, et al. Dissociation of angiogenesis and osteoclastogenesis during endochondral bone formation in neonatal mice. J Bone Miner Res. 2002;17(6):998–1007. doi: 10.1359/jbmr.2002.17.6.998. [DOI] [PubMed] [Google Scholar]
- 62.Cackowski FC, Anderson JL, Patrene KD, et al. Osteoclasts are important for bone angiogenesis. Blood. 2010;115(1):140–9. doi: 10.1182/blood-2009-08-237628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Jones PA. Functions of DNA methylation: islands, start sites, gene bodies and beyond. Nat Rev Genet. 2012;13(7):484–92. doi: 10.1038/nrg3230. [DOI] [PubMed] [Google Scholar]
- 64.Ball MP, Li JB, Gao Y, et al. Targeted and genome-scale strategies reveal gene-body methylation signatures in human cells. Nat Biotechnol. 2009;27(4):361–8. doi: 10.1038/nbt.1533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Yang X, Han H, De Carvalho DD, Lay FD, Jones PA, Liang G. Gene body methylation can alter gene expression and is a therapeutic target in cancer. Cancer Cell. 2014;26(4):577–90. doi: 10.1016/j.ccr.2014.07.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Weber M, Hellmann I, Stadler MB, et al. Distribution, silencing potential and evolutionary impact of promoter DNA methylation in the human genome. Nat Genet. 2007;39(4):457–66. doi: 10.1038/ng1990. [DOI] [PubMed] [Google Scholar]
- 67.Pamnani M, Sinha P, Singh A, Nara S, Sachan M. Methylation of the Sox9 and Oct4 promoters and its correlation with gene expression during testicular development in the laboratory mouse. Genet Mol Biol. 2016;39(3):452–8. doi: 10.1590/1678-4685-GMB-2015-0172. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.