

Abstract
The cellular context that integrates upstream signaling and downstream nuclear response dictates the oncogenic behaviour and shapes treatment responses in distinct cancer types. Here, we uncover that in GIST, the forkhead family member, FOXF1, directly controls the transcription of two master regulators, KIT and ETV1, both required for GIST precursor-interstitial cells of Cajal (ICC) lineage-specification and GIST tumorigenesis. Further, FOXF1 co-localizes with ETV1 at enhancers and functions as a pioneer factor that regulates the ETV1-dependent GIST-lineage specific transcriptome through modulation of the local chromatin context, including chromatin accessibility, enhancer maintenance and ETV1 binding. Functionally, FOXF1 is required for human GIST cell growth in vitro and murine GIST tumor growth and maintenance in vivo. The simultaneous control of the upstream signaling and nuclear response sets up a unique regulatory paradigm and highlights the critical role of FOXF1 in enforcing the GIST cellular context for highly lineage-restricted clinical behaviour and treatment response.
Introduction
Gastrointestinal stromal tumor (GIST) is one of the most common subtypes of human soft tissue sarcoma. GIST arises from the interstitial cells of Cajal (ICCs), a cell lineage that requires KIT- the principal signaling regulator, and ETV1- a lineage-specific master transcription factor, for lineage specification and survival (1–3). Physiologically, normal levels of KIT activation by the KIT ligand stabilizes the ETV1 protein through active downstream MAPK signaling, and results in physiological transcriptional output critical for ICC lineage specification and development. GIST is characterized by frequent activating mutations in KIT. Mutant KIT aberrantly activates downstream MAPK signaling, which stabilizes the ETV1 protein, and stabilized ETV1 protein in turn enhances mutant KIT expression. Therefore, mutant KIT and ETV1 form a positive feedback loop and cooperate in GIST oncogenesis (4). The lineage-specific expression of KIT and ETV1 and their interplay in GIST underline the exquisite therapeutic sensitivity and clinical success of targeting the lineage-dependence on KIT and ETV1 (5–8). However, how KIT and ETV1 are regulated and what defines the cellular context in GIST remain unclear.
In addition to GIST, ETV1 is involved in the tumorigenesis of multiple cancer types, including prostate cancer and melanoma, where it regulates distinct transcriptional programs (1,9–11). The enhancer landscape of accessible chromatin defines cellular lineage and the distinct cistrome and transcriptional output of individual transcription factor in different cell types. We thus speculate that additional master regulator(s) may function as “pioneer factor(s)” that modulate chromatin accessibility and help define and maintain the cistrome of ETV1, analogous to the pioneer function of FOXA1 to androgen receptor (AR) in prostate cancer and estrogen receptor-α (ERα) in breast cancer (12–17). Here, we describe the discovery of FOXF1, as a novel ICC/GIST lineage-specific master regulator that directly regulates KIT, ETV1 expression and the ICC/GIST lineage-specific transcriptome. Moreover, FOXF1 functions as a pioneer factor required to maintain open chromatin and ETV1 binding at many lineage-specific ETV1 binding sites. We further demonstrate that FOXF1 functionally is required for GIST cell growth and survival in vitro and GIST tumor growth and maintenance in genetically engineered mouse models. Overall, our data demonstrate a unique regulatory hierarchy of FOXF1 that distinguishes itself from other pioneer factors, e.g. FOXA1, in that beyond chromatin context modulation and active recruitment of ETV1, it also directly controls the expression of ETV1 and the cooperative signaling factor KIT.
Results
FOXF1 is nearly universally and uniquely expressed in human GISTs
To identify critical factor(s) that regulate the lineage-specific cellular context for oncogenic transformation, we focused our initial analyses on ETV1, a transcription factor that drives tumorigenesis in two distinct cancer types: prostate cancer and GIST (1,9,10). We generated genome-wide localization of ETV1 by ChIP-seq in two human GIST cell lines (GIST-T1 and GIST48) and two prostate cancer cell lines that harbor aberrant expression of full-length ETV1 due to translocation of its entire coding locus (LNCaP and MDA-PCa2b) (1,9,10,18–20). ETV1 cistrome analyses demonstrated that the majority of the ETV1 promoter binding sites (TSS±1kb) were shared between prostate cancer and GIST, whereas the majority of non-promoter (referred as “enhancer” hereafter) binding sites were distinct between the two cancer types (Fig. 1A and B). Unsupervised k-means clustering divided enhancer ETV1 binding sites into three distinct clusters of GIST-specific (C1), prostate-specific (C2) and shared sites (C3). This is consistent with previous observation that enhancer landscape is more lineage-specific than promoter (12,14,15,17,21–24). The observation that ETV1 binds to distinct enhancer regions in prostate cancer and GIST suggests that additional factors are involved in lineage enhancer specification and maintenance. To identify potential lineage-specific transcription factors that co-localize with ETV1 at enhancer sites, we performed de novo motif analysis. We identified the FOX motif as the second most enriched motif, behind the ETS motif, at both the prostate cancer-specific (P=1×10−198) and GIST-specific (P=1×10−153) ETV1-bound enhancer sites, and to a lesser significance at the shared enhancer regions (P=1×10−23), but not at ETV1-bound promoters (Fig. 1A and Supplementary Tables S1-S4). Other enriched motifs at the GIST-specific ETV1-bound enhancer sites included RUNX (P=1×10−39), HOXA9/B9/C9 (P=1×10−29), bHLH (P=1×10−22) and HOXD9 (P=1×10−20) (Supplementary Table S1). Hence, we focused on the most significantly enriched FOX motif.
Figure 1. ETV1 cistrome analysis identifies FOXF1 as a uniquely and highly expressed transcription factor in GIST.
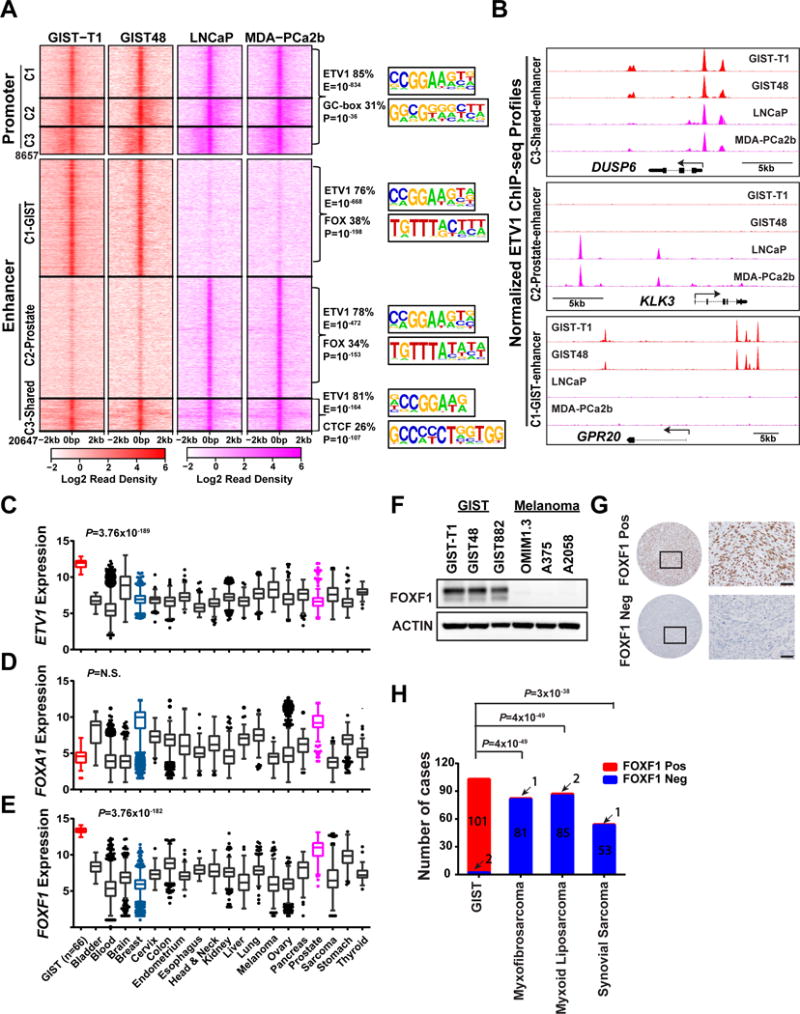
A, Left panel: Heatmap of ETV1 binding sties centered on peak summit at the promoter (Transcriptional start site [TSS] ± 1kb) and enhancer (non-promoter) regions in GIST-T1 and GIST48 GIST cells (red) and LNCaP and MDA-PCa2b ETV1-translocated prostate cancer cells (magenta). Promoter and Enhancer sites were each clustered into 3 clusters (C1, C2, C3) using K-means. Right panel: De novo motif analysis of shared and distinct ETV1 binding sites in the promoter and enhancer regions. Top 2 most enriched motifs by significance are shown as motif sequence logo, percentage of peaks with the motif, and significance value, corresponding to different genomic regions. B, Representative ETV1 ChIP-seq profiles at DUSP6 (C3-shared enhancer), KLK3 (C2-prostate-specific enhancer) and GPR20 (C1-GIST-specific enhancer) gene loci in GIST and prostate cancer cells. C-E, Tukey plots of gene expression of ETV1 (C), FOXA1 (D), and FOXF1 (E) in different cancer types (Red: GIST; Magenta: Prostate cancer) in the Gene Expression across Normal and Tumor tissue (GENT) publically available pan-cancer dataset. P value is from two-tailed unpaired t-test of GIST vs all other tumors. F, Representative immunoblots of FOXF1 and ACTIN control in human GIST (GIST-T1, GIST48, GIST882) and melanoma (OMIM1.3, A375, A2058) cell lines. G, Representative IHC images of FOXF1 in FOXF1-positive (FOXF1 Pos, top panels) GIST and FOXF1-negative (FOXF1 Neg, bottom panels) sarcoma clinical samples. H, Distribution of FOXF1 IHC in tissue microarray of GIST and other sarcoma subtypes, Myxofibrosarcoma, Myxoid Liposarcoma, Synovial Sarcoma. P value is from Fisher’s exact test.
In the prostate lineage, FOXA1 is a well-known pioneer factor that has the ability to modulate chromatin accessibility and regulate the binding of other transcription factors such as AR (12–14,25). We examined ETV1 and FOXA1 expression in multiple cancer types from the Gene Expression across Normal and Tumor Tissue (GENT) database (26) and confirmed high ETV1 expression level in GIST and a subset of prostate cancer (Fig. 1C), and high FOXA1 expression in prostate cancer and breast cancer (Fig. 1D). However, FOXA1 expression is low in GIST tumors as well as cell lines (Fig. 1D and Supplementary Fig. S1A-B).
We thus speculate that a different FOX family transcription factor is involved in the modulation of ETV1 cistrome in GIST. We examined the expression of all FOX factors and uncovered that FOXF1 is the highest in both absolute expression and significance of differential expression in GIST compared to other cancer types (Fig. 1E, Supplementary Fig. S1A and Supplementary Table S5). We further examined RNA-seq profiles of GIST48 and GIST882 cells and observed that FOXF1 was the highest expressing FOX family member (Supplementary Fig. S1B). We confirmed the presence of FOXF1 protein in all three human GIST cell lines, but not in the negative control melanoma cell lines (OMIM1.3, A375 and A2058) (Fig. 1F). Furthermore, we examined FOXF1 protein expression in tissue microarrays of GIST and several other sarcoma subtypes from MSK archived tumor specimens by immunohistochemistry (IHC). Independent review by two sarcoma pathologists confirmed positive FOXF1 staining in >98% of all human GIST samples regardless of KIT/PDGFRA-mutational status, but rarely in other sarcoma subtypes, including myxofibrosarcoma, myxoid liposarcoma and synovial sarcoma (Fig. 1G and H). These data demonstrate that FOXF1 is nearly universally and uniquely expressed in human GISTs and it can be used as a novel sensitive and specific diagnostic marker for GIST.
FOXF1 directly regulates the expression of KIT, ETV1 and ETV1-dependent ICC/GIST lineage-specific transcriptome through enhancer binding
To determine whether FOXF1 is enriched at ETV1 enhancer sites in GIST, we performed FOXF1 ChIP-seq in GIST48 and GIST-T1 cells. FOXF1 binding sites primarily localized to enhancers (~94% of all FOXF1 peaks) and were highly concordant between the two GIST cell lines. Approximately 35% of high-confidence ETV1 enhancer binding sites overlapped with high-confidence FOXF1 binding sites (P=0), including ICC/GIST-lineage specific enhancers such as KIT, GPR20, ETV1 (Fig. 2A and B, and Supplementary Fig. S2A). Among “ETV1-only” peaks, many exhibited modest FOXF1 binding, but the FOXF1 ChIP-seq signals did not meet the peak calling significance threshold (q < 10−3). Additionally, these ETV1-only peaks also contained the FOX motif that correlated with FOXF1 binding strength (Fig. 2A, and Supplementary Fig. S2B), suggesting that the actual co-occupancy of ETV1 and FOXF1 at GIST enhancers may be underestimated. De novo motif analysis of high-confidence ETV1/FOXF1 co-bound sites revealed expected high frequency of FOX motif (71%) and ETV1 motif (71%). Further, we found combined FOX/ETS motifs with no orientation preference but with a gap preference of either directly adjacent (no gap) or 11 base-pairs, corresponding to one turn of helical DNA (Fig. 2A, and Supplementary Fig. S2C).
Figure 2. FOXF1 is a master-regulator that directly regulates KIT, ETV1 and the GIST-lineage specific transcriptome through enhancer binding.
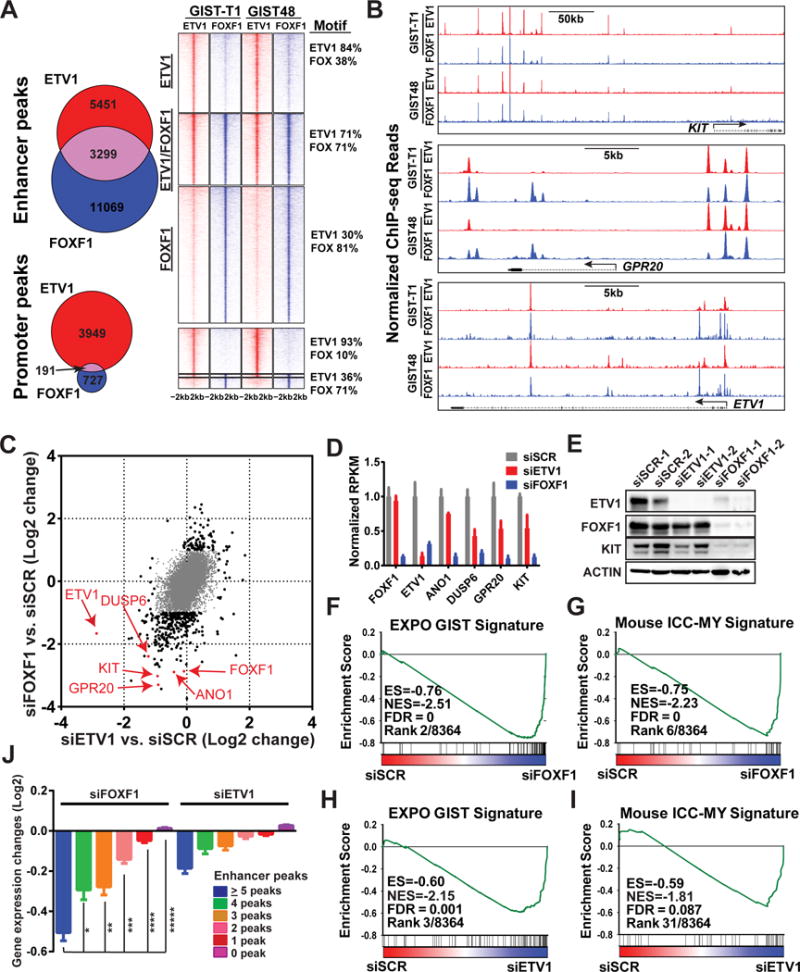
A, Venn Diagram and density plots of FOXF1 (blue) and ETV1 (red) global binding sites by ChIP-seq in human GIST-T1 and GIST48 cells. Promoter: TSS±1kb, Enhancer: non-promoter. B, Representative FOXF1 and ETV1 ChIP-seq profiles in GIST cells at KIT, GPR20 and ETV1 loci. C, Scatter plot of gene expression changes by siRNA-mediated downregulation of FOXF1 (y-axis) and ETV1 (x-axis) compared to scrambled controls in GIST48 cells. Genes with ≥ 2-fold (Log2=1) change by either perturbation are marked in black dots, < 2-fold change in gray dots. D, Bar graph of gene expression changes by siRNA-mediated downregulation of FOXF1 and ETV1 compared to scrambled controls in GIST48 cells. N=2 distinct siRNA. Mean ± SD. E, Immunoblots of ETV1, FOXF1, KIT and ACTIN control with siRNA-mediated downregulation of FOXF1 and ETV1 in GIST48 cells. F-I, GSEA of transcriptome changes in GIST48 as a result of siRNA-mediated downregulation of FOXF1 (siFOXF1) (F-G) or ETV1 (siETV1) (H-I) compared to scramble controls (siSCR), demonstrating that the EXPO GIST Signature and Mouse ICC-MY Signature are among the most negatively enriched gene set signatures. ES: Enrichment Score; NES: Normalized Enrichment Score; FDR: False Discovery Rate. Rank: by FDR and ES amongst 8,364 gene sets. J, Mean expression change of all genes grouped by total number of ETV1 or FOXF1 enhancer peaks, induced by si-mediated downregulation of FOXF1 (left) and ETV1 (right). Error bars: Mean ± SEM. P value is from two-tailed unpaired t-test. *: P=0.0012; **-*****: P<0.0001.
Given their significant co-localization at lineage-specific enhancers, we next performed whole transcriptome analyses of the FOXF1-regulated and the ETV1-regulated transcriptomes in GIST48 cells using siRNA-mediated downregulation of FOXF1 and ETV1, respectively. To minimize individual siRNA off-target effects, we averaged two independent siRNA specific for ETV1, FOXF1 or scramble controls (SCR) (Supplementary Fig. S3A). Exploring the interplay between FOXF1 and ETV1, we found that siETV1 decreased ETV1 RNA expression by ~8-fold without significantly affecting FOXF1 RNA levels, while siFOXF1 decreased FOXF1 RNA expression by ~8-fold and ETV1 RNA levels by ~3-fold (Fig. 2C and D, and Supplementary Fig. S3B and C). There was a highly significant correlation between transcriptome changes induced by siFOXF1 and siETV1, respectively (R=0.58, P=3.8×10−37) (Fig. 2C). Consistent with prior observations, ETV1 downregulation caused downregulation of ICC/GIST lineage-specific genes, including DUSP6, GPR20, ANO1 and KIT (1,4). Notably, FOXF1 knockdown led to a greater magnitude of downregulation of these genes. In particular, KIT expression level was reduced by ~2-fold with ETV1 knockdown and by ~8-fold with FOXF1 knockdown (Fig. 2C and D), suggesting that FOXF1 is a more robust regulator of KIT RNA expression than ETV1 in GIST. The differential effects of FOXF1 and ETV1 on each other and on KIT expression were confirmed by immunoblot (Fig. 2E). Gene Set Enrichment Analysis (GSEA) using >8,300 gene sets from the Molecular Signatures Database (MSigDB) (27) and custom ICC and GIST signatures (1) revealed that the human GIST (EXPO GIST Signature) and the myenteric ICC (Mouse ICC-MY Signature) were among the most negatively enriched gene sets with both siFOXF1 and siETV1-mediated perturbations in GIST (Fig. 2F-I, and Supplementary Table S6-S8). Additional highly negatively enriched gene sets with FOXF1 knockdown included significantly downregulated genes with shRNA-mediated ETV1 knockdown in GIST cells (ETV1 KD GIST48 DN), significantly downregulated genes with imatinib (a KIT inhibitor) treatment in GIST882 cells, as well as cell cycle regulated genes (Supplementary Fig. S3D-F). Integrative analyses of FOXF1 and ETV1-cistrome with corresponding transcriptome revealed that the genes with more FOXF1 or ETV1 enhancer peaks were significantly correlated with expression changes in FOXF1 knockdown (Fig. 2J).
Since KIT and downstream MAPK signaling stabilize ETV1 protein levels, we examined their effect on FOXF1 protein levels in imatinib-sensitive GIST882 and GIST-T1 cell lines. Consistent with prior observations, short-term (2-hour) and relatively long-term (24-hour) treatment with imatinib resulted in effective inhibition of the KIT and its downstream MAPK pathway signaling, which lead to ETV1 protein degradation. Treatment with single agent MEK162 (a MEK inhibitor) resulted in short-term inhibition of MAPK pathway signaling and gradual re-activation of MAPK signaling by 24 hours, with corresponding recovery of ETV1 protein levels (Supplementary Fig. S3G). In contrast, FOXF1 protein levels were not significantly perturbed under these conditions.
These data demonstrate that FOXF1 directly transcriptionally regulates ETV1, KIT and the ICC/GIST lineage-specific genes mainly through enhancer binding, while KIT signaling and ETV1 transcriptional activity does not affect FOXF1, thus placing FOXF1 at the top of the lineage regulatory hierarchy.
FOXF1 maintains the local chromatin context and ETV1 cistrome
To explore mutual requirements between ETV1 and FOXF1 chromatin binding and the role of each transcription factor in the maintenance of enhancer chromatin landscape, we examined the effect of FOXF1 and ETV1 depletion on the FOXF1 and ETV1 cistrome and enhancer chromatin landscape in GIST. Compared to siSCR control, siETV1 led to a similar decrease of ETV1 binding at all ETV1 binding sites including ETV1-only and ETV1/FOXF1 co-localized (both) enhancers, and ETV1-bound promoters, without significant perturbation of FOXF1 binding (Fig. 3A). On the contrary, FOXF1 downregulation by siFOXF1 resulted in not only decreased FOXF1 binding, but also significant reduction of ETV1 binding. At ETV1-bound enhancer sites, we quantified the change in ETV1 binding induced by either siETV1 or siFOXF1 and correlated with the level of FOXF1 binding. As expected, siETV1 resulted in global reduction of ETV1 RNA and protein levels, which resulted in reduction of ETV1 binding at both ETV1-only and ETV1/FOXF1 co-localized (both) enhancer sites without significant difference (Fig. 3B). As a result of its direct transcriptional effect on ETV1 RNA in GIST, siFOXF1 led to reduction of total ETV1 protein levels and corresponding global reduction of ETV1 chromatin binding at all ETV1-binding sites, including ETV1-bound promoters and enhancers. In contrast to siETV1, there is significantly more reduction of ETV1 binding at the ETV1/FOXF1 co-localized enhancers than ETV1-only enhancers (effect size d=0.71, P=10−104) (Fig. 3B), suggesting that FOXF1 regulates ETV1 chromatin binding beyond simple transcriptional regulation of ETV1 dosage. Moreover, we compared the ETV1 enhancer binding signal changes induced by siETV1 and siFOXF1 in reference to FOXF1 binding strength measured by FOXF1 ChIP-signal. siFOXF1-mediated reduction in ETV1 enhancer binding was highly correlated with the signal strength of FOXF1 binding (Fig. 3C), indicating that FOXF1 binding to chromatin may influence ETV1 binding. Examination of representative ETV1/FOXF1 co-bound peaks (e.g., KIT and GPR20 loci) and ETV1 only enhancer and promoter peaks (e.g., HES1, ACTB, SNAP20) illustrates the differential effect of FOXF1 knockdown on ETV1 binding (Fig. 3D, Supplementary Fig. S4A and B).
Figure 3. FOXF1 modulates enhancer landscape and ETV1 binding to GIST-lineage specific enhancers.
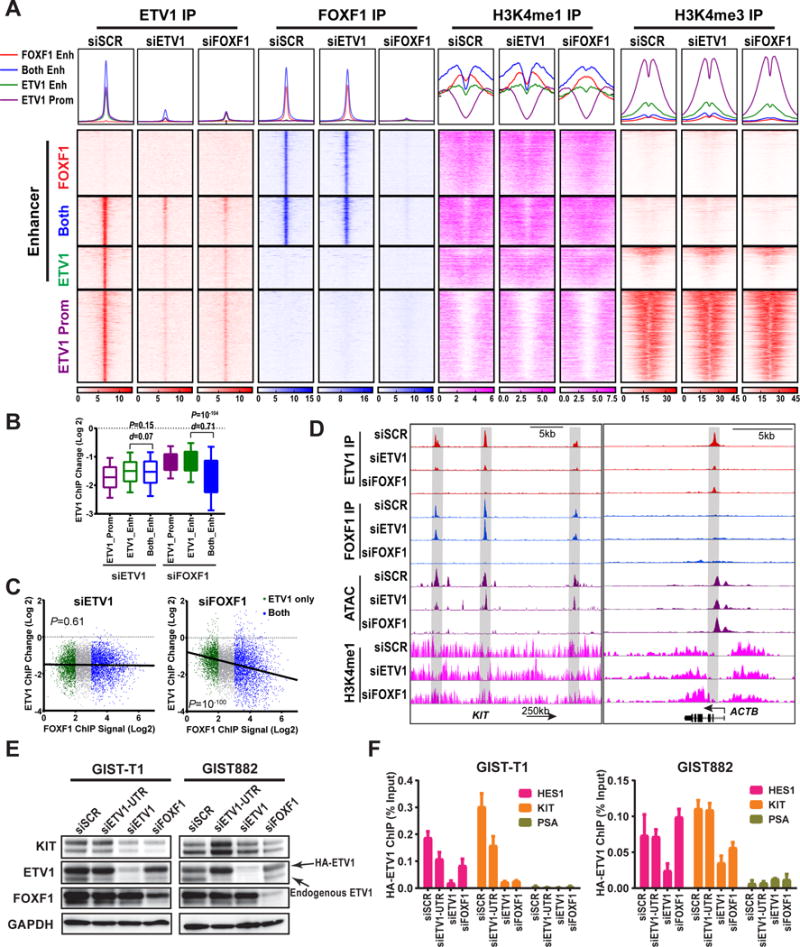
A, ChIP-seq profile (top) and heatmap (bottom) of ETV1, FOXF1, H3K4me1 and H3K4me3 signal around peak center at enhancers (FOXF1 only: red, FOXF1/ETV1-shared [both]: blue and ETV1-only: green) and at ETV1-bound promoters (purple) with siETV1, siFOXF1, or scramble (siSCR) controls in GIST48 cells. B, Box and whisker plots showing change of ETV1-ChIP-seq (Log 2) by siRNA-mediated downregulation of ETV1 or FOXF1 in GIST48 cells. Box 75%, whiskers 90%. P: Mann-Whitney test d: Cohen size-effect. C, Scatter plots of ETV1-ChIP-seq (Log 2) correlated with FOXF1 ChIP-seq signal (Log 2) by siRNA-mediated downregulation of ETV1 or FOXF1 in GIST48 cells. The ETV1-ChIP-signal change is marked green for ETV1-only, and blue for FOXF1and ETV1-shared (both) enhancer peaks. P: Fisher exact test. D, Representative FOXF1, ETV1 and H3K4me1 ChIP-seq and ATAC-seq profiles at the indicated gene loci with siRNA-mediated perturbation of ETV1 and FOXF1 in GIST48 cells. Gray highlighted the enhancers with appreciable changes in ATAC-seq, and H3K4me1 ChIP-seq signals at the FOXF1-regulated KIT locus, but not in the ETV1-regulated and FOXF1-independent HES1 locus. E, Immunoblots of GIST-T1 and GIST882 cells with exogenous expression of Flag-HA-tagged-ETV1 (HA-ETV1) independent of endogenous FOXF1-mediated transcriptional control under experimental perturbations as indicated. F, ChIP-qRT-PCR signals of HA-ETV1 (α-HA ChIP) at the HES1 enhancer (an ETV1-, but not FOXF1-regulated gene), KIT enhancer (a FOXF1- regulated gene) and PSA control (neither ETV1- nor FOXF1-regulated gene) loci under different conditions as indicated in GIST-T1 and GIST882 cells with exogenous expression of HA-ETV1 as in E. N=3, Mean ± SEM.
To further determine if FOXF1 is required for ETV1 binding to co-bound enhancer sites beyond its regulation of ETV1 protein level, we examined the enhancer binding of exogenously expressed HA-tagged ETV1 (HA-ETV1) that is not under FOXF1 transcriptional control in two human GIST cell lines (Fig. 3E). While siFOXF1 did not significantly affect the HA-ETV1 protein level, it resulted in significant reduction of HA-ETV1 binding at the ETV1/FOXF1 co-localized KIT enhancers, but not in ETV1-only HES1 enhancers (Fig. 3F). These data suggest that FOXF1, in addition to direct transcriptional regulation of ETV1, also regulates the ETV1 cistrome by recruiting ETV1 to bind to the ETV1/FOXF1 co-localized enhancers.
To address the mechanistic basis for the requirement of FOXF1 on ETV1 enhancer binding, we first examined whether FOXF1 directly interacts with ETV1 endogenously. Co-immunoprecipitation of FOXF1 and ETV1 in two human GIST cell lines revealed no evidence of direct binding (Supplementary Fig. S4C). We next assessed characteristic chromatin marks of enhancers and promoters, H3K4me1 and H3K4me3, respectively. As expected, ETV1-bound promoters exhibited a bimodal H3K4me3 enrichment with a central depleted nucleosome, and depletion of H3K4me1 marks (Fig. 3A and Supplementary Fig. S4B). This promoter chromatin landscape was not affected by either siETV1 or siFOXF1. All three classes of enhancers defined by ETV1 and FOXF1 binding exhibited a bimodal H3K4me1 signal indicating central depleted nucleosome that allow binding of transcription factors, with ETV1/FOXF1 co-bound sites showing strongest H3K4me1 enrichment. ETV1 knockdown did not significantly affect the H3K4me1 landscape. However, FOXF1 knockdown changed the H3K4me1 to a more unimodal profile suggesting loss of the nucleosome depleted region at the FOXF1-only and ETV1/FOXF1 co-bound enhancers (Fig. 3A).
To directly examine whether FOXF1 can modulate chromatin accessibility and help guide binding of other transcription factors, we performed assay for transposase-accessible chromatin coupled with next generation sequencing (ATAC-seq) (28,29) in GIST48, GIST882, and GIST-T1 cells (21,22,30–32). Knockdown of ETV1 caused a modest median decrease in chromatin accessibility, especially in ETV1/FOXF1 co-bound sites. Knockdown of FOXF1 caused a more dramatic reduction in ATAC signal preferentially at FOXF1-only and ETV1/FOXF1 co-bound enhancers in all three GIST cells lines (Fig. 4A and B, and Supplementary Fig. S4A and B). Among enhancers bound by ETV1, the FOXF1 binding strength correlated with changes in ATAC signal induced by FOXF1 knockdown (Fig. 4C). These data posit FOXF1 as a pioneer factor that has the ability to modulate chromatin accessibility and regulate and maintain enhancer chromatin characteristics, as well as recruiting the lineage-specific master regulator, ETV1, in GIST.
Figure 4. FOXF1 modulates chromatin accessibility in GIST.
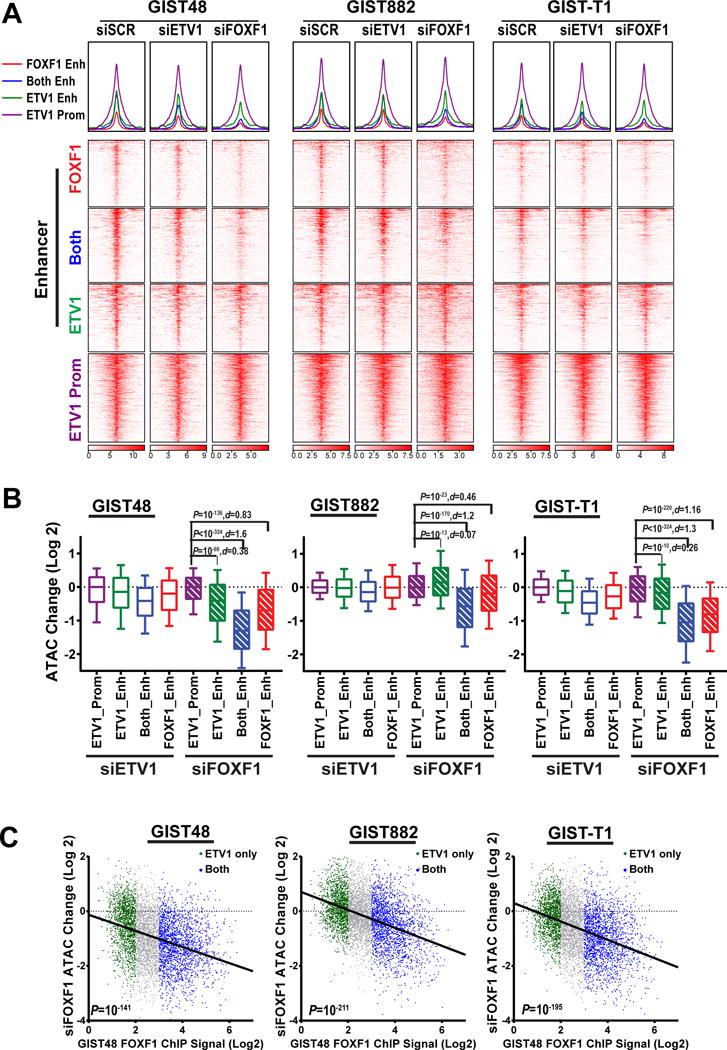
A, ATAC-seq profile (top) and heatmap (bottom) around ETV1 or FOXF1 peak center at enhancers (FOXF1 only: red, FOXF1/ETV1-shared [both]: blue and ETV1-only: green) and at ETV1-bound promoters (purple) with siRNA mediated downregulation of ETV1, FOXF1, or scramble (SCR) controls in GIST48, GIST882 and GIST-T1 cells. B, Box and whiskers plot showing change of ATAC-seq (Log 2) signal by siRNA-mediated downregulation of ETV1 or FOXF1 in GIST48, GIST882 and GIST-T1 cells. Box 75%, whiskers 90%. P: Mann-Whitney test d: Cohen size-effect. C, Scatter plots of ATAC-seq signal change (Log 2) correlated with FOXF1 ChIP-seq signal (Log 2) by siRNA-mediated downregulation of ETV1 or FOXF1 in GIST48, GIST882 and GIST-T1 cells. ETV1-only is marked green, and blue for ETV1 and FOXF1 shared binding sites. P: Fisher exact test.
FOXF1 is required for human GIST cell growth in vitro
To evaluate the functional significance of FOXF1 in GIST pathogenesis, we established cells stably expressing doxycycline-inducible GFP with mirE-based hairpins specific for FOXF1 (shFOXF1) or Renilla luciferase control (shREN) in GIST48, GIST882, and GIST-T1 cells. We confirmed doxycycline inducible FOXF1 knockdown and consequent downregulation of KIT and ETV1 protein (Fig. 5A, and Supplementary Fig. S5A). We further assessed the effect of FOXF1 downregulation on KIT expression at individual cell level using fluorescence-activated cell sorting analysis (FACS) of GFP and KIT immunostaining of shRNA-expressing GFP-positive cells in the presence of parental control cells (Fig. 5B). Since the mutant KIT proteins have previously been shown to preferentially localize to the endoplasmic reticulum (ER) and the Golgi apparatus, and the wild-type KIT to the plasma membrane (33–35), we analyzed the cell-surface and total (including both intracellular and surface) KIT protein levels. We observed a dramatic reduction of both the cell surface KIT and total KIT protein levels with FOXF1 downregulation (Fig. 5C, and Supplementary Fig. S5B and C), suggesting that FOXF1 regulates both the wild-type and mutant KIT expression levels. The shFOXF1-mediated reduction of total KIT and phospho-KIT proteins in GIST also led to consistent reduction of the known KIT downstream signaling pathways in all GIST cell lines, including the MAPK and AKT pathways; but the STAT3 signaling pathway was less affected (Fig. 5A, and Supplementary Fig. S5A).
Figure 5. FOXF1 is required for growth and survival of GIST in vitro.
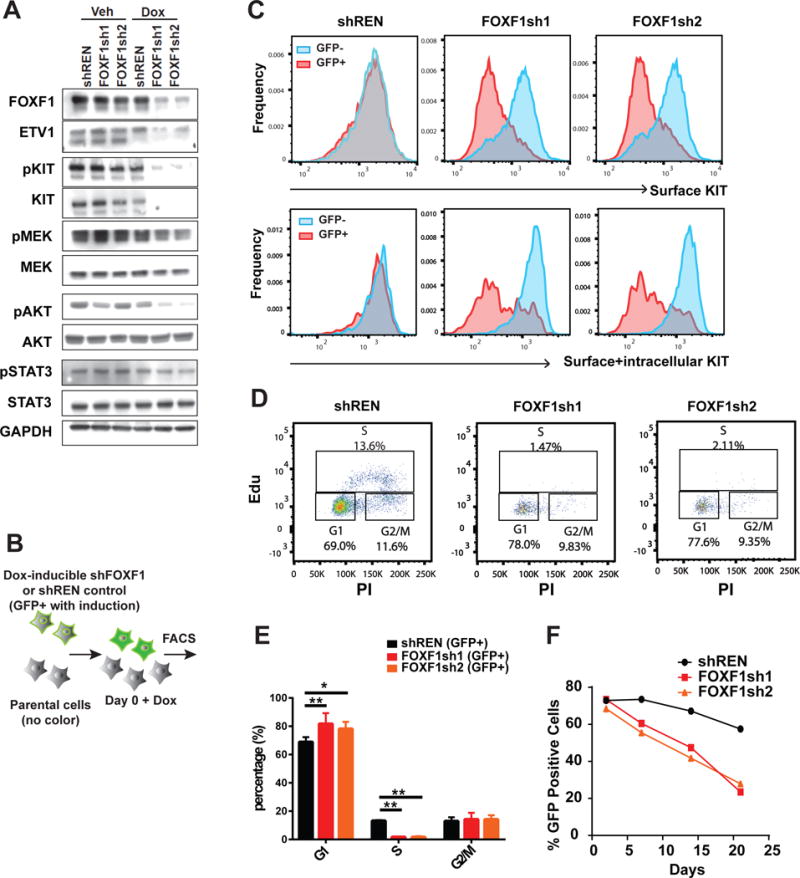
A, Validation of doxycycline inducible shRNA-mediated downregulation of FOXF1 and target proteins, ETV1 and KIT in GIST48 cells as well as components of MAPK, AKT and STAT3 pathways by immunoblots. B, A schematic of co-culture of parental unlabelled cells with GFP-labeled doxycycline-inducible shRNA expressing-GIST cells. Co-cultures are used for KIT FACS, cell cycle analysis, and growth competition assays each comparing GFP-positive and GFP-negative cells. C, FACS analysis of cell surface KIT protein (upper) and total KIT protein (surface+intracellular) levels with shRNA-mediated FOXF1 downregulation in GIST48 cells. D–E, The effect of shRNA-mediated FOXF1 downregulation on cell cycle using DNA content and EdU labeling in GIST 48 cells using FACS plots (D) and bar graphs (E) demonstrating percentages of GFP-positive cells in G1, S, and G2/M cycle phase. N=3, Mean ± SD, Student t-test. *P<0.05, **P<0.01. F, Representative growth curves of GIST48 cells with doxycycline inducible shFOXF1 compared to controls in growth competition assay.
We next assessed the effect of FOXF1 downregulation on cell cycle regulation and cell growth by quantification of shRNA-expressing GFP-positive cells in the presence of parental control cells over time using FACS (Fig. 5B). In all three GIST cell lines, after doxycycline administration, there was an increased percentage of cells in G1-phase and decreased percentage of cells in S-phase (Fig. 5D and E, and Supplementary Fig. S6A-D). These data indicates that FOXF1 is required for GIST cell cycle progression in vitro. The changes in cell cycle progression were accompanied by a steady decrease in the percentage of GFP-positive shFOXF1-expressing GIST cells but not that of GFP-positive shREN control-expressing cells in competition cell growth assays (Fig. 5F, and Supplementary Fig. S6E and F). These effects were confirmed using siRNA-mediated downregulation of FOXF1 and independent cell growth assays (Supplementary Fig. S6G-J). These data indicate that FOXF1 is required for GIST cell growth and fitness, and its effect is likely mediated through regulation of the signaling and transcriptional survival factors, KIT and ETV1.
FOXF1 is required for GIST tumor growth and maintenance in vivo
To examine the functional significance of FOXF1 in GIST pathogenesis in vivo, we generated a conditional HA-tagged Foxf1 knock-in mouse model, where exon 1 containing the forkhead DNA-binding domain of Foxf1 were flanked by LoxP sites and an HA-tag was added to the amino-terminus of Foxf1 (Fig. 6A, and Supplementary Fig. S7A and B). Homozygous Foxf1f-HA-Foxf1/f-HA-Foxf1mice were viable, fertile, and born with Mendelian ratios, indicating that the HA-tag did not perturb the normal function of Foxf1. HA-tag and Kit co-immunofluorescence of the GI tract showed that HA-Foxf1 is expressed in all ICC subclasses, including Etv1-high myenteric and intramuscular ICCs that give rise to GIST (Fig. 6B, arrow heads) and Etv1-low submucosal ICCs (Fig. 6B, arrows), as well as smooth muscle cells, consistent with prior gene expression data of murine GI tract (36) (Supplementary Fig. S8A). These data suggest that Foxf1 has a broader spectrum of expression pattern than Etv1 in Kit-positive ICCs, and is well-positioned at the top of the regulatory hierarchy to regulate Kit and Etv1 in GIST and its precursor ICCs.
Figure 6. FOXF1 is required for GIST tumor growth and maintenance in vivo.
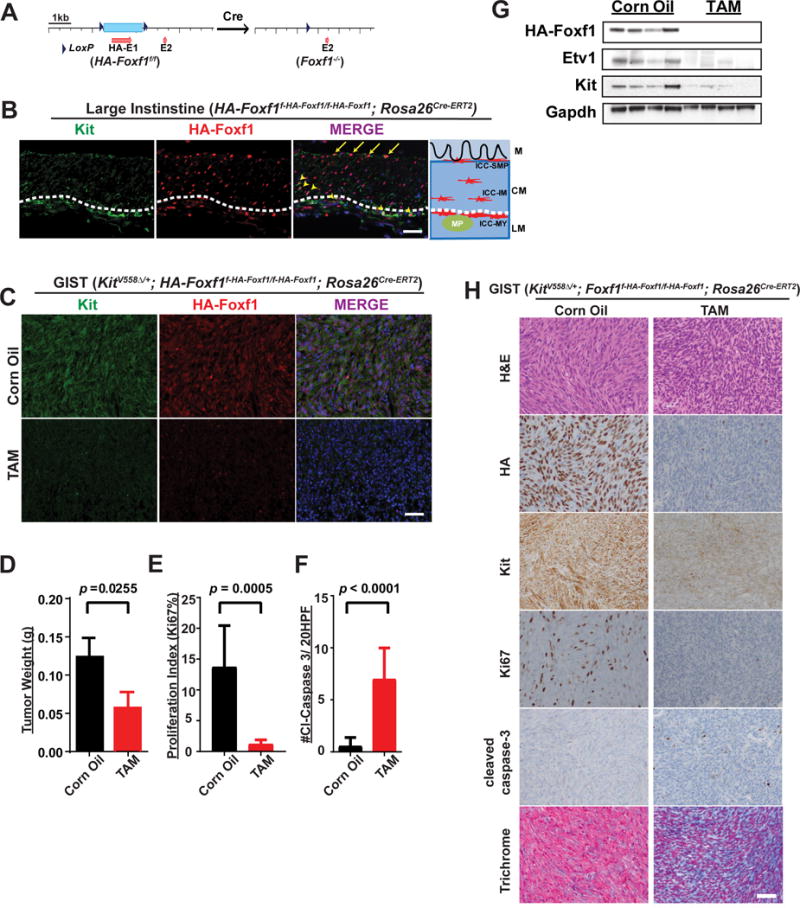
A, Schematic of HA tagged-Foxf1 conditional allele (Foxf1f-HA-Foxf1/f-HA-Foxf1). B, Schematic and representative immunofluorescence images of Kit (green), HA-Foxf1 (red), and DAPI (nuclei, blue in the merged image) in the large intestine of a mouse harbouring the Foxf1f-HA-Foxf1/f-HA-Foxf1 conditional allele. Scale bar: 25μm. White dotted line marks the border of longitudinal muscle and circular muscle. M: mucusa; CM: circular muscle layer; LM: longitudinal muscle layer; ICC-SMP: submucosal ICCs; ICC-IM: intramuscular ICCs; ICC-MY: myenteric ICCs. C, Representative immunofluorescence images of Kit (green), HA-Foxf1 (red) and DAPI (nuclei, blue in the merged image) of GIST-like tumor from mice with KitV558Δ/+; Foxf1f-HA-Foxf1/f-HA-Foxf1; Rosa26Cre-ERT2 genotype with either coil oil control or tamoxifen treatment (TAM). Scale bar: 25μm. D–F, Comparison of tumor weight (D), proliferation index by percent of Ki67 IHC (E), and number of cleaved-caspase 3 (#Cl-Caspase 3)/20 high power fields (HPF) (F) from age-matched (~5 week old) KitV558Δ/+; Foxf1f-HA-Foxf1/f-HA-Foxf1; Rosa26Cre-ERT2 mice tumors with either corn oil control or tamoxifen treatment (TAM). Error bars: Mean ± SD. n=3 for each cohort (Corn Oil or TAM). All P values are as indicated and significant by two-tailed and unpaired t test. G, Immunoblots of the indicated proteins in mouse cecal GIST-like tumors derived from KitV558Δ/+; Foxf1f-HA-Foxf1/f-HA-Foxf1; Rosa26Cre-ERT2 mice with either coil oil control or tamoxifen treatment (TAM). H, Representative images of H&E, IHC of indicated markers and trichrome staining of mouse GIST tumor derived from KitV558Δ/+; Foxf1f-HA-Foxf1/f-HA-Foxf1; Rosa26Cre-ERT2 treated with either corn oil control or TAM. Scale bar: 50 μm.
Next, we studied the role of Foxf1 in a genetically engineered mouse model of GIST driven by the germline KitV558Δ/+ mutation. These mice develop GIST-like tumors and ICC hyperplasia of large intestine with 100% penetrance (4,37). We generated the compound KitV558Δ/+; Foxf1f-HA-Foxf1/f-HA-Foxf1f; Rosa26Cre-ERT2 mice and conditionally deleted the HA-Foxf1 by tamoxifen (TAM) administration in adult mice with already developed GIST-like tumors. Compared to corn oil control, TAM administration led to expected deletion of HA-Foxf1 in cecal GIST, and resulted in significant reduction of Kit protein levels by immunofluorescence and IHC (Fig. 6C and H). HA-Foxf1 deletion in murine GIST tumors also resulted in profound reduction of Etv1 and Kit levels by immunoblots, significant reduction of tumor weight and proliferation index by Ki67, as well as a significant increase in apoptosis measured by cleaved-caspase 3 IHC (Fig. 6D-H). In addition, these effects are accompanied by increased fibrosis by trichrome stain (Fig. 6H, blue). Furthermore, HA-Foxf1 ablation also resulted in reduction of the ICC hyperplasia of the large intestine of the murine models (Supplementary Fig. S8B). These data demonstrate that FOXF1 is required for GIST cell growth and survival in vitro and Foxf1 is required for GIST tumor growth and maintenance in vivo.
Discussion
GIST is highly resistant to conventional systemic chemotherapy and radiotherapy, yet, highly sensitive to therapeutics targeting lineage-specific master regulators, KIT and ETV1 (5–8,38), pointing to the critical role of lineage-specific cellular context.
Here, we uncovered that FOXF1 is at the top of a unique regulatory hierarchy for the GIST lineage-specific cellular context through direct transcriptional regulation of the key survival factors, KIT and ETV1; FOXF1 also functions as a pioneer factor that can modulate the chromatin accessibility and maintain lineage-specific enhancers, and recruits ETV1 to lineage-specific enhancers to regulate ETV1-dependent GIST lineage-specific transcriptome. The pioneering function of FOXF1 in guiding ETV1 in GIST resembles that of FOXA1 in guiding ER and AR localization in the context of breast cancer and prostate cancer, respectively (12–14,25). Importantly in GIST pathogenesis, FOXF1 differs from FOXA1 in prostate cancer in that FOXA1 does not regulate AR expression, and that FOXA1 loss result in redistribution of AR chromatin binding, while FOXF1 loss results in decreased ETV1 protein, and global loss of ETV1 chromatin binding.
Although both ETV1 and FOXF1 directly regulate KIT expression through enhancer binding in GIST, it is important to note that the strength of FOXF1-mediated regulation of KIT is significantly stronger than that of ETV1 (Fig. 2C and D). This is likely due to a combination of FOXF1’s ability to directly regulate the KIT enhancer chromatin accessibility and expression, as well as its indirect effect on KIT through regulation of ETV1 dosage and cistrome. The direct regulation of KIT by FOXF1 is also inferred from our observations in murine GI tract: Foxf1 is expressed in all ICC subclasses, including the myenteric ICCs (ICC-MY), intramuscular ICCs (ICC-IM), and submucosal plexus ICCs (ICC-SMP) in large intestine (Fig. 6B), whereas Etv1 is expressed only in the ICC-MY and ICC-IM, but not in ICC-SMP (1). These data indicate that the regulatory circuitry hierarchy in GIST may be pre-determined in its precursor ICCs.
How FOXF1 is regulated in GIST remains unclear. During development, hedgehog signaling pathways have been indicated to regulate Foxf1 expression in murine GI development (39). More recently, LINC01081 lncRNA has been described to regulate the expression of FOXF1 in fetal lung fibroblasts (40). Whether these signaling pathways continue to remain functionally significant in FOXF1 regulation beyond development remain unclear. A recent report has described hedgehog signaling pathway dysregulation through Gli-mediated regulation of KIT expression in human GISTs (41). Whether the hedgehog pathway dysregulation also directly impacts on FOXF1 regulation in GIST pathogenesis remains to be investigated.
We had previously described the KIT and MAPK signaling-dependent regulation of ETV1 protein stability (1,4). Here, we observed that in contrast to ETV1, the FOXF1 protein levels were not significantly affected with either KIT or MAPK pathway perturbations. Nevertheless, whether the KIT and/or MAPK signaling activity modulate the FOXF1 transcriptional activity on the chromatin template remains an open question. It is conceivable that signaling can crosstalk with FOXF1 at multiple levels, including signaling dependent modification of FOXF1 itself that may affect FOXF1 binding affinity and/or specificity to chromatin and its transcriptional activity, signaling-dependent modification of the chromatin template that FOXF1 operates as recently demonstrated in breast cancer (42), or signaling-dependent modification of FOXF1 co-activator and other cofactor binding to chromatin. These areas shall be the focus of future investigations.
The forkhead domain is structurally similar to linker H1 histones and thus FOX family transcription factors have the ability to bind compacted chromatin and establish enhancers de novo. During early development, FOXF1/2 are expressed in the mesoderm and is involved in early mesoderm specification, extra-embryonic and lateral plate mesoderm differentiation, whereas FOXA1/2/3 is involved in endoderm specification (43–49). How two pioneer transcription factors that bind to very similar primary DNA sequence can have such disparate cistromes and drive opposing developmental processes represent a fundamental question.
In summary, our data indicate that FOXF1 not only represents a novel diagnostic biomarker, but also a potential novel therapeutic target in GIST. We speculate that the unique regulatory logic set forth by the FOXF1-KIT/ETV1 hierarchy provides a highly enforced dependence on lineage-specific context for GIST pathogenesis and creates a special therapeutic opportunity to target the cellular context for all GISTs, including those that do not have druggable mutations, such as SDH-deficient GIST. Traditionally, transcription factors are difficult to target with the exception of nuclear hormone receptors. However, new development in Proteolysis Targeting Chimeras (PROTACs) that uses bifunctional small molecules to link targeted proteins to the E3 ubiquitin ligase system for targeted protein degradation have provided new promise for targeting transcription factors (50–55).
Methods
Cell lines, antibodies and reagents
The GIST48 and GIST882 cell lines were obtained from Dr. Jonathan A. Fletcher (Dana-Farber Cancer Institute) in 2009. The GIST-T1 cell line was obtained from Dr. Takahiro Taguchi (Kochi University) in 2010 (56). All GIST cell lines were maintained as previously described (1). The A2058 and A375 cell lines were obtained from Dr. Joan Massagué (MSKCC) in 2014, OMIM1.3 cell line was obtained from Dr. Boris C. Bastian (UCSF) in 2011. All media were supplemented with L-glutamine (2 mM), penicillin (100 U/ml), and streptomycin (100 μg/ml), and 10% heat-inactivated fetal bovine serum (FBS) except the medium used for GIST882 cells was supplemented with 15% FBS. All cell lines were cultured at 37 °C in 5% CO2 and were biochemically tested negative for mycoplasma contamination by the MycoAlert Plus MycoPlasma Detection Kit (Lonza), most recently in October, 2016. To authenticate cell lines, all next-generation sequencing data were analyzed to confirm known SNPs.
The following primary antibodies were used: rabbit anti-human FOXF1(Abcam, ab168383) for immunoblot, immunoprecipitation, IHC, and ChIP; rabbit anti-ETV1(Abcam; ab184120) for immunoblot, immunoprecipitation and ChIP; rabbit anti-H3K4me3 for ChIP (active motif, 39159); rabbit anti-H3K4me1 for ChIP (Abcam, ab8895); HRP-conjugated anti-beta ACTIN (Abcam, ab49900) for immunoblot; GAPDH (Abcam, ab9385) for immunoblot; rabbit anti-KIT (Cell Signaling Technology, #3074) for immunoblot and IHC; rabbit anti-phospho-c-Kit (Tyr703) (Cell Signaling Technology, #3073); rabbit anti-Akt (pan) (Cell Signaling Technology, #4691); rabbit anti-phospho-Akt (Ser473) (Cell Signaling Technology, #4060); rabbit anti-MEK1/2 (Cell Signaling Technology, #9122), rabbit anti-phospho-MEK1/2 (Ser217/221) (Cell Signaling Technology, #9154); Phospho-Akt (Ser473) (D9E) XP® Rabbit mAb #4060; rabbit anti-Stat3 (Cell Signaling Technology, #12640); rabbit anti-phospho-Stat3 (Tyr705)) (Cell Signaling Technology, #9145); rabbit anti- p44/42 MAPK (Erk1/2) (Cell Signaling Technology, #4695), rabbit anti-phospho-p44/42 MAPK (Erk1/2) (Thr202/Tyr204) Cell Signaling Technology, #4370) for immunoblot; rat anti-mouse Kit (Cedarlane; CL8936ap) for immunofluorescence; APC-conjugated anti-human CD117 (c-kit) (Biolegend, 313205) for FACS; rabbit anti-HA tag (Cell Signaling Technology, #3724) for immunoblot; rabbit anti-HA tag (Abcam, ab9110) for ChIP; rabbit anti-cleaved caspase 3 (Asp175) (Cell Signaling Technology, #9661) for IHC; rabbit anti-Ki67 (Abcam, ab16667) for IHC.
Human tumor samples
Clinical samples from patients with GIST and other sarcomas were obtained according to Memorial Sloan Kettering Cancer Center (MSKCC) Institutional Review Board (IRB) protocol, and frozen and paraffin embedded tissue samples were banked, and tissue microarrays (TMAs) were generated. All GIST and other sarcomas were pathologically reviewed and confirmed by a sarcoma expert (C.R.A.) at MSKCC. The FOXF1 IHC staining were reviewed and scored independently by two independent sarcoma pathologists (C.R.A., MSKCC and K.D., MSKCC and Erciyes University, Kayseri, Turkey).
siRNA transfection
GIST cells were transfected with siRNAs using DharmaFECT2 (GE healthcare) according to the manufacture’s protocol. In brief, transfection was performed under serum free conditions, using 20nM siRNA in Opti-MEM. Two independent siRNAs targeting the ETV1 coding region (catalog #: J-003801-06-0002 and J-003801-07-0002) and one siRNA customized siRNA targeting the ETV1 UTR (Targeting sequence: CGUCAAAGAAUAUGAGGAAUU) were purchased from GE Healthcare Dharmacon. Two independent siRNAs targeting FOXF1 (Catalog#: 4392420 s5220 and s5221) and two non-targeting control siRNAs (Catalog#: 4390843 and 4390846) were purchased from Thermo Fisher Scientific.
RNA isolation and qRT–PCR
For tissue culture cells, RNA was isolated using E.Z.N.A total RNA kit (Omega). For qRT-PCR, RNA was reverse transcribed using High-Capacity cDNA Reverse Transcription Kit (ABI) and PCR was run using Power SYBR Master Mix (ABI) on a Realplex machine (Eppendorf). Expression was normalized to the ribosomal protein RPL27.
The following primer pairs were used:
hETV1-Exon67: F: CTACCCCATGGACCACAGATTT, R: CTTAAAGCCTTGTGGTGGGAAG;
hKIT: F: GGGATTTTCTCTGCGTTCTG, R: GATGGATGGATGGTGGAGAC;
hFOXF1: F: AGCCCCTGTCCCCCTGTAACCC, R: CTGGGCGACTGCGAGTGATACC
hGPR20: F: TCCCATCTCCAGCCTGCCCG, R: CGCTGGCATTGGTCCGCACT
hDUSP6: F: TGCCGGGCGTTCTACCTGGA, R: GGCGAGCTGCTGCTACACGA;
hRPL27: F: CATGGGCAAGAAGAAGATCG, R: TCCAAGGGGATATCCACAGA;
Transcriptome analysis
Total RNA was isolated from tissue culture cells using QIAGEN RNeasy kits (#74104, Qiagen) after designed experimental perturbation. Transcriptional profiles were generated by RNA-sequencing in the MSKCC Integrative Genomics Operation (IGO) facility using poly-A capture. The RNA-seq libraries were sequenced on the Illumina HiSeq2500 platform with 50-bp single reads to obtain at least 30 million reads for each sample. The sequence data were processed and mapped to the human reference genome (hg19) using STAR v2.3 (57). Gene expression was quantified to reads-per-kilobase mapped (RPKM) using the Cufflinks (58) and Log 2 transformed.
Gene set enrichment analysis was performed using the JAVA program (http://www.broadinstitute.org/gsea) as described (59). We performed GSEA on 2 transcriptional profiles: GIST48 cells treated with siETV1 vs. siSCR control; and GIST48 cells treated with siFOXF1 vs. siSCR control. To identify gene sets enriched among siETV1 and siFOXF1 downregulated genes in GIST48, we used >8,3000 gene sets from the Molecular Signatures Database (MSigDB) as well as custom gene sets of ETV1-dependent, KIT signaling-dependent and GIST-signatures (Supplementary Tables S6-8).
Heatmaps were generated using GENE-E software (http://www.broadinstitute.org/cancer/software/GENE-E).
Chromatin immunoprecipitation (ChIP) and ChIP-qPCR
Chromatin isolation from GIST48, GIST882 and GIST-T1 cells was performed as previously described (1). For siRNA knockdown experiments, chromatin was isolated 72 h after siRNA transfection.
The human ChIP-qPCR primers pairs were:
KIT enhancer: F: GGGGAAGCACGAAAAACACC, R: TCGAAGACTTGTCCCTTGGC;
DUSP6 enhancer: F: TTGTTTGCACTGGGGCTTAT, R:GCTGGAACAGGTTGTGTTGA;
HES1 enhancer: F: TAATTATACGGCCTCGGGCA, R: ATCCGGACGCAGTTTGGAG;
PSA promoter: F: TGGGCGTGTCTCCTCTGC, R: CCTGGATGCACCAGGCC;
GPR20 enhancer: F: CCCTCCCAGGCTCTCCCCAC, R: TCCGGGCCTGCTCTCTGTCC.
ChIP-seq and analysis
Chromatin isolation was performed as previously described (1). Next-generation sequencing was performed on an Illumina HiSeq2500 platform with 50-bp single reads. Reads were aligned to the human genome (hg 19) using the Bowtie alignment software (60), and duplicate reads were eliminated for subsequent analysis. Peak calling was performed using MACS 2.1 (61) comparing immunoprecipitated chromatin with input chromatin, using a false-discovery rate cutoff of q<10−3. We discarded peaks mapped to blacklisted genomic regions identified by ENCODE (62,63).
For each analysis, we merged peaks called for each condition or cell line using Homer mergePeaks (17) and considered overlap if peak summit were within 250-bp. For overlapping peaks, the summit of the merged peak is the center of the summit in the individual experiments. We used Homer annotatePeaks to categorize each peak as promoter (TSS±1kb) or enhancer (non-promoter), quantify number of reads in each condition at each peak (summit±250 bp), identify the nearest gene, and identify the presence of ETS and FOX motif within the peak (summit±250 bp). For de novo motif analysis, we employed two software suites, Homer and MEME-ChIP (64), using summit±250 bp as input. For visualization, we generated coverage “bigwig” files using bamCoverage command from deepTools2 (65), normalizing to total reads and human genome size. The ChIP-seq density plots of Bigwig files were generated using either SeqPlots (66) or deepTools2 and ChIP-seq profiles of Bigwig files were generated using Integrated Genome Browser software (67).
Analysis of ETV1 peaks between two GIST and two prostate cancer cell lines was performed as above. The merged promoter and non-promoter peaks were log2 transform and separately clustered using K-means algorithm (n=3 groups) and plotted using SeqPlots. Analysis of ETV1 and FOXF1 peaks in GIST48 and GIST-T1 cell lines was performed as above. ETV1-only peaks were defined as MACS2 called peaks in either cell line and no FOXF1 peaks in both cell lines. Similarly, FOXF1-only peaks were called in either cell line, and no ETV1 peaks in both cell lines. ETV1/FOXF1 peaks were called for ETV1 in either cell line and for FOXF1 in either cell line. Peaks were log2 transformed and plotted using SeqPlots.
For analysis of ETV1, FOXF1, H4K3me1, and H4K3me3 ChIP-seq and ATAC-seq in GIST48 cells after siSCR, siETV1 (siETV1-1) and siFOXF1 (siFOXF1-2) transfection, we used ETV1 and FOXF1 peaks at baseline condition (siSCR) and merged and annotated the peaks as above. By using MACS2 peak caller with cutoff of q<10−3, we noted a number of ETV1 enhancer peaks that was not called as a FOXF1 peak but exhibited modest FOXF1 binding and contained FOX motif (Fig. 2A, Supplementary Fig. 2B). Thus, to determine the effect of FOXF1 on ETV1 binding and chromatin accessibility, we restricted “ETV1-only peaks” to those with FOXF1 ChIP signal < 4 (log2=2) and “ETV1/FOXF1 both peaks” to those with FOXF1 ChIP signal > 8 (log2=3) (see Fig. 3A–C and Fig. 4A–C). Integrative Chip-seq and ATAC-seq profile and density plot was generated using deepTools2 in linear scale.
For integrative analysis of ChIP-seq and RNA-seq, we annotated each expressed gene (FKPM >4) by the number of FOXF1 or ETV1 enhancer peaks mapped to the gene and grouped them by the number of peaks. We calculated the mean change by FOXF1 knockdown (siFOXF1-1 and siFOXF2-2 vs siSCR-1 and siSCR-2) and ETV1 knockdown for all genes in each group.
Assay for transposase-accessible chromatin using sequencing (ATAC-seq) and analysis
ATAC-seq was performed as previously described (28). To examine the initial changes in chromatin, we harvested nuclei from GIST48, GIST-T1 and GIST882 cells with siRNA-mediated downregulations of FOXF1, ETV1 or scramble controls at a time point (48-72 hours after siRNA transfection) before any remarkable growth suppression or cell death was observed. For each sample, cell nuclei were prepared from 50,000 cells, and incubated with 2.5 μl of transposase (Illumina) in a 50-μl reaction for 30 min at 37 °C. After purification of transposase-fragmented DNA, the library was amplified by PCR and subjected to paired-end 50 base-pair high-throughput sequencing on an Illumina HiSeq2500 platform.
ATAC-seq reads were quality and adapter trimmed using ‘trim_galore’ before aligning to human genome assembly hg19 with bowtie2 using the default parameters. Aligned reads with the same start position and orientation were collapsed to a single read before subsequent analysis. Density profiles were created by extending each read to the average library fragment size and then computing density using the BEDTools suite, with subsequent normalization to a sequencing depth of ten million reads for each library. Subsequent data analysis and display is as described in the ChIP-seq analysis section.
Generation of stable cell lines
Two miRE-based shRNA constructs against FOXF1 and scrambled controls were purchased from Mirimus, Inc. shRNA sequences were subcloned into doxycycline-inducible LT3GEPIR (pRRL) vector. LT3GEPIR is a single lentiviral vector with doxycycline inducible GFP and mirE with constitutive puromycin and rTTA (68). Targeting sequences: FOXF1sh1: AGGAGTTTGTCTTCTCTTTCA; FOXF1sh2: TCCTTCCTCACTCCTTTCCTTCCTCACTCCTT. ETV1 cDNA (Open Biosystems) cloned into MSCV-flag-HA-IRES-GFP vector (Addgene). All constructs were confirmed by Sanger sequencing. Viruses were generated and infected as previously described (1). Stable cell lines were validated by immunoblot.
FACS-based growth competition assay
For growth competition assay by fluorescence-activated cell sorting analysis (FACS), cells (a mixture of parental GIST cells and the GIST cells containing doxycycline-inducible GFP and shRNA construct) were plated at 2 × 106 cells per well of 6-well plate on day 0 in triplicates and treated with 1μg/ml doxycycline to induce GFP and concomitant shRNA expression. GFP and KIT surface protein were analyzed by FACS twice weekly after doxycycline treatment and the percentage of GFP-positive cells were followed overtime. Total (surface plus intracellular) KIT proteins of cells were detected similarly by FACS after fixing and permeabilizing the cells with Foxp3/Transcription Factor Staining Buffer Set (eBioscience). For all competition assays, media was replaced every 3-4 days and the GFP-positive cells were tracked using LSRFortessa cytometer (BD Biosciences).
Cell cycle analysis
For cell cycle analysis, cells were prepared by click-iT EdU pacific blue flow cytometry assay kit (Thermo Fisher Scientific) before FACS. Cell cycle analysis was performed using FlowJo software.
Cell viability assay
The number of viable cells was measured using CellTiter-Glo 2.0 Luminescent Cell Viability Assay (Promega) according to manufacturer’s instructions. GIST-T1, GIST882 and GIST48 cells were plated at 10,000, 40,000 and 50,000 cells, respectively, per well in triplicates on a 96-well plate on day 0 and transfected with 2 individual siRNA targeting FOXF1 along with scrambled siRNA controls. The cells were cultured and their viability was assessed on day1, 3, 6 post transfection with siRNAs. RNA was collected on day 3 to check for knockdown efficiency.
Immunoblot and Immunoprecipitation
Cell lysates were prepared in RIPA buffer supplemented with proteinase/phosphatase inhibitor. Proteins were resolved in NuPAGE Novex 4–12% Bis-Tris Protein Gels (#NP0321BOX, Life Technologies) and transferred electrophoretically onto a nitrocellulose 0.45 μm membrane (#162-0115, BioRad). Membranes were blocked for 1 hour at room temperature in Blocking Buffer and were incubated overnight at 4 °C with the primary antibodies as described in the reagent section. Signal was visualized either with secondary HRP conjugated antibodies and ECL or secondary antibodies (IRDye 800CW goat anti-Rabbit #926-32211, 1:20,000, LI-COR; IRDye 680RD goat anti-mouse #926-68070, 1:20,000, LI-COR) in 50% Odyssey Blocking Buffer in PBS plus 0,1% Tween 20 and a LI-COR Odyssey CLx scanner and adjusted using LI-COR Image Studio. Immunoblots were independently performed at least twice and a representative experiment is shown.
For ETV1 and FOXF1 co-immunoprecipitation (co-IP), GIST48 and GIST-T1 cells were lysed in 20 mM Tris-HCl (pH 7.5), 1% Triton X-100, 150 mM NaCl, 1 mM EDTA, 1 mM DTT, 1 mM PMSF, and proteinase/phosphatase inhibitors. After incubation and centrifugation, 120 μl supernatant was used as input, and 1000 μl for immunoprecipitation using the following antibodies: 0.5 μg of anti-ETV1 antibody, 0.5ug of anti-FOXF1 antibody and 0.5ug of rabbit IgG as control. 20μl of Protein A/G UltraLink Resin (#53133, Thermo Scientific) were used for immunoprecipitation. The immunoprecipitated material was eluted in 4 × SDS loading buffer for immunoblotting. Co-Immunoprecipitation was independently performed at least three times and a representative immunoblot is shown.
Generation of compound genetically engineered mouse models
All mouse studies are approved by Memorial Sloan Kettering Cancer Center (MSKCC) Institutional Animal Care and Use Committee under protocol 11-12-029.
We used the pEZ-Frt-lox-DT (Addgene #11736) that contain cloning site for 5′ and 3′ homology arms, and a central targeting region flanked by LoxP sites, an neomycin cassette flanked by FRT sites, and the Diphtheria toxin gene outside the homology region. To generate Foxf1-targeting vector, we cloned mouse Foxf1 with an amino-terminal HA-tag using the SalI site, the 3′ arm between ClaI and NotI sites, and the 5′ arm between XhoI and HindIII sites. The Foxf1f-HA-Foxf1-targeting vector was used for electroporation of mouse ES cells, which were selected for neo (G418) resistance. Correct targeting was confirmed using Southern blotting with 5′ Probe digested of HindIII digested DNA and 3′ Probe of BglII digested DNA. ES cells with the appropriate Foxf1f-HA-Foxf1 -targeted locus were used to generate chimeric mice by injecting Foxf1f-HA-Foxf1 ES cells into mouse blastocysts. To produce Foxf1f-HA-Foxf1/+ mice, chimeric mice were bred with C57Bl/6 mice in the MSKCC animal facility. The Neo cassette was deleted by breeding of HA-Foxf1fl/+ mice with Actb-FlpE mice (Jackson Lab).
Genotyping of 5′ LoxP site and 3′ LoxP site were performed using the following primers:
Foxf1a-5′ LoxP: F: CGGGTCCAGGTCGGCAGAGG, R: TGCAGTGTCCGATCCCCCGT
Foxf1a-3′LoxP: F: AGCAAAGGCCCTGTGTATCTA, R: GGCTTGGAGGCTGAAAGCTA
The Kit Δ558V/+ knockin mouse was a generous gift from Dr. Peter Besmer (Memorial Sloan Kettering Cancer Center)(37), the Rosa26Cre-ERT2 mice were a generous gift from Dr. Andrea Ventura (Memorial Sloan Kettering Cancer Center). Kit Δ558V/+;Foxf1f-HA-Foxf1/f-HA-Foxf1; Rosa26 Cre-ERT2/+ and KitΔ558V/+; Foxf1f-HA-Foxf1/f-HA-Foxf1; Rosa26 Cre-ERT2/Cre-ERT2 mice were generated through standard mouse breeding within the MSKCC animal facility.
For tamoxifen or corn oil treatment, tamoxifen (TAM) Toronto Research Chemicals) was dissolved at 20 mg/mL in corn oil and TAM (at a dose of 2 mg) or corn oil was injected intraperitoneally to 5-6 week-old age matched Kit Δ558V/+;Foxf1f-HA-Foxf1/f-HA-Foxf1; Rosa26 Cre-ERT2 mice every other day for two doses to induce HA-Foxf1 ablation. The GI tract and GIST tumors were harvested 7 days post TAM or corn coil administration for analyses.
Immunofluorescence(IF), Immunohistochemistry(IHC) and Histology
For IF of cryostat sections of the mouse gastrointestinal tract, mouse stomach, small intestine, large intestine and cecum (or cecal tumor) were dissected and fixed in 4% paraformaldehyde for 2 hours followed by an overnight incubation in 30% sucrose. They were then embedded in optimal cutting temperature compound, flash-frozen, and cut into 5-μm sections using a cryostat. Tissue sections were blocked for 1 hour using 5% goat serum, and incubated with primary antibodies at 4°C overnight and secondary antibody for 2 hours at room temperature. Slides were mounted using Prolong Gold (Invitrogen), and images were taken on a Nikon Eclipse TE2000-E microscope using a Photometric Coolsnap HQ camera. Images were taken with ×20 (numerical aperture, 0.75) objectives. Monochrome images taken with DAPI, FITC, and Texas Red filter sets were pseudocolored blue, green, and red, respectively, and merged using ImageJ. The exposure, threshold, and maximum were identical between tamoxifen-treated and corn oil–treated controls of Foxf1f-HA-Foxf1/f-HA-Foxf1; Rosa26 CreERT2/+ or Kit Δ558V/+; Foxf1f-HA-Foxf1/f-HA-Foxf1; Rosa26 CreERT2/CreERT2 images.
Tissue paraffin embedding, sectioning, and H&E staining were performed by the Histoserv, Inc. IHC of mouse FFPE and human tissue microarray (TMA) tumor samples was performed by the MSKCC Human Oncology and Pathogenesis Program automatic staining facility using a Ventana BenchMark ULTRA automated stainer.
Statistics
All statistical comparisons between two groups were performed by GraphPad Prism software 6.0 using a two-tailed unpaired t-test, unless otherwise noted in the figure legend. The variance between the statistically compared groups was similar.
Gene Expression Omnibus (GEO) Accession numbers of datasets generated or used
GSE22852: ETV1 ChIP-seq in steady-state GIST48 cells from our previous study (1)
GSE47120: ETV1 ChIP-seq in steady-state LNCaP cells from our previous study (10).
- GSE106626:
-
○RNA-seq expression profile of ETV1 or FOXF1 knockdown (siSCR vs. siETV1 vs. siFOXF1) in GIST48 cells;
-
○ETV1 ChIP-seq in steady-state MDA-PCa2b cells;
-
○ETV1 ChIP-seq and FOXF1 ChIP-seq in steady-state GIST-T1 cells;
-
○FOXF1 ChIP-seq in steady-state GIST48 cells;
-
○ETV1 ChIP-seq, FOXF1 ChIP-seq, H3K4Me1 ChIP-seq and H3K4Me3 ChIP-seq of ETV1 or FOXF1 knockdown (siSCR vs. siETV1 vs. siFOXF1) in GIST48 cells;
-
○ATAC-seq of ETV1 or FOXF1 knockdown (siSCR vs. siETV1 vs. siFOXF1) in GIST48 cells, GIST882 and GIST-T1 cells.
-
○
Supplementary Material
Statement of Significances.
We uncover that FOXF1 defines the core regulatory circuitry in GIST through both direct transcriptional regulation and pioneer factor function. The unique and simultaneous control of signaling and transcriptional circuitry by FOXF1 sets up an enforced transcriptional addiction to FOXF1 in GIST, which can be exploited diagnostically and therapeutically.
Acknowledgments
We thank the following Memorial Sloan Kettering Cancer Center (MSKCC) core facilities: Tri-institutional Gene Targeting (C. Yang), Mouse Genetics Core (W. Mark and P. Romanienko), Integrated Genomics Operation (A. Viale), Molecular Cytogenetics (M. Leversha), and Molecular Cytology (K. Manova). We thank Drs. Massague, Fletcher, Bastian and Taguchi for cell lines. Next generation sequencing for RNA-seq, ChIP-seq and ATAC-seq were done at the MSKCC Integrated Genomics Operation Facility.
Grant Support
This work was supported by grants from the NCI (K08CA140946, Y. Chen; R01CA193837, Y. Chen; P50CA092629, Y. Chen; P50CA140146, P. Chi, C.R. Antonescu; K08CA151660, P. Chi; DP2 CA174499, P. Chi), US DOD (W81XWH-10-1-0197, P. Chi), Prostate Cancer Foundation (Y. Chen), Geoffrey Beene Cancer Research Center (Y. Chen, PC), Gerstner Family Foundation (Y. Chen), Bressler Scholars Fund (Y. Chen), GIST Cancer Research Fund (P. Chi, C.R. Antonescu), Shuman Fund (P. Chi, C.R. Antonescu) and GIST Cancer Awareness Fund (P. Chi).
Footnotes
Conflict of Interest: none.
References
- 1.Chi P, Chen Y, Zhang L, Guo X, Wongvipat J, Shamu T, et al. ETV1 is a lineage survival factor that cooperates with KIT in gastrointestinal stromal tumours. Nature. 2010;467:849–53. doi: 10.1038/nature09409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hirota S, Isozaki K, Moriyama Y, Hashimoto K, Nishida T, Ishiguro S, et al. Gain-of-function mutations of c-kit in human gastrointestinal stromal tumors. Science. 1998;279:577–80. doi: 10.1126/science.279.5350.577. [DOI] [PubMed] [Google Scholar]
- 3.Huizinga JD, Thuneberg L, Kluppel M, Malysz J, Mikkelsen HB, Bernstein A. W/kit gene required for interstitial cells of Cajal and for intestinal pacemaker activity. Nature. 1995;373:347–9. doi: 10.1038/373347a0. [DOI] [PubMed] [Google Scholar]
- 4.Ran L, Sirota I, Cao Z, Murphy D, Chen Y, Shukla S, et al. Combined inhibition of MAP kinase and KIT signaling synergistically destabilizes ETV1 and suppresses GIST tumor growth. Cancer discovery. 2015;5:304–15. doi: 10.1158/2159-8290.CD-14-0985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Blanke CD, Demetri GD, von Mehren M, Heinrich MC, Eisenberg B, Fletcher JA, et al. Long-term results from a randomized phase II trial of standard- versus higher-dose imatinib mesylate for patients with unresectable or metastatic gastrointestinal stromal tumors expressing KIT. J Clin Oncol. 2008;26:620–5. doi: 10.1200/JCO.2007.13.4403. [DOI] [PubMed] [Google Scholar]
- 6.Chi P, Qin L, D’Angelo SP, Dickson MA, Gounder MM, Keohan ML, Shoushtari AN, Condy MM, Konen T, Fruauff A, DeMatteo RP, Singer S, Hwang S, Antonescu CR, Tap WD. A phase Ib/II study of MEK162 (binimetinib [BINI]) in combination with imatinib in patients with advanced gastrointestinal stromal tumor (GIST) J Clin Oncol. 2015;33(suppl) abstr 10507. [Google Scholar]
- 7.Demetri GD, von Mehren M, Blanke CD, Van den Abbeele AD, Eisenberg B, Roberts PJ, et al. Efficacy and safety of imatinib mesylate in advanced gastrointestinal stromal tumors. N Engl J Med. 2002;347:472–80. doi: 10.1056/NEJMoa020461. [DOI] [PubMed] [Google Scholar]
- 8.Verweij J, Casali PG, Zalcberg J, LeCesne A, Reichardt P, Blay JY, et al. Progression-free survival in gastrointestinal stromal tumours with high-dose imatinib: randomised trial. Lancet. 2004;364:1127–34. doi: 10.1016/S0140-6736(04)17098-0. [DOI] [PubMed] [Google Scholar]
- 9.Tomlins SA, Rhodes DR, Perner S, Dhanasekaran SM, Mehra R, Sun XW, et al. Recurrent fusion of TMPRSS2 and ETS transcription factor genes in prostate cancer. Science. 2005;310:644–8. doi: 10.1126/science.1117679. [DOI] [PubMed] [Google Scholar]
- 10.Chen Y, Chi P, Rockowitz S, Iaquinta PJ, Shamu T, Shukla S, et al. ETS factors reprogram the androgen receptor cistrome and prime prostate tumorigenesis in response to PTEN loss. Nature medicine. 2013;19:1023–1029. doi: 10.1038/nm.3216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jane-Valbuena J, Widlund HR, Perner S, Johnson LA, Dibner AC, Lin WM, et al. An oncogenic role for ETV1 in melanoma. Cancer Res. 2010;70:2075–84. doi: 10.1158/0008-5472.CAN-09-3092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hurtado A, Holmes KA, Ross-Innes CS, Schmidt D, Carroll JS. FOXA1 is a key determinant of estrogen receptor function and endocrine response. Nat Genet. 2011;43:27–33. doi: 10.1038/ng.730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jozwik KM, Carroll JS. Pioneer factors in hormone-dependent cancers. Nature reviews Cancer. 2012;12:381–5. doi: 10.1038/nrc3263. [DOI] [PubMed] [Google Scholar]
- 14.Lupien M, Eeckhoute J, Meyer CA, Wang Q, Zhang Y, Li W, et al. FoxA1 translates epigenetic signatures into enhancer-driven lineage-specific transcription. Cell. 2008;132:958–70. doi: 10.1016/j.cell.2008.01.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zaret KS, Mango SE. Pioneer transcription factors, chromatin dynamics, and cell fate control. Curr Opin Genet Dev. 2016;37:76–81. doi: 10.1016/j.gde.2015.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zaret K. Developmental competence of the gut endoderm: genetic potentiation by GATA and HNF3/fork head proteins. Dev Biol. 1999;209:1–10. doi: 10.1006/dbio.1999.9228. [DOI] [PubMed] [Google Scholar]
- 17.Heinz S, Benner C, Spann N, Bertolino E, Lin YC, Laslo P, et al. Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol Cell. 2010;38:576–89. doi: 10.1016/j.molcel.2010.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tomlins SA, Laxman B, Dhanasekaran SM, Helgeson BE, Cao X, Morris DS, et al. Distinct classes of chromosomal rearrangements create oncogenic ETS gene fusions in prostate cancer. Nature. 2007;448:595–9. doi: 10.1038/nature06024. [DOI] [PubMed] [Google Scholar]
- 19.Tomlins SA, Mehra R, Rhodes DR, Smith LR, Roulston D, Helgeson BE, et al. TMPRSS2:ETV4 gene fusions define a third molecular subtype of prostate cancer. Cancer Res. 2006;66:3396–400. doi: 10.1158/0008-5472.CAN-06-0168. [DOI] [PubMed] [Google Scholar]
- 20.Gasi D, van der Korput HA, Douben HC, de Klein A, de Ridder CM, van Weerden WM, et al. Overexpression of full-length ETV1 transcripts in clinical prostate cancer due to gene translocation. PLoS One. 2011;6:e16332. doi: 10.1371/journal.pone.0016332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Calo E, Wysocka J. Modification of enhancer chromatin: what, how, and why? Mol Cell. 2013;49:825–37. doi: 10.1016/j.molcel.2013.01.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Heintzman ND, Hon GC, Hawkins RD, Kheradpour P, Stark A, Harp LF, et al. Histone modifications at human enhancers reflect global cell-type-specific gene expression. Nature. 2009;459:108–12. doi: 10.1038/nature07829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Visel A, Blow MJ, Li Z, Zhang T, Akiyama JA, Holt A, et al. ChIP-seq accurately predicts tissue-specific activity of enhancers. Nature. 2009;457:854–8. doi: 10.1038/nature07730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Iwafuchi-Doi M, Donahue G, Kakumanu A, Watts JA, Mahony S, Pugh BF, et al. The Pioneer Transcription Factor FoxA Maintains an Accessible Nucleosome Configuration at Enhancers for Tissue-Specific Gene Activation. Mol Cell. 2016;62:79–91. doi: 10.1016/j.molcel.2016.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zaret KS, Carroll JS. Pioneer transcription factors: establishing competence for gene expression. Genes Dev. 2011;25:2227–41. doi: 10.1101/gad.176826.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Shin G, Kang TW, Yang S, Baek SJ, Jeong YS, Kim SY. GENT: gene expression database of normal and tumor tissues. Cancer informatics. 2011;10:149–57. doi: 10.4137/CIN.S7226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Subramanian S, West RB, Marinelli RJ, Nielsen TO, Rubin BP, Goldblum JR, et al. The gene expression profile of extraskeletal myxoid chondrosarcoma. J Pathol. 2005;206:433–44. doi: 10.1002/path.1792. [DOI] [PubMed] [Google Scholar]
- 28.Buenrostro JD, Giresi PG, Zaba LC, Chang HY, Greenleaf WJ. Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin, DNA-binding proteins and nucleosome position. Nat Methods. 2013;10:1213–8. doi: 10.1038/nmeth.2688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Buenrostro JD, Wu B, Chang HY, Greenleaf WJ. ATAC-seq: A Method for Assaying Chromatin Accessibility Genome-Wide. In: Ausubel Frederick M, et al., editors. Current protocols in molecular biology. Vol. 109. 2015. pp. 21.9.1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Creyghton MP, Cheng AW, Welstead GG, Kooistra T, Carey BW, Steine EJ, et al. Histone H3K27ac separates active from poised enhancers and predicts developmental state. Proc Natl Acad Sci U S A. 2010;107:21931–6. doi: 10.1073/pnas.1016071107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Heintzman ND, Stuart RK, Hon G, Fu Y, Ching CW, Hawkins RD, et al. Distinct and predictive chromatin signatures of transcriptional promoters and enhancers in the human genome. Nat Genet. 2007;39:311–8. doi: 10.1038/ng1966. [DOI] [PubMed] [Google Scholar]
- 32.Rada-Iglesias A, Bajpai R, Swigut T, Brugmann SA, Flynn RA, Wysocka J. A unique chromatin signature uncovers early developmental enhancers in humans. Nature. 2011;470:279–83. doi: 10.1038/nature09692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tabone-Eglinger S, Subra F, El Sayadi H, Alberti L, Tabone E, Michot JP, et al. KIT mutations induce intracellular retention and activation of an immature form of the KIT protein in gastrointestinal stromal tumors. Clin Cancer Res. 2008;14:2285–94. doi: 10.1158/1078-0432.CCR-07-4102. [DOI] [PubMed] [Google Scholar]
- 34.Kim WK, Yun S, Park CK, Bauer S, Kim J, Lee MG, et al. Sustained Mutant KIT Activation in the Golgi Complex Is Mediated by PKC-theta in Gastrointestinal Stromal Tumors. Clin Cancer Res. 2017;23:845–56. doi: 10.1158/1078-0432.CCR-16-0521. [DOI] [PubMed] [Google Scholar]
- 35.Obata Y, Horikawa K, Takahashi T, Akieda Y, Tsujimoto M, Fletcher JA, et al. Oncogenic signaling by Kit tyrosine kinase occurs selectively on the Golgi apparatus in gastrointestinal stromal tumors. Oncogene. 2017;36:3661–72. doi: 10.1038/onc.2016.519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chen H, Ordog T, Chen J, Young DL, Bardsley MR, Redelman D, et al. Differential gene expression in functional classes of interstitial cells of Cajal in murine small intestine. Physiol Genomics. 2007;31:492–509. doi: 10.1152/physiolgenomics.00113.2007. [DOI] [PubMed] [Google Scholar]
- 37.Sommer G, Agosti V, Ehlers I, Rossi F, Corbacioglu S, Farkas J, et al. Gastrointestinal stromal tumors in a mouse model by targeted mutation of the Kit receptor tyrosine kinase. Proc Natl Acad Sci U S A. 2003;100:6706–11. doi: 10.1073/pnas.1037763100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Dematteo RP, Heinrich MC, El-Rifai WM, Demetri G. Clinical management of gastrointestinal stromal tumors: before and after STI-571. Human pathology. 2002;33:466–77. doi: 10.1053/hupa.2002.124122. [DOI] [PubMed] [Google Scholar]
- 39.Madison BB, McKenna LB, Dolson D, Epstein DJ, Kaestner KH. FoxF1 and FoxL1 link hedgehog signaling and the control of epithelial proliferation in the developing stomach and intestine. J Biol Chem. 2009;284:5936–44. doi: 10.1074/jbc.M808103200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Szafranski P, Dharmadhikari AV, Wambach JA, Towe CT, White FV, Grady RM, et al. Two deletions overlapping a distant FOXF1 enhancer unravel the role of lncRNA LINC01081 in etiology of alveolar capillary dysplasia with misalignment of pulmonary veins. Am J Med Genet A. 2014;164A:2013–9. doi: 10.1002/ajmg.a.36606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tang CM, Lee TE, Syed SA, Burgoyne AM, Leonard SY, Gao F, et al. Hedgehog pathway dysregulation contributes to the pathogenesis of human gastrointestinal stromal tumors via GLI-mediated activation of KIT expression. Oncotarget. 2016;7:78226–41. doi: 10.18632/oncotarget.12909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Toska E, Osmanbeyoglu HU, Castel P, Chan C, Hendrickson RC, Elkabets M, et al. PI3K pathway regulates ER-dependent transcription in breast cancer through the epigenetic regulator KMT2D. Science. 2017;355:1324–30. doi: 10.1126/science.aah6893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Fleury M, Eliades A, Carlsson P, Lacaud G, Kouskoff V. FOXF1 inhibits hematopoietic lineage commitment during early mesoderm specification. Development. 2015;142:3307–20. doi: 10.1242/dev.124685. [DOI] [PubMed] [Google Scholar]
- 44.Ormestad M, Astorga J, Landgren H, Wang T, Johansson BR, Miura N, et al. Foxf1 and Foxf2 control murine gut development by limiting mesenchymal Wnt signaling and promoting extracellular matrix production. Development. 2006;133:833–43. doi: 10.1242/dev.02252. [DOI] [PubMed] [Google Scholar]
- 45.Kalinichenko VV, Lim L, Stolz DB, Shin B, Rausa FM, Clark J, et al. Defects in pulmonary vasculature and perinatal lung hemorrhage in mice heterozygous null for the Forkhead Box f1 transcription factor. Dev Biol. 2001;235:489–506. doi: 10.1006/dbio.2001.0322. [DOI] [PubMed] [Google Scholar]
- 46.Mahlapuu M, Ormestad M, Enerback S, Carlsson P. The forkhead transcription factor Foxf1 is required for differentiation of extra-embryonic and lateral plate mesoderm. Development. 2001;128:155–66. doi: 10.1242/dev.128.2.155. [DOI] [PubMed] [Google Scholar]
- 47.Levinson-Dushnik M, Benvenisty N. Involvement of hepatocyte nuclear factor 3 in endoderm differentiation of embryonic stem cells. Mol Cell Biol. 1997;17:3817–22. doi: 10.1128/mcb.17.7.3817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Duncan SA, Navas MA, Dufort D, Rossant J, Stoffel M. Regulation of a transcription factor network required for differentiation and metabolism. Science. 1998;281:692–5. doi: 10.1126/science.281.5377.692. [DOI] [PubMed] [Google Scholar]
- 49.Sekiya S, Suzuki A. Direct conversion of mouse fibroblasts to hepatocyte-like cells by defined factors. Nature. 2011;475:390–3. doi: 10.1038/nature10263. [DOI] [PubMed] [Google Scholar]
- 50.Deshaies RJ. Protein degradation: Prime time for PROTACs. Nat Chem Biol. 2015;11:634–5. doi: 10.1038/nchembio.1887. [DOI] [PubMed] [Google Scholar]
- 51.Winter GE, Buckley DL, Paulk J, Roberts JM, Souza A, Dhe-Paganon S, et al. DRUG DEVELOPMENT. Phthalimide conjugation as a strategy for in vivo target protein degradation. Science. 2015;348:1376–81. doi: 10.1126/science.aab1433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Bondeson DP, Mares A, Smith IE, Ko E, Campos S, Miah AH, et al. Catalytic in vivo protein knockdown by small-molecule PROTACs. Nat Chem Biol. 2015;11:611–7. doi: 10.1038/nchembio.1858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Lu G, Middleton RE, Sun H, Naniong M, Ott CJ, Mitsiades CS, et al. The myeloma drug lenalidomide promotes the cereblon-dependent destruction of Ikaros proteins. Science. 2014;343:305–9. doi: 10.1126/science.1244917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kronke J, Udeshi ND, Narla A, Grauman P, Hurst SN, McConkey M, et al. Lenalidomide causes selective degradation of IKZF1 and IKZF3 in multiple myeloma cells. Science. 2014;343:301–5. doi: 10.1126/science.1244851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Petzold G, Fischer ES, Thoma NH. Structural basis of lenalidomide-induced CK1alpha degradation by the CRL4(CRBN) ubiquitin ligase. Nature. 2016;532:127–30. doi: 10.1038/nature16979. [DOI] [PubMed] [Google Scholar]
- 56.Taguchi T, Sonobe H, Toyonaga S, Yamasaki I, Shuin T, Takano A, et al. Conventional and molecular cytogenetic characterization of a new human cell line, GIST-T1, established from gastrointestinal stromal tumor. Lab Invest. 2002;82:663–5. doi: 10.1038/labinvest.3780461. [DOI] [PubMed] [Google Scholar]
- 57.Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013;29:15–21. doi: 10.1093/bioinformatics/bts635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Trapnell C, Williams BA, Pertea G, Mortazavi A, Kwan G, van Baren MJ, et al. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nature biotechnology. 2010;28:511–5. doi: 10.1038/nbt.1621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Morgensztern D, Waqar S, Subramanian J, Trinkaus K, Govindan R. Prognostic impact of malignant pleural effusion at presentation in patients with metastatic non-small-cell lung cancer. Journal of thoracic oncology : official publication of the International Association for the Study of Lung Cancer. 2012;7:1485–9. doi: 10.1097/JTO.0b013e318267223a. [DOI] [PubMed] [Google Scholar]
- 60.Langmead B, Trapnell C, Pop M, Salzberg SL. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009;10:R25. doi: 10.1186/gb-2009-10-3-r25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Zhang Y, Liu T, Meyer CA, Eeckhoute J, Johnson DS, Bernstein BE, et al. Model-based analysis of ChIP-Seq (MACS) Genome Biol. 2008;9:R137. doi: 10.1186/gb-2008-9-9-r137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Consortium EP. An integrated encyclopedia of DNA elements in the human genome. Nature. 2012;489:57–74. doi: 10.1038/nature11247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Carroll TS, Liang Z, Salama R, Stark R, de Santiago I. Impact of artifact removal on ChIP quality metrics in ChIP-seq and ChIP-exo data. Frontiers in genetics. 2014;5:75. doi: 10.3389/fgene.2014.00075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Machanick P, Bailey TL. MEME-ChIP: motif analysis of large DNA datasets. Bioinformatics. 2011;27:1696–7. doi: 10.1093/bioinformatics/btr189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Ramirez F, Ryan DP, Gruning B, Bhardwaj V, Kilpert F, Richter AS, et al. deepTools2: a next generation web server for deep-sequencing data analysis. Nucleic Acids Res. 2016;44:W160–5. doi: 10.1093/nar/gkw257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Stempor P, Ahringer J. SeqPlots - Interactive software for exploratory data analyses, pattern discovery and visualization in genomics. Wellcome open research. 2016;1:14. doi: 10.12688/wellcomeopenres.10004.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Freese NH, Norris DC, Loraine AE. Integrated genome browser: visual analytics platform for genomics. Bioinformatics. 2016;32:2089–95. doi: 10.1093/bioinformatics/btw069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Fellmann C, Hoffmann T, Sridhar V, Hopfgartner B, Muhar M, Roth M, et al. An optimized microRNA backbone for effective single-copy RNAi. Cell reports. 2013;5:1704–13. doi: 10.1016/j.celrep.2013.11.020. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.