

Abstract
Understanding neuroadaptations involved in obesity is critical for developing new approaches to treatment. Diet-induced neuroadaptations within the dorsal striatum have the capacity to drive excessive food seeking and consumption. Five week old C57BL/6J mice consumed a high-fat, high-sugar ‘western diet’ (WD) or a control ‘standard diet’ (SD) for 16 weeks. Weight gain, glucose tolerance, and insulin tolerance were measured to confirm an obese-like state. Following these 16 weeks, electrophysiological recordings were made from medium spiny neurons (MSNs) in the medial (DMS) and lateral (DLS) portions of dorsal striatum to evaluate diet effects on neuronal excitability and synaptic plasticity. In addition, fast-scan cyclic voltammetry evaluated dopamine transmission in these areas. WD mice gained significantly more weight and consumed more calories than SD mice and demonstrated impaired glucose tolerance. Electrophysiology data revealed that MSNs from WD mice demonstrated increased AMPA to NMDA receptor current ratio and prolonged spontaneous glutamate-mediated currents, specifically in the DLS. Evoked dopamine release was also significantly greater and reuptake slower in both subregions of WD striatum. Finally, dorsal striatal MSNs from WD mice were significantly less likely to demonstrate mu-opioid receptor-mediated synaptic plasticity. Neuronal excitability and GABAergic transmission were unaffected by diet in either striatal subregion. Our results demonstrate that a high-fat, high-sugar diet alters facets of glutamate, dopamine, and opioid signaling within the dorsal striatum, with some subregion specificity. These alterations within a brain area known to play a role in food motivation/consumption and habitual behavior are highly relevant for the clinical condition of obesity and its treatment.
Keywords: Obesity, dorsal striatum, dopamine, electrophysiology, glutamate, mice
Introduction
Obesity and its related health issues (e.g. diabetes) are associated with increased mortality and morbidity and the cost to society (e.g. healthcare, lost work days, etc.) is great (Flegal, 2005; Flegal et al., 2013; Haslam and James, 2005; Hedley et al., 2004; Jia and Lubetkin, 2010; Stice et al., 2008b). It is believed that the ease of access to and the low cost of highly palatable, high calorie foods is largely responsible for the ‘obesity-epidemic’ (Swinburn et al., 2011). It is estimated that over 70% of U.S. adults meet criteria for being overweight or obese (Fryar et al., 2016). The tremendous extent of this issue necessitates further understanding of how brain function may be altered in obese individuals to more accurately assess risk factors and explore novel treatment options.
The dorsal striatum is a brain region with known involvement in habitual and compulsive behaviors, including food seeking and binge-like eating (Balleine, 2005; Furlong et al., 2014; Wang et al., 2011; Yin et al., 2004). Furthermore, the dorsal striatum plays a role in homeostatic energy consumption (Palmiter, 2008), thus, dysfunction may contribute to the continuation of food consumption when already sated. Clinical research has demonstrated that the motivation to consume and hedonic properties of food are associated with increased DA signaling in the dorsal striatum of healthy, hungry subjects (Small et al., 2003; Volkow et al., 2002), implying that the rewarding value of food is encoded by dorsal striatal DA signaling. Studies directly examining neuronal functioning in the dorsal striatum of obese individuals, however, have yielded mixed findings. Data from imaging studies are apparently equivocal, providing evidence of both increases (Rothemund et al., 2007; Wang et al., 2011; Yokum et al., 2014) and decreases (Babbs et al., 2013; Stice et al., 2008a; Volkow et al., 2011; Wang et al., 2001) in the activity of the dorsal striatum related to obesity/overeating disorders. It is important to bear in mind, however, that obesity may be distinct in its neural mechanisms relative to other overeating disorders. Preclinical studies in rodent models also demonstrate significant alterations in various aspects of glutamate and DA signaling in the brain (Auer et al., 2015; Berridge et al., 2010; Brown et al., 2017; Cano et al., 2014; Charles et al., 2015; Fordahl and Jones, 2017; Johnson and Kenny, 2010; Sickmann et al., 2010; Speed et al., 2011; Stamatakis et al., 2016; Xu et al., 2013). However, very little is known about effects within the dorsal striatum, specifically.
Although the data are not currently clear, the available evidence generally suggests that obese individuals exhibit altered neurotransmission, in some capacity, in the dorsal striatum relative to healthy controls. The nature and mechanisms of these functional differences, however, are poorly understood. In the current study, we set out to evaluate the electrophysiological and neurochemical effects of sustained consumption of a high-fat, high-sugar ‘western diet’ in the mouse dorsal striatum. Alterations in these measures may offer potential mechanisms of neurocircuitry adaptations relevant to obesity. As the medial (DMS) and lateral (DLS) portions of the dorsal striatum exhibit differential control over goal-directed versus habitual food seeking in rodents respectively (Yin et al., 2004), we examined these parameters separately in each subregion.
Methods
Animals
Male C57BL/6J mice (N=23; subset of 15 mice used for electrophysiology/neurochemistry) were ordered from the Jackson Laboratory (Bar Harbor, ME, USA) at 4 weeks of age and allowed 7 days to habituate to the vivarium space prior to experimentation. All mice were group housed 3-4 per cage and maintained on a standard 12 hr light/dark cycle (lights ON at 0700). The temperature and humidity of the vivarium space were held constant at 20°C and 50%, respectively. Following the 7 day acclimation period, all animals had ad libitum access to water and either a high-fat, high sugar ‘western diet’ (WD) or a control ‘standard diet’ (SD) (3 cages SD, 3 cages WD). All procedures reported herein were approved by the Indiana University School of Medicine Institutional Animal Care and Use Committee and closely followed the guidelines for the care and use of laboratory animals as promulgated by the National Institutes of Health.
Diets
Mouse diets were purchased from Envigo (Madison, WI, USA). The key metrics of each diet are presented in Table 1. Mice were fed either SD (Envigo TD.08485) or WD (Envigo TD.88137) for 16 weeks.
Table 1.
Content of standard and ‘western’ diets.
| Standard Diet | ‘Western’ Diet | |
|---|---|---|
|
| ||
| Kcal/g | 3.6 | 4.5 |
| Protein (% kcal) | 19.1 | 15.2 |
| Carbohydrate (%kcal) | 67.9 | 42.7 |
| Fat (%kcal) | 13.0 | 42 |
| Sucrose (%kcal) | 120.0 | 341.5 |
Obesity model monitoring
Mice were weighed weekly to determine whether WD animals were gaining significantly more weight than SD mice. Once a week, a food intake was measured over a specific 24 hr period. Food intake was determined by the weight difference between the amount of food initially in the cage and the remaining food after the 24 hr period. Using these data, energy consumption was recorded as kcal/mouse/cage. Given that obesity has become increasingly associated with the development of type 2 diabetes, we also evaluated common early assessments of pre-diabetic risk: glucose tolerance and insulin tolerance.
Glucose tolerance
Animals were fasted for 16 hours prior to assessment the following morning. All mice were weighed and a baseline (Time ‘0’) blood glucose reading was taken via a glucometer sampling from a small cut to the tail vein. Animals were then injected intraperitoneally with 1 g/kg D-glucose (Sigma Aldrich, St. Louis, MO, USA) at a volume of 4 ml/kg. Blood glucose readings were then taken at 10, 20, 30, 60, 90, and 120 minutes post-injection.
Insulin tolerance
Animals were fasted for 2 hrs prior to assessment. All mice were weighed and a baseline (Time ‘0’) blood glucose reading was taken via a glucometer sampling from a small cut to the tail vein. Animals were then injected intraperitoneally with 0.75 U/kg human insulin Humulin-R (Eli Lilly, Indianapolis, IN, USA) at a volume of 3 ml/kg. Blood glucose readings were then taken at 15, 30, 45, and 60 minutes post-injection.
Brain slice preparation
Mice were sacrificed via decapitation under deep isoflurane anesthesia and the brain was rapidly excised and placed into ice-cold, continuously 95% CO2/5% O2 bubbled cutting solution containing: 30 mM NaCl, 4.5 mM KCl, 1 mM MgCl2, 26 mM NaHCO3, 1.2 mM NaH2PO4, 10 mM glucose and 194 mM sucrose. 300 μm coronal brain slices containing the striatum were prepared on a Leica VT1200S vibratome. For electrophysiology experiments, sections were rapidly transferred to 32°C, continuously 95% CO2/5% O2 bubbled artificial cerebrospinal fluid (aCSF) containing: 124 mM NaCl, 4.5 mM KCl, 2 mM CaCl2, 1 mM MgCl2, 26 mM NaHCO3, 1.2 mM NaH2PO4 and 10 mM glucose. For fast scan cyclic voltammetry, the aCSF contained: 126 mM NaCl, 1.2 mM NaH2PO4, 2.4 mM CaCl2, 1.2 mM MgCl2, 2.5 mM KCl, 0.4 mM L-ascorbic acid, 20 mM HEPES, 11 mM D-glucose, 25 mM NaHCO3. After incubating for ~1h, slices were moved to a 21°C environment until recording.
Electrophysiology
Whole-cell voltage and current clamp recordings were made using a Multiclamp 700B and Digidata 1550B amplifier (Molecular Devices, Sunnyvale, CA, USA). For recording, brain slices were moved to the recording chamber which was held at 32°C and continuously perfused with 95% CO2/5% oxygenated aCSF at a rate of ~1.5ml/min. Slices were visualized on an Olympus BX51WI microscope (Olympus Corporation of America). Recordings were made from GABAergic medium spiny neurons (MSNs) in medial and lateral regions of the dorsal striatum. MSNs make up the vast majority (~ 95%) of neurons in the rodent striatum (Tepper et al., 2007) and are the region’s sole output neuron type. MSNs were confirmed by their membrane resistance and capacitance. Drugs were prepared as stock solutions and diluted in aCSF to their final concentrations and used on the same day. Drugs were administered to brain slices via bath application, in which normal aCSF was replaced with aCSF containing drug. Borosilicate glass recording pipettes of 2-4 MΩ were filled with specific internal solutions (see below) for whole cell recordings. Internal solutions were adjusted to 295–310 mOsm. All whole-cell recordings were filtered at 2 kHz and digitized at 10 kHz. Data were acquired using Clampex 10 (Molecular Devices). Series resistance was monitored and only cells with a stable series resistance (less than 25 MΩ and that did not change more than 15%) were included for data analysis.
Excitability
In order to determine whether diet consumption influenced the excitability of MSNs in the DMS and DLS, increasing current steps (−200 pA to 400 pA in 50 pA increments) were applied at 500 ms intervals and action potentials were recorded in current clamp mode. The K-gluconate internal solution used in these experiments consisted of: 4 mM KCl, 10mM HEPES, 4 mM MgATP, 0.3 mM NaGTP, 10 mM phosphocreatine, 126 mM K-gluconate. The excitability parameters recorded included: resting membrane potential, action potential threshold, action potential peak amplitude, action potential half-width, input resistance, and action potential frequency. Calculations of the action potential threshold, peak amplitude, and peak half-width were generated with data collected from the first current step that produced action potentials.
Excitatory transmission
In order to isolate excitatory transmission in these experiments, the aCSF contained 50 μM picrotoxin (potent antagonist of the GABAA receptor). All of these experiments were conducted in voltage clamp mode (−60 mV holding potential, unless otherwise noted). A Teflon-coated bipolar stimulating electrode (PlasticsONE, Roanoke, VA) was placed at the border of the corpus callosum and the DLS or DMS. The internal solution contained: 120 mM CsMeSO3, 5 mM NaCl, 10 mM TEA-Cl, 10 mM HEPES, 5 mM lidocaine bromide, 1.1 mM EGTA, 0.3 mM Na-GTP and 4 mM Mg-ATP. Once a cell was successfully patched, stimulation was produced via a DS3 Isolated Current Stimulator (Digitimer, Ft. Lauderdale, FL, USA) and the intensity was adjusted until a stable excitatory postsynaptic current (EPSC) response between −200 to −400 pA was achieved. At this point, 3 different assessments of excitatory transmission commenced with each cell.
Spontaneous EPSCs (sEPSCs) were measured over the course of 5 min of gap-free recording in the absence of any form of electrical stimulation. Measured sEPSC parameters included: frequency, amplitude, rise time, and the decay constant. AMPA to NMDA receptor-mediated current ratios were measured by first holding the cell at −80 mV and electrically-evoking an AMPAR-mediated EPSC. The NMDAR current was determined by holding the membrane potential at +40 mV and electrically-evoking an EPSC. Since the AMPAR component of EPSCs at −80 mV was not apparent 100 ms following the electrical stimulus (i.e. the measured current returned to baseline), we calculated the NMDAR-mediated portion of the EPSC at +40 mV as the average of the measured current over the following 25 ms (i.e. 100-125 ms post-stimulus). Finally, paired-pulse ratios (25-200 ms between pulses) were also measured to determine if the probability of presynaptic neurotransmitter release differed between treatment groups. sEPSCs were measured first, so that repeated stimulation required by the AMPAR/NMDAR ratio and paired-pulse ratio experiments did not influence sEPSC parameters.
Inhibitory transmission
In different cells, spontaneous inhibitory GABAA receptor-mediated currents (sIPSCs) were recorded in the absence of any electrical stimulation over the course of 5 min. To block excitatory transmission, the aCSF contained 5μM NBQX (AMPAR antagonist) and 50 μM AP5 (NMDAR antagonist). In addition, the internal solution contained: 60 mM CsMeSO3, 60 mM CsCl2, 10 mM HEPES, 0.2 mM EGTA, 8 mM NaCl, 2 mM MgCl2, 3 mM MgATP, 0.3 mM NaGTP, 5 mM lidocaine, 10 mM phosphocreatine. sIPSC parameters included: frequency, amplitude, rise time, and the decay constant.
Mu-opioid receptor-mediated plasticity
A role for the mu-opioid receptor (MOPr) in binge-like, compulsive eating has been identified (Colantuoni et al., 2002; Cooper and Turkish, 1989; Cottone et al., 2008; Doyle et al., 1993; Drewnowski et al., 1992; Ferraro et al., 2002; Katsuura and Taha, 2010; Kirkham and Cooper, 1988). Experiments in rodents demonstrate that the DMS and DLS subregions of the dorsal striatum contribute to goal-directed and habitual food seeking, respectively (Yin et al., 2004). Therefore, MOPr function in dorsal striatum is likely an important factor in various food consumption phenotypes. Similar to our previous work (Atwood et al., 2014), we examined MOPr-mediated long term synaptic plasticity in DMS and DLS MSNs. In order to do this, EPSCs (at −60 mV) were electrically evoked every 20 s as described above. After a stable 10-min baseline, the selective MOPr agonist [D-Ala2, NMe-Phe4, Gly-ol5]-enkephalin (DAMGO; Bachem, Bubendorf, Switzerland) was washed on to the slice for a 5-min period at a concentration of 0.3 μM. EPSCs continued to be recorded following DAMGO application for a total of 40 minutes of recording. Plasticity was assessed by comparing the average EPSC peak amplitude for the final 10 min of recording to the average for the 10 min baseline (Atwood et al., 2014).
Neurochemistry
Electrically-evoked DA release in DLS and DMS was measured via in vitro fast-scan cyclic voltammetry (FSCV). For a detailed description of the technique, see Heien et al. (2004). After brain slice preparation and incubation as described above, slices were transferred to the FSCV recording chamber which was continuously perfused (~1 ml/min) with 95% CO2/5% oxygenated FSCV aCSF. All slices were from brains also used in electrophysiology experiments. FSCV electrodes were constructed by aspirating a single carbon fiber in a glass capillary tube. The tube was then tapered by a pipette puller, thus sealing the glass tightly around the fiber. The protruding fiber was cut to a length between 100-150 μM. The fiber of the electrode was inserted ~75 μM into the DMS or DLS. A bipolar stimulating electrode locally applied a 4 ms, monophasic, 350 μA current every 5 min using a DS4 Bi-phasic Stimulus Isolator (Digitimer). Recordings were made using Demon Voltammetry software (Wake Forest University, Wake Forest, NC, USA) and custom-built recording equipment. Electrically-evoked DA was measured by applying a triangular voltage waveform to the recording electrode: −0.4 to +1.2 to −0.4 V relative to a Ag/AgCl reference electrode at a rate of 400 V/s every 100 ms. Once a stable response was achieved, an average of 3 evoked DA peaks per recording site was calculated. Parameters measured included: peak amplitude (quantification of peak DA levels), tau (measure of peak decay duration), peak half-life, peak half-width, T20 (time at which measured DA is 20% lower than the measured peak), and T80 (time at which measured DA is 80% lower than the measured peak). FSCV recording electrodes were calibrated after experimental data collection by adding 1 μM dopamine to the perfusate. Measured DA during data collection was inferred on the basis of this post-hoc calibration peak.
Statistical Analysis
Unless otherwise noted, all data graphing and analysis was conducted with GraphPad Prism 7.0 software (GraphPad Software, Inc., La Jolla, CA, USA). Weight and in kcal consumption were analyzed by repeated measures ANOVAs with diet as between subjects factor and week as the within subjects factor. Glucose tolerance test (GTT) and insulin tolerance test (ITT) data were analyzed via unpaired t-tests of the area under of the curve. With the exception of synaptic plasticity data, all other data were analyzed via 2-factor ANOVAs with diet and brain region as between subjects factors. For opioid plasticity data, a categorical analysis of the distribution of 3 different classifications of plasticity was calculated. Group assignment of individual cells was initially done visually, then confirmed via t-tests vs. baseline: long-term depression (average EPSC amplitude of final 10 min < baseline average amplitude), long-term potentiation (average EPSC amplitude of final 10 min was > baseline average amplitude), or no change. The observed distribution of cells into these classifications within the WD and SD groups was compared to an expected distribution, given that the null hypothesis is true. The statistical method of comparison used calculates the degree to which the observed values differ from the expected values and these are quantified as ‘adjusted residuals’ (Agresti, 2007; Sharpe, 2015). Adjusted residual values in excess of 3.0 are considered statistically significant. Where appropriate, Sidak’s multiple comparisons test was used for post-hoc comparisons and the level of significance for all analyses was set at p < 0.05. Due to technical issues, 4 WD, 4 SD mice were not tested for insulin tolerance and were therefore not included in ITT analyses.
Results
Obesity model monitoring
Mice fed the WD gained significantly more weight (F1,21 = 31.74, p < 0.001; Figure 1A) over the course of the experiment and a significant interaction with time (F14,294 = 24.8, p < 0.001) indicated that this was true at week 7 and beyond (post hoc p’s < 0.01). WD mice also consumed significantly more kilocalories than mice fed the SD over the 16-week monitoring period (F1,64 = 29.21, p < 0.001; Figure 1B). SD mice also gained weight and increased their caloric intake, however this was expected as all mice began the study at ~5 weeks of age. The ITT conducted at week 12, however, found no difference between the diet groups (p = 0.323; Figure 1C,D). Insulin tolerance/resistance is associated with a more advanced type 2 diabetic phenotype (Reaven, 1988). Thus, 12 weeks of WD exposure may not have been sufficient to influence insulin sensitivity. Nevertheless, significantly impaired glucose tolerance, an important pre-diabetic correlate of obesity (Sinha et al., 2002), was observed in WD mice at week 16 (t21 = 2.84, p < 0.001; Figure 1E,F). Impaired glucose tolerance is considered a reliable correlate of insulin resistance (Winzell and Ahrén, 2004). Unfortunately, we were unable to evaluate insulin resistance at week 16 due to technical issues.
Figure 1. WD consumption produces an obese-like state.
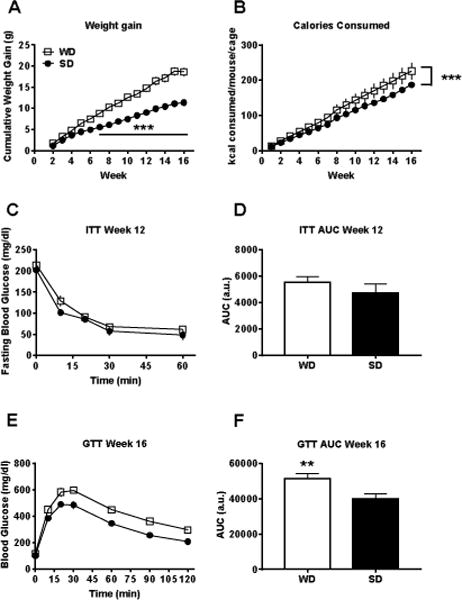
Mice fed the ‘western’ diet (WD) A) gained significantly more weight and B) consumed significantly more calories than mice fed the control standard diet (SD). C,D) Although the insulin tolerance test (ITT) at week 12 found no effect of diet, E,F) the glucose tolerance test (GTT) at week 16 revealed significantly impaired glucose tolerance in WD mice. n = 7-12; p** < 0.01, p*** < 0.001 vs. SD.
Electrophysiology
Excitability
No significant differences in excitability parameters were detected between SD and WD mice in either dorsal striatal subregion (Figure 2A–E). Resting membrane potential, action potential threshold, action potential half-width, action potential peak amplitude, and input resistance were all similar in MSNs from the DLS and DMS in WD and SD mice (all main effects and interactions p’s > 0.09). Furthermore, action potential frequency during increasing current steps was not different between SD and WD MSNs in both the DMS and DLS (p’s > 0.46; Figure 2F–H).
Figure 2. Dorsal striatal medium spiny neurons (MSNs) from WD and SD mice do not differ in excitability parameters.
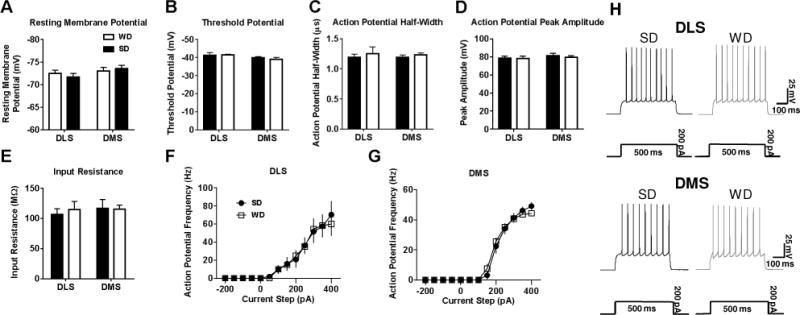
Current clamp electrophysiological recordings revealed no differences between WD and SD MSNs in A) resting membrane potential, B) threshold potential, C) action potential half-width, D) action potential peak amplitude, E) input resistance. F,G) Increasing steps of injected current also revealed no differences in action potential frequency in either striatal subregion. H) Representative traces for 200pA of injected current in both dorsal striatal subregions. n = 5-12 cells per diet × subregion; 2-3 mice per condition.
Excitatory transmission
Statistical analysis of the AMPAR/NMDAR current ratio found a significant interaction of diet and brain region (F1,31 = 7.2, p < 0.05; Figure 3A,B), with post-hoc testing indicating that WD MSNs exhibited a significantly greater AMPAR/NMDAR current ratio than those from SD mice specifically in the DLS (p < 0.05). Paired-pulse ratio (PPR) analysis did not reveal any significant differences between WD and SD cells in the likelihood of glutamate release in either the DLS or DMS (p’s > 0.63; Figure 3C–E). The analysis of sEPSCs indicated a significant interaction of diet and brain region for the decay constant (F1,29 = 7.57, p < 0.05; Figure 4A,E), with post-hoc testing revealing that the decay of spontaneous EPSCs back to baseline was significantly slower in WD relative to SD recordings, specifically in DLS (p < 0.01). Other sEPSC parameters (rise time, frequency, amplitude) were not different between WD and SD cells (all p’s > 0.2; Figure 4B–D). Main effects of region for sEPSC frequency (F1,27 = 4.7, p < 0.05) and amplitude (F1,29 = 11.83, p < 0.051), however, indicated significantly larger and more frequent sEPSCs in DMS relative to DLS. Cumulative probability plots for all sEPSC parameters in Figure 5 demonstrate the relative distribution of values for all cells.
Figure 3. WD consumption increases the AMPAR/NMDAR specifically in the DLS, with no effect on the probability of presynaptic glutamate release.
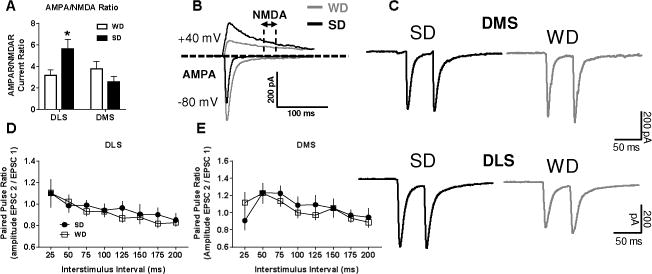
Voltage clamp recordings revealed A) elevated AMPAR to NMDAR current ratio within the DLS of WD relative to SD mice. B) Representative AMPAR and NMDAR traces from WD and SD cells. C) Representative PPR traces for 50 ms interstimulus interval in both dorsal striatal subregions. Paired pulse ratio analysis of presynaptic glutamate release probability in D) DLS and E) DMS also revealed no group differences. n = 5-10 cells per diet × subregion; 2-3 mice per condition. p* < 0.05, p** < 0.01 vs. SD.
Figure 4. WD consumption prolongs glutamategic sEPSCs specifically in the DLS.
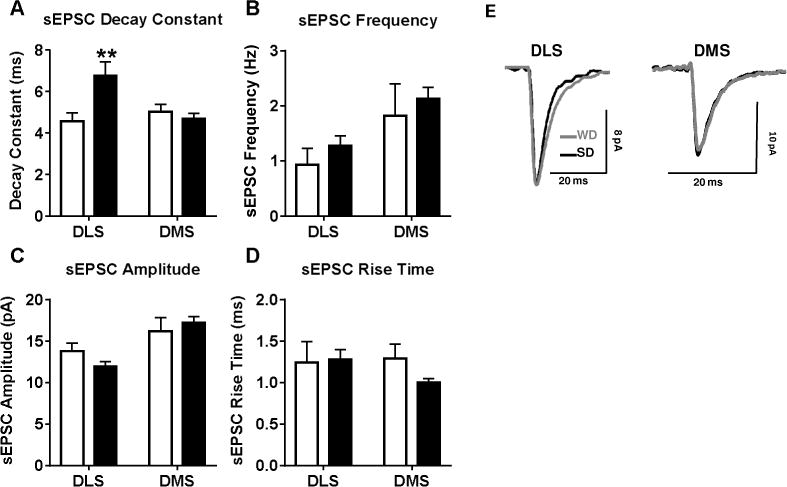
A) Recordings of spontaneous EPSCs (sEPSCs) demonstrated a prolonged decay period for sEPSCs within the DLS. Measures of B) sEPSC frequency, C) amplitude, D) and rise time revealed no diet-induced differences. E) Representative sEPSC traces from WD and SD cells. n = 5-10 cells per diet × subregion; 2-3 mice per condition. p* < 0.05, p** < 0.01 vs. SD.
Figure 5. Cumulative probability plots for sEPSC parameters in DLS and DMS.
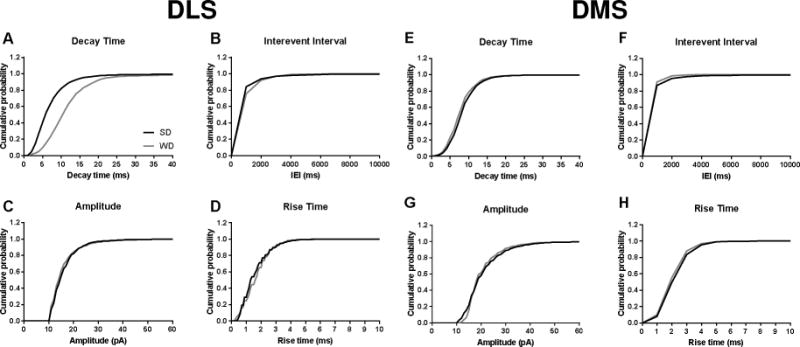
The noted increase in decay time in WD mice is apparent in the A) DLS, but not the E) DMS. No group differences were observed in B,F) interevent interval, C,G) amplitude, D,H) or rise time in either region. n = 5-10 cells per diet × subregion; 2-3 mice per condition.
Inhibitory Transmission
Spontaneous IPSC parameters (frequency, amplitude, rise time, decay constant) were not statistically different between SD and WD cells in both the DLS and DMS (all p’s > 0.23; Figure 6A–E). A main effect of region for amplitude (F1,21 = 4.57, p < 0.05) and decay constant (F1,21 = 8.0, p < 0.05), however, indicated that these parameters were significantly greater in DLS. Cumulative probability plots for all sIPSC parameters in Figure 7 demonstrate the relative distribution of values for all cells.
Figure 6. Diet did not influence spontaneous GABAergic inhibitory transmission in the dorsal striatum.
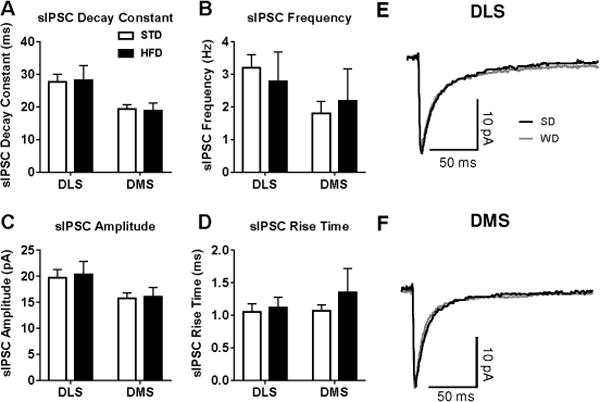
Recordings of spontaneous IPSCs (sIPSCs) revealed no differences between diet groups for the average A) decay constant, B) sEPSC frequency, C) amplitude, or D) rise time in either region. E) Representative sIPSC traces in both dorsal striatal subregions. n = 5-8 cells per diet × subregion; 1 mouse per condition.
Figure 7. Cumulative probability plots for sIPSC parameters in DLS and DMS.
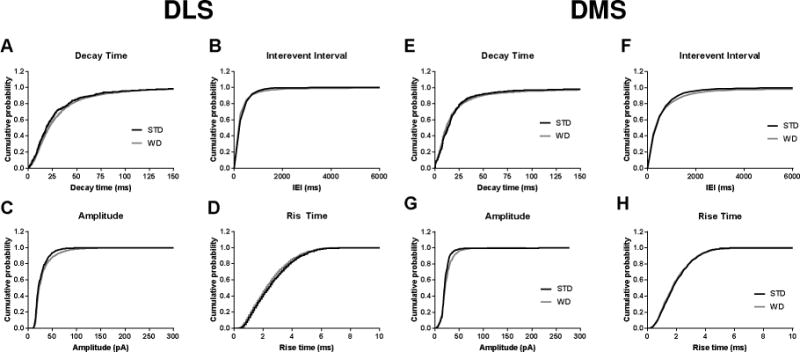
No group differences were observed in A,E) decay time, B,F) interevent interval, C,G) amplitude, D,H) or rise time in either region. n = 5-8 cells per diet × subregion; 1 mouse per condition.
Mu-opioid receptor-mediated plasticity
Although the overall analysis revealed no difference between SD and WD cells in the response to DAMGO, a MOPr agonist, (Figure 8A,B), the distribution of outcomes revealed a clear difference (Figure 8C). Across both dorsal striatal subregions, collectively, the WD condition exhibited a substantial number of cells (6/11 cells) demonstrating no change from ESPC baseline amplitude following DAMGO application, whereas no SD cells (0/13) received this designation. A categorical analysis of adjusted residuals of expected values revealed that this was indeed a significant difference between SD and WD conditions (absolute value of adjusted residual was > 3.0). Conversely, all SD cells were determined to demonstrate some form of plasticity, either long-term depression or long-term potentiation. Therefore, WD cells demonstrated a significant reduced responsivity/plasticity in response to MOPr activation via DAMGO in the dorsal striatum.
Figure 8. WD consumption disrupts mu-opiod receptor (MOPr) plasticity in the dorsal striatum.
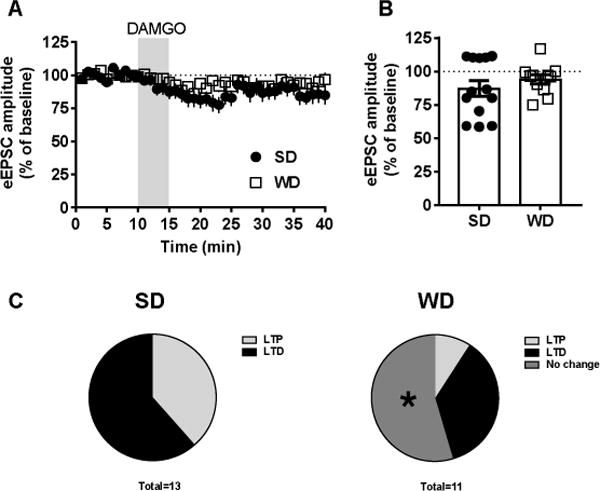
A) Time series of electrically-evoked EPSC (eEPSC) averages over the 40 min recording session. B) Application of the MOPr agonist DAMGO (0.3 μM) did not reveal overall diet group differences in response to DAMGO. However, an examination of cell groupings C) revealed that there was a significant lack of response to DAMGO in WD cells. n = 11-13 cells; 2 mice per condition. p* < 0.05 for adjusted residual analysis relative to expected distribution.
Electrochemistry
Electrically-evoked DA release was significantly higher across the dorsal striatal subregions in WD compared to SD slices (F1,20 = 26.01, p < 0.001; Figure 9A,B). Furthermore, measures of DA uptake/clearance kinetics indicated that DA clearance was significantly less efficient in WD dorsal striatum. Tau (measures of peak decay duration) values were significantly larger for WD recordings (F1,20 = 22.88, p < 0.001; Figure 9C), indicating slower rate of DA peak decay. Additionally, tau values were generally larger in the DMS relative to the DLS (F1,20 = 14.12, p < 0.01). The same pattern of results (WD and DMS > than SD and DLS) was consistent in the other uptake/clearance measures (Figure 9D–F): half-life (Diet: F1,20 = 23.06, p < 0.001; Region: F1,20 = 13.15, p < 0.01), half-width (Diet: F1,20 = 17.64, p < 0.001; Region: F1,20 = 14.02, p < 0.01), and T80 (Diet: F1,20 = 30.09, p < 0.001; Region: F1,20 = 12.57, p < 0.01). No difference between groups/regions was detected for T20 (p’s > 0.2; not shown). Overall, these data indicate that evoked DA release was significantly greater and uptake was significantly less efficient in WD relative to SD dorsal striatum.
Figure 9. WD mice demonstrate a robust increase in evoked dopamine release and impaired reuptake across the dorsal striatum.
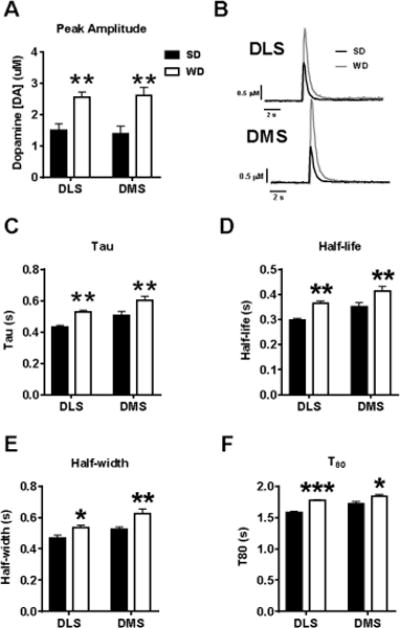
A) Fast scan cyclic voltammetry (FSCV) quantification of electrically evoked dopamine (DA) release was significantly elevated in the dorsal striatum of WD mice. B) Representative FSCV traces in SD and WD slices. C-F) Measures of DA clearance/reuptake also demonstrate a significantly prolonged DA signal, indicating a reduced DA clearance capacity. n = 6 slice recordings per diet × subregion (3 recordings in rostral striatum slice and 3 recordings in caudal striatum slice × subregion × condition); 1 mouse per condition. p* < 0.05, p** < 0.01, p*** < 0.001 vs. SD.
Discussion
In the present study, an obese-like phenotype was observed in mice exposed to a high-fat, high-sugar diet (WD). These animals gained significantly more weight and consumed more kilocalories than mice fed the control (SD) diet. Importantly, WD mice also demonstrated impaired glucose tolerance after 16 weeks of diet exposure; an important pre-diabetic correlate of obesity (Sinha et al., 2002). When evaluating synaptic function in the dorsal striatum of these animals, we observed that the obese-like phenotype was associated with altered glutamate transmission specifically in the DLS as well as greatly enhanced DA transmission throughout the dorsal striatum. These findings offer novel insight into the neural mechanisms affected by high-fat, high-sugar diet consumption and their potential role in excessive, non-homeostatic feeding behavior.
We found that basal excitability parameters of dorsal striatal MSNs were unaffected by diet. Although unlikely, it is possible that smaller current steps (i.e. < 50 pA) could have uncovered some subtle differences in excitability parameters. Nevertheless, we observed significantly altered glutamate function in the DLS of WD mice. The decay time for presynaptic glutamate-driven sEPSCs was significantly prolonged in the DLS of WD MSNs. The lack of difference in the amplitude of these currents (in addition to null PPR data) suggests that presynaptic glutamate release dynamics were unaffected. Therefore, the most parsimonious explanation for these data is that glutamate uptake is slower in the DLS of WD mice. As GLT-1 is the primary glutamate transporter in dorsal striatum (Rothstein et al., 1994), GLT-1 function may have been decreased as a consequence of WD consumption. GLT-1 is the predominant glutamate transporter in the CNS, and ~80-90% of its expression in the brain is found on astrocytes (Murphy-Royal et al., 2017; Rimmele and Rosenberg, 2016; Rothstein et al., 1994). A loss of GLT-1 function prolongs the presence of glutamate in the synaptic cleft, thus producing a longer duration of glutamate synaptic activity and increases the probability of glutamate-induced synaptic adaptations. Some previous work has demonstrated that high fat diet consumption in mice produces an increase in GLT-1 expression and function at hippocampal synapses which alters basal synaptic transmission and subsequently reduces glutamate receptor-mediated synaptic plasticity (Cano et al., 2014; Valladolid-Acebes et al., 2012). On the other hand, many neurological disorders produce a down-regulation of GLT-1 expression (Soni et al., 2014). One indication of glutamate transporter functional changes is in the kinetics of glutamate-mediated synaptic currents (Murphy-Royal et al., 2015). A decrease in transporter function results in slower EPSC decay (Marcaggi et al., 2003; Weng et al., 2007), as was observed in the current study. GLT-1 dysfunction has been implicated in many diseases including substance use disorders, neurodegenerative and neuroinflammatory diseases, and epilepsy (Petr et al., 2015; Roberts-Wolfe and Kalivas, 2015; Soni et al., 2014).
Additionally, the glutamate-driven AMPAR/NMDAR current ratio was significantly reduced in DLS MSNs of WD mice relative to SD mice. Given the lack of group differences in sEPSC amplitude, this effect is likely not due to increased AMPAR expression. An alternative explanation is that AMPAR subunit composition may have been altered by prolonged WD diet consumption. Indeed, AMPAR subunit composition substantially influences the functional dynamics (including EPSC decay time and peak amplitude) of AMPARs (Lomeli et al., 1994). Therefore, altered AMPAR subunit composition may explain the observation of increased sEPSC decay time, however this is not a likely explanation for the observed increase in AMPAR/NMDAR ratio given the lack of difference in sEPSC amplitude. An alternative may be that NMDAR expression or function is reduced. It is important to note that AMPAR and NMDAR currents were not pharmacologically isolated (i.e. use of antagonists NBQX and APV). As such, we cannot rule out that our measured currents are purely AMPAR- or NMDAR-mediated.
NMDARs are involved in controlling feeding behavior, body weight, and glucose homeostasis (Campos and Ritter, 2015; Doane et al., 2007; Guard et al., 2009a, b; Khan et al., 1999; Resch et al., 2014; Ritter, 2011; Stamatakis et al., 2016; Stanley et al., 1996; Uner et al., 2015; Wright et al., 2011; Wu et al., 2013). NMDARs are also important for developing a preference for high fat diet (Buttigieg et al., 2014). The NMDAR blocker memantine, normally used to treat Alzheimer’s disease, reduces palatable food consumption in rodents and non-human primates (Bisaga et al., 2008; Popik et al., 2011; Smith et al., 2015) and may positively impact binge eating in humans, although its effectiveness on weight loss has been inconsistent (Brennan et al., 2008; Hermanussen and Tresguerres, 2005). Memantine also reduces compulsive food seeking behavior in rodents, suggesting dorsal striatal circuits could be involved in addition to mesolimbic circuits (Smith et al., 2015). This is consistent with its efficacy in treating compulsive behavior in humans as well (Ghaleiha et al., 2013). However, the synaptic mechanisms underlying NMDAR-mediated regulation of compulsive eating have not been elucidated.
Our electrochemical assessment of DA signaling revealed that DA transmission was greatly enhanced in WD mice across both subregions of the dorsal striatum. Clinical PET studies have observed that DA receptor binding in the dorsal striatum is positively associated with the motivation to obtain food and subjective ratings of ‘meal pleasantness’ in healthy individuals (Wang et al., 2001; Yokum et al., 2014). Findings from studies evaluating the response of the dorsal striatum to food cues or reward in obese individuals are much less clear, however. Imaging studies note both increases (Rothemund et al., 2007; Wang et al., 2011; Yokum et al., 2014) and decreases (Babbs et al., 2013; Stice et al., 2008a; Volkow et al., 2011; Wang et al., 2001) in striatal activation in response to food reward or cues in obese individuals, or individuals that would later develop obesity. There is tremendous methodological variability in these studies (i.e. studying only women versus men and women, different levels of subject obesity, subjects were hungry or sated, cues tapping different sensory modalities, distinction of dorsal striatum or whole striatum) which is likely a key factor in the large degree of study discordance. A fairly recent study benefiting from methodological strengths (clear differentiation between dorsal and ventral striatum and control over previous energy intake in all subjects), demonstrated significantly greater D2 receptor binding potential in the dorsal striatum, but lower binding potential in the ventral striatum, of obese individuals relative to healthy controls (Guo et al., 2014). Collectively, these findings resemble a dominant theory in the extant drug and alcohol literature that a shift in activation from ventral to dorsal striatum governs an individual’s transition from recreational to habitual use (Everitt and Robbins, 2013).
To our knowledge, this study is the first to address whether an obesogenic diet alters evoked DA release and uptake kinetics in the subregions of the dorsal striatum. We found that evoked DA release in both the DLS and DMS of WD mice was nearly twice that of SD mice. In addition, DA uptake parameters indicated that uptake was significantly slower across the dorsal striatal subregions of these mice. Thus, prolonged consumption of the WD diet resulted in enhanced DA transmission throughout the dorsal striatum. Impaired uptake/clearance is likely indicative of reduced DA transporter (DAT) function or expression. Indeed, prior studies have observed a reduced DA clearance capacity in the nucleus accumbens core (Fordahl and Jones, 2017) and reduced membrane-bound DAT levels (Fordahl et al., 2016; Hryhorczuk et al., 2016; Speed et al., 2011) within the striatum and nucleus accumbens of mice fed a high-fat diet. Although not quantified, it is likely that our observations in the current study were also due to reduced DAT expression. While DA reuptake mechanisms were clearly affected, this previous voltammetry work in the nucleus accumbens core also observed that 6 weeks of high-fat diet consumption in mice had no effect on evoked DA release (Fordahl and Jones, 2017). The accumbens is commonly deemed analogous to the ventral striatum in humans and is thought to be a primary regulator of hedonic, reward processes (Carlezon and Thomas, 2009). The clinical observations of Guo and colleagues (2014) suggest that obesity is associated with elevated DA signaling in the dorsal striatum, but reduced DA signaling ventral striatum. Although Fordahl and Jones (2017) observed no alteration in evoked accumbal DA in mice fed a high fat diet, it is possible that the diet exposure period was not long enough and with more protracted exposure, DA levels may actually decrease. The current study did not evaluate accumbal DA dynamics, however we observed a robust increase in evoked DA release in the dorsal striatum of mice fed WD for 16 weeks. Enhanced non-hedonic motivation to obtain food is a correlate of enhanced dorsal striatal DA tone in clinical populations (Volkow et al., 2002). Future work should evaluate whether a longer exposure period to obesogenic diets models the ventral-to-dorsal shift in DA signaling observed in clinical work (Guo et al., 2014).
In our final experiment, we evaluated whether MOPr-mediated synaptic plasticity was altered by WD consumption. We found that dorsal striatal MSNs of WD mice were significantly lacking in MOPr plasticity relative to SD controls. The majority of MSNs from SD mice display typically observed MOPr-LTD (Atwood et al., 2014), while in a smaller proportion the MOPr agonist DAMGO produced long-term potentiation (MOR-LTP). This is a previously unobserved phenomenon with an unknown mechanism. In mice that consumed WD for 16 weeks, MSN responses to DAMGO were significantly more heterogeneous. About 50% of cells displayed mOP-LTD or -LTP, but a group of neurons emerged that did not respond to DAMGO. These data indicate a subset of MSNs in WD-fed mice lose MOPr regulation of glutamate input. One explanation for both the heterogeneous response to WD as well as the two types of MOPr-mediated plasticity observed is that glutamate synapses are heterogeneous in both their originating cortical brain regions as well their target MSN type (direct or indirect pathway of striatal output) (Hunnicutt et al., 2016; Voorn et al., 2004; Wall et al., 2013). Direct pathway MSNs reinforce and promote behaviors, whereas indirect pathway neurons inhibit behavior. The balance of activity between the two determines behavioral output (Cui et al., 2013; Freeze et al., 2013; Kravitz et al., 2012). Direct pathway activation promotes feeding whereas indirect pathway activation inhibits feeding (Kenny et al., 2013). Each type of MSN is approximately equally represented in dorsal striatum (Bateup et al., 2010). Some forms of glutamatergic synaptic plasticity occur at synapses on to only one class of these output pathways (Kreitzer and Malenka, 2007; Lerner et al., 2010; Nazzaro et al., 2012). A plausible explanation is that WD impairs plasticity at glutamate input selectively, in one of the pathways. Perhaps a simpler explanation is that sustained WD consumption reduced MOPr levels in both the DMS and DLS. Prior work in rats demonstrated the MOPr mRNA is reduced across both subregions of the dorsal striatum after 3 months of a high-fat and high-sugar diet (Robinson et al., 2015). A reduction in MOPr expression in the current experiment could translate to the observed lack of MOPr-mediated plasticity throughout the dorsal striatum.
MORs have been implicated in the consumption of fatty foods and binge-like eating (Colantuoni et al., 2002; Cooper and Turkish, 1989; Cottone et al., 2008; Doyle et al., 1993; Drewnowski et al., 1992; Ferraro et al., 2002; Katsuura and Taha, 2010; Kirkham and Cooper, 1988). Rodent work has also demonstrated that endogenous opioids surge in the dorsal striatum during the consumption of highly palatable food (DiFeliceantonio et al., 2012). We have previously observed that pretreatment with oxycodone (Atwood et al., 2014) similarly ablates dorsal striatal MOPr plasticity. This is intriguing considering the growing body of evidence that there are significant overlaps in the neurocircuitry underlying addiction and obesity/overeating (Kenny, 2011). The lack of a neural response to MOPr activation may be indicative of inflexible neurocircuitry. Given the known role for the dorsal striatum in goal-directed and habitual food/drug seeking (Corbit et al., 2012; Yin et al., 2004), this neural inflexibility may be associated with behavioral inflexibility disorders (i.e. compulsive consumption/seeking).
Although the clinical condition of obesity is very complex and evidence suggests that individual differences in a variety of neurobehavioral parameters are critically important (Vainik et al., 2013), a number of theories characterizing the pathological shift in motivation to overeat have been developed. One theory is a dysregulation in dopamine (DA) signaling, suggesting that obese individuals exhibit a reduced DA response to food consumption and reduced D2 receptor levels in reward circuitry (Stice et al., 2010). This theory argues that overconsumption of food is a compensatory mechanism to elevate the low level of DA signaling, thus manifesting in obesity. Another theory suggests that a dysregulation of energy balance (caloric) homeostasis makes it difficult for an individual to determine when sufficient calories have been consumed, resulting in pathological overeating (Dagher, 2010). Finally, a hedonic-shift theory suggests that the initial rewarding properties of highly palatable/caloric foods that are initially ‘liked’ in high-risk individuals lose their rewarding value over time and instead recruit neural mechanisms driving food ‘wanting’ (i.e. craving) (Berridge et al., 2010). While our findings here do not directly buttress any of these particular theories, the data do demonstrate that multiple neurotransmitter systems in dorsal striatum, including DA, are impacted by long-term western diet consumption and obesity. Given the key role of the dorsal striatum in the control of action, this dysfunction could be part of an underlying mechanism for each theory. As previously mentioned, the clinical condition of obesity is very complex and the degree to which a particular theory applies likely varies on a case-by-case basis.
In conclusion, the obese-like phenotype produced by prolonged WD consumption in mice resulted in focally prolonged sEPSCs, likely as a result of decreased glutamate buffering, and blunted NMDAR signaling within the DLS. DA transmission was also robustly elevated throughout the dorsal striatum of WD mice. These findings were accompanied by reduced presynaptic MOPr-mediated plasticity. Future work should address the therapeutic potential for targeting glutamate signaling within the DLS and/or dorsal striatal DA and opioid signaling.
Highlights.
16 weeks of high-sugar, high-fat ‘western’ diet (WD) consumption generated an obese-like mouse phenotype
WD mice demonstrated altered glutamate transmission in medium spiny neurons in dorsolateral, but not dorsomedial striatum
Evoked dopamine release was augmented and reuptake impaired in medial and lateral regions of the dorsal striatum of WD mice
Dorsal striatal medium spiny neurons of WD mice were less likely to express mu-opioid receptor-mediated synaptic plasticity
Acknowledgments
This work was supported by NIDDK Grant P30 DK097512 and NIAAA grant R00 AA023507. The authors would also like to thank Dr. Jordan Yorgason (Oregon Health and Science University) for his assistance with voltammetry setup and troubleshooting, Dr. Sarah Tersey (Indiana University School of Medicine’s Center for Diabetes and Metabolic Diseases; CDMD) for her recommendations about the design of the western diet model, and Kara Benninger (CDMD) for her assistance with performing the metabolic testing.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Agresti A. An introduction to categorical data analysis. 2nd. Hoboken NJ: Wiley-Interscience; 2007. [Google Scholar]
- Atwood BK, Kupferschmidt DA, Lovinger DM. Opioids induce dissociable forms of long-term depression of excitatory inputs to the dorsal striatum. Nature neuroscience. 2014;17:540–548. doi: 10.1038/nn.3652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Auer MK, Sack M, Lenz JN, Jakovcevski M, Biedermann SV, Falfan-Melgoza C, Deussing J, Steinle J, Bielohuby M, Bidlingmaier M, et al. Effects of a high-caloric diet and physical exercise on brain metabolite levels: a combined proton MRS and histologic study. J Cereb Blood Flow Metab. 2015;35:554–564. doi: 10.1038/jcbfm.2014.231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Babbs RK, Sun X, Felsted J, Chouinard-Decorte F, Veldhuizen MG, Small DM. Decreased caudate response to milkshake is associated with higher body mass index and greater impulsivity. Physiology & behavior. 2013;121:103–111. doi: 10.1016/j.physbeh.2013.03.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balleine BW. Neural bases of food-seeking: affect, arousal and reward in corticostriatolimbic circuits. Physiology & behavior. 2005;86:717–730. doi: 10.1016/j.physbeh.2005.08.061. [DOI] [PubMed] [Google Scholar]
- Bateup HS, Santini E, Shen W, Birnbaum S, Valjent E, Surmeier DJ, Fisone G, Nestler EJ, Greengard P. Distinct subclasses of medium spiny neurons differentially regulate striatal motor behaviors. Proceedings of the National Academy of Sciences of the United States of America. 2010;107:14845–14850. doi: 10.1073/pnas.1009874107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berridge KC, Ho CY, Richard JM, DiFeliceantonio AG. The tempted brain eats: pleasure and desire circuits in obesity and eating disorders. Brain research. 2010;1350:43–64. doi: 10.1016/j.brainres.2010.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bisaga A, Danysz W, Foltin RW. Antagonism of glutamatergic NMDA and mGluR5 receptors decreases consumption of food in baboon model of binge-eating disorder. European neuropsychopharmacology: the journal of the European College of Neuropsychopharmacology. 2008;18:794–802. doi: 10.1016/j.euroneuro.2008.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brennan BP, Roberts JL, Fogarty KV, Reynolds KA, Jonas JM, Hudson JI. Memantine in the treatment of binge eating disorder: an open-label, prospective trial. Int J Eat Disord. 2008;41:520–526. doi: 10.1002/eat.20541. [DOI] [PubMed] [Google Scholar]
- Brown RM, Kupchik YM, Spencer S, Garcia-Keller C, Spanswick DC, Lawrence AJ, Simonds SE, Schwartz DJ, Jordan KA, Jhou TC, et al. Addiction-like Synaptic Impairments in Diet-Induced Obesity. Biological psychiatry. 2017;81:797–806. doi: 10.1016/j.biopsych.2015.11.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buttigieg A, Flores O, Hernandez A, Saez-Briones P, Burgos H, Morgan C. Preference for high-fat diet is developed by young Swiss CD1 mice after short-term feeding and is prevented by NMDA receptor antagonists. Neurobiol Learn Mem. 2014;107:13–18. doi: 10.1016/j.nlm.2013.10.018. [DOI] [PubMed] [Google Scholar]
- Campos CA, Ritter RC. NMDA-type glutamate receptors participate in reduction of food intake following hindbrain melanocortin receptor activation. Am J Physiol Regul Integr Comp Physiol. 2015;308:R1–9. doi: 10.1152/ajpregu.00388.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cano V, Valladolid-Acebes I, Hernandez-Nuno F, Merino B, Del Olmo N, Chowen JA, Ruiz-Gayo M. Morphological changes in glial fibrillary acidic protein immunopositive astrocytes in the hippocampus of dietary-induced obese mice. Neuroreport. 2014 doi: 10.1097/WNR.0000000000000180. [DOI] [PubMed] [Google Scholar]
- Carlezon WA, Thomas MJ. Biological substrates of reward and aversion: a nucleus accumbens activity hypothesis. Neuropharmacology. 2009;56:122–132. doi: 10.1016/j.neuropharm.2008.06.075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charles JR, Hernandez E, Winter A, Yang CR, Stanley BG. Site selective activation of lateral hypothalamic mGluR1 and R5 receptors elicits feeding in rats. Physiology & behavior. 2015;139:261–266. doi: 10.1016/j.physbeh.2014.10.039. [DOI] [PubMed] [Google Scholar]
- Colantuoni C, Rada P, McCarthy J, Patten C, Avena NM, Chadeayne A, Hoebel BG. Evidence that intermittent, excessive sugar intake causes endogenous opioid dependence. Obesity. 2002;10:478–488. doi: 10.1038/oby.2002.66. [DOI] [PubMed] [Google Scholar]
- Cooper SJ, Turkish S. Effects of naltrexone on food preference and concurrent behavioral responses in food-deprived rats. Pharmacology Biochemistry and Behavior. 1989;33:17–20. doi: 10.1016/0091-3057(89)90422-x. [DOI] [PubMed] [Google Scholar]
- Corbit LH, Nie H, Janak PH. Habitual alcohol seeking: time course and the contribution of subregions of the dorsal striatum. Biological psychiatry. 2012;72:389–395. doi: 10.1016/j.biopsych.2012.02.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cottone P, Sabino V, Steardo L, Zorrilla EP. Opioid-dependent anticipatory negative contrast and binge-like eating in rats with limited access to highly preferred food. Neuropsychopharmacology. 2008;33:524. doi: 10.1038/sj.npp.1301430. [DOI] [PubMed] [Google Scholar]
- Cui G, Jun SB, Jin X, Pham MD, Vogel SS, Lovinger DM, Costa RM. Concurrent activation of striatal direct and indirect pathways during action initiation. Nature. 2013;494:238–242. doi: 10.1038/nature11846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dagher A. Obesity Prevention. San Diego: Academic Press; 2010. Chapter 2 - The Neurobiology of Appetite: Hunger as Addiction; pp. 15–22. [Google Scholar]
- DiFeliceantonio AG, Mabrouk OS, Kennedy RT, Berridge KC. Enkephalin surges in dorsal neostriatum as a signal to eat. Current Biology. 2012;22:1918–1924. doi: 10.1016/j.cub.2012.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doane DF, Lawson MA, Meade JR, Kotz CM, Beverly JL. Orexin-induced feeding requires NMDA receptor activation in the perifornical region of the lateral hypothalamus. Am J Physiol Regul Integr Comp Physiol. 2007;293:R1022–1026. doi: 10.1152/ajpregu.00282.2007. [DOI] [PubMed] [Google Scholar]
- Doyle TG, Berridge KC, Gosnell BA. Morphine enhances hedonic taste palatability in rats. Pharmacology Biochemistry and Behavior. 1993;46:745–749. doi: 10.1016/0091-3057(93)90572-b. [DOI] [PubMed] [Google Scholar]
- Drewnowski A, Krahn DD, Demitrack MA, Nairn K, Gosnell BA. Taste responses and preferences for sweet high-fat foods: evidence for opioid involvement. Physiology & Behavior. 1992;51:371–379. doi: 10.1016/0031-9384(92)90155-u. [DOI] [PubMed] [Google Scholar]
- Everitt BJ, Robbins TW. From the ventral to the dorsal striatum: Devolving views of their roles in drug addiction. Neuroscience & Biobehavioral Reviews. 2013;37:1946–1954. doi: 10.1016/j.neubiorev.2013.02.010. [DOI] [PubMed] [Google Scholar]
- Ferraro FM, Hill KG, Kaczmarek HJ, Coonfield DL, Kiefer SW. Naltrexone modifies the palatability of basic tastes and alcohol in outbred male rats. Alcohol. 2002;27:107–114. doi: 10.1016/s0741-8329(02)00220-3. [DOI] [PubMed] [Google Scholar]
- Flegal KM. Epidemiologic aspects of overweight and obesity in the United States. Physiology & behavior. 2005;86:599–602. doi: 10.1016/j.physbeh.2005.08.050. [DOI] [PubMed] [Google Scholar]
- Flegal KM, Kit BK, Orpana H, Graubard BI. Association of all-cause mortality with overweight and obesity using standard body mass index categories: a systematic review and meta-analysis. JAMA. 2013;309:71–82. doi: 10.1001/jama.2012.113905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fordahl SC, Jones SR. High-Fat-Diet-Induced Deficits in Dopamine Terminal Function Are Reversed by Restoring Insulin Signaling. ACS chemical neuroscience. 2017;8:290–299. doi: 10.1021/acschemneuro.6b00308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fordahl SC, Locke JL, Jones SR. High fat diet augments amphetamine sensitization in mice: role of feeding pattern, obesity, and dopamine terminal changes. Neuropharmacology. 2016;109:170–182. doi: 10.1016/j.neuropharm.2016.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freeze BS, Kravitz AV, Hammack N, Berke JD, Kreitzer AC. Control of basal ganglia output by direct and indirect pathway projection neurons. The Journal of neuroscience: the official journal of the Society for Neuroscience. 2013;33:18531–18539. doi: 10.1523/JNEUROSCI.1278-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fryar CD, Carroll MD, Ogden CL. Prevalence of overweight, obesity, and extreme obesity among adults aged 20 and over: United States, 1960–1962 through 2013–2014. National Center for Health Statistics Division of Health and Nutrition Examination Surveys 2016 [Google Scholar]
- Furlong TM, Jayaweera HK, Balleine BW, Corbit LH. Binge-like consumption of a palatable food accelerates habitual control of behavior and is dependent on activation of the dorsolateral striatum. Journal of Neuroscience. 2014;34:5012–5022. doi: 10.1523/JNEUROSCI.3707-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghaleiha A, Entezari N, Modabbernia A, Najand B, Askari N, Tabrizi M, Ashrafi M, Hajiaghaee R, Akhondzadeh S. Memantine add-on in moderate to severe obsessive-compulsive disorder: randomized double-blind placebo-controlled study. J Psychiatr Res. 2013;47:175–180. doi: 10.1016/j.jpsychires.2012.09.015. [DOI] [PubMed] [Google Scholar]
- Guard DB, Swartz TD, Ritter RC, Burns GA, Covasa M. Blockade of hindbrain NMDA receptors containing NR2 subunits increases sucrose intake. Am J Physiol Regul Integr Comp Physiol. 2009a;296:R921–928. doi: 10.1152/ajpregu.90456.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guard DB, Swartz TD, Ritter RC, Burns GA, Covasa M. NMDA NR2 receptors participate in CCK-induced reduction of food intake and hindbrain neuronal activation. Brain research. 2009b;1266:37–44. doi: 10.1016/j.brainres.2009.02.003. [DOI] [PubMed] [Google Scholar]
- Guo J, Simmons WK, Herscovitch P, Martin A, Hall KD. Striatal dopamine D2-like receptor correlation patterns with human obesity and opportunistic eating behavior. Molecular psychiatry. 2014;19:1078–1084. doi: 10.1038/mp.2014.102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haslam DW, James WP. Obesity. Lancet. 2005;366:1197–1209. doi: 10.1016/S0140-6736(05)67483-1. [DOI] [PubMed] [Google Scholar]
- Hedley AA, Ogden CL, Johnson CL, Carroll MD, Curtin LR, Flegal KM. Prevalence of overweight and obesity among US children, adolescents, and adults, 1999–2002. JAMA. 2004;291:2847–2850. doi: 10.1001/jama.291.23.2847. [DOI] [PubMed] [Google Scholar]
- Heien ML, Johnson MA, Wightman RM. Resolving neurotransmitters detected by fast-scan cyclic voltammetry. Analytical chemistry. 2004;76:5697–5704. doi: 10.1021/ac0491509. [DOI] [PubMed] [Google Scholar]
- Hermanussen M, Tresguerres JA. A new anti-obesity drug treatment: first clinical evidence that, antagonising glutamate-gated Ca2+ ion channels with memantine normalises binge-eating disorders. Econ Hum Biol. 2005;3:329–337. doi: 10.1016/j.ehb.2005.04.001. [DOI] [PubMed] [Google Scholar]
- Hryhorczuk C, Florea M, Rodaros D, Poirier I, Daneault C, Des Rosiers C, Arvanitogiannis A, Alquier T, Fulton S. Dampened Mesolimbic Dopamine Function and Signaling by Saturated but not Monounsaturated Dietary Lipids. Neuropsychopharmacology. 2016;41:811–821. doi: 10.1038/npp.2015.207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunnicutt BJ, Jongbloets BC, Birdsong WT, Gertz KJ, Zhong H, Mao T. A comprehensive excitatory input map of the striatum reveals novel functional organization. Elife. 2016;5 doi: 10.7554/eLife.19103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jia H, Lubetkin EI. Obesity-related quality-adjusted life years lost in the U.S. from 1993 to 2008. American journal of preventive medicine. 2010;39:220–227. doi: 10.1016/j.amepre.2010.03.026. [DOI] [PubMed] [Google Scholar]
- Johnson PM, Kenny PJ. Dopamine D2 receptors in addiction-like reward dysfunction and compulsive eating in obese rats. Nature neuroscience. 2010;13:635–641. doi: 10.1038/nn.2519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katsuura Y, Taha SA. Modulation of feeding and locomotion through mu and delta opioid receptor signaling in the nucleus accumbens. Neuropeptides. 2010;44:225–232. doi: 10.1016/j.npep.2009.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kenny PJ. Common cellular and molecular mechanisms in obesity and drug addiction. Nature reviews Neuroscience. 2011;12:638. doi: 10.1038/nrn3105. [DOI] [PubMed] [Google Scholar]
- Kenny PJ, Voren G, Johnson PM. Dopamine D2 receptors and striatopallidal transmission in addiction and obesity. Current opinion in neurobiology. 2013;23:535–538. doi: 10.1016/j.conb.2013.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan AM, Curras MC, Dao J, Jamal FA, Turkowski CA, Goel RK, Gillard ER, Wolfsohn SD, Stanley BG. Lateral hypothalamic NMDA receptor subunits NR2A and/or NR2B mediate eating: immunochemical/behavioral evidence. Am J Physiol. 1999;276:R880–891. doi: 10.1152/ajpregu.1999.276.3.R880. [DOI] [PubMed] [Google Scholar]
- Kirkham TC, Cooper SJ. Naloxone attenuation of sham feeding is modified by manipulation of sucrose concentration. Physiology & Behavior. 1988;44:491–494. doi: 10.1016/0031-9384(88)90310-1. [DOI] [PubMed] [Google Scholar]
- Kravitz AV, Tye LD, Kreitzer AC. Distinct roles for direct and indirect pathway striatal neurons in reinforcement. Nature neuroscience. 2012;15:816–818. doi: 10.1038/nn.3100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kreitzer AC, Malenka RC. Endocannabinoid-mediated rescue of striatal LTD and motor deficits in Parkinson’s disease models. Nature. 2007;445:643–647. doi: 10.1038/nature05506. [DOI] [PubMed] [Google Scholar]
- Lerner TN, Horne EA, Stella N, Kreitzer AC. Endocannabinoid signaling mediates psychomotor activation by adenosine A2A antagonists. The Journal of neuroscience: the official journal of the Society for Neuroscience. 2010;30:2160–2164. doi: 10.1523/JNEUROSCI.5844-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lomeli H, Mosbacher J, Melcher T, Hoger T, Geiger JR, Kuner T, Monyer H, Higuchi M, Bach A, Seeburg PH. Control of kinetic properties of AMPA receptor channels by nuclear RNA editing. Science-AAAS-Weekly Paper Edition. 1994;266:1709–1712. doi: 10.1126/science.7992055. [DOI] [PubMed] [Google Scholar]
- Marcaggi P, Billups D, Attwell D. The role of glial glutamate transporters in maintaining the independent operation of juvenile mouse cerebellar parallel fibre synapses. The Journal of physiology. 2003;552:89–107. doi: 10.1113/jphysiol.2003.044263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy-Royal C, Dupuis J, Groc L, Oliet SH. Astroglial glutamate transporters in the brain: Regulating neurotransmitter homeostasis and synaptic transmission. J Neurosci Res. 2017 doi: 10.1002/jnr.24029. [DOI] [PubMed] [Google Scholar]
- Murphy-Royal C, Dupuis JP, Varela JA, Panatier A, Pinson B, Baufreton J, Groc L, Oliet SH. Surface diffusion of astrocytic glutamate transporters shapes synaptic transmission. Nature neuroscience. 2015;18:219–226. doi: 10.1038/nn.3901. [DOI] [PubMed] [Google Scholar]
- Nazzaro C, Greco B, Cerovic M, Baxter P, Rubino T, Trusel M, Parolaro D, Tkatch T, Benfenati F, Pedarzani P, et al. SK channel modulation rescues striatal plasticity and control over habit in cannabinoid tolerance. Nature neuroscience. 2012;15:284–293. doi: 10.1038/nn.3022. [DOI] [PubMed] [Google Scholar]
- Palmiter RD. Dopamine Signaling in the Dorsal Striatum Is Essential for Motivated Behaviors. Annals of the New York Academy of Sciences. 2008;1129:35–46. doi: 10.1196/annals.1417.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petr GT, Sun Y, Frederick NM, Zhou Y, Dhamne SC, Hameed MQ, Miranda C, Bedoya EA, Fischer KD, Armsen W, et al. Conditional deletion of the glutamate transporter GLT-1 reveals that astrocytic GLT-1 protects against fatal epilepsy while neuronal GLT-1 contributes significantly to glutamate uptake into synaptosomes. The Journal of neuroscience: the official journal of the Society for Neuroscience. 2015;35:5187–5201. doi: 10.1523/JNEUROSCI.4255-14.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Popik P, Kos T, Zhang Y, Bisaga A. Memantine reduces consumption of highly palatable food in a rat model of binge eating. Amino Acids. 2011;40:477–485. doi: 10.1007/s00726-010-0659-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reaven GM. Role of insulin resistance in human disease. Diabetes. 1988;37:1595–1607. doi: 10.2337/diab.37.12.1595. [DOI] [PubMed] [Google Scholar]
- Resch JM, Maunze B, Phillips KA, Choi S. Inhibition of food intake by PACAP in the hypothalamic ventromedial nuclei is mediated by NMDA receptors. Physiology & behavior. 2014;133:230–235. doi: 10.1016/j.physbeh.2014.05.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rimmele TS, Rosenberg PA. GLT-1: The elusive presynaptic glutamate transporter. Neurochemistry international. 2016;98:19–28. doi: 10.1016/j.neuint.2016.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ritter RC. A tale of two endings: modulation of satiation by NMDA receptors on or near central and peripheral vagal afferent terminals. Physiology & behavior. 2011;105:94–99. doi: 10.1016/j.physbeh.2011.02.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts-Wolfe DJ, Kalivas PW. Glutamate Transporter GLT-1 as a Therapeutic Target for Substance Use Disorders. CNS & neurological disorders drug targets. 2015;14:745–756. doi: 10.2174/1871527314666150529144655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson MJ, Burghardt PR, Patterson CM, Nobile CW, Akil H, Watson SJ, Berridge KC, Ferrario CR. Individual differences in cue-induced motivation and striatal systems in rats susceptible to diet-induced obesity. Neuropsychopharmacology. 2015;40:2113–2123. doi: 10.1038/npp.2015.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rothemund Y, Preuschhof C, Bohner G, Bauknecht HC, Klingebiel R, Flor H, Klapp BF. Differential activation of the dorsal striatum by high-calorie visual food stimuli in obese individuals. NeuroImage. 2007;37:410–421. doi: 10.1016/j.neuroimage.2007.05.008. [DOI] [PubMed] [Google Scholar]
- Rothstein JD, Martin L, Levey AI, Dykes-Hoberg M, Jin L, Wu D, Nash N, Kuncl RW. Localization of neuronal and glial glutamate transporters. Neuron. 1994;13:713–725. doi: 10.1016/0896-6273(94)90038-8. [DOI] [PubMed] [Google Scholar]
- Sharpe D. Your Chi-Square Test is Statistically Significant: Now What? Practical Assessment, Research, & Evaluation. 2015;20 [Google Scholar]
- Sickmann HM, Waagepetersen HS, Schousboe A, Benie AJ, Bouman SD. Obesity and type 2 diabetes in rats are associated with altered brain glycogen and amino-acid homeostasis. J Cereb Blood Flow Metab. 2010;30:1527–1537. doi: 10.1038/jcbfm.2010.61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sinha R, Fisch G, Teague B, Tamborlane WV, Banyas B, Allen K, Savoye M, Rieger V, Taksali S, Barbetta G, et al. Prevalence of Impaired Glucose Tolerance among Children and Adolescents with Marked Obesity. New England Journal of Medicine. 2002;346:802–810. doi: 10.1056/NEJMoa012578. [DOI] [PubMed] [Google Scholar]
- Small DM, Jones-Gotman M, Dagher A. Feeding-induced dopamine release in dorsal striatum correlates with meal pleasantness ratings in healthy human volunteers. Neuroimage. 2003;19:1709–1715. doi: 10.1016/s1053-8119(03)00253-2. [DOI] [PubMed] [Google Scholar]
- Smith KL, Rao RR, Velazquez-Sanchez C, Valenza M, Giuliano C, Everitt BJ, Sabino V, Cottone P. The uncompetitive N-methyl-D-aspartate antagonist memantine reduces binge-like eating, food-seeking behavior, and compulsive eating: role of the nucleus accumbens shell. Neuropsychopharmacology: official publication of the American College of Neuropsychopharmacology. 2015;40:1163–1171. doi: 10.1038/npp.2014.299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soni N, Reddy BV, Kumar P. GLT-1 transporter: an effective pharmacological target for various neurological disorders. Pharmacology, biochemistry, and behavior. 2014;127:70–81. doi: 10.1016/j.pbb.2014.10.001. [DOI] [PubMed] [Google Scholar]
- Speed N, Saunders C, Davis AR, Owens WA, Matthies HJ, Saadat S, Kennedy JP, Vaughan RA, Neve RL, Lindsley CW. Impaired striatal Akt signaling disrupts dopamine homeostasis and increases feeding. PloS one. 2011;6:e25169. doi: 10.1371/journal.pone.0025169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stamatakis AM, Van Swieten M, Basiri ML, Blair GA, Kantak P, Stuber GD. Lateral Hypothalamic Area Glutamatergic Neurons and Their Projections to the Lateral Habenula Regulate Feeding and Reward. The Journal of neuroscience: the official journal of the Society for Neuroscience. 2016;36:302–311. doi: 10.1523/JNEUROSCI.1202-15.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanley BG, Willett VL, 3rd, Donias HW, Dee MG, 2nd, Duva MA. Lateral hypothalamic NMDA receptors and glutamate as physiological mediators of eating and weight control. Am J Physiol. 1996;270:R443–449. doi: 10.1152/ajpregu.1996.270.2.R443. [DOI] [PubMed] [Google Scholar]
- Stice E, Spoor S, Bohon C, Small D. Relation between obesity and blunted striatal response to food is moderated by TaqIA A1 allele. Science. 2008a;322:449–452. doi: 10.1126/science.1161550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stice E, Spoor S, Bohon C, Small DM. Relation between obesity and blunted striatal response to food is moderated by TaqIA A1 allele. Science. 2008b;322:449–452. doi: 10.1126/science.1161550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stice E, Yokum S, Zald D, Dagher A. Behavioral neurobiology of eating disorders. Springer; 2010. Dopamine-based reward circuitry responsivity, genetics, and overeating; pp. 81–93. [DOI] [PubMed] [Google Scholar]
- Swinburn BA, Sacks G, Hall KD, McPherson K, Finegood DT, Moodie ML, Gortmaker SL. The global obesity pandemic: shaped by global drivers and local environments. The Lancet. 2011;378:804–814. doi: 10.1016/S0140-6736(11)60813-1. [DOI] [PubMed] [Google Scholar]
- Tepper J, Abercrombie E, Bolam J. Basal ganglia macrocircuits. Progress in brain research. 2007;160:3–7. doi: 10.1016/S0079-6123(06)60001-0. [DOI] [PubMed] [Google Scholar]
- Uner A, Goncalves GH, Li W, Porceban M, Caron N, Schonke M, Delpire E, Sakimura K, Bjorbaek C. The role of GluN2A and GluN2B NMDA receptor subunits in AgRP and POMC neurons on body weight and glucose homeostasis. Mol Metab. 2015;4:678–691. doi: 10.1016/j.molmet.2015.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vainik U, Dagher A, Dubé L, Fellows LK. Neurobehavioural correlates of body mass index and eating behaviours in adults: A systematic review. Neuroscience & Biobehavioral Reviews. 2013;37:279–299. doi: 10.1016/j.neubiorev.2012.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valladolid-Acebes I, Merino B, Principato A, Fole A, Barbas C, Lorenzo MP, Garcia A, Del Olmo N, Ruiz-Gayo M, Cano V. High-fat diets induce changes in hippocampal glutamate metabolism and neurotransmission. Am J Physiol Endocrinol Metab. 2012;302:E396–402. doi: 10.1152/ajpendo.00343.2011. [DOI] [PubMed] [Google Scholar]
- Volkow ND, Wang GJ, Baler RD. Reward, dopamine and the control of food intake: implications for obesity. Trends in cognitive sciences. 2011;15:37–46. doi: 10.1016/j.tics.2010.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volkow ND, Wang GJ, Fowler JS, Logan J, Jayne M, Franceschi D, Wong C, Gatley SJ, Gifford AN, Ding YS. “Nonhedonic” food motivation in humans involves dopamine in the dorsal striatum and methylphenidate amplifies this effect. Synapse. 2002;44:175–180. doi: 10.1002/syn.10075. [DOI] [PubMed] [Google Scholar]
- Voorn P, Vanderschuren LJ, Groenewegen HJ, Robbins TW, Pennartz CM. Putting a spin on the dorsal-ventral divide of the striatum. Trends in neurosciences. 2004;27:468–474. doi: 10.1016/j.tins.2004.06.006. [DOI] [PubMed] [Google Scholar]
- Wall NR, De La Parra M, Callaway EM, Kreitzer AC. Differential innervation of direct- and indirect-pathway striatal projection neurons. Neuron. 2013;79:347–360. doi: 10.1016/j.neuron.2013.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang GJ, Volkow ND, Logan J, Pappas NR, Wong CT, Zhu W, Netusll N, Fowler JS. Brain dopamine and obesity. The Lancet. 2001;357:354–357. doi: 10.1016/s0140-6736(00)03643-6. [DOI] [PubMed] [Google Scholar]
- Wang GJ, Geliebter A, Volkow ND, Telang FW, Logan J, Jayne MC, Galanti K, Selig PA, Han H, Zhu W. Enhanced striatal dopamine release during food stimulation in binge eating disorder. Obesity. 2011;19:1601–1608. doi: 10.1038/oby.2011.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weng HR, Chen JH, Pan ZZ, Nie H. Glial glutamate transporter 1 regulates the spatial and temporal coding of glutamatergic synaptic transmission in spinal lamina II neurons. Neuroscience. 2007;149:898–907. doi: 10.1016/j.neuroscience.2007.07.063. [DOI] [PubMed] [Google Scholar]
- Winzell M, Ahrén B. The high-fat diet-fed mouse: a model for studying mechanisms and treatment of impaired glucose tolerance and type 2 diabetes. Diabetes. 2004;53(Suppl 3):S215–S219. doi: 10.2337/diabetes.53.suppl_3.s215. [DOI] [PubMed] [Google Scholar]
- Wright J, Campos C, Herzog T, Covasa M, Czaja K, Ritter RC. Reduction of food intake by cholecystokinin requires activation of hindbrain NMDA-type glutamate receptors. Am J Physiol Regul Integr Comp Physiol. 2011;301:R448–455. doi: 10.1152/ajpregu.00026.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Q, Zheng R, Srisai D, McKnight GS, Palmiter RD. NR2B subunit of the NMDA glutamate receptor regulates appetite in the parabrachial nucleus. Proceedings of the National Academy of Sciences of the United States of America. 2013;110:14765–14770. doi: 10.1073/pnas.1314137110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Y, Wu Z, Sun H, Zhu Y, Kim ER, Lowell BB, Arenkiel BR, Xu Y, Tong Q. Glutamate mediates the function of melanocortin receptor 4 on Sim1 neurons in body weight regulation. Cell metabolism. 2013;18:860–870. doi: 10.1016/j.cmet.2013.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin HH, Knowlton BJ, Balleine BW. Lesions of dorsolateral striatum preserve outcome expectancy but disrupt habit formation in instrumental learning. European journal of neuroscience. 2004;19:181–189. doi: 10.1111/j.1460-9568.2004.03095.x. [DOI] [PubMed] [Google Scholar]
- Yokum S, Gearhardt AN, Harris JL, Brownell KD, Stice E. Individual differences in striatum activity to food commercials predict weight gain in adolescents. Obesity. 2014;22:2544–2551. doi: 10.1002/oby.20882. [DOI] [PMC free article] [PubMed] [Google Scholar]