

Abstract
Background
LIS1 (aka PAFAH1B1), a critical mediator of neuronal migration in developing brain, is expressed throughout life. However, relatively little is known about LIS1 function in the mature brain. We previously demonstrated LIS1 involvement in the formation and turnover of synaptic protrusions and synapses of young brain, after neuronal migration is complete. Here we examine the requirement for LIS1 to maintain hippocampal circuit function in adulthood.
Methods
Effects of conditional Lis1 inactivation in excitatory pyramidal neurons, starting in juvenile mouse brain, were probed using high-resolution approaches combining mouse genetics, designer receptor (DREADD) technology to specifically manipulate CA1 pyramidal neuron excitatory activity, electrophysiology, hippocampus selective behavioral testing and MRI tractography to examine connectivity of LIS1 deficient neurons.
Results
We found progressive excitatory and inhibitory postsynaptic dysfunction as soon as 10 days following conditional inactivation of Lis1 targeting CA1 pyramidal neurons. Surprisingly, by P60, it also caused CA1 histological disorganization with a selective decline in parvalbumin expressing interneurons and further reduction in inhibitory neurotransmission. Accompanying these changes were behavioral and cognitive deficits that could be rescued by either DREADD-directed specific increases in CA1 excitatory transmission or pharmacological enhancement of GABA transmission. Lagging behind electrophysiological changes was a progressive, selective decline in neural connectivity, affecting hippocampal efferent pathways documented by MRI tractography.
Conclusions
LIS1 supports synaptic function and plasticity of mature CA1 neurons. Post-juvenile loss of LIS1 there disrupts the structure and cellular make-up of the hippocampus, its connectivity with other brain regions, and cognition dependent on hippocampal circuits.
Keywords: LIS1, synaptic excitation-inhibition, synaptic homeostasis, plasticity, DREADD, cognitive behavior
INTRODUCTION
Both during development and in aging, neuronal circuits must maintain stability and integrity but also have great capacity for plasticity (1). An intriguing molecular candidate for supporting synaptic integrity is LIS1, associated with lissencephaly, a neuronal migration disorder affecting the cerebral, hippocampal and cerebellar cortices(2, 3). LIS1 continues to be present in synaptosomal fractions and post-migratory neurons in adulthood. We previously demonstrated an important role for LIS1 in regulating dendritic filopodia and spine dynamics indicative of altered neural plasticity in conventional Lis1+/– mice and in juvenile conditional knockouts that lose LIS1 after neurons have completed migration and initial connectivity (4). Interestingly, LIS1 deficits in specific hippocampal neuron populations significantly alters excitatory synaptic transmission in adult-born Lis1+/– dentate gyrus projection neurons (5) and dendritic spine density and clustering of excitatory synapses on hippocampal CA1 projection neurons that lose LIS1 expression starting at P20 (4). Supporting a role for LIS1 involvement in neuropsychiatric disorders is LIS1 binding to DISC1, which is associated with schizophrenia. Moreover, reduced LIS1 levels were reported in brain of adult schizophrenic patients (6) and in a mouse model expressing inducible mutant hDISC1 (7).
In this study, we examined the effects of inactivating LIS1 expression in post-migratory hippocampal CA1 projection neurons from the late juvenile period through adulthood. We focused on hippocampus because it is particularly sensitive to Lis1 gene dosage (8). Highly selective genetic manipulation of Lis1 in CA1 glutamatergic neurons is achievable using a CaMKIICre driver for which recombination is > 95% selective for CA1 glutamatergic projection neurons starting on postnatal day 20 (P20) (9). Moreover, several classical behavioral tests of hippocampal-dependent cognition, and electrophysiological correlates of learning and memory including LTP are well characterized. We therefore used a high-resolution, targeted approach, combining mouse genetics, viral vector-mediated selective gene transfer, electrophysiology, a powerful technique--designer receptor exclusively activated by designer drug (DREADD) (10), 3 MRI tractography and cognitive tests to probe the effects of post-juvenile loss of LIS1 in hippocampal CA1 projection neurons on synaptic integrity and hippocampal circuit function in adulthood. We show that, distinct from its role in neuronal migration in early brain development, LIS1 acts in mature (post-migratory) CA1 neuron excitatory and inhibitory synaptic transmission, and is necessary for plasticity of the excitatory synaptic response to a major input to these neurons. Moreover, behavioral data show that these functions “at the synapse” have functional relevance to cognitive processes mediated by hippocampal circuits.
METHODS AND MATERIALS
All methods and materials are detailed in Supplemental information
Mice
Mouse protocols were reviewed and approved by the Institutional Animal Care and Use Committee of Weill Cornell Medicine. Lis1flox/flox mice (8) crossed with the CaMKII-Cre (T29-1) driver line (9) inactivated Lis1 in post-migratory hippocampal pyramidal cells starting in CA1 at postnatal day (P) 20 as described in the Supplemental Materials. The day of birth was postnatal day 0 (P0). Throughout, Lis1cko refers to the CaMKIICre;Lis1fl/fl genotype, Cre-negative Lis1fl/+ littermates were the primary control group, while Cre-negative Lis1fl/fl and CaMKIICre;Lis1fl/+ (Cre;Lis1fl/+) mice were separate comparison groups in selected experiments.
Histology
Brains were fixed and processed as described in Supplemental materials and serially sectioned at intervals of 40μm. Standard immunohistochemical methods were used to reveal NeuN, LIS1, parvalbumin (PV), somatostatin (SS), and calretinin (CR). Images were acquired using a compound microscope and processed in Adobe Photoshop to optimize visualization of specific labeling. Apparent interneuron densities were determined by counting cells within a grid of 200 μm2 placed over CA1 at the mid dorsal hippocampal level. Counts were averaged across 3–5 sections with ROI location and section numbers yoked across genotype groups.
Electrophysiology
Transverse brain slices (350 μm thick) were prepared from 30–60-day-old, avertin-anesthetized mice as described in Supplemental materials. Briefly, brains were cut while immersed in a phosphate buffer with relatively high magnesium ion, high glucose, and low calcium ion concentrations, and supplemented with choline. Slices were then transferred to a standard (physiological) artificial cerebral spinal fluid (ACSF), bubbled with 95% O2 and 5% CO2, and incubated at 32°C for 1 h. Recordings were obtained from slices incubating in 34°C ASCF. Miniature postsynaptic currents were isolated by adding 1 μM tetrodotoxin to the bath. CA1 pyramidal cells were clamped at −70 mV. An infrared-DIC microscope (Olympus BX51) with epifluorescence illumination, a charge-coupled device camera, and water immersion lenses were used to identify CA1 pyramidal cells. Whole cell recordings used glass electrodes (7–9 MΩ resistance) containing an intracellular solution (in mM) of 126 potassium-gluconate, 2 KCl, 2 MgCl2, 0.2 EGTA, 10 HEPES, 4 Na2ATP, 0.4 Na2GTP and 0.5% neurobiotin (Invitrogen) (pH 7.25, 295 mOsm/kg). An internal solution for mIPSC recording contained 130 mM CsCl, 4 mM NaCl, 5 mM Na2phosphocreatine, 2 mM MgCl2, 2 mM Na2ATP, 0.6 mM Na3GTP, 1.1 mM EGTA, and 5 mM Hepes (pH 7.3, 295 mOsm/kg).
Excitatory and inhibitory events in the same cell were distinguished by distinct reversal potentials. Membrane voltage was clamped at −70 and 0 mV to isolate EPSCs and IPSCs, respectively. At −70 mV holding potential, the inward sEPSCs exhibited the fast kinetics of AMPA events. At a holding potential of 0 mV, outward currents were mainly sIPSCs, exhibiting relatively slower kinetics of GABA-A events. In some experiments EPSCs were pharmacologically isolated adding 10 μM bicuculline methiodide (BMI) to the bath to block GABAA receptor-mediated inhibitory synaptic currents; while IPSCs were isolated with a cocktail of 50 μM NBQX and 20 μM D-AP5 to block AMPA and NMDA glutamate receptors.
Synaptic responses were evoked by square-pulse stimulation (1~10V, duration 0.1 ms) at 0.05 Hz of Schaffer collaterals. Evoked EPSC amplitude was defined as maximum current measured within 5 msec of the stimulus. LTP was induced with 200 synaptic stimuli at 2 Hz with a 3-min depolarization to 0 mV, then compared 30 min post-stimulation (11, 12). EPSCs were recorded in ACSF with BMI. Paired-pulse ratios (PPRs) of EPSC amplitude were determined across an interstimulus interval (ISI) range of 20–200 msec. Low-pass filtered at 2 kHz and acquired at 5–10 kHz, recordings were analyzed using an Axopatch 700B amplifier and pCLAMP10 software (Molecular Devices). The series resistance (Rs) varied between 4 to 6 MΩ, continuously monitored during recording to document that no re-sealing of the ruptured membrane occurred. Cells in which the Rs or capacitance deviated by >20 % from initial values were excluded from the analysis. Also, cells with Rs > 20 MΩ at any time during the recording were excluded from the analysis.
Behavior
Full descriptions of methods for behavioral assays can be found in Supplemental Methods.
Spontaneous Locomotion
Mice were acclimated to the room for 1 hour, then individually placed in a polycarbonate test chamber (27.3 cm2) equipped with grids of 10×10 infrared beams. Horizontal movement was automatically quantified from beam breaks every 10 min for 2 hrs.
Spontaneous Novel Object or Object-Place Recognition
Methods were adapted from previous studies (13, 14), and fully described in Supplemental Methods. On the first two-days, mice were allowed to acclimate to the white-walled, open arena (65 × 40 × 20 cm) for 10 minutes. On Day 3 two identical objects were placed in predetermined locations on the chamber floor. The mouse was placed at the wall opposite the two objects and then allowed to explore the arena freely for 10 minutes. Novel object or object-place recognition was tested on Day 4. Novel object recognition: - one from Day 3, the other a novel object of equivalent size - were placed in the same locations used on Day 3. Novel object-place recognition: on Day 4 the same objects used on Day 3 were placed in the arena: one at one of the locations used on Day 3, the other in a new location equidistant from the other object. The mouse was allowed to freely explore for 5 minutes. Time spent exploring each object was quantified from video and a discrimination ratio, defined as the proportion of total exploration spent on the novel object or place, was used for analysis.
Fear conditioning
Methods were adapted from previous studies (15, 16). The training apparatus had a shock grid floor placed within a sound-attenuating chamber with a distinctive combination of visuospatial, tactile, and odor cues, defining the “training context”. On Day 1, after 1 min acclimation in this chamber, mice received 5 conditioning trials of a 30 sec, 70 dB tone cue (CS), followed by 1 sec, 0.7-mA shock (US) through the grid floor (inter-trial interval (ITI) ranging from 90–160 s). Behavioral freezing was quantified using a software-automated system (Etho Vision XT 8.5, Noldus Technology). On Day 2, cue conditioning was tested in a novel chamber configuration. Following the 1-min habituation period, 5 tone CS’s were presented. On Day 3 mice were placed back in the training context for 6 min to test the strength of context conditioning. “Percentage time freezing”, the primary outcome measure, was calculated for the habituation period, each tone, and each ITI on Days 1 (training) and 2 (cue test), and for minutes 2–6 of the context conditioning test on Day 3.
Stereotaxic injections
Recombinant AAV vector serotype 2 particles (2 ×109/2 L in PBS) were stereotactically injected bilaterally into the CA1. Mice were tested after 20 days. Only cases with the injection site within dorsal CA1 (confirmed with immunohistochemistry) were included in analysis.
Adenoviral DREADD production
DREADD allows for time and target specific neuronal activation (17). An evolved G-protein coupled receptor (GPCR), hM3Dq, is specifically activated by the synthetic ligand, CNO, which has no physiological action in the absence of the mutant GPCR. The hM3Dq receptor was delivered using an adenoviral vector (AAV2) that requires Cre-recombinase to express its hM3Dq insert (18). Virus stocks (AAV2.loxP.hM3Dq-mCherry) were packaged in a plasmid transfection system, purified with heparin chromatography, and titers determined by qPCR.
Clonazepam treatment
Clonazepam in PBS was administered by intraperitoneal injection in a volume of 0.01ml/kg 30 minutes before the behavioral tests.
Magnetic Resonance and Diffusion Tensor Imaging (MRI and DTI)
Diffusion-weighted MR 7 T Bruker BioSpin scanner images were acquired with 16 isotropically distributed diffusion-encoding directions at b=1000 sec/mm2 and three at b=0 sec/mm2. Image Post-Processing and Tractography was done using the eighth version of Statistical Parametric Mapping (version 8) (SPM) (19), implemented with MATLAB (Mathworks, Inc., Natick, MA). Using B0 images, tissues were segmented into cerebrospinal fluid, white matter (WM) and gray matter (GM). Manual editing was used when in-image tissue contrast was insufficient for accurate automated segmentation. Tissue was segregated according to the IBASPM mouse atlas, and the GM was parcellated into 148 different ROIs ((20–22)), The segmented gray and white matter masks established the interface boundary and seeding areas for Bayesian formulation for fiber tracking (23). Tracts were seeded in voxels lining the gray-white matter boundary. Fibers were tracked through white matter regions, terminating when the fiber reached a different tissue layer of gray matter or CSF, the edge of an image volume, or when the angle between subsequent steps exceeded π/3.
Statistical analyses
All data were collected and analyzed with the operator blinded to genotype. At least three independent sets of animals were used for each experiment. Statistical analyses, including numbers of observations, are summarized in figures or figure legends and fully described in Supplemental Material. significance threshold was set p<0.05. Error bars indicate standard error of the mean (S.E.M.)
RESULTS
Post-juvenile loss of LIS1 in CA1 affects hippocampal lamination and interneuron density
In the Lis1cko model used here, Lis1 inactivation in CaMKII-expressing CA1 neurons starts around postnatal day (P) 20, a time when LTP can be induced in hippocampal excitatory circuits (9, 24–26), At this time point, gross hippocampal morphology and PV+ interneuron density were not different between Lis1fl/+ and Lis1cko brains (Sup Fig S1). At P30, LIS1 protein was reduced in CA1 of Lis1cko mice as shown with anti-LIS1 immunohistochemistry (Sup Fig S2A, B) and western blot (Sup Fig S2E), while LIS1 in the dentate gyrus (DG) was spared (Sup Fig S2C,D). We have previously shown that neuritic spine density of CA1 pyramidal neurons in the P30 Lis1cko is also reduced (4). By P60 Lis1cko mice showed a significantly thinner CA1 pyramidal layer (Fig 1A, B), accompanied by cell displacement into the stratum oriens, and a small gene dose-dependent decrease in hippocampal size (area)(Sup Fig S3). We also performed a systematic quantification of the apparent densities of PV+, CR+ and SS+ interneurons within CA1. For region of interest (ROI) we used a subregion within dorsal CA1 that could be defined independently of staining using gross anatomical landmarks and equated across cases (see Supplemental Methods). We found a significant decrease in the density of PV+ neurons in Lis1cko animals (Fig 1C–E), with no change in CR+ and SS+ interneuron densities (Fig 1E; Sup Fig S4).
Figure 1. Reduced apparent density of parvalbumin-positive (PV+) interneurons and disorganization of the CA1 in adult Lis1cko mice.
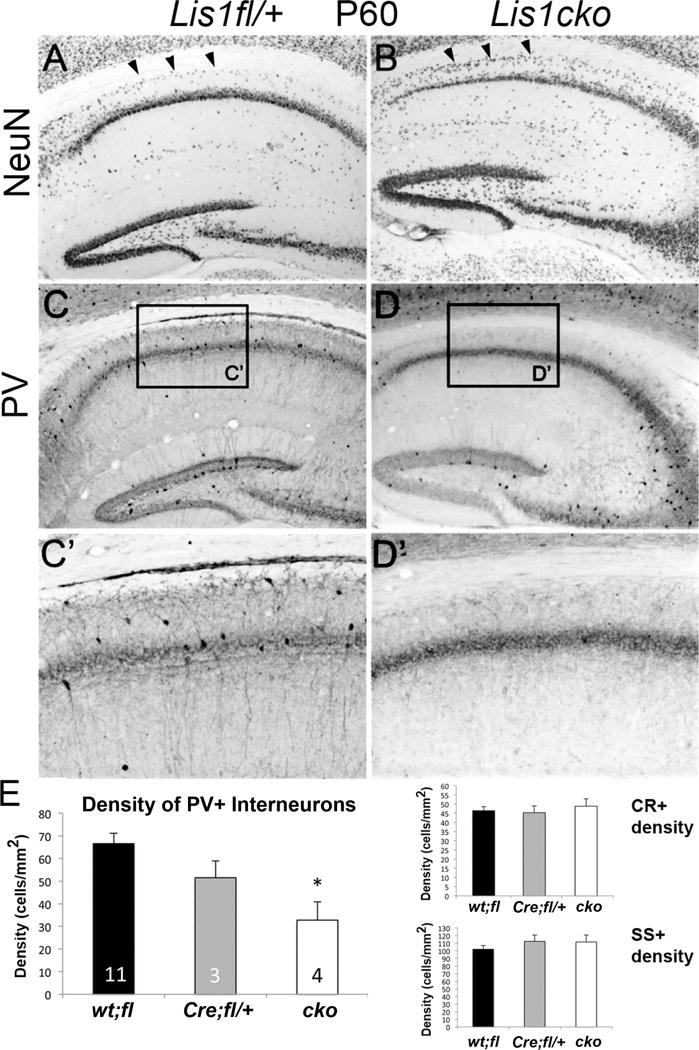
(A, B) Brain sections immunostained for NeuN. Relative to hippocampi from Lis1fl/+ littermates (A), Lis1cko hippocampi appeared to have a thinner pyramidal cell layer in the CA1 region and increased cell density in the stratum oriens (arrowheads). (C,D) PV immunohistochemistry in CA1. Lis1cko mice showed a decrease in PV+ neuron cell body density in CA1. (C’, D’) Higher-magnification of boxes in C and D. Unlike the thinning of the pyramidal layer NeuN+ neuronal nuclei, the PV+ band reflecting neurite staining is of similar width between Lis1fl/+ and Lis1cko CA1 fields. (E) CA1 interneuron densities from brains stained for PV+, SS+ and CR+ were compared across genotype groups using a MANOVA co-varying for hippocampal ROI area. This showed a decrease in the PV+ interneuron density across genotypes (Left panel; F[1,2,14]=5.0, p<0.05); PV+ cell density in Lis1cko mice was lower than in other genotypes (p < 0.05, K-matrix post-hoc; no difference between Cre;fl/+ and wt:fl). CR+ and SS+ interneuron densities were not different across Lis1 genotypes (Right panels; F’s[1,2,14] <0.6, p’s >0.55). Number of cases shown in bars on left.
LIS1 loss in juvenile and adult brain impairs synaptic function and plasticity
Whole-cell and patch-clamp recordings of Lis1fl/+, and Lis1cko hippocampal slices (including CaMKIICre;Lis1fl/+ slices in FigS6) compared basic membrane properties, and spontaneous synaptic events as well as evoked responses and LTP in Schaffer collateral input to CA1. At P16, prior to expression of CaMKII-driven Cre, all physiological properties were comparable in control and Lis1cko mice (Sup Fig S5). At P60, CA1 pyramidal neurons from Lis1cko mice showed decreases in the frequencies of miniature (spike-independent) EPSCs and IPSCs, with mPSC amplitudes unchanged (Fig 2A–D). On the other hand, spontaneous EPSCs and IPSCs, which depend more on exocytotic neurotransmitter release, were decreased in both frequency and amplitude, with most of these deficits observed as early as P30 (Sup Fig S6D–I). Taken together, these results are consistent with deficits in synaptic release from at least a subset of glutamatergic and GABAergic inputs to CA1 pyramidal neurons, but may also reflect abnormalities in the post-synaptic compartments of these synapses. Intrinsic membrane properties, excitability, and action potential waveforms were not altered in the Lis1cko hippocampus (Sup Fig S6 A–C).
Figure 2. Electrophysiological characterization of hippocampal CA1 pyramidal neurons 30 to 40 days after Lis1 inactivation.
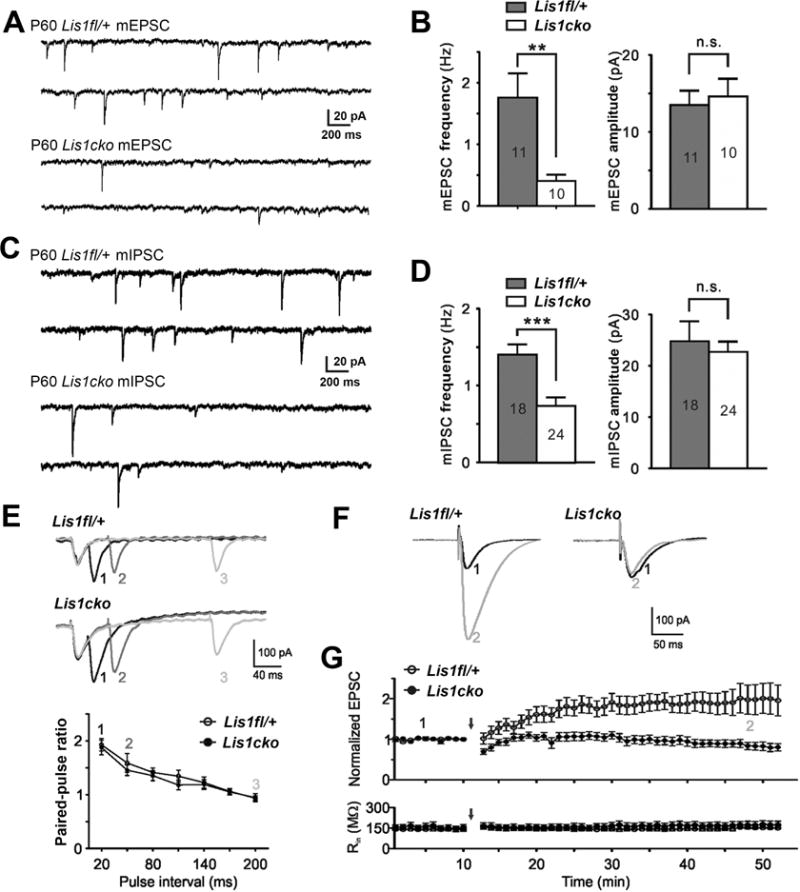
(A) Representative traces of miniature(m) EPSCs in CA1 pyramidal neurons at P60 in Cre-negative Lis1fl/+ and Lis1cko mice. (B) Lis1cko neurons show lower mEPSC frequency (**p<0.01) but not amplitude (n.s.). (C, D) Traces (C) and quantitation (D) show mIPSC frequency is also decreased in Lis1cko mice (***p<0.001). Numbers of neurons recorded are indicated inside bars in B and D. (E) Dependence of paired-pulse ratios (PPR) of evoked EPSC amplitudes in CA1 projection neurons on inter-stimulus time interval (ISI) is similar in Lis1fl/+ (top trace; n=20) and Lis1cko (n=19) mice. (Top) Representative traces from Lis1fl/+ and Lis1cko neurons of EPSCs evoked by successive stimuli delivered with ISIs of 20 (1), 50 (2) and 200 (3) ms. (Bottom) Summary data show no difference between genotypes in PPRs across ISIs. (F–G) LTP expression in CA1 projection neurons in acute hippocampal slices from Lis1fl/+ and Lis1cko P50 mice (n=10 each genotype). (F) Example traces of normalized EPSC amplitudes before (1) and after (2) paired stimulation (classical pairing protocol used for induction; EPSC amplitude 30 min post-induction). (G) EPSC amplitude over time; arrow indicates LTP induction. (Top) Compared to Lis1fl/+ mice, Lis1cko CA1 neurons show reduced LTP expression (p<0.01). (Bottom) Input resistance (Rin) was identical between groups throughout the procedure.
We examined synaptic responses evoked by stimulation of Schaffer collateral inputs to CA1 (Fig 2E), as well as LTP (Fig 2F–G) in this pathway. The paired pulse amplitude ratio (PPR) for evoked EPSCs was not different between Lis1cko and control hippocampal slices (ISI range 20–200 msec; Fig 2E) suggesting intact spike-dependent glutamate release in the CA3 inputs to CA1 pyramidal neurons in the Lis1cko hippocampus. Induction of long-term potentiation (LTP), using a classical pairing protocol (11, 12)(Fig 2F–G), revealed markedly diminished LTP in the Schaeffer collateral-CA1 circuit of Lis1cko mice, while input resistances of recorded neurons remained constant over time (Fig 2G, bottom panel). Together the electrophysiological data indicate that a post-juvenile loss of LIS1 in CA1 projection neurons produces marked deficits in the fundamental components of synaptic transmission at these neurons, and a loss of plasticity in synaptic activity driven by the Schaffer collaterals.
Post-juvenile LIS1 ablation in hippocampal pyramidal neurons alters psychomotor behavior and hippocampus-mediated cognition in adulthood
Deficits in cognitive functions that recruit the hippocampus are evident in schizophrenia, dementias, and other neuropsychiatric disorders (27–29). Notably, hippocampal dysfunction is a common feature of mouse models of these diseases (30–32). We hypothesized that ablation of Lis1 in hippocampal CA1 neurons would affect cognitive and behavioral domains that are sensitive to the integrity of the hippocampus and/or downstream limbic motor circuits. For example, hippocampal inputs to the basal ganglia modulate spontaneous locomotor activity (33). Indeed, in a novel open field, Lis1cko mutant animals showed sustained increased spontaneous locomotor activity (Fig 3A,B, *p<0.01, **p<0.001).
Figure 3. Increased locomotion and deficits in object location and contextual fear conditioning in adult Lis1cko mice.
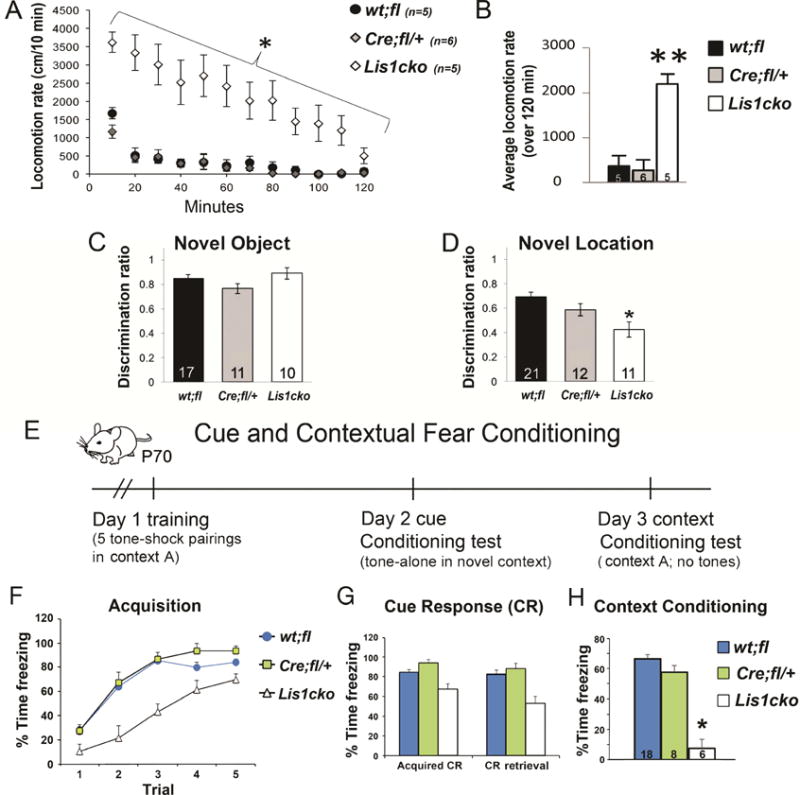
(A) Locomotor activity measured as distance over time. The repeated measures ANOVA showed group differences across genotypes in the habituation rate (genotype × time interaction: F[22,154]=5.0, p<0.001) with Lis1cko showing less habituation than other genotypes (interaction F[1,14]=66.7, *p<0.01, corrected for multiple comparisons). (B) Significant increase in average locomotion in Lis1cko relative to both wt;fl and Cre;fl/+ groups (overall F[2,14]= 23, p < 0.001; Bonferroni-corrected pairwise comparisons; **p’s < 0.001)). (C) Novel object recognition did not differ across genotype groups (F[1,2,34] = 2.2, p > 0.1). (D) In contrast, new location discrimination, which requires spatial encoding and memory mediated by hippocampal circuits (13), was significantly affected (F[1,2,40]. Bonferroni-corrected comparisons showed that Lis1cko mice were impaired relative to wt;fl/+ mice (*p <0.01), with Cre;fl/+ mice showing intermediate place discrimination (Lis1cko < Cre;fl/+, p < 0.08). (E) Time line for the cue/contextual fear conditioning paradigm (protocol in Supplemental Materials). (F) The repeated measures ANOVA covarying for baseline (spontaneous) freezing showed an increase in the freezing across trials (F[1,4,112]=29.1; p < 0.001), reflecting acquisition of the conditioned freezing response (CR). The effect of genotype (F[1,2,28] = 16.0, p<0.01) and the interaction of genotype × trial (F[1,8,112] = 2.1, p< 0.05) were also significant. Lis1cko mice showed less freezing that continues to increase by trial 4 but asymptotes in wt;fl and Cre;fl/+ groups. (G) Cue memory, reflected in the strength of CR to the tone in a novel context on Day 2 (CR Retrieval) relative acquisition on Day 1 (Acquired CR). A repeated measures ANOVA showed a small but significant decrement in the tone CR across days in all groups. There was an effect of genotype (F1,2,28]=14.7, p < 0.01), but no genotype × day interaction (F[1,2,28]= 2.1, n.s.), so that retention and retrieval of the cue CR was not significantly different between genotypes. (H) Retrieval of the CR when placed in Context A on Day 3. There was a highly significant effect of genotype (F[1,2,27] = 37.8, *p<0.001). Bonferonni-corrected comparisons showed Lis1cko mice to be impaired relative to both other genotype groups (p’s < 0.05). N’s appear in the bars.
Hippocampus-mediated cognitive processes in Lis1cko animals were tested in several paradigms. Spontaneous novel object and object-place recognition paradigms measure non-associative object or place memory, exploiting innate tendency to habituate and orient relatively more to novel objects or objects moved to a new location (13, 34). Novel object recognition, a process dependent upon the perirhinal cortex (13), appeared intact in CaMKII-Cre;Lis1fl/fl animals (Fig 3C). On the other hand, novel object-place recognition, mediated by hippocampal circuits (13), was decreased in the CaMKII-Cre;Lis1fl/+ or Lis1cko animals relative to Lis1fl/+ or Lis1fl/fl mice. (Fig 3D, p<0.05). The intermediate place discrimination of heterozygous CaMKII-Cre;Lis1fl/+ mice suggested a LIS1 dose sensitivity for this form of implicit memory.
Pavlovian cue- and context-associational learning and memory was tested in adult (P70) animals (Fig 3E–H). Acquisition of the conditioned freezing response (CR) to the cue was delayed in Lis1cko mice (change from trial (t) 1-t3; Fig 3F, p<0.05), but the relative increase in the CR was similar across groups by trial 5 (p>0.4). Twenty-four hours later, the tone CR was largely retained and retrieved similarly across genotypes (Fig 3G). However, Lis1cko mice showed markedly reduced CR to the training context (Fig 3H, p<0.001). Together these results indicate a deficit in hippocampus-mediated associational context memory in Lis1cko mice.
Rescue of cognitive function in CaMKII-Cre;Lis1cko animals by specific enhancement of CA1 excitatory neuronal activity via hM3Dq-DREADD
We selectively transfected CaMKII-expressing (Lis1–inactivated) CA1 neurons with DREADD hM3Dq receptors, mutated muscarinic GPCRs, which bind a pharmacologically inert ligand clozapine-N-oxide (CNO) (17). The hM3Dq receptor is inactive under physiological conditions, but when bound by CNO acts to depolarize the neuron. Stereotactic injection of Cre-dependent AAV (AAV2.loxP.hM3Dq-mCherry) vector was used to target hM3Dq expression to the CaMKII-Cre+ pyramidal neurons (Fig 4A). At P70, voltage clamp recording in hippocampal slices confirmed that bath application of CNO (20 μM, 5 min) depolarized mCherry positive, but not mCherry negative, pyramidal cells (Fig 4B). Next we tested the behavioral effects of this selective activation of Cre+ (LIS-deficient) hippocampal neurons. P70 mice received CNO 15 minutes prior to either locomotion assessment or fear conditioning (Day 1) (10, 35). DREADD activation did not affect the relative hyperlocomotion of the Lis1cko mice (Fig 4C). Remarkably, Lis1cko-DREADD mice receiving CNO prior to fear conditioning on Day 1 (Fig 4D) showed normalized learning and memory of the context tested after administration of a single CNO dose. Specifically, expression of the context CR at 48 hrs post CNO dose in Lis1cko-DREADD mice was not different from virus-injected Lis1fl/+ controls (Fig 4F), in contrast to the marked deficit observed in the non-treated Lis1cko mice (Fig 3H).
Figure 4. Rescue of hippocampally mediated cognitive function in Lis1cko animals by increasing excitability selectively in CA1 pyramidal neurons (A–F) or with pharmacological augmentation of GABA-mediated synaptic inhibition (G–K).
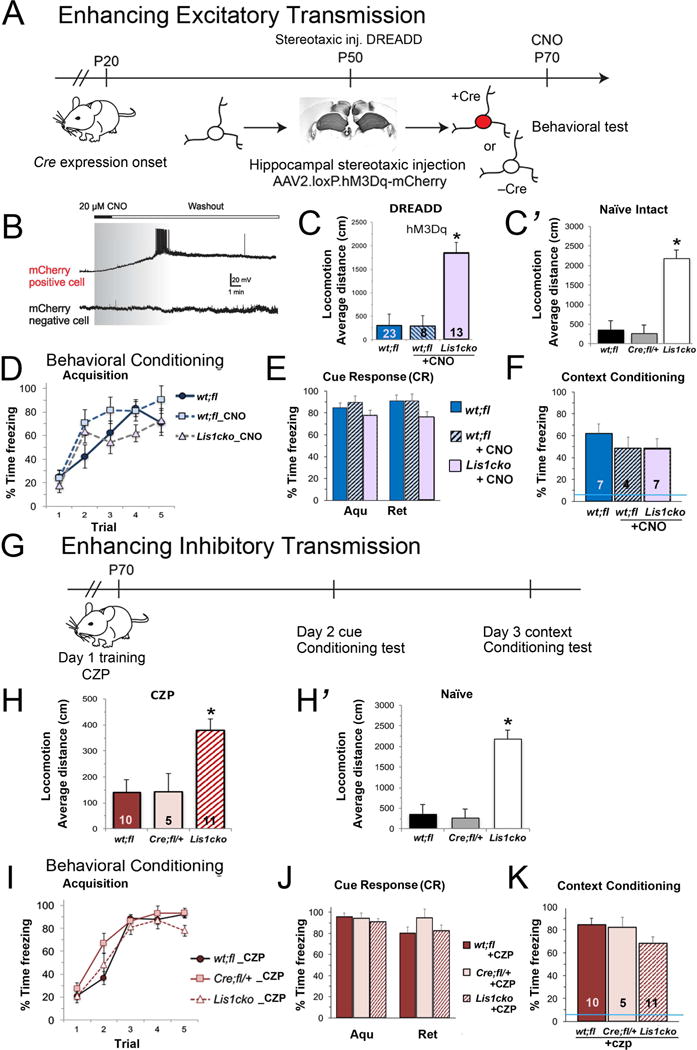
(A) Experimental design for DREADD rescue experiments. In CaMKII-Cre;Lis1fl/fl (Lis1cko) mice, hippocampal glutamatergic neurons lose LIS1 starting at P20. At P50, AAV2.loxP.hM3Dq-mCherry virus was injected bilaterally into hippocampus of Lis1cko or Cre-negative Lis1fl/+ controls. Behavioral testing started on P70. (B) Bath application of CNO to hippocampal P70 slices triggered depolarization and burst firing only in mCherry-positive, hM3Dq-expressing pyramidal cells. (C) Open field locomotion in hM3Dq-transfected mice. CNO (0.3mg/kg of body weight, i.p.) was administered to wt;fl and Lis1cko mice 15 min prior to testing. Untreated hM3Dq wt;fl mice were run in parallel. hM3Dq-expressing Lis1cko mice showed higher locomotor activity than untreated hM3Dq-wt;fl or CNO-treated hM3Dq-wt;fl mice (overall F[2,41] =71.6, *p<0.001; Bonferroni-corrected comparisons p’s < 0.05)(wt;fl vs. wt;fl-CNO, n.s.). (C’) Data from naïve wt;fl/+ and Lis1cko mice (Fig 3B) shown for reference. Lis1cko mice show hyper-locomotion irrespective of the DREADD-CA1 manipulation (Effect sizes: Cohen’s d for Lis1cko versus respective controls 1.6 – 3.7). (D–F) Effects of the DREADD-CA1 manipulation on contextual fear learning and memory. (D) CNO treatment as above on Day 1. The rate of CR acquisition in CNO-treated Lis1cko mice was not different than in untreated or CNO-treated wt;fl controls (genotype × trial interaction F[1,8,56] = 1.8, n.s.). (E) Retrieval memory. The relative ‘retention/retrieval’ of the CR to tone was similar across groups (lack of genotype × day interaction: F[1,2,14] = 2.4, n.s.). (F) Context retrieval on Day 3. In contrast to freezing levels in naïve Lis1cko mice (Fig 3H, blue line in Fig 4F), there were no significant differences across CNO-treated hM3Dq-Lis1cko and hM3Dq-wt;fl/+ CNO-treated and untreated groups. (F[1,2,13] = 0.6, n.s.), indicating rescue of context learning and/or memory in Lis1cko mice. (G–K) Augmentation of GABAA receptor function with clonazepam (CPZ) (0.0625 mg/kg) administered 30 min prior to Day 1 acquisition. (G) Schematic for testing effect of CPZ on cue and context fear conditioning. (H, H’) Locomotion in wt;fl, Cre;fl/+, and Lis1cko mice treated with CPZ (H) and reference data from untreated mice (H’). CZP reduced locomotion generally, but Lis1cko mice remained significantly hyperactive relative to wt;fl (F[1,24] = 15.7, *p<0.005; Bonferroni pairwise comparisons p’s < 0.05; average effect size (Cohen’s d) = 1.6). (I) Under the acute effects of CZP on Day 1, CR acquisition is similar in Lis1cko mice and controls (genotype × trial interaction F[1,8,88] = 0.69, n.s.). (J) Retention of the CR to tone is the same across genotype groups in average strength of the CR (F[1,2,22] = 0.82, n.s.) or the relative change in CR across Days 1 and 2 (genotype × day interaction F[1,2,22] = 1.2, n.s.). (K) Context retrieval test, Day 3. The CR to Context A in Lis1cko mice is restored by CPZ treatment to wt;fl and Cre;fl/+ levels F[1,2,21] = 1.9, n.s.). Blue line indicates performance of naïve Lis1cko mice (from Fig 3H).
Enhanced GABA transmission improves contextual learning and memory in Lis1cko animals
Lis1cko and control mice received the benzodiazepine receptor (BZR) agonist, clonazepam (CZP), a positive allosteric modulator of the GABAA receptor that amplifies GABA signal in part by increasing the affinity of the receptor for GABA (36). A non-sedating dose of CZP was administered 30 min prior to testing locomotion in an open field or prior cue/contextual fear conditioning (Fig 4G). While CZP had an overall suppressing effect on locomotion, Lis1cko mice remained significantly more active than controls (Fig 4H). Remarkably, however, this single low dose of CZP prior to conditioning on Day 1 reversed deficits in acquisition of conditioned fear and context learning and memory (Fig 4I,K).
MRI tractography shows reduced structural connectivity in hippocampal projections of Lis1cko mice
We examined how Lis1cko CA1 pyramidal layer thinning and synaptic dysfunction relates to hippocampal fiber-connections and integrity of the terminal region connections. High-resolution images obtained with a specialized structural and diffusion MRI protocol were processed to produce connectome maps. Connection strength between pairs of brain regions was estimated by the number of tracts between the regions normalized by the total number of fibers detected in the same brain (Fig 5; Sup Fig S7) (23, 37, 38). At P30, when LIS1 is absent in pyramidal neurons (Sup fig S2) and electrophysiological changes are evident in the Lis1cko hippocampus (Sup Fig S6), hippocampal connectivity was not different than control mice (Fig 5A,B). However, by P60, the Lis1cko hippocampus showed aberrant connectivity affecting interhemispheric connections, ipsilateral/associational pathways and in pathways connecting hippocampus to subcortical regions (Fig 5A,C). Specifically, there were connectivity losses in tracts between left and right hippocampi, as well as ipsilateral connectivity between the hippocampus and striatum, septal nuclei, interpeduncular nucleus, substantia nigra and inferior colliculus (Fig 5C, Sup Fig S7). Next, connectivity loss selectivity at P60 was tested using the striatum as the seed region, finding significant connectivity reductions between striatum and hypothalamus, thalamus, substantia nigra, medial geniculate, and hippocampus (Fig 5C). However, tracts between these seed regions and amygdala, globus pallidus, superior colliculus, cerebellum and specific brainstem nuclei (Fig 5C, Sup Fig S7) were relatively intact. Considered with the electrophysiological data, we postulate that loss of LIS1 in post-migrational hippocampal pyramidal neurons leads to a weakening of synaptic function in these neurons followed by a progressive loss of inter-regional connectivity.
Figure 5. Structural connectivity in P30 and P60 Lis1cko and control brains.
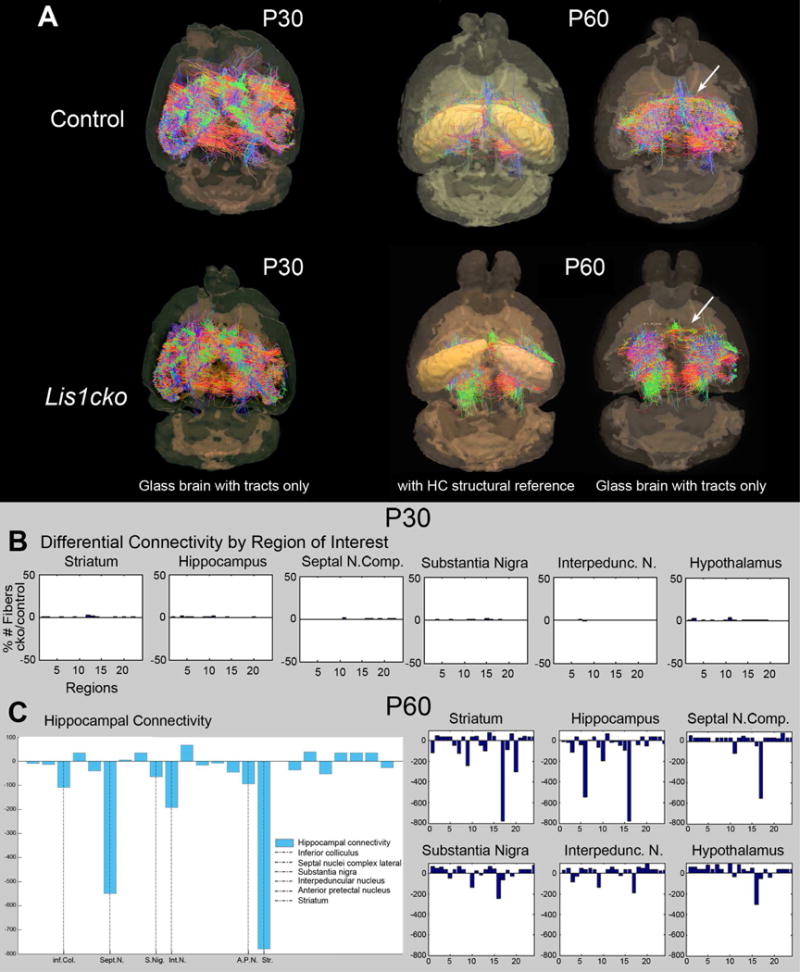
(A) Structural mouse brain networks were obtained from 7T diffusion MRI scans by performing whole-brain tractography. The nodes of the derived network correspond to the cortical and subcortical regions of the labeled mouse brain atlas which was parcellated using SPM5 and SPMMouse. The strength of the edges (connections) in this network is represented as the number of calculated tracts between two nodes, normalized by the total number of tracts found in the brain. (B, C) Connectivity comparisons between Lis1cko and control (Lis1fl/+) mice at P30 in B and at P60 in C. The x-axis in the tractography graphs show numbers assigned to the 24 brain regions to which the axon fibers are tracked from each seed (e.g. hippocampus, septal nucleus, thalamus, etc.), hence the connectivity. The y-axis is the % difference from control in the number of Lis1cko fiber connections between the seed to the region. Regions are labeled by a numerical index going from 1 to 24 (x-axis); the region names are shown as ordered in supplementary Figure S7. The more extensive comparisons of 24 regions of interest at P60 further demonstrate specificity in supplementary Figure S7. (n=3 each genotype).
DISCUSSION
Here, the demonstrated importance of LIS1 in post-migrational neurons to hippocampal synaptic integrity and circuit function is consistent with the hypothesis that LIS1 is needed for CA1 cytoarchitecture maintenance. Moreover, Lis1 inactivation in glutamatergic neurons reduces both excitatory and inhibitory synaptic activity at these neurons. Reduced GABA- mediated inhibitory synaptic responses in mature CA1 pyramidal neurons corresponds with, and is likely driven in part by decreased GABAergic input from PV+ interneurons, as indicated by the selective reduction of PV+ neuron density in Lis1cko mice. These data are consistent with the idea that glutamatergic inputs from LIS1-expressing pyramidal neurons are important for the maintenance of PV+ inhibitory interneurons.
It is plausible that with these marked changes in both excitatory and inhibitory synaptic transmission, accompanied by loss of inputs from PV+ interneurons, there is a disruption of synaptic homeostasis, an aspect of plasticity in which a balance between excitatory and inhibitory transmission at a neuron is maintained. This homeostasis depends on both spontaneous and evoked neurotransmitter release, NMDA and AMPA receptor activity at synapses, and appropriate dendritic morphology and structural stability of synapses (reviewed in (39–41)). Structural stability is, in turn, strengthened by coordinated activation of pre- and post-synaptic voltage-gated and NMDA-receptor-coupled calcium channels, such as that which occurs during sustained excitatory input. Synaptic homeostasis deficits could stem from altered synapse number and post-synaptic spine density, unbalanced excitatory and inhibitory inputs or NMDA/AMPA receptor malfunction. Such abnormalities have been reported in animal models of schizophrenia, autism spectrum disorders, Alzheimer’s disease and other neuropsychiatric disorders, implicating altered synaptic homeostasis as a major factor in these diseases (reviewed in (42)).
Glutamate-mediated spontaneous and miniature excitatory synaptic responses in CA1 pyramidal neurons were weakened following post-juvenile loss of LIS1 function in these neurons. Moreover, there was a marked deficit in activity-dependent plasticity in the excitatory inputs from CA3 (Schaffer collaterals). We postulate that these deficits underlie the impairment in encoding, retention and/or retrieval context-shock association in the Lis1cko mice, as these inputs have been shown to be necessary for object location and context memory (43, 44). By contrast, “baseline” excitatory synaptic responses evoked by direct single-pulse stimulation of the Schaffer collaterals were intact in Lis1cko CA1 neurons. These findings would be consistent with a downregulation of excitatory input from the entorhinal cortex to the distal dendrites of CA1 pyramidal neurons, given that the activity of these inputs can potently regulate plasticity of CA3-CA1 synapses (45). The alteration to other glutamatergic inputs such as the subiculum (46), and/or post-synaptic mechanisms intrinsic to the LIS1-deficient CA1 neurons (47) must be considered.
Importantly, we also found evidence for secondary and/or non-hippocampal-specific effects of the post-juvenile suppression of LIS1 function in CaMKII-expressing neurons. First, both the decrease in PV+ neuron density and the changes in tractography were delayed relative to the relatively rapid (10 days) emergence of the synaptic transmission deficits in CA1 neurons. Based on this timing, we postulate that changes to PV+ neurons and hippocampal projection systems are secondary to the synaptic transmission deficits in CA1 projection neurons. Notably, the post-juvenile loss of LIS1 function in CA1 projection neurons led to abnormal connectivity patterns in subcortical pathways and increased locomotion, a behavior mediated by basal ganglia and brainstem circuits that receive hippocampal input (48, 49). However, augmenting excitability specifically in CA1 projection neurons did not reverse this phenotype. Further elucidation of LIS1 function may yield new insights into the regulation of this highly conserved complex behavior (50, 51), and a greater understanding of psychomotor disturbances in neuropsychiatric disorders (14).
Unexpectedly, systemic augmentation of GABAA receptor dependent inhibitory neurotransmission in Lis1cko mice also improved associational learning/memory of context in the fear conditioning paradigm. It is plausible that both the selective increase in pyramidal neuron excitability produced by the CaMKII-DREADD manipulation or the non-sedating enhancement of GABA inhibitory tone can improve “signal-to-noise” during learning and/or retrieval of the context. In one model, contextual information relayed from sensory cortex is encoded by CA1 neurons via excitatory synaptic inputs. In turn, this excitatory input to CA1 projection neurons regulates output to the amygdala and other nodes in the fear-learning circuit. The CNO-induced depolarization of the DREADD-expressing CA1 neurons would, via a Hebbian mechanism, increase probability of potentiation of excitatory inputs relaying contextual information to these neurons (52, 53). Increasing pyramidal neuron excitability would also amplify the local inhibitory feedback PV+ neurons, thus limiting the temporal window for excitatory feed-forward signal output from the hippocampus. In another model, systemic low-dose CZP would tonically upregulate the effects of GABA at GABAA receptors on CA1 neurons, raising the activation threshold and biasing the drive of these neurons toward the most synchronous excitatory input, possibly that carrying context-related information (54). This calls for further examination of the role(s) of LIS1 in optimizing inhibitory and excitatory synaptic transmission in the hippocampus, and the impact thereof on information/experience-specific circuit activity (55).
This study demonstrates the importance of LIS1 in synaptic transmission and plasticity, not only during development but also in adulthood; and marks LIS1 protein as a potential novel target in the treatment of neuropsychiatric disorders. Further elucidation of LIS1 action in the adult brain will open new avenues toward pharmacological strategies that correct excitatory-inhibitory transmission balance and mitigate cognitive impairment associated with developmental and aging-related brain disorders.
Supplementary Material
Acknowledgments
MRI data were acquired in collaboration with Dr. Henning U. Voss at the Citigroup Biomedical Imaging Center at Weill Cornell Medicine.
FINANCIAL DISCLOSURES
Supported by the following grants from NIH: F32-MH931262 to AS, P01-NS048120 to MER, SShi and HM.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
The authors report no biomedical financial interests or potential conflicts of interest.
AUTHOR CONTRIBUTIONS:
AS, MER, SShi, HM, AR designed the study and experiments;
AS, SSingh, and YT bred, genotyped and provided all mice and tissues used in the experiments;
XZ, AS and SShi performed and evaluated electrophysiological experiments;
HV, EL-C, and AR performed and evaluated the magnetic resonance imaging;
AS, LB, HM and MER performed and evaluated behavioral experiments;
AS, SSingh, YT and MER performed and evaluated immunohistological experiments;
AS, MER, XZ, EL-C, prepared figures;
AS, MER, SShi, AR, HM wrote the manuscript and all authors provided comments and edits.
References
- 1.Emes RD, Grant SG. Evolution of synapse complexity and diversity. Annu Rev Neurosci. 2012;35:111–131. doi: 10.1146/annurev-neuro-062111-150433. [DOI] [PubMed] [Google Scholar]
- 2.Reiner O, Carrozzo R, Shen Y, Wehnert M, Faustinella F, Dobyns WB, et al. Isolation of a Miller-Dieker lissencephaly gene containing G protein beta-subunit-like repeats. Nature. 1993;364:717–721. doi: 10.1038/364717a0. [DOI] [PubMed] [Google Scholar]
- 3.Lo Nigro C, Chong CS, Smith AC, Dobyns WB, Carrozzo R, Ledbetter DH. Point mutations and an intragenic deletion in LIS1, the lissencephaly causative gene in isolated lissencephaly sequence and Miller-Dieker syndrome. Hum Mol Genet. 1997;6:157–164. doi: 10.1093/hmg/6.2.157. [DOI] [PubMed] [Google Scholar]
- 4.Sudarov A, Gooden F, Tseng D, Gan WB, Ross ME. Lis1 controls dynamics of neuronal filopodia and spines to impact synaptogenesis and social behaviour. EMBO Mol Med. 2013;5:591–607. doi: 10.1002/emmm.201202106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hunt RF, Dinday MT, Hindle-Katel W, Baraban SC. LIS1 deficiency promotes dysfunctional synaptic integration of granule cells generated in the developing and adult dentate gyrus. J Neurosci. 2012;32:12862–12875. doi: 10.1523/JNEUROSCI.1286-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lipska BK, Peters T, Hyde TM, Halim N, Horowitz C, Mitkus S, et al. Expression of DISC1 binding partners is reduced in schizophrenia and associated with DISC1 SNPs. Hum Mol Genet. 2006;15:1245–1258. doi: 10.1093/hmg/ddl040. [DOI] [PubMed] [Google Scholar]
- 7.Pletnikov MV, Ayhan Y, Nikolskaia O, Xu Y, Ovanesov MV, Huang H, et al. Inducible expression of mutant human DISC1 in mice is associated with brain and behavioral abnormalities reminiscent of schizophrenia. Mol Psychiatry. 2008;13:173–186. 115. doi: 10.1038/sj.mp.4002079. [DOI] [PubMed] [Google Scholar]
- 8.Hirotsune S, Fleck MW, Gambello MJ, Bix GJ, Chen A, Clark GD, et al. Graded reduction of Pafah1b1 (Lis1) activity results in neuronal migration defects and early embryonic lethality. Nat Genet. 1998;19:333–339. doi: 10.1038/1221. [DOI] [PubMed] [Google Scholar]
- 9.Tsien JZ, Chen DF, Gerber D, Tom C, Mercer EH, Anderson DJ, et al. Subregion- and cell type-restricted gene knockout in mouse brain. Cell. 1996;87:1317–1326. doi: 10.1016/s0092-8674(00)81826-7. [DOI] [PubMed] [Google Scholar]
- 10.Alexander GM, Rogan SC, Abbas AI, Armbruster BN, Pei Y, Allen JA, et al. Remote control of neuronal activity in transgenic mice expressing evolved G protein-coupled receptors. Neuron. 2009;63:27–39. doi: 10.1016/j.neuron.2009.06.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chen G, Kolbeck R, Barde YA, Bonhoeffer T, Kossel A. Relative contribution of endogenous neurotrophins in hippocampal long-term potentiation. J Neurosci. 1999;19:7983–7990. doi: 10.1523/JNEUROSCI.19-18-07983.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mack V, Burnashev N, Kaiser KM, Rozov A, Jensen V, Hvalby O, et al. Conditional restoration of hippocampal synaptic potentiation in Glur-A-deficient mice. Science. 2001;292:2501–2504. doi: 10.1126/science.1059365. [DOI] [PubMed] [Google Scholar]
- 13.Barker GR, Warburton EC. When is the hippocampus involved in recognition memory? J Neurosci. 2011;31:10721–10731. doi: 10.1523/JNEUROSCI.6413-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Perry W, Minassian A, Paulus MP, Young JW, Kincaid MJ, Ferguson EJ, et al. A reverse-translational study of dysfunctional exploration in psychiatric disorders: from mice to men. Archives of general psychiatry. 2009;66:1072–1080. doi: 10.1001/archgenpsychiatry.2009.58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gilani AI, Chohan MO, Inan M, Schobel SA, Chaudhury NH, Paskewitz S, et al. Interneuron precursor transplants in adult hippocampus reverse psychosis-relevant features in a mouse model of hippocampal disinhibition. Proc Natl Acad Sci U S A. 2014;111:7450–7455. doi: 10.1073/pnas.1316488111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fanselow MS. Contextual fear, gestalt memories, and the hippocampus. Behavioural brain research. 2000;110:73–81. doi: 10.1016/s0166-4328(99)00186-2. [DOI] [PubMed] [Google Scholar]
- 17.Armbruster BN, Li X, Pausch MH, Herlitze S, Roth BL. Evolving the lock to fit the key to create a family of G protein-coupled receptors potently activated by an inert ligand. Proc Natl Acad Sci U S A. 2007;104:5163–5168. doi: 10.1073/pnas.0700293104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Huang ZJ, Taniguchi H, He M, Kuhlman S. Cre-dependent adeno-associated virus preparation and delivery for labeling neurons in the mouse brain. Cold Spring Harb Protoc. 2014;2014:190–194. doi: 10.1101/pdb.prot080382. [DOI] [PubMed] [Google Scholar]
- 19.SPM. Statistical Parametric Mapping: The Analysis of Functional Brain Images. Elsevier; 2006. [Google Scholar]
- 20.Sawiak SJ, Wood NI, Williams GB, Morton AJ, Carpenter TA. SPMMouse: A new toolbox for SPM in the animal brain. Proc Int’l Soc Mag Res Med. 2009;1086 [Google Scholar]
- 21.Sawiak SJ, Wood NI, Williams GB, Morton AJ, Carpenter TA. Voxel-based morphometry in the R6/2 transgenic mouse reveals differences between genotypes not seen with manual 2D morphometry. Neurobiology of disease. 2009;33:20–27. doi: 10.1016/j.nbd.2008.09.016. [DOI] [PubMed] [Google Scholar]
- 22.Alemán-Gómez Y, Melie-García L, Valdés-Hernandez P. IBASPM: Toolbox for automatic parcellation of brain structures. IBASPM: Individual Brain Atlases using Statistical Parametric Mapping. 2006 Available at http://wwwthomaskoenigch/Lester/ibaspmhtm.
- 23.Iturria-Medina Y, Perez Fernandez A, Valdes Hernandez P, Garcia Penton L, Canales-Rodriguez EJ, Melie-Garcia L, et al. Automated discrimination of brain pathological state attending to complex structural brain network properties: the shiverer mutant mouse case. PloS one. 2011;6:e19071. doi: 10.1371/journal.pone.0019071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Basu K, Gravel C, Tomioka R, Kaneko T, Tamamaki N, Sik A. Novel strategy to selectively label excitatory and inhibitory neurons in the cerebral cortex of mice. Journal of neuroscience methods. 2008;170:212–219. doi: 10.1016/j.jneumeth.2008.01.016. [DOI] [PubMed] [Google Scholar]
- 25.Alberi L, Liu S, Wang Y, Badie R, Smith-Hicks C, Wu J, et al. Activity-induced Notch signaling in neurons requires Arc/Arg3.1 and is essential for synaptic plasticity in hippocampal networks. Neuron. 2012;69:437–444. doi: 10.1016/j.neuron.2011.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yuste R, Bonhoeffer T. Morphological changes in dendritic spines associated with long-term synaptic plasticity. Annu Rev Neurosci. 2001;24:1071–1089. doi: 10.1146/annurev.neuro.24.1.1071. [DOI] [PubMed] [Google Scholar]
- 27.Daselaar SM, Fleck MS, Dobbins IG, Madden DJ, Cabeza R. Effects of healthy aging on hippocampal and rhinal memory functions: an event-related fMRI study. Cereb Cortex. 2006;16:1771–1782. doi: 10.1093/cercor/bhj112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sperling RA, Dickerson BC, Pihlajamaki M, Vannini P, LaViolette PS, Vitolo OV, et al. Functional alterations in memory networks in early Alzheimer’s disease. Neuromolecular medicine. 2010;12:27–43. doi: 10.1007/s12017-009-8109-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Barch D, Pagliaccio D, Luking K. Mechanisms Underlying Motivational Deficits in Psychopathology: Similarities and Differences in Depression and Schizophrenia. Springer; Berlin Heidelberg: 2015. pp. 1–39. [DOI] [PubMed] [Google Scholar]
- 30.Chao HT, Chen H, Samaco RC, Xue M, Chahrour M, Yoo J, et al. Dysfunction in GABA signalling mediates autism-like stereotypies and Rett syndrome phenotypes. Nature. 2010;468:263–269. doi: 10.1038/nature09582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.McGraw CM, Samaco RC, Zoghbi HY. Adult neural function requires MeCP2. Science. 2011;333:186. doi: 10.1126/science.1206593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bhattacharya A, Kaphzan H, Alvarez-Dieppa AC, Murphy JP, Pierre P, Klann E. Genetic removal of p70 S6 kinase 1 corrects molecular, synaptic, and behavioral phenotypes in fragile X syndrome mice. Neuron. 2012;76:325–337. doi: 10.1016/j.neuron.2012.07.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bast T, Feldon J. Hippocampal modulation of sensorimotor processes. Progress in neurobiology. 2003;70:319–345. doi: 10.1016/s0301-0082(03)00112-6. [DOI] [PubMed] [Google Scholar]
- 34.Ennaceur A. One-trial object recognition in rats and mice: methodological and theoretical issues. Behavioural brain research. 2010;215:244–254. doi: 10.1016/j.bbr.2009.12.036. [DOI] [PubMed] [Google Scholar]
- 35.Krashes MJ, Koda S, Ye C, Rogan SC, Adams AC, Cusher DS, et al. Rapid, reversible activation of AgRP neurons drives feeding behavior in mice. J Clin Invest. 2011;121:1424–1428. doi: 10.1172/JCI46229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zorumski CF, Isenberg KE. Insights into the structure and function of GABA-benzodiazepine receptors: ion channels and psychiatry. Am J Psychiatry. 1991;148:162–173. doi: 10.1176/ajp.148.2.162. [DOI] [PubMed] [Google Scholar]
- 37.Iturria-Medina Y, Canales-Rodriguez EJ, Melie-Garcia L, Valdes-Hernandez PA, Martinez-Montes E, Aleman-Gomez Y, et al. Characterizing brain anatomical connections using diffusion weighted MRI and graph theory. NeuroImage. 2007;36:645–660. doi: 10.1016/j.neuroimage.2007.02.012. [DOI] [PubMed] [Google Scholar]
- 38.Sawiak SJ, Wood NI, Williams GB, Morton AJ, Carpenter TA. Voxel-based morphometry with templates and validation in a mouse model of Huntington’s disease. Magnetic resonance imaging. 2013;31:1522–1531. doi: 10.1016/j.mri.2013.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Davis GW. Homeostatic signaling and the stabilization of neural function. Neuron. 2013;80:718–728. doi: 10.1016/j.neuron.2013.09.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kavalali ET. The mechanisms and functions of spontaneous neurotransmitter release. Nat Rev Neurosci. 2015;16:5–16. doi: 10.1038/nrn3875. [DOI] [PubMed] [Google Scholar]
- 41.Yin J, Yuan Q. Structural homeostasis in the nervous system: a balancing act for wiring plasticity and stability. Front Cell Neurosci. 2014;8:439. doi: 10.3389/fncel.2014.00439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ganguly K, Poo MM. Activity-dependent neural plasticity from bench to bedside. Neuron. 2013;80:729–741. doi: 10.1016/j.neuron.2013.10.028. [DOI] [PubMed] [Google Scholar]
- 43.Kesner RP, Lee I, Gilbert P. A behavioral assessment of hippocampal function based on a subregional analysis. Reviews in the neurosciences. 2004;15:333–351. doi: 10.1515/revneuro.2004.15.5.333. [DOI] [PubMed] [Google Scholar]
- 44.Lopez AJ, Kramar E, Matheos DP, White AO, Kwapis J, Vogel-Ciernia A, et al. Promoter-Specific Effects of DREADD Modulation on Hippocampal Synaptic Plasticity and Memory Formation. J Neurosci. 2016;36:3588–3599. doi: 10.1523/JNEUROSCI.3682-15.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Han EB, Heinemann SF. Distal dendritic inputs control neuronal activity by heterosynaptic potentiation of proximal inputs. J Neurosci. 2013;33:1314–1325. doi: 10.1523/JNEUROSCI.3219-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sun Y, Nguyen AQ, Nguyen JP, Le L, Saur D, Choi J, et al. Cell-type-specific circuit connectivity of hippocampal CA1 revealed through Cre-dependent rabies tracing. Cell Rep. 2014;7:269–280. doi: 10.1016/j.celrep.2014.02.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sanhueza M, Fernandez G. CaMKII: A master functional and structural molecule in synaptic plasticity and memory Novel Mechanisms of Memory. Springer International Publishing; 2015. pp. 43–66. [Google Scholar]
- 48.Grillner S, Wallen P, Saitoh K, Kozlov A, Robertson B. Neural bases of goal-directed locomotion in vertebrates--an overview. Brain Res Rev. 2008;57:2–12. doi: 10.1016/j.brainresrev.2007.06.027. [DOI] [PubMed] [Google Scholar]
- 49.Roseberry TK, Lee AM, Lalive AL, Wilbrecht L, Bonci A, Kreitzer AC. Cell-Type-Specific Control of Brainstem Locomotor Circuits by Basal Ganglia. Cell. 2016;164:526–537. doi: 10.1016/j.cell.2015.12.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Tzschentke TM, Schmidt WJ. Functional relationship among medial prefrontal cortex, nucleus accumbens, and ventral tegmental area in locomotion and reward. Crit Rev Neurobiol. 2000;14:131–142. [PubMed] [Google Scholar]
- 51.Robertson B, Kardamakis A, Capantini L, Perez-Fernandez J, Suryanarayana SM, Wallen P, et al. The lamprey blueprint of the mammalian nervous system. Prog Brain Res. 2014;212:337–349. doi: 10.1016/B978-0-444-63488-7.00016-1. [DOI] [PubMed] [Google Scholar]
- 52.Bliss TV, Collingridge GL. A synaptic model of memory: long-term potentiation in the hippocampus. Nature. 1993;361:31–39. doi: 10.1038/361031a0. [DOI] [PubMed] [Google Scholar]
- 53.Shors TJ, Matzel LD. Long-term potentiation: what’s learning got to do with it? Behav Brain Sci. 1997;20:597–614. doi: 10.1017/s0140525x97001593. discussion 614–555. [DOI] [PubMed] [Google Scholar]
- 54.Sanchez PE, Zhu L, Verret L, Vossel KA, Orr AG, Cirrito JR, et al. Levetiracetam suppresses neuronal network dysfunction and reverses synaptic and cognitive deficits in an Alzheimer’s disease model. Proc Natl Acad Sci U S A. 2012;109:E2895–2903. doi: 10.1073/pnas.1121081109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Chevaleyre V, Piskorowski R. Modulating excitation through plasticity at inhibitory synapses. Front Cell Neurosci. 2014;8:93. doi: 10.3389/fncel.2014.00093. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.