

Abstract
Autosomal dominant leukodystrophy (ADLD) is a rare adult-onset demyelinating disease caused by overexpression of lamin B1, a nuclear lamina filament. Early autonomic dysfunction involving the cardiovascular system before progressive somatic motor dysfunction is a striking feature of most cases of ADLD. In the Plp-FLAG-LMNB1 transgenic mouse model, lamin B1 overexpression in oligodendrocytes elicits somatic motor dysfunction and neuropathology akin to ADLD. Here, we investigate whether Plp-FLAG-LMNB1 mice also develop autonomic cardiovascular dysfunctions before or after somatic motor dysfunction. We find that Plp-FLAG-LMNB1 mice have preserved cardiovascular responses to changes in wake-sleep state and ambient temperature and normal indexes of autonomic modulation at 37-42 weeks of age despite a progressive somatic motor dysfunction, which includes impairments of walking ability (the ability to walk on a narrow path was impaired in 80% of mice at 34-38 weeks of age) and subtle breathing derangements. Only late in the development of the disease phenotype did Plp-FLAG-LMNB1 mice develop a structural deficit of sympathetic noradrenergic fibers, with a 38% decrease of fiber profiles in the kidneys at 44-47 weeks of age. We demonstrate that while the Plp-FLAG-LMNB1 mouse model recapitulates the age-dependent motor dysfunction of ADLD, it does not show signs of early autonomic cardiovascular dysfunction, raising the possibility that oligodendrocyte dysfunction may not be sufficient to cause the full spectrum of clinical features present in ADLD.
Keywords: Sleep, Lamin, Oligodendrocyte, Mice, Leukodystrophy, Gait, Breathing, Heart rate, Arterial Pressure, Sympathetic
Introduction2
Autosomal dominant leukodystrophy (ADLD; OMIM #169500) is a rare adult-onset demyelinating disease, which shows some clinical overlap with multiple sclerosis (Eldridge et al., 1984; Nahhas et al., 2016). ADLD starts clinically in the 4th-5th decades of life with 100% penetrance and no gender variation, and is fatal after a generally slow progression (Nahhas et al., 2016). Autonomic derangements, which may include constipation, bladder symptoms, erectile dysfunction, and orthostatic hypotension (Guaraldi et al., 2011; Terlizzi et al., 2016), are the first symptoms in most patients with ADLD. Orthostatic hypotension is particularly disabling, as it contributes to impair walking ability (Finnsson et al., 2015; Guaraldi et al., 2011; Terlizzi et al., 2016). These symptoms are accompanied by a loss of noradrenergic sympathetic innervation (Guaraldi et al., 2011; Terlizzi et al., 2016), and followed by progressive motor dysfunction, with impaired walking ability because of leg spasticity, weakness, and ataxia, and with pseudobulbar palsy in the terminal stages (Finnsson et al., 2015). Nonetheless, exceptions to this disease progression pattern have been documented: in some atypical cases, significant autonomic dysfunctions either are detected after somatic motor dysfunction, or are not detected at all (Finnsson et al., 2015; Potic et al., 2013; Quattrocolo et al., 1997).
ADLD is caused by genomic mutations, i.e., duplications involving the lamin B1 (LMNB1) gene or deletions upstream of the gene, both leading to overexpression of lamin B1 (Giorgio et al., 2015; Padiath et al., 2006). A key role of oligodendrocytes was supported by the demonstration that Plp-FLAG-LMNB1 transgenic mice (TG) overexpressing lamin B1 selectively in oligodendrocytes develop walking deficits at 40 weeks of age, and progress rapidly (in two months) to a terminal disease stage, when they become moribund (Heng et al., 2013). These mice also showed seizures, brainstem demyelination, and axonal degeneration (Heng et al., 2013). A later study on an independently-derived TG mouse strain established a molecular link between lamin B1 overexpression and lipid synthesis in oligodendrocytes (Rolyan et al., 2015). Autonomic cardiovascular derangements were not investigated by either of these studies.
Considering that autonomic dysfunction is a cardinal symptom in most ADLD cases, we carried out an extensive characterization of autonomic cardiovascular deficits in TG mice in order to better understand the mechanisms underlying ADLD disease pathology. In particular, we tested the hypothesis that autonomic cardiovascular dysfunction precedes the occurrence of somatic motor dysfunction in TG mice, as it is found in most patients with ADLD.
Material and Methods
Animal research ethics
The study protocol complied with the EU Directive 2010/63/EU for animal experiments and was approved by the Committees on the Ethics of Animal Experiments of the University of Bologna and of the Italian Ministry of Education, University, and Research. Surgery was performed under isoflurane anesthesia, and all efforts were made to minimize suffering.
Mice
Experiments were performed on TG (Plp-FLAG-LMNB1) mice (Rolyan et al., 2015) and on their wild-type (WT) control littermates on a FVB/N background. TG mice overexpress the lamin B1 gene under control of the murine proteolipid protein 1 (Plp1) promoter, which is preferentially expressed in oligodendrocytes. A mouse colony was established at the University of Bologna (Department of Veterinary Medical Sciences, Ozzano dell'Emilia, Italy and Department of Biomedical and Neuromotor Sciences, Bologna, Italy) from founder mice from the colony of Prof. Q. Padiath at the Department of Human Genetics, University of Pittsburgh, PA, USA, and from FVB/N mice purchased from Charles River, Italy (Calco, Italy). Breeding was performed with hemizygote x WT mating and TG mice were identified by genotyping with standard polymerase chain reactions (Rolyan et al., 2015). Mice were housed under a 12:12-h light–dark cycle with ambient temperature set at 23°C and free access to water and food (4RF21diet; Mucedola, Settimo Milanese, Italy).
Overview of the experimental protocol
The experiments were performed on mice of 3 age groups. The age groups I and III corresponded to 24-27 and 44-47 (range) weeks of age, respectively. The age group II corresponded to 34-38 weeks of age for walking and water licking tests. A subset of mice of age group II was surgically instrumented after these tests to undergo respiratory phenotyping at 36-40 weeks of age, cardiovascular phenotyping at 37-42 weeks of age, and euthanasia immediately afterwards with tissue explant for immunohistochemistry. Based on the previous reports on TG mice (Heng et al., 2013; Rolyan et al., 2015), somatic motor deficits were expected to be absent for age group I, mild for age group II, and severe for age group III. It was thus expected that TG mice of age group II would be the focus of the work, aimed at investigating whether these mice had autonomic defects before somatic motor defects became severe in age group III. At present, there is no evidence indicating gender variation of ADLD (Nahhas et al., 2016). We performed all experiments on female mice. In addition, we replicated on male mice the tests of walking ability for each age group and of sympathetic innervation for age group III.
Tests of walking ability
Mice were tested for their walking ability on a narrow path (ledge test) and in an open field using a structured assessment (Guyenet et al., 2010). For the ledge test, mice were gently lifted and positioned on the 5-mm ledge of a rectangular cage. Mice were assigned the following scores: 0, walk on the cage ledge without hind paw slips and/or descent into the cage landing on paws; 1, repeated hind-paw slips; 2, ineffective use of the hind legs, or uncoordinated descent into the cage not landing on paws; 3, falls off the ledge and/or refusal to move despite encouragement by gentle nudges. For the open field walking test, mice were left free to walk on a large flat surface and assigned the following scores: 0, coordinated walk with body weight support on all limbs, the abdomen not touching the ground, and even participation of both hind limbs; 1, mild tremor and/or limp; 2, severe tremor and/or limp, lowered pelvis, or feet pointing away from the long axis of the body; 3, difficulty moving forward, with the abdomen dragging along the ground. These evaluations were performed by trained investigators blind to the mouse genotype, and were repeated 3 times, with the final scores computed as the median values of the 3 trial scores for each test. The third trial was omitted for mice that received scores of 0 at the ledge and open field tests for the first and second trial. The sample sizes for the tests of walking ability are reported in Table S1.
Test of water licking
Water licking was recorded during the 12-h dark period in the mouse home cage by measuring the junction potential between the steel spout of a water bottle and an aluminum foil on the cage bottom every time the mouse licked the spout, thereby closing the electrical circuit between the foil and the spout with its body (Hayar et al., 2006). The junction potential voltage signals were manually reviewed to reject artefacts and identify trains of ≥ 4 licks. The inter-lick time interval (onset to onset) was computed for each lick, and its median value over the whole recording was retained for each mouse. The sample sizes for the test of water licking are reported in Table S2.
Surgery
Mice underwent surgery under isoflurane anesthesia (1.8-2.4% in O2) with intra-operative analgesia (Carprofen 0.1 mg subcutaneously, Pfizer Italy, Latina) as previously described in detail (Silvani et al., 2009). Briefly, mice were instrumented with 2 screw electrodes for electroencephalogram (EEG) recordings (frontal-parietal differential lead) and 2 wire electrodes in the neck muscles for electromyogram (EMG) recordings. A calibrated telemetric arterial pressure transducer (TA11-PAC10, DSI, Tilburg, The Netherlands) was implanted subcutaneously, with the catheter tip advanced via the femoral artery until below the renal arteries.
Estral phase determination
Before respiratory phenotyping and again after cardiovascular phenotyping, female mice of age group II underwent vaginal lavage and cytology to discriminate the estral phase following published criteria (Byers et al., 2012) as previously described (Silvani et al., 2015). Vaginal cytology did not evidence significant differences in estral phase between TG and WT either before or after cardiovascular recordings (Table S3).
Assessment of the respiratory phenotype
The EEG, EMG and breathing of mice unrestrained except for the electrode tether were recorded simultaneously inside a modified 2-chamber whole-body plethysmograph (PLY4223, Buxco, Wilmington, NC, USA) for the first 8 hours of the light period with ambient temperature set at 25 °C, as previously described (Silvani et al., 2014b). The mouse chamber was fitted with an electrical swivel (Plastics One, Roanoke, VA, USA) to prevent the electrode tether from twisting, and continuously purged with 1.5 L/min air to prevent CO2 build-up. The system was calibrated dynamically with a 100 μL syringe (Hamilton, Reno, NV, USA) at the termination of each recording. The states of wakefulness, non-rapid-eye-movement sleep (N), and rapid-eye-movement sleep (R) were scored visually with 4-s time resolution based on the EEG and EMG signals using published criteria (Silvani et al., 2009). The breathing analysis was performed on stable sleep episodes lasting ≥ 12 s as previously described in detail (Silvani et al., 2014b). Episodes of wakefulness were excluded from the analysis because of the artefacts caused by body movements on the plethysmography signal.
Total breath duration (TTOT), tidal volume (VT) and minute volume (VE =Vt/TTOT) were calculated, and volumes were expressed per gram body weight. Augmented breaths (sighs) and breathing pauses (apneas) were defined as VT and TTOT > 3 times their average values, respectively, for each mouse and sleep state. The short-term and long-term variability of TTOT was estimated from Poincaré plots of the TTOT of consecutive breaths by computing the standard deviations of TTOT with respect to new axes oriented with (SD1) or orthogonal to (SD2) the line of identity of the Poincaré plots, as previously described (Silvani et al., 2014b). The sample sizes for the assessment of the respiratory phenotype were 10 TG and 11 WT mice.
Assessment of the cardiovascular phenotype
The EEG, EMG, and arterial pressure signals of mice unrestrained except for the electrode tether were recorded simultaneously inside the home cage for 48 hours with ambient temperature at 21.2 ± 0.5 °C, then, after 24 hours' habituation to 31.1 ± 0.2 °C ambient temperature, for 24 hours at the latter temperature, as previously described (Lo Martire et al., 2012). A rotating electrical swivel (Plastics One) and a balanced suspensor arm prevented the electrode tether from twisting and balanced its weight, allowing unhindered movement to the mice. The states of wakefulness, N, and R were scored visually with 4-s time resolution based on the EEG and EMG signals using published criteria (Silvani et al., 2009). For each heartbeat, values of systolic arterial pressure (SAP) and heart period (HP, the reciprocal of heart rate) were computed from the raw arterial pressure signal as previously described (Silvani et al., 2009) and averaged over spontaneous full-blown episodes of wakefulness, N, and R with duration ≥ 60 s (Silvani et al., 2010). Furthermore, the changes (Δ) in SAP and HP during spontaneous transitions between spontaneous episodes of N with duration ≥ 36 s and episodes of either wakefulness or R with duration ≥ 96 s were analyzed as previously described (Lo Martire et al., 2012; Silvani et al., 2014a). HP was analyzed instead of heart rate, which is its reciprocal, because HP is linearly related to the frequency of cardiac sympathetic and parasympathetic discharge (Silvani et al., 2016) and is required for the computation of the indexes pNN8 and BRS (cf. below). For each wake-sleep state, the parasympathetic modulation of HP, the sympathetic modulation of arterial pressure, and the cardiac baroreflex sensitivity were estimated with validated indexes. In particular, the parasympathetic modulation of HP was estimated in the time domain with the index pNN8, which is the % of HP values that differ from the following ones by > 8 ms (Laude et al., 2008b), and in the frequency domain with the index RSA, which quantifies respiratory sinus arrhythmia based on the spectral power of HP in the frequency range 2.5-5.0 Hz (Baudrie et al., 2007). The index of sympathetic modulation of arterial pressure (SYM) was computed based on the spectral power of SAP in the frequency range 0.15–0.6 Hz (Baudrie et al., 2007). The cardiac baroreflex sensitivity (BRS) was estimated with the sequence technique (Laude et al., 2008a). The indexes pNN8, RSA, SYM, and BRS were computed based on beat-to-beat values of SAP and HP in wake-sleep episodes with duration ≥ 60 s, as previously described (Silvani et al., 2010). The sample sizes for the assessment of the cardiovascular phenotype were 8 TG and 10 WT mice.
Analysis of the sympathetic innervation of the heart and kidneys
An immunofluorescence analysis of the noradrenergic sympathetic innervation of the heart and kidneys was performed post-mortem using an antibody against the enzyme tyrosine hydroxylase, which catalyzes the first reaction (hydroxylation of tyrosine to dihydroxyphenylalanine) in the synthesis of noradrenaline. The heart and kidneys were explanted immediately after euthanasia by 4% isoflurane in O2, and fixed by immersion in 4% paraformaldehyde for 24 hours. After fixation, samples were washed 3 times over 8 hours in phosphate buffered saline (PBS) with pH 7.4 at 4 °C, then stored at 4 °C in a PBS-sodium azide with 30% saccharose solution until they sank. Samples were then included in OCT resin (Killik, Bio-Optica, Milano, Italy), frozen at -196 °C in liquid N2, and cut at -20 °C in 15 μm sections. The heart and kidneys were sectioned along their short axes, the former at the level of the ventricles. Sections were transferred on slides pre-coated with 0.1% gelatin (Merck, Darmstadt, Germany), dried, and stored at -80 °C. For further processing, slides were thawed, rehydrated by washing 3 times in PBS 0.01 M for 10 minutes, and incubated for 2 hours at room temperature with 10% normal donkey serum in PBS 0.01 M with 0.5% Triton. Slides were then incubated with the primary antibody against tyrosine hydroxylase raised in sheep (Life Technologies, Temecula, CA; 100 μL diluted 1:5000 in PBS 0.01 M with 1% normal donkey serum and 0.5% Triton) for 24 hours at 4 °C, washed 3 times in PBS 0.01 M to remove excess primary antibody, incubated with the secondary antibody (donkey anti-sheep, Alexa Fluor 594, Thermo Fisher Scientific, Italy; 2 mg/mL, diluted 1:400 in PBS 0.01 M with 1% normal donkey serum and 0.3% Triton) for 90 minutes at room temperature in the dark, and washed three times again in PBS 0.01 M. Slides were finally cover-slipped with glycerol buffer and stored at -20°C in the dark. Fiber profiles positive for tyrosine hydroxylase were counted at fluorescence microscopy by two investigators (CB and VLM) blind to the mouse genotype. The sample sizes and number of histological fields analyzed to quantify sympathetic innervation are detailed in Table S4. Data from the heart of 1 male WT mouse of age group III were excluded from the analysis as an outlier value (estimated 12.6 fiber profiles/mm2 vs. a group average of 3.1 profiles/mm2) possibly due to a technical issue with tissue processing and immunohistochemistry.
Data analysis and statistics
Labview (National Instruments, Austin, TX, USA), Matlab (Mathworks, Natick, MA, USA) and SPSS (SPSS, Armonk, NY, USA) were employed for data acquisition, data analysis, and statistical analysis, respectively. The categorical scores of the tests of walking ability were analyzed with Mann-Whitney tests. The other results were analyzed with mixed-model analysis of variance (general linear model procedure, with Huyn-Feldt correction) and t-tests. For the sake of clarity, since the focus of this study was on differences between TG and WT, we reported on the significance of the main effect (ANOVA) or simple effect (t-test) of TG vs. WT. Statistical significance was at P < 0.05. Data are reported as means ± SD.
Results
Somatic motor deficits
In order to test whether autonomic cardiovascular deficits precede the occurrence of somatic motor deficits in TG mice, we first assessed somatic motor deficits, extending previous studies (Heng et al., 2013; Rolyan et al., 2015). We tested the time course of the impairments of walking ability of TG mice both on a narrow path (ledge test) and in the open field. The results of these tests indicated that the walking ability of TG was significantly impaired compared to that of WT mice even at 24-27 weeks of age in the relatively demanding conditions of the ledge test. Impairments at the ledge test persisted in TG vs. WT mice at 34-38 weeks of age, when these impairments occurred in 80% of TG mice, and at 44-47 weeks of age, when walking ability became impaired also in the less demanding conditions of the open field test (Figure 1).
Figure 1. Walking ability.

The bars show the % of the total number of mice in each group with test scores ranging from 0 (no impairment, white) to 3 (maximal impairment, black) at the ledge test (walk on a narrow path, panels A-B) or at the open field walking test (panels C-D). WT and TG, wild-type and Plp-FLAG-LMNB1 transgenic mice, respectively. ♂, male; ♀, female. Age groups I, II, and II correspond to 24-27, 34-38, and 44-47 weeks of age, respectively (cf. methods). Sample sizes are reported in Table S1. *, P < 0.05, TG vs. WT of the same sex and age group (Mann-Whitney test).
In addition to gait impairment due to leg spasticity and weakness, signs of cerebellar ataxia are a frequent finding in patients with ADLD. Pseudobulbar palsy may also occur in the terminal stages of the disease (Finnsson et al., 2015). A lengthening of the time intervals between the successive licks of a water bottle spout (inter-lick intervals) is a sensitive sign of cerebellar dysfunction in mice (Heck et al., 2008). The same sign may plausibly reflect also somatic motor bulbar defects in mice. We therefore measured the inter-lick interval of TG vs. WT mice. The results of these measurements did not reveal any significant age-related lengthening of the inter-lick interval of TG vs. WT mice from 24-27 to 44-47 weeks of age (Figure 2). However, the inter-lick interval was significantly prolonged in the TG mice with the most severe impairment of walking ability at 44-47 weeks of age compared with that of the TG mice of the same age, which had less severe walking impairments (Figure 2).
Figure 2. Water licking.
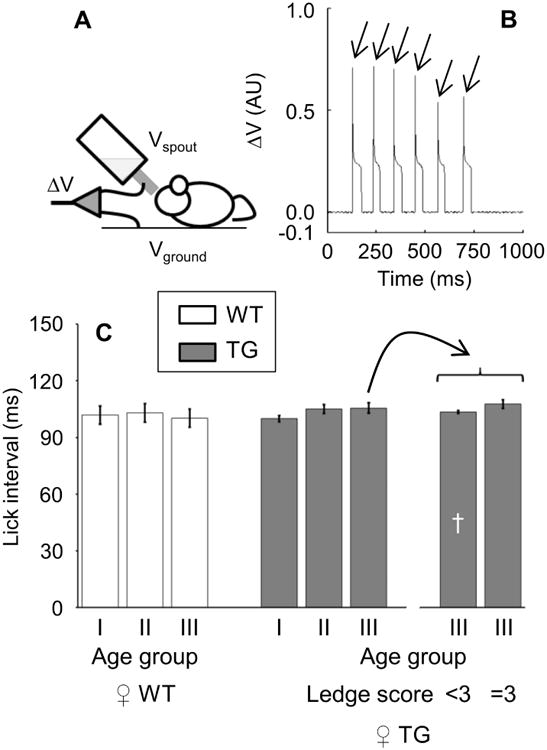
Water licking was measured by recording the voltage difference (ΔV) between the steel spout of a water bottle (Vspout) and an aluminum foil on the ground (Vground) as the electrical circuit was closed by the mouse licking (panel A). Panel B shows an example of ΔV recordings (arbitrary units, AU) in a Plp-FLAG-LMNB1 transgenic (TG) mouse of the 46-week age group. For each mouse, the median inter-lick interval was computed between the onset (arrows) of successive ΔV spikes. Panel C shows values of the inter-lick interval in female (♀) TG (filled bars) and wild-type mice (WT, open bars) as a function of the age group and, for TG mice of age group III, of the occurrence of maximal impairment (score = 3) at the ledge test of walking ability (cf. Figure 1A). Age groups I, II, and II correspond to 24-27, 34-38, and 44-47 weeks of age, respectively (cf. methods). Data are mean ± SD, with sample sizes reported in Table S2. There were no significant differences between TG and WT. †, P < 0.05, TG of age group III with ledge test score < 3 vs. TG of age group III with ledge test score = 3 (t-test).
To our knowledge, a detailed assessment of respiratory motor function has never been performed either in patients with ADLD or in TG mice. However, brainstem (Heng et al., 2013) and spinal cord (Rolyan et al., 2015) neuropathological changes have been well described in TG mice, and may alter breathing by affecting respiratory control centers or respiratory motor neurons, respectively. To better assess the extent of somatic motor dysfunction in TG, we therefore performed a careful assessment of their respiratory activity by means of whole-body plethysmography. This assessment was restricted to sleep states (N and R), scored objectively based on EEG and EMG recordings, to avoid breathing artifacts due to mouse movements during wakefulness. The recordings were performed on mice at 36-40 weeks of age, i.e., intermediate between that of the youngest mice in which we found impairments at the ledge test (24-27 weeks of age) and that of mice with the most severe impairments of walking ability and slowing of the inter-lick interval (44-47 weeks of age). These recordings revealed subtle alterations of breathing in TG mice at 36-40 weeks of age, which consisted of greater tidal volume during N (Figure 3A) and greater short-term variability of the breathing period during N and R (Figure 3E) compared with age-matched WT mice.
Figure 3. Breathing during sleep.
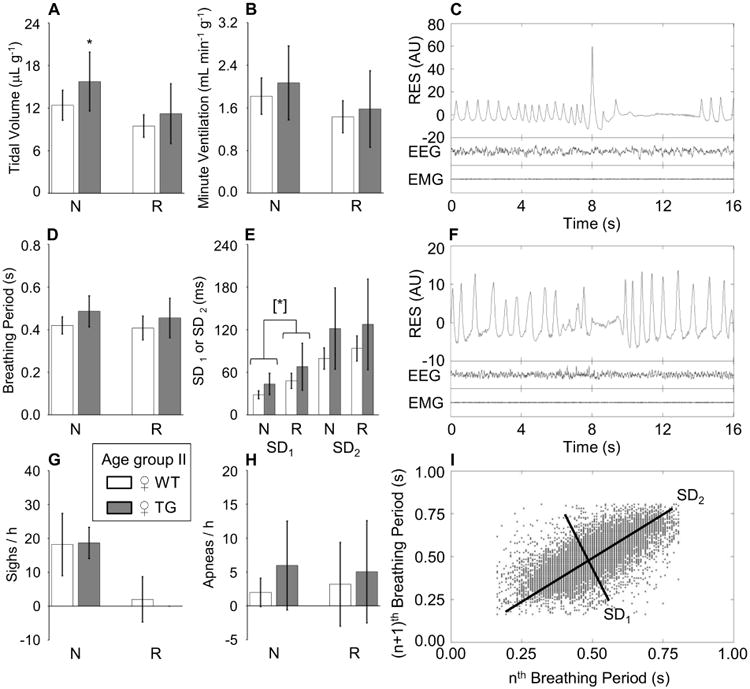
Panel A and panel B: tidal volume and minute ventilation, respectively, per unit body weight. SD1 and SD2 (panel E): standard deviations of the breathing period around a new set of axes oriented with (SD1) or orthogonal to (SD2) the line of identity of the Poincaré plots of breathing periods of each breath (n) versus the following breath (n+1). The occurrence rate of augmented breaths (sighs) and breathing pauses (apneas) is shown in panels G and H, respectively, per hour of sleep time. N and R, non-rapid-eye-movement and rapid-eye-movement sleep, respectively. Data are means ± SD for 10 female (♀) Plp-FLAG-LMNB1 transgenic mice (TG, filled bars) and 11 wild-type (WT, open bars) at 36-40 weeks of age (age group II). [*]: P < 0.05, main effect of TG vs. WT across sleep states (ANOVA). *, P < 0.05, TG vs. WT within sleep state (t-test). Panels C and F show examples of raw recordings of the electroencephalogram (EEG), neck muscle electromyogram (EMG) and respiratory activity (RES, in arbitrary units, AU) during N (C, including a post-sigh apnea) and R (F, including an apnea) in a ♀ TG mouse of age group II. Panel I shows an example of Poincaré plots of breathing periods of each breath (n) versus the following breath (n+1) during N in a ♀ TG mouse of age group II.
Autonomic cardiovascular function
Orthostatic hypotension is common and disabling in patients with ADLD (Guaraldi et al., 2011; Terlizzi et al., 2016), but the cardiovascular compensation of the standing posture is not biologically relevant for mice. We thus investigated cardiovascular activation during cold exposure and during spontaneous transitions from N to either wakefulness or R. These endogenous stimuli both entail sympathetic activation (Silvani, 2008; Swoap et al., 2008; Swoap et al., 2004), as the standing posture does in humans. These experiments were performed on mice aged 37-42 weeks, when the somatic motor dysfunction is already well developed (Figure 1). Since one of the endogenous stimuli of cardiovascular activation that we employed was a change in the wake-sleep state, we first determined the wake-sleep structure of TG and WT at 37-42 weeks of age based on EEG and EMG recordings. We found that the wake-sleep structure was similar in TG and WT mice, except for a significantly longer time spent in N by TG than by WT (Table 1). This is a novel finding in TG mice and not previously analyzed in patients with ADLD. On the other hand, despite extensive recordings, we did not find any evidence of seizures in TG mice, which had been reported in previous work (Heng et al., 2013).
Table 1.
Wake-sleep structure as a function of ambient temperature.
| Age group II | 21 °C | 31 °C | ANOVA | |||
|---|---|---|---|---|---|---|
| ♀ WT | ♀ TG | ♀ WT | ♀ TG | |||
| W | % | 41 ± 7 | 37 ± 3 | 33 ± 4 | 31 ± 6 | |
| F (h-1) | 225 ± 66 | 263 ± 61 | 244 ± 92 | 231 ± 56 | ||
| D (s) | 169 ± 86 | 120 ± 33 | 118 ± 46 | 111 ± 41 | ||
| N | % | 48 ± 5 | 54 ± 4 | 57 ± 3 | 60 ± 6 | [*] |
| F (h-1) | 537 ± 149 | 590 ± 214 | 671 ± 183 | 640 ± 138 | ||
| D (s) | 82 ± 24 | 85 ± 29 | 79 ± 33 | 84 ± 20 | ||
| R | % | 6 ± 2 | 5 ± 1 | 7 ± 3 | 6 ± 2 | |
| F (h-1) | 117 ± 51 | 92 ± 31 | 141 ± 70 | 109 ± 36 | ||
| D (s) | 44 ± 12 | 53 ± 14 | 46 ± 13 | 51 ± 14 | ||
| latency (s) | 894 ± 208 | 777 ± 231 | 847 ± 258 | 849 ± 215 | ||
| IND | % | 5 ± 3 | 4 ± 2 | 3 ± 2 | 3 ± 2 | |
W, N, and R: wakefulness, non-rapid-eye-movement sleep, and rapid-eye-movement sleep, respectively; IND, indeterminate state. Time in each wake-sleep state is expressed as percentage (%) of total recording time. F, number of wake-sleep episodes per hour of recording time. D, episode duration. R latency indicates time elapsed from the onset of each R episode to the end of the previous W episode. Data are means ± SD in 10 Plp-FLAG-LMNB1 transgenic (TG) female (♀) mice aged 37-42 weeks (age group II) and 12-13 wild-type (WT) controls.
P< 0.05, main effect of TG vs. WT across ambient temperatures (ANOVA).
We then analyzed the mean values of SAP and HP (the reciprocal of heart rate) over the whole recordings as a function of the wake-sleep state (wakefulness, N, and R) at different ambient temperatures (21 °C or 31 °C) (Figure 4). The results of these analyses did not reveal any significant difference in SAP or HP between TG and WT mice in any wake-sleep state at either ambient temperature. The cold-induced increases in SAP and HP at 21 °C compared to 31 °C ambient temperature also did not differ significantly between TG and WT in any wake-sleep state. We then performed a coherent averaging of spontaneous transitions from N to wakefulness (i.e., awakenings from N, Figure 5) and from N to R (Figure 6) at 21 °C and 31 °C ambient temperature. Again, this analysis did not indicate any significant weakening of the cardiovascular activation caused by state transitions in TG with respect to WT mice at either ambient temperature. As expected based on previous work (Lo Martire et al., 2012), we found that in WT mice, HP decreased from N to R at 31 °C ambient temperature, whereas it increased at 21 °C ambient temperature. Even this rather subtle feature of the physiological control of the heart as a function of sleep and ambient temperature was perfectly preserved in TG mice (Figure 6).
Figure 4. Systolic arterial pressure and heart period during full-blown wake-sleep episodes at different ambient temperatures.
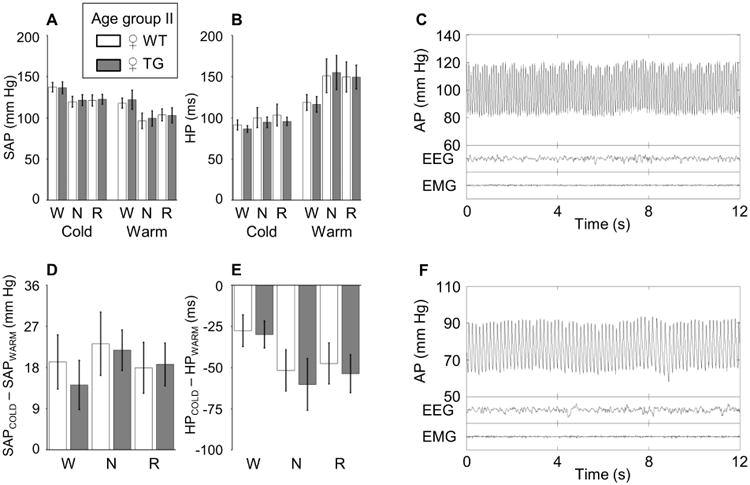
Panels A and B show values of systolic arterial pressure (SAP) and heart period (HP), respectively, during full-blown (> 60 s) episodes of wakefulness (W), non-rapid-eye-movement sleep (N) and rapid-eye-movement sleep (R) at ambient temperatures of 21 °C and 31 °C. Panels D and E show differences in SAP and HP, respectively, between ambient temperatures of 21 °C and 31 °C. Data are means ± SD for 8 female (♀) Plp-FLAG-LMNB1 transgenic mice (TG, filled bars) and 10 wild-type controls (WT, open bars) at 37-42 weeks of age (age group II). There were no significant differences between TG and WT. Panels C and F show examples of raw recordings of the electroencephalogram (EEG), neck muscle electromyogram (EMG) and arterial pressure (AP) during N in a ♀ TG mouse of age group II at 21 °C and 31 °C, respectively.
Figure 5. Systolic arterial pressure and heart period upon awakening from non-rapid-eye-movement sleep at different ambient temperatures.

Panels A and C show values of systolic arterial pressure (SAP) and heart period (HP) during spontaneous transitions from non-rapid-eye-movement sleep (N) to wakefulness (W) at ambient temperature of 21 °C. Panels B and D show the corresponding values at ambient temperature of 31 °C. Values are reported as mean ± SD of differences (Δ) from baseline for 8 female (♀) Plp-FLAG-LMNB1 transgenic mice (TG, filled squares) and 10 wild-type controls (WT, open squares) at 37-42 weeks of age (age group II). The average values over the last 20 s of W, indicated by the black horizontal bar, were employed for the analysis. There were no significant differences between TG and WT.
Figure 6. Systolic arterial pressure and heart period upon entering rapid-eye-movement sleep at different ambient temperatures.

Panels A and C show values of systolic arterial pressure (SAP) and heart period (HP) during spontaneous transitions from non-rapid-eye-movement sleep (N) to rapid-eye-movement sleep (R) at ambient temperature of 21 °C. Panels B and D show the corresponding values at ambient temperature of 31 °C. Values are reported as mean ± SD of differences (Δ) from baseline for 8 female (♀) Plp-FLAG-LMNB1 transgenic mice (TG, filled bars) and 10 wild-type controls (WT, open bars) at 37-42 weeks of age (age group II). The average values over the last 20 s of R, indicated by the black horizontal bar, were employed for the analysis. There were no significant differences between TG and WT.
Finally, we asked whether the parasympathetic (vagal) and particularly the sympathetic modulation of spontaneous cardiovascular variability and cardiac baroreflex sensitivity were impaired in TG mice by computing validated indexes based on the spontaneous fluctuations of SAP and HP. The analysis of these indexes (Figure 7) did not show any significant impairment of vagal and sympathetic cardiovascular modulation and of cardiac baroreflex sensitivity in TG vs. WT mice.
Figure 7. Indexes of vagal and sympathetic modulation and of cardiac baroreflex sensitivity at different ambient temperatures.

The panels show the values of indexes of cardiac vagal modulation (pNN8, panel A; RSA, panel B), vascular sympathetic modulation (SYM, panel C), and cardiac baroreflex sensitivity (BRS, panel D) computed on full-blown (> 60 s) episodes of wakefulness (W), non-rapid-eye-movement sleep (N) and rapid-eye-movement sleep (R) at ambient temperatures of 21 °C and 31 °C. Data are means ± SD for 8 female (♀) Plp-FLAG-LMNB1 transgenic mice (TG, filled bars) and 10 wild-type controls (WT, open bars) at 37-42 weeks of age (age group II). There were no significant differences between TG and WT.
Sympathetic innervation of the heart and kidneys
A partial deficit of noradrenergic sympathetic innervation has been detected with immunohistochemistry of skin biopsies in 4 patients with ADLD (Guaraldi et al., 2011; Terlizzi et al., 2016). Conversely, non-invasive indirect assessment of cardiac sympathetic innervation with I123-metaiodobenzylguanidine scintigraphy did not reveal significant impairments in 3 patients with ADLD (Guaraldi et al., 2011; Terlizzi et al., 2016). Cardiac sympathetic innervation has never been assessed at autopsy with immunohistochemistry in ADLD patients. We therefore completed our assessment of autonomic cardiovascular deficits with a direct assessment of noradrenergic sympathetic innervation by immunohistochemistry on heart and kidney samples. These organs receive a prominent sympathetic innervation, which is more critical for the hemodynamic and salt balance than that of the skin. In these experiments, we studied TG and WT mice at 37-42 weeks of age, for direct comparison with experiments on autonomic cardiovascular function, and extended the analysis to mice at 44-47 weeks of age, to investigate whether structural sympathetic deficits are restricted to the terminal stage of the disease. At the cardiac level, the results indicated a significant reduction of sympathetic noradrenergic innervation in TG vs. WT mice at 37-42 weeks of age, but the difference was not statistically significant in mice at 44-47 weeks of age (Figure 8 A,B). At the renal level, conversely, we found that sympathetic noradrenergic innervation did not differ significantly between TG and WT mice at 37-42 weeks of age, whereas it was significantly reduced (-38% fiber profiles) in TG vs. WT mice at 44-47 weeks of age (Figure 8 C,D).
Figure 8. Sympathetic noradrenergic innervation of the heart and kidneys.

The left panels show the number of profiles of sympathetic noradrenergic fibers counted per mm2 area of heart (panel A) and kidney (panel C) tissue sections. Data are means ± SD in Plp-FLAG-LMNB1 transgenic mice (TG, filled bars) and wild-type controls (WT, open bars) with sample sizes detailed in Table S4. ♂, male; ♀, female. Age groups II and III correspond to 37-42 and 44-47 weeks of age, respectively (cf. methods). The right panels show examples of microscopic fields (scale bar: 100 μm) of heart (panel B) and kidney (panel D), with magnification of selected details in insets, demonstrating the persistence of sympathetic fiber profiles (arrows) in TG of age group III. *, P < 0.05, TG vs. WT within age group II (t-test). [*]: P < 0.05, main effect of TG vs. WT across sexes within age group III (ANOVA).
Discussion
We tested the hypothesis that autonomic cardiovascular dysfunction precedes the occurrence of somatic motor dysfunction in TG mice, as it is found in most cases of ADLD. We found a progressive impairment of walking ability in TG mice. This impairment was detectable in TG mice as early as 24-27 weeks of age in the demanding conditions of the ledge test, and was accompanied by subtle derangements in the control of breathing during sleep at 36-40 weeks of age and by a significant slowing of water licking in the most severe cases at 44-47 weeks of age. On the other hand, we did not detect any significant autonomic cardiovascular dysfunction in TG mice at 37-42 weeks of age in response to changes in wake-sleep state and ambient temperature that elicit sympathetic cardiovascular activation. Validated indexes of sympathetic and vagal modulation and of cardiac baroreflex sensitivity were also normal in TG mice at 37-42 weeks of age. However, we found evidence of a structural deficit of sympathetic noradrenergic innervation in the heart of TG mice at 37-42 weeks of age and in the kidney of TG mice at 44-47 weeks of age.
Our experiments confirmed and extended previous reports of somatic motor dysfunction in TG mice (Heng et al., 2013; Rolyan et al., 2015). Our findings with the ledge test, which, similarly to the balance beam test employed in the first study on TG (Heng et al., 2013), assesses the ability to walk on a narrow path, anticipate the first observed somatic motor deficit of TG mice from 40 weeks of age (Heng et al., 2013) to 24-27 weeks of age (Figure 1A-B). This corresponded to the youngest age group that we tested. Therefore, we cannot exclude that impairments of walking ability ensue even earlier in TG mice. On the other hand, our finding that motor deficits in a plain, open field became significant only at 44-47 weeks of age (Figure 1C-D) supports the age-related, progressive nature of the somatic motor dysfunction of TG mice. Incidentally, our finding that walking ability was significantly impaired both in male and female TG agrees with the reported lack of gender variation in ADLD (Nahhas et al., 2016).
Cerebellar ataxia is a frequent finding in patients with ADLD (Finnsson et al., 2015). The ledge test is considered a measure of coordination directly comparable to human signs of cerebellar ataxia (Guyenet et al., 2010). A lengthening of the inter-lick interval may also be a sensitive sign of cerebellar dysfunction in mice (Heck et al., 2008). However, no pathological changes were detected in the cerebellum of TG mice in a previous study, whereas the white matter of the cervical spinal cord was substantially affected (Rolyan et al., 2015). We also did not find any significant age-related lengthening of the inter-lick interval in TG vs. WT mice (Figure 2C). Taken together, these data do not support an interpretation of our findings in terms of cerebellar ataxia. Our results rather suggest that impairments at the ledge test in TG mice were due to leg spasticity and weakness, which also occur in patients with ADLD (Finnsson et al., 2015), and that the late lengthening of the inter-lick interval in the TG mice with severe impairments at the ledge test was due to bulbar motor dysfunction, akin to pseudobulbar palsy in the terminal stages of ADLD (Finnsson et al., 2015). Nonetheless, significant slowing of water licking occurred only for TG mice with the most severe walking impairment at the ledge test, so it was a subtle difference at best. Similarly, the subtle alterations of breathing that we detected in TG mice at 36-40 weeks of age may reflect the mild brainstem pathology that was previously reported in TG (Heng et al., 2013; Rolyan et al., 2015). On the other hand, our finding that TG spent more time in N than WT did (Table 1) was unexpected, and hitherto unreported in patients with ADLD. The investigation of the mechanisms of this sleep alteration in TG mice was beyond the scope of the present study, but is worth pursuing in future work to map possible links between lamin B1, oligodendrocytes, and sleep.
Our study was the first to investigate the effects of lamin B1 overexpression on autonomic deficits. Despite a careful investigation (Figures 4-7), we did not find evidence of significant autonomic cardiovascular dysfunction in TG mice in terms of the responses to changes in wake-sleep state and ambient temperature. Dopamine beta-hydroxylase knockout mice, which lack sympathetic noradrenergic transmission, show dramatically weaker cardiovascular activation at colder vs. warmer ambient temperatures (Swoap et al., 2008) and in the dark vs. the light period (Swoap et al., 2004). In mice, the cardiovascular differences between the dark and the light periods mainly reflect the cardiovascular activation from N to wakefulness (Bastianini et al., 2012). Our negative findings therefore do not support the existence of any significant functional deficit in sympathetic noradrenergic control in TG mice at 37-42 weeks of age, which is well after the time (24-27 weeks of age) of the first observed motor deficit. We also found that cardiac parasympathetic modulation (Figure 7A-B) and cardiac baroreflex sensitivity (Figure 7C) were well preserved in TG mice at 37-42 weeks of age. This has translational relevance because early autonomic cardiovascular dysfunction causing orthostatic hypotension is a cardinal feature of most patients with ADLD and is particularly disabling, as it may contribute to impair walking ability (Finnsson et al., 2015; Guaraldi et al., 2011; Terlizzi et al., 2016). Orthostatic hypotension in patients with ADLD is not due to primary baroreflex dysfunction, but, rather, to a defective sympathetic noradrenergic control of blood vessels, while cardiac parasympathetic control is relatively preserved (Guaraldi et al., 2011; Terlizzi et al., 2016).
The available evidence indicates a selective loss of noradrenergic sympathetic innervation of the skin in patients with ADLD, whereas the noradrenergic sympathetic innervation of the heart appeared preserved at an indirect evaluation with myocardial scintigraphy (Guaraldi et al., 2011; Terlizzi et al., 2016). With a direct immunohistochemical analysis, we found a significant reduction of the sympathetic noradrenergic innervation of the heart of female TG mice at 37-42 weeks of age. However, this reduction was not associated with any evidence of a deficit of sympathetic cardiac control (Figure 4-7), possibly because of functional compensation by the remaining fibers, and was not replicated in TG mice of either sex at 44-47 weeks of age (Figure 8A). At this stage, it may be fair to conclude that the reduction in sympathetic noradrenergic innervation of the heart in TG mice is not a robust phenomenon, which would be in broad agreement with the negative results of myocardial scintigraphy on patients with ADLD (Terlizzi et al., 2016). On the other hand, we found a robust reduction of sympathetic noradrenergic innervation of the kidneys of TG mice at 44-47 weeks of age. This reduction was not present at 37-42 weeks of age (Figure 8C) and may, therefore, indicate a late-onset deficit ensuing in the terminal stages of the disease.
Taken together, our results indicate that the onset of somatic motor deficits in TG mice is neither accompanied nor preceded by significant deficits of autonomic cardiovascular control, and is followed by structural deficits of the sympathetic noradrenergic innervation, at least at the level of the kidneys in the terminal stages of the disease. This time course is not consistent with that of the clinical signs and symptoms found in most patients with ADLD, whose autonomic cardiovascular dysfunction precedes the onset of somatic motor dysfunction (Guaraldi et al., 2011; Terlizzi et al., 2016). However, the available evidence indicates that the clinical picture of ADLD is heterogeneous. In approximately one out of four patients with ADLD included in a recently published series, autonomic dysfunction included bladder dysfunction, obstipation and/or erectile dysfunction, but not orthostatic hypotension (Finnsson et al., 2015). In approximately 10% of the patients of that series (Finnsson et al., 2015) and in few other well-documented cases (Potic et al., 2013; Quattrocolo et al., 1997), significant autonomic dysfunction occurred well after somatic motor dysfunction or was not detected at all. Regardless, our results indicate that TG mice are adequate to model the somatic motor deficits of ADLD, but not its autonomic features, highlighting the difficulties of employing mutant mice to reproduce human diseases. It is worth remarking that the absence of evidence cannot be taken as evidence for the absence of anything. We strived to perform a thorough physiological evaluation of cardiovascular autonomic control in response to stimuli that entail sympathetic activation (spontaneous transitions from N to either W or R, exposure to colder ambient temperature), and analyzed indexes of cardiovascular autonomic modulation (Figures 4-7). Nonetheless, this evaluation did not include all possible tests of autonomic cardiovascular control in mice. It is possible that the tests that we performed, however thorough, were not optimal to assess autonomic dysfunctions in TG mice, particularly given the histologic evidence of reductions in sympathetic innervation of the heart in TG mice of age group II and of the kidneys in TG mice of age group III (Figure 8). In sum, more study and evaluation will be needed to ascertain whether the TG mouse model fully recapitulates all the clinical features of at least some of the patients with ADLD.
In conclusion, our study confirms the validity of TG mice as a model of the age-related deficits of somatic motor dysfunction of ADLD patients, and shows for the first time that TG mice also have late-onset structural deficits of noradrenergic sympathetic innervation. However, we were not able to identify in TG mice the early-onset sympathetic cardiovascular dysfunction, which is a cardinal feature of most cases of ADLD.
In perspective, our data may provide insights into the ADLD disease mechanism because of the nature of the transgenic construct used to generate TG mice. TG mice overexpress a transgene coding for human lamin B1 in oligodendrocytes under the control of the Plp1 promoter, which leads to the greatest lamin B1 overexpression in the spinal cord (Rolyan et al., 2015). Robust lamin B1 overexpression in other regions of the central nervous system, such as the brainstem, and/or in other cell populations, such as neurons or astrocytes, may be key for the early development of autonomic cardiovascular dysfunctions in patients with ADLD. This would be in line with previous findings suggesting that lamin B1 overexpression is associated with altered neuronal differentiation (Giacomini et al., 2016; Mahajani et al., 2017) and with derangements of genes involved in neural development (Bartoletti-Stella et al., 2015). Information related to the consequences of cell-specific lamin B1 overexpression in transgenic murine models of ADLD is critical since we know so little of the mechanisms that underlie ADLD and demyelinating disorders in general.
Supplementary Material
Highlights.
Lamin B1 overexpression causes adult-onset autosomal dominant leukodystrophy (ADLD).
Age-dependent motor dysfunction and early autonomic cardiovascular dysfunction are cardinal features of ADLD.
Here, we show that mice overexpressing lamin B1 in oligodendrocytes develop early motor dysfunction in the face of preserved cardiovascular control, and a late loss of sympathetic noradrenergic fibers.
These mice thus recapitulate the age-dependent motor dysfunction, but not the early autonomic cardiovascular dysfunction of ADLD.
Lamin B1 overexpression in cell types other than oligodendrocytes, such as neurons or astrocytes may be key for the early development of autonomic cardiovascular dysfunction in patients with ADLD.
Acknowledgments
This work was supported by the National Institutes of Health [R01NS095884 to Q.P.]; the National Multiple Sclerosis Society [RG 5045A1 to Q.P.]; the Telethon foundation [GGP10184 to L.G. and P.C.]; the Fondazione del Monte di Bologna e Ravenna bank foundation [FdM/3954 to P.C.], and the University of Bologna [RFO to C.B., A.S., and G.Z.].
Footnotes
Abbreviations: ADLD, Adult-onset autosomal dominant leukodystrophy; BRS, Index of cardiac baroreflex sensitivity; EEG, Electroencephalogram; EMG, Electromyogram; HP, Heart period; LMNB1, Lamin B1 gene; N, Non-rapid-eye-movement sleep; PBS, Phosphate buffered saline; Plp1, Proteolipid protein 1; pNN8, Index of parasympathetic modulation of heart period computed in the time domain; R, Rapid-eye-movement sleep; RSA, Index of parasympathetic modulation of heart period computed in the frequency domain; SAP, Systolic arterial pressure; SD1 and SD2, Indexes of short-term and long-term variability of total breath duration, respectively; SYM, Index of sympathetic modulation of arterial pressure computed in the frequency domain; TG, Mice hemizygote for the Plp-FLAG-LMNB1 transgene; TTOT, Total breath duration; VE, Minute volume; VT, Tidal volume
Conflict of interest statement: The authors declare no competing financial interests.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Bartoletti-Stella A, Gasparini L, Giacomini C, Corrado P, Terlizzi R, Giorgio E, Magini P, Seri M, Baruzzi A, Parchi P, Brusco A, Cortelli P, Capellari S. Messenger RNA processing is altered in autosomal dominant leukodystrophy. Hum Mol Genet. 2015;24:2746–2756. doi: 10.1093/hmg/ddv034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bastianini S, Silvani A, Berteotti C, Lo Martire V, Zoccoli G. Mice show circadian rhythms of blood pressure during each wake-sleep state. Chronobiol Int. 2012;29:82–86. doi: 10.3109/07420528.2011.635231. [DOI] [PubMed] [Google Scholar]
- Baudrie V, Laude D, Elghozi JL. Optimal frequency ranges for extracting information on cardiovascular autonomic control from the blood pressure and pulse interval spectrograms in mice. Am J Physiol Regul Integr Comp Physiol. 2007;292:R904–912. doi: 10.1152/ajpregu.00488.2006. [DOI] [PubMed] [Google Scholar]
- Byers SL, Wiles MV, Dunn SL, Taft RA. Mouse estrous cycle identification tool and images. PLoS One. 2012;7:e35538. doi: 10.1371/journal.pone.0035538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eldridge R, Anayiotos CP, Schlesinger S, Cowen D, Bever C, Patronas N, McFarland H. Hereditary adult-onset leukodystrophy simulating chronic progressive multiple sclerosis. N Engl J Med. 1984;311:948–953. doi: 10.1056/NEJM198410113111504. [DOI] [PubMed] [Google Scholar]
- Finnsson J, Sundblom J, Dahl N, Melberg A, Raininko R. LMNB1-related autosomal-dominant leukodystrophy: Clinical and radiological course. Ann Neurol. 2015;78:412–425. doi: 10.1002/ana.24452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giacomini C, Mahajani S, Ruffilli R, Marotta R, Gasparini L. Lamin B1 protein is required for dendrite development in primary mouse cortical neurons. Mol Biol Cell. 2016;27:35–47. doi: 10.1091/mbc.E15-05-0307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giorgio E, Robyr D, Spielmann M, Ferrero E, Di Gregorio E, Imperiale D, Vaula G, Stamoulis G, Santoni F, Atzori C, Gasparini L, Ferrera D, Canale C, Guipponi M, Pennacchio LA, Antonarakis SE, Brussino A, Brusco A. A large genomic deletion leads to enhancer adoption by the lamin B1 gene: a second path to autosomal dominant adult-onset demyelinating leukodystrophy (ADLD) Hum Mol Genet. 2015;24:3143–3154. doi: 10.1093/hmg/ddv065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guaraldi P, Donadio V, Capellari S, Contin M, Casadio MC, Montagna P, Liguori R, Cortelli P. Isolated noradrenergic failure in adult-onset autosomal dominant leukodystrophy. Auton Neurosci. 2011;159:123–126. doi: 10.1016/j.autneu.2010.07.011. [DOI] [PubMed] [Google Scholar]
- Guyenet SJ, Furrer SA, Damian VM, Baughan TD, La Spada AR, Garden GA. A simple composite phenotype scoring system for evaluating mouse models of cerebellar ataxia. J Vis Exp. 2010;21:1787. doi: 10.3791/1787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayar A, Bryant JL, Boughter JD, Heck DH. A low-cost solution to measure mouse licking in an electrophysiological setup with a standard analog-to-digital converter. J Neurosci Methods. 2006;153:203–207. doi: 10.1016/j.jneumeth.2005.10.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heck DH, Zhao Y, Roy S, LeDoux MS, Reiter LT. Analysis of cerebellar function in Ube3a-deficient mice reveals novel genotype-specific behaviors. Hum Mol Genet. 2008;17:2181–2189. doi: 10.1093/hmg/ddn117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heng MY, Lin ST, Verret L, Huang Y, Kamiya S, Padiath QS, Tong Y, Palop JJ, Huang EJ, Ptacek LJ, Fu YH. Lamin B1 mediates cell-autonomous neuropathology in a leukodystrophy mouse model. J Clin Invest. 2013;123:2719–2729. doi: 10.1172/JCI66737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laude D, Baudrie V, Elghozi JL. Applicability of recent methods used to estimate spontaneous baroreflex sensitivity to resting mice. Am J Physiol Regul Integr Comp Physiol. 2008a;294:R142–150. doi: 10.1152/ajpregu.00319.2007. [DOI] [PubMed] [Google Scholar]
- Laude D, Baudrie V, Elghozi JL. Effects of atropine on the time and frequency domain estimates of blood pressure and heart rate variability in mice. Clin Exp Pharmacol Physiol. 2008b;35:454–457. doi: 10.1111/j.1440-1681.2008.04895.x. [DOI] [PubMed] [Google Scholar]
- Lo Martire V, Silvani A, Bastianini S, Berteotti C, Zoccoli G. Effects of ambient temperature on sleep and cardiovascular regulation in mice: the role of hypocretin/orexin neurons. PLoS One. 2012;7:e47032. doi: 10.1371/journal.pone.0047032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahajani S, Giacomini C, Marinaro F, De Pietri Tonelli D, Contestabile A, Gasparini L. Lamin B1 levels modulate differentiation into neurons during embryonic corticogenesis. Sci Rep. 2017;7:4897. doi: 10.1038/s41598-017-05078-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nahhas N, Sabet Rasekh P, Vanderver A, Padiath Q. Autosomal Dominant Leukodystrophy with Autonomic Disease. In: Pagon RA, et al., editors. GeneReviews(R) University of Washington; Seattle: 2016. [Google Scholar]
- Padiath QS, Saigoh K, Schiffmann R, Asahara H, Yamada T, Koeppen A, Hogan K, Ptacek LJ, Fu YH. Lamin B1 duplications cause autosomal dominant leukodystrophy. Nat Genet. 2006;38:1114–1123. doi: 10.1038/ng1872. [DOI] [PubMed] [Google Scholar]
- Potic A, Pavlovic AM, Uziel G, Kozic D, Ostojic J, Rovelli A, Sternic N, Bjelan M, Sarto E, Di Bella D, Taroni F. Adult-onset autosomal dominant leukodystrophy without early autonomic dysfunctions linked to lamin B1 duplication: a phenotypic variant. J Neurol. 2013;260:2124–2129. doi: 10.1007/s00415-013-6958-3. [DOI] [PubMed] [Google Scholar]
- Quattrocolo G, Leombruni S, Vaula G, Bergui M, Riva A, Bradac GB, Bergamini L. Autosomal dominant late-onset leukoencephalopathy. Clinical report of a new Italian family Eur Neurol. 1997;37:53–61. doi: 10.1159/000117406. [DOI] [PubMed] [Google Scholar]
- Rolyan H, Tyurina YY, Hernandez M, Amoscato AA, Sparvero LJ, Nmezi BC, Lu Y, Estecio MR, Lin K, Chen J, He RR, Gong P, Rigatti LH, Dupree J, Bayir H, Kagan VE, Casaccia P, Padiath QS. Defects of lipid synthesis are linked to the age-dependent demyelination caused by Lamin B1 overexpression. J Neurosci. 2015;35:12002–12017. doi: 10.1523/JNEUROSCI.1668-15.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silvani A. Physiological sleep-dependent changes in arterial blood pressure: central autonomic commands and baroreflex control. Clin Exp Pharmacol Physiol. 2008;35:987–994. doi: 10.1111/j.1440-1681.2008.04985.x. [DOI] [PubMed] [Google Scholar]
- Silvani A, Bastianini S, Berteotti C, Cenacchi G, Leone O, Lo Martire V, Papa V, Zoccoli G. Sleep and cardiovascular phenotype in middle-aged hypocretin-deficient narcoleptic mice. J Sleep Res. 2014a;23:98–106. doi: 10.1111/jsr.12081. [DOI] [PubMed] [Google Scholar]
- Silvani A, Bastianini S, Berteotti C, Franzini C, Lenzi P, Lo Martire V, Zoccoli G. Sleep modulates hypertension in leptin-deficient obese mice. Hypertension. 2009;53:251–255. doi: 10.1161/HYPERTENSIONAHA.108.125542. [DOI] [PubMed] [Google Scholar]
- Silvani A, Bastianini S, Berteotti C, Franzini C, Lenzi P, Lo Martire V, Zoccoli G. Dysregulation of heart rhythm during sleep in leptin-deficient obese mice. Sleep. 2010;33:355–361. doi: 10.1093/sleep/33.3.355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silvani A, Berteotti C, Bastianini S, Cohen G, Lo Martire V, Mazza R, Pagotto U, Quarta C, Zoccoli G. Cardiorespiratory anomalies in mice lacking CB1 cannabinoid receptors. PLoS One. 2014b;9:e100536. doi: 10.1371/journal.pone.0100536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silvani A, Lo Martire V, Salvade A, Bastianini S, Ferri R, Berteotti C, Baracchi F, Pace M, Bassetti CL, Zoccoli G, Manconi M. Physiological time structure of the tibialis anterior motor activity during sleep in mice, rats and humans. J Sleep Res. 2015;24:695–701. doi: 10.1111/jsr.12319. [DOI] [PubMed] [Google Scholar]
- Silvani A, Calandra-Buonaura G, Dampney RAL, Cortelli P. Brain-heart interactions: physiology and clinical implications. Philos Trans A Math Phys Eng Sci. 2016;374 doi: 10.1098/rsta.2015.0181. [DOI] [PubMed] [Google Scholar]
- Swoap SJ, Li C, Wess J, Parsons AD, Williams TD, Overton JM. Vagal tone dominates autonomic control of mouse heart rate at thermoneutrality. Am J Physiol Heart Circ Physiol. 2008;294:H1581–1588. doi: 10.1152/ajpheart.01000.2007. [DOI] [PubMed] [Google Scholar]
- Swoap SJ, Weinshenker D, Palmiter RD, Garber G. Dbh(-/-) mice are hypotensive, have altered circadian rhythms, and have abnormal responses to dieting and stress. Am J Physiol Regul Integr Comp Physiol. 2004;286:R108–113. doi: 10.1152/ajpregu.00405.2003. [DOI] [PubMed] [Google Scholar]
- Terlizzi R, Calandra-Buonaura G, Zanigni S, Barletta G, Capellari S, Guaraldi P, Donadio V, Cason E, Contin M, Poda R, Tonon C, Sambati L, Gallassi R, Liguori R, Lodi R, Cortelli P. A longitudinal study of a family with adult-onset autosomal dominant leukodystrophy: Clinical, autonomic and neuropsychological findings. Auton Neurosci. 2016;195:20–26. doi: 10.1016/j.autneu.2016.02.005. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.