

Abstract
Aim
The aim of the present study was to predict olanzapine (OLZ) exposure in individual patients using physiologically based pharmacokinetic modelling and simulation (PBPK M&S).
Methods
A ‘bottom‐up’ PBPK model for OLZ was constructed in Simcyp® (V14.1) and validated against pharmacokinetic studies and data from therapeutic drug monitoring (TDM). The physiological, demographic and genetic attributes of the ‘healthy volunteer population’ file in Simcyp® were then individualized to create ‘virtual twins’ of 14 patients. The predicted systemic exposure of OLZ in virtual twins was compared with measured concentration in corresponding patients. Predicted exposures were used to calculate a hypothetical decrease in exposure variability after OLZ dose adjustment.
Results
The pharmacokinetic parameters of OLZ from single‐dose studies were accurately predicted in healthy Caucasians [mean‐fold errors (MFEs) ranged from 0.68 to 1.14], healthy Chinese (MFEs 0.82 to 1.18) and geriatric Caucasians (MFEs 0.55 to 1.30). Cumulative frequency plots of trough OLZ concentration were comparable between the virtual population and patients in a TDM database. After creating virtual twins in Simcyp®, the R2 values for predicted vs. observed trough OLZ concentrations were 0.833 for the full cohort of 14 patients and 0.884 for the 7 patients who had additional cytochrome P450 2C8 genotyping. The variability in OLZ exposure following hypothetical dose adjustment guided by PBPK M&S was twofold lower compared with a fixed‐dose regimen – coefficient of variation values were 0.18 and 0.37, respectively.
Conclusions
Olanzapine exposure in individual patients was predicted using PBPK M&S. Repurposing of available PBPK M&S platforms is an option for model‐informed precision dosing and requires further study to examine clinical potential.
Keywords: dose prediction, olanzapine, PBPK, personalized medicine, physiologically based pharmacokinetics
What is Already Known about this Subject
Physiologically based pharmacokinetic modelling and simulation (PBPK M&S) is widely used in drug development to estimate pharmacokinetics in support of drug applications to regulatory agencies.
Olanzapine (OLZ) is an antipsychotic drug that exhibits large interindividual variability in pharmacokinetics (up to 10‐fold), the sources of which include ethnicity, smoking status, gender, age and comedication.
What this Study Adds
A validated PBPK model was constructed describing the PK of OLZ and its variability.
OLZ steady state trough concentrations were predicted in individual patients using PBPK M&S, including accurate identification of an ‘outlier’ patient.
A hypothetical decrease in drug exposure variability was demonstrated following dose adjustment based on ‘virtual twin’ predictions.
Proof‐of‐concept was demonstrated for model‐informed precision dosing after repurposing a widely available PBPK M&S platform.
Introduction
The need for individualized drug dosing as part of ‘personalized medicine’ has been recognized for many years 1. In this context, the traditional pharmacokinetic (PK)‐based approach to dose adjustment has utilized therapeutic drug monitoring (TDM), the direct measurement of plasma drug concentration 2. Disadvantages of this approach include its retrospective nature, in that the dose is adjusted after initial drug administration, the requirement for at least one time‐sensitive blood sample and the need for specific assays to measure the concentrations of each drug. Another approach is population PK ± pharmacodynamic (PD) modelling (‘population PKPD’), where the dose is predicted based on individual values of covariates that have been shown mathematically to influence PK and/or PD in the population. Examples include the prediction of metformin doses in patients with renal impairment 3 and the optimization of antibiotic dosing in the critically ill 4. However, some disadvantages of population PKPD for precision dosing include the tedious, labour‐intensive and time‐consuming nature of model building, lack of confidence in models built on small sample sizes, and difficulties in handling missing data, such as when a value for an important covariate in a particular patient is unknown 5. Recently, several platforms have become available that combine population PKPD models and TDM – a Bayesian approach is then used to predict subsequent dose from inputs to the model that include a recent drug concentration (e.g. DoseMe™, PK‐PD Compass™, TCI Works®, etc).
An alternative, albeit complementary, approach is based on the concept of matching the attributes of the real patient to those of his or her ‘virtual twin’, using a generic physiologically based PK (PBPK) model that accommodates for patient demographics, genetics, disease and comedication 6, 7. This so‐called ‘bottom‐up’ procedure requires the prior development and validation of specific compound files, constructed using physicochemical properties and the in vitro–in vivo extrapolation (IVIVE) of information on drug metabolism and transport in association with existing in vivo PK data 8. Application of the model to the prediction of drug exposure in specific patients then requires a means of marking the activity of specific enzymes and transporters in an individual that can be done rapidly, cheaply and as non‐invasively as possible. This can be achieved by prior phenotyping, with exogenous or endogenous marker compounds, and by genotyping that is calibrated to enzyme/transporter activity. The use of a PBPK model then allows diverse patient information to be integrated in predicting drug concentrations in both plasma and target organs, circumventing the deficiencies of using a single genotype or biomarker to evaluate net drug exposure. Prediction of the extent of specific PK drug–drug interactions (PK‐DDIs) in individual patients is based on the prior development of validated compound files for specific enzyme/transporter inhibitors and inducers 6. The influence of ‘special populations’, such as paediatrics, hepatic disease, renal impairment and pregnancy, is accounted for in the physiological model 9.
Olanzapine (OLZ) is a commonly prescribed antipsychotic drug with large interindividual variability in pharmacokinetics (up to 10‐fold). The intrinsic and extrinsic factors shown to contribute to this variability include ethnicity, smoking status, gender, age and comedication 10, 11, 12, 13. Patients identified as black or African‐American eliminate OLZ more rapidly than patients of other ethnic backgrounds, and the systemic clearance of OLZ in regular tobacco smokers is about twofold higher than in nonsmokers 14. In terms of age and gender, the clearance of OLZ appears to be lower in the elderly and in women 14, 15. Fluvoxamine, a strong cytochrome P450 (CYP) 1A2 inhibitor, can double the systemic exposure of OLZ 16, whereas inducers of CYP1A2, such as carbamazepine, decrease exposure by approximately half 17. Several population PK models for OLZ have been published recently, but they are specific for certain populations (e.g. adolescents, Chinese, patients who have overdosed, patients taking sertraline as comedication), and have not been applied to predict OLZ exposure in patients 14, 18, 19, 20, 21, 22.
As proof‐of‐concept for PBPK model‐informed precision dosing (MIPD) in individual patients, OLZ was selected as a prototype drug. The reasons for this were:
OLZ is eliminated almost entirely by metabolism (the fraction excreted unchanged in urine is <0.09), predominantly by CYP1A2, CYP2C8 and UDP‐glucuronosyltransferase (UGT) 1A4 23. CYP1A2 activity can be marked by a non‐invasive phenotyping test based on the administration of caffeine and the collection of a single saliva sample 24, and CYP2C8 activity can be estimated by genotyping 25, 26.
The lack of significant OLZ metabolism by CYP3A4 (<10%) simplifies matters, as ways of marking the activity of this enzyme easily in individual patients are currently limited. There are no informative and sufficiently frequent genetic variants associated with functional differences of the enzyme 27, and endogenous markers such as 6β‐hydroxycortisol and 4β‐hydroxycholesterol are insufficiently correlated with CYP3A4 activity 28. Midazolam micro‐dosing is available but is invasive and may be not be feasible in many clinical institutions 29.
Olanzapine is a Biopharmaceutical Classification System class 2 compound 30, and although theoretically affected by solubility, the pharmacokinetics of OLZ are not expected to be rate limited by transporters [note that changes in gastric/intestinal pH and transit time do not alter the oral bioavailability of OLZ [Zyprexa®, Prescribing Information], so any changes in OLZ solubility in vivo are unlikely to be clinical important].
A therapeutic concentration range of 20–80 μg l−1 has been established 31.
The response to OLZ is highly variable, with many patients discontinuing treatment owing to lack of efficacy and/or adverse effects 10. Thus, a rapid and non‐invasive approach to improve OLZ dose selection could have considerable clinical and health‐economic benefits.
In the present study, a comprehensive PBPK model for OLZ was constructed in Simcyp®, the most common PBPK M&S platform used in drug development. The model was validated by predicting the pharmacokinetics of OLZ after single oral doses and by predicting the distribution of OLZ steady state trough concentrations in a TDM database of patient samples. The PBPK model was then applied successfully to predict the systemic exposure of OLZ in individual patients, together with a hypothetical decrease in the variability of OLZ exposure following dose adjustment.
Methods
Development of the PBPK model for OLZ
There are several reviews that describe in detail how physiological processes and their variability are modelled in PBPK to predict the population‐level between‐subject variability in pharmacokinetics following IVIVE 6, 9, 32, 33, 34. In short, the ‘system’ components (e.g. haematocrit, tissue composition, body size, and enzyme and transporter abundances) are assigned coefficients of variation (CVs) based on known values (i.e. they are nonfixed), and a Monte Carlo approach is used to generate virtual individual subjects for simulations. In the present study, a minimal PBPK model comprising gut, liver and a lumped compartment for all other organs was utilized in Simcyp® (V14.1) (Figure 1). The differential equations in Simcyp® to describe drug concentrations in these compartments over time have been described previously 32. Table 1 shows the physicochemical, blood‐binding, absorption, distribution and elimination parameters used to construct the compound file for OLZ. Parameters were taken from the published literature or predicted using the validated prediction tools in Simcyp®.
Figure 1.
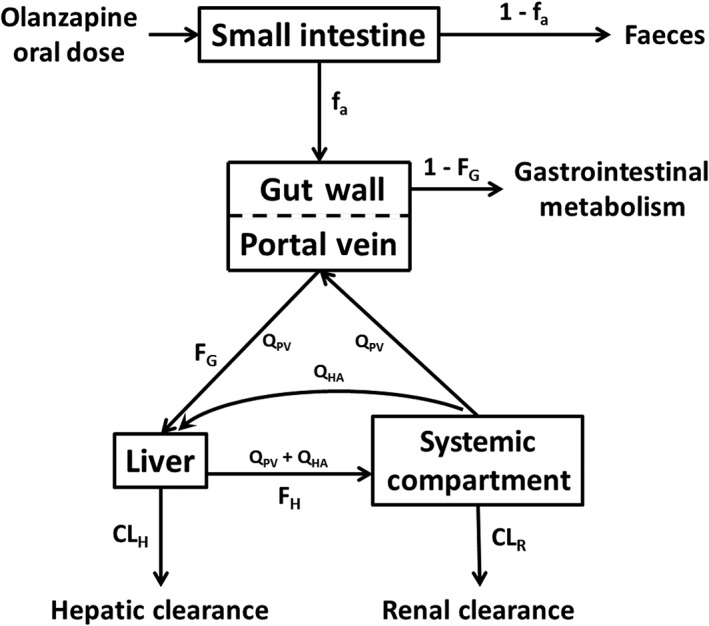
Minimal physiologically based pharmacokinetic model used to predict the pharmacokinetics of olanzapine. CLH, hepatic clearance; CLR, renal clearance; fa, fraction absorbed; 1 – fa, fraction excreted unchanged in the faeces; FG, fraction escaping gastrointestinal metabolism; 1 – FG, fraction metabolized in the gut; FH, fraction escaping hepatic metabolism
Table 1.
Inputs for the olanzapine (OLZ) compound file in Simcyp®
| Parameter | Value | Reference |
|---|---|---|
| Physicochemical a | ||
| Molecular weight | 312.43 | |
| log P o:w | 3.0 | |
| pK a (monoprotic base) | 7.24 | |
| Blood binding | ||
| B : P | 0.83 | Simcyp predictedb |
| fu p | 0.07 | 65 |
| Absorption (first‐order model) | ||
| f a | 0.6 | 65 |
| k a (r −1 ) | 0.52c | |
| fu gut | 1.0 | |
| Q gut (l h −1 ) | 15.02 | Simcyp predictedb |
| P eff ,man (10 −4 cm s −1 ) | 8.02 | Simcyp predictedb |
| Distribution (minimal PBPK model) | ||
| V ss (l kg −1 ) | 4.14 | Simcyp predictedb |
| Elimination | ||
| N ‐demethylation, rCYP1A2 | ||
| V max (pmol min −1 pmol P450 −1) | 1.34 | 23 |
| K m (μM) | 61 | 23 |
| 2‐hydroxylation, rCYP1A2 | ||
| V max (pmol min −1 pmol P450 −1 ) | 1.92 | 23 |
| K m (μM) | 592 | 23 |
| 7‐hydroxylation, rCYP1A2 | ||
| CL int (pmol min −1 pmol P450 −1 ) | 0.00324 | 23 |
| N ‐demethylation, rCYP2C8 | ||
| V max (pmol min −1 pmol P450 −1 ) | 1.37 | 23 |
| K s (μM) | 30 | 23 |
| 10‐ N ‐glucuronidation, rUGT1A4 | ||
| V max (pmol min −1 mg −1 ) | 216 | 23 |
| K s (μM) | 183 | 23 |
| rUGT scalar d | 2.24 | 23 |
| Additional liver clearance, FMO3 | ||
| HLM CL int (μL min −1 mg −1 ) | 0.439 | 23 |
| Biliary clearance | ||
| CL int (μl min −1 mg −1 ) | 0.0 | 23 |
| CL R for 20–30‐year‐old healthy male (l h −1 ) e | 1.8 | |
B : P, blood‐to‐plasma partition ratio; CLint, intrinsic clearance; CLR, renal clearance; fa, fraction absorbed from dosage form; FMO3, flavin‐containing monooxygenase 3; fugut, fraction unbound in the gut; fup, fraction unbound in the plasma; HLM, human liver microsomes; ka, first‐order absorption rate constant; Km, michaelis‐menten constant; PBPK, physiologically based pharmacokinetic; Peff,man, effective permeability in man; pKa acid dissociation constant; Po:w, neutral species octanol : buffer partition coefficient; Qgut, gut blood flow; rCYP1A2, recombinant cytochrome P450 1A2; rUGT, recombinant UDP‐glucuronosyltransferase; Vmax, maximum rate of metabolism; Vss, volume of distribution at steady state
Physicochemical data were obtained from the ChEMBL database (https://www.ebi.ac.uk)
These parameters were predicted using previously validated functions in Simcyp®. The B : P was predicted from the log Po:w and pKa (monoprotic base) of OLZ. The value of Qgut was predicted from the equation CLperm × Qent/Qent + CLperm, where Qent is the average enterocytic blood flow (18 l h–1) and CLperm is the product of intestinal surface area (defined in Simcyp®) and apparent permeability 66. The value for Peff,man was predicted using physicochemical data and the equation Log (Peff,man) = 4–2.546 – 0.011PSA – 0.278HBD, where PSA is polar surface area and HBD is hydrogen bond donors. The predicted Vss for OLZ was determined using the corrected Poulin and Theil method 67
Back calculated based on the clinically observed time to maximum concentration in Shirley et al. [13], according to the method described by Yamazaki et al. 68
Based on differences in Vmax between user recombinant expression system [HEK293T cells; 216 (pmol min−1 mg−1) and HLM (486 pmol min−1 mg−1)]
fe, fraction excreted unchanged in urine (0.09); CLiv, intravenous clearance (20 l h−1)
Validation of the PBPK model for OLZ
The pharmacokinetics of OLZ in single‐dose healthy volunteer studies was predicted (2.5, 5 and 10 mg oral doses). Virtual subjects were matched to the volunteers for age, gender and ethnicity. For each clinical study 10 virtual trials were run. The mean of each predicted PK parameter, taken as the overall mean of the mean values from each virtual trial, were then compared with the reported mean in the corresponding clinical study.
A TDM database of patients treated with OLZ at the Flinders Medical Centre (a tertiary hospital in Adelaide, South Australia) was searched to catalogue trough OLZ concentrations at steady state. Patients were grouped into three cohorts according to OLZ dose of 5, 10 and 15 mg daily. Simcyp® was then used to predict the steady state plasma concentrations and to examine intersubject variability. Virtual subjects were matched to the patients for age, gender, ethnicity and OLZ dose as follows: cohort 1 (n = 24; Caucasian; 21–65 years; 38% female; 5 mg), cohort 2 (n = 160; Caucasian; 18–65 years; 36% female; 10 mg) and cohort 3 (n = 80; Caucasian; 18–65 years; 36% female; 15 mg). Simulations were run with oral dosing daily for 15 days: three trials with eight subjects (n = 24) at 5 mg; 20 trials with eight subjects (n = 160) at 10 mg; and 10 trials with eight subjects (n = 80) at 15 mg. Virtual subjects were allocated to separate trials, to replicate what might occur clinically (i.e. small studies conducted by different groups), although simulations that allocate all subjects to one trial give equivalent summary PK outputs in Simcyp®. The mean predicted steady‐state trough OLZ concentration at 24 h after the day 14 dose was compared with the measured concentration from patients in samples taken between 12 and 26 h after OLZ dosing (mean = 20 h).
Simulations of covariates that influence the clearance of OLZ
The OLZ PBPK model was used to examine the influence of covariates thought to alter OLZ exposure. The covariates were age, gender, ethnicity, CYP1A2 phenotype, CYP2C8 genotype and liver/kidney function, and they were assessed relative to a 20–50‐year‐old healthy Caucasian cohort with a 1:1 male to female distribution of extensive metabolizer (EM) CYP1A2 phenotype and wild‐type CYP2C8 genotype. Ten virtual trials with 10 subjects were conducted for each subgroup analysis. Previously defined and validated population profiles in Simcyp® were used to assess outcomes in Chinese, Japanese, geriatric (66–90 years) and impaired liver/kidney function cohorts.
Prediction of OLZ exposure in individual patients
A clinical study was conducted to determine whether the validated OLZ PBPK model in Simcyp® could be applied to predict OLZ exposure in patients. A cohort of inpatients recently commenced on OLZ were recruited at the Flinders Medical Centre (ethics approval number HREC/13/SAC/181). Inclusion criteria were: >18 years of age, taking OLZ for >7 days without dose adjustment, no evidence of significant liver or renal dysfunction [liver function tests <2 × upper limit of normal and an estimated glomerular filtration rate (GFR) >60 ml min–1] and the ability to consent, as judged by the treating psychiatrist. To facilitate recruitment and generate a diverse study population, no exclusions were placed on indication for OLZ, ethnicity, smoking status, weight or comedications.
Following consent, the weight and height of each patient were measured, current smoking status was recorded and their consumption of caffeine (CAF), alcohol, cruciferous vegetables and chargrilled meat was assigned a ranking of either low, moderate or high. Each patient then ingested a 100 mg CAF tablet (NoDoz Tablets, Key Pharmaceutical, Sydney, NSW, Australia), having refrained from consuming CAF‐containing products for the previous 24 h. Saliva samples for the analysis of CAF and paraxanthine (PXT) were collected 5 min before CAF dosing and 4 h after. The samples were centrifuged to remove sediment, and stored at −20°C until analysis (see below). Venous blood (5 ml) was collected into K3 ethylenediamine tetraacetic acid tubes at the end of the OLZ dosage interval, centrifuged (4000 g for 5 min), and the plasma was frozen at −20°C until analysis of the OLZ plasma concentration (see below). In those patients who consented, 1 ml of the blood sample was stored separately for CYP2C8 genotyping (see below).
A ‘virtual twin’ profile for each study participant was constructed by individualizing the Simcyp® healthy volunteer population file for ethnicity, gender, age, height, weight, CYP2C8 genotype and CYP1A2 phenotype. The methods used to assign CYP2C8 genotype and CYP1A2 phenotype are described in subsequent sections. Ten simulations with each virtual twin were run at the OLZ dose taken by the corresponding real patient (either 10 or 15 mg OLZ). The individualizing input parameters were therefore ‘fixed’ for all 10 simulations, while values for the remaining ‘nonfixed’ system components were generated using the Monte Carlo approach in Simcyp® based on the known population variability for each parameter. The mean predicted steady state trough OLZ concentration at 24 h after the day 14 dose from 10 simulations with a virtual twin was then compared with the measured concentration from patients taken at the end of the OLZ dosing interval (approximately 22–24 h).
Assays
Authentic standards for OLZ and d3‐OLZ were purchased from Toronto Research Chemicals (Toronto, ON, Canada). Authentic standards for benzotriazole (BTZ), CAF and PXT were purchased from Sigma‐Aldrich (Sydney, NSW, Australia). High‐purity water was obtained using a MilliQ Synergy UV Ultrapure water system (Merck Millipore, Sydney, NSW, Australia). Acetonitrile [liquid chromatography–mass spectrometry (LC–MS) grade] and methanol (LC–MS grade) was obtained from Merck Millipore (Melbourne, VIC, Australia). All other solvents and reagents were of analytical reagent grade.
The PXT : CAF ratio in saliva was determined by adapting a previously published method 24. Saliva (100 μl) was vortexed for 30 s with 50 μl of BTZ (5 μg ml–1) in water (assay internal standard) followed by further vortexing (2 × 30 s) with 4 ml of ethyl acetate and centrifugation (5000 g, 10 min). The organic layer was collected and evaporated to dryness under a stream of nitrogen gas. Dried eluates were reconstituted in 100 μl of acetic acid (1%) in water and transferred to high‐performance liquid chromatography (HPLC) vials containing a plastic insert for analysis. Chromatographic separations were performed on a Waters NovaPak C18 column (150 × 3.9 mm, 4 μm; Waters, Milford, MA, USA) using an Agilent 1100 series HPLC. The column temperature was maintained at 25°C. CAF, PXT and BTZ were separated from matrix components by isocratic elution at a flow rate of 1 ml min–1 using a mobile phase comprising acetic acid (1%) in water. The retention times of PXT, BTZ and CAF under these conditions were 6.2, 12.1 and 15.6 min, respectively. Column eluent was monitored by ultraviolet absorbance at 270 nm. Quantification of CAF and PXT in patient samples was carried out by comparison of peak areas with those of authentic standards. Calibration curves (0.05–5 μg ml–1) and quality control samples (0.75 μg ml–1) were prepared by spiking CAF‐free saliva with CAF and PXT.
To measure OLZ concentration, plasma (100 μl) was vortexed for 30 s with 100 μl of a solution of d3‐OLZ (50 ng ml–1) in acetonitrile (assay internal standard). Carbonate buffer (50 μl; 1 M, pH 9.5) was added followed by 1 ml of methyl tert‐butyl ether, and the mixture was vortexed (2 × 1 min) and centrifuged (13 000 g, 5 min). The organic layer was collected and evaporated to dryness using a MiVac centrifugal vacuum concentrator (GeneVac, Sydney, NSW, Australia). Dried eluates were reconstituted in 100 μl of ammonium formate (10 mM, pH 3.0) with 50% v/v acetonitrile and transferred to Ultra Performance LC™ (UPLC) vials containing a spring‐loaded low volume glass insert. Chromatographic separations were performed on a Waters ACQUITY™ BEH C18 analytical column (100 mm × 2.1 mm, 1.7 μm; Waters Corporation, Milford, MA, USA) using a Waters ACQUITY UPLC system. The column temperature was maintained at 40°C, and the sample compartment at 15°C. OLZ and d3‐OLZ were separated from matrix components by isocratic elution at a flow rate of 0.25 ml min–1 using a mobile phase comprising ammonium formate (10 mM, pH 3.0) with 30% v/v acetonitrile. The total run time for each sample was 2.5 min. The retention time for OLZ and d3‐OLZ under these conditions was 1.8 min. Column elutant was monitored by mass spectrometry, performed on a Waters Q‐ToF Premier™ quadrupole, orthogonal acceleration time‐of‐flight tandem (ToF) mass spectrometer (Q‐ToF‐MS) operated in positive ion mode with electrospray ionization (ESI+). ToF data were collected in wide pass (MS) mode, with the resolving quadrupole acquiring data between m/z 100 and 500. Selected ion data were extracted at a precursor m/z of 313.14 (OLZ) and 316.14 (d3‐OLZ). Resulting pseudo multiple reaction monitoring (MRM) spectra were analysed using Waters QuanLynx™ software (Waters Corporation, Sydney, NSW, Australia). Quantification of OLZ in patient samples was accomplished by comparison of peak areas with those of authentic standards. Calibration curves (10–200 ng ml–1) and quality control samples (75 ng ml–1) were prepared by spiking blank plasma with OLZ. The limit of quantification (LOQ) and limit of determination (LOD) values for OLZ in plasma were 0.5 ng ml−1 and 0.1 ng ml−1, respectively, and the CV for the LOQ was 12.8%.
Determination of CYP2C8 genotype
Genomic DNA was extracted from patient whole‐blood samples. The concentration and purity of the samples were determined using a Nanodrop (Thermo Fisher Scientific, Waltham, MA, USA), and DNA was stored at −20°C until required. The CYP2C8*3 (rs11572080) and CYP2C8*4 (rs1058930) single nucleotide polymorphisms (SNPs) were determined by TaqMan® SNP genotyping assays using an ABI 7500 Fast Real‐Time polymerase chain reaction system (Applied Biosystems, Carlsbad, CA, USA). Assays were performed according to the manufacturer's instructions.
Assignment of CYP1A2 activity from PXT : CAF ratio
CYP phenotypes are defined in Simcyp® as a function of enzyme abundance (pmol mg–1 protein) and the turnover rate constant (1 h–1). Individualized CYP1A2 activity in each virtual twin was assigned from the measured PXT : CAF ratio as a function of ‘relative enzyme abundance’ using the equation:
where 52 pmol mg–1 protein is the population mean hepatic enzyme abundance for CYP1A2 (Simcyp® in‐built parameter), and 0.549 is the population mean 4‐h PXT : CAF ratio in healthy Caucasians of both genders, reported across three studies, weighted to account for study sample sizes 13, 35, 36.
Assignment of CYP2C8 enzyme abundance and turnover rate from genotype
The most common alleles observed in Caucasians that affect CYP2C8 activity are *3 and *4 26. Simcyp® model inputs defining the enzyme activity (relative enzyme abundance and turnover rate constant) of CYP2C8 genotypes are shown in Table 2. Model inputs were determined from pooled in vitro enzyme abundance 25, 37 and kinetic [in human liver microsomes (HLM), hepatocytes or recombinant CYP (rCYP)] 26, 37, 38, 39, 40, 41, 42, 43 data for each genotype with multiple substrates. Additional studies have reported reduced catalytic activity for carriers of one or more CYP2C8*3 and CYP2C8*4 alleles 44, 45, 46, 47; however, as these studies did not provide quantitative data, they were not incorporated when defining model inputs. In the absence of in vitro kinetic data describing the pharmacokinetics in carriers of CYP2C8*1/*3 and CYP2C8*1/*4 genotypes, turnover rate constants were assigned based on the relative in vivo impact of these genotypes (accounting for differences in enzyme abundance) determined in studies that assessed both CYP2C8 genotype in vitro and in vivo.
Table 2.
Simcyp® inputs for cytochrome P450 (CYP) 2C8 genotypes
| Parameter | Value | Reference |
|---|---|---|
| *1 / *1 | ||
| Relative enzyme abundance (pmol mg protein−1) | 1 | 25, 37 |
| Turnover rate constant (h−1) | 0.0301 | 26, 37, 38, 39, 40, 41, 42, 43 |
| *3 / *3 | ||
| Relative enzyme abundance (pmol mg protein−1) | 21 | 25, 37 |
| Turnover rate constant (h−1) | 0.0143 | 26, 37, 38, 40, 42, 43 |
| *4 / *4 | ||
| Relative enzyme abundance (pmol mg protein−1) | 8 | 37 |
| Turnover rate constant (h−1) | 0.0101 | 26, 37, 39, 40, 41, 43 |
| *1 / *3 | ||
| Relative enzyme abundance (pmol mg protein−1) | 37 | 25 |
| Turnover rate constant (h−1) | 0.0222 | |
| *1 / *4 | ||
| Relative enzyme abundance (pmol mg protein−1) | 16 | 25 |
| Turnover rate constant (h−1) | 0.0201 |
Analysis of model performance
The performance of simulations was assessed by the mean‐fold error (MFE):
Predicted PK parameters were directly extracted from Simcyp® [area under the plasma concentration‐time curve, peak plasma concentration and time to maximum concentration (Tmax)] or determined by regression of the mean predicted semi‐logarithmic concentration vs. time data for the elimination phase (t½). The model was accepted if all predicted PK parameters were within twofold of the corresponding observed values from the single‐dose PK studies (MFE 0.5 to 2.0). Pooled data for each PK parameter and subgroup were also subjected to Bland–Altman plot analysis, with statistical significance set at P < 0.05. Differences between predicted and observed values for single‐dose simulations were determined by paired sample Student's t‐test (SPSS v 22; IBM Corporation, Armonk, NY, USA). Multiple comparison testing (one‐way analysis of variance with Tukey post hoc analysis) was conducted to assess the statistical significance of covariate effects for age, gender, ethnicity, CYP1A2 phenotype, CYP2C8 genotype and liver/kidney function (SPSS v 22).
Analysis of hypothetical decrease in OLZ exposure variability after dose adjustment
Individualized drug dosing requires decreasing the variability in overall response based on individual attributes of the patient which define the sensitivity of that patient to a given systemic exposure. Therefore, the predicted systemic exposure was used to calculate a hypothetical decrease in variability had the OLZ dose been adjusted based on outputs from virtual twins. The following steps were taken to compare the CVs for the fixed‐dosing regimen and hypothetical adjusted‐dosing regimens:
The CV for observed OLZ exposure (OLZ trough concentration) was calculated.
The target for desired OLZ trough concentration was set at 50 μg l−1, which is in the middle of the therapeutic window.
The ratio of predicted OLZ exposure relative to the target value in step 2 was established.
The ratio in step 3 was then used as a measure of hypothetical dose adjustment in each patient.
In theory, and assuming linear kinetics, each patient would have the exact target exposure (50 μg l−1) had the dose been adjusted according to step 4 and the model from the virtual twins was 100% accurate and predictive. However, using the ratio of observed exposures to predicted exposures under the fixed‐dose regimen, measures of deviations from a perfect model were obtained and then used to calculate hypothetical values of OLZ trough concentration under the dose‐adjustment scenario.
The CV for observed OLZ trough concentration under the fixed‐dose regimen was compared with the CV of the hypothetical OLZ trough concentrations after dose adjustment.
Results
Validation of the PBPK model for OLZ
The predicted and observed PK parameters of OLZ after single oral doses are summarized in Table 3. The differences between predicted and observed PK parameters were not statistically significant (P > 0.05). Time to maximum concentration was systematically underpredicted in all simulations (MFEs = 0.55–0.82), whereas other simulated PK parameters were more consistent with the observed values (MFEs ranged from 0.84 to 1.55).
Table 3.
Mean predicted (pred) and observed (obs) pharmacokinetic parameters of olanzapine (OLZ) after single oral doses
| Population | Study reference | OLZ dose | AUC a | CL | t½ b | C max | T max | |
|---|---|---|---|---|---|---|---|---|
| (mg) | (μg l h −1 ) | (l h −1 ) | (h) | (μg l −1 ) | (h) | |||
| Healthy volunteers | Shirley et al. 2003 13 | 10 | Observed | 501 | 20.0 | 32.0 | 15.0 | 4.0 |
| Predicted | 621 | 18.5 | 30.2 | 14.5 | 4.2 | |||
| Pred/obs ratio | 1.24 | 0.93 | 0.94 | 0.97 | 1.06 | |||
| Callaghan et al. 1999 11 #BY | 10 | Observed | 512 | 20.6 | 32.4 | 12.6 | 6.1 | |
| Predicted | 645 | 18.7 | 29.5 | 15.4 | 4.2 | |||
| Pred/obs ratio | 1.26 | 0.91 | 0.91 | 1.22 | 0.69 | |||
| Sathirakul et al. 2003 12 | 10 | Observed | 578 | 18.5 | 29.6 | 15.3 | 7.0 | |
| Predicted | 648 | 18.3 | 29.4 | 15.5 | 4.1 | |||
| Pred/obs ratio | 1.12 | 0.99 | 0.99 | 1.01 | 0.59 | |||
| Callaghan et al. 1999 11 #CH | 7.5 | Observed | 500 | 15.6 | 46.7 | 9.2 | 8.0 | |
| Predicted | 492 | 18.2 | 30.1 | 11.4 | 4.3 | |||
| Pred/obs ratio | 0.98 | 1.17 | 0.64 | 1.24 | 0.54 | |||
| Sathirakul et al. 2003 12 | 5 | Observed | 293 | 18.5 | 31.2 | 6.9 | 6.0 | |
| Predicted | 324 | 18.3 | 29.4 | 7.7 | 4.1 | |||
| Pred/obs ratio | 1.11 | 0.99 | 0.94 | 1.11 | 0.68 | |||
| Sathirakul et al. 2003 12 | 2.5 | Observed | 146 | 18.0 | 29.5 | 3.9 | 8.0 | |
| Predicted | 162 | 18.3 | 29.4 | 3.9 | 4.2 | |||
| Pred/obs ratio | 1.11 | 1.02 | 1.00 | 1.01 | 0.53 | |||
| Healthy volunteer simulation MFE | 1.14 | 1.00 | 0.91 | 1.09 | 0.68 | |||
| Chinese healthy volunteers | Sathirakul et al. 2003 12 | 10 | Observed | 712 | 14.6 | 28.7 | 17.3 | 4.0 |
| Predicted | 836 | 13.9 | 31.6 | 18.7 | 4.2 | |||
| Pred/obs ratio | 1.17 | 0.95 | 1.10 | 1.08 | 1.05 | |||
| Sathirakul et al. 2003 12 | 5 | Observed | 351 | 15.0 | 29.1 | 8.0 | 6.0 | |
| Predicted | 418 | 13.9 | 31.6 | 9.4 | 4.2 | |||
| Pred/obs ratio | 1.19 | 0.93 | 1.09 | 1.17 | 0.70 | |||
| Sathirakul et al. 2003 12 | 2.5 | Observed | 179 | 14.9 | 30.0 | 4.5 | 6.0 | |
| Predicted | 209 | 13.9 | 31.6 | 4.7 | 4.2 | |||
| Pred/obs ratio | 1.17 | 0.93 | 1.05 | 1.05 | 0.70 | |||
| Chinese healthy simulation MFE | 1.18 | 0.94 | 1.08 | 1.10 | 0.82 | |||
| Geriatric North European Caucasians | Callaghan et al. 1999 11 #BE | 10 | Observed | 574 | 18.5 | 48.0 | 9.2 | 8.2 |
| Predicted | 748 | 15.9 | 40.3 | 14.3 | 4.5 | |||
| Pred/obs ratio | 1.30 | 0.9 | 0.8 | 1.5 | 0.5 | |||
| Geriatric NEC simulation MFE | 1.30 | 0.86 | 0.84 | 1.55 | 0.55 | |||
AUC, area under the plasma concentration‐time curve; Cmax, maximum concentration; CL, clearance; t½, half‐life; MFE, mean‐fold error; NEC, North European Caucasians; Tmax, time to maximum concentration; #BY, study B [young (20–41‐year‐old) cohort]; #BE; study B [elderly (65–79‐year‐old) cohort]; #CH, study C [healthy (nonhepatic cirrhosis) cohort]
0–120 h for simulations and 0–∞ for in vivo studies
Predicted t1/2 determined by regression of the mean predicted semi‐logarithmic concentration vs. time data for the elimination phase (12–120 h)
The predicted and observed cumulative frequency plots of trough OLZ concentrations at steady state are compared in Figure 2. While the recovery of the observed distributions was generally good, there was a trend to overpredict the range of concentrations for the 10 and 15 mg doses (Figure 2B and 2C). With the exception of the 5 mg dose (P = 0.986), differences between predicted and observed trough OLZ concentrations were statistically significant.
Figure 2.
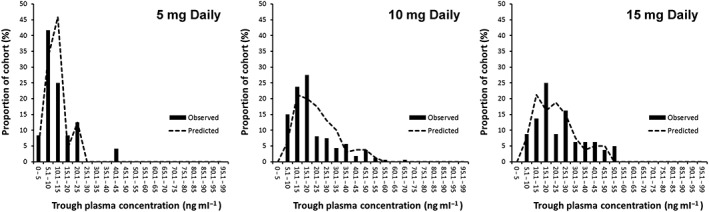
Predicted (lines) and observed (columns) frequency distributions of steady state trough olanzapine (OLZ) concentrations in patients monitored by the Flinders Medical Centre therapeutic drug monitoring service
Simulations of covariates that influence the clearance of OLZ
The box and whisker plots showing the impact of covariates on predicted OLZ clearance are shown in Figure 3. As CYP1A2 phenotype was considered as a continuous variable, subgroup analysis was not performed. Virtual subjects in subgroups aged over 41 years had a significantly decreased OLZ clearance compared with younger subjects. Chinese and Japanese subjects had a significantly decreased OLZ clearance compared with Caucasians. The CYP2C8*4/*4 genotype was associated with a statistically significant lower OLZ clearance compared with the CYP2C8*1/*1 genotype (P < 0.05). All degrees of liver disease (Child–Pugh A, B and C) and kidney impairment (defined as GFR <60 ml min−1) were associated with statistically significant decreases in clearance (P < 0.05). Although the mean clearance of OLZ was lower in females compared with males by about 1 l h−1, this was not statistically significant.
Figure 3.
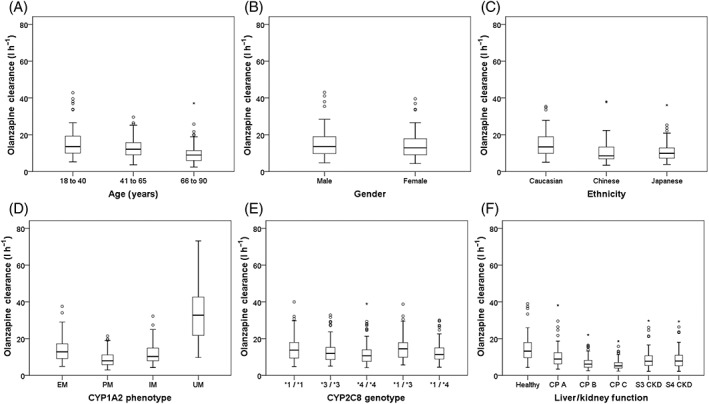
Box and whisker plots showing the impact of the following covariates on predicted olanzapine (OLZ) clearance: (A) Age; (B) Gender; (C) Ethnicity; (D) CYP1A2 phenotype; (E) CYP2C8 genotype; (F) Disease states. The bars represent the upper and lower quartiles, the mean (lines in the bars) and the 95% confidence intervals (whiskers) of data from simulations. * P < 0.05 compared with control group (20–50‐year‐old Caucasian male, extensive metabolizer, cytochrome P450 (CYP) 1A2 phenotype, CYP2C8*1/*1, normal liver and kidney function). CKD, chronic kidney disease; CP, Child–Pugh; S, stage
Prediction of OLZ exposure in individual patients
Fourteen patients were recruited for the clinical study (Table 4). Nine were female (65%) and seven consented for CYP2C8 genotyping. All patients were Caucasian and had normal liver and kidney function, defined as liver function tests <2 × upper limit of normal and estimated GFR >60 ml min−1. Half of the patients were current smokers. Based on self‐report, almost all patients had moderate to high CAF intake (13/14), 65% considered themselves as moderate‐ to high‐level drinkers of alcohol (9/14), and the consumption of cruciferous vegetable and chargrilled meat was generally low. There was significant heterogeneity in measured trough OLZ concentrations at steady state, with observed values ranging from 7.8 to 76.1 μg l−1 (mean = 46.1 μg l−1). With respect to CYP1A2 phenotype, the mean PXT : CAF ratio was 0.511 (range 0.019–2.978). Of the seven patients who consented to CYP2C8 genotyping, the genotypes were CYP2C8*1//*1 (n = 4), CYP2C8*1/*3 (n = 2) and CYP2C8*1/*4 (n = 1).
Table 4.
Characteristics of patients in the clinical study
| Gender | Age (years) | Smoking status | CYP1A2 phenotype (PXT : CAF ratio) | CYP2C8 genotype | Caffeine intake | Alcohol intake | Cruciferous vegetable intake | Chargrilled meat intake | Height (m) | Weight (kg) | BMI | Current comedication |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| F | 53 | Nonsmoker | 0.497 | *1/*4 | High | Low | High | Low | 1.59 | 94 | 37.2 | None |
| F | 27 | Smoker | 0.492 | *1/*1 | High | Low | Low | Low | 1.65 | 65 | 23.8 | None |
| F | 40 | Nonsmoker | 0.019 | Unknown | Moderate | Low | Low | High | 1.83 | 126 | 37.6 | Lithium, naltrexone, acamprosate |
| F | 27 | Smoker | 0.451 | Unknown | Moderate | High | Low | High | 1.80 | 95 | 29.3 | Clozapine, risperidone |
| F | 57 | Nonsmoker | 0.281 | Unknown | High | High | High | Low | 1.62 | 95 | 36.2 | Lithium, valproate, oxycodone, salbutamol, fluticasone, salmeterol |
| M | 22 | Smoker | 0.034 | Unknown | Moderate | Low | Low | Low | 1.70 | 78 | 27.0 | None |
| M | 23 | Smoker | 0.194 | Unknown | Moderate | Low | Low | Low | 1.85 | 69 | 20.2 | Escitalopram |
| F | 47 | Nonsmoker | 0.493 | *1/*1 | Low | Moderate | High | Low | 1.66 | 67 | 24.3 | Gingko, vitamin C |
| M | 46 | Nonsmoker | 0.038 | *1/*1 | High | Moderate | Low | Low | 1.81 | 90 | 38.5 | Valproate, fluticasone, salbutamol, aspirin |
| M | 43 | Smoker | 0.176 | *1/*1 | Moderate | Moderate | Low | Low | 1.74 | 96 | 31.7 | Valproate, clozapine |
| F | 39 | Smoker | 2.978 | *1/*3 | High | Moderate | Low | Low | 1.72 | 78 | 26.4 | None |
| F | 22 | Nonsmoker | 0.493 | *1/*3 | Moderate | Moderate | Moderate | High | 1.68 | 68 | 24.1 | Amoxicillin, paracetamol, valproate |
| M | 27 | Nonsmoker | 0.472 | Unknown | Moderate | High | Low | High | 1.84 | 83 | 24.5 | Valproate |
| F | 34 | Smoker | 0.533 | Unknown | High | Moderate | Low | Low | 1.59 | 51 | 20.2 | None |
BMI, body mass index; CAF, caffeine; CYP, cytochrome P450; PXT, paraxanthine
Figure 4 shows the predicted vs. observed trough OLZ concentrations for the full cohort (n = 14; panel A), with a subgroup analysis of subjects also genotyped for CYP2C8 (n = 7; panel B). The corresponding Bland–Altman plots are provided in Figure 4C and D. These panels suggest systematic bias resulting in underprediction of trough OLZ concentrations by about 10 μg l−1. The prediction accuracy was similar between the full cohort and CYP2C8 genotyped subgroup (assessed as mean difference between simulated vs. observed OLZ concentrations). Precision was greater for the CYP2C8 genotyped subgroup, with correlation of determination (R2) values for predicted vs. observed trough OLZ concentrations of 0.884 (genotyped subgroup) and 0.833 (full cohort), respectively.
Figure 4.
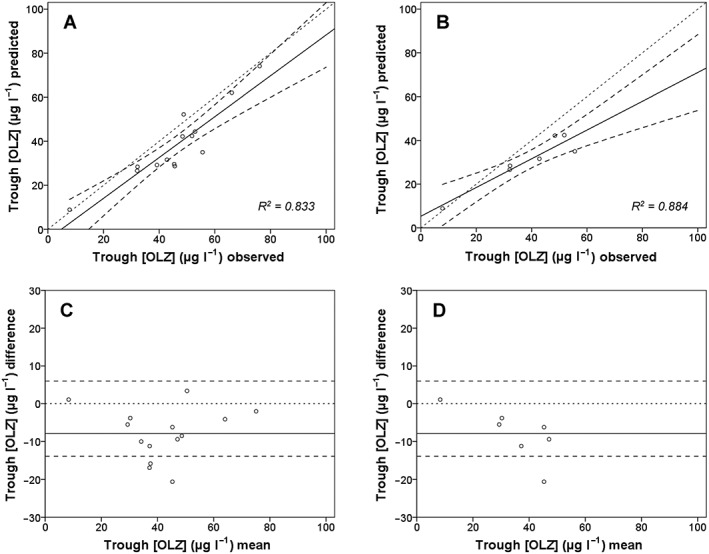
Correlations of predicted and observed steady state trough olanzapine (OLZ) concentrations in subjects phenotyped for cytochrome P450 (CYP) 1A2 (all subjects) and those also genotyped for CYP2C8. (A) All subjects (n = 14); (B) Only the subjects genotyped for CYP2C8 (n = 7). The corresponding Bland–Altman plots are shown in (C) and (D). Small dotted lines, line of unity; thick black lines, regression line for the data; larger dotted lines, 95% CIs
Hypothetical decrease in OLZ exposure variability after dose adjustment
Hypothetical dose adjustments for each patient were determined. In 11/14 patients, the dose of OLZ would need to be increased to attain the target concentration of 50 μg l−1 – that is, Figure 4A shows that the predicted OLZ trough concentrations are below the target concentration, indicating a systematic underprediction in most virtual twins. The variability in OLZ exposure was decreased after adjusting the OLZ dose based on predicted OLZ exposures. The CV of observed OLZ trough concentration (range 7.8–76.1 μg l−1) with a fixed‐dose regimen was 0.37. The CV of hypothetical OLZ trough concentration (range 31.5–57.1 μg l−1) with a dose‐adjusted regimen was 0.18. This translates to a decrease in exposure variability of about twofold using hypothetical dose adjustment based on virtual twin predictions.
Discussion
This is the first study to predict drug exposure in patients after individualizing a population file in Simcyp® to create virtual twins based on covariates known to influence pharmacokinetics. A ‘bottom‐up’ approach generated a PBPK model of OLZ that was validated by comparison with single oral‐dose PK studies (MFE 0.55–1.30) and by recapturing reasonably well the distribution of trough OLZ concentrations in a TDM database of patient samples (Table 3 and Figure 2). After considering the ethnicity, gender, age, height, weight, liver and kidney functions, the CYP1A2 phenotype, as determined by the PXT : CAF ratio in saliva, and the CYP2C8 genotype, in patients who consented, the OLZ PBPK model was applied successfully to predict steady state OLZ concentration in individual patients (Figure 4). A hypothetical decrease in OLZ exposure variability after dose adjustment based on predictions was also demonstrated. These data are proof‐of‐concept for MIPD after repurposing a widely available PBPK M&S platform.
The last 15 years have seen a dramatic increase in PBPK M&S to support drug applications to regulatory agencies 48, 49. Approximately two‐thirds of studies are for predicting potential PK‐DDIs, which can mitigate the need for clinical studies 9, 49, 50. Other important applications include predicting age‐ and ethnic‐related changes in pharmacokinetics and the assessment of pharmacokinetics in pathophysiological conditions such as liver and renal disease, and heart failure 9, 51. Although the role of PBPK M&S in drug development is universally accepted, with regulatory guidance now available 52, 53, there is growing interest in clinical medicine to harness this approach for better patient care. Areas of initial focus include infectious diseases, transplant medicine, oncology and paediatrics, as these disciplines most often have the prerequisites necessary for high‐impact MIPD – that is, the patient, disease and drug characteristics that make MIPD worthwhile. Examples of prerequisites include: a patient group with minimal clinical data; a disease with a high unmet medical need; and a drug with a well‐defined narrow therapeutic window (see Darwich et al., 2017 for further detail 54). Although there are many examples of how model‐informed approaches have been used successfully to optimize dose in individual patients, most use population PKPD models in local collaborative efforts between academia and healthcare 54.
The potential benefits of repurposing a widely available PBPK M&S platform for MIPD include:
A user‐friendly interface for which extensive training in pharmacometrics is not essential to build models and run simulations (i.e. suitable for clinicians).
Rapid application to novel clinical scenarios, as there is no need to generate an individual model each time that is only valid for a certain population. An example is the simulation of pharmacokinetics in a novel population (e.g. ethnic group), in which the compound file for that drug has been previously validated, typically using the healthy volunteer population file, and where the physiological characteristics of the novel population are well defined and the performance of the population file is validated.
Flexibility to change as additional information becomes available, such as new in vitro data for a disposition process or updating the platform to include validated compound file(s) of potential interacting drugs.
Finally, an ability to simulate the effects of intrinsic and/or extrinsic factors (e.g. comedications, genetic polymorphisms in drug metabolizing enzymes and transporters, and so forth) outside the range found in the clinical data used to build population PKPD models 54, 55.
Simulations were performed to examine the impact of covariates considered to influence OLZ clearance 11, 12, 13 (Table 2 and Figure 3). The CYP2C8 genotype was also included, given recent evidence showing the contribution of CYP2C8 to OLZ metabolism in vitro 23, a discovery that may explain the poor correlations between CYP1A2 phenotype or genotype and plasma OLZ and N‐desmethyl‐OLZ concentrations in a few studies 13, 56, 57. A series of small clinical studies by Callaghan et al. showed trends for lower OLZ clearance in various subgroups that were not statistically significant (elderly, non‐Caucasians, females, those with renal impairment and nonsmokers 11). When simulation conditions were matched to the clinical study designs, significant differences in OLZ clearance between subgroups were undetectable (data not shown). However, simulating a larger number of trials to include more subjects overall (10 × 10 = 100 subjects), powered to reject a bioequivalence null hypothesis with 80% power (i.e. to detect subgroup differences), did simulate a statistically significant lower OLZ clearance for age (≥ 66 years of age), Chinese and Japanese ethnicity, and all degrees of liver disease (Child–Pugh A, B and C) and kidney impairment (GFR <60 ml min−1) (Figure 3). These data highlight how small clinical studies (n ≤ 15) with substantial heterogeneity (standard deviation ranging from 20% to 100% in Callaghan et al. 11) have limited capacity to identify subtle differences in pharmacokinetics 58. Consistent with relative low enzyme abundance 37 and decreased metabolic clearance of other CYP2C8 substrates in vivo 59, the CYP2C8*4/*4 genotype was associated with a statistically significant lower OLZ clearance compared with CYP2C8*1/*1. This finding for OLZ requires further investigation in vivo. Interestingly, gender difference in OLZ clearance was not recovered in simulations, a finding at odds with some clinical data, the largest such difference being found in the Clinical Antipsychotic Trials for Intervention Effectiveness (CATIE) trial, which reported approximately one‐third lower OLZ clearance in women compared with men 60. Gender difference in CYP1A2 activity is controversial, with decreased activity observed during pregnancy or treatment with oral contraceptives, but not during different times of a woman's menstrual cycle, and for some ethnic groups (Spanish, Turkish, Chinese, South Asian and African‐American) but not others (those with European ancestry) (see Perera et al. 10 for further detail). Once again, the clinical studies on which to base comparisons are highly variable, so in the absence of superior clinical data it is difficult to access accurately the performance of the PBPK model 58.
Only 14 patients were eligible for the clinical study (Table 4). The reason for this poor recruitment is because OLZ use at Flinders Medical Centre is predominantly for the management of acute psychosis and agitation ‘pro re nata’ (p.r.n., as required), so it was difficult to recruit inpatients taking a stable dose for >7 days. However, even with this small cohort, the interindividual variabilities in OLZ trough concentration (range 7.8–76.1 μg l−1, mean = 46.1 μg l−1) and CYP1A2 phenotype (PXT : CAF ratio range 0.019–2.978, mean 0.511) were consistent with those seen previously in Caucasians 13, 35, 36. The twofold higher mean PXT : CAF ratio in smokers compared with nonsmokers (0.694 vs. 0.328) also aligned well with findings in the literature 10, although this resulted from a particularly high value in one participant, a 39‐year‐old female smoker in whom the PXT : CAF ratio was 2.978 (Table 4). Other extrinsic factors that contribute to variability in CYP1A2 activity did not show obvious relationships to the CYP1A2 phenotype and were not analysed independently (e.g. consumption of CAF, alcohol, cruciferous vegetables and chargrilled meat, and current comedications). It should be noted that all extrinsic factors were considered together when individualizing the PBPK model of OLZ to create virtual twins, as assignment of CYP1A2 activity in Simcyp® was based on CYP1A2 phenotype. While recognizing the limitations of the small number, additional consideration of the CYP2C8 genotype in seven participants only slightly improved the correlation between predicted and observed OLZ concentrations (Figure 4). This may further support the putative role of CYP2C8 in OLZ clearance 23. Importantly, the approach identified the outlier patient with an OLZ trough concentration well below the therapeutic range (7.8 μg l−1). Such patients are less likely to respond to OLZ and may require longer hospitalization, with increased costs to healthcare systems.
An important objective of MIPD is to reduce variability in response by adjusting dose. There are many sources of variability in response, such as adherence, pharmacokinetics, pharmacodynamics and disease phenotype 61. As the focus of this research was on the PK component, a hypothetical scenario was tested, whereby the OLZ dose could be adjusted after virtual twin predictions to attain a target exposure (set at an OLZ steady state trough concentration of 50 μg l−1). In reality, such dose adjustment would be hampered by the availability of dose units – that is, it is not possible to prescribe 12 mg of OLZ based on currently available formulations. However, the exercise was undertaken to demonstrate the potential of PBPK M&S in reducing variability that might be exerted through PK variation in patients. Importantly, variability in OLZ exposure was decreased when a hypothetical dose‐adjustment regimen was applied to the virtual twin predictions – the CV for exposure decreased from 0.37 for the fixed‐dose regimen to 0.18 for the hypothetical adjusted‐dose regimen. As the science underpinning PBPK‐guided MIPD develops, it will be necessary to test whether this hypothetical improvement in exposure variability occurs in reality.
There were some limitations to the present study. Firstly, there was no record in the TDM database of the extrinsic factors that influence CYP1A2 activity, such as the smoking status of patients, raising the possibility of confounding in the model validation based on multiple doses (Figure 2). Sensitivity analyses could be conducted to examine this further. Secondly, comedications that may alter enzymes other than CYP1A2 involved in OLZ clearance were not considered in the modelling (e.g. valproate, oxycodone, risperidone or fluticasone). As PBPK M&S becomes increasingly sophisticated with the validation of more compound files, the impact of comedications on OLZ exposure could be assessed. Thirdly, adherence to OLZ treatment was not observed directly, so it is not possible to guarantee that the dose simulated was the dose that was taken. Fourthly, UGT1A4 is the enzyme responsible for generating OLZ‐10‐N‐glucuronide and OLZ‐4′‐N‐glucuronide, and may contribute up to a quarter of OLZ clearance 23. Lower OLZ concentrations in heterozygous carriers of UGT1A4*3 have been reported in Caucasian 62 but not in Japanese 63 individuals, raising the possibility of a substrate‐specific rapid‐metabolizer phenotype associated with this allele in some ethnicities 64. Therefore, another limitation of the present study was that the UGT1A4 genotype was not considered. (5) Finally, the authors recognize that only OLZ exposure was predicted, a surrogate endpoint for response, and that there was no intention to investigate the PKPD relationship. For MIPD to gain traction in healthcare and become a clinical reality, considerable work is required to investigate robust clinical endpoints around efficacy and safety, and to demonstrate health economic advantages at the point of care.
Conclusions
In conclusion, the present study generated a PBPK model for OLZ in Simcyp® that was validated by predicting the pharmacokinetics of OLZ after single oral doses and by predicting the distribution of OLZ steady state trough concentrations in a TDM database. The PBPK model was then applied successfully to predict with reasonable accuracy the systemic exposure of OLZ in individual patients. Importantly, this approach identified an outlier patient with an OLZ trough concentration well below the therapeutic range and could hypothetically be used to decrease the variability in drug exposure via individualized dose adjustment. Repurposing of available PBPK M&S platforms is an option for MIPD that requires further study to examine clinical potential.
Competing Interests
M.J.S, M.D.W, T.M. and A.R. declare no competing interests. T.M.P. (d3 Medicine) and A.R.H. are currently employees of Certara; however, T.M.P. only commenced employment with d3 Medicine during the final stages of manuscript preparation prior to submission. G.T.T. is a cofounder of Simcyp Ltd, but now acts only in an advisory role.
This study was funded by the Simcyp Grant and Partnership Scheme (2012–2013), Simcyp Ltd, A Certara Company, Sheffield, UK.
Contributors
T.M.P., V.P. and A.R. contributed to study design. T.M. and M.D.W. contributed to experiments. G.T.T., P.K. and A.R. contributed to simulations. T.M.P., M.J.S., G.T.T., P.K., A.R.H. and A.R. contributed to data analysis. T.M.P., G.T.T., M.J.S., M.D.W., T.M., A.R.H., P.K., V.P. and A.R. contributed to the preparation of the manuscript.
Polasek, T. M. , Tucker, G. T. , Sorich, M. J. , Wiese, M. D. , Mohan, T. , Rostami‐Hodjegan, A. , Korprasertthaworn, P. , Perera, V. , and Rowland, A. (2018) Prediction of olanzapine exposure in individual patients using physiologically based pharmacokinetic modelling and simulation. Br J Clin Pharmacol, 84: 462–476. doi: 10.1111/bcp.13480.
References
- 1. Dawson WT. Relations between age and weight and dosage of drugs. Ann Intern Med 1940; 13: 1594–1615. [Google Scholar]
- 2. Jelliffe RW, Schumitzky A, Bayard D, Milman M, Van Guilder M, Wang X, et al Model‐based, goal‐oriented, individualised drug therapy. Linkage of population modelling, new 'multiple model' dosage design, Bayesian feedback and individualised target goals. Clin Pharmacokinet 1998; 34: 57–77. [DOI] [PubMed] [Google Scholar]
- 3. Duong JK, Kroonen M, Kumar SS, Heerspink HL, Kirkpatrick CM, Graham GG, et al A dosing algorithm for metformin based on the relationships between exposure and renal clearance of metformin in patients with varying degrees of kidney function. Eur J Clin Pharmacol 2017; 73: 981–990. [DOI] [PubMed] [Google Scholar]
- 4. Roberts JA, Abdul‐Aziz MH, Lipman J, Mouton JW, Vinks AA, Felton TW, et al Individualised antibiotic dosing for patients who are critically ill: challenges and potential solutions. Lancet Infect Dis 2014; 14: 498–509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Charles B. Population pharmacokinetics: an overview. Aust Prescr 2014; 37: 210–213. [Google Scholar]
- 6. Rowland M, Peck C, Tucker G. Physiologically‐based pharmacokinetics in drug development and regulatory science. Annu Rev Pharmacol Toxicol 2011; 51: 45–73. [DOI] [PubMed] [Google Scholar]
- 7. Tucker GT. Personalized drug dosage – closing the loop. Pharm Res 2017; 34: 1539–1543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Polasek TM, Polak S, Doogue MP, Rostami‐Hodjegan A, Miners JO. Assessment of inter‐individual variability in predicted phenytoin clearance. Eur J Clin Pharmacol 2009; 65: 1203–1210. [DOI] [PubMed] [Google Scholar]
- 9. Jamei M. Recent advances in development and application of physiologically‐based pharmacokinetic (PBPK) models: a transition from academic curiosity to regulatory acceptance. Curr Pharmacol Rep 2016; 2: 161–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Perera V, Gross AS, Polasek TM, Quin Y, Rao G, Forest A, et al Considering CYP1A2 phenotype and genotype for optimizing the dose of olanzapine in the management of schizophrenia. Expert Opin Drug Metab Toxicol 2013; 9: 1115–1137. [DOI] [PubMed] [Google Scholar]
- 11. Callaghan JT, Bergstrom RF, Ptak LR, Beasley CM. Olanzapine. Pharmacokinetic and pharmacodynamic profile. Clin Pharmacokinet 1999; 37: 177–193. [DOI] [PubMed] [Google Scholar]
- 12. Sathirakul K, Chan C, Teng L, Bergstrom RF, Yeo KP, Wise SD. Olanzapine pharmacokinetics are similar in Chinese and Caucasian subjects. Br J Clin Pharmacol 2003; 56: 184–187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Shirley KL, Hon YY, Penzak SR, Lam YW, Spratlin V, Jann MW. Correlation of cytochrome P450 (CYP) 1A2 activity using caffeine phenotyping and olanzapine disposition in healthy volunteers. Neuropsychopharmacology 2003; 28: 961–966. [DOI] [PubMed] [Google Scholar]
- 14. Bigos KL, Pollock BG, Coley KC, Miller DD, Marder SR, Aravagiri M, et al Sex, race, and smoking impact olanzapine exposure. J Clin Pharmacol 2008; 48: 157–165. [DOI] [PubMed] [Google Scholar]
- 15. Kelly DL, Conley RR, Tamminga CA. Differential olanzapine plasma concentrations by sex in a fixed‐dose study. Schizophr Res 1999; 40: 101–104. [DOI] [PubMed] [Google Scholar]
- 16. Hiemke C, Peled A, Jabarin M, Hadjez J, Weigmann H, Härtter S, et al Fluvoxamine augmentation of olanzapine in chronic schizophrenia: pharmacokinetic interactions and clinical effects. J Clin Psychopharmacol 2002; 22: 502–506. [DOI] [PubMed] [Google Scholar]
- 17. Haslemo T, Olsen K, Lunde H, Molden E. Valproic acid significantly lowers serum concentrations of olanzapine‐an interaction effect comparable with smoking. Ther Drug Monit 2012; 34: 512–517. [DOI] [PubMed] [Google Scholar]
- 18. Davies SJ, Mulsant BH, Flint AJ, Meyers BS, Rothschild AJ, Whyte EM, et al The impact of sertraline co‐administration on the pharmacokinetics of olanzapine: a population pharmacokinetic analysis of the STOP‐PD. Clin Pharmacokinet 2015; 54: 1161–1168. [DOI] [PubMed] [Google Scholar]
- 19. Lobo ED, Robertson‐Plouch C, Quinlan T, Hong Q, Bergstrom RF. Oral olanzapine disposition in adolescents with schizophrenia or biopolar I disorder: a population pharmacokinetic model. Paediatr Drugs 2010; 12: 201–211. [DOI] [PubMed] [Google Scholar]
- 20. Pilla Reddy V, Kozielska M, Suleiman AA, Johnson M, Vermeulen A, Liu J, et al Pharmacokinetic–pharmacodynamic modeling of antipsychotic drugs in patients with schizophrenia part I: the use of PANSS total score and clinical utility. Schizophr Res 2013; 146: 144–152. [DOI] [PubMed] [Google Scholar]
- 21. Tylutki Z, Jawien W, Ciszowski K, Wilimowska J, Anand JS. Abnormal olanzapine toxicokinetic profiles – population pharmacokinetic analysis. Toxicol Mech Methods 2015; 25: 1–12. [DOI] [PubMed] [Google Scholar]
- 22. Yin A, Shang D, Wen Y, Li L, Zhou T, Lu W. Population pharmacokinetics analysis of olanzapine for Chinese psychotic patients based on clinical therapeutic drug monitoring data with assistance of meta‐analysis. Eur J Clin Pharmacol 2016; 72: 933–944. [DOI] [PubMed] [Google Scholar]
- 23. Korprasertthaworn P, Polasek TM, Sorich MJ, McLachlan AJ, Miners JO, Tucker GT, et al In vitro characterization of the human liver microsomal kinetics and reaction phenotyping of olanzapine metabolism. Drug Metab Dispos 2015; 43: 1806–1814. [DOI] [PubMed] [Google Scholar]
- 24. Perera V, Gross AS, Xu H, McLachlan AJ. Pharmacokinetics of caffeine in plasma and saliva, and the influence of caffeine abstinence on CYP1A2 metrics. J Pharm Pharmacol 2011; 63: 1161–1168. [DOI] [PubMed] [Google Scholar]
- 25. Naraharisetti SB, Lin YS, Rieder MJ, Marciante KD, Psaty BM, Thummel KE, et al Human liver expression of CYP2C8: gender, age, and genotype effects. Drug Metab Dispos 2010; 38: 889–893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Bahadur N, Leathart JB, Mutch E, Steimel‐Crespi D, Dunn SA, Gilissen R, et al CYP2C8 polymorphisms in Caucasians and their relationship with paclitaxel 6alpha‐hydroxylase activity in human liver microsomes. Biochem Pharmacol 2002; 64: 1579–1589. [DOI] [PubMed] [Google Scholar]
- 27. Wang D, Sadee W. The making of a CYP3A biomarker panel for guiding drug therapy. J Pers Med 2012; 2: 175–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Shin KH, Choi MH, Lim KS, Yu KS, Jang IJ, Cho JY. Evaluation of endogenous metabolic markers of hepatic CYP3A activity using metabolic profiling and midazolam clearance. Clin Pharmacol Ther 2013; 94: 601–609. [DOI] [PubMed] [Google Scholar]
- 29. Hohmann N, Kocheise F, Carls A, Burhenne J, Haefeli WE, Mikus G. Midazolam microdose to determine systemic and pre‐systemic metabolic CYP3A activity in humans. Br J Clin Pharmacol 2015; 79: 278–285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Kumar S, Bhargava D, Thakkar A, Arora S. Drug carrier systems for solubility enhancement of BCS class II drugs: a critical review. Crit Rev Ther Drug Carrier Syst 2013; 30: 217–256. [DOI] [PubMed] [Google Scholar]
- 31. Baumann P, Hiemke C, Ulrich S, Eckermann G, Gaertner I, Gerlach M, et al The AGNP‐TDM expert group consensus guidelines: therapeutic drug monitoring in psychiatry. Pharmacopsychiatry 2004; 37: 243–265. [DOI] [PubMed] [Google Scholar]
- 32. Jamei M, Dickinson GL, Rostami‐Hodjegan A. A framework for assessing inter‐individual variability in pharmacokinetics using virtual human populations and integrated general knowledge of physical chemistry, biology, anatomy, physiology and genetics: a tale of ‘bottom‐up’ vs ‘top‐down’ recognition of covariates. Drug Metab Pharmacokinet 2009; 24: 53–75. [DOI] [PubMed] [Google Scholar]
- 33. Jamei M, Marciniak S, Feng K, Barnett A, Tucker G, Rostami‐Hodjegan A. The Simcyp® population‐based ADME simulator. Expert Opin Drug Metab Toxicol 2009; 5: 1–13. [DOI] [PubMed] [Google Scholar]
- 34. Rostami‐Hodjegan A, Tucker GT. Simulation and prediction of in vivo drug metabolism in human populations from in vitro data. Nat Rev Drug Discov 2007; 6: 140–148. [DOI] [PubMed] [Google Scholar]
- 35. Perera V, Gross AS, McLachlan AJ. Influence of environmental and genetic factors on CYP1A2 activity in individuals of south Asian and European ancestry. Clin Pharmacol Ther 2012; 92: 511–519. [DOI] [PubMed] [Google Scholar]
- 36. Perera V, Gross AS, McLachlan AJ. Diurnal variation in CYP1A2 enzyme activity in south Asians and Europeans. J Pharm Pharmacol 2013; 65: 264–270. [DOI] [PubMed] [Google Scholar]
- 37. Gao Y, Liu D, Wang H, Zhu J, Chen C. Functional characterization of five CYP2C8 variants and prediction of CYP2C8 genotype‐dependent effects on in vitro and in vivo drug‐drug interactions. Xenobiotica 2010; 40: 467–475. [DOI] [PubMed] [Google Scholar]
- 38. Dai D, Zeldin DC, Blaisdell JA, Chanas B, Coulter SJ, Ghanayem BI, et al Polymorphisms in human CYP2C8 decrease metabolism of the anticancer drug paclitaxel and arachidonic acid. Pharmacogenetics 2001; 11: 597–607. [DOI] [PubMed] [Google Scholar]
- 39. Jiang H, Zhong F, Sun L, Feng W, Huang ZX, Tan X. Structural and functional insights into polymorphic enzymes of cytochrome P450 2C8. Amino Acids 2011; 40: 1195–1204. [DOI] [PubMed] [Google Scholar]
- 40. Rowbotham SE, Boddy AV, Redfern CP, Veal GJ, Daly AK. Relevance of nonsynonymous CYP2C8 polymorphisms to 13‐cis retinoic acid and paclitaxel hydroxylation. Drug Metab Dispos 2010; 38: 1261–1266. [DOI] [PubMed] [Google Scholar]
- 41. Singh R, Ting JG, Pan Y, Teh LK, Ismail R, Ong CE. Functional role of Ile264 in CYP2C8: mutations affect haem incorporation and catalytic activity. Drug Metab Pharmacokinet 2008; 23: 165–174. [DOI] [PubMed] [Google Scholar]
- 42. Soyama A, Hanioka N, Saito Y, Murayama N, Ando M, Ozawa S, et al Amiodarone N‐deethylation by CYP2C8 and its variants, CYP2C8*3 and CYP2C8 P404A. Pharmacol Toxicol 2002; 91: 174–178. [DOI] [PubMed] [Google Scholar]
- 43. Yu L, Shi D, Ma L, Zhou Q, Zeng S. Influence of CYP2C8 polymorphisms on the hydroxylation metabolism of paclitaxel, repaglinide and ibuprofen enantiomers in vitro . Biopharm Drug Dispos 2013; 34: 278–287. [DOI] [PubMed] [Google Scholar]
- 44. de Morais SM, Wilkinson GR, Blaisdell J, Meyer UA, Nakamura K, Goldstein JA. Identification of new genetic defect responsible for the polymorphism of S‐mephenytoin metabolism in Japanese. Mol Pharmacol 1994; 46: 594–598. [PubMed] [Google Scholar]
- 45. Parikh S, Ouedraogo JB, Goldstein JA, Rosenthal PJ, Kroetz DL. Amodiaquine metabolism is impaired by common polymorphisms in CYP2C8: implications for malaria treatment in Africa. Clin Pharmacol Ther 2007; 82: 197–203. [DOI] [PubMed] [Google Scholar]
- 46. Smith HE, Jones JP 3rd, Kalhorn TF, Farin FM, Stapleton PL, Davis CL, et al Role of cytochrome P450 2C8 and 2J2 genotypes in calcineurin inhibitor‐induced chronic kidney disease. Pharmacogenet Genomics 2008; 18: 943–953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Taniguchi R, Kumai T, Matsumoto N, Watanabe M, Kamio K, Suzuki S, et al Utilization of human liver microsomes to explain individual differences in paclitaxel metabolism by CYP2C8 and CYP3A4. J Pharmacol Sci 2005; 97: 83–90. [DOI] [PubMed] [Google Scholar]
- 48. Luzon E, Blake K, Cole S, Nordmark A, Versantvoort C, Berglund EG. Physiologically based pharmacokinetic modeling in regulatory decision‐making at the European Medicines Agency. Clin Pharmacol Ther 2016; 102: 98–105. [DOI] [PubMed] [Google Scholar]
- 49. Wagner C, Zhao P, Pan Y, Hsu V, Grillo J, Huang SM, et al Application of physiologically based pharmacokinetic (PBPK) modeling to support dose selection: report of an FDA public workshop on PBPK. CPT Pharmacometrics Syst Pharmacol 2015; 4: 226–230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Jadhav PR, Cook J, Sinha V, Zhao P, Rostami‐Hodjegan A, Sahasrabudhe V, et al A proposal for scientific framework enabling specific population drug dosing recommendations. J Clin Pharmacol 2015; 55: 1073–1078. [DOI] [PubMed] [Google Scholar]
- 51. Jones HM, Chen Y, Gibson C, Heimbach T, Parrott N, Peters SA, et al Physiologically based pharmacokinetic modeling in drug discovery and development: a pharmaceutical industry perspective. Clin Pharmacol Ther 2015; 97: 247–262. [DOI] [PubMed] [Google Scholar]
- 52. US Food and Drug Administration . Physiologically based pharmacokinetic analyses – format and content. Rockville, MD: Guidance for Industry, 2016. [Google Scholar]
- 53. European Medicines Agency . Guideline on the qualification and reporting of physiologically based pharmacokinetic (PBPK) modelling and simulation. London, UK, 2016.
- 54. Darwich AS, Ogungbenro K, Vinks AA, Powell JR, Reny JL, Marsousi N, et al Why has model‐informed precision dosing not yet become common clinical reality? Lessons from the past and a roadmap for the future. Clin Pharmacol Ther 2017; 101: 646–656. [DOI] [PubMed] [Google Scholar]
- 55. Vieira MD, Kim MJ, Apparaju S, Sinha V, Zineh I, Huang SM, et al PBPK model describes the effects of comedication and genetic polymorphism on systemic exposure of drugs that undergo multiple clearance pathways. Clin Pharmacol Ther 2014; 95: 550–557. [DOI] [PubMed] [Google Scholar]
- 56. Carrillo JA, Herraiz AG, Ramos SI, Gervasini G, Vizcaino S, Benitez J. Role of the smoking‐induced cytochrome P450 (CYP)1A2 and polymorphic CYP2D6 in steady‐state concentration of olanzapine. J Clin Psychopharmacol 2003; 23: 119–127. [DOI] [PubMed] [Google Scholar]
- 57. Hagg S, Spigset O, Lakso HA, Dahlqvist R. Olanzapine disposition in humans is unrelated to CYP1A2 and CYP2D6 phenotypes. Eur J Clin Pharmacol 2001; 57: 493–497. [DOI] [PubMed] [Google Scholar]
- 58. Polasek TM, Patel F, Jensen BP, Sorich MJ, Wiese MD, Doogue MP. Predicted metabolic drug clearance with increasing adult age. Br J Clin Pharmacol 2013; 75: 1019–1028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Lai XS, Yang LP, Li XT, Liu JP, Zhou ZW, Zhou SF. Human CYP2C8: structure, substrate specificity, inhibitor selectivity, inducers and polymorphisms. Curr Drug Metab 2009; 10: 1009–1047. [DOI] [PubMed] [Google Scholar]
- 60. Manschreck TC, Boshes RA. The CATIE schizophrenia trial: results, impact, controversy. Harv Rev Psychiatry 2007; 15: 245–258. [DOI] [PubMed] [Google Scholar]
- 61. Peck RW. The right dose for every patient: a key step for precision medicine. Nat Rev Drug Discov 2016; 15: 145–146. [DOI] [PubMed] [Google Scholar]
- 62. Ghotbi R, Mannheimer B, Aklillu E, Suda A, Bertilsson L, Eliasson E, et al Carriers of the UGT1A4 142T>G gene variant are predisposed to reduced olanzapine exposure – an impact similar to male gender or smoking in schizophrenic patients. Eur J Clin Pharmacol 2010; 66: 465–474. [DOI] [PubMed] [Google Scholar]
- 63. Nozawa M, Ohnuma T, Matsubara Y, Sakai Y, Hatano T, Hanzawa R, et al The relationship between the response of clinical symptoms and plasma olanzapine concentration, based on pharmacogenetics: Juntendo University schizophrenia projects (JUSP). Ther Drug Monit 2008; 30: 35–40. [DOI] [PubMed] [Google Scholar]
- 64. Gulcebi MI, Ozkaynakci A, Goren MZ, Aker RG, Ozkara C, Onat FY. The relationship between UGT1A4 polymorphism and serum concentration of lamotrigine in patients with epilepsy. Epilepsy Res 2011; 95: 1–8. [DOI] [PubMed] [Google Scholar]
- 65. Kassahun K, Mattiuz E, Nyhart E, Obermeyer B, Gillespie T, Murphy A, et al Disposition and biotransformation of the antipsychotic agent olanzapine in humans. Drug Metab Dispos 1997; 25: 81–93. [PubMed] [Google Scholar]
- 66. Gertz M, Harrison A, Houston JB, Galetin A. Prediction of human intestinal first‐pass metabolism of 25 CYP3A substrates from in vitro clearance and permeability data. Drug Metab Dispos 2010; 38: 1147–1158. [DOI] [PubMed] [Google Scholar]
- 67. Poulin P, Theil F‐R. Prediction of pharmacokinetics prior to in vivo studies. 1. Mechanism‐based prediction of volume of distribution. J Pharm Sci 2002; 91: 129–156. [DOI] [PubMed] [Google Scholar]
- 68. Yamazaki S, Johnson TR, Smith BJ. Prediction of drug‐drug interactions with crizotinib as the CYP3A substrate using a physiologically based pharmacokinetic model. Drug Metab Dispos 2015; 43: 1417–1429. [DOI] [PubMed] [Google Scholar]