

Abstract
Tissue fibrosis, the development of fibrous connective tissue as a result of injury or damage, is associated with many common diseases and cannot be effectively treated. The complex biological processes accompanying fibrosis often involve aberrant signaling through the transforming growth factor beta (TGF-β) pathway. In the search for mechanisms to repress this signaling, microRNAs have emerged as a novel class of molecules capable of targeting single members of the TGF-β pathway, or the pathway as a whole. We previously identified miR-1343 as a potent repressor of TGF-β signaling and fibrosis through the direct attenuation of both canonical TGF-β receptors. Here, we build upon our previous findings to better characterize the function of endogenous miR-1343 in normal biology and examine the potential role of exogenous miR-1343 as a repressor of TGF-β signaling. CRISPR/Cas9-mediated deletion of miR-1343 from A549 lung epithelial cells impacts several processes and genes implicated in fibrosis and known to be TGF-β pathway effectors. Moreover, the responses are opposite to those we observed previously when miR-1343 was overexpressed in the same cell type. We also show that miR-1343 can be shuttled into exosomes, a type of extracellular vesicle, which are exported by cells into the surrounding medium and can absorbed by distant target cells. miR-1343 delivered into primary lung fibroblasts by exosomes has a measurable function in reducing TGF-β signaling and markers of fibrosis. These results highlight a role for miR-1343 in fine-tuning the TGF-β pathway and propose its use as a therapeutic in fibrotic disease.
Keywords: microRNA, TGF-β, fibrosis, extracellular vesicle, exosome
INTRODUCTION
Fibrotic diseases are characterized by the pathological accumulation of connective tissue resulting from inappropriate inflammatory activation. In turn this promotes excessive wound healing which often results in organ failure (Wynn 2011). The causes of fibrosis are generally a consequence of physical insult to the tissue, either resulting from genetic conditions (such as cystic fibrosis) or environmental factors (for example radiation-induced injury), or both. There are currently no treatments for fibrosis, and most therapeutic interventions only target symptoms of the disease. A pivotal mediator of fibrosis is transforming growth factor beta (TGF-β), which is normally released in response to injury to stimulate repair processes, and is often observed at increased levels in disease (Leask and Abraham 2004; Akhurst and Hata 2012). In its canonical signaling pathway, TGF-β functions by binding to cell-surface receptors TGF-β receptor 1 and 2 (TGFBR1, TGFBR2), which activate a signaling cascade via Mothers against decapentaplegic homolog proteins, SMAD2 and SMAD3, to alter gene expression (Shi and Massagué 2003; Derynck and Zhang 2003). TGF-β signaling causes the proliferation and differentiation of fibroblasts into myofibroblasts, marked by smooth muscle actin (αSMA) expression, which then deposit collagens and other extracellular matrix proteins into the surrounding tissue matrix (Leask and Abraham 2004; Akhurst and Hata 2012).
In previous work we identified microRNA-1343 (miR-1343) as a strong repressor of TGF-β signaling and fibrotic processes in the lung (Stolzenburg et al. 2016). MicroRNAs are small 21-25 nucleotide non-coding RNAs capable of post-transcriptional gene repression that promote the destabilization and/or degradation of protein-coding mRNAs. Several miRNAs, including miR-29, miR-21, and miR-155, are reported to regulate fibrotic processes, particularly via members of the TGF-β signaling pathway (reviewed in (O’Reilly 2016)). Using exogenous miR-1343, gene reporter assays and RNA-sequencing (RNA-seq), we showed that miR-1343 directly targets and represses TGFBR1 and TGFBR2 expression (Stolzenburg et al. 2016). Thus uniquely to date, miR-1343 directly inhibits both canonical TGF-β receptors. Furthermore, miR-1343 transient overexpression resulted in decreased activation of SMAD2/3 in response to TGF-β, and attenuated markers of fibrosis in both lung epithelial cell lines and primary lung fibroblasts. The genomic location of miR-1343 adjacent to a modifier locus for cystic fibrosis lung disease severity is of note (Wright et al. 2011; Corvol et al. 2015).
Due to the low abundance of miR-1343 in human cells, evaluating its mechanisms of action ex vivo is challenging. Moreover, this miR is not conserved in rodents, which hampers in vivo experiments. Our previous work on miR-1343 characterized its role in TGF-β signaling and fibrosis exclusively through transient overexpression (Stolzenburg et al. 2016). Here our goal was to determine the functions of endogenous miR-1343 in both normal and diseased tissue as a prelude to the development of miR-1343-based therapeutics.
Current approaches to microRNA therapies rely on two main strategies to alter miRNA function: the introduction of miRNA mimics to increase miRNA expression, or delivery of anti-miRNA oligos to repress miRNA action (Christopher et al. 2016). Though both are effective in vitro, the unstable nature of small RNA oligos in vivo necessitates chemical modifications to the miR, such as locked nucleic acids or the addition of 2′-O-methyl groups (Yoo et al. 2004; Krützfeldt et al. 2005; Zhang et al. 2013). Correct tissue targeting is also problematic. The observation that miRNAs are found circulating in extracellular vesicles, and may be recruited for intercellular communication, has gained increasing attention (Zhou et al. 2012; Gallo et al. 2012; Lv et al. 2013). The microRNA-containing vesicles are a special class of 40-100 nm structures, named exosomes. These originate as multivesicular endosomes that fuse with the plasma membrane and were first documented as a pathway for cell receptor exocytosis (Pan and Johnstone 1983; Harding et al. 1984). Exosomes may be used to transfer both endogenous and exogenous microRNAs between physically distant cells, where they have functional roles (Valadi et al. 2007; Pegtel et al. 2010; Meckes et al. 2010; Chiba et al. 2012; Umezu et al. 2013). Therefore, utilizing exosomes as a decoy delivery mechanism for microRNAs has great potential.
In the current study, we build upon our previous work to better characterize the role of miR-1343 in normal biology. Using CRISPR/Cas9 technology, we generate cell lines in which the miR-1343 locus is deleted and validate its role in the fibrosis-associated processes of cell growth and division, and in reducing the expression of genes downstream of TGF-β signaling. Furthermore, we investigate the utility of exosomes to deliver miR-1343 to target cells. HL-60 neutrophil-like cells are known to produce exosomes (Huan et al. 2013) and express endogenous miR-1343 (Stolzenburg et al. 2016). We show that the miR is packaged into HL-60-derived exosomes and is taken up by A549 lung epithelial cells and primary lung fibroblasts, where it appears to inhibit TGF-β signaling and myofibroblast differentiation. These findings confirm the role of miR-1343 in protecting against processes of fibrosis and encourage its use in developing microRNA-based therapies for fibrotic diseases.
METHODS
Cell culture and transfection
A549 lung adenocarcinoma cells (Giard et al. 1973) were cultured in Dulbecco’s modified Eagle’s medium (DMEM, low glucose) supplemented with 10% fetal bovine serum (FBS). HL-60 promyelocytic leukemia cells (Collins et al. 1977) were grown in Roswell Park Memorial Institute (RPMI) 1640 medium with 10% FBS. Primary lung fibroblasts were cultured in DMEM supplemented with 4.5 g/L glucose, 4 mM L-glutamine, sodium pyruvate, and 10% FBS for up to 15 passages.
The miR-1343 locus was removed from A549 cells by clustered regularly interspaced short palindromic repeat (CRISPR)/Cas9-mediated genomic editing (Jinek et al. 2012; Cong et al. 2013; Mali et al. 2013). Guide RNAs were designed using the CRISPR design program (http://crispr.mit.edu) and synthesized as 455 bp gBlocks from Integrated DNA Technologies (according to the Church lab protocol, Addgene). These were then blunt cloned into the pSCB vector (Agilent). The gBlock sequence and guide RNAs used for each region are shown in Suppl. Table 1. The pMJ920-Cas9-GFP plasmid (Jinek et al. 2013), and two cloned guide RNAs flanking the miR-1343 locus were transfected into A549 cells using Lipofectamine 2000 (Life Technologies), according to the manufacturer’s instructions. Cas9-GFP positive cells were selected by fluorescence activated sell sorting (FACS) 48 hrs after transfection and grown as single clones. miR-1343 locus deletion and PDHX (host-gene) integrity were determined by PCR of genomic DNA and total RNA isolated from clones, with primers listed in Suppl. Table 2.
Transient transfection of pre-miR-1343 and negative control miRNA precursor #2 (both from Life Technologies) was performed as described previously (Stolzenburg et al. 2016).
Electroporation of pCMV-MIR vectors (Origene) into HL-60 cells was completed by standard methods. Briefly, 1.5 × 106 cells were resuspended in 0.5 ml of ice-cold HEPES-buffered saline and mixed with 10 μg of plasmid DNA in a 0.4 cm electrode gap cuvette. Cells were pulsed with the Gene Pulser System (Biorad) at 950 μF, 220 mV (time constant = ~27 msec) and transferred to flasks containing regular growth medium to recover.
RNA-sequencing
RNA-sequencing was performed and analyzed as previously described (Fossum et al. 2014). All data are deposited on the GEO database (GEO99209). Due to significant variability between non-targeted (NT) control clones, differentially expressed genes in miR-1343 deletion clones were determined by a three-way analysis compared to both NT controls and non-clonal A549 cells.
Quantitative RT-PCR
RNA was isolated from cells using TRIzol (Life Technologies). Standard cDNA reactions were performed using the TaqMan Reverse Transcription Kit (Life Technologies) according to the manufacturer’s instructions. qPCR assays were completed using Power SYBR Green (Life Technologies) as previously described (Stolzenburg et al. 2016) and Ct values were normalized against beta-2-microglobulin (β2M). See Suppl. Table 3 for primers used.
MicroRNA cDNA was made from DNase I-treated total RNA using the TaqMan Advanced miRNA Reverse Transcription Kit (Life Technologies) following the manufacturer’s instructions. TaqMan Fast qPCR assays were performed using the hsa-miR-1343-3p TaqMan Advanced Assay (Life Technologies). Ct values were normalized against the geometric mean of hsa-miR-186-5p and hsa-miR-423-3p (both Advanced assays from Life Technologies), or against a standard curve. The standard curve was generated using dilutions of synthetic mature miR-1343 (Invitrogen) as starting material for the TaqMan Advanced Assay.
TGF-β treatment
Cells were serum starved in growth media containing 0.5% FBS for 6 – 16 hrs prior to treatment. Human recombinant TGF-β1 (R&D Systems) was added to serum-depleted media at a final concentration of 5 ng/ml. The vehicle control was 1 mg/ml bovine serum albumin (BSA) in 4 mM HCl. Cells were incubated with TGF−β for 1 – 48 hrs, depending on each experiment.
Western blot
Cell lysates were analyzed by standard Western blot methods (Stolzenburg et al. 2016). Primary antibodies were against: pSMAD3 (phosphorylated SMAD3, 9520), SMAD2/3 (8828), GAPDH (glyceraldehyde-3-phosphate dehydrogenase, 5174), all from Cell Signaling Technology; TGFBR2 (TGF-β receptor 2, sc-400), PAI-1 (plasminogen activator inhibitor type 1, sc-5297), both from Santa Cruz Biotechnology; TGFBR1 (TGF-β receptor 1, ab31013) from Abcam; αSMA (alpha smooth muscle actin, M0851) from Dako; and CD81 (10630D) from Invitrogen. Secondary antibodies were against mouse (P0447) or rabbit (P0448), both from Dako. Western bots were scanned by densitometry and quantified using ImageJ software (NIH).
Exosome-free media preparation
Exosome-free media was prepared by ultracentrifugation. RPMI 1640 media containing 20% FBS was centrifuged at 100,000 × g for 16 hrs at 4°C to pellet microvesicles present in the FBS. Immediately prior to use, the exosome-free media was diluted with media containing no FBS to obtain a final concentration of 10% FBS.
Exosome purification
Exosomes were purified using the exoEasy Maxi Kit (Qiagen) according to the manufacturer’s protocol. HL-60 cells were incubated in exosome-free media for 48 hrs to condition exosome release. Cells and cell debris were cleared from media by centrifugation at 300 × g for 5 min, followed by centrifugation at 4200 × g for 15 min. Briefly, exosomes were bound to the exoEasy spin column, washed, and eluted in 400-450 μl of Buffer XE (Qiagen).
Exosome labeling and microscopy
Exosomes were labeled with the membrane-intercalating dye, BODIPY TR Ceramide (Fisher). BODIPY TR dye was added to HL-60 cells in pre-conditioned exosome-free media at a final concentration of 1 μM and incubated in the dark for 1-3 hrs at 37°C. Exosomes were then purified as above. Following purification, excess unincorporated dye was removed using Exosome Spin Columns (MW 3,000; Fisher) according to the manufacturer’s instructions.
A549 cells were cultured in Nunc Lab-Tek 4-well glass chamber slides (Fisher) or grown on round glass coverslips in exosome-free media. Labeled exosomes were added directly to the cells together with Hoescht 33258 and Calcein AM dyes (both from Fisher) and incubated at 37°C for 2 hrs. Cells were then washed, fixed in 4% paraformaldehyde, and mounted. Microscopy was performed on a Leica DMR-HC upright microscope with a QImaging Retiga 4000R camera or a Zeiss 880 Confocal microscope with ZEN software.
Exosome treatment
Following purification, exosomes to be treated for 3 hrs or less were directly added to cells in exosome-free growth media at an appropriate concentration (typically 10 – 30 μg, equivalent to 50 – 100 μl of eluted exosomes in 500 μl media). For exosomes to be treated for longer than 3 hrs, a buffer exchange step was performed due to elution buffer (Buffer XE, Qiagen) toxicity. Briefly, exosomes were added to the top of a sterile 25 mm type-VS membrane (0.025 μm pore size; Millipore) floating on sterile PBS and dialyzed for ~2 hrs. An appropriate amount of exosomes (typically 10 – 30 μg; 50 – 100 μl in 500 μl media) were added to cells in exosome-free media and incubated for 6-48 hrs.
Statistical analysis and graphs
Results are shown as mean +/− standard error of the mean (n≥3) or mean +/− the standard deviation (n<3). Statistics were performed using the appropriate test (as described in each figure legend) in Prism software (GraphPad).
RESULTS
Deletion of miR-1343 has a subtle effect on gene expression
We previously reported that miR-1343 directly targets both canonical TGF-β receptors, TGFBR1 and TGFBR2, via their 3′ untranslated regions (UTR) to reduce their expression (Stolzenburg et al. 2016). As a result, we found decreased activation of the TGF-β effector proteins, SMAD2 and SMAD3, and corresponding reductions in their nuclear translocation. Furthermore, markers of fibrosis in primary lung fibroblasts, such as Alpha smooth muscle actin (αSMA) and Collagen type I A1 (COL1A1), were attenuated by miR-1343 following TGF-β treatment, while indicators of epithelial-to-mesenchymal transition in A549 lung epithelial cells, such as Epithelial-cadherin (E-cad) expression and wound healing, were maintained. However, as all these data were derived utilizing transient overexpression of miR-1343, the current experiments were designed to examine the role of endogenous miR-1343 in human cells.
First, we examined the impact of loss of endogenous miR-1343 on the biology of airway epithelial cells. CRISPR/Cas9–mediated technology was used to remove a ~150 bp fragment encompassing miR-1343 with two flanking guide RNAs. Cas9-GFP positive cells were isolated by FACS, delivering one cell per well into 96 well plates, and grown as individual clones. PCR assays (Fig. 1a) confirmed homozygous deletion of miR-1343 in 9 clones out of 18 generated, with no apparent changes to PDHX expression or splicing (Fig. 1a’, 3 non-targeted and 3 deletion clones shown). Furthermore, miR-1343 was undetectable in the deletion (Δ) clones using a Taqman qRT-PCR assay specific for the mature miRNA (Fig. 1b). Total RNA was isolated from 3 independent deletion clones and 3 non-targeted (NT) clones from the same experiment (shown in Fig. 1a,b), to evaluate clonal variability, and RNA-seq was performed on libraries generated from them. Tophat and Cufflinks software were used to generate fragments per kilobase per million mapped reads (FPKM) values and CuffDiff was used to identify differentially expressed genes (DEGs) (Trapnell et al. 2012). A total of 367 DEGs were identified when miR-1343Δ clones were compared to NT clones. However, multidimensional scaling analysis of the RNA-seq data (Suppl. Fig. 1a), showed significant variability between the 3 NT clones, indicating that many DEGs were likely the result of clonal variation and not the deletion of miR-1343. Therefore, we expanded our analysis to also include 3 replicates of non-clonal A549 cells. We found 903 genes in miR-1343Δ compared to NT clones, 1226 genes in miR-1343Δ compared to A549 cells, and 2145 genes in NT clones compared to A549 cells that showed significant changes in expression (Fig. 1c, Suppl. Fig. 1b). Figure 1d shows a Venn diagram illustrating the numbers of DEGs that were identified in more than one comparison, where 97 were differentially expressed in miR-1343Δ clones compared to both the non-targeted clones and A549 cells. The large number of DEGs identified in NT clones compared to A549 cells supports the existence of significant variability within the different NT clonal populations. We therefore excluded any gene that was differentially expressed between NT clones and A549 cells and compiled a list of 383 high-confidence DEGs most likely to be altered by miR-1343 deletion, with 287 showing increased expression and 95 showing decreased expression in miR-1343Δ clones (see Suppl. Table 4 for gene list). In contrast to the more than four thousand DEGs in miR-1343 over-expression studies (Stolzenburg et al. 2016), this relatively small number of genes demonstrates that loss of miR-1343 has a more subtle effect on the transcriptome. About one third (137/383) of the high-confidence genes altered by miR-1343 deletion were also differentially expressed by miR-1343 overexpression in A549 cells (Stolzenburg et al. 2016), with 26 of these also appearing as TargetScan-predicted (version 7.1) miR-1343 targets (Suppl. Fig. 2). Among these shared DEGs was TGFBR1, which exhibited increased expression in miR-1343Δ clones consistent with its down-regulation following miR-1343 over-expression.
Fig. 1. RNA-seq identifies pathways altered by miR-1343 deletion.
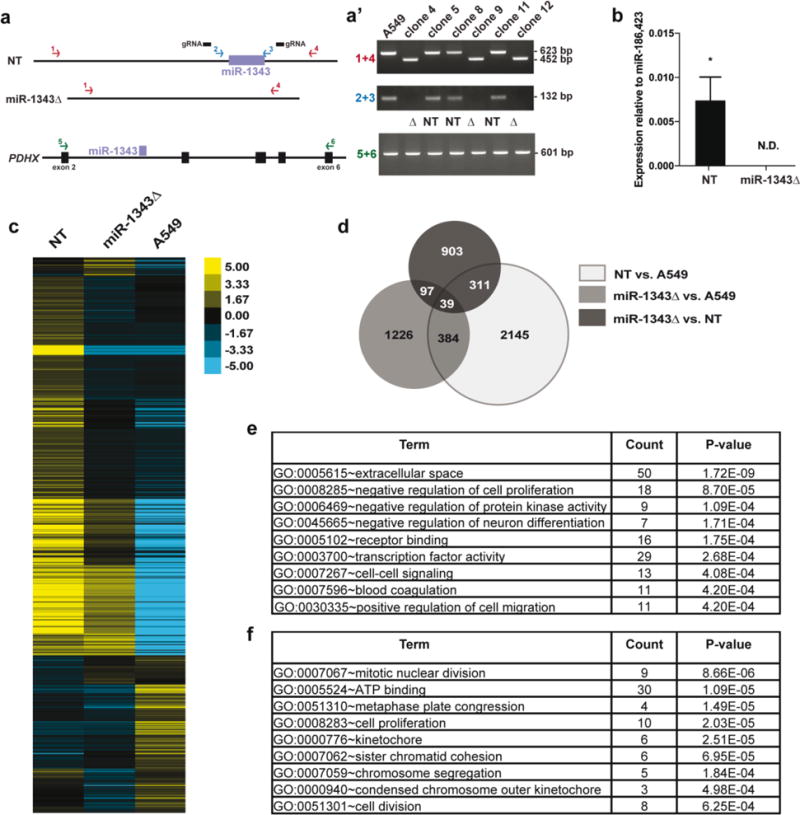
a. Schematic showing PCR assays used to test for miR-1343 deletion and confirm PDHX integrity following CRISPR/Cas9 genome editing. Two primer sets located external (1 + 4, in red) and internal (2 + 3, in blue) to the guide RNAs (gRNAs, black bars) were used to identify non-targeted (NT) and miR-1343 deletion (miR-1343Δ) clones. PCR primers spanning the deletion (5+6, in green) were used to confirm PDHX mRNA integrity. a’. Clones 4, 9, and 12 show a product at 452 bp (and no wild type product at 623 bp) with the external primers, and lack a product with the internal primers, indicating homozygous deletion. RT-PCR reactions show normal splicing of PDHX exons 2 – 6 in all clones (figure not to scale). b. miR-1343 expression levels measured by TaqMan Advanced qRT-PCR assay in 3 NT clones and 3 miR-1343Δ clones. Values are normalized to the geometric mean of miR-186 and miR-423 values. N.D.=not detected, *p≤0.05 by unpaired Student’s t test. c. Heatmap illustrating the relative expression of differentially expressed genes (DEGs) in NT clones, miR-1343Δ clones, or non-clonal A549 cells. Each line represents a different gene. d. Venn diagram of DEGs identified by RNA-seq between NT clones and A549 cells (light grey), miR-1343Δ clones and A549 cells (medium grey), and miR-1343Δ clones and NT clones (dark grey). The overlapping regions represent the number of DEGs shared between groups. DEGs identified between NT clones and A549 cells were excluded from further analysis. e.,f. DAVID gene ontology analysis of down-regulated genes (e) and up-regulated genes (f) identified by RNA-seq following miR-1343 deletion.
Next, gene ontology analysis was completed on the 383 high-confidence miR-1343Δ DEGs using the Database for Annotation, Visualization and Integrated Discovery (DAVID, (Huang et al. 2009a, b). The most significant processes in our analyses were confirmed using the g:Profiler database (Suppl Table 5) (Reimand et al. 2016). Of the genes down-regulated after miR-1343 deletion, processes involving cell growth, signaling, and migration were enriched (Fig. 1e). This finding is consistent with processes that were overrepresented in miR-1343 over-expression studies (Stolzenburg et al. 2016). Conversely, processes related to cell division, including nuclear division, kinetochore, and chromosome segregation, were enriched among DEGs up-regulated after miR-1343 deletion (Fig. 1f). Together with data in miR-1343 overexpressing cells (Stolzenburg et al. 2016), these results posit cell growth and division as potential fibrosis-associated cellular functions that are decreased by miR-1343 action. It is also possible that clonal differences are a confounding variable in this interpretation.
Endogenous miR-1343 regulates genes in the TGF-β signaling pathway
Since transient miR-1343 overexpression reduced TGF-β signaling and downstream phenotypes (Stolzenburg et al. 2016), we next assessed whether miR-1343 also regulated TGF-β target genes. A549 cells and three miR-1343Δ clones were grown to similar confluence and then transiently transfected with precursor (pre)-miR-1343 or negative control (NC) miR for 24 hours. Cells were then treated for 24 hours with TGF-β or vehicle control and RNA or protein extracted. Since receptor levels are known to regulate TGF-β signaling activity (Shi and Massagué 2003; Derynck and Zhang 2003), we first performed RT-qPCR to measure TGFBR2 (Fig. 2a,a’) and TGFBR1 (Fig. 2b,b’) with the prediction that these would be repressed. TGFBR2 levels were substantially reduced following TGF-β treatment in NC cells and miR-1343 caused a further decrease in transcript levels. Conversely, TGF-β treatment increased TGFBR1 expression in NC cells, though the repressive effect of miR-1343 was still evident. Our data suggest that the two receptors may be regulated by TGF-β through independent mechanisms. miR-1343 deletion had no impact on TGF-β induced TGFBR1 transcript levels, and appeared to reduce TGFBR2 expression, suggesting that additional compensatory mechanisms for loss of the miR may exist.
Fig. 2. Expression of genes downstream of TGF-β are altered by loss of miR-1343.
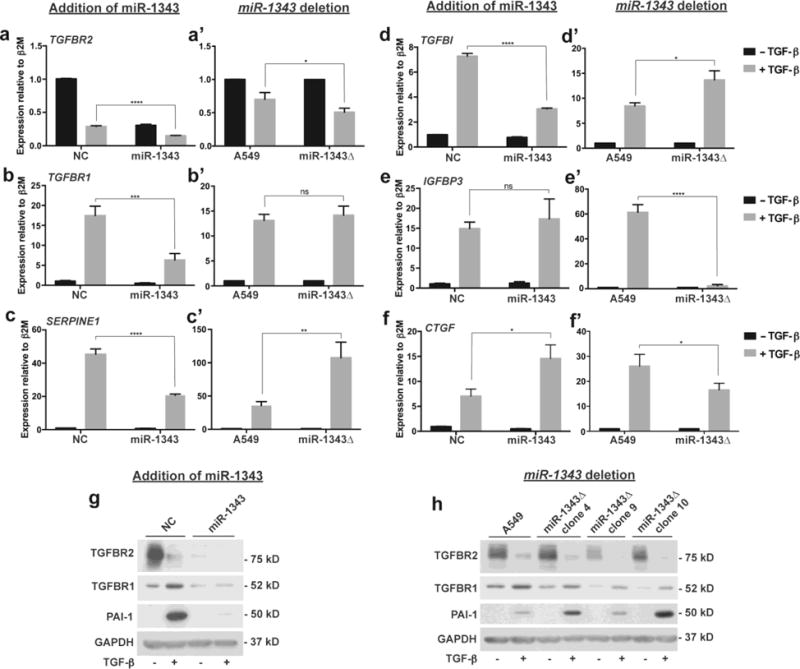
a-f’. Each pair of panels show gene expression levels in A549 cells measured by qRT-PCR assays following treatment with TGF-β (5 ng/mL, +; grey bars) or vehicle control (−; black bars) for 24 hours. The left panel (a-f) shows expression levels following transient transfection of miR-1343 or negative control (NC) miRNA, n=3. The right panel (a’-f’) shows expression levels in A549 cells versus 3 independent miR-1343 deletion (miR-1343Δ) clones, each assayed in triplicate (n=3). Ct values were normalized to beta-2-microglobulin (β2M). (a,a’) TGFBR2. (b,b’) TGFBR1. (c,c’) SERPINE1. (d,c’) TGFBI. (e,e’) IGFBP3. (f,f’) CTGF. For all panels, *p≤0.05, **p≤0.01, ***p≤0.001, ****p≤0.0001, ns=not significant by ANOVA and Tukey-Kramer post hoc test between TGF-β treatment groups. g. Western blots of A549 lysates transiently transfected with miRs and treated with TGF-β as above and probed with antibodies specific to TGFBR2, TGFBR1, and PAI-1 (protein encoded by SERPINE1) or GAPDH as the loading control. h. Western blots of A549 cells and 3 independent miR-1343Δ clones treated with TGF-β as above and probed with antibodies as in (g).
Next, RT-qPCR was performed to measure expression of Serpin family E member 1 (SERPINE1, Fig. 2c,c’) and Transforming growth factor beta induced (TGFBI, Fig. 2d,d’), both genes that are TGF-β-responsive (Skonier et al. 1992; Boehm et al. 1999). Also assayed were Insulin-like growth factor binding protein 3 (IGFBP3, Fig. 2e,e’) and Connective tissue growth factor (CTGF, Fig. 2f,f’), which were differentially expressed following miR-1343 overexpression and deletion in our RNA-seq data. These two proteins are similarly implicated in TGF-β signaling and fibrosis (Cohen et al. 2000; Gore-Hyer et al. 2002; Grotendorst et al. 2004; Flynn et al. 2011). As expected, expression of SERPINE1, TGFBI, IGFBP3, and CTGF were all significantly up-regulated by TGF-β in NC-transfected cells. The induction of SERPINE1 and TGFBI was attenuated by miR-1343. Conversely, the increase in SERPINE1 and TGFBI expression was enhanced by loss of miR-1343, indicating a direct role of the miR in their regulation. Interestingly, miR-1343 overexpression had no apparent effect on IGFBP3 stimulation by TGF-β, yet this induction was abolished by loss of the miR. This result suggests that the effects of miR-1343 deletion on gene expression may be further enhanced by stimulation with TGF-β. Induction of CTGF by TGF-β, in contrast, was increased by miR-1343 overexpression and decreased by deletion of the miR.
To confirm that changes in the transcript levels of miR-1343 target genes were evident in their encoded proteins, we next performed western blots on protein lysates from cells treated as before and separated by SDS-PAGE. Blots were probed with antibodies specific for TGFBR2, TGFBR1, and Plasminogen activator inhibitor type 1 (PAI-1, the protein encoded by SERPINE1) and normalized to GAPDH as a loading control (Fig. 2g-h, Suppl. Fig. 3a-c). Stimulation with TGF-β decreased levels of TGFBR2 and increased TGFBR1 and PAI-1 abundance in cells treated with NC miRNA, and consistent with the RT-qPCR data, these levels were attenuated by miR-1343. Also in agreement with transcript expression, TGFBR2 levels were slightly reduced, TGFBR2 levels were unchanged, and PAI-1 levels were increased in miR-1343 deletion clones compared to non-clonal A549 cells. Overall, these results confirm our previous report on the impact of miR-1343 on protein expression and reveal the potent effects of miR-1343 in reducing activity of canonical TGF-β signaling. Further, they demonstrate an important role for miR-1343 in A549 cells, and identify several key genes involved in fibrosis that are regulated downstream of the TGF-β-miR-1343 axis.
Utilizing extracellular vesicles for miR-1343 delivery
Since our results reinforced an important function for miR-1343 in lung epithelial cells, our next goal was to determine mechanisms whereby it could be used as a therapeutic against fibrotic lung disease. The absence of miR-1343 and low conservation of its seed sites within target genes in rodents excludes a small animal model for these experiments. Hence we concentrated on protocols for use in human tissue culture or ex vivo-based methods. As a first step, we examined whether miR-1343 was amenable to use in a common microRNA delivery platform: extracellular vesicles, specifically exosomes. It is known that microRNAs can act as signaling molecules, often traveling between distant cell types and exerting their negative regulatory effects independently from the cell they were synthesized within (reviewed in (Zhang et al. 2015). Since our previous data indicated that miR-1343 is most highly expressed in human neutrophils (Stolzenburg et al. 2016), we first tested whether this cell type could generate and release the miR via exosomes. We chose the HL-60 neutrophil-like cell line for these experiments, since it was shown previously to produce exosomes (Huan et al. 2013). A miR-1343 expression vector (in which ~300 bp encompassing the miR-1343 stem-loop is driven by the cytomegalovirus (CMV) promoter (pCMV-MIR-1343, Origene)) or the pCMV-MIR empty vector (vector control, VC) were electroporated into HL-60 cells. After a 48 hour incubation of these cells in exosome-depleted media, secreted exosomes were purified by membrane affinity spin column. Exosome lysates were separated by non-reducing SDS-PAGE and probed with an antibody specific for CD81, an exosomal marker (Fig. 3a). CD81 was detected in exosomes released by both vector control and miR-1343 over-expressing cells. Next, RNA was extracted from the exosomes and the donating HL-60 cells, DNase I digested, and miR-1343 expression was measured by an RT-qPCR TaqMan assay specific for the mature form of the miR. Data were normalized either against a standard curve (Fig. 3b), or relative to the geometric mean of miR-186 and miR-423, which are exosome-associated miRs that show stable expression levels (Fig. 3c). In both analyses, pCMV-MIR-1343-transfected HL-60 cells and exosomes released by them contained substantially more miR-1343 than did VC cells and their exosomes. The two analysis methods differ in the observed relative levels of the miR, but both indicate that HL-60 cells can process and release high levels of mature miR-1343 into exosomes. Notably, overexpression of miR-1343 had no measurable effect on transcript levels of the target genes TGFBR1 or TGFBR2 within the transfected HL-60 cells, suggesting that its function was directed to an extracellular compartment (Fig. 3d).
Fig. 3. miR-1343 is shuttled into exosomes produced by HL-60 cells.
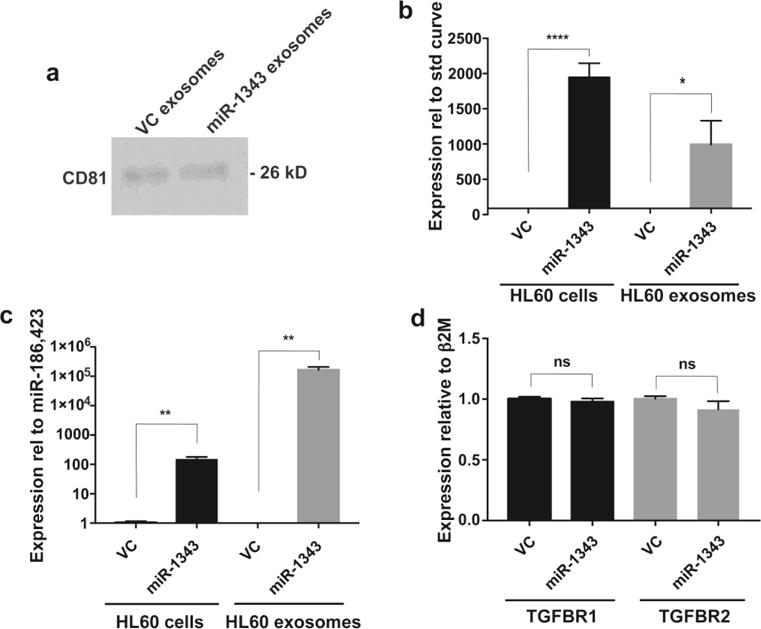
a. Western blot of lysates from HL-60 exosomes probed with an antibody specific for CD81. Cells were electroporated with either the pCMV-MIR vector control (VC) or pCMV-MIR-1343 (miR-1343) and exosomes were collected from the media after 48 hours. b.,c. miR-1343 levels measured by TaqMan qRT-PCR assay in HL-60 cells electroporated with VC or pCMV-MIR-1343 and corresponding exosomes. n=3. Values are normalized to a standard curve (see methods) in (b) or to the geometric mean of miR-183 and miR-423 in (c). d. TGFBR1 (black bars) and TGFBR2 (grey bars) gene expression levels measured by qRT-PCR assay in HL-60 cells following electroporation with VC or pCMV-MIR-1343 for 48 hours. Ct values were normalized to beta-2-microglobulin (β2M). n=6. For all panels, *p≤0.05, **p≤0.01, ****p≤0.0001, ns=not significant by unpaired Student’s t test.
miR-1343-containing exosomes can be transferred between cell types
We next asked whether miR-1343-containing exosomes from over-expressing HL-60 cells could be transferred to A549 lung epithelial cells or primary lung fibroblasts, as we showed previously that these cells were highly responsive to miR-1343 (Stolzenburg et al. 2016). HL-60 cells were electroporated with pCMV-MIR-1343 or empty VC plasmids and incubated for exosome release as above. Three hours prior to purification, the culture media was labeled with a membrane-intercalating dye, BODIPY TR Ceramide. Media without HL-60 cells was also labeled as a negative control. Exosomes were then purified, excess dye was removed, and A549 cells were directly treated with the labeled exosomes for 2 hours. Cells were simultaneously counterstained with Calcein AM and Hoechst 33258 to label the cytoplasmic and nuclear compartments, respectively, and then washed, fixed, and examined by fluorescence microscopy (Fig. 4a-c’”). The A549 cells treated with both VC and miR-1343 exosomes exhibited abundant BODIPY TR Ceramide labeling throughout the cytoplasm (Fig. 4b”,c”), which is the expected cellular compartment for exosome uptake. Confocal images show the Ceramide staining to be localized within the cell, particularly in the same plane as the nucleus, indicating that exosomes were taken up by the cell and not simply retained on the cell surface (Fig. 4d). In addition, an absence of staining was evident in the negative control cells (Fig. 4a”), indicating the BODIPY TR Ceramide labeling was due to uptake of exosomes and not unincorporated dye.
Fig. 4. Exosomes from HL-60 cells can be delivered to A549 cells.
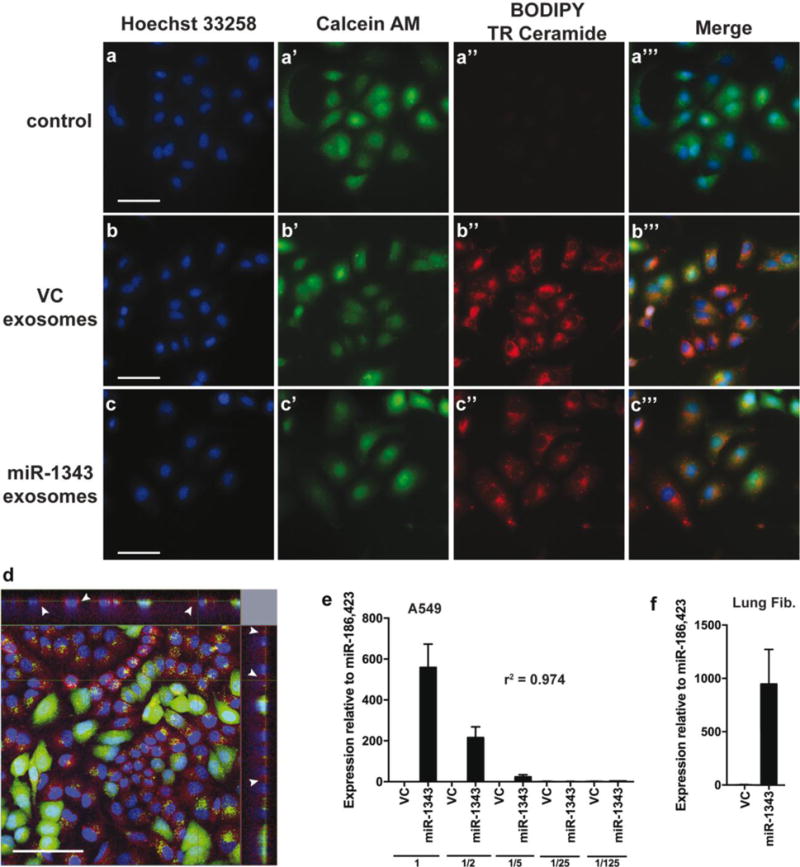
a-c’”. A549 cells treated with labeled exosomes. HL-60 cells were electroporated with pCMV-MIR vector control (VC) or pCMV-MIR-1343 (miR-1343). Media was collected after 48 hours and stained with BODIPY TR Ceramide (a”,b”,c”; red) for 3 hours prior to exosome isolation. A549 cells were incubated with labeled exosomes and counterstained with Hoechst 33258 (a,b,c; nucleus, blue) and Calcein AM (a’,b’,c’; cytoplasm, green) for 2 hours. The Merge panels (a’”,b’”,c’”) show Hoechst, Calcein AM, and BODIPY TR staining. Scale bar = 50 μm. d. Representative confocal image of A549 cells treated with labeled exosomes (red, shown by white arrows) as described in (a-c’”). The top and right panels show cross-sectional views. Scale bar = 100 μm. Colors as in (a-c’”). e.,f. miR-1343 levels measured by TaqMan Advanced qRT-PCR assay (e) in A549 cells treated with HL-60 exosomes. Following purification, exosomes were serially diluted with elution buffer (1=undiluted, 1/2=50% diluted, 1/5=80% diluted, etc.), directly added to A549 cells, and incubated for 48 hours. Values are normalized to the geometric mean of miR-186 and miR-423. R-squared (r2) value indicates the correlation coefficient. n=3. (f) In primary lung fibroblasts treated with an undiluted dose of HL-60 exosomes as in (e). n=2.
Next, to determine whether miR-1343 from exosomes could be transferred to recipient cell types, HL-60 cells were electroporated and exosomes purified as above. Exosomes were then serially diluted (to 1/125 fold) and incubated with A549 cells for 48 hours. RNA was isolated from the recipient cells, and levels of miR-1343 were measured via TaqMan assay (Fig. 4e). We found that miR-1343 expression strongly correlated with exosome dose (r2 = 0.974), with the highest dose exhibiting ~600-fold more miR-1343 compared to VC exosomes. Similarly, primary lung fibroblasts treated with miR-1343-containing exosomes also showed high levels of miR-1343 compared to the VC exosomes (Fig. 4f). These results indicate that miR-1343 can be transferred between cell types via exosome secretion and subsequent uptake.
Exosomes containing miR-1343 can repress fibrotic processes in recipient cells
To determine whether exosomal-miR-1343 is functional in recipient cells, we next assayed A549 cells and primary lung fibroblasts for changes in miR-1343 targets following exosome treatment. Exosomes were purified from VC- or pCMV-MIR-1343-electroporated HL-60 cells as above, and then dialyzed in phosphate buffered saline (PBS). This buffer exchange was necessary to circumvent an observed down-regulation of TGFBR2 protein expression associated with the exosome elution Buffer XE (Suppl. Fig. 4a), and did not result in significant loss of exosomes (Suppl. Fig. 4b) or miR-1343 levels (Suppl. Fig. 4c). Following dialysis, exosomes were immediately added to A549 cells in exosome-free medium and incubated for 48 hours prior to RNA or protein extraction. RT-qPCR assays for TGFBR1 and TGFBR2, two known miR-1343 target genes (Stolzenburg et al. 2016), did not show significant decreases in transcript levels (Fig. 5a). Similarly, miR-1343 exosomes had no significant effect on TGFBR2 levels compared to VC exosomes in either cell type when lysates from exosome-treated A549 cells and primary lung fibroblasts were separated by SDS-PAGE and western blots probed with antibodies against TGFBR2 or GAPDH as a control (Fig. 5b,b’).
Fig. 5. miR-1343-containing exosomes reduce TGF-β signaling and markers of fibrosis.
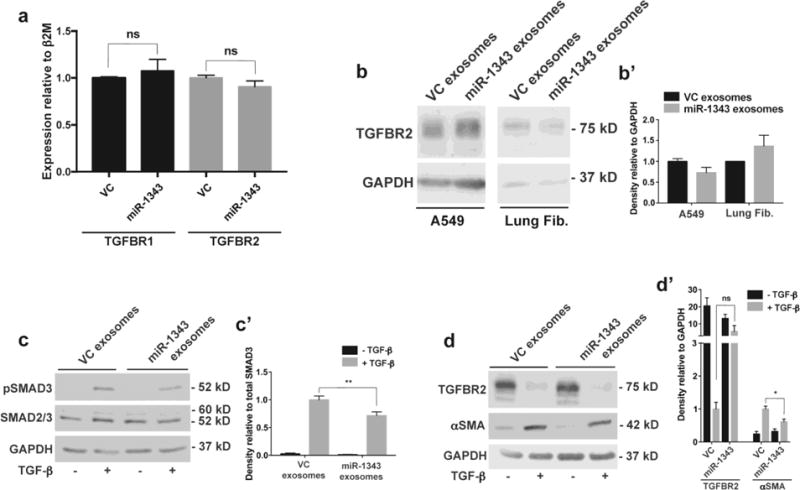
a. TGFBR1 (black bars) and TGFBR2 (grey bars) mRNA measured by qRT-PCR in A549 cells treated for 48 hours with exosomes purified from pCMV-MIR (VC) or pCMV-MIR-1343 (miR-1343) electroporated HL-60 cells. Ct values were normalized to beta-2-microglobulin (β2M). n=4. ns=not significant by unpaired Student’s t test. b. Western blot of lysates from A549 cells (left) and primary lung fibroblasts (right) treated with VC or miR-1343 exosomes as in (a). Blots were probed with antibodies specific for TGFBR2 or GAPDH as the loading control. b’. Proteins were quantified relative to GAPDH and are expressed as fold change compared to VC, n=2. c-c’. Western blot of lysates from primary lung fibroblasts treated with VC or miR-1343 exosomes as in (a) and treated with TGF-β1 (5 ng/mL, +; grey bars) or vehicle control (−; black bars) for 1 hr. (c) Blots were probed with antibodies specific for pSMAD3 (phosphorylated [active] SMAD3), total SMAD2/3 (SMAD2 larger form and SMAD3 below) or GAPDH. (c’) Proteins were quantified relative to total SMAD3 and shown as fold change compared to VC +TGF-β, n=4. **p≤0.01 by ANOVA and Tukey-Kramer post hoc test between TGF-β treatment groups. d-d’. Blots as in (cc’) treated with TGF-β1 for 48 hrs, and (d) probed with antibodies specific for TGFBR2, αSMA, or GAPDH. (d’) Proteins were quantified relative to GAPDH and shown as fold change compared to VC +TGF-β. n=4. *p≤0.05, ns=not significant by ANOVA and Tukey-Kramer post hoc test between TGF-β treatment groups.
Since miR-1343 delivered in exosomes could have a more subtle effect on target gene expression than transfection of non-physiological levels of the miR, we predicted that stimulation with TGF-β might be required to measure an effect. To investigate this possibility, primary lung fibroblasts were treated with dialyzed exosomes as described above and, approximately 20 hours later, TGF-β was added to the culture. Following 1 hr of TGF-β treatment, cell lysates were resolved by SDS-PAGE and probed with antibodies specific for pSMAD3 or total SMAD2/3 (Fig. 5c,c’). Levels of pSMAD3 relative to total SMAD3 were significantly diminished in fibroblasts treated with TGF-β and miR-1343 exosomes compared to the VC exosomes. Similarly, cell lysates treated with exosomes and TGF-β for 48 hrs were resolved by SDS-PAGE and probed with antibodies specific for TGFBR2 or αSMA (Fig. 5d,d’). Induction of αSMA relative to GAPDH was significantly reduced following TGF-β and miR-1343 exosome treatment. TGFBR2 levels trended toward an increase with TGF-β and miR-1343 exosome treatment, though this did not reach statistical significance. Though pSMAD3 and αSMA were not reduced to the same extent compared to miR-1343 transient overexpression, these data are consistent with our results reported previously (Stolzenburg et al. 2016), and suggest that miR-1343 delivered by exosomes can diminish TGF-β signaling and induction of fibrotic phenotypes in recipient cells.
DISCUSSION
The role of miR-1343 in reducing TGF-β signaling and phenotypes associated with fibrosis was demonstrated previously (Stolzenburg et al. 2016). However, previous experiments relied on synthetic miR-1343 to study its function, and the contribution of the endogenous miR was not investigated. Through deletion of the miR-1343 locus in A549 cells, we demonstrate by RNA-seq that endogenous miR-1343 directly regulates multiple genes involved in TGF-β signaling and fibrosis. These include TGFBR1, which is repressed by miR-1343 overexpression (Stolzenburg et al. 2016) and increased by miR-1343 deletion and conversely IGFBP3 and CTGF, which are both upregulated by overexpression (Stolzenburg et al. 2016) and decreased by deletion. As a direct target of miR-1343, the TGFBR1 result is consistent with predictions (Stolzenburg et al. 2016). Mutations in TGFBR1 are evident in Loeys-Dietz syndrome, which presents with skin, cardiovascular, and connective tissue defects that are the reverse of fibrotic phenotypes (Loeys et al. 2005). IGFBP3 functions by binding to insulin-like growth factors to regulate their activity, either promoting or restricting cell growth based on context (Martin and Baxter 1986). Its expression is known to be induced by TGF-β and is upregulated in Crohn’s disease and idiopathic pulmonary fibrosis (Pilewski et al. 2005; Flynn et al. 2011; Schedlich et al. 2013). Our finding that IGFBP3 stimulation by TGF-β is abolished in the absence of miR-1343 suggests a more extensive role for the miR in regulating this gene, which likely occurs through indirect mechanisms. CTGF is also induced by TGF-β and has a broad spectrum of roles in fibrosis (Duncan et al. 1999; Xie et al. 2004; Brigstock 2010). Though CTGF is critically important in development, as evidenced by the failure of null mice to survive, its overexpression promotes fibrotic disease in a variety of organ systems (Ivkovic et al. 2003; Sonnylal et al. 2010). Contradictory to our predicted function of miR-1343 in reducing fibrosis and our RNA-seq data, we find the miR to upregulate CTGF expression by RT-qPCR. Data showing that CTGF stimulation by TGF-β is mediated through the ERK and JNK pathways (Xie et al. 2004) suggests that this effect may be due to parallel pro-fibrotic pathways that are upregulated in response to downregulation of SMAD signaling.
RNA-seq of miR-1343Δ clones only identified a fraction of the number of DEGs seen in our previous overexpression experiment (Stolzenburg et al. 2016). It is likely that some off-target effects arose during over-expression of the miR. However, the substantial variation in expression profiles of the NT clones, necessitating an additional comparison to non-clonal A549 cells, may have further reduced the number of DEGs. Of note, others have also shown that most microRNA deletions have a more subtle phenotype, or none at all (Miska et al. 2007). The relatively low expression of miR-1343 in human cells and its low conservation across species predict a modest effect for this miR. Molecular redundancy of microRNAs (families sharing similar targeting sequences, for example) and their apparent function in fine-tuning (rather than complete abolition) of complex regulatory networks may explain this phenomenon (Park et al. 2010; Lai 2015). Nonetheless, many miRNA knockouts exhibit phenotypes under stress, suggesting that network buffering may mask the phenotype (Li et al.; van Rooij et al. 2007; Simone et al. 2009; Park et al. 2010). In agreement, we find that stimulation with TGF-β was required to reveal certain functions of miR-1343, as measured through effects on SERPINE1 (PAI-1), TGFBI, IGFBP3, and CTGF expression. This result illustrates that useful study of the effect of miR-1343 deletion requires an appropriate stimulus. Additional studies utilizing TGF-β treatment and other stressors may be required to determine the full impact of miR-1343Δ on a cell.
Consistent with the importance of this miR in a disease-relevant cellular process, we find that mature miR-1343 is processed by HL-60 cells and can be delivered to distinct cell types via exosomes. These data describe the cellular fate of many molecules, including microRNAs, mRNAs, and peptides, following their synthesis. This concept was proposed by Valadi and colleagues, who found that exosomes, containing RNA from as many as 1300 genes, were secreted by one cell type and taken up by another (Valadi et al. 2007). Many of the exosomal-RNAs were not found in the donor cell cytoplasm and were only translated upon uptake. Similarly, the trafficking of miR-1343 into exosomes could prevent its ability to function as a gene regulator in HL-60 cells. Gaining a better understanding of the types of molecules that are shuttled into exosomes in different cell types will facilitate their development as a medical tool for assaying biomarkers of disease (Properzi et al. 2013; Sandfeld-Paulsen et al. 2016). Furthermore, learning how to manipulate the contents and destinations of exosomes could have additional consequences for future therapies, not only for fibrosis, but a variety of diseases.
Expression of miR-1343 from the pCMV-MIR vector did not appear to have an effect on its previously documented targets, TGFBR1 and TGFBR2 in the host HL-60 cells. Furthermore, we did not initially observe a significant effect of the miR following exosome delivery to A549 cells and primary lung fibroblasts under resting conditions. However, when stimulated with TGF-β, we detected significant changes in markers of fibrosis (pSMAD3 and αSMA) in the lung fibroblasts, though this effect was more subtle than found through transient overexpression (Stolzenburg et al. 2016). Several theories may explain this. First, the abundance of miR-1343 expressed from pCMV-MIR is substantially lower than is achieved by transient overexpression of the pre-miR. Second, the pre-miRs synthesized by Life Technologies are probably chemically modified to increase their stability and functionality (though this information is proprietary), while miR-1343 in this study was expressed in its native form. Lipid-based transfection tends to force RNA into a cell, while vector-based expression and exosome-mediated delivery is a more gentle process that may affect miR compartmentalization and signaling capabilities. Tapering the amount of transfected microRNA may solve some of these problems, though our results demonstrate that this method is largely not ideal for assessing in vitro miR function. Further refinement of the exosome production, purification, and treatment methods described in this study are necessary to fully address these questions.
Despite the current lack of a small animal model for studying the role of miR-1343 in fibrosis, our data posit the miR as a compelling candidate for future therapeutic approaches. Since miR-1343 is well-conserved in pigs (and was identified in exosomes from porcine breast milk), a large animal model may be available to accurately study its role in TGF-β signaling and pathways of fibrosis (Chen et al. 2012, 2014). Proper formulation of miR-1343, as well as the identification of a robust delivery mechanism, will be required for its use as a treatment. Several exosome- and nanoparticle-based delivery platforms are currently under review in clinical applications (Alvarez-Erviti et al. 2011; Ohno et al. 2013; Wang et al. 2016). As these novel treatment technologies are advanced, miR-1343 stands out as a promising candidate for both preventative and targeted therapies against diseases involving overactive TGF-β signaling and fibrosis.
Supplementary Material
Acknowledgments
We thank Dr. Scott Randell, Dr. Wanda O’Neal and Lisa Jones (University of North Carolina Marsico Lung Institute) for the primary lung fibroblast cultures and A549 RNA-seq.
FUNDING
This work was supported by the National Institutes of Health (R01HL117843 to AH; F31HL126458 to LRS) and the Children’s Research Fund.
References
- Akhurst RJ, Hata A. Targeting the TGFβ signalling pathway in disease. Nat Rev Drug Discov. 2012;11:790–811. doi: 10.1038/nrd3810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alvarez-Erviti L, Seow Y, Yin H, Betts C, Lakhal S, Wood MJA. Delivery of siRNA to the mouse brain by systemic injection of targeted exosomes. Nat Biotechnol. 2011;29:341–345. doi: 10.1038/nbt.1807. [DOI] [PubMed] [Google Scholar]
- Boehm JR, Kutz SM, Sage EH, Staiano-Coico L, Higgins PJ. Growth state-dependent regulation of plasminogen activator inhibitor type-1 gene expression during epithelial cell stimulation by serum and transforming growth factor-beta1. J Cell Physiol. 1999;181:96–106. doi: 10.1002/(SICI)1097-4652(199910)181:1<96::AID-JCP10>3.0.CO;2-I. [DOI] [PubMed] [Google Scholar]
- Brigstock DR. Connective tissue growth factor (CCN2, CTGF) and organ fibrosis: lessons from transgenic animals. J Cell Commun Signal. 2010;4:1–4. doi: 10.1007/s12079-009-0071-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen C, Deng B, Qiao M, Zheng R, Chai J, Ding Y, Peng J, Jiang S. Solexa sequencing identification of conserved and novel microRNAs in backfat of Large White and Chinese Meishan pigs. PLoS One. 2012;7:e31426. doi: 10.1371/journal.pone.0031426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen T, Xi Q-Y, Ye R-S, Cheng X, Qi Q-E, Wang S-B, Shu G, Wang L-N, Zhu X-T, Jiang Q-Y, Zhang Y-L. Exploration of microRNAs in porcine milk exosomes. BMC Genomics. 2014;15:100. doi: 10.1186/1471-2164-15-100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiba M, Kimura M, Asari S. Exosomes secreted from human colorectal cancer cell lines contain mRNAs, microRNAs and natural antisense RNAs, that can transfer into the human hepatoma HepG2 and lung cancer A549 cell lines. Oncol Rep. 2012;28:1551–8. doi: 10.3892/or.2012.1967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christopher AF, Kaur RP, Kaur G, Kaur A, Gupta V, Bansal P. MicroRNA therapeutics: Discovering novel targets and developing specific therapy. Perspect Clin Res. 2016;7:68–74. doi: 10.4103/2229-3485.179431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen P, Rajah R, Rosenbloom J, Herrick DJ. IGFBP-3 mediates TGF-beta1-induced cell growth in human airway smooth muscle cells. Am J Physiol Lung Cell Mol Physiol. 2000;278:L545–51. doi: 10.1152/ajplung.2000.278.3.L545. [DOI] [PubMed] [Google Scholar]
- Collins SJ, Gallo RC, Gallagher RE. Continuous growth and differentiation of human myeloid leukaemic cells in suspension culture. Nature. 1977;270:347–9. doi: 10.1038/270347a0. [DOI] [PubMed] [Google Scholar]
- Cong L, Ran FA, Cox D, Lin S, Barretto R, Habib N, Hsu PD, Wu X, Jiang W, Marraffini LA, Zhang F. Multiplex Genome Engineering Using CRISPR/Cas Systems. Science (80-) 2013;339:819–823. doi: 10.1126/science.1231143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corvol H, Blackman SM, Boëlle P-Y, Gallins PJ, Pace RG, Stonebraker JR, Accurso FJ, Clement A, Collaco JM, Dang H, Dang AT, Franca A, Gong J, Guillot L, Keenan K, Li W, Lin F, Knowles MR. Genome-wide association meta-analysis identifies five modifier loci of lung disease severity in cystic fibrosis. Nat Commun. 2015;6:8382. doi: 10.1038/ncomms9382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Derynck R, Zhang YE. Smad-dependent and Smad-independent pathways in TGF-β family signalling. Nature. 2003;425:577–584. doi: 10.1038/nature02006. [DOI] [PubMed] [Google Scholar]
- Duncan MR, Frazier KS, Abramson S, Williams S, Klapper H, Huang X, Grotendorst GR. Connective tissue growth factor mediates transforming growth factor beta-induced collagen synthesis: down-regulation by cAMP. FASEB J. 1999;13:1774–86. [PubMed] [Google Scholar]
- Flynn RS, Mahavadi S, Murthy KS, Grider JR, Kellum JM, Akbari H, Kuemmerle JF. Endogenous IGFBP-3 regulates excess collagen expression in intestinal smooth muscle cells of Crohn’s disease strictures. Inflamm Bowel Dis. 2011;17:193–201. doi: 10.1002/ibd.21351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fossum SL, Mutolo MJ, Yang R, Dang H, O’Neal WK, Knowles MR, Leir S-H, Harris A. Ets homologous factor regulates pathways controlling response to injury in airway epithelial cells. Nucleic Acids Res. 2014;42:13588–98. doi: 10.1093/nar/gku1146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallo A, Tandon M, Alevizos I, Illei GG. The majority of microRNAs detectable in serum and saliva is concentrated in exosomes. PLoS One. 2012;7:e30679. doi: 10.1371/journal.pone.0030679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giard DJ, Aaronson SA, Todaro GJ, Arnstein P, Kersey JH, Dosik H, Parks WP. In vitro cultivation of human tumors: establishment of cell lines derived from a series of solid tumors. J Natl Cancer Inst. 1973;51:1417–23. doi: 10.1093/jnci/51.5.1417. [DOI] [PubMed] [Google Scholar]
- Gore-Hyer E, Shegogue D, Markiewicz M, Lo S, Hazen-Martin D, Greene EL, Grotendorst G, Trojanowska M. TGF-beta and CTGF have overlapping and distinct fibrogenic effects on human renal cells. Am J Physiol Renal Physiol. 2002;283:F707–16. doi: 10.1152/ajprenal.00007.2002. [DOI] [PubMed] [Google Scholar]
- Grotendorst GR, Rahmanie H, Duncan MR. Combinatorial signaling pathways determine fibroblast proliferation and myofibroblast differentiation. FASEB J. 2004;18:469–479. doi: 10.1096/fj.03-0699com. [DOI] [PubMed] [Google Scholar]
- Harding C, Heuser J, Stahl P. Endocytosis and intracellular processing of transferrin and colloidal gold-transferrin in rat reticulocytes: demonstration of a pathway for receptor shedding. Eur J Cell Biol. 1984;35:256–63. [PubMed] [Google Scholar]
- Huan J, Hornick NI, Shurtleff MJ, Skinner AM, Goloviznina NA, Roberts CT, Kurre P. RNA trafficking by acute myelogenous leukemia exosomes. Cancer Res. 2013;73:918–29. doi: 10.1158/0008-5472.CAN-12-2184. [DOI] [PubMed] [Google Scholar]
- Huang DW, Sherman BT, Lempicki RA. Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res. 2009a;37:1–13. doi: 10.1093/nar/gkn923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang DW, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc. 2009b;4:44–57. doi: 10.1038/nprot.2008.211. [DOI] [PubMed] [Google Scholar]
- Ivkovic S, Yoon BS, Popoff SN, Safadi FF, Libuda DE, Stephenson RC, Daluiski A, Lyons KM. Connective tissue growth factor coordinates chondrogenesis and angiogenesis during skeletal development. Development. 2003;130:2779–91. doi: 10.1242/dev.00505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jinek M, Chylinski K, Fonfara I, Hauer M, Doudna JA, Charpentier E. A Programmable Dual-RNA-Guided DNA Endonuclease in Adaptive Bacterial Immunity. Science (80-) 2012;337:816–821. doi: 10.1126/science.1225829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jinek M, East A, Cheng A, Lin S, Ma E, Doudna J. RNA-programmed genome editing in human cells. Elife. 2013;2:e00471. doi: 10.7554/eLife.00471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krützfeldt J, Rajewsky N, Braich R, Rajeev KG, Tuschl T, Manoharan M, Stoffel M. Silencing of microRNAs in vivo with “antagomirs”. Nature. 2005;438:685–689. doi: 10.1038/nature04303. [DOI] [PubMed] [Google Scholar]
- Lai EC. Two decades of miRNA biology: lessons and challenges. RNA. 2015;21:675–7. doi: 10.1261/rna.051193.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leask A, Abraham DJ. TGF-beta signaling and the fibrotic response. FASEB J. 2004;18:816–27. doi: 10.1096/fj.03-1273rev. [DOI] [PubMed] [Google Scholar]
- Li G, Luna C, Qiu J, Epstein DL, Gonzalez P. Alterations in microRNA expression in stress-induced cellular senescence. Mech Ageing Dev. 130:731–41. doi: 10.1016/j.mad.2009.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loeys BL, Chen J, Neptune ER, Judge DP, Podowski M, Holm T, Meyers J, Leitch CC, Katsanis N, Sharifi N, Xu FL, Myers LA, Spevak PJ, Cameron DE, Backer J De, Hellemans J, Chen Y, Dietz HC. A syndrome of altered cardiovascular, craniofacial, neurocognitive and skeletal development caused by mutations in TGFBR1 or TGFBR2. Nat Genet. 2005;37:275–281. doi: 10.1038/ng1511. [DOI] [PubMed] [Google Scholar]
- Lv L-L, Cao Y, Liu D, Xu M, Liu H, Tang R-N, Ma K-L, Liu B-C. Isolation and quantification of microRNAs from urinary exosomes/microvesicles for biomarker discovery. Int J Biol Sci. 2013;9:1021–31. doi: 10.7150/ijbs.6100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mali P, Yang L, Esvelt KM, Aach J, Guell M, DiCarlo JE, Norville JE, Church GM. RNA-Guided Human Genome Engineering via Cas9. Science (80-) 2013;339:823–826. doi: 10.1126/science.1232033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin JL, Baxter RC. Insulin-like growth factor-binding protein from human plasma. Purification and characterization. J Biol Chem. 1986;261:8754–60. [PubMed] [Google Scholar]
- Meckes DG, Shair KHY, Marquitz AR, Kung C-P, Edwards RH, Raab-Traub N. Human tumor virus utilizes exosomes for intercellular communication. Proc Natl Acad Sci USA. 2010;107:20370–5. doi: 10.1073/pnas.1014194107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miska EA, Alvarez-Saavedra E, Abbott AL, Lau NC, Hellman AB, McGonagle SM, Bartel DP, Ambros VR, Horvitz HR. Most Caenorhabditis elegans microRNAs Are Individually Not Essential for Development or Viability. PLoS Genet. 2007;3:e215. doi: 10.1371/journal.pgen.0030215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Reilly S. MicroRNAs in fibrosis: opportunities and challenges. Arthritis Res Ther. 2016;18:11. doi: 10.1186/s13075-016-0929-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohno S, Takanashi M, Sudo K, Ueda S, Ishikawa A, Matsuyama N, Fujita K, Mizutani T, Ohgi T, Ochiya T, Gotoh N, Kuroda M. Systemically Injected Exosomes Targeted to EGFR Deliver Antitumor MicroRNA to Breast Cancer Cells. Mol Ther. 2013;21:185–191. doi: 10.1038/mt.2012.180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan BT, Johnstone RM. Fate of the transferrin receptor during maturation of sheep reticulocytes in vitro: selective externalization of the receptor. Cell. 1983;33:967–78. doi: 10.1016/0092-8674(83)90040-5. [DOI] [PubMed] [Google Scholar]
- Park CY, Choi YS, McManus MT. Analysis of microRNA knockouts in mice. Hum Mol Genet. 2010;19:R169–75. doi: 10.1093/hmg/ddq367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pegtel DM, Cosmopoulos K, Thorley-Lawson DA, van Eijndhoven MAJ, Hopmans ES, Lindenberg JL, de Gruijl TD, Wurdinger T, Middeldorp JM. Functional delivery of viral miRNAs via exosomes. Proc Natl Acad Sci. 2010;107:6328–6333. doi: 10.1073/pnas.0914843107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pilewski JM, Liu L, Henry AC, Knauer AV, Feghali-Bostwick CA. Insulin-like growth factor binding proteins 3 and 5 are overexpressed in idiopathic pulmonary fibrosis and contribute to extracellular matrix deposition. Am J Pathol. 2005;166:399–407. doi: 10.1016/S0002-9440(10)62263-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Properzi F, Logozzi M, Fais S. Exosomes: the future of biomarkers in medicine. Biomark Med. 2013;7:769–778. doi: 10.2217/bmm.13.63. [DOI] [PubMed] [Google Scholar]
- Reimand J, Arak T, Adler P, Kolberg L, Reisberg S, Peterson H, Vilo J. g:Profiler-a web server for functional interpretation of gene lists (2016 update) Nucleic Acids Res. 2016;44:W83–9. doi: 10.1093/nar/gkw199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandfeld-Paulsen B, Jakobsen KR, Bæk R, Folkersen BH, Rasmussen TR, Meldgaard P, Varming K, Jørgensen MM, Sorensen BS. Exosomal Proteins as Diagnostic Biomarkers in Lung Cancer. J Thorac Oncol. 2016;11:1701–1710. doi: 10.1016/j.jtho.2016.05.034. [DOI] [PubMed] [Google Scholar]
- Schedlich LJ, Yenson VM, Baxter RC. TGF-β-induced expression of IGFBP-3 regulates IGF1R signaling in human osteosarcoma cells. Mol Cell Endocrinol. 2013;377:56–64. doi: 10.1016/j.mce.2013.06.033. [DOI] [PubMed] [Google Scholar]
- Shi Y, Massagué J. Mechanisms of TGF-beta signaling from cell membrane to the nucleus. Cell. 2003;113:685–700. doi: 10.1016/s0092-8674(03)00432-x. [DOI] [PubMed] [Google Scholar]
- Simone NL, Soule BP, Ly D, Saleh AD, Savage JE, Degraff W, Cook J, Harris CC, Gius D, Mitchell JB. Ionizing radiation-induced oxidative stress alters miRNA expression. PLoS One. 2009;4:e6377. doi: 10.1371/journal.pone.0006377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skonier J, Neubauer M, Madisen L, Bennett K, Plowman GD, Purchio AF. cDNA cloning and sequence analysis of beta ig-h3, a novel gene induced in a human adenocarcinoma cell line after treatment with transforming growth factor-beta. DNA Cell Biol. 1992;11:511–22. doi: 10.1089/dna.1992.11.511. [DOI] [PubMed] [Google Scholar]
- Sonnylal S, Shi-Wen X, Leoni P, Naff K, Van Pelt CS, Nakamura H, Leask A, Abraham D, Bou-Gharios G, de Crombrugghe B. Selective expression of connective tissue growth factor in fibroblasts in vivo promotes systemic tissue fibrosis. Arthritis Rheum. 2010;62:1523–1532. doi: 10.1002/art.27382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stolzenburg LR, Wachtel S, Dang H, Harris A. miR-1343 attenuates pathways of fibrosis by targeting the TGF-β receptors. Biochem J. 2016;473:245–56. doi: 10.1042/BJ20150821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapnell C, Roberts A, Goff L, Pertea G, Kim D, Kelley DR, Pimentel H, Salzberg SL, Rinn JL, Pachter L. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat Protoc. 2012;7:562–78. doi: 10.1038/nprot.2012.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Umezu T, Ohyashiki K, Kuroda M, Ohyashiki JH. Leukemia cell to endothelial cell communication via exosomal miRNAs. Oncogene. 2013;32:2747–2755. doi: 10.1038/onc.2012.295. [DOI] [PubMed] [Google Scholar]
- Valadi H, Ekström K, Bossios A, Sjöstrand M, Lee JJ, Lötvall JO. Exosome-mediated transfer of mRNAs and microRNAs is a novel mechanism of genetic exchange between cells. Nat Cell Biol. 2007;9:654–659. doi: 10.1038/ncb1596. [DOI] [PubMed] [Google Scholar]
- van Rooij E, Sutherland LB, Qi X, Richardson JA, Hill J, Olson EN. Control of Stress-Dependent Cardiac Growth and Gene Expression by a MicroRNA. Science (80-) 2007;316:575–579. doi: 10.1126/science.1139089. [DOI] [PubMed] [Google Scholar]
- Wang X, Hao L, Bu H-F, Scott AW, Tian K, Liu F, De Plaen IG, Liu Y, Mirkin CA, Tan X-D. Spherical nucleic acid targeting microRNA-99b enhances intestinal MFG-E8 gene expression and restores enterocyte migration in lipopolysaccharide-induced septic mice. Sci Rep. 2016;6:31687. doi: 10.1038/srep31687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright FA, Strug LJ, Doshi VK, Commander CW, Blackman SM, Sun L, Berthiaume Y, Cutler D, Cojocaru A, Collaco JM, Corey M, Dorfman R, Goddard K, Green D, Kent JW, Lange EM, Lee S, Cutting GR. Genome-wide association and linkage identify modifier loci of lung disease severity in cystic fibrosis at 11p13 and 20q13.2. Nat Genet. 2011;43:539–46. doi: 10.1038/ng.838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wynn TA. Integrating mechanisms of pulmonary fibrosis. J Exp Med. 2011;208:1339–50. doi: 10.1084/jem.20110551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie S, Sukkar MB, Issa R, Oltmanns U, Nicholson AG, Chung KF. Regulation of TGF- 1- induced connective tissue growth factor expression in airway smooth muscle cells. AJP Lung Cell Mol Physiol. 2004;288:L68–L76. doi: 10.1152/ajplung.00156.2004. [DOI] [PubMed] [Google Scholar]
- Yoo BH, Bochkareva E, Bochkarev A, Mou T-C, Gray DM. 2′-O-methyl-modified phosphorothioate antisense oligonucleotides have reduced non-specific effects in vitro. Nucleic Acids Res. 2004;32:2008–2016. doi: 10.1093/nar/gkh516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Li S, Li L, Li M, Guo C, Yao J, Mi S. Exosome and Exosomal MicroRNA: Trafficking, Sorting, and Function. Genomics Proteomics Bioinformatics. 2015;13:17–24. doi: 10.1016/j.gpb.2015.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Wang Z, Gemeinhart RA. Progress in microRNA delivery. J Control Release. 2013;172:962–74. doi: 10.1016/j.jconrel.2013.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Q, Li M, Wang X, Li Q, Wang T, Zhu Q, Zhou X, Wang X, Gao X, Li X. Immune-related microRNAs are abundant in breast milk exosomes. Int J Biol Sci. 2012;8:118–23. doi: 10.7150/ijbs.8.118. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.