

Abstract
Ex vivo systems that incorporate features of the tumor microenvironment (TME) and model the dynamic response to immune checkpoint blockade (ICB) may facilitate efforts in precision immuno-oncology and the development of effective combination therapies. Here, we demonstrate the ability to interrogate ex vivo response to ICB using murine- and patient-derived organotypic tumor spheroids (MDOTS/PDOTS). MDOTS/PDOTS isolated from mouse and human tumors retain autologous lymphoid and myeloid cell populations, and respond to ICB in short-term 3-dimensional microfluidic culture. Response and resistance to ICB was recapitulated using MDOTS derived from established immunocompetent mouse tumor models. MDOTS profiling demonstrated that TBK1/IKKε inhibition enhanced response to PD-1 blockade, which effectively predicted tumor response in vivo. Systematic profiling of secreted cytokines in PDOTS captured key features associated with response and resistance to PD-1 blockade. Thus, MDOTS/PDOTS profiling represents a novel platform to evaluate ICB using established murine models as well as clinically relevant patient specimens.
Keywords: PD-1, CTLA-4, immunotherapy, tumor microenvironment, organotypic tumor spheroids, 3D culture, microfluidics, cytokines
INTRODUCTION
Monoclonal antibodies targeting co-inhibitory immune checkpoints (e.g. PD-1, CTLA-4) have demonstrated unprecedented clinical activity in several malignances, and are revolutionizing the practice of medical oncology (1–3). Reinvigoration of anti-tumor immune responses by ICB can result in durable clinical responses suggestive of long-lasting immunologic memory (4,5). Despite the remarkable success of ICB in melanoma and other cancers, overcoming innate (primary) resistance remains a major challenge and robust biomarkers to guide treatment are lacking (4,6). Combination immunotherapy, or dual immune checkpoint blockade (anti-PD-1 + anti-CTLA-4), has shown improved response rates in patients with metastatic melanoma, however survival benefit for this approach over PD-1 monotherapy has yet to be demonstrated and over half of patients experienced significant toxicity from the treatment regimen (7,8). Clinical trials are underway evaluating ‘rational’ combination strategies to overcome innate resistance (3,9,10), but given the increasing number of novel therapies and combination trials, there is an unmet need for development of precision immuno-oncology techniques and methods for pre-clinical and clinical use.
There is growing interest in understanding the dynamic interactions between the tumor and the immune system that influence response and resistance to ICB therapy using patient-derived materials (11–13), however appropriate model systems are lacking. Current approaches to study anti-tumor immune responses in patients (14–16) are limited by remote measurements in whole blood or plasma, or static assessment of biopsies (15,17,18). Given the emerging importance of the TME in modulating T cell function, particularly in response PD-1 blockade (19), more sophisticated tumor models that incorporate features of the TME are needed to elucidate cellular, molecular, and immunologic mechanisms of response and resistance. Existing patient-derived cancer models, including circulating tumor cells (CTCs), organoid cultures, and patient-derived xenografts (PDXs) can guide precision cancer therapy (20,21), but take weeks to months to generate and lack the native tumor immune microenvironment. Novel ex vivo systems that retain key features of the native immune TME and analyzed in real-time may accelerate identification of novel predictive and/or prognostic biomarkers, facilitate identification of therapies to combat anti-PD-1/PD-L1 resistance, and drive translational research efforts to ultimately guide optimal selection and sequencing of ICB therapy in the clinic (10,22). Here, we describe a novel platform to evaluate tumor-immune interactions in 3D culture using organotypic tumor spheroids.
RESULTS
The Immune Contexture of the Tumor Microenvironment is Preserved in Murine-Derived Organotypic Tumor Spheroids
To model ICB ex vivo we adapted a 3D microfluidic device (23,24) to the short-term culture of murine- and patient-derived organotypic tumor spheroids (MDOTS/PDOTS). Following limited collagenase digestion of fresh tumor specimens, multicellular organotypic spheroids with autologous immune cells were isolated. MDOTS/PDOTS were analyzed by flow cytometry (Supplementary Fig. 1) or loaded in collagen into the central channel of the device for exposure to anti-PD-1 or anti-CTLA-4 antibodies (Fig. 1a). After confirming that enzymatic digestion did not alter surface antigen expression (Supplementary Fig. 2a–c), we evaluated the lymphoid and myeloid immune cell composition of bulk tumor with different spheroid populations (S1 >100 μm; S2 40–100 μm; and S3 <40 μm) from MC38 and B16F10 tumors (25–27). Flow cytometric analysis revealed similar populations across all immune cell fractions examined (Fig. 1b–c, Supplementary Fig. 2d–e, Table S1). MDOTS (S2) from B16F10 demonstrated fewer CD45+ cells than MC38 and another model, CT26 (Supplementary Fig. 2f), although immune sub-populations were consistently maintained across MC38, B16F10, and CT26 models (Supplementary Fig. 2g) and reflected immune infiltration of explanted tumors by immunohistochemical analysis (Supplementary Fig. 2h). Since S2 MDOTS (40–100μm) were optimally sized for culture in the microfluidic device, we utilized this fraction for subsequent studies. 3D microfluidic culture of MDOTS resulted in growth and expansion over time (Fig. 1d) as well as cytokine elaboration in conditioned medium (Fig. 1e–f). MC38 MDOTS measured after six days in 3D microfluidic culture demonstrated interval growth with final spheroid diameter averaging 138.31 +/− 42.94 μm (range 68.17–210.77 μm) (Supplementary Fig. 2i). We further demonstrated that tumor/immune cell intermixture in spheroids was preserved within the device by immunofluorescence microscopy (Fig. 1g). Thus, MDOTS retain autologous immune cells, and short-term culture and cytokine profiling of MDOTS is feasible using this 3D microfluidic device.
Figure 1. Immune profiling and ex vivo culture of murine-derived organotypic tumor spheroids.
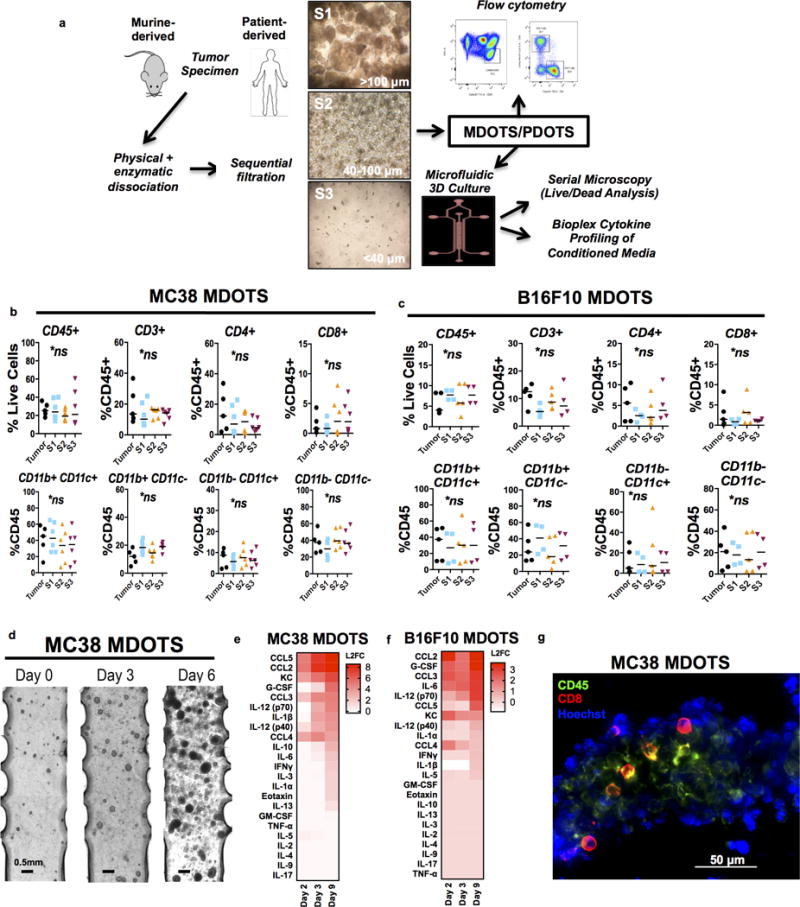
a, Schematic for preparation and analysis of MDOTS/PDOTS (S2 fraction) from murine or patient-derived tumor specimens. b, MC38 immune profiling by flow cytometry comparing bulk tumor (n=5) to S1, S2, S3 (n=6) spheroid fractions (Kruskal-Wallis with Dunn’s multiple comparisons test, α=0.05; ns=not significant). c, B16F10 immune profiling by flow cytometry comparing bulk tumor (n=5) to S1 (n=4), S2 (n=5), and S3 (n=4) evaluated by flow cytometry (Kruskal-Wallis test with Dunn’s multiple comparisons test, α=0.05; p<.05; ns=not significant). d, Phase-contrast imaging (4×) of MC38 MDOTS in 3D microfluidic culture. e–f, Heatmaps of secreted cytokine profiles from cultured (e) MC38 and (f) B16F10 MDOTS expressed as log-2 fold change relative to Day 1. g, Immunofluorescence staining of CD45+ and CD8+ immune cells in MC38 MDOTS.
Murine-Derived Organotypic Tumor Spheroids Recapitulate Sensitivity and Resistance to PD-1 Blockade in Ex Vivo 3D Microfluidic Culture
To evaluate ex vivo response to PD-1 blockade, MDOTS were treated with anti-PD-1 antibody (or isotype control) for 3 days or 6 days in the device, and dual labeling de-convolution fluorescence microscopy using acridine orange (AO, live cells) and propidium iodide (PI, dead cells) was performed (Fig. 2a). Starting with the MC38 syngeneic model (27) that responds to anti-PD-1 treatment in vivo (Fig. 2b), we demonstrated baseline viability of MC38 MDOTS (>90% at Day 0) dose- and time-dependent killing of MC38 MDOTS in response to anti-PD-1 treatment (Fig. 2c). This result was confirmed by multiple independent replicates across different laboratories (Supplementary Fig. 3a). The effect of PD-1 blockade on cell viability required immune cells, as cell line-derived MC38 spheroids lacking autologous immune/stromal cells were insensitive to anti-PD-1 treatment (Fig. 2d). Importantly, MDOTS derived from the PD-1 resistant B16F10 (26) (Fig. 2e,f) and Lewis lung carcinoma (LLC) (Supplementary Fig. 3b) models exhibited little cell death compared to MC38 MDOTS despite identical treatment (Fig. 2f). Modest killing was evident in the intermediately sensitive CT26 model (28) (Fig. 2g). Moreover, anti-PD-1 induced killing of CT26 MDOTS was disrupted by co-treatment with an anti-CD8α Ab, demonstrating a specific requirement for CD8+ T cells (Fig. 2h), which we also confirmed using MC38 MDOTS (Supplementary Fig. 3c). To examine ex vivo killing in an orthotopic anti-PD-1 sensitive model, we also prepared MDOTS from explanted GL261 glioma tumors (29), which similarly retained sensitivity to ex vivo PD-1 blockade in a CD8+ T cell-dependent manner (Supplementary Fig. 3d). We further confirmed survival of CD45+ immune cells and CD8+ T cells ex vivo in the device even after tumor cell death following PD-1 blockade (Supplementary Fig. 3e). These data demonstrate the ability to recapitulate sensitivity and resistance to PD-1 blockade ex vivo using well-defined mouse models.
Figure 2. Ex vivo profiling of PD-1 blockade using MDOTS.
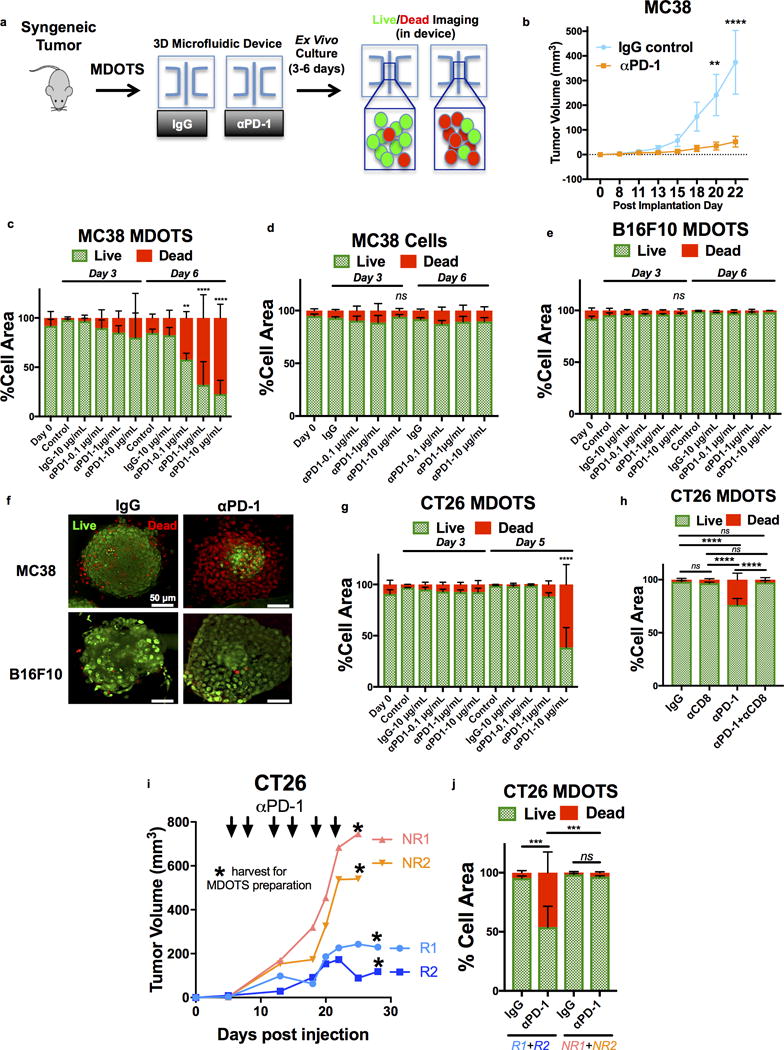
a, Schematic of MDOTS Live/Dead Imaging workflow. b, MC38 implanted tumor volume following isotype control IgG (n=10) or rat-anti-mouse anti-PD-1 antibody (n=10) treatment (mean ± s.e.m., 2-way ANOVA, Sidak’s multiple comparison test, **p<.01, ****p<0.0001). c, Live (AO = green)/dead (PI = red) quantification of MC38 MDOTS Day 0 (immediately after loading), Day 3, and Day 6 following IgG control or indicated anti-PD-1 antibody doses (n=4, biological replicates, 2-way ANOVA with Dunnett’s with multiple comparisons test, **p<.01, ****p<0.0001). d, Live/dead analysis of MC38 spheroids lacking immune cells ± anti-PD1 (n=4, biological replicates). e, Live/dead analysis of B16F10 MDOTS ± anti-PD1 (n=3, biological replicates). f, Deconvolution fluorescence microscopy of MC38 and B16F10 MDOTS Day 6 ± anti-PD1 (representative images shown). g, Live/dead analysis of CT26 MDOTS ± anti-PD1 (n=3, biological replicates, 2-way ANOVA with Dunnett’s with multiple comparisons test; ****p<0.0001). h, Live/dead analysis of CT26 MDOTS performed on Day 6 following treatment with isotype IgG control (10 μg/mL) or anti-PD-1 (10 μg/mL) ± anti-CD8 (10 μg/mL) (n=6, biological replicates; 2-way ANOVA with Tukey’s multiple comparisons test; ****p<.0001, ns = not significant). i, CT26 tumor volumes for responder (R1+R2) and non-responder (NR1+NR2) Balb/c mice treated with anti-PD-1 (10mg/kg twice weekly × 6 doses, starting at Day 5) with time of tumor harvest for MDOTS preparation indicated (*). j, Live/dead analysis (Day 6) of CT26 MDOTS from responder and non-responder mice following ex vivo treatment with isotype IgG control (10 μg/mL) or anti-PD-1 (10 μg/mL) (n=3, biological replicates; 2-way ANOVA with Tukey’s multiple comparisons test; ***p<.001, ns = not significant).
Despite implantation of identical cell numbers into syngeneic mice, stochastic tumor growth and response to ICB is frequently observed. To evaluate the impact of inter- and intra-tumoral heterogeneity on sensitivity to PD-1 blockade using MDOTS, we prepared MDOTS from explanted CT26 tumors of different sizes. Diminished CD8 T cell infiltration and diminished ex vivo sensitivity to PD-1 blockade was observed in MDOTS from larger CT26 tumors compared to MDOTS prepared from smaller tumors (Supplementary Fig. 3f). Importantly, CD8 T cell infiltration and ex vivo killing also correlated in MDOTS prepared from distinct regions of a single explanted CT26 tumor (Supplementary Fig. 3f). We next asked if in vivo sensitivity (and resistance) to PD-1 blockade was preserved during ex vivo testing. CT26 MDOTS were prepared from mice responding (R) or not responding (NR) to in vivo treatment with PD-1 blockade and re-challenged with anti-PD-1 therapy in 3D microfluidic culture (Fig. 2i). CT26 MDOTS prepared from PD-1 responsive tumors (R1+R2) retained sensitivity to PD-1 blockade ex vivo, whereas MDOTS prepared from PD-1 non-responsive tumors (NR1+NR2) maintained resistance to PD-1 blockade (Fig. 2j). These results demonstrate that heterogeneity in tumor growth and response to ICB observed in vivo is preserved ex vivo in MDOTS.
Murine-Derived Organotypic Tumor Spheroids Facilitate Testing of Novel Therapeutic Combinations
Because of its partial sensitivity to anti-PD1, we focused on the CT26 model to determine whether MDOTS profiling could identify novel combination therapies that overcome intrinsic resistance to PD-1 blockade. Furthermore, we noted that CT26 MDOTS elaborated particularly high levels of CCL2 (Supplementary Fig. 4a), an immunosuppressive chemokine associated with resistance to PD-1 blockade (30). However, neutralization of CCL2 alone failed to enhance PD-1 mediated killing of CT26 MDOTS (Supplementary Fig. 4b–c), suggesting the need for alternative strategies that more broadly inhibit immune suppressive signaling within the TME and reactivate T cells (31,32). To determine if the MDOTS platform could identify novel compounds to enhance efficacy of PD-1 blockade, we evaluated the impact of combination with a novel potent/selective TBK1/IKKε inhibitor, Compound 1 (Cmpd1) (Supplementary Fig. 4d). The homologous innate immune signaling kinases TBK1 and IKKε not only promote autocrine/paracrine cytokine signaling (24), but also restrain T cell activation (33,34), suggesting that TBK1/IKKε inhibition could enhance tumor control in response to PD-1 blockade via multiple mechanisms to overcome an immunosuppressive TME (Fig. 3a). We confirmed the potency and selectivity of Cmpd1 (Fig. 3b, Supplementary Fig. 4e, Table S2), including the absence of JAK inhibitory activity (Fig. 3c), in contrast to the multi-targeted inhibitor momelotinib (CYT387) (24). Cmpd1 effectively blocked immune suppressive cytokine elaboration by CT26 cell line spheroids, without cytotoxic effects (Fig. 3d, Supplementary Fig. 4f), and enhanced secretion of IL-2 and IFN-γ from purified CD4+ and CD8+ T cells from healthy human donors (Fig. 3e–f) and IL-2 from Jurkat human T-cell leukemia cells (Supplementary Fig. 4g). Ex vivo addition of Cmpd1 to PD-1 blockade enhanced killing of CT26 MDOTS (Fig. 4a–b), associated with decreased levels of CCL4, CCL3, and IL-1β and induction of cytokines involved in activated innate immune responses (e.g. G-CSF) (Fig. 4c). To determine if MDOTS profiling predicted in vivo response to dual TBK1/IKKε inhibition, Balb/c mice bearing CT26 tumors were treated with Cmpd 1 ± anti-PD-L1 (Fig. 4d–f). Consistent with MDOTS profiling data, greater tumor control and longer survival was evident with Cmpd1 + anti-PD-L1 than with either Cmpd1 or anti-PD-L1 alone (Fig. 4d–f). Reimplantation of CT26 into mice with exceptional responses to combination therapy showed no growth, whereas EMT-6 implanted tumors grew normally, suggesting induction of immunologic memory of CT26 cells in mice treated with Cmpd 1 + anti-PD-L1 (Supplementary Fig. 4h). Therefore, MDOTS profiling effectively recapitulated the in vivo response to PD-1 blockade +/− TBK1/IKKε inhibition, highlighting the potential of ex vivo screening in MDOTS to develop combination immunotherapies.
Figure 3. Characterization of Compound 1, a Novel TBK1/IKKε inhibitor.
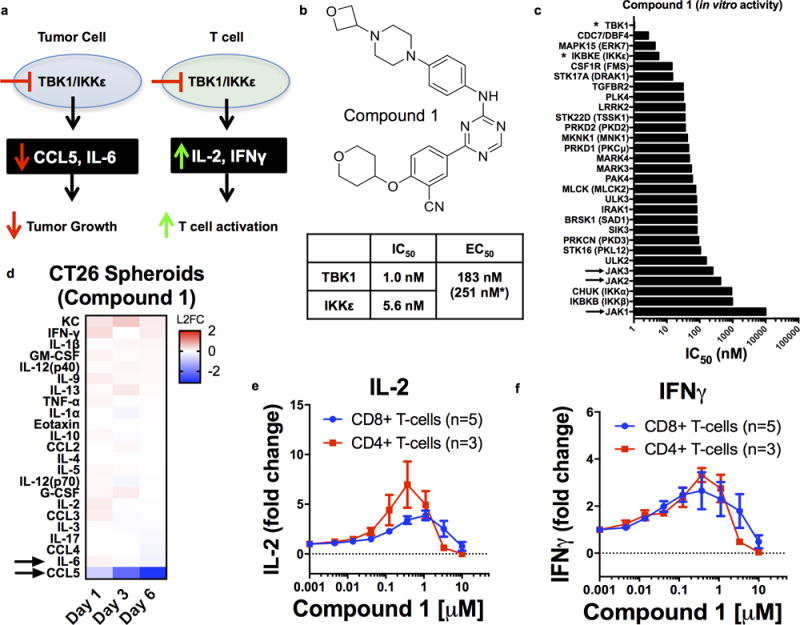
a, Scheme of impact of TBK1/IKKε inhibition on cytokine production from tumor cells and T cells. b, Compound 1 chemical structure with IC50 towards TBK1/IKKε, and EC50 in HCT116 cells. c, IC50 values for indicated enzymes treated with Compound 1. d, Cytokine heatmaps for CT26 spheroids (lacking immune cells) on Day 1, 3, and 6 ± Compound 1 (n=3, biological replicates) expressed as log2 fold-change (L2FC) relative to vehicle control. e–f, Dose-response curves for Compound 1 on IL-2 (e) and IFNγ (f) in human CD4 (n=3) and CD8 (n=5) T cells.
Figure 4. TBK1/IKKε inhibition enhances response to PD-1 blockade.
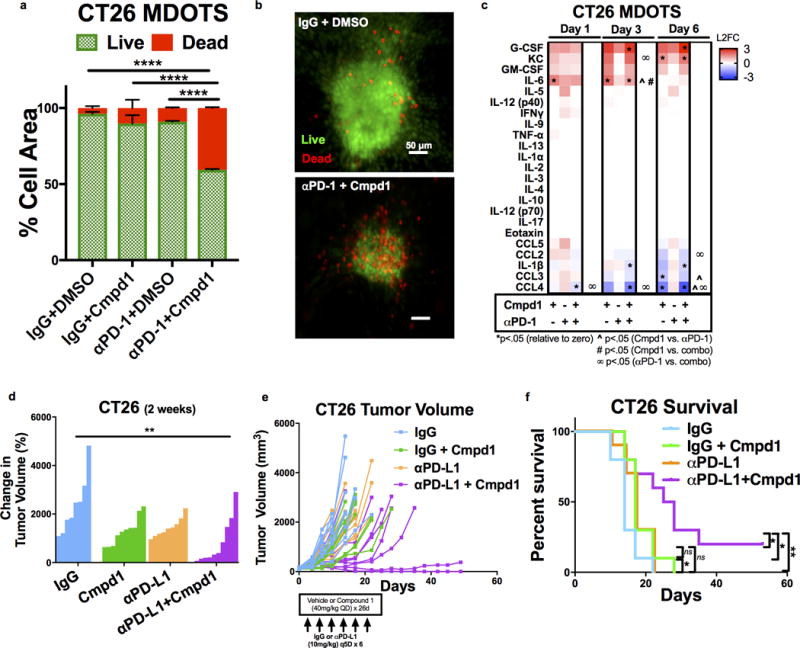
a–b, Live (AO = green)/dead (PI = red) quantification of CT26 MDOTS after 6 days treated with IgG-DMSO, Cmpd1 (1μM), αPD-1, and αPD-1+ Cmpd1 (*p<0.05, Kruskal-Wallis ANOVA with multiple comparisons; n=3). c, Cytokine heatmaps for CT26 MDOTS treated with IgG+Cmpd1 (1μM), αPD-1 (10 μg/mL), or αPD-1+Cmpd1 (1μM) from the mean of n=3 biological replicates, plotted as L2FC relative to isotype control IgG with vehicle control. 2-sided Welch’s 2-sample t-test with unequal variance (α=.05). d-f, CT26 implanted tumor volume (d-e) and percent survival (f) following IgG+vehicle, IgG+Cmpd1, αPD-L1+vehicle, and αPD-L1+Cmpd1 (n=10 per group, **p<.01, 1-way ANOVA with Tukey’s multiple comparison’s test for tumor volume, log-rank Mantel-Cox test for Kaplan-Meier analysis for entire group and pairwise comparisons).
Immunophenotyping and Secreted Cytokine Profiling Using Patient-Derived Organotypic Tumor Spheroids
We next immunophenotyped a large panel of PDOTS (n=40) by flow cytometry, enriching for cancers responsive to PD-1 blockade, such as melanoma (4) and Merkel cell carcinoma (35), to establish feasibility of ex vivo PDOTS profiling using patient-derived tumors samples (Fig. 5a, Table S3a). We consistently detected a range of lymphoid (CD19+ B cells, CD4+ and CD8+ T cells) and myeloid (CD15+ granulocytic, CD14+ monocytic lineages, CD123+ dendritic cells) populations in PDOTS (Fig. 5a). Immunofluorescence staining of PDOTS confirmed presence of CD45+ and CD8+ cells interspersed with EpCAM+ tumor cells (Fig. 5b–c), and live imaging revealed dynamic cellular interactions (Supplementary Movies 1–2). We also detected variable surface expression of exhaustion markers (PD-1, CTLA-4, TIM-3) on CD4+ and CD8+ T cells (Supplementary Fig. 5a–b), and PD-1 ligands (PD-L1 and PD-L2) on myeloid populations, including dendritic cells, myeloid-derived suppressor cells (MDSCs), and tumor-associated macrophages (TAMs) (Supplementary Fig. 5c). We confirmed strong correlation of T-cell profiles between PDOTS (S2) and S3 fractions, including antigen-experienced (CD45RO+) (Fig. 5d) and exhausted CD4 and CD8 T cells (Fig. 5e), and overall conservation of immunophenotype regardless of spheroid size (Supplementary Fig. 5d–f). Taken together, these results confirm that PDOTS retain autologous immune cells, including key tumor-infiltrating T lymphocyte populations.
Figure 5. Immune profiling of patient-derived organotypic tumor spheroids.
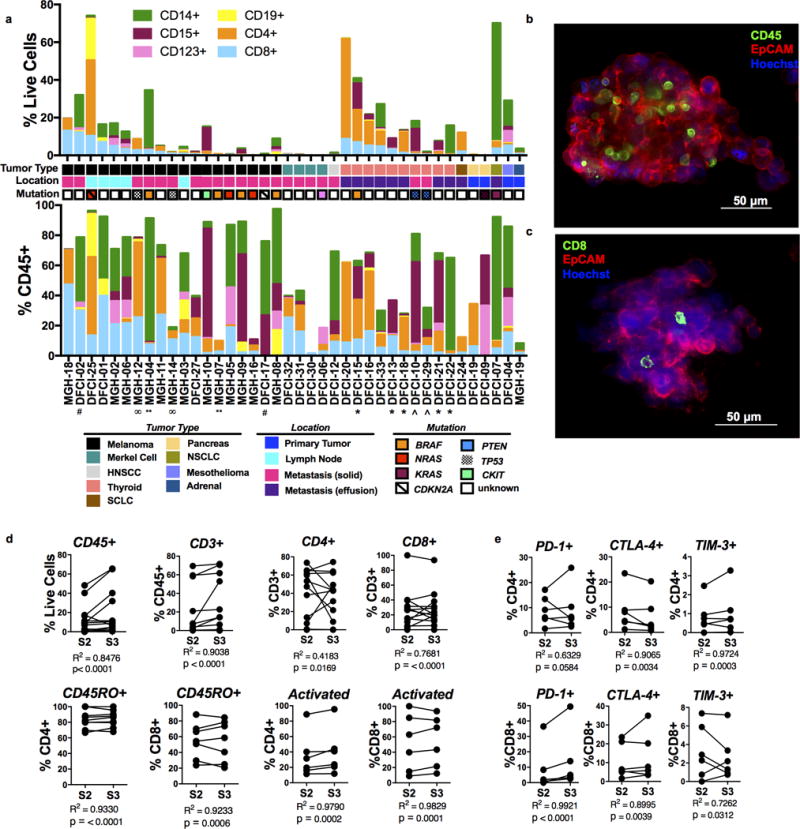
a, Immune profiling of PDOTS (S2; n=40) (upper panel = % live cells, lower panel = %CD45+ cells) with indicated patient/tumor characteristics, grouped by tumor type and ranked by %CD8+ T cells. b–c, Immunofluorescence staining identifying (b) CD45+ immune cells and (c) CD8+ T cells with EpCAM+ cancer cells NSCLC PDOTS. d, Immune cell correlation of S2/S3 fractions (CD45, n=14; CD3, n=15; CD4/CD8, n=13; CD4+CD45RO+, n=9; CD8+CD45RO+, n=8; activated = CD38+ and/or CD69+, n=6), R2 significant for all comparisons. e, PD-1, CTLA-4, TIM-3 expression on CD4 and CD8 T cell populations in S2/S3 fractions (n=6), R2 significant for all comparisons.
We next evaluated responses to PD-1 blockade in PDOTS. While ex vivo killing was evident in specific PDOTS samples, including those with robust T cell infiltration (Supplementary Fig. 5g–k), we focused on acute cytokine production as a systematic quantitative measure of early immune activation, given the more heterogeneous time course of clinical response in patients relative to well-defined syngeneic mouse tumor models. We analyzed Day 3 cytokine release from PDOTS (n=28) ± PD-1 blockade, and observed upregulation of CCL19 and CXCL13 in the majority of samples (23/28) (Fig. 6a–b Supplementary Fig. 6a–c). CCL19/CXCL13 induction was not observed with isotype IgG control (Supplementary Fig. 6d), and was comparatively minimal following CTLA-4 blockade (Fig. 6c–d). CCL19/CXCL13 generation was also preserved across a range of spheroid numbers (Supplementary Fig. 6e), with little intra-assay variability (Supplementary Fig. 6f–g). CCL19 correlated with CXCL13 across PDOTS samples, and required the microfluidic device for robust induction (Supplementary Fig. 6h–i). CCL19/CXCL13 upregulation was also evident following dual PD-1 + CTLA-4 blockade (Fig. 6e–f), and accompanied by induction of additional effector cytokines (e.g. IFN-γ, IL-2, and TNF-α) in select samples (Supplementary Fig. 7a–d). Induction of CCL19/CXCL13 was independent of total number of CD45+ cells or other immune subpopulations in PDOTS (Supplementary Fig. 8a–c), and qRT-PCR of sorted cell populations (Supplementary Fig. 8d) and immunohistochemical staining on tumor sections (Supplementary Fig. 8e) confirmed expression of CCL19 and CXCL13 in stromal and immune cells. Furthermore, CCL19 was preferentially expressed at high levels in tumor-draining lymph nodes in the CT26 model (Supplementary Fig. 8f–g) and not in subcutaneously implanted tumors, consistent with a known relationship to lymph node architecture (36), further highlighting an important distinction of PDOTS models which are derived from autochthonous tumors.
Figure 6. Cytokine profiling of PD-1 blockade in PDOTS reveals CCL19/CXCL13 upregulation.
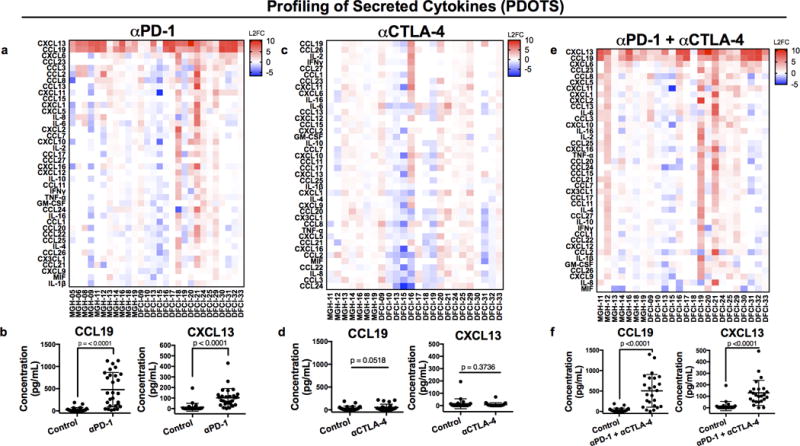
Cytokine heatmaps Day 3 ± anti-PD-1 (a, n=28), anti-CTLA-4 (c, n=24), or anti-PD-1 + anti-CTLA-4 (e, n=24) expressed as log2 fold-change (L2FC) relative to untreated control. Absolute CCL19/CXCL13 levels (pg/mL) observed at Day 3 ± anti-PD-1 (b, n=28), anti-CTLA-4 (d, n=24), or anti-PD-1 + anti-CTLA-4 (f, n=24) (2-sided, paired, t-test, α= 0.05).
Cytokine/Chemokine Effectors Identified by PDOTS Profiling and Patterns of Response and Resistance to PD-1 Blockade
To confirm CCL19/CXCL13 upregulation in patients treated with PD-1 blockade, we evaluated paired biopsy specimens from patients with melanoma before and after treatment with ICB. In consonance with PDOTS profiling results, CCL19/CXCL13 mRNA expression increased in patients treated with PD-1 blockade (Fig. 7a–b, Table S3b–c). As PD-1 blockade promotes immune cell infiltration in vivo (13), we also performed single sample gene set enrichment analysis (ssGSEA) using published immune signatures (37), which revealed enrichment of diverse immune cell populations in a subset of patients (group A) treated with ICB (Fig. 7c). Although fold induction of CCL19/CXCL13 did not clearly correlate with immune infiltration in this subset of samples (Supplementary Fig. 9a), higher absolute expression of CCL19 and CXCL13, and their receptors (CCR7 and CXCR5), was evident in immune-infiltrated samples (Group A, Fig. 7d–e), in contrast to other cytokines/cytokine receptors (Supplementary Fig. 9b–c). Immune infiltration (Group A) was associated with improved overall survival (OS), with a trend towards improved progression-free survival (PFS) (Supplementary Fig. 9d, Table S3d). To evaluate the clinical significance of CCL19/CXCL13, we analyzed Cancer Genome Atlas melanoma (SKCM) data (38). Improved patient survival was evident in melanoma specimens with higher expression of CCL19/CXCL13 (Fig. 7f, Supplementary Fig. 9e, Table S4). Immune ssGSEA using melanoma TCGA data confirmed enrichment of diverse immune cell gene sets in melanoma patients with high levels of both CCL19 and CXCL13 (Fig. 7g), consistent with their established roles as chemoattractants. These data confirm that dynamic induction of CCL19/CXCL13 is an important feature of the immune response to PD-1 blockade, which is captured ex vivo in PDOTS.
Figure 7. CCL19/CXCL13 induction following PD-1 treatment and association with immune infiltration.
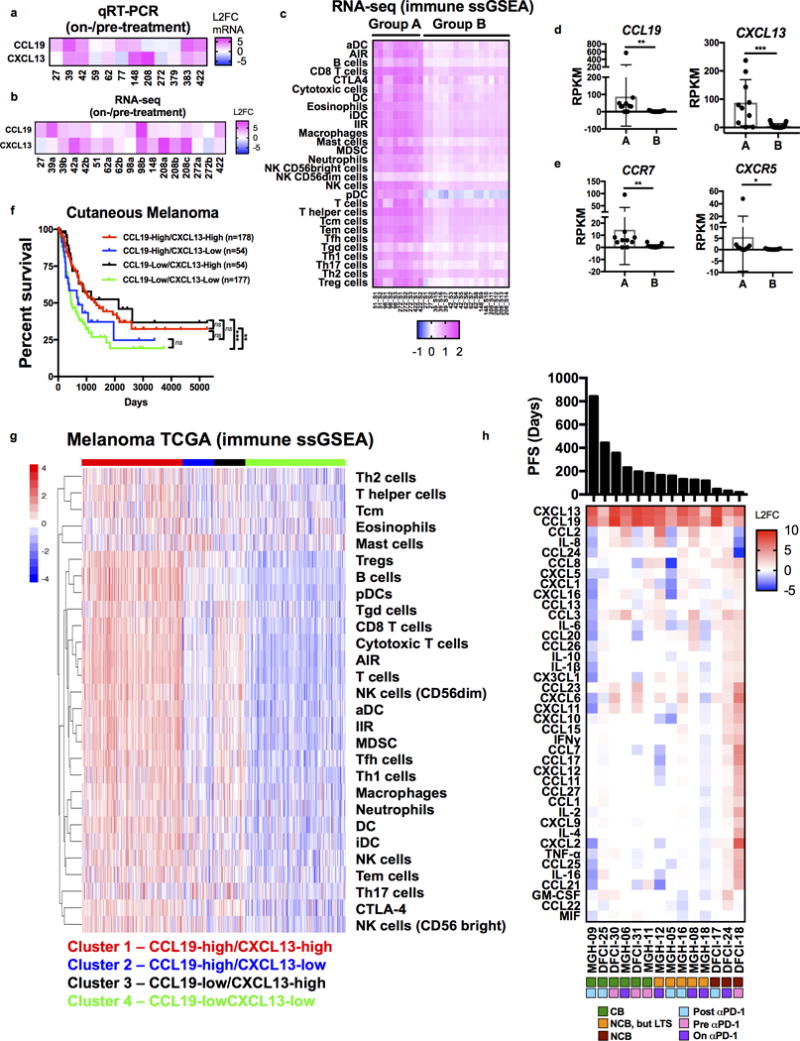
a–b, CCL19/CXCL13 mRNA levels from melanoma biopsy samples on anti-PD1 treatment relative to pre-PD-1 (L2FC) by (a) qRT-PCR (n=12) and (b) RNA-seq (n=17 from 10 patients). c, Immune signatures (ssGSEA) in melanoma biopsy specimens (pre- and on-treatment) define immune-infiltrated (Group A, n=10 samples from 4 patients) and immune-poor tumor samples (Group B, n=17 samples from 6 patients). d–e, absolute expression (RPKM) for (d) CCL19 and CXCL13 and (e) their respective receptors, CCR7 and CXCR5 in melanoma biopsy specimens (pre- and on-treatment) in indicated sets of patient samples (group A - immune infiltrated; group B – immune poor by ssGSEA, Fig. 7c). f, Kaplan-Meier survival curve by four-way sorting of CCL19/CXCL13 expression using cutaneous melanoma (SKCM) TCGA data (38) (log-rank Mantel-Cox test). g, immune signatures (ssGSEA) in melanoma biopsy specimens (pre- and on-treatment) in clusters of patients with varying expression of CCL19 and CXCL13 in cutaneous melanoma (SKCM) TCGA. h, Heatmap of Day 3 PDOTS anti-PD1 induced cytokines (L2FC; n=14), ranked by progression-free survival (PFS) and annotated by response to anti-PD-1 therapy (CB, NCB/LTS, or NCB) and timing of sample collection.
To better understand the relationship between anti-PD1 induced PDOTS cytokine profiles and clinical benefit to ICB, we ranked cytokine profiles from the subset of patients specifically treated with anti-PD-1 therapy by progression-free survival (PFS), annotating each sample (Fig. 7h). PDOTS from patients with the shortest PFS in particular elaborated multiple immune suppressive cytokines/chemokines in addition to CCL19/CXCL13 (Fig. 7h, Supplementary Fig. 10a–c). Several of these immune suppressive cytokines/chemokines induced in PDOTS were also components of the recently reported innate PD-1 resistance (IPRES) gene expression signature (14), which correlated with non-response but missed statistical significance (Supplementary Fig. 10d). Serial PDOTS profiling from an individual patient with thyroid carcinoma and a malignant pleural effusion revealed that induction of multiple granulocytic and monocytic chemoattractants predicted infiltration of these respective myeloid populations following disease progression on anti-PD-1 therapy in vivo (Supplementary Fig. 10e–f). These data highlight the potential of this assay to identify cytokines changes associated with ineffective anti-tumor immune responses in addition to CCL19/CXCL13 induction following ICB. Taken together, these data provide the first demonstration that ex vivo functional immune profiling using MDOTS as well as PDOTS is feasible and recapitulates key features of in vivo response and resistance to ICB.
DISCUSSION
Immune checkpoint blockade (ICB) therapy targeting the PD-1/PD-L1 axis has demonstrated clinical activity against diverse tumor types, in some cases resulting in durable disease control lasting years. The evidence of clinical responses to ICB across tumor types suggests a fundamental role for the immune system in restraining cancer growth. Despite rapidly expanding interest in ICB therapy, the molecular, cellular, and immunologic determinants of response to ICB therapy remain incompletely characterized, and novel therapies and therapeutic combinations are needed to overcome primary resistance to PD-1/PD-L1 blockade. In this study, we describe a novel approach for functional evaluation of ICB using organotypic tumor spheroids grown in collagen hydrogels in a three-dimensional microfluidic culture system. Patient- and murine-derived tumor spheroids (MDOTS/PDOTS) retain relevant tumor infiltrating lymphoid and myeloid sub-populations, and respond to PD-1 blockade in short-term ex vivo culture. Using well-studied syngeneic murine tumor models and patient derived samples, we recapitulate features of ex vivo sensitivity and resistance to PD-1 blockade, providing the first evidence of a functional assay to evaluate and quantify response to PD-1 blockade in a model TME using tumor cells and autologous stromal and immune cells from explanted tumors.
Functional assays evaluate drug effects through one or more methods, including dynamic evaluation of target engagement and pathway activation that can be measured rapidly, avoiding the complications inherent in longer term ex vivo culture (17,39). The ability to measure functional ex vivo responses to PD-1 blockade in MDOTS/PDOTS provides a distinct advantage over organoids and patient-derived xenografts, which take months to develop and lack the native features of the immune TME. Unlike most existing functional assays, the MDOTS/PDOTS platform permits testing of non-cell autonomous processes and tumor-immune interactions, which facilities evaluation of ICB and ICB-based combination therapies. Three-dimensional culture of MDOTS/PDOTS may provide a more realistic environment in which to evaluate tumor-immune interactions (40). Recent studies have begun to evaluate the interaction of multicellular tumor spheroids with immune cells (41–43), highlighting growing interest in evaluating tumor-immune interactions in more relevant culture systems. However, there are several important differences between the MDOTS/PDOTS platform and previously published systems. First, these other model systems evaluated tumor-immune interactions using standard 96 well culture and were not performed in 3D microfluidic culture. Second, these spheroid models are primarily comprised of cultured tumor cells and lack the diversity and complexity of the stromal and immune components evident in PDOTS/MDOTS. The differences in cytokine profiles between CT26 spheroids and CT26 MDOTS highlights the importance and relevance of the native tumor-infiltrating immune cells. Lastly, these studies relied on cultured or engineered T cell lines (43) or T cells from derived from peripheral blood mononuclear cells (PBMCs) from healthy donors that are phenotypically and functionally distinct from tumor-infiltrating T cells with absent or low expression of immune checkpoints (44).
The PDOTS/MDOTS platform was also used to profile secreted cytokines and nominate novel effectors of ICB. PDOTS cytokine profiling represents an acute, dynamic readout for response to PD-1 blockade used in the present study to identify candidate early effector cytokines. We hypothesized that evaluation of specimens from autochthonous tumors and metastases may differ substantially from our well-defined murine models and MDOTS, particularly with respect to their differential tumor evolution as well as infiltration of CD8+ effector memory T cells, which are enriched in earlier stage tumors relative to more advanced stage III/IV tumors (45). Given our relatively small cohort of primarily metastatic (stage IV) samples from mixed tumor types with heterogeneous timing of response, we instead focused our efforts on the identification of shared acute cytokine responses rather than Live/Dead imaging. Further work is required to evaluate the relationship between short-term cytokine production and clinical response, and larger prospective studies are also needed to assess prediction of clinical benefit by PDOTS cytokine profiling and/or ex vivo killing.
From our analysis of both treated and untreated melanoma patients, there is a clear relationship between CCL19/CXCL13 production and immune infiltration as inferred from transcriptomic data. CCL19 and CXCL13 facilitate recruitment of naïve T cells and dendritic (CCR7+) and specific B and T cell subsets (CXCR5+) to the sites of chronic inflammation to coordinate both humoral and cell-mediated adaptive anti-tumor immune responses (46–48). While reported sources of CCL19 and CXCL13 include stromal, immune, and tumor cells, a recent high-resolution study using single-cell RNA-seq of melanoma specimens demonstrated CCL19 expression in cancer-associated fibroblasts (CAFs) and CXCL13 with CD8+ exhausted T cells (46). CCL19 is also strongly expressed in lymph node high endothelial venules (49), and another recent study demonstrated endothelial-specific induction of CCL19 and CXCL13 in cancer-associated endothelial cells following combined anti-angiogenic therapy with PD-L1 blockade (50). Interestingly, both CCL19 and CXCL13 have established roles in lymphoid neogenesis (51). Importantly, CCL19 and CXCL13 have no direct cytotoxic activity and act indirectly to recruit additional immune cells to the TME, and indeed our studies in PDOTS identified co-induction of cytokines/chemokines that recruit immune suppressive cells as a potential mediator if intrinsic resistance to PD-1 blockade. These data also highlight important differences with implantable syngeneic models, which form tumors over a matter of weeks and may especially rely on tumor-draining lymph nodes as site of T cell priming (Supplementary Fig. 8g), in contrast to autochthonous tumors or metastatic implants that form over months to years representing sources of chronic antigen exposure. Future studies will be required to address the role of CCL19/CXCL13 in both patient derived and murine tumor models, both within the tumor bed and the draining lymph nodes.
Despite the benefits and advantages of MDOTS/PDOTS profiling, there are several important limitations. First, MDOTS/PDOTS profiling is presently limited to pre-existing tumor-infiltrating immune cells, and does not reflect recruitment of additional immune cells into the model TME. More sophisticated adaptations are under development using this microfluidic platform to model anti-PD1 induced recruitment of immune cells (e.g. following CCL19/CXCL13 induction), evaluate T cell priming and/or recruitment using naïve immune cells derived from peripheral blood, tumor-draining lymph nodes, antigen-experienced tumor-infiltrating T cell clones, or even genetically-engineered T cells. Second, comprehensive evaluation of all stromal and immune components is currently not feasible. Immunofluorescence imaging can evaluate 2–4 cell types at a time, but more comprehensive and sensitive methods (e.g. single cell RNA-seq) will be required to provide a more complete picture of the evolution of the immune contexture of MDOTS/PDOTS over time. Third, the importance and impact of biophysical (e.g. interstitial flow, shear stress) and metabolic parameters (e.g. glucose levels, hypoxia) on PD-1 response and resistance remain unclear. For example, previous studies evaluating hypoxia in multicellular tumor spheroids have demonstrated that spheroids measuring <200 μm in size do not exhibit signs of hypoxia, whereas growth to diameters of approximately 200–300 μm results in formation of central hypoxic regions in the spheroid core and in spheroids 500 μm diameter or larger, central necrosis develops (52,53). We suspect hypoxia is of minimal significance early in culture as all spheroids range in size from 40–100 μm at Day 0 of ex vivo spheroid culture, and based on our evaluation of MDOTS (Supplementary Fig. 2h), average spheroid size was <200 μm on Day 6 with no evidence of central necrosis, although focal hypoxia may develop if MDOTS/PDOTS expand to a size to >200–300 μm. Future studies to model the impact of hypoxia and other physiologic parameters on MDOTS/PDOTS immune responses may provide important additional biologic insights.
Innate resistance to PD-1 blockade remains a major challenge, and strategies to overcome immune suppression and render the TME more permissive to T cell infiltration and function are under pre-clinical and clinical evaluation (3,9,10). After successfully demonstrating ex vivo response to PD-1 blockade in MDOTS, we explored novel therapeutic combinations to enhance response to PD-1 blockade by targeting immune suppressive cytokine signaling. TANK Binding Kinase-1 (TBK1) is a Ser/Thr kinase involved in innate immune signaling, autophagy, and xenophagy. TBK1 promotes growth of KRAS-mutant lung adenocarcinoma cells via autocrine cytokine signaling involving CCL5 and IL-6 (24), which can be disrupted with momelotinib (CYT387), a multitargeted inhibitor of JAK1/2, TBK1 and its homolog IKKε. Genetic deletion of TBK1 (33) and IKKε (34) in T cells promotes T cell activation, suggesting that pharmacologic inhibition of TBK1/IKKε could enhance response to PD-1 blockade by inhibiting immune suppressive cytokines/chemokines and re-activating immune cells. We confirmed that TBK1/IKKε inhibition enhanced response to PD-1 blockade in MDOTS with an immunosuppressive microenvironment, and established in vivo efficacy of this combination. Importantly, recent orthogonal data from an in vivo CRISPR screen identified TBK1 as one of several genes in the NF-κB pathway whose deletion enhanced response to PD-1 blockade (54), providing additional evidence supporting TBK1 as a target to enhance response to PD-1 blockade. Another recent report demonstrated that TBK1 deletion specifically in dendritic cells also enhanced responsiveness to ICB therapy (55). Thus, additional studies will be required to fully understand the impact of TBK1/IKKε inhibition on the immune TME. Regardless, these findings demonstrate concordance between ex vivo MDOTS responses and in vivo responses, thereby highlighting the immense potential of ex vivo testing in MDOTS to identify effective therapeutic combinations to overcome intrinsic resistance to PD-1 blockade.
In summary, we provide here the first evidence that short-term organotypic tumor spheroid culture can model response to PD-1 blockade and identify specific interventions that counteract resistance, a much needed advance given the challenges of testing the massive number of potential combination therapies in vivo. This system and future adaptations may thus provide a functional precision medicine approach to drive clinical-translational efforts to develop novel combinations and ultimately personalized immunotherapy.
MATERIALS AND METHODS
Patient Samples
A cohort of patients (Table S3a) treated at Massachusetts General Hospital and Dana-Farber Cancer Institute was assembled for PDOTS profiling and culture between 8/2015 and 8/2016. Informed consent was obtained from all subjects. Tumor samples were collected and analyzed according to Dana-Farber/Harvard Cancer Center IRB-approved protocols. These studies were conducted according to the Declaration of Helsinki and approved by the MGH and DFCI IRBs. De-identified archival matched plasma and tissue samples from the MGH Melanoma Tissue Bank were obtained from patients pre- or on-treatment with anti-PD-1 therapy as indicated. Mutational analysis performed on clinically validated next-generation sequencing (NGS) platforms at MGH and DFCI. Clinical benefit (CB) was defined as overall survival (OS) >360 days and progression-free survival (PFS) >180 days. No clinical benefit (NCB) was defined as OS <360 days. A third sub-group was defined as no clinical benefit, but long-term survival (NCB, but LTS) defined as OS >360 days + PFS <180 days. These designations were adapted from Van Allen et al. (56). OS and PFS data are shown in Supplementary Fig. 9d, Supplementary Fig. 10a–c, and Table S3d using both PDOTS samples and RNA-seq data unless otherwise indicated.
Syngeneic Murine Models
All animal experiments were performed in compliance with established ethical regulations and were approved by the Dana-Farber Animal Care and Use Committee. MC38 murine colon adenocarcinoma cells were generously provided by Dr. Gordon Freeman (DFCI) received under an MTA from Dr. Jeffrey Schlom of NCI (Bethesda, MD). MC38 cells were acquired from Dr. Freeman in 2015. B16F10 melanoma cells, CT26 colon carcinoma cells, Lewis lung carcinoma (LLC) cells, and EMT-6 breast mammary carcinoma cells were purchased from ATCC (2015). GL261-luc2 cells were obtained from Perkin Elmer (obtained in 2013). Cells were expanded, tested free for mycoplasma by PCR as part of Rodent Cell Line Examination And Report (CLEAR) panel testing (Charles River Laboratories). Independent mycoplasma testing of EMT-6 was performed (IDEXX, last tested 4/2017) without formal viral pathogen testing. Murine pathogen testing performed on MC38 (last tested 12/2015), CT26 (last tested 5/2014), LLC (last tested 5/2014), B16F10 (last tested 8/2014), and GL261-luc2 (last tested 1/2016) by Rodent CLEAR panel testing (Charles River Laboratories). Thawed cells were cultured for up to three passages in DMEM (MC38, B16F10, and GL261) or RPMI 1640 (CT26, LLC) supplemented with 10% heat-inactivated FBS at 37°C in a humidified incubator maintained at 5% CO2. Cell counts were performed prior to implantation by both hemocytometer & Invitrogen Countess Cell Counter. MC38, CT26, B16F10, and LLC cells (5 × 105 cells/mouse in 100μL), re-suspended in sterile PBS (Ca+, Mg+ free), were injected into 8 week old female C57BL/6 albino mice or Balb/c (Jackson) and tumors were collected 2–3 weeks post-implantation or on reaching 2000 mm3 in size (or if there were any humane reason, including decreased BW >15% for 1 week or moribund) and MDOTS were prepared as described below. Implantation of CT26 colon carcinoma cells was performed using BALB/c mice in identical fashion. GL261-luc2 cells (Perkin Elmer) were grown in DMEM with 100μg/mL G418. The tumors were harvested 3 weeks after cell implantation orthotopically in the brain, as described (29). For in vivo treatment studies, mice were randomized (using the deterministic method) and then injected with 10mg/kg isotype control IgG (clone 2A3, BioXCell) or rat-anti-mouse PD-1 (clone RMP1-14, BioXCell) every 3 days × 7 doses and tumor volume was measured as shown (up to Day 22). Investigators were not blinded to treatment groups. Tumor volume (TV) was monitored on a weekly basis after the initial tumor volume is about 100mm3. TV was measured twice weekly during the exponential tumor growth phase, and body weight was monitored on a weekly basis after implantation. For CT26/Balb-C responder/non-responder studies, studies were performed as above and tumors were harvested for MDOTS preparation at the indicated time points. Treatment with anti-PD-1 clone RMP 1–14 (10 mg/kg) was performed twice weekly (Monday/Thursday) starting on Day 5 for 3 weeks total (6 total treatments).
Spheroid Preparation and Microfluidic Culture
Fresh tumor specimens (murine and human patients) were received in media (DMEM) on ice and minced in a 10cm dish (on ice) using sterile forceps and scalpel. Minced tumor was resuspended in DMEM (4.5 mM glucose, 100 mM Na pyruvate, 1:100 penicillin-streptomycin) (Corning CellGro, Manassas, VA) +10% FBS (Gemini Bio-Products, West Sacramento, CA), 100 U/mL collagenase type IV (Life Technologies, Carlsbad, CA), and 15 mM HEPES (Life Technologies, Carlsbad, CA), except for CT26 tumors that were prepared in RPMI. Samples were pelleted and resuspended in 10–20 mL media. Red blood cells were removed from visibly bloody samples using RBC Lysis Buffer (Boston Bio-Products, Ashland, MA). Samples were pelleted and then resuspended in fresh DMEM +10% FBS and strained over 100 μm filter and 40 μm filters to generate S1 (>100 μm), S2 (40–100 μm), and S3 (<40 μm) spheroid fractions, which were subsequently maintained in in ultra low-attachment tissue culture plates. S2 fractions were used for ex vivo culture. An aliquot of the S2 fraction was pelleted and resuspended in type I rat tail collagen (Corning, Corning, NY) at a concentration of 2.5 mg/mL following addition of 10× PBS with phenol red with pH adjusted using NaOH. pH 7.0–7.5 confirmed using PANPEHA Whatman paper (Sigma-Aldrich, St. Louis, MO). The spheroid-collagen mixture was then injected into the center gel region of the 3D microfluidic culture device. Collagen hydrogels containing PDOTS/MDOTS were hydrated with media with or without indicated therapeutic monoclonal antibodies after 30 minutes at 37°C. MDOTS were treated with isotype control IgG (10 μg/mL, clone 2A3) or anti-PD-1 (0.1, 1.0, 10 μg/mL, clone RMP1-14). Monoclonal rat-anti-mouse-CCL2 (5 μg/mL, clone 123616, R&D Systems) was used for CCL2 neutralization in MDOTS. PDOTS were treated with anti-PD-1 (pembrolizumab, 250 μg/mL), anti-CTLA-4 (ipilimumab, 50 μg/mL), or combination (250 μg/mL pembrolizumab + 50 μg/mL ipilimumab). For indicated PDOTS studies, anti-human PD-L1 (atezolizumab) was used at 600 μg/mL (1:100) alongside recombinant human interferon-gamma (200 ng/mL) obtained from R&D Systems (285-IF). Doses were selected (1:100 dilutions of stock concentrations used clinically) to correspond to reported peak plasma concentrations of each drug following administration of 10mg/kg (FDA CDER application). In select experiments, PDOTS were treated with InVivoMAb human IgG isotype control (BioXCell). For spheroid cultures lacking immune cells, MC38 or CT26 cells (1 × 106) were seeded in low attachment conditions for 24 hours and were filtered (as above). The S2 fraction was pelleted and resuspended in collagen (as above) prior to microfluidic culture (see Supplementary Video 3).
Flow Cytometric Immune Profiling of Murine Tumors and MDOTS
Tumors from MC38 and B16F10 syngeneic murine models were procured as described above. Cells were incubated for 20 minutes in the dark at room temperature using the Zombie NIR Fixable Viability Kit (Biolegend, 423105) at a dilution of 1:500 in PBS. FcR were blocked by incubation with the anti-mouse CD16/CD32 clone 2.4G2 blocking Ab (Fisher Scientific) for 15 min. at 4C at a 1:100 dilution in flow cytometry staining buffer (PBS + 5% FBS). Cell surface staining was performed by incubation for 20 min. at 4C using the following Abs diluted in flow cytometry staining buffer (total staining volume of 100 ul): Lymphocyte Staining Panel - CD45 AF488 (BioLegend 103122), CD25 PE (BioLegend 101904), CD19 PE-Dazzle (115554), CD49b PE-Cy7 (BioLegend, 108922), CD3 BV421 (BioLegend, 100228), CD8 BV510 (BioLegend 100752), CD4 BV786 (Fisher Scientific # BDB563331); Myeloid Staining Panel - F4/80 AF488 (BioLegend # 123120), MHCII PE (BioLegend # 107608), CD11c BV421 (BioLegend # 117330), Ly6G BV510 (BioLegend # 127633), CD11b BV650 (BioLegend # 101239), Ly6C BV711 (BioLegend # 128037), CD45 BV786 (Fisher Scientific # BDB564225), CD19 APC-Cy7 (BioLegend # 115530), and CD49b APC-Cy7 (BioLegend # 108920) were included with the myeloid staining panel to be used as a dump channel along with the dead cells as determined by the Zombie NIR viability stain. After cell surface staining, cells were fixed by incubating in 200 μL IC Fixation Buffer (eBioscience # 00-8222-49) for 10 min. at room temp. Cells were washed and resuspended in flow cytometry staining buffer and read the following day on a BD LSR Fortessa flow cytometer. Data were analyzed using FlowJo software version 10.0.8.
Flow Cytometric Immune Profiling of PDOTS and Human Tumor Samples
Cells were incubated with Live/Dead Fixable Yellow Dead Cell Stain Kit (Life Technologies, Carlsbad, CA) for 8 min in the dark at room temperature or Live/Dead Fixable Zombie NIR™ (Biolegend, San Diego, CA) for 5 min in the dark at room temperature in FACS buffer (PBS +2% FBS) at a ratio of 250 μL L/D 1X dilution per 100 mg of original sample weight. Surface marker and intracellular staining were performed according to the manufacturer’s protocol (eBioscience, San Diego, CA). FcR were blocked prior to surface antibody staining using Human FcR Blocking Reagent (Miltenyi, San Diego, CA). Cells were fixed in 1% PBS +2% FBS and washed prior to analysis on a BD LSRFortessa with FACSDiva software (BD Biosciences, San Jose, CA). Data were analyzed using FlowJo (Ashland, OR) software version 10.0.8. Cell viability was determined by negative live/dead staining. Antibodies were specific for the following human markers: CD3 (HIT3a; UCHT1), CD8 (RPA-T8), CD14 (M5E2; MphiP9), CD45 (HI30), CD56 (B159), CCR7 (150503), EpCAM (EBA-1), HLA-DR (G46-6), PD-1 (EH12.1), and IgG1 isotype control (MOPC-21) from BD Biosciences (San Jose, CA); CD3 (UCHT1), CD4 (RPA-T4), CD14 (M5E2), CD15 (W6D3), CD16 (3G8), CD19 (HIB19), CD25 (BC96), CD33 (WM53), CD38 (HIT2), CD40L (24–31), CD45 (HI30), CD45RA (HI100), CD45RO (UCHL1), CD56 (HCD56; 5.1H11), CD66b (G10F5), CD69 (FN50), CD123 (6H6), CD163 (GHI/61), CTLA-4 (L3D10), CXCR5 (J252D4), EpCAM (9C4), Ki-67 (Ki-67), PD-1 (EH12.2H7), PD-L1 (29E.2A3), PD-L2 (24F.10C12), TIM-3 (F38-2E2), IgG2a isotype control (MOPC-173), IgG2b isotype control (MPC-11), and IgG1 isotype control (MOPC-21) from BioLegend (San Diego, CA); Pan-cytokeratin (C11) and PD-L1 (E1L3N) from Cell Signaling Technologies (Danvers, MA); CD45 (2D1), FOXP3 (236A/E7), and IL-10 (236A/E7) from Affymetrix/eBioscience (San Diego, CA). Four-way flow sorting of immune cells (CD45+), tumor cells (CD45−CD31−CD90−), cancer-associated fibroblasts (CD45−CD31−CD90+), and endothelial cells (CD45−CD31+CD144+) was conducted on a BD Aria II SORP with gates set using single stain controls and manual compensation using the following antibodies: CD31−APC (Biolegend, 303115), CD45−BV711 Biolegend, 304050), CD90-PE/Cy7 (Biolegend, 328123), and CD144-PE (Biolegend, 348505). Cells were sorted into cold PBS and stored on ice before mRNA extraction using established techniques. For PBMC studies, human PBMCs isolated from healthy donors were cultured for 45min either in plain media (DMEM + 10% FBS + pen/strep) or dissociation media (plain media + 100U/mL collagenase type IV + 50μg/mL DNase I) and analyzed by flow cytometry for quantification of surface antigen expression, as previously described (44).
Microfluidic Device Design and Fabrication
Microfluidic device design and fabrication was performed as described(23), with modifications of device dimensions to accommodate larger volumes of media. MDOTS were also evaluated using DAX-1 3D cell culture chip (AIM Biotech, Singapore), for select studies.
Live/Dead Staining
Dual labeling was performed by loading microfluidic device with Nexcelom ViaStain™ AO/PI Staining Solution (Nexcelom, CS2-0106). Following incubation with the dyes (20 minutes at room temperature in the dark), images were captured on a Nikon Eclipse 80i fluorescence microscope equipped with Z-stack (Prior) and CoolSNAP CCD camera (Roper Scientific). Image capture and analysis was performed using NIS-Elements AR software package. Image deconvolution was done using AutoQuant Module. Whole device images were achieved by stitching in multiple captures. Live and dead cell quantitation was performed by measuring total cell area of each dye. Three different laboratories verified immune-mediated cell death of MC38 MDOTS following PD-1 blockade. To inhibit CD8+ T cell cytotoxicity, CT26 MDOTS were treated with 10 μg/mL anti-CD8α Ab (clone 53-6.72, BioXCell). Inter-tumoral and intra-tumoral heterogeneity experiments were performed using CT26 implanted tumors, as described above. MDOTS were prepared using separate pieces of a larger tumor alongside MDOTS prepared from a smaller implanted tumor. MDOTS were processed, treated, and profiled as described above. Immunofluorescence for CD8+ T cells was performed as described below.
Immunofluorescence and Time-Lapse Imaging
For immunofluorescence studies PDOTS and MDOTS were washed with PBS, and blocked with FcR blocking reagent (PDOTS – Miltenyi, Cambridge, MA, MDOTS – BioLegend, San Diego, CA) for 30 min at room temp. Directly conjugated antibodies for PDOTS were CD326 EpCAM-PE (clone 9C4), CD45-AlexaFluor-488 (HI30), CD8a-AlexaFluor488 (RPA-T8); for MDOTS – CD45-AlexaFluor488 or 647 (30-F11), CD8a-PE (53–6.7) (BioLegend, San Diego, CA). Antibodies were diluted 1:50 in 10ug/mL solution of Hoechst 33342 (Thermo Fisher Scientific, Waltham, MA) in PBS and loaded into microfluidic devices for 1-hour incubation at room temp in the dark. Spheroids were washed twice with PBS with 0.1%Tween20 followed by PBS. For viability assessment microfluidic devices were loaded with 1:1000 solution of calcein AM (Thermo Fisher Scientific, Waltham, MA) in PBS. Images were captured on a Nikon Eclipse 80i fluorescence microscope equipped with Z-stack (Prior) and CoolSNAP CCD camera (Roper Scientific). Image capture and analysis was performed using NIS-Elements AR software package. Brightfield time-lapse images were captured with a 10× NA 0.3 objective and cooled CCD camera (Orca R2, Hamamatsu) in a humidified, temperature-controlled chamber. Illumination was with a CoolLED pe-100 white light LED. Time lapse imaging of several fields of view over time was controlled by NIS-Elements software of a Prior motorized stage along with the LED and camera.
Cytokine Profiling
Two multiplex assays were performed utilizing a bead-based immunoassay approach, the Bio-Plex Pro™ Human Cytokine 40-plex Assay (Cat# 171AK99MR2) and Bio-Plex Pro™ Mouse Cytokine Panel 1, 23-plex (Cat# M60009RDPD) on a Bio-plex 200 system (Cat# 171000201). MDOTS/PDOTS conditioned media concentration levels [pg/mL] of each protein were derived from 5-parameter curve fitting models. Fold changes relative to the MDOTS/PDOTS control were calculated and plotted as log2FC. Lower and upper limits of quantitation (LLOQ/ULOQ) were imputed from standard curves for cytokines above or below detection. Conditioned media from PDOTS were assayed neat. Mouse CCL19 (Abcam, ab100729) and mouse CXCL13 (R&D Systems, MCX130) ELISAs were used according to manufacturer instructions.
RNA-seq
Freshly isolated patient tumor samples (from patients consented to DF/HCC protocol 11–181) were snap frozen in liquid nitrogen and RNA was collected using the Qiagen RNeasy Mini kit. RNA libraries were prepared from 250 ng RNA per sample using standard Illumina protocols. RNA sequencing was performed at the Broad Institute (Illumina HiSeq2000) and the Wistar Institute (Illumina NextSeq 500). RNA samples were ribo-zero treated and then subjected to library prep. using Epicentre’s ScriptSeq Complete Gold kit. Quality check was done on the Bioanalyzer using the High Sensitivity DNA kit and quantification was carried out using KAPA Quantification kit. Raw RNA-Seq data (BAM files) read counts were summarized by featureCounts (57) with parameters that only paired-ended, not chimeric and well mapped (mapping quality >= 20) reads are counted. Then normalization was applied to eliminate bias from sequencing depths and gene lengths by edgeR (58), thus RPKMs (Reads Per Kilobase of transcript per Million mapped reads) (Table S3c).
Quantitative Real-Time PCR
Analysis of expression levels of CCL19 and CXCL13 by quantitative reverse transcription polymerase chain reaction (qRT-PCR) was performed using tissue samples obtained from patients with metastatic melanoma. Samples were selected from patients treated with anti-PD-1 therapy with available tissue pre- and on- treatment. All patients provided written consent to DF/HCC protocol 11–181 (Melanoma Tissue and Blood Collection). Tissue samples were snap frozen in liquid nitrogen and processed to yield RNA, which was stored at −80°C after extraction. Normal lymph node tissue was used as a positive control for expression of CCL19 and CXCL13. Primers were designed for CXCL13 (Fwd: 5′-GAGGCAGATGGAACTTGAGC-3′, Rev: 5′ CTGGGGATCTTCGAATGCTA-3′) and CCL19 (Fwd: 5′-CCAACTCTGAGTGGCACCAA-3′, Rev: 5′-TGAACACTACAGCAGGCACC-3′). Total RNA was extracted via the QIAGEN RNeasy Mini Kit after being ground with the QIAGEN TissueRuptor. The extraction process was automated via the QIAGEN QIAcube. RNA was stored in 1.5 mL RNAse-free EP tubes and then quantified using the QIAGEN Qubit. cDNA was reverse-transcribed from RNA using the Invitrogen Superscript VILO kit run on an Applied Biosystems 2720 Thermo Cycler and then stored in 1.5 mL Ep tubes in a −40°C freezer until later use. Samples were run on a Roche LightCycler 96 using Bio-Rad’s SsoAdvanced Universal SYBR Green Supermix in a total volume of 20 μL per well. β-tubulin (Fwd: 5′-CGCAGAAGAGGAGGAGGATT-3′, Rev: 5′-GAGGAAAGGGGCAGTTGAGT-3′) were employed to normalize the expression of target genes. 4 runs were performed. RT-PCR was performed in triplicate and values averaged. Effect of PD-1 blockade depicted as log2 fold change (L2FC) in CCL19 or CXCL13 expression (normalized to β-tubulin) from on-treatment samples relative to pre-treatment samples. For analysis of CCL19/CXCL13 expression in sorted cell populations (Supplementary Fig. 8d), qRT-PCR was performed as previously described (24) using the following primers: and CXCL13 (Fwd: 5′-CTCTGCTTCTCATGCTGCTG-3′, Rev: 5-TGAGGGTCCACACACACAAT-3′) and CCL19 (Fwd: 5-ATCCCTGGGTACATCGTGAG-3′, Rev: 5′- GCTTCATCTTGGCTGAGGTC-3′), using 36B4 (Fwd: 5′-CAGATTGGCTACCCAACTGTT-3′, Rev: 5′- GGAAGGTGTAATCCGTCTCCAC-3′) to normalize gene expression.
Immunohistochemistry
Immunohistochemistry staining was performed on 4μm formalin-fixed paraffin-embedded sections. MC38 tumor staining was performed as previously described (59) using anti-CD45 (BD Bioscience, 550539) and anti-CD8 (Synaptic System, 361 003) antibodies employing a citrate buffer pressure cooker for antigen retrieval. For human tumor staining, all procedures were done on the automated Ventana Discovery Ultra staining system. Sections were first deparaffinized using EZ prep solution and antigen retrieval was achieved using Cell Conditioning solution 1. Sections were blocked with Discovery Inhibitor (all from Ventana). Sections were incubated with primary antibodies for 16 minutes for population markers and 12 hours for CXCL13 and CCL19 then washed and incubated with OmniMap anti-Mouse or anti-Rabbit conjugated with horseradish peroxidase (HRP) (Ventana, cat# 760-4310 and 760-4311) for additional 16 minutes. Discovery Purple or OmiMAP DAB chromogen kits (cat# 760-229 or 760-159) was then applied to generate a color reaction. Slides were then counterstained with hematoxylin II followed by bluing reagent (Ventana, cat# 790-2208 and cat# 760-2037). Primary antibodies used for staining were: anti-CCL19 (RD Systems, cat# MAB361-100; 1:200) and anti-CXCL13 (Abcam, cat# ab112521; 1:150), CD31 (Cell Marque, cat# 131M-94; 1:500); αSMA (Abcam, cat# ab5694; 1:400).
Source Data
For TCGA analysis, raw RNA-Seq data (BAM files) of TCGA SKCM samples was downloaded from Genomic Data Commons and read counts were summarized (featureCounts) and normalized using edgeR, to generate RPKMs. CCL19 and CXCL13, samples were separated into two groups by k-means clustering of RPKM values: high group and low group, value of center of each group was used to label high or low (Table S4a). The survival curves were constructed according to Kaplan-Meier method on these two groups (high; n=205 and low; n=257) and survival was compared between groups using the log-rank (Mantel-Cox) test (α=0.05). Four-way grouping was performed using median cutoff to define high and low expression (Table S4c). Single sample GSEA (ssGSEA) was performed using immune cell signatures as described (37). Multivariate cox regression analysis was performed using SKCM TCGA data (Table S4b).
Synthesis of Compound 1, 5-(4-((4-(4-(oxetan-3-yl)piperazin-1-yl)phenyl)amino)-1,3,5-triazin-2-yl)-2-((tetrahydro-2H-pyran-4-yl)oxy)benzonitrile
Step 1
To a solution of 2,4-dichloro-1,3,5-triazine (9.5 g, 63 mmol) in N,N-dimethylformamide (150 mL) at 0 °C (flushed with argon balloon) was added solution of 4-(4-(oxetan-3-yl)piperazin-1-yl)aniline (14.1 g, 60.2 mmol) in N,N-dimethylformamide (100 mL) over 5 min via cannula and stirred in an ice-bath for 1h. A solution of 40% Methanol/CH2Cl2 (200 mL) was added to the reaction mixture and stirred at room temperature. After 1 h, the solid formed were filtered washed twice with diethyl ether. Solids were collected to provide the first lot of product. To the filtrate, diethyl ether (200 mL) was added and stirred overnight at room temperature. The solids were separated by filtration to provide a second lot of product. Both lots were combined to provide a total yield of 20 g (91 %) of 4-chloro-N-(4-(4-(oxetan-3-yl)piperazin-1-yl)phenyl)-1,3,5-triazin-2-amine which was used without purification. LCMS-ESI+ (m/z): calcd for C16H19ClN6O: 346.1; found: 347.1 (M+H).
Step 2
To a mixture of 4-chloro-N-(4-(4-(oxetan-3-yl)piperazin-1-yl)phenyl)-1,3,5-triazin-2-amine (4.6 g, 13.2 mmol), 2-fluoro-5-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)benzonitrile (3.5 g, 14.5 mmol), Pd(dppf)Cl2CH2Cl2 (1.2 g, 1.6 mmol) and potassium carbonate (3.6 g, 26.4 mmol) under argon was added a mixture of de-gassed solvents (1,2-dimethoxyethane (53 mL)/water (27 mL)) and sonicated until all solids went into solution (~5 min). The mixture was stirred under argon at 100 °C in a heating block for 1 hr. After cooling to room temperature, water (200 mL) was poured into the reaction mixture and the solids were filtered off and washed with diethyl ether (100 mL). The resulting dark brown solids were suspended in Acetonitrile (20 mL) and stirred at reflux (~2 min) and then stirred at room temperature for 2 hrs. To this suspension di-ethyl ether (20 mL) was added and the mixture was stirred at room temperature overnight. Solids were taken by filtration to yield crude dark brown product. In 1 g batches the crude product was suspended in Dichloromethane (150ml) in a separatory funnel. To the suspension Trifluoroacetic acid was added until all solids had gone into solution. Water was added (150 mL) and mixture was shaken vigorously until black precipitates appeared. The black solids were filtered off. To the filtrate, a saturated aqueous solution of NaHCO3 was added slowly to fully neutralize the mixture. No solids precipitated during this process. The organic phase was dried over MgSO4 and evaporated under reduced pressure to yield a bright yellow material (Total yield 3.1 g, 55 % yield). LCMS-ESI+ (m/z): calcd for C23H22FN7O: 431.1; found: 432.2 (M+H)
Step 3
To a solution of Tetrahydro-2H-pyran-4-ol (4.7 mL, 49 mmol) in THF (200 mL) at 0 °C, Potassium t-butoxide (3.8 g, 51 mmol) was added and the reaction mixture was allowed to warm to room temperature. After 30 min, solid 2-fluoro-5-[4-([4-[4-(oxetan-3-yl)piperazin-1-yl]phenyl]amino)-1,3,5-triazin-2-yl]benzonitrile (10 g, 23.2 mmol) was added and stirred overnight at 60 °C. The reaction mixture was then cooled to 0 °C with a water/ice bath and slowly diluted with water (1.2 L) over 30 min and stirred at room temperature for 45 min. The solids formed were filtered and dried to give 5-(4-((4-(4-(oxetan-3-yl)piperazin-1-yl)phenyl)amino)-1,3,5-triazin-2-yl)-2-((tetrahydro-2H-pyran-4-yl) oxy)benzonitrile as a yellow solid (10.6 g, 90% yield) LCMS-ESI+ (m/z): calcd for C28H31N7O3: 513.3; found: 514.5 (M+H) 1H NMR (400 MHz, DMSO-d6) δ 10.23 (d, J = 19.4 Hz, 1H), 8.80 (s, 1H), 8.74 – 8.50 (m, 2H), 7.68 (br, 2H), 7.60 (d, J = 9.0 Hz, 1H), 7.11 (br, 2H), 4.99 (m, 1H), 4.91 – 4.74 (m, 4H), 4.52 (m, 1H), 3.91 (m, 2H), 3.59 (ddd, J = 11.6, 8.5, 3.0 Hz, 2H), 3.80 – 3.30 (m, 4H), 3.30 – 2.90 (m, 4H), 2.09 (m, 2H), 1.87 – 1.64 (m, 2H).
In vitro characterization of Compound 1
Biochemical single point inhibition and IC50 concentrations for TBK1, IKKε (IKBKE), and off-target kinases were determined at ThermoFisher Scientific using their SelectScreen Kinase Profiling Services (Madison, WI, USA) (Table S2). To determine cellular potency, the human colorectal cancer carcinoma cell line HCT116 (ATCC, Manassas, VA) was maintained in T175 flasks in complete RPMI medium; RPMI 1640 supplemented with 10% FBS, 1X Penicillin-Streptomycin solution and 1X MEM (non-essential amino acids). HCT116 cells were grown to 90–95% confluency in T175 flasks containing complete RPMI medium and transfected in bulk using Lipofectamine 2000 (Invitrogen, Carlsbad, CA) with 70 μg of ISG54-luciferase reporter plasmid (Elim Biopharmaceuticals Inc., Hayward, CA). The reporter plasmid contains a luciferase gene expression cassette under the transcriptional regulation of the promoter of the human interferon stimulated gene 54 (ISG54). Transfection of the cells was allowed to take place for 6 hours, after which the cells were harvested by treatment with 0.25% trypsin EDTA (Corning Inc., Corning, NY). Trypsinized cells were added to 384-well poly-d-lysine treated black clear bottom tissue culture assay plates (Greiner Bio-One GmbH, Kremsmünster, Austria) at a density of 20,000 cells/well in 80 μl of complete RPMI medium and incubated overnight. After 16–18 hours post-transfection, the assay plates were washed with PBS (Corning Inc., Corning, NY), followed by addition of 80 μl/well of serum-free RPMI 1640 medium containing 1X Penicillin-Streptomycin solution, 1X MEM and 350 nl of DMSO or titrations of Compound 1. Compound 1 titrations were generated by 1.5-fold dilution steps in two overlapping serial dilution series to generate a 40-point compound dose range. After incubation at 37 °C for 1 hour, the cells were stimulated with Poly(I:C) (InvivoGen, San Diego, CA) at a final concentration of 15 μg/ml in Optimem media (Life Technologies, Rockville, MD). The assay plates were incubated for 5 hours at 37 °C, followed by addition of One-Glo luciferase firefly reagent (Promega, Madison, WI) at 1:1 volume/well and luminescence was measured in an EnVision Multilabel Plate Reader (PerkinElmer, Santa Clara, CA). The EC50 values were calculated from the fit of the dose–response curves to a four-parameter equation. All EC50 values represent geometric mean values of a minimum of four determinations.
Interleukin-2 and interferon gamma analysis
Freshly isolated human CD4+ and CD8+ T cells were obtained from AllCells (Alameda, CA, USA). Cells were spun down and resuspended in serum free X-vivo15 media (Lonza Walkersville, Inc., Chicacgo, IL, USA) supplemented with 5 ng/mL IL-17 and incubated overnight at 37°C. Cells were plated on anti-CD3 coated plates (5 μg/ml OKT3, eBioscience, Dallas, TX, USA, overnight) with 2 μg/mL anti-CD28 (eBioscience, Dallas, TX, USA). Cells are treated in replicate plates with a dose-titration of COMPOUND 1 for 24 hours for IL-2 and 96 hours for IFNγ. IL-2 and IFNγ in the supernatant were measured using single-or multi-plex immunoassay (Mesoscale Discovery, Rockville, MD, USA) Jurkat T cell leukemia cells (clone E61 obtained from ATCC) plated on anti-CD3 coated plates were treated with a dose titration of COMPOUND 1 for 24 hours. IL-2 in the supernatant was measured as described above.
In vivo Compound 1 combination treatments
Combination studies were performed by vivoPharm (Hummelstown, PA, USA). All procedures used in the performance of these studies were carried out in accordance with vivoPharm’s Standard Operating Procedures, with particular reference to US_SOPvP_EF0314 “General Procedures for Efficacy Studies.” CT26 colon carcinoma cells (1 × 106 cells/mouse in 100 μL, passage 2–3) were re-suspended in serum-free DMEM and implanted in the upper right flank of 11–12 week old female Balb/c mice (Charles River Laboratories). Mice were randomized into four groups of 10 using a matched pair distribution method based on tumor size for CT26. Treatment was initiated 12 days post-inoculation with mean tumor volume at start of dosing of 125.85 mm3. Vehicle or Compound 1 (40 mg/kg, to cover protein adjusted (pa) EC90 for ~4 hours and paEC50 for ~12 hours) was administered by oral gavage daily for 26 days and isotype control or a reverse chimera anti-PD-L1 cloned from literature reports and placed into a mouse IgG1 framework(60) (10mg/kg) was administered every 5 days for a total of six doses. Investigators were not blinded to treatment groups. Mice bearing CT26 tumors with exceptional responses to combination therapy with αPD-L1 and Compound 1 were re-implanted with CT26 cells (1×106, lower left flank) and EMT6 cells (0.5×106, upper left flank) to evaluate development of immunologic memory.
Statistical Methods and Data Analysis
All graphs depict mean ± s.d. unless otherwise indicated. Graphs were generated and statistical analysis performed using GraphPad/Prism (v7.0) and R statistical package. Pearson correlation matrix using 21 cell surface markers for MDOTS were calculated with R across tumors and different sized spheroids.
Supplementary Material
STATEMENT OF SIGNIFICANCE.
Resistance to PD-1 blockade remains a challenge for many patients, and biomarkers to guide treatment are lacking. Here we demonstrate feasibility of ex vivo profiling of PD-1 blockade to interrogate the tumor immune microenvironment, develop novel therapeutic combinations, and facilitate precision immuno-oncology efforts.
Acknowledgments
We thank the patients for the use of clinical specimens without which this study would not have been possible. We also thank the clinical and support staff at Massachusetts General Hospital, Brigham & Women’s Hospital, and the Dana-Farber Cancer Institute for their assistance in these efforts. In particular, we thank the clinical research coordinators who were instrumental in procuring and transporting clinical specimens, especially Michael Manos, Phillip Groden, Sarah Garcia, and Shauna Blackmon. We thank Mei Zhang from the Pathology Department at Brigham and Women’s Hospital for immunohistochemical staining. Financial support was provided: by the Robert A. and Renée E. Belfer Foundation; NCI Training Grant T32CA009172-41 (R.W.J.), John R. Svenson Fellowship (R.W.J.), NCI-R01 CA190394-01 (D.A.B.), the Gloria T. Maheu and Heerwagen Family Funds for Lung Cancer Research, (D.A.B.), Rising Tide Foundation (D.A.B), Expect Miracles Foundation (C.P.P.) and the Robert A. and Renée E. Belfer Foundation. NIGMS T32 GM008313 (M.X.H.), NSF GRFP DGE1144152 (M.X.H.), Caja Navarra Foundation, Excellence Doctoral Mobility Grant (M.A.), Association of Friends of the University of Navarra, Doctoral Mobility Grant (M.A.), NIH/NCI P01CA120964 (K.K.W.), 5R01CA163896-04 (K.K.W), 5R01CA140594-07 (K.K.W), 5R01CA122794-10 (K.K.W), 5R01CA166480-04 (K.K.W), the Gross-Loh Family Fund for Lung Cancer Research (K.K.W), Susan Spooner Family Lung Cancer Research Fund at Dana-Farber Cancer Institute (K.K.W), 1R01CA149738-01 (S.P.) and the Elizabeth and Michael Ruane Fund (S.P.). Additional funding provided by NIH grants P01 CA114046, P01 CA025874, P30 CA010815, and R01 CA047159 and by the Dr. Miriam and Sheldon G. Adelson Medical Research Foundation and the Melanoma Research Foundation. The support for Shared Resources used in this study was provided by Cancer Center Support Grant (CCSG) CA010815 (to The Wistar Institute). Additional support from a Stand Up To Cancer-American Cancer Society Lung Cancer Dream Team Translational Research Grant (SU2C-AACR-DT1715). Stand Up to Cancer is a program of the Entertainment Industry Foundation. Research grants are administered by the American Association for Cancer Research, the Scientific Partner of SU2C
Footnotes
The authors declare no competing interests.
References
- 1.Topalian SL, Drake CG, Pardoll DM. Immune checkpoint blockade: a common denominator approach to cancer therapy. Cancer Cell. 2015;27:450–61. doi: 10.1016/j.ccell.2015.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Pardoll DM. The blockade of immune checkpoints in cancer immunotherapy. Nat Rev Cancer. 2012;12:252–64. doi: 10.1038/nrc3239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sharma P, Hu-Lieskovan S, Wargo JA, Ribas A. Primary, Adaptive, and Acquired Resistance to Cancer Immunotherapy. Cell. 2017;168:707–23. doi: 10.1016/j.cell.2017.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ribas A, Hamid O, Daud A, Hodi FS, Wolchok JD, Kefford R, et al. Association of Pembrolizumab With Tumor Response and Survival Among Patients With Advanced Melanoma. JAMA. 2016;315:1600–9. doi: 10.1001/jama.2016.4059. [DOI] [PubMed] [Google Scholar]
- 5.Schadendorf D, Hodi FS, Robert C, Weber JS, Margolin K, Hamid O, et al. Pooled Analysis of Long-Term Survival Data From Phase II and Phase III Trials of Ipilimumab in Unresectable or Metastatic Melanoma. J Clin Oncol. 2015;33:1889–94. doi: 10.1200/JCO.2014.56.2736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Festino L, Botti G, Lorigan P, Masucci GV, Hipp JD, Horak CE, et al. Cancer Treatment with Anti-PD-1/PD-L1 Agents: Is PD-L1 Expression a Biomarker for Patient Selection? Drugs. 2016;76:925–45. doi: 10.1007/s40265-016-0588-x. [DOI] [PubMed] [Google Scholar]
- 7.Larkin J, Hodi FS, Wolchok JD. Combined Nivolumab and Ipilimumab or Monotherapy in Untreated Melanoma. N Engl J Med. 2015;373:1270–1. doi: 10.1056/NEJMc1509660. [DOI] [PubMed] [Google Scholar]
- 8.Postow MA, Chesney J, Pavlick AC, Robert C, Grossmann K, McDermott D, et al. Nivolumab and ipilimumab versus ipilimumab in untreated melanoma. N Engl J Med. 2015;372:2006–17. doi: 10.1056/NEJMoa1414428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.O’Donnell JS, Long GV, Scolyer RA, Teng MW, Smyth MJ. Resistance to PD1/PDL1 checkpoint inhibition. Cancer Treat Rev. 2017;52:71–81. doi: 10.1016/j.ctrv.2016.11.007. [DOI] [PubMed] [Google Scholar]
- 10.Pitt JM, Vetizou M, Daillere R, Roberti MP, Yamazaki T, Routy B, et al. Resistance Mechanisms to Immune-Checkpoint Blockade in Cancer: Tumor-Intrinsic and -Extrinsic Factors. Immunity. 2016;44:1255–69. doi: 10.1016/j.immuni.2016.06.001. [DOI] [PubMed] [Google Scholar]
- 11.Taube JM, Young GD, McMiller TL, Chen S, Salas JT, Pritchard TS, et al. Differential Expression of Immune-Regulatory Genes Associated with PD-L1 Display in Melanoma: Implications for PD-1 Pathway Blockade. Clin Cancer Res. 2015;21:3969–76. doi: 10.1158/1078-0432.CCR-15-0244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Van Allen EM, Miao D, Schilling B, Shukla SA, Blank C, Zimmer L, et al. Genomic correlates of response to CTLA-4 blockade in metastatic melanoma. Science. 2015;350:207–11. doi: 10.1126/science.aad0095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ribas A, Shin DS, Zaretsky J, Frederiksen J, Cornish A, Avramis E, et al. PD-1 Blockade Expands Intratumoral Memory T Cells. Cancer Immunol Res. 2016;4:194–203. doi: 10.1158/2326-6066.CIR-15-0210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hugo W, Zaretsky JM, Sun L, Song C, Moreno BH, Hu-Lieskovan S, et al. Genomic and Transcriptomic Features of Response to Anti-PD-1 Therapy in Metastatic Melanoma. Cell. 2016;165:35–44. doi: 10.1016/j.cell.2016.02.065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Smyth MJ, Ngiow SF, Ribas A, Teng MW. Combination cancer immunotherapies tailored to the tumour microenvironment. Nat Rev Clin Oncol. 2016;13:143–58. doi: 10.1038/nrclinonc.2015.209. [DOI] [PubMed] [Google Scholar]
- 16.Chen PL, Roh W, Reuben A, Cooper ZA, Spencer CN, Prieto PA, et al. Analysis of Immune Signatures in Longitudinal Tumor Samples Yields Insight into Biomarkers of Response and Mechanisms of Resistance to Immune Checkpoint Blockade. Cancer Discov. 2016;6:827–37. doi: 10.1158/2159-8290.CD-15-1545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Friedman AA, Letai A, Fisher DE, Flaherty KT. Precision medicine for cancer with next-generation functional diagnostics. Nat Rev Cancer. 2015;15:747–56. doi: 10.1038/nrc4015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Huang AC, Postow MA, Orlowski RJ, Mick R, Bengsch B, Manne S, et al. T-cell invigoration to tumour burden ratio associated with anti-PD-1 response. Nature. 2017;545:60–5. doi: 10.1038/nature22079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Anderson KG, Stromnes IM, Greenberg PD. Obstacles Posed by the Tumor Microenvironment to T cell Activity: A Case for Synergistic Therapies. Cancer Cell. 2017;31:311–25. doi: 10.1016/j.ccell.2017.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yu M, Bardia A, Aceto N, Bersani F, Madden MW, Donaldson MC, et al. Cancer therapy. Ex vivo culture of circulating breast tumor cells for individualized testing of drug susceptibility. Science. 2014;345:216–20. doi: 10.1126/science.1253533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gao D, Vela I, Sboner A, Iaquinta PJ, Karthaus WR, Gopalan A, et al. Organoid cultures derived from patients with advanced prostate cancer. Cell. 2014;159:176–87. doi: 10.1016/j.cell.2014.08.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zitvogel L, Pitt JM, Daillere R, Smyth MJ, Kroemer G. Mouse models in oncoimmunology. Nat Rev Cancer. 2016;16:759–73. doi: 10.1038/nrc.2016.91. [DOI] [PubMed] [Google Scholar]
- 23.Aref AR, Huang RY, Yu W, Chua KN, Sun W, Tu TY, et al. Screening therapeutic EMT blocking agents in a three-dimensional microenvironment. Integr Biol (Camb) 2013;5:381–9. doi: 10.1039/c2ib20209c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhu Z, Aref AR, Cohoon TJ, Barbie TU, Imamura Y, Yang S, et al. Inhibition of KRAS-driven tumorigenicity by interruption of an autocrine cytokine circuit. Cancer Discov. 2014;4:452–65. doi: 10.1158/2159-8290.CD-13-0646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Peggs KS, Quezada SA, Chambers CA, Korman AJ, Allison JP. Blockade of CTLA-4 on both effector and regulatory T cell compartments contributes to the antitumor activity of anti-CTLA-4 antibodies. J Exp Med. 2009;206:1717–25. doi: 10.1084/jem.20082492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Curran MA, Montalvo W, Yagita H, Allison JP. PD-1 and CTLA-4 combination blockade expands infiltrating T cells and reduces regulatory T and myeloid cells within B16 melanoma tumors. Proc Natl Acad Sci U S A. 2010;107:4275–80. doi: 10.1073/pnas.0915174107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Woo SR, Turnis ME, Goldberg MV, Bankoti J, Selby M, Nirschl CJ, et al. Immune inhibitory molecules LAG-3 and PD-1 synergistically regulate T-cell function to promote tumoral immune escape. Cancer Res. 2012;72:917–27. doi: 10.1158/0008-5472.CAN-11-1620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Duraiswamy J, Kaluza KM, Freeman GJ, Coukos G. Dual blockade of PD-1 and CTLA-4 combined with tumor vaccine effectively restores T-cell rejection function in tumors. Cancer Res. 2013;73:3591–603. doi: 10.1158/0008-5472.CAN-12-4100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Reardon DA, Gokhale PC, Klein SR, Ligon KL, Rodig SJ, Ramkissoon SH, et al. Glioblastoma Eradication Following Immune Checkpoint Blockade in an Orthotopic, Immunocompetent Model. Cancer Immunol Res. 2016;4:124–35. doi: 10.1158/2326-6066.CIR-15-0151. [DOI] [PubMed] [Google Scholar]
- 30.Peng W, Chen JQ, Liu C, Malu S, Creasy C, Tetzlaff MT, et al. Loss of PTEN Promotes Resistance to T Cell-Mediated Immunotherapy. Cancer Discov. 2016;6:202–16. doi: 10.1158/2159-8290.CD-15-0283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Benci JL, Xu B, Qiu Y, Wu TJ, Dada H, Twyman-Saint Victor C, et al. Tumor Interferon Signaling Regulates a Multigenic Resistance Program to Immune Checkpoint Blockade. Cell. 2016;167:1540–54 e12. doi: 10.1016/j.cell.2016.11.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Vanneman M, Dranoff G. Combining immunotherapy and targeted therapies in cancer treatment. Nat Rev Cancer. 2012;12:237–51. doi: 10.1038/nrc3237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yu J, Zhou X, Chang M, Nakaya M, Chang JH, Xiao Y, et al. Regulation of T-cell activation and migration by the kinase TBK1 during neuroinflammation. Nat Commun. 2015;6:6074. doi: 10.1038/ncomms7074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhang J, Feng H, Zhao J, Feldman ER, Chen SY, Yuan W, et al. IkappaB Kinase epsilon Is an NFATc1 Kinase that Inhibits T Cell Immune Response. Cell Rep. 2016;16:405–18. doi: 10.1016/j.celrep.2016.05.083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Nghiem PT, Bhatia S, Lipson EJ, Kudchadkar RR, Miller NJ, Annamalai L, et al. PD-1 Blockade with Pembrolizumab in Advanced Merkel-Cell Carcinoma. N Engl J Med. 2016;374:2542–52. doi: 10.1056/NEJMoa1603702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Forster R, Davalos-Misslitz AC, Rot A. CCR7 and its ligands: balancing immunity and tolerance. Nat Rev Immunol. 2008;8:362–71. doi: 10.1038/nri2297. [DOI] [PubMed] [Google Scholar]
- 37.Bindea G, Mlecnik B, Tosolini M, Kirilovsky A, Waldner M, Obenauf AC, et al. Spatiotemporal dynamics of intratumoral immune cells reveal the immune landscape in human cancer. Immunity. 2013;39:782–95. doi: 10.1016/j.immuni.2013.10.003. [DOI] [PubMed] [Google Scholar]
- 38.Cancer Genome Atlas N. Genomic Classification of Cutaneous Melanoma. Cell. 2015;161:1681–96. doi: 10.1016/j.cell.2015.05.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Letai A. Functional precision cancer medicine-moving beyond pure genomics. Nat Med. 2017;23:1028–35. doi: 10.1038/nm.4389. [DOI] [PubMed] [Google Scholar]
- 40.Hirt C, Papadimitropoulos A, Mele V, Muraro MG, Mengus C, Iezzi G, et al. “In vitro” 3D models of tumor-immune system interaction. Adv Drug Deliv Rev. 2014;79–80:145–54. doi: 10.1016/j.addr.2014.05.003. [DOI] [PubMed] [Google Scholar]
- 41.Chang CH, Wang Y, Li R, Rossi DL, Liu D, Rossi EA, et al. Combination Therapy with Bispecific Antibodies and PD-1 Blockade Enhances the Antitumor Potency of T Cells. Cancer Res. 2017;77:5384–94. doi: 10.1158/0008-5472.CAN-16-3431. [DOI] [PubMed] [Google Scholar]
- 42.Hoffmann TK, Schirlau K, Sonkoly E, Brandau S, Lang S, Pivarcsi A, et al. A novel mechanism for anti-EGFR antibody action involves chemokine-mediated leukocyte infiltration. Int J Cancer. 2009;124:2589–96. doi: 10.1002/ijc.24269. [DOI] [PubMed] [Google Scholar]
- 43.Zboralski D, Hoehlig K, Eulberg D, Froemming A, Vater A. Increasing tumor-infiltrating T cells through inhibition of CXCL12 with NOX-A12 synergizes with PD-1 blockade. Cancer Immunol Res. 2017 doi: 10.1158/2326-6066.CIR-16-0303. [DOI] [PubMed] [Google Scholar]
- 44.Lizotte PH, Jones RE, Keogh L, Ivanova E, Liu H, Awad MM, et al. Fine needle aspirate flow cytometric phenotyping characterizes immunosuppressive nature of the mesothelioma microenvironment. Sci Rep. 2016;6:31745. doi: 10.1038/srep31745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Charoentong P, Finotello F, Angelova M, Mayer C, Efremova M, Rieder D, et al. Pan-cancer Immunogenomic Analyses Reveal Genotype-Immunophenotype Relationships and Predictors of Response to Checkpoint Blockade. Cell Rep. 2017;18:248–62. doi: 10.1016/j.celrep.2016.12.019. [DOI] [PubMed] [Google Scholar]
- 46.Tirosh I, Izar B, Prakadan SM, Wadsworth MH, 2nd, Treacy D, Trombetta JJ, et al. Dissecting the multicellular ecosystem of metastatic melanoma by single-cell RNA-seq. Science. 2016;352:189–96. doi: 10.1126/science.aad0501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Yu H, Shahsafaei A, Dorfman DM. Germinal-center T-helper-cell markers PD-1 and CXCL13 are both expressed by neoplastic cells in angioimmunoblastic T-cell lymphoma. Am J Clin Pathol. 2009;131:33–41. doi: 10.1309/AJCP62WRKERPXDRT. [DOI] [PubMed] [Google Scholar]
- 48.Gu-Trantien C, Loi S, Garaud S, Equeter C, Libin M, de Wind A, et al. CD4(+) follicular helper T cell infiltration predicts breast cancer survival. J Clin Invest. 2013;123:2873–92. doi: 10.1172/JCI67428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Onder L, Danuser R, Scandella E, Firner S, Chai Q, Hehlgans T, et al. Endothelial cell-specific lymphotoxin-beta receptor signaling is critical for lymph node and high endothelial venule formation. J Exp Med. 2013;210:465–73. doi: 10.1084/jem.20121462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Allen E, Jabouille A, Rivera LB, Lodewijckx I, Missiaen R, Steri V, et al. Combined antiangiogenic and anti-PD-L1 therapy stimulates tumor immunity through HEV formation. Sci Transl Med. 2017;9 doi: 10.1126/scitranslmed.aak9679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Pitzalis C, Jones GW, Bombardieri M, Jones SA. Ectopic lymphoid-like structures in infection, cancer and autoimmunity. Nat Rev Immunol. 2014;14:447–62. doi: 10.1038/nri3700. [DOI] [PubMed] [Google Scholar]
- 52.Khaitan D, Chandna S, Arya MB, Dwarakanath BS. Establishment and characterization of multicellular spheroids from a human glioma cell line; Implications for tumor therapy. J Transl Med. 2006;4:12. doi: 10.1186/1479-5876-4-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Mueller-Klieser W, Freyer JP, Sutherland RM. Influence of glucose and oxygen supply conditions on the oxygenation of multicellular spheroids. Br J Cancer. 1986;53:345–53. doi: 10.1038/bjc.1986.58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Manguso RT, Pope HW, Zimmer MD, Brown FD, Yates KB, Miller BC, et al. In vivo CRISPR screening identifies Ptpn2 as a cancer immunotherapy target. Nature. 2017 doi: 10.1038/nature23270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Xiao Y, Zou Q, Xie X, Liu T, Li HS, Jie Z, et al. The kinase TBK1 functions in dendritic cells to regulate T cell homeostasis, autoimmunity, and antitumor immunity. J Exp Med. 2017;214:1493–507. doi: 10.1084/jem.20161524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Van Allen EM, Miao D, Schilling B, Shukla SA, Blank C, Zimmer L, et al. Genomic correlates of response to CTLA-4 blockade in metastatic melanoma. Science. 2015;350:207–11. doi: 10.1126/science.aad0095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Liao Y, Smyth GK, Shi W. FeatureCounts: An efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics. 2014;30:923–30. doi: 10.1093/bioinformatics/btt656. [DOI] [PubMed] [Google Scholar]
- 58.Robinson MD, McCarthy DJ, Smyth GK. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics. 2009;26:139–40. doi: 10.1093/bioinformatics/btp616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Lizotte PH, Ivanova EV, Awad MM, Jones RE, Keogh L, Liu H, et al. Multiparametric profiling of non-small-cell lung cancers reveals distinct immunophenotypes. JCI Insight. 2016;1:e89014. doi: 10.1172/jci.insight.89014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Deng R, Bumbaca D, Pastuskovas CV, Boswell CA, West D, Cowan KJ, et al. Preclinical pharmacokinetics, pharmacodynamics, tissue distribution, and tumor penetration of anti-PD-L1 monoclonal antibody, an immune checkpoint inhibitor. MAbs. 2016;8:593–603. doi: 10.1080/19420862.2015.1136043. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.