

Abstract
Aims
Congenital hyperinsulinism (HI) is the most common cause of persistent hypoglycaemia in infants and children. Exendin‐(9‐39), an inverse glucagon‐like peptide 1 (GLP‐1) agonist, is a novel therapeutic agent for HI that has demonstrated glucose‐raising effect. We report the first population pharmacokinetic (PopPK) model of the exendin‐(9‐39) in patients with HI and propose the optimal dosing regimen for future clinical trials in neonates with HI.
Methods
A total of 182 pharmacokinetic (PK) observations from 26 subjects in three clinical studies were included for constructing the PopPK model using first order conditional estimation (FOCE) with interaction method in nonlinear mixed‐effects modelling (NONMEM). Exposure metrics (area under the curve [AUC] and maximum plasma concentration [C max]) at no observed adverse effect levels (NOAELs) in rats and dogs were determined in toxicology studies.
Results
Observed concentration–time profiles of exendin‐(9‐39) were described by a linear two‐compartmental PK model. Following allometric scaling of PK parameters, age and creatinine clearance did not significantly affect clearance. The calculated clearance and elimination half‐life for adult subjects with median weight of 69 kg were 11.8 l h−1 and 1.81 h, respectively. The maximum recommended starting dose determined from modelling and simulation based on the AUC0‐last at the NOAEL and predicted AUC0‐inf using the PopPK model was 27 mg kg−1 day−1 intravenously.
Conclusions
This is the first study to investigate the PopPK of exendin‐(9‐39) in humans. The final PopPK model was successfully used with preclinical toxicology findings to propose the optimal dosing regimen of exendin‐(9‐39) for clinical studies in neonates with HI, allowing for a more targeted dosing approach to achieve desired glycaemic response.
Keywords: congenital disorders, modelling and simulation, NONMEM, pharmacokinetics, population analysis
What is Already Known about this Subject
Exendin‐(9‐39) is an inverse agonist of the GLP‐1 receptor that has been shown to inhibit GLP‐1‐induced insulin release and increase peak postprandial glucose levels in healthy volunteers.
PK of exendin‐(9‐39) were only investigated in a small number of healthy subjects and have never been studied in patients with HI.
What this Study Adds
The first PopPK model of exendin‐(9‐39) was developed to describe the distribution and elimination of exendin‐(9‐39) and to characterize the effects of patient characteristics on PK parameters.
The PopPK model was used with preclinical toxicology findings to guide the optimal dose selection in future clinical studies in neonates with HI.
Introduction
Persistent hypoglycaemia in infants and children is most commonly caused by congenital hyperinsulinism (HI), a rare genetic condition in which pancreatic β‐cells fail to suppress insulin secretion when plasma glucose concentrations are low 1. If inadequately treated, HI can result in permanent brain damage 2, 3, 4. Therefore, it is imperative to promptly diagnose and initiate appropriate therapy in these children. The estimated incidence of HI is about 1 in 50 000 live births, though it could be significantly higher in regions with high rates of consanguinity 5.
HI results from mutations in genes that encode proteins that play an important role in the regulation of insulin secretion 4, 6. The most common and severe form of HI is caused by inactivating mutations in the ATP‐sensitive potassium channel (KATP channel) genes ABCC8 and KCNJ11, which encode SUR1 and Kir6.2 subunits of the channel, respectively 7, 8. Children affected by this form of HI (KATP HI) do not respond to the first‐line drug therapy, diazoxide, which activates the intact KATP channel and reduces insulin secretion 1, 9. In children with diazoxide‐unresponsive HI, somatostatin analogues such as octreotide can be used as second‐line drug therapy; however, their use in neonates is limited because of potentially severe side effects, particularly, necrotizing enterocolitis 3, 10, 11. Pancreatectomy should be considered for children who do not respond to drug therapy. Children with focal lesions can be cured by complete excision of the lesion 12, 13. On the other hand, near‐total pancreatectomy performed in diazoxide‐refractory patients with the diffuse form of HI as the last resort is associated with high risks of exocrine pancreatic insufficiency and postsurgical diabetes 3, 14.
Due to the complexity of disease management and the lack of optimal treatment for diazoxide‐unresponsive children with diffuse HI, there is a need to develop an alternative drug therapy that is safe and effective. We have strong preclinical and clinical data supporting a novel therapeutic approach employing exendin‐(9‐39) in the treatment of HI 15, 16. Glucagon‐like peptide 1 (GLP‐1) is an incretin hormone produced by L‐cells of the small intestine and is a potent stimulant of postprandial insulin secretion 17. Exendin‐(9‐39), a truncated form of exendin‐4 (exenatide), is an inverse agonist of the GLP‐1 receptor that has been shown to inhibit insulin release induced by GLP‐1 and increase peak postprandial glucose levels in healthy volunteers 17, 18. Data on the pharmacokinetics (PK) of exendin‐(9‐39) in human subjects are limited. In a study conducted in six healthy volunteers, Edwards et al. 17 reported that the elimination of exendin‐(9‐39) was characterized by first‐order kinetics with a half‐life around 33 min, mean clearance of 2.3 ml kg−1 min−1, and apparent volume of distribution of 111 ml kg−1. No data is available on the metabolism of exendin‐(9‐39), but it is expected that elimination of exendin‐(9‐39) would be similar to that of its parent peptide, exenatide, which is primarily eliminated through glomerular filtration followed by proteolytic degradation 19. Furthermore, PK of exendin‐(9‐39) have not been previously studied and reported in children with HI.
In this study, PK data from three clinical studies of exendin‐(9‐39) in patients with HI were used to develop the first population pharmacokinetic (PopPK) model to (1) describe the distribution and elimination of exendin‐(9‐39), and (2) identify and quantify the effects of patient characteristics on the PK of exendin‐(9‐39). In addition, the final PopPK model was used with toxicological findings from preclinical studies to propose the optimal dosing regimen for future clinical trials in neonates with HI.
Methods
Preclinical studies
Juvenile toxicology studies were conducted in Sprague Dawley rats (7 ± 1 days of age) and beagle dogs (14 ± 2 days of age) in compliance with the US Food and Drug Administration Good Laboratory Practice Regulations. Exendin‐(9‐39) was administered via three times daily subcutaneous dosing. A total of 152 rats were assigned to receive 0, 20, 80 or 300 mg kg−1 dose−1 (equivalent to 0, 60, 240 or 900 mg kg−1 day−1), and 40 dogs were assigned to receive 0, 10, 30 or 100 mg kg−1 dose−1 (equivalent to 0, 30, 90 or 300 mg kg−1 day−1) for 28 days consecutively, followed by a 14‐day recovery period. Toxicological endpoints including mortality, clinical observations, body weights, food consumption, ophthalmic examinations, clinical/microscopic pathology, urinalysis, gross necropsy, functional observational battery assessments (only in rats), and electrocardiography (only in dogs) were assessed. In the rat study, blood samples for toxicokinetics (TK) analysis were taken from the TK subgroup rats on Day 28 at 15, 45, 90 min and 4, 8 and 12 h following a single dose administration. In the dog study, blood samples for TK analysis were taken on Days 1 and 28 at 0, 15, 45, 90 min and 4, 6 and 12 h following the third dose administration. TK analysis of the two studies was conducted using noncompartmental analysis in the WinNonlin software (Version 6.2, Pharsight Corporation, Mountain View, CA). TK parameters evaluated for dose selection included maximum plasma concentrations (C max) and area under the curve from zero to the last time point (AUC0‐last). The human equivalent dose (HED) was then determined from the no observed adverse effect levels (NOAELs) identified in the rat and dog toxicology studies using the following equations 20:
| (1) |
| (2) |
where k in Equation (1) is a conversion factor with standard values of 0.162 and 0.541 for rat and dog species, respectively. In Equation (2), the reference animal weights were 0.15 kg and 10 kg for rat and dog species, respectively; the mean body weight of neonates with HI (3.7 kg) was used as the human weight 21. The maximum recommended starting dose (MRSD) for neonatal clinical studies was then derived by dividing the HED from the most sensitive species (i.e., the species from which the lowest HED was identified) by a safety factor of 10 20. This default safety factor was chosen because there are no additional safety concerns from animal studies (e.g., steep dose response curve, severe/nonmonitorable toxicities, nonlinear pharmacokinetics, etc.) that justify an increase in the safety factor 20.
Clinical studies
Three open‐label, two‐period crossover pilot clinical studies were conducted with several exploratory doses of exendin‐(9‐39) in different populations of patients with HI at The Children's Hospital of Philadelphia. All three studies were approved by the human subjects committee of The Children's Hospital of Philadelphia and the U.S. Food and Drug Administration. Informed consent was obtained from all subjects or their legally authorized representatives. Assent was obtained from subjects under 18 when indicated. The three studies were registered on clinicaltrials.gov with the following identifying numbers: NCT00571324, NCT00897676, NCT00835328.
The Effect of Exendin‐(9‐39) on Glycaemic Control in Subjects with Congenital Hyperinsulinism (later referred to as the Adult Study) was a completed clinical trial in older adolescents and adults with KATPHI 16. Subjects were excluded if they had acute diseases, a history of systemic chronic conditions, or were treated with medications that alter glucose metabolism (e.g., glucocorticoids, β‐agonists, diazoxide and octreotide) 16. The study was aimed at examining the effect of exendin‐(9‐39) on glucose metabolism. Fasted subjects received an IV infusion of exendin‐(9‐39) at 0.02, 0.06, 0.1 mg kg−1 h−1 for 2 h each or vehicle (0.9% NaCl) for 6 h in 2 consecutive days in random order. The primary outcomes were blood glucose and exendin‐(9‐39) levels. Secondary outcomes were insulin, GLP‐1 and glucagon levels. Plasma samples for exendin‐(9‐39) concentration determination were obtained at 60 and 120 min after initiation of each dose and hourly for 3 h post infusion.
The Effect of Exendin‐(9‐39) on Fasting Adaptation and Protein Sensitivity (later referred to as the Children Study) was a clinical trial in children aged 6 months to 18 years with KATPHI. The goal of the study was to examine the effect of exendin‐(9‐39) on phenotypic characteristics in HI, including fasting hypoglycaemia and protein‐induced hypoglycaemia. For the investigation of the effect of exendin‐(9‐39) on fasting blood glucose levels, fasted subjects either received one of the two dosing regimens of exendin‐(9‐39) IV infusion (0.06, 0.1, 0.06 mg kg−1 h−1 for 2 h each, or 0.06, 0.02, 0.06 mg kg−1 h−1 for 2 h each) or vehicle (0.9% NaCl) for 6 h in 2 consecutive days in random order. The primary outcome of the study was blood glucose level, and secondary outcomes were insulin, C‐peptide, GLP‐1, glucagon, beta‐hydroxybutyrate, and exendin‐(9‐39) levels. Plasma samples for exendin‐(9‐39) concentration determination were obtained hourly during the infusion and for 3 h post infusion.
The Effect of Exendin‐(9‐39) on Glucose Requirements (later referred to as the Neonate Study) was a clinical trial in infants less than 12 months old with HI who did not respond to medical therapy, with the goal of studying the effect of exendin‐(9‐39) on glucose requirements to maintain euglycaemia. Subjects either received one of the three dosing regimens of exendin‐(9‐39) IV infusion (0.02 mg kg−1 h−1 for 12 h, 0.04 mg kg−1 h−1 for 12 h, or 0.1 mg kg−1 h−1 for 6 h) or vehicle (0.9% NaCl) in 2 consecutive days in random order. The primary outcome was glucose infusion rate needed to maintain euglycaemia. Secondary outcomes were insulin, glucagon and exendin‐(9‐39) levels. Plasma samples for exendin‐(9‐39) concentration determination were obtained at half the infusion time, at the end of infusion, and hourly for 3 h post infusion.
Bioanalytical methods
The concentration of exendin‐(9‐39) in plasma samples collected from the clinical studies was quantified by liquid chromatography tandem mass spectrometry (LC–MS/MS) in the same laboratory. The LC–MS/MS method has been validated for quantification of exendin‐(9‐39) plasma concentrations as described previously 22. Briefly, 95 μl of thawed plasma sample out of 1 ml in total that was withdrawn from the subjects was combined with 5 μl of 20‐fold internal standard stock solution. Twenty‐five μl of the resulting mixture was used for solid‐phase extraction and the subsequent chromatographic analysis. Sample processing time was limited to 1 h to minimize degradation. Single reaction monitoring of m/z transitions 842.9 → 991.8 and 848.2 → 998.8 was used to quantify exendin‐(9‐39) in plasma samples and the added isotopically labelled internal standard, respectively. The calibration curve had a linear range of 10–1390 ng ml−1, and the limit of detection was 1.3 ng ml−1.
PopPK model development
The PopPK model was constructed using nonlinear mixed‐effects modelling (NONMEM version 7.3, ICON Development Solutions, Hanover, Maryland) with first order conditional estimation (FOCE) with interaction method. Exendin‐(9‐39) concentration–time data from clinical studies were fitted to one‐ and two‐compartmental linear PK models for the base model development. The interindividual variability was described using a log‐normal distribution model. Residual error was initially modelled using a proportional, a combined additive and proportional error, or a Poisson error model.
Following selection of the base model, the covariate models were built using a stepwise approach as previously described 23, 24. First, the effects of weight (body size) on all PK parameters was investigated using an allometric model:
| (3) |
where TVP is the typical value of PK parameters in individuals with WT i; θ TVP describes typical PK parameters for an individual with weight equal to the reference weight WT ref; and θ a is an allometric power parameter. The standard adult weight of 70 kg was used as WT ref, and θ a was either estimated or fixed to 0.75 for clearance and 1 for volumes based on physiologic consideration of the impact of size on metabolic rate 25. After the effects of weight were accounted for in the model, a model for other biological covariates (i.e., age, creatinine clearance [CrCL], and gender) was built using a stepwise selection approach. CrCL in ml min−1 1.73 m−2 was calculated using the MDRD equation (Equation (4)) in the adult group and the Shull et al. equation (Equation (5)) in the paediatric group, respectively 26, 27.
| (4) |
| (5) |
where Scr stands for serum creatinine in mg dl−1. After the effects of biological covariates were incorporated in the model, the effects of patient groups (paediatric vs. adult) on PK parameters were tested to produce the final model. In addition, a sigmoidal maturation model was used to explore the effect of age on CrCL. For the patient groups covariate, the paediatric group included patients from the Children Study and the Neonate Study, and the adult group included patients from the Adult Study. Additionally, the patients were re‐assigned to separate neonates (i.e., subjects from the Neonate Study) from the rest of the subjects to explore the effects of patient groups (neonates vs. others) on PK parameters. This sequential approach was used to distinguish changes in PK parameters related to weight and other biological covariates from those related to different patient groups.
The serum drug concentrations below 10 ng ml−1 (the lower limit of quantification, LLOQ) were handled as fixed‐point censored observations, and the maximum likelihood estimation approach was used to fit the model to all including the censored observations based on the M3 approach proposed by Beal et al. 28. The likelihoods for all data were maximized with respect to the model parameters, and the likelihood for a censored concentration was taken to be the likelihood that this observation was truly below the LLOQ.
The likelihood ratio test and typical diagnostic plots were used to compare goodness‐of‐fit for the nested models and select statistically significant covariates 29. A difference in objective function values between models greater than 6.64 was selected as the critical value, which corresponded to a significance level of 0.01 at one degree of freedom in the χ2 distribution. A bootstrap resampling technique was used to estimate the confidence interval of parameters and assess the stability of the final model 23, 29. Due to the presence of two distinct study populations in this analysis, a stratified sampling approach was used to obtain bootstrap samples in order to match the numbers of adult and paediatric subjects from the original data 23. The results from 1000 successful runs were recorded; the mean and 95% confidence interval for population parameters were obtained and compared with the final model estimates from the original data. The posterior predictive model check was used to evaluate the ability of the final model to describe the observed exposure parameters 30, 31, 32. The 10th, 50th (median) and 90th percentile of the C max and area under the curve from zero to infinity (AUC0‐inf) of observed data were computed and selected as the test statistics for the posterior predictive model check. The posterior predictive distribution of test statistics from 1000 simulated data sets using the final PopPK model was computed and compared with observed test statistics, and the P‐value (P PPC) was estimated by calculating the proportion of cases in which test statistics from the simulated data sets were above or below realized values of observed test statistics using the following equation 31, 33:
| (6) |
where I(.) is the indicator function which takes the value 1 when its argument is true and 0 when the argument is false. T(y, θ) is the realized value of observed test statistics, and is the test statistic from simulated data sets i (i ranging from 1 to 1000) 31, 33. A P‐value of <0.05 or >0.95 would indicate significant differences between observed test statistics and the posterior distribution of the test statistics in the simulated data sets, and that the model did not adequately describe the C max or AUC0‐inf of the observed data. In addition, prediction‐corrected visual predictive check was used to evaluate the performance of the final model 34.
The final PopPK model was then used to estimate C max and AUC0‐inf following the administration of different exendin‐(9‐39) regimens in neonates with HI. The dosage regimens tested in the simulation were 0.02, 0.04, 0.1, 0.2, 0.3, 0.4, 0.5, 0.6, 0.8, 1, 2, 3, 5 and 6 mg kg−1 h−1 infused intravenously over 6 or 9 h. A total of 1000 simulated subjects with the reported mean body weight of 3.7 kg in neonates with HI were included in the analysis.
Nomenclature of targets and ligands
Key protein targets and ligands in this article are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY 35, and are permanently archived in the Concise Guide to PHARMACOLOGY 2017/18 36.
Results
Preclinical studies
In the rat study, the only statistically significant findings were decreased red cell mass values in female rats, increased serum glucose concentrations in female rats, and decreased serum triglyceride concentrations in male rats at the end of the recovery period on Day 43. In the dog study, there were sporadic occurrences of statistically significant changes in haematology, serum chemistry and coagulation parameters, but many of them were small in magnitude without the demonstration of dose dependency. These findings in rats and dogs were considered non‐adverse with no or unclear association with exendin‐(9‐39) administration. Therefore, NOAELs in rats and dogs were the highest doses tested, i.e., 900 mg kg−1 day−1 and 300 mg kg−1 day−1, respectively. At NOAELs, based on the TK parameters obtained following the administration of exendin‐(9‐39) on Day 28, the average AUC0‐last at steady‐state was 757 500 ng h−1 ml−1 in rats (747 000 ng h−1 ml−1 in males and 768 000 ng h−1 ml−1 in females) and 1122 000 ng h−1 ml−1 in dogs (1 218 000 ng h−1 ml−1 in males and 1026 000 ng h−1 ml−1 in females). C max values were 63 200 ng ml−1 and 65 100 ng ml−1 in male and female rats; 56 900 ng ml−1 and 49 500 ng ml−1 in male and female dogs, respectively. Using 70% as the bioavailability of subcutaneous administration and the standard conversion factors (Equation (1)), the HED was calculated to be 102 mg kg−1 day−1 and 117 mg kg−1 day−1 based on NOAELs from rat and dog species, respectively. Using the HED from the rat as the more sensitive species and a safety factor of 10, the MRSD was calculated to be 10.2 mg kg−1 day−1. On the other hand, using the mean body weight of neonates with HI in Equation (2) and accounting for the bioavailability of subcutaneous administration, the HED was 219 mg kg−1 day−1 and 292 mg kg−1 day−1 based on NOAELs of rat and dog species, respectively. After the application of a safety factor of 10, the MRSD was calculated to be 21.9 mg kg−1 day−1, which was higher than the result obtained by using standard conversion factors.
PopPK model
A total of 182 plasma levels of exendin‐(9‐39) from 26 subjects were included in the development of the PopPK model, of which 16 observations below the LLOQ were included in the analysis. Demographic data of the paediatric and adult groups are summarized in Table 1, and the concomitant medications of all subjects are described in Table S1. In the paediatric group including ten children and seven neonates, the median age was 3 years (range: 0.06–15). In the adult group including nine older adolescents and adults, the median age was 19 years (range: 15–47).
Table 1.
Demographic data of the paediatric and adult groups for population pharmacokinetic model development. The paediatric group is further divided into neonates and children with subjects in the Neonate Study and the Children Study, respectively. Data are presented as median (range)
| Paediatric group (n = 17) | Adult group (n = 9) | ||
|---|---|---|---|
| Neonates (n = 7) | Children (n = 10) | ||
| Age (years) | 0.11 (0.060–0.41) | 5 (2–15) | 19 (15–47) |
| Weight (kg) | 6.25 (3.92–6.60) | 21.0 (11.8–69.2) | 69.1 (58.2–130) |
| Creatinine clearance (ml min −1 1.73 m −2 ) | 80.0 (60.0–122) | 108 (85.3–127) | 107 (85.0–113) |
| Gender | |||
| Female | 4 | 4 | 6 |
| Male | 3 | 6 | 3 |
| Race | |||
| Caucasian | 6 | 9 | 9 |
| Others | 1 (unknown) | 1 (unknown) | 0 |
| Number of exendin‐(9‐39) plasma levels | 34 | 67 | 81 |
Based on the difference in objective function values (P < 0.01), PK of exendin‐(9‐39) were best described by a two‐compartmental linear PK model using the NONMEM subroutine ADVAN3 TRANS4. Four PK parameters were described in the subroutine, including clearance 3, central volume (V1), intercompartmental clearance 25, and peripheral volume (V2). Including interindividual variability terms for V1, Q and V2 failed to achieve model convergence and/or generated poorly parameter estimates, with percent coefficient of variation (%CV) over 50%. Therefore, only the interindividual term of CL was retained in the model. Among the different residual error models tested, the Poisson error model was the most stable with lower objective function value, and therefore it was used to describe the residual variability in the analysis. After the effects of weight on PK parameters (CL, V1, Q, V2) were accounted for using allometric scaling, none of the other covariates tested (age, CrCL, gender and patient groups) had significant effects on CL (P > 0.01). The final PopPK model was described as following:
| (7) |
| (8) |
| (9) |
| (10) |
where PK parameters with the ‘TV’ subscript represent typical population values in reference adult subjects with body weight of 70 kg; WT represents body weight in kilograms.
The final PopPK model parameter estimates are presented in Table 2. All parameters were estimated with good precision (%CV < 50%). CLTV, V1TV, QTV, and V2TV were 11.6 l h−1, 9.59 l, 2.20 l h−1 and 8.89 l, respectively. Based on these parameter estimates, the calculated CL and elimination half‐life for the paediatric group with median weight of 15 kg were 3.65 l h−1 and 2.34 h, respectively; the calculated CL and elimination half‐life for the adult group with median weight of 69 kg were 11.5 l h−1 and 3.42 h, respectively.
Table 2.
Parameter estimates of the final population pharmacokinetic model and the model stability using bootstrap resampling approach
| Original data set | 1000 bootstrap replicates | |||
|---|---|---|---|---|
| Parameter | Estimate | SE | Mean | 95% CI |
| Structural model | ||||
| CL TV (l h −1 ) | 11.6 | 0.728 | 11.7 | 9.53–13.7 |
| V1 TV (l) | 9.59 | 0.449 | 9.36 | 6.36–10.5 |
| Q TV (l h −1 ) | 2.20 | 0.576 | 2.72 | 1.57–7.33 |
| V2 TV (l) | 8.89 | 0.996 | 8.24 | 3.55–42.5 |
| Interindividual variability | ||||
| ω 2 CL | 0.0572 | 0.0262 | 0.0548 | 0.0121–0.148 |
| Covariate model | ||||
| WT effect on CL and Q | 0.750 (fixed) | — | — | — |
| WT effect on V1 and V2 | 1.00 (fixed) | — | — | — |
| Residual variability | ||||
| Σ poisson | 3.66 | 0.508 | 3.59 | 2.73–4.47 |
SE, standard error; 95% CI, 95% confidence interval; CLTV, V1TV, QTV, and V2TV represent typical population values of clearance, central volume, intercompartmental clearance, and peripheral volume in reference adult subjects with weight of 70 kg, respectively. WT, weight; ω2 CL, interindividual variance of clearance; σpoisson, residual error using a Poisson error model
For the final model with covariates, the standard model diagnostic plots are shown in Figure 1. The observed concentrations were generally in accordance with the predicted concentrations with the exception of the concentration range below 20 ng ml−1, in which the model underestimated the observed concentrations possibly due to biases introduced by the M3 method in handling the PK observation below the LLOQ. From the original data set, 1500 replicate data sets were generated and used to evaluate the stability of the final model. NONMEM achieved successful minimization steps for 1261 out of 1500 (84.1%) replicate data sets, suggesting that the final PopPK developed in this study was relatively stable. The results from the first 1000 runs are shown in Table 2. The mean population parameters obtained from the bootstrap procedure were similar to the parameter estimates of the original data set, again indicating the stability of the final PopPK model 23. However, there were relatively large 95% confidence intervals for Q and V2, and the interindividual variance of CL, ηCL, from the bootstrap procedure, which could be due to the limited number of subjects included in this analysis and/or the presence of influencing subjects with PK parameters and observations that significantly deviated from population reference values. A posterior predictive model check was used to evaluate the ability of the final PopPK model to describe exposure parameters of observed data for dose selection in future clinical studies. Test statistics were computed for each of the 1000 simulated data sets from the final PopPK model. As shown in Figure 2, the observed values were within posterior predictive distributions with estimated P‐values between 0.05 and 0.95 for each test statistic, suggesting that the model was able to describe and predict the C max and AUC0‐inf of observed data reasonably well. Posterior predictive check for adult subjects and paediatric subjects separately is presented in Figure S1, and again all P PPC values were between 0.05 and 0.95. The result from prediction‐corrected visual predictive check is presented in Figure S2, which indicates the lack of random effects misspecification, as there is no apparent discrepancy between observations and model predictions.
Figure 1.
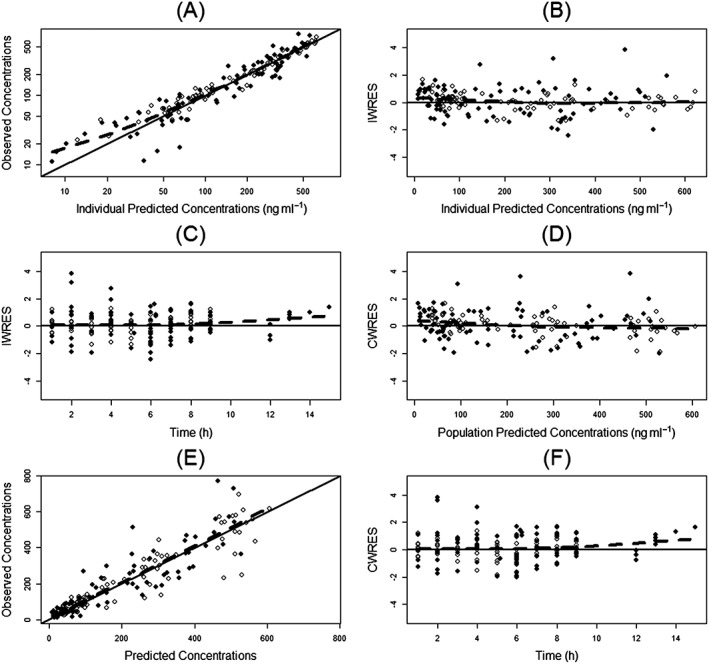
Diagnostic plots. (A) Observed vs. individual predicted concentrations; (B) individual weighted residuals (IWRES) vs. individual predicted concentrations; (C) IWRES vs. time; (D) conditional weighted residuals (CWRES) vs. population predicted concentrations; (E) observed vs. population predicted concentrations; (F) CWRES vs. time. The broken line in each panel is the loess smoothing line. Open circles represent the adult subjects, and closed circles represent the paediatric subjects
Figure 2.
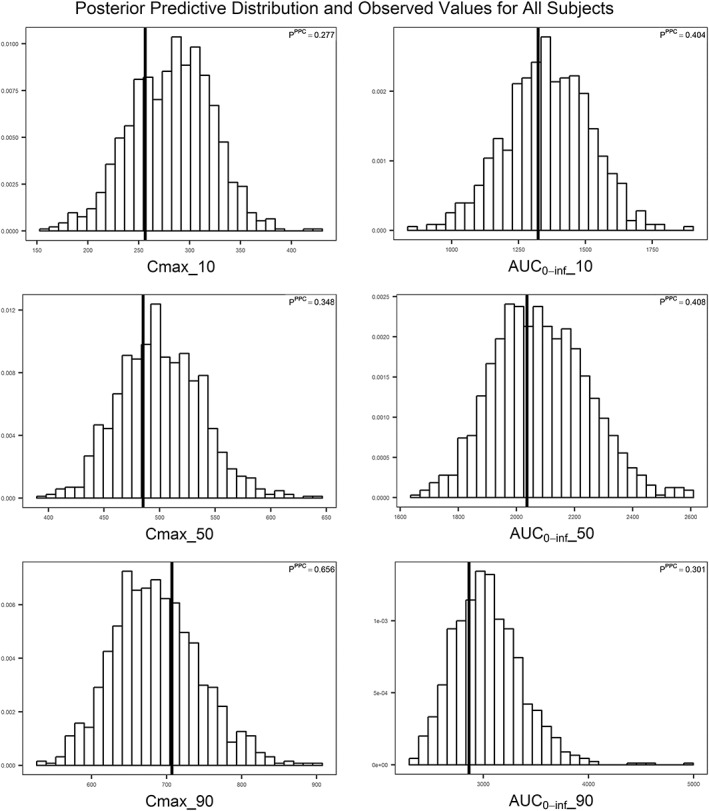
Model evaluation of exendin‐(9‐39) population pharmacokinetic model using the posterior predictive check for all subjects. Posterior predictive distributions of test statistics from 1000 simulated data sets are presented. The vertical line on each histogram represents the observed value of the test statistic. C max_10, C max_50, and C max_90 represent C max values at 10th, 50th (median), and 90th percentile, respectively. AUC0‐inf_10, AUC0‐inf_50, and AUC0‐inf_90 represent AUC0‐inf values at 10th, 50th (median), and 90th percentile, respectively
Dose determination in neonatal clinical studies
C max and AUC0‐inf values simulated from the final PopPK model were used with the AUC0‐last at the NOAEL to determine the MRSD, which was then compared with the results from the HED conversion. The simulation results of different exendin‐(9‐39) regimens in neonates based on the final PopPK model are shown in Figure 3. The median AUC0‐inf of 0.02, 0.04, 0.1, 0.2, 0.3, 0.4, 0.5, 0.6, 0.8, 1, 2, 3, 5, 6 mg kg−1 h−1 infused over 6 h were 343, 686, 1.71 × 103, 3.43 × 103, 5.14 × 103, 6.86 × 103, 8.57 × 103, 1.03 × 104, 1.37 × 104, 1.71 × 104, 3.42 × 104, 5.14 × 104, 8.57 × 104, 1.03 × 105 ng h−1 ml−1, respectively. The median AUC0‐inf of 0.02, 0.04, 0.1, 0.2, 0.3, 0.4, 0.5, 0.6, 0.8, 1, 2, 3, 5, 6 mg kg−1 h−1 infused over 9 h were 514, 1.03 × 103, 2.57 × 103, 5.14 × 103, 7.72 × 103, 1.03 × 104, 1.29 × 104, 1.54 ×104, 2.06 × 104, 2.57 × 104, 5.14 × 104, 7.72 × 104, 1.29 × 105, 1.54 × 105 ng h−1 ml−1, respectively. The median C max of 0.02, 0.04, 0.1, 0.2, 0.3, 0.4, 0.5, 0.6, 0.8, 1, 2, 3, 5, 6 mg kg−1 h−1 infused over 6 h were 15.2, 30.4, 75.9, 152, 228, 304, 380, 456, 607, 759, 1.52 × 103, 2.28 × 103, 3.80 × 103, 4.56 × 103 ng ml−1, respectively. The median C max of 0.02, 0.04, 0.1, 0.2, 0.3, 0.4, 0.5, 0.6, 0.8, 1, 2, 3, 5, 6 mg kg−1 h−1 infused over 9 h were 15.4, 30.7, 76.9, 154, 231, 307, 384, 461, 615, 769, 1.54 × 103, 2.31 × 103, 3.84 × 103, 4.61 × 103 ng ml−1, respectively. The C max values following 6 and 9 h IV infusion with the same infusion rate were very similar, probably due to the relatively short half‐life of the exendin‐(9‐39) and the achievement of steady‐state condition at 6 h. Using a factor of 10 as the safety margin based on the AUC0‐last value at steady‐state in rats at the NOAEL, the AUC0‐inf for the MRSD was calculated to be (757 500 ng h−1 ml−1)/10 = 75 750 ng h−1 ml−1, which approximated the AUC0‐inf produced by 3 mg kg−1 h−1 infused over 9 h or 27 mg kg−1 day−1 (7.72 × 104 ng h−1 ml−1). With this dosage regimen, the simulated C max value was 2.31 × 103 ng ml−1, and the safety margin based on the C max value in male rats at the NOAEL was (63 200 ng ml−1)/(2310 ng ml−1) ≈ 27. In comparison, the MRSD obtained from the HED conversion was 10 mg kg−1 day−1 and 21.9 mg kg−1 day−1 using (Equations (1) and (2)), respectively, and both values were more conservative compared to that obtained using the AUC0‐last at the NOAEL and PopPK model predictions (27 mg kg−1 day−1) (Table 3).
Figure 3.
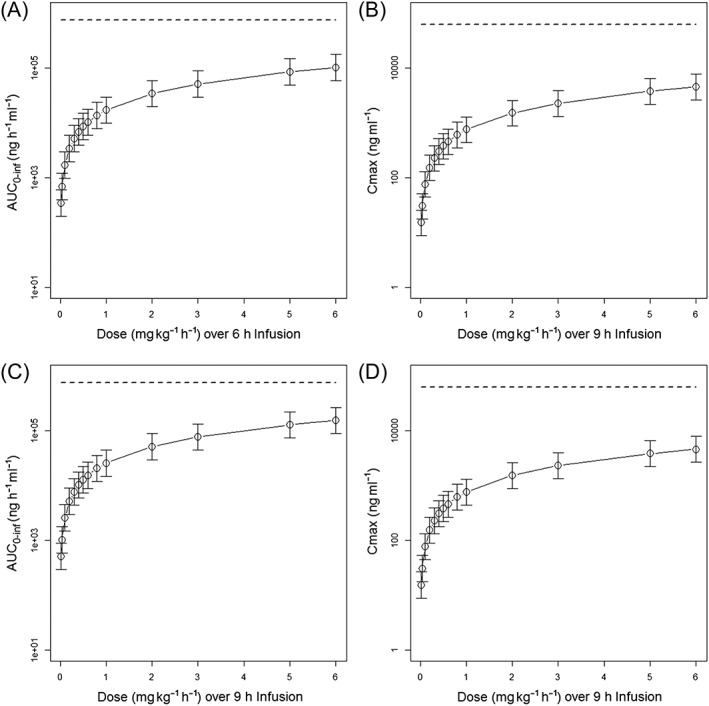
Simulated exendin‐(9‐39) AUC0–inf and C max for subjects from the Neonate Study for 0.02, 0.04, 0.1, 0.2, 0.3, 0.4, 0.5, 0.6, 0.8, 1, 2, 3, 5, 6 mg kg−1 h−1 infused over 6 (A and B) and 9 (C and D) hours. The dashed line represents the exposure levels at no observed adverse effect level in rats. Open circles represent the median value, and error bar represents the 95% percentile. AUC0‐inf = area under the plasma concentration–time curve from zero to infinity, C max = maximum plasma concentration
Table 3.
Comparisons of the maximum recommended starting dose (MRSD) determined by different approaches
| Approach | MRSD | |
|---|---|---|
| Conversion of NOAELs to HEDs | Conversion factor = 0.162 (rats) or 0.541 (dogs) | 10.2 mg kg−1 day−1 |
| Conversion factor = (Wanimal/Whuman)0.33 | 21.9 mg kg−1 day−1 | |
| Pharmacokinetic modelling guided approach | 27 mg kg−1 day−1 | |
NOAEL, no observed adverse effect level; HED, human equivalent dose; Wanimal, reference animal weight (0.150 kg for rats and 10 kg for dogs); Whuman, reference human weight (3.7 kg for mean body weight of neonates with congenital hyperinsulinism)
Discussion
Poorly treated HI could result in delays in development and permanent neurological impairment in infants and children. Diazoxide is the preferred medical treatment in HI, but it is often ineffective for patients with KATPHI. Patients with diffuse HI who are refractory to first‐ and second‐line pharmacotherapy may need a near‐total pancreatectomy, which could produce serious long‐term complications 3. Given that HI is such a complicated disease that is challenging to manage, several novel treatments are being developed in order to improve therapeutic outcomes for these patients. Among the investigational agents, exendin‐(9‐39), an inverse GLP‐1 receptor agonist, has been shown to decrease insulin secretion and increase blood glucose levels in preclinical and human studies 15, 16, 17, 18, 37.
The first objective of this study was to construct a PopPK model using PK observations from three clinical studies. PopPK analysis with NONMEM showed that exendin‐(9‐39) concentration–time profiles were well characterized by a two‐compartmental linear PK model. The model estimates of CL, V1, Q and V2 for a typical 70‐kg adult were 11.6 l h−1, 9.59 l, 2.20 l h−1 and 8.89 l, respectively. The PK characteristics of exendin‐(9‐39) reported in this study were similar to those typically observed with therapeutic peptides. Many peptides are rapidly cleared from systemic circulation primarily due to enzymatic proteolysis and/or renal elimination 38. Hydrophilic peptides with molecular weight < 2–25 kDa are likely to undergo rapid filtration through renal glomeruli and have high renal clearance due to limited reabsorption through the renal tubule 38. However, no data on renal elimination and metabolism of exendin‐(9‐39) is available. The parent peptide of exendin‐(9‐39), exenatide, is resistant to rapid proteolytic cleavage by peptidases and is predominately eliminated by glomerular filtration and proteolytic degradation in the renal tubule 39. The elimination half‐life of exendin‐(9‐39) was relatively shorter compared to that of exenatide (~2.5 h) 40, and the CL of exendin‐(9‐39) is relatively higher than the normal glomerular filtration rate in humans. These observations indicated the presence of low proteolytic activity in other tissues in addition to renal elimination (i.e., glomerular filtration and the subsequent intraluminal metabolism) 41. In the final PopPK model, following the allometric scaling of PK parameters, CrCL was not shown to significantly affect the CL of exendin‐(9‐39). This lack of effect may seem unexpected, but it could be explained by relatively narrow ranges of CrCL in the original data set due to lack of subjects with moderate to severe renal impairment. In addition, a PK study on therapeutic doses of exenatide found that mild renal impairment did not produce significant reduction in CL, while moderate renal impairment and end‐stage renal disease significantly decreased CL of exenatide 40. As exenatide and exendin‐(9‐39) share similar PK characteristics, it is reasonable to infer that the effects of renal impairment on the PK of the two therapeutic agents are likely to be similar. Nevertheless, more studies are needed in order to determine the PK of exendin‐(9‐39) in patients with moderate to severe renal impairment.
As the size of peptides is between that of small molecule drugs and large proteins, the volume of distribution of peptides is relatively small, with typical V1 values of ~3–8 l that slightly exceed the plasma volume, and typical steady‐state volume of distribution (Vss) in the range of extracellular fluid volume (~15 l in 70‐kg adults) 39. In 70‐kg adults, V1 and Vss of exendin‐(9‐39) were estimated to be 8.57 l and 15.6 l, respectively; both values were consistent with reported values of peptides. Sixteen out of the 182 PK samples included in the final analysis were below the LLOQ. It is well known that omission of the PK samples below LLOQ causes substantial bias to parameter estimates in the population PK analysis 42, and in this study excluding these observations from the analysis produced poorly estimated parameter estimates for V2 with CV% ≈ 61%. As it was very difficult to obtain sufficient PK samples in paediatric subjects with HI, a rare orphan disease, it would be very useful to include the PK samples with values below the LLOQ in the analysis to create a population PK model that can best describe the disposition of exendin‐(9‐39) in these patients. Among the modelling methods that handle PK observations below LLOQ in PopPK analysis, a likelihood‐based approach or the M3 method proposed by Beal et al. had been shown to produce consistent model parameter estimates 28, 42, 43, and this method has been successfully used to develop PopPK models using PK data with many observations below LLOQ to generate reliable model parameter estimates by fitting the developed models to the complete data sets 44, 45.
The secondary objective of this study was to select a safe and potentially more effective dosing regimen of exendin‐(9‐39) in future clinical studies involving neonates with HI based on the final PopPK model and preclinical toxicity data. According to the published results of the Adult Study, the exendin‐(9‐39) dose studied (0.02, 0.06 and 0.1 mg kg−1 h−1 for 2 h each) significantly increased fasting glucose levels compared to vehicle 16. Because neonates have a more severe phenotype of HI compared to adolescents and adults 9, they are likely to require higher exendin‐(9‐39) doses due to potentially different exposure–response relationships. Thus, a PK and efficacy approach with no extrapolation from adult efficacy data would be required for the development of exendin‐(9‐39) in neonates 46, 47. It is therefore, reasonable to include doses that produce higher exposure than that associated with the studied adult dose in future neonate studies 47. To determine a dosage regimen in neonates that could achieve adequate exposure for the evaluation of efficacy while maintaining acceptable safety margins, this study used a comparative approach to select the MRSD based on the traditional HED conversion method and the pharmacokinetically guided method. The study analysis showed that the MRSD determined from modelling and simulation based on the AUC0‐last at the NOAEL and predicted AUC0‐inf using the developed PopPK model was higher than that derived from the HED using standard conversion factors, allowing higher doses to be considered, as the pharmacokinetically guided method appeared to be a more rational approach in the presence of adequate preclinical toxicology data and clinical PK data. Nevertheless, this assumption would still need to be validated in future clinical trials in neonates, and the MRSDs determined by all approaches will be incorporated into the dose escalation scheme for future clinical studies in neonates with HI. The efficacy of the new dosing scheme determined from this study would be evaluated by the glycaemic response it produces in neonates (i.e., its ability to maintain euglycaemia without supraphysiological supplementation of glucose) in future safety/efficacy trials.
In summary, this was the first study to examine the population pharmacokinetics of exendin‐(9‐39) in HI patients of various age groups. This study showed that the final PopPK model could reasonably describe the observed plasma concentration–time profiles of exendin‐(9‐39). One limitation of the study was the small sample size, which restricted the ability of the model to estimate interindividual variability of PK parameters other than CL with good precision. Further studies are needed to improve the current PopPK model with more PK observations. Nevertheless, this study should serve as an important starting point to enable a more targeted dosing approach to link dose exposure to desired glycaemic response to exendin‐(9‐39).
Conclusion
HI is the most common cause of persistent hypoglycaemia in infants and children, and the management of the disease is often challenging, especially in patients with KATPHI who are resistant to available medical therapy. Exendin‐(9‐39) is a novel pharmacologic therapy of HI and has demonstrated efficacy in several animal and human studies. This study constructed a PopPK model of exendin‐(9‐39) using PK data from three clinical studies in various age groups of HI patients. The final model was used along with preclinical toxicology findings to guide the optimal dose selection in future clinical studies in neonates, which could potentially produce the desired pharmacologic response while maintaining considerable margins of safety.
Competing Interests
There are no competing interests to declare. This study was funded by grants from the National Institute of Health: R03DK078535, 1R56DK083670, 1X01NS064876, R01FD004095 to DDDL. In addition, this project was supported by the Goldsmith Foundation, and by the Children's Hospital of Philadelphia (CHOP) Clinical and Translational Research Center (CTRC): Grant Number UL1TR000003 from the NIH/NCATS (National Center for Advancing Translational Sciences).
The authors thank Pramod S. Terse, PhD, DABT, DABVT; NCATS/NIH, Bethesda, MD for designing and monitoring non‐clinical toxicology studies. The preclinical toxicology studies were supported by NCI‐Liedos Contract No. HHSN261200800001E, NICHD and NINDS under BrIDGs Program; and performed by Battelle, Columbus, OH under the contract. The authors would like to acknowledge the families of children with congenital hyperinsulinism for their support and participation in this study, as well as the staff of the CHOP Hyperinsulinism Center and the CHOP CTRC. The authors are also grateful for the expert contributions of Drs. Hossein Fazelinia and Hua Ding.
Contributors
C.M.N. designed and conducted the population pharmacokinetics study, analysed and interpreted study results, and drafted and revised the manuscript. F.T. drafted and revised the manuscript, and contributed to interpretation of study results. S.H.S. developed and performed the assay to measure exendin‐(9‐39) plasma concentrations, and reviewed the manuscript. Y.Z. contributed to the statistical analysis and generated results for model evaluation and data visualization. D.D.D.L. is the principal investigator and holds the IND for the investigational use of exendin‐(9‐39). She designed the clinical studies, acquired data, and supervised all aspects of the research. She reviewed and edited the manuscript. All authors reviewed and approved the final manuscript.
Supporting information
Table S1 Concomitant medications that all the subjects received during the study period
Figure S1 Model evaluation of exendin‐(9‐39) population pharmacokinetic model using the posterior predictive check in the adult group (A) and the paediatric group (B), respectively. Posterior predictive distributions of test statistics from a total of 1000 simulated data sets are presented. The vertical line on each histogram represents the observed value of the test statistic. C max_10, C max_50, and C max_90 represent C max values at 10th, 50th (median), and 90th percentile, respectively. AUC0‐inf_10, AUC0‐inf_50, and AUC0‐inf_90 represent AUC0‐inf values at 10th, 50th (median), and 90th percentile, respectively
Figure S2 Model evaluation of exendin‐(9‐39) population pharmacokinetic model using prediction‐corrected visual predictive check. The solid red line represents the median prediction‐corrected observed plasma concentration in ng ml−1, and the simulation‐based 95% confidence interval for the median is represented by the semitransparent red area. The dashed red lines represent the observed 5% and 95% percentiles, and the semitransparent blue fields show the 95% confidence intervals for the model predicted percentiles. Prediction‐corrected observed plasma concentrations are shown in blue circles
Ng, C. M. , Tang, F. , Seeholzer, S. H. , Zou, Y. , and De León, D. D. (2018) Population pharmacokinetics of exendin‐(9‐39) and clinical dose selection in patients with congenital hyperinsulinism. Br J Clin Pharmacol, 84: 520–532. doi: 10.1111/bcp.13463.
References
- 1. Lord K, De Leon DD. Monogenic hyperinsulinemic hypoglycemia: current insights into the pathogenesis and management. Int J Pediatr Endocrinol 2013; 2013: 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Meissner T, Wendel U, Burgard P, Schaetzle S, Mayatepek E. Long‐term follow‐up of 114 patients with congenital hyperinsulinism. Eur J Endocrinol 2003; 149: 43–51. [DOI] [PubMed] [Google Scholar]
- 3. Lord K, Radcliffe J, Gallagher PR, Adzick NS, Stanley CA, De Leon DD. High risk of diabetes and neurobehavioral deficits in individuals with surgically treated hyperinsulinism. J Clin Endocrinol Metab 2015; 100: 4133–4139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Stanley CA. Perspective on the genetics and diagnosis of congenital hyperinsulinism disorders. J Clin Endocrinol Metab 2016; 101: 815–826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Huopio H, Shyng SL, Otonkoski T, Nichols CG. K(ATP) channels and insulin secretion disorders. Am J Physiol Endocrinol Metab 2002; 283: E207–E216. [DOI] [PubMed] [Google Scholar]
- 6. Mohamed Z, Arya VB, Hussain K. Hyperinsulinaemic hypoglycaemia:genetic mechanisms, diagnosis and management. J Clin Res Pediatr Endocrinol 2012; 4: 169–181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. James C, Kapoor RR, Ismail D, Hussain K. The genetic basis of congenital hyperinsulinism. J Med Genet 2009; 46: 289–299. [DOI] [PubMed] [Google Scholar]
- 8. Dunne MJ, Cosgrove KE, Shepherd RM, Aynsley‐Green A, Lindley KJ. Hyperinsulinism in infancy: from basic science to clinical disease. Physiol Rev 2004; 84: 239–275. [DOI] [PubMed] [Google Scholar]
- 9. Rozenkova K, Guemes M, Shah P, Hussain K. The diagnosis and management of hyperinsulinaemic hypoglycaemia. J Clin Res Pediatr Endocrinol 2015; 7: 86–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. McMahon AW, Wharton GT, Thornton P, De Leon DD. Octreotide use and safety in infants with hyperinsulinism. Pharmacoepidemiol Drug Saf 2017; 26: 26–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Pinney SE, MacMullen C, Becker S, Lin YW, Hanna C, Thornton P, et al Clinical characteristics and biochemical mechanisms of congenital hyperinsulinism associated with dominant KATP channel mutations. J Clin Invest 2008; 118: 2877–2886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Lord K, Dzata E, Snider KE, Gallagher PR, De Leon DD. Clinical presentation and management of children with diffuse and focal hyperinsulinism: a review of 223 cases. J Clin Endocrinol Metab 2013; 98: E1786–E1789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Snider KE, Becker S, Boyajian L, Shyng SL, MacMullen C, Hughes N, et al Genotype and phenotype correlations in 417 children with congenital hyperinsulinism. J Clin Endocrinol Metab 2013; 98: E355–E363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Vora S, Chandran S, Rajadurai VS, Hussain K. Hyperinsulinemic hypoglycemia in infancy: current concepts in diagnosis and management. Indian Pediatr 2015; 52: 1051–1059. [DOI] [PubMed] [Google Scholar]
- 15. De Leon DD, Li C, Delson MI, Matschinsky FM, Stanley CA, Stoffers DA. Exendin‐(9‐39) corrects fasting hypoglycemia in SUR‐1−/− mice by lowering cAMP in pancreatic beta‐cells and inhibiting insulin secretion. J Biol Chem 2008; 283: 25786–25793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Calabria AC, Li C, Gallagher PR, Stanley CA, De Leon DD. GLP‐1 receptor antagonist exendin‐(9‐39) elevates fasting blood glucose levels in congenital hyperinsulinism owing to inactivating mutations in the ATP‐sensitive K+ channel. Diabetes 2012; 61: 2585–2591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Edwards CM, Todd JF, Mahmoudi M, Wang Z, Wang RM, Ghatei MA, et al Glucagon‐like peptide 1 has a physiological role in the control of postprandial glucose in humans: studies with the antagonist exendin 9‐39. Diabetes 1999; 48: 86–93. [DOI] [PubMed] [Google Scholar]
- 18. Schirra J, Sturm K, Leicht P, Arnold R, Goke B, Katschinski M. Exendin(9‐39)amide is an antagonist of glucagon‐like peptide‐1(7‐36)amide in humans. J Clin Invest 1998; 101: 1421–1430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Gedulin BR, Smith PA, Jodka CM, Chen K, Bhavsar S, Nielsen LL, et al Pharmacokinetics and pharmacodynamics of exenatide following alternate routes of administration. Int J Pharm 2008; 356: 231–238. [DOI] [PubMed] [Google Scholar]
- 20. FDA . Guidance for industry: estimating the maximum safe starting dose in initial clinical trials for therapeutics in adult healthy volunteers, July 2005.
- 21. de Lonlay‐Debeney P, Poggi‐Travert F, Fournet JC, Sempoux C, Dionisi Vici C, Brunelle F, et al Clinical features of 52 neonates with hyperinsulinism. N Engl J Med 1999; 340: 1169–1175. [DOI] [PubMed] [Google Scholar]
- 22. Lasaosa M, Patel P, Givler S, De Leon DD, Seeholzer SH. A liquid chromatography‐mass spectrometry assay for quantification of exendin[9‐39] in human plasma. J Chromatogr B Analyt Technol Biomed Life Sci 2014; 947–948: 186–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Ng CM, Dombrowsky E, Lin H, Erlich ME, Moody DE, Barrett JS, et al Population pharmacokinetic model of sublingual buprenorphine in neonatal abstinence syndrome. Pharmacotherapy 2015; 35: 670–680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Shellhaas RA, Ng CM, Dillon CH, Barks JD, Bhatt‐Mehta V. Population pharmacokinetics of phenobarbital in infants with neonatal encephalopathy treated with therapeutic hypothermia. Pediatr Crit Care Med 2013; 14: 194–202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. West GB, Brown JH, Enquist BJ. The fourth dimension of life: fractal geometry and allometric scaling of organisms. Science 1999; 284: 1677–1679. [DOI] [PubMed] [Google Scholar]
- 26. Levey AS, Coresh J, Greene T, Marsh J, Stevens LA, Kusek JW, et al Expressing the modification of diet in renal disease study equation for estimating glomerular filtration rate with standardized serum creatinine values. Clin Chem 2007; 53: 766–772. [DOI] [PubMed] [Google Scholar]
- 27. Shull BC, Haughey D, Koup JR, Baliah T, Li PK. A useful method for predicting creatinine clearance in children. Clin Chem 1978; 24: 1167–1169. [PubMed] [Google Scholar]
- 28. Beal SL. Ways to fit a PK model with some data below the quantification limit. J Pharmacokinet Pharmacodyn 2001; 28: 481–504. [DOI] [PubMed] [Google Scholar]
- 29. Ng CM, Bruno R, Combs D, Davies B. Population pharmacokinetics of rituximab (anti‐CD20 monoclonal antibody) in rheumatoid arthritis patients during a phase II clinical trial. J Clin Pharmacol 2005; 45: 792–801. [DOI] [PubMed] [Google Scholar]
- 30. Yano Y, Beal SL, Sheiner LB. Evaluating pharmacokinetic/pharmacodynamic models using the posterior predictive check. J Pharmacokinet Pharmacodyn 2001; 28: 171–192. [DOI] [PubMed] [Google Scholar]
- 31. Gelman A, Meng XL. Model checking and model improvement In: Markov Chain Monte Carlo in Practice, eds Gilks WR, Richardson S, Spiegelhalter DJ. Boca Raton, FL: Chapman & Hall/CRC, 1996; 189–202. [Google Scholar]
- 32. Gelman A, Carlin JB, Stern HS, Rubin DB. Bayesian Data Analysis. Chapman & Hall/CRC: Boca Raton, FL, 2004. [Google Scholar]
- 33. Ng CM, Lum BL, Gimenez V, Kelsey S, Allison D. Rationale for fixed dosing of pertuzumab in cancer patients based on population pharmacokinetic analysis. Pharm Res 2006; 23: 1275–1284. [DOI] [PubMed] [Google Scholar]
- 34. Bergstrand M, Hooker AC, Wallin JE, Karlsson MO. Prediction‐corrected visual predictive checks for diagnosing nonlinear mixed‐effects models. AAPS J 2011; 13: 143–151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Southan C, Sharman JL, Benson HE, Faccenda E, Pawson AJ, Alexander SPH, et al The IUPHAR/BPS guide to PHARMACOLOGY in 2016: towards curated quantitative interactions between 1300 protein targets and 6000 ligands. Nucl Acids Res 2016; 44: D1054–D1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Alexander SPH, Christopoulos A, Davenport AP, Kelly E, Marrion NV, Peters JA, et al The Concise Guide to Pharmacology 2017/18: G protein‐coupled receptors. Br J Pharmacol 2017; 174 (Suppl 1): S17–S129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. De Leon DD, Stanley CA. Congenital hypoglycemia disorders: new aspects of etiology, diagnosis, treatment and outcomes: highlights of the Proceedings of the Congenital Hypoglycemia Disorders Symposium, Philadelphia, April 2016. Pediatr Diabetes 2017; 18: 3–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Di L. Strategic approaches to optimizing peptide ADME properties. AAPS J 2015; 17: 134–143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Diao L, Meibohm B. Pharmacokinetics and pharmacokinetic‐pharmacodynamic correlations of therapeutic peptides. Clin Pharmacokinet 2013; 52: 855–868. [DOI] [PubMed] [Google Scholar]
- 40. Linnebjerg H, Kothare PA, Park S, Mace K, Reddy S, Mitchell M, et al Effect of renal impairment on the pharmacokinetics of exenatide. Br J Clin Pharmacol 2007; 64: 317–327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Meibohm B, Zhou H. Characterizing the impact of renal impairment on the clinical pharmacology of biologics. J Clin Pharmacol 2012; 52 (1 Suppl): 54s–62s. [DOI] [PubMed] [Google Scholar]
- 42. Bergstrand M, Karlsson MO. Handling data below the limit of quantification in mixed effect models. AAPS J 2009; 11: 371–380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Ahn JE, Karlsson MO, Dunne A, Ludden TM. Likelihood based approaches to handling data below the quantification limit using NONMEM VI. J Pharmacokinet Pharmacodyn 2008; 35: 401–421. [DOI] [PubMed] [Google Scholar]
- 44. Ng CM, Joshi A, Dedrick RL, Garovoy MR, Bauer RJ. Pharmacokinetic‐pharmacodynamic‐efficacy analysis of efalizumab in patients with moderate to severe psoriasis. Pharm Res 2005; 22: 1088–1100. [DOI] [PubMed] [Google Scholar]
- 45. Ng CM, Loyet KM, Iyer S, Fielder PJ, Deng R. Modeling approach to investigate the effect of neonatal Fc receptor binding affinity and anti‐therapeutic antibody on the pharmacokinetic of humanized monoclonal anti‐tumor necrosis factor‐alpha IgG antibody in cynomolgus monkey. Eur J Pharm Sci 2014; 51: 51–58. [DOI] [PubMed] [Google Scholar]
- 46. Dunne J, Rodriguez WJ, Murphy MD, Beasley BN, Burckart GJ, Filie JD, et al Extrapolation of adult data and other data in pediatric drug‐development programs. Pediatrics 2011; 128: e1242–e1249. https://doi.org/10.1542/peds.2010‐3487. [DOI] [PubMed] [Google Scholar]
- 47. FDA . Guidance for industry: general clinical pharmacology considerations for pediatric studies for drugs and biological products, December 2014.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1 Concomitant medications that all the subjects received during the study period
Figure S1 Model evaluation of exendin‐(9‐39) population pharmacokinetic model using the posterior predictive check in the adult group (A) and the paediatric group (B), respectively. Posterior predictive distributions of test statistics from a total of 1000 simulated data sets are presented. The vertical line on each histogram represents the observed value of the test statistic. C max_10, C max_50, and C max_90 represent C max values at 10th, 50th (median), and 90th percentile, respectively. AUC0‐inf_10, AUC0‐inf_50, and AUC0‐inf_90 represent AUC0‐inf values at 10th, 50th (median), and 90th percentile, respectively
Figure S2 Model evaluation of exendin‐(9‐39) population pharmacokinetic model using prediction‐corrected visual predictive check. The solid red line represents the median prediction‐corrected observed plasma concentration in ng ml−1, and the simulation‐based 95% confidence interval for the median is represented by the semitransparent red area. The dashed red lines represent the observed 5% and 95% percentiles, and the semitransparent blue fields show the 95% confidence intervals for the model predicted percentiles. Prediction‐corrected observed plasma concentrations are shown in blue circles