

Abstract
Clinical use of multipotent Mesenchymal Stromal Cell (MSC)-based medicinal products requires their production in compliance with Good Manufacturing Practices, thus ensuring that the final drug product meets specifications consistently from batch to batch in terms of cell viability, identity, purity and potency. Potency relates to the efficacy of the medicine in its target clinical indication, so adequate release tests need to be defined and validated as quality controls. Herein we report the design and optimisation of parameters affecting the performance of an in vitro cell-based assay for assessing immunomodulatory potential of clinical grade MSC for human use, based on their capacity to inhibit proliferation of T lymphocytes under strong polyclonal stimuli. The resulting method was demonstrated to be reproducible and relatively simple to execute. Two case studies using clinical grade MSC are presented as examples to illustrate the applicability of the methodology described in this work.
Keywords: Multipotent Mesenchymal Stromal Cell, Immunomodulative potential, Potency assay, Cellular therapy, Cell culture, Lymphocyte, Cell-based assay, Co-culture
Introduction
Potency assays are legally required in the production of cell-based Advanced Therapy Medicinal Products (ATMP) and provide critical information on the quality attributes needed in their characterisation, in addition to other product qualities including physicochemical and biological characteristics, purity, and stability (Schneider et al. 2010). The ultimate aim of potency assessment is to identify and measure those parameters critically related to efficacy, so their proper control brings consistency that ATMP will meet the expected clinical effects (Bravery et al. 2013). Ideally, potency tests should be accurate, specific, suitable and robust, and correspond to the product’s mechanism of action (MoA). These objectives have been addressed by developing quantitative biological assays that measure the activity of the product related to its biologic function. However, most ATMP have complex and/or not fully characterized MoA, making it difficult to determine which of the product’s attributes is most relevant to measuring potency (Galipeau et al. 2015). Both the European Medicines Agency (EMA) and the United States Food and Drug Administration (FDA) have provided general guidance for testing the potency of ATMP, but no cell type-specific or condition-specific tests have yet been proposed. Therefore the adequacy of potency assays to be used as quality control is usually evaluated on a case-by-case basis (de Wolf et al. 2017).
From all ATMP currently under development, Mesenchymal Stromal Cells (MSC) are probably the most frequently used cell type (Prockop 2017). MSC are multipotent cells that can be isolated from many tissue sources (i.e. bone marrow, umbilical cord and adipose tissue) and can differentiate into several specialised cell types (i.e. bone, cartilage and fat) (Nombela-Arrieta et al. 2011). Prospective isolation of MSC or characterisation of cell cultures is challenging due to the lack of a unique marker (Bianco et al. 2013). In 2006, the International Society for Cellular Therapy (ISCT) proposed three minimal criteria to define human MSC, namely: (1) MSC should be plastic-adherent when maintained in standard culture conditions; (2) MSC should express CD105, CD73 and CD90 and should lack expression of CD45, CD34, CD14/CD11b, CD79α/CD19 and Human Leukocyte Antigen (HLA)-DR; and (3) MSC should be able to differentiate in vitro into osteoblasts, adipocytes and chondroblasts (Dominici et al. 2006; Wang et al. 2014). However, MSC show other features of enormous interest in regenerative medicine (Keating 2012). In this sense, it is well known that MSC display marked immunomodulatory activity in addition to low immunogenicity and multipotency, which confer these cells a great potential for therapeutic applications, such as tissue regeneration and the management of immunological disorders (Reis et al. 2016). The main immunomodulatory role of MSC appears to involve the participation on the immunologic tolerance. To do this, MSC directly suppress the activation, proliferation, and effector functions of pro-inflammatory cells; and stimulate anti-inflammatory cells to indirectly augment immunoregulatory responses (de Wolf et al. 2017), cells from both the innate (dendritic cells [DC], natural killer cells, neutrophils, monocytes, macrophages) and adaptive (T and B cells) immune system (Reis et al. 2016).
Existing potency assays for evaluating the immunomodulatory ability of MSC-based products involve the inhibition of T cell activation or proliferation in co-culture (de Wolf et al. 2017). Responder cells used in immune cell inhibition assays can correspond to a whole population of isolated Peripheral Blood Mononuclear Cells (PBMC) or a specific purified population as for instance CD3+ T cells. Despite PBMC being representative of the in vivo environment in which MSC are expected to exert their immunomodulatory function, the interaction between different cell types leads to variability and this may impact on assay robustness and reproducibility (de Wolf et al. 2017; Galipeau et al. 2015). Proliferation of T cells (either purified or present in PBMC population) is usually triggered by mitogens (i.e., phorbol myristate acetate, PMA, combined with ionomycin; phytohemagglutinin, PHA), memory antigens, T cell receptor cross-linking and co-stimulation (anti-CD3:anti-CD28) or via the effect of allogeneity (e.g., allogeneic PBMC or DC in a mixed lymphocyte reaction [MLR]) (de Wolf et al. 2017). In all cases, assay variability and read-out parameters are the major concerns when testing potency. Read-outs commonly rely either on (1) the inhibition of activation markers (e.g. CD25, CD69 or CD154); or (2) parameters related to functioning of responder cells (e.g. cytokine production, chemotaxis or proliferation). Both types of read-outs show advantages and concerns. Perhaps the main concern with the first type of read-outs is that the anti-proliferative effect of MSC may not be due to an impaired T cell activation, but other negative effects in their proliferation. As reviewed by de Wolf and collaborators, MSC can induce cell cycle arrest in activated T cells without affecting the expression of early activation markers CD25 and CD69. Besides, the effect on T cell activation markers could be dependent on the MSC culture conditions and the presence of other cells in the co-culture with T cells (de Wolf et al. 2017). Regarding the second type of read-outs, these are considered surrogate markers for MSC functionality that can be influenced by a number of parameters. For instance, cytokine release can be influenced by timing of production and duration of exposure influence (de Wolf et al. 2017). Among these surrogate markers, the most common is determining the suppression of T cell proliferation, which can be easily measured by several techniques including 3H-Thymidine (3H-dT) or Bromodeoxyuridine (BrdU) incorporation assays and carboxyfluorescein diacetate succinimidyl ester (CFSE) dilution. This later assay offers several advantages, as it can resolve multiple successive generations using flow cytometry and allows the use of fluorochrome-conjugated antibodies to be used to immunophenotype the dividing cells. It requires the staining of PBMC with. CFSE, which binds covalently to intracellular proteins. Fluorescence intensity equitably distributes into daughter cells upon cell division, so proliferation can be readily measured up to 8 cell doublings, by measuring the drop in fluorescence intensity (Lyons and Parish 1994).
In the present work, we studied the critical aspects mentioned previously in order to (1) provide detailed description of a robust and relatively simple method for isolation and CFSE labelling of PBMC, and (2) their use in lymphocyte proliferation assays in co-culture with MSC, thus contributing to standardise a potency assay for use with MSC. Furthermore, the applicability of such cell-based assay is demonstrated in two examples that illustrate its robustness and versatility with practical cases.
Materials and methods
MSC cultures
MSC derived from bone marrow (BM) were produced for clinical use following a Good Manufacturing Practice (GMP)-compliant bioprocess with appropriate donor informed consent, as described elsewhere (Codinach et al. 2016). MSC were further expanded in vitro by using “expansion medium”, which was composed of Dulbecco’s Modified Eagle’s Medium (DMEM; Gibco, Grand Island, NY, USA) containing 2 mM glutamine and supplemented with 10% (v/v) human Serum B (hSerB; Banc de Sang i Teixits, Barcelona, Spain). Batches of sera are validated internally following GMP compliant procedures by using control MSC lines. All cultures were maintained at + 37 °C and 5% CO2 in humidified incubators and whole medium replacement was performed every 3–4 days.
Based on the minimal criteria established by the International Society for Cellular Therapy (Dominici et al. 2006), MSC used in this work tested negative for the expression of the surface markers CD31 (WM59 clone; BD Biosciences, San Diego, USA) and CD45 (HI30 clone; BD Biosciences, USA); whereas they expressed CD90 (5E10 clone; BD Biosciences), CD73 (AD2 clone; BD Biosciences), and CD105 (43A4E1 clone; Miltenyi Biotec, Bergisch Gladbach, Germany), as determined by flow cytometry using a FACS Calibur flow cytometer (Becton–Dickinson, San Jose, CA, USA).
In the experiment shown in Fig. 1E, MSC were irradiated when indicated at 20 Gy for 574 s using an IBL-437 Cs 137 Irradiator (CIS Bio International, Saclay, France).
Fig. 1.

Optimisation of conditions affecting a cell-based assay design for the assessment of the effect of MSC on the proliferation of stimulated lymphocytes. a Schematic view of the design of the assay, in which PBMC are first isolated from blood samples and stained with CFSE, then CFSE-labelled PBMC are co-cultured with MSC in the presence of proliferative stimuli; b viability of PBMC 24 h after labelling with increasing concentrations of CFSE. BM represents 10% human Serum B supplemented medium; c histogram plot allowing for the tracking of labelled PBMC by the detection of sequential halving of CFSE fluorescence intensity (stimulated population in grey overlaid with the non-stimulated control); d bright field microscopy revealing clumping of PBMC after 5 days in presence of 25 ng/mL PMA and 0.5 µM ionomycin (right), as opposed to non-stimulated control (left); e effect of MSC irradiation and MSC:PBMC ratio on the inhibition of PBMC proliferation percentage. Statistical significance was set at: **p < 0.005; and ***p < 0.001
MSC cryopreservation and thawing
MSC were cryopreserved in a solution composed of Dulbecco’s Phosphate-Buffered Saline (DPBS; Gibco) supplemented with 10% (v/v) dimethyl sulfoxide (DMSO; OriGen Biomedical, Austin, TX, USA) and 2% (w/v) Human Serum Albumin (HSA; Grífols, Barcelona, Spain), by applying a controlled freezing rate of 1 °C/min in a Mr. Frosty device (Nalgene, Rochester, NY, USA) kept in a − 80 °C freezer for 24 h. Then cryovials were transferred to a liquid nitrogen tank for long-term storage until further use. Cryopreserved cells were rapidly thawed in a 37 °C water bath, then slowly diluted 1:10 using pre-cooled “expansion medium” and plated at high density (4 × 103 – 6 × 103 cells/cm2).
MSC sorting by HLA-DR expression
Prior to sorting, cells were labelled with 25 μL of anti-HLA-DR-FITC antibody (TU36 clone; BD Biosciences) in 250 μL of a solution made of DPBS supplemented with 1% hSerB, and 100 units/mL penicillin and 100 µg/mL streptomycin (P/S; Sigma-Aldrich, St. Louis, MO, USA). The resulting cell suspension was incubated for 15 min in the dark, at room temperature (RT). The excess of antibody was washed out by centrifugation at 340 g, 10 min, RT and the cell pellet was resuspended in DPBS supplemented with 1% hSerB and P/S. HLA-DR+ and HLA-DR- populations were isolated using a BD FACSAria II sorter device (Becton–Dickinson), and then cells were resuspended in “expansion medium” supplemented with P/S.
PBMC isolation
The immunomodulation potential of MSC was analysed with PBMC obtained by density gradient centrifugation (Histopaque®-1077; Sigma-Aldrich) from 24 to 48 h old buffy-coats or peripheral blood of healthy blood donors, which were confirmed negative for hepatitis B virus (HBV), hepatitis C virus (HCV), human immunodeficiency virus (HIV) and syphilis, both by serology and viral Nucleic Acid Detection (NAD).
PBMC were isolated following either one of the two methodologies described next: (1) classical density gradient method, or (2) SepMate™ Tubes. In both cases, prior to the density gradient, blood was diluted with DPBS, either 1:10 if the starting material was buffy coat blood or 1:2 if peripheral blood was used.
Classical density gradient method 25 mL of diluted blood were layered on top of 15 mL Histopaque®-1077, which were previously added to 50 mL conical tubes. To avoid mixing the two phases, diluted blood was dispensed slowly and the tube was handled with extreme care. Tubes were centrifuged at 400×g for 30 min at RT without brake. After the centrifugation, a cellular ring was formed in the interphase (white colour) containing mononucleated cells that were collected with a Pasteur pipette and then transferred into new 50 mL conical tubes. PBMC were then washed twice with DPBS by centrifugation at 340×g for 10 min at RT. Cellular pellets were resuspended and collected in a single tube, washed again with DPBS by centrifugation at 340×g for 10 min at RT, and finally resuspended in 5–10 mL of DPBS.
Isolation with SepMate tubes Histopaque®-1077 was added into SepMate™ tubes (STEMCELL Technologies Inc., Vancouver, British Columbia, Canada) until filling completely the total volume available (approximately 15 mL). 32 mL of diluted blood were layered on top of Histopaque volume. A centrifugation step was performed at 1200×g for 20 min at RT, with the brake on. After centrifugation, PBMC were collected by pouring supernatants into new 50 mL conical tubes (each supernatant in a new tube). The following steps were the same as described previously for the classical isolation method.
Assessment of PBMC counting and viability
The determination of cell concentration was performed by flow cytometry using a microbead-based single-platform system for absolute counts (Perfect-Count Microspheres™; Cytognos, Salamanca, Spain). Criterion for selection of cell population was based on size and complexity.
The percentage of cell viability was determined by 7-aminoactinomycin D (7AAD; BD Biosciences USA) staining, which binds specifically to DNA when the cell membrane is damaged by necrotic processes. Cells were incubated with 20 µL of 7AAD for 5 min at RT in the dark, and then washed with 3 mL of FACS Flow sheath fluid (BD Biosciences) by centrifugation at 340×g, 10 min, RT, prior to cytometric acquisition.
Labelling of PBMC
PBMC were labelled with CFSE (CellTrace™ CFSE Cell Proliferation Kit; Invitrogen, Waltham, MA, USA) in order to monitor their proliferation by flow cytometry. Briefly, cells were washed with DPBS, then resuspended at 2.5 – 5 × 106 cells/mL in either DPBS or “staining medium” [composed of Roswell Park Memorial Institute medium (RPMI) supplemented with 10% hSerB] and labelled with 0.625, 2, 8 and 30 µM CFSE for 5–10 min. Then, PBMC were washed with 5–10 volumes of medium, resuspended at 1 – 2 × 107 cells/mL and incubated for 12–15 min either at + 37 °C or at RT with pre-warmed medium. A final wash with 5–10 volumes of medium was performed and CFSE-labelled PBMC were resuspended at 2 – 4 × 106 cells/mL and maintained overnight at + 37 °C, 5% CO2, until their use for the Lymphocyte proliferation assay. Media used for the washing steps and pellet resuspensions consisted either in “staining” or “expansion” medium.
Lymphocyte proliferation assays
CFSE labelled PBMC were seeded onto flat-bottomed 24-well plates in either presence or absence of MSC. Proliferation of PBMC was determined by measuring the reduction of fluorescence intensity at day 5 after stimulation with either: (1) PMA and ionomycin (both from Sigma-Aldrich); (2) PHA (Sigma-Aldrich); or antibodies against CD3 and CD28 (Biolegend, San Diego, CA, USA) (Fig. 1a). Basal fluorescence was determined approximately 24 h after CFSE staining, hence allowing for the stabilization of fluorescence signal. FlowJo VX software (FlowJo LLC, Ashland, OR, USA) was used for the quantification of proliferation. All conditions were tested in duplicates. Proliferation threshold was set-up with non-stimulated PBMC (Eqs. 1–3). To determine the effect of MSC on the inhibition of PBMC proliferation, data were normalised considering the stimulated condition of single PBMC culture (without MSC) as 100% of proliferation. Calculations were made following Eqs. 1, 2 and 3.
| 1 |
| 2 |
| 3 |
Data analysis
Statistical significance was set at: * = p < 0.05; ** = p < 0.005; *** = p < 0.001 and assessed by using the t test using GraphPad Prism program (GraphPad Software, Inc., San Diego, CA, USA).
Results
Isolation of PBMC
Successful isolation of PBMC from peripheral blood and buffy coats was achieved either by using density gradients in standard 50 mL or with SepMate™ tubes, resulting in both cases in similar yields with no remarkable differences in terms of cell recovery (8.70 × 106 ± 1.8 × 106 vs. 7.53 × 106 ± 2.07 × 106 of total cell population, 5.44 × 106 ± 6.93 × 105 vs. 4.83 × 106 ± 1.17 × 106 of lymphocyte population) and viability (94.55 ± 0.12 vs. 94.53 ± 0.03%). For convenience, SepMate™ tubes were used subsequently, therefore mitigating risk of manipulation error and simplifying the experimental workflow.
PBMC labelling with CFSE
Optimal CFSE labelling reaction setup was sought by testing different conditions (0.625, 2, 8 and 30 µM CFSE) using 2.5 − 5 × 106 cells/mL PBMC, and reaction time set at 5–10 min at RT and quenched by adding 5–10 volumes of either “staining” or “expansion” medium before spinning the cells down and resuspension in pre-warmed “staining” or “expansion” medium. Reactions were left to stand for 12–15 min at + 37 °C or RT, before performing an additional wash. For each concentration, the labelling reaction was performed in DPBS and “staining medium”. Conditions in which the cells were labelled in “staining medium” exhibited lower fluorescent levels compared to the ones labelled in DPBS, consistent with the presence of proteins in the “staining medium” that can couple with a fraction of the dye added. We also found that optimal visualisation of CFSE fluorescent signal was observed when plotted between the third and fourth logarithm of the FL1 axis. This allowed sufficient separation between labelled samples and non-labelled controls. For instance, the mean fluorescence intensity (MFI) obtained in “staining medium” (5.73 with 2 µM CFSE, 5.13 with 8 µM CFSE, and 11.87 with 30 µM CFSE) was too low for all conditions to be considered a feasible option. Conditions having concentrations higher than 2 µM CFSE in DPBS showed fluorescent intensity so high that a fraction of the cell counts fell outside the plotting area of the graphic (MFI of 3161.48 for 8 µM and 4191.20 for 30 µM). Therefore conditions consisting of 0.625 and 2 µM CFSE in DPBS gave the best results (MFI: 317.90 and 675.42, respectively).
In order to rule out potential toxicity of CFSE on the cells, viability of CFSE-labelled PBMC was analysed by 7AAD staining. Interestingly, values of cell viability were similar in all conditions tested (81.8 ± 3.71%) (Fig. 1b).
Stimulation of PBMC
First, PBMC were stimulated with PMA:ionomycin as mitogens at 0.062:0.625 µg/mL (Condition A), 0.125:1.250 µg/mL (Condition B), and 0.250:2.500 µg/mL (Condition C); and a control reaction was performed without stimuli (Fig. 1c). Different concentrations of CFSE-labelled PBMC (namely, 2 × 105, 1 × 106 and 2 × 106 PBMC/mL per well in 24-well plates) were tested in order to establish whether proliferation correlates with the initial number of cells seeded. Upon stimulation, characteristic clumps of activated cells were observed (Fig. 1d), denoting successful PBMC proliferation in high cell density conditions. At day 5, non-stimulated cells hardly proliferated, so this condition was considered a proper negative control for setting up the proliferation threshold. Interestingly, 2 × 105 PBMC/mL was a suboptimal concentration for observing clearly proliferation by flow cytometry (46.50% ± 3.46 for Condition A; 49.57% ± 0.55 for Condition B and 65.93% ± 6.48 for Condition C; percentages of proliferation from n = 3). On the contrary, higher number of PBMC (that is 1 × 106 and 2 × 106 PBMC/mL) resulted in measurable proliferation (78.17% ± 3.63 for Condition A; 77.40% ± 5.03 for Condition B and 82.87% ± 3.18 for Condition C in case of 1 × 106 PBMC/mL; and 80.60% ± 0.53 for Condition A; 77.83% ± 1.44 for Condition B and 76.20% ± 1.92 for Condition C in case of 2 × 106 PBMC/mL. Percentages of proliferation from n = 3 in all cases), regardless of the stimuli used and without significant differences among them. Hence, proliferation of PBMC was affected by the starting cell dose, which had to be no less than 1 × 106 cell/mL.
Next, a series of experiments were performed to define optimal conditions for the stimulation of proliferation of CFSE-labelled PBMC. Stimuli used were PMA:ionomycin, PHA and anti-CD3:anti-CD28 IgG (Table 1). For the setup of the proliferation threshold, non-stimulated PBMC were used in PMA:ionomycin and PHA conditions; and non-specific IgG binding for anti-CD3:anti-CD28 stimulation. Altogether, PMA:ionomycin resulted in the strongest proliferation conditions, above 70% with the two highest conditions showing a percentage of proliferation of 78.55% ± 1.20 and 80.54 ± 3.18% using 0.250 µg/mL PMA:0.5 µM ionomycin and 0.250 µg/mL PMA:0.33 µM ionomycin, respectively. On the other hand, addition of PHA or anti-CD3:anti-CD28 resulted in lower proliferation rates (maximum of 42.34 ± 2.87% using 20 µg/mL PHA, 59.84 ± 0.75% using 1 µg/mL of each antibody, respectively). Consequently and in order to homogenize our data with the majority of published studies, 0.250 µg/mL PMA and 0.5 µM ionomycin were set as optimal concentrations for reliable stimulation of proliferation of PBMC.
Table 1.
Proliferation of PBMC upon stimulation under different conditions. Proliferation of PBMC is expressed as a percentage increase in proliferation (%) with respect to non-induced controls
| Stimuli (µg/mL) | Mean (%) | Standard deviation (%) | ||||
|---|---|---|---|---|---|---|
| PMA | Ionomycin | PHA | Anti-CD3 | Anti-CD28 | ||
| 0.062 | 0.625 (0.08 µM) | 75.84 | 3.63 | |||
| 0.083 | 0.833 (0.11 µM) | 73.71 | 3.40 | |||
| 0.100 | 1.000 (0.13 µM) | 73.07 | 4.16 | |||
| 0.125 | 1.250 (0.17 µM) | 75.07 | 5.03 | |||
| 0.250 | 2.500 (0.33 µM) | 80.54 | 3.18 | |||
| 0.250 | 3.735 (0.50 µM) | 78.55 | 1.20 | |||
| 5 | 1.57 | 0.30 | ||||
| 10 | 12.27 | 2.41 | ||||
| 20 | 42.34 | 2.87 | ||||
| 0.1 | 0.1 | 30.57 | 0.53 | |||
| 0.4 | 0.4 | 53.90 | 3.20 | |||
| 1 | 1 | 59.84 | 0.75 | |||
PBMC peripheral blood mononuclear cells; PHA phytohemagglutinin; PMA phorbol myristate acetate
Co-cultures with MSC
MSC were cultured in 24-well plates at different densities (1 × 105, 5 × 104 and 5 × 103 MSC/well) in the presence of a fixed number of PBMC (5 × 105 PBMC/well), which corresponded to MSC:PBMC ratios of 1:5, 1:10 and 1:100, respectively. In order to determine whether proliferation of MSC can interfere in the readout of the assay, the same conditions were tested using MSC irradiated at 20 Gy for 574 s. MSC had an inhibitory effect on the proliferation of PBMC stimulated with 0.250 μg/mL PMA and 0.5 μM ionomycin for 5 days as a function of the MSC:PBMC ratio (Fig. 1e). The higher the number of MSC with respect to PBMC is, the greater is the inhibitory effect on proliferation. At 1:5 MSC:PBMC, an inhibition of 73.09 ± 2.52% was observed using non-irradiated MSC and 57.12 ± 3.49% for irradiated MSC. At 1:10, the inhibition with non-irradiated MSC was 31.93 ± 3.85 and 13.84 ± 5.28% for irradiated MSC. In contrast, inhibition was not observed in neither control nor MSC:PBMC co-culture at a ratio of 1:100, no matter the irradiation status.
Optimization of the potency assay and application in two practical cases
The key steps of the procedure that have been optimised in this work are listed next and described in Table 2. First, the use of SepMate™ tubes was found more convenient for isolation of PBMC to save time and reduce variability of yields; high yields of staining and low cell death rates were observed when using 0.625–2 µM CFSE for 5 min at RT, followed by a 15 min incubation in + 37 °C pre-warmed medium; strongest stimulation of PBMC was obtained at 1 × 106 cells/mL using 0.250 µg/mL PMA and 0.5 µM ionomycin; irradiation of MSC was not required; and the optimal MSC:PBMC ratio was 1:5 (Fig. 2).
Table 2.
Optimised conditions for PBMC isolation and labelling, and assessment of immunomodulative properties of MSC
| Step | Conditions/comments |
|---|---|
| PBMC isolation | 80 mL of peripheral blood (diluted 1/2 with DPBS) or 10 mL of buffy coat blood (diluted 1/10 with DPBS) SepMate™ Tubes with 15 mL of Histopaque®-1077 and 32 mL of diluted blood |
| CFSE labelling | Staining of PBMC with 0.625–2 µM CFSE for 5 min at RT, followed by 15 min stabilisation at RT in + 37 °C pre-warmed medium PBMC concentration for the staining: 5E6 cells/mL |
| PBMC stimulation | PBMC stimulation (1E6 cells/mL) with 0.250 µg/mL PMA and 0.5 µM ionomycin |
| Co-culture with MSC | Non-irradiated MSC:PBMC at a ratio of 1:5 in a cell–cell contact co-culture |
| Proliferation assessment | 5-day stimulation Read-out by flow cytometry |
CFSE carboxyfluorescein diacetate succinimidyl ester; DPBS phosphate-buffered saline solution; MSC mesenchymal stromal cells; PMA phorbol myristate acetate; PBMC peripheral blood mononuclear cells; RT room temperature
Fig. 2.
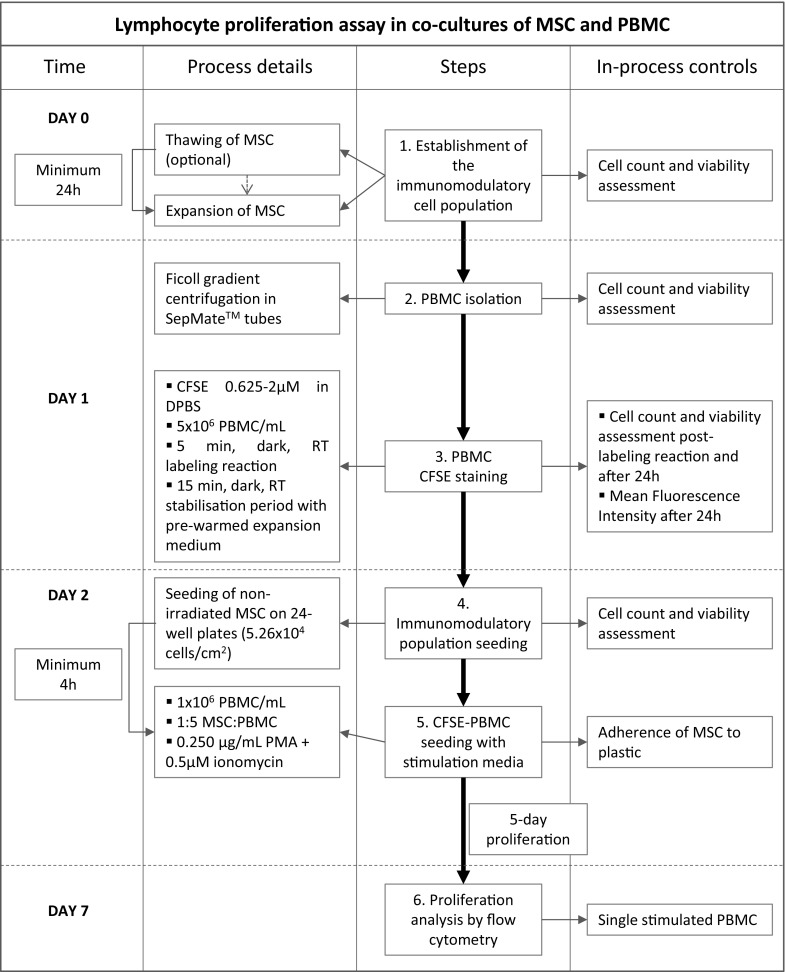
Flowchart of the optimised procedure for assessing lymphocyte proliferation in co-cultures of PBMC and MSC. Provided that MSC for potency testing can be fresh or cryopreserved, the first step of thawing cells is only required if cells are cryopreserved. CFSE carboxyfluorescein diacetate succinimidyl ester; DPBS phosphate-buffered saline solution; MSC mesenchymal stromal cells; PMA phorbol myristate acetate; PBMC peripheral blood mononuclear cells; RT room temperature. Day 0 is applicable only for cryopreserved MSC
Two examples are shown below describing practical uses of the optimized procedure to assess the immunomodulatory potency of MSC populations.
Example 1
Immunomodulative potential of cryopreserved MSC.
Retention of the immunomodulatory capacity of cryopreserved BM-derived MSC was assessed immediately after thawing, in suspension, and compared to control samples of MSC which were maintained in adherent cell culture for 24 h. Under these conditions, the same MSC:PBMC ratio of 1:5 exhibited 28.94% less inhibition of PBMC proliferation in test samples than the control condition. By increasing the ratio of thawed MSC:PBMC, a progressive increase in the inhibition of proliferation was also observed. However, a ratio of 1:2 was required to reach the same immunomodulatory capacity as the control condition (Fig. 3a).
Fig. 3.

Examples to illustrate practical uses of the assessment of lymphocyte proliferation in co-cultures of PBMC and MSC. a Example 1: inhibition of PBMC proliferation by thawed MSC at different concentrations, which were compared to a control condition consisting of PBMC (initial number: 5 × 105) proliferation inhibition by 1 × 105 cultured MSC, showing that equal numbers of thawed MSC had lower capacity to inhibit proliferation of PBMC and therefore higher number of cells are necessary to reach the same levels of immunomodulation activity as in the control conditions; b Example 2: inhibition of PBMC proliferation inhibition of in co-culture with MSC that were sorted by HLA-DR expression using a cell line initially expressing 19.5% HLA-DR. Positive (HLA-DR+)., showing that HLA-DR positive cells (HLA-DR+) cells inhibited PBMC proliferation significantly less (p = 0.0250) than HLA-DR− MSC although all populations displayed high ability to inhibit polyclonally stimulated PBMC (over 70%). Statistical significance was set at: *p < 0.05; and **p < 0.005
Example 2
Immunomodulative potential of HLA-DR+ and HLA-DR− MSC populations.
A BM-derived MSC cell line expressing HLA-DR was sorted by its expression of HLA-DR resulting in 19.5% of the cells being HLA-DR+. Then, the immunomodulatory potential of the two cell populations (that is HLA-DR+ and HLA-DR− MSC) was determined. Interestingly, no significant differences were found between the two populations (p = 0.0250) regarding their capacity to inhibit proliferation of stimulated PBMC (79.26 ± 0.74% PBMC proliferation inhibition for HLA-DR− population and 71.50 ± 1.44% PBMC proliferation inhibition for HLA-DR+ population, respectively) (Fig. 3b). Although not statistically significant, a control condition of unsorted cells showed lower immunomodulatory capacity than the HLA-DR− population, but higher than the HLA-DR+ population (73.41 ± 2.59% PBMC proliferation inhibition). Despite the differences observed between HLA-DR+ and HLA-DR− MSC, both populations displayed high immunomodulatory potential, regardless of its HLA-DR expression.
Discussion
In the present report, we described the optimisation of a methodology for assessing the immunomodulatory potential of MSC, providing a suitable and relatively simple potency test for use as quality control of MSC-based medicinal products in compliance with current quality and regulatory requirements in the cell therapy field (Vives et al. 2015). So far, this methodology has been proven useful in the characterisation of MSC for use in two clinical trials (EudraCT No.’s 2012-000734-19 and 2015-005786-23) for the treatment of multiple sclerosis and spinal cord injury, respectively. In these two studies, the mode of action is not thought to involve MSC’s multi-potentiality to differentiate into other specialised cell types but their capacity to modulate the immune system thus highlighting the importance of using suitable immunopotency assays, rather than differentiation assays. Preliminary data seem to confirm batch-to-batch consistency in the percentage of inhibition of lymphocyte proliferation in addition to the characteristic MSC phenotype (data not shown).
Besides experimental treatments, immunomodulatory potential of MSC is already commercially used in the treatment of refractory acute graft-versus-host disease (GvHD) with two ATMP: Prochymal® (Canada and New Zealand) and TEMCELL® HS Inj. (Japan) (Guadix et al. 2017). Despite the great potential of MSC for therapeutic use, no standardised assay determining their immunomodulatory capacity has been established yet. Instead, developers use variants of existing methods and diverse detection platforms to measure immunomodulation properties of MSC-based products (Bloom et al. 2015). Perhaps the most accepted type of immunomodulation potency assays to date are those based on the capacity of MSC to inhibit T cell proliferation, which can be used for both in-process control and as batch release tests. However, other methods to determine MSC ability to suppress T cell proliferation should also be explored, as well as the analysis of critical parameters affecting the results observed in such assays, principally: donor source variability of responder cells, mitogenic stimuli, read-out parameters, duration of the assay, ratio of MSC:PBMC in co-culture, irradiation of MSC, and cell to cell-contact. In the present study, we addressed all these concerns towards the definition of a cell-based assay that fulfils both technical and regulatory requirements for use as suitable potency assay in the production of clinical grade MSC.
Concerning variability of donor responder cells, the first action we took was setting the same source of donor cells in terms of time from blood extraction (that is 24–48 h) and storage (that is + 4 °C), in order to bring consistency between assays. Then PBMC isolation was optimised by using SepMate™ tubes, instead of classical home-made density gradients in conical tubes. In this way, results from both techniques were very similar, but the latter requires well-trained staff for consistent results, so simpler methods are proposed in order to simplify the experimental workflow.
Instead of fresh PBMC, some authors have proposed the use of cryopreserved cells, either in pools from ten random donors (Ketterl et al. 2015), or aphaeresis of healthy donors from which large numbers of cells were stored, so the same batch of PBMC was used in several experimental runs (Bloom et al. 2015). The same study by Bloom and collaborators performed a selective gating of CD4+ T cells by flow cytometry to standardise the read-out parameter regarding responder cells. However PBMC represent a heterogeneous sample and cell populations other than T cells may also influence in the response. Therefore, we decided to avoid mixing different donors in order not to have MLR and chose individual total PBMC population, considering donor inter-variability as part of the assay, and taking measures, such as standardization of techniques and data normalization to mitigate it. Also related to read-out parameters, one of the most common techniques for measuring T cell proliferation consists in determining the reduction of fluorescence intensity of CFSE-labelled PBMC. One of the major concerns over this option is the high mortality rate reported in the literature, which has been attributed to the toxicity resulting from excessive surface protein substitution after CFSE coupling (Quah and Parish 2012). A potential solution to overcome this issue might be the use of medium with high content of proteins, such as DMEM supplemented with 10% hSerB, which would compensate higher concentrations of CFSE. Based on this rationale, we tested a range of CFSE concentrations and compared the yields of the staining reactions either in saline solution or 10% human serum B-supplemented medium. Best results were found after labelling PBMC with 0.625–2 µM CFSE in DPBS, as all the stainings performed in serum-supplemented medium showed very low fluorescence intensity. Remarkably, when the labelling was carried out in DPBS, fluorescence intensity with 8 and 30 µM of CFSE was too high to correctly discriminate by flow cytometry. Regarding viability, values were found similar in all conditions tested, suggesting that mortality observed by other authors may be associated with the procedure itself rather than toxicity of CFSE under these experimental conditions.
Induction of T cell proliferation is also a subject of intense discussion, provided that MLR has been one of the most used assays, as it is considered to mimic an in vivo response like the one found with e.g. GvHD. However this assay has some drawbacks, reviewed in de Wolf et al. (2017). Apart from the effect of allogeneity induced by MLR, T cells activation is also usually achieved by mitogens and T cell receptor (TCR) cross-linking and co-stimulation. In these sense we tried different polyclonal stimuli including PMA:ionomycin, PHA as mitogens and monoclonal antibodies anti-CD3:anti-CD28 for the cross-linking and co-stimulation of TCR, respectively. This kind of activation is technically easier than the MLR assay and it is also more suitable for standardisation, as it avoids stimulator cells variability, which is an important issue in terms of a potency assay. Normalisation of data is key for proper comparison of results, therefore values of absolute proliferation (subtracting non stimulated average values) and normalised proliferation (considering single PBMC culture absolute proliferation as 100%) were calculated for each condition, thus reducing the impact of donor-dependent proliferation variability of PBMC. From all stimuli tested, PMA:ionomycin triggered the strongest response, over 70% of proliferation in all conditions. Similarly, the combination of monoclonal antibodies anti-CD3:anti-CD28 at the minimal concentration of 1 µg/mL each resulted also in a strong proliferation.
It is important to highlight that MSC-based products are subjected to inherent heterogeneity as a result of differences between donors and tissue sources (Schimke et al. 2015; Wagner et al. 2005). Lack of cell product consistency is further amplified because of the lack of standardisation of ex vivo expansion and cell manufacturing methods (Schnitzler et al. 2016; Sensebé et al. 2011). As a result, cellular products generated for each clinical study are likely to show differences in their biological properties, with the potential for significant product-to-product and batch-to-batch variability. In addition, pre-treatment of MSC (i.e. irradiation and/or the used of cryopreserved products) can introduce more confusion to the interpretation of results from the immunopotency assay. Therefore, there is a strong need to invest efforts in the design of robust and reproducible potency assays for MSC characterisation, which would ideally involve the use of product-specific reference material and/or control samples thus ensuring that the assay is performed within established limits (Galipeau et al. 2015). To increase robustness of the assay, Bloom and collaborators tested each MSC product at five different titrations/ratios to render a titration curve that was used to calculate what they called the immunopotency assay value (IPAv), which corresponded to the “mean suppression value” for all the titrations. This way, they can assign a robust IPAv for each MSC product, corresponding to the “suppression area” represented as a single value used to evaluate each MSC line’s PBMC-inhibitory capabilities or potency (Bloom et al. 2015).
Our results seem to indicate that the inhibitory effect of non-irradiated MSC was higher than irradiated MSC, although differences were low and not statistically significant (p = 0.2). It is possible that irradiation caused MSC to adhere or survive less efficiently than non-irradiated cells. Thus, the reduction in the immunomodulatory ability of irradiated MSC may be caused because they are found in fewer numbers, meaning lower effective ratio, which was found as one of the most important parameters to consider. This could be an explanation on why other groups have found that irradiation itself does not influence MSC immunosuppressive potency (Ketterl et al. 2015).
Finally, in order to demonstrate the applicability of our assay design to different practical cases, two examples are provided. In the first one, we demonstrated that thawed MSC have impaired immunosuppressive properties which made necessary to increase their ratio in co-culture with PBMC in order to obtain similar inhibition of proliferation to control cultured MSC. The successful commercialization of ATMP will require the extension of their shelf-life beyond shipping “fresh” at ambient or chilled temperatures for “off-the-shelf”, “just in time” infusion. In this sense, cryopreservation offers potential advantages, such as storing and retaining patient samples in case of a relapse, banking large quantities of allogeneic cells for broader distribution and use, and retaining testing samples for further verifications. However, cryopreservation is only useful if cells can be reanimated to physiological life with negligible loss of viability and functionality (Woods et al. 2016). In our case, our findings are in accordance with the results of some other groups showing that cryopreservation and subsequent freeze–thaw procedure impair MSCs immunomodulatory properties (François et al. 2012; Galipeau 2013; Moll et al. 2014). Given the importance for banking MSC, allowing for an “off-the shelf” available product to treat acute diseases, the design of future studies should aim to improve the therapeutic efficacy of cryopreserved cells (Moll et al. 2014).
The second example exposed in this article is related with HLA class II expression (HLA-DR). HLA-DR is a controversial issue when referring to mesenchymal stromal cells, as these cells express negligibly low levels of HLA-DR, although this receptor was readily detectable with Western blot on whole-cell lysates of unstimulated MSC, suggesting that MSC contain intracellular deposits of HLA-DR class II alloantigens (Le Blanc et al. 2003). Some factors can induce HLA-DR expression. Bocelli-Tyndall and collaborators described that induction of functional HLA-DR in proliferating progenitor MSC is a property of human MSC that have been expanded with mitogenic growth factors. However, this induction is different from the one exerted by IFNγ which affects both differentiated and non-differentiated progenitor MSC and differentiated cells, without requiring cell proliferation (Bocelli-Tyndall et al. 2010). Le Blanc and collaborators also found that stimulation with IFN-γ for 48 h led to prompt upregulation of these receptors on the cell surface (Le Blanc et al. 2003). Our methodology allowed determining the immunomodulatory potential of MSC regardless of HLA-DR expression. To do so, HLA-DR+ and HLA-DR− populations of MSC were used as test items, being HLA-DR+ slightly less inhibitory than HLA-DR−, although both efficiently inhibited T cell proliferation. Our results seem to be in accordance with those found by Le Blanc and collaborators and Bloom and collaborators, who have also not been able to find a strong correlation between MSC suppression of PBMC proliferation and HLA-DR expression (Bloom et al. 2015; Le Blanc et al. 2003).
Conclusions
The present work contributes to the definition of robust, reproducible and versatile methods for determining immunomodulation potency of MSC by the optimisation of parameters affecting the performance of lymphocyte proliferation assays. We believe that the protocol described here is of practical relevance and provides the means to assess immunomodulatory potential of MSC for use in a range of cell and tissue engineering therapeutic strategies in both preclinical and clinical settings. Further work in the clinical setting would help to correlate quantitative values of in vitro potency assays and clinical outcomes in specific pathologies.
Acknowledgements
The authors would like to acknowledge J. García-López, A. Pla, J. A. Marco, for technical support and advice; E. Valdivia and N. Martínez, for supplying blood samples; and M. Blanco and M. Codinach, for supplying MSC. We also want to thank Dr. Sergi Querol for critical review of the manuscript. This work was supported by “Fundació la Marató de TV3” (expedient No. 122831) and the Spanish Cell Therapy Network (TerCel, expedient No. RD16/0011/0028); and developed in the context of AdvanceCat with the support of ACCIÓ (Catalonia Trade & Investment; Generalitat de Catalunya) under the Catalonian ERDF operational program (European Regional Development Fund) 2014–2020. Figure 1 has been produced using Servier Medical Art (http://smart.servier.com).
Contributor Information
Irene Oliver-Vila, Phone: +34 93 5573500, Email: irolvila@gmail.com.
Joaquim Vives, Phone: +34 93 5573500, Email: jvives@bst.cat.
References
- Bianco P, Cao X, Frenette PS, Mao JJ, Robey PG, Simmons PJ, Wang CY. The meaning, the sense and the significance: translating the science of mesenchymal stem cells into medicine. Nat Med. 2013;19:35–42. doi: 10.1038/nm.3028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bloom DD, Centanni JM, Bhatia N, Emler CA, Drier D, Leverson GE, McKenna DH, Gee AP, Lindblad R, Hei DJ, Hematti P. A reproducible immunopotency assay to measure mesenchymal stromal cell-mediated T-cell suppression. Cytotherapy. 2015;17:140–151. doi: 10.1016/j.jcyt.2014.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bocelli-Tyndall C, Zajac P, Di Maggio N, Trella E, Benvenuto F, Iezzi G, Scherberich A, Barbero A, Schaeren S, Pistoia V, Spagnoli G, Vukcevic M, Martin I, Tyndall A. Fibroblast growth factor 2 and platelet-derived growth factor, but not platelet lysate, induce proliferation-dependent, functional class II major histocompatibility complex antigen in human mesenchymal stem cells. Arthritis Rheum. 2010;62:3815–3825. doi: 10.1002/art.27736. [DOI] [PubMed] [Google Scholar]
- Bravery CA, Carmen J, Fong T, Oprea W, Hoogendoorn KH, Woda J, Burger SR, Rowley JA, Bonyhadi ML, Van’t Hof W. Potency assay development for cellular therapy products: an ISCT review of the requirements and experiences in the industry. Cytotherapy. 2013;15:9–19. doi: 10.1016/j.jcyt.2012.10.008. [DOI] [PubMed] [Google Scholar]
- Codinach M, Blanco M, Ortega I, Lloret M, Reales L, Coca MI, Torrents S, Doral M, Oliver-Vila I, Requena-Montero M, Vives J, Garcia-López J. Design and validation of a consistent and reproducible manufacture process for the production of clinical-grade bone marrow-derived multipotent mesenchymal stromal cells. Cytotherapy. 2016;18:1197–1208. doi: 10.1016/j.jcyt.2016.05.012. [DOI] [PubMed] [Google Scholar]
- de Wolf C, van de Bovenkamp M, Hoefnagel M. Regulatory perspective on in vitro potency assays for human mesenchymal stromal cells used in immunotherapy. Cytotherapy. 2017;19:784–797. doi: 10.1016/j.jcyt.2017.03.076. [DOI] [PubMed] [Google Scholar]
- Dominici M, Le Blanc K, Mueller I, Slaper-Cortenbach I, Marini F, Krause D, Deans R, Keating A, Prockop D, Horwitz E. Minimal criteria for defining multipotent mesenchymal stromal cells. The International Society for Cellular Therapy position statement. Cytotherapy. 2006;8:315–317. doi: 10.1080/14653240600855905. [DOI] [PubMed] [Google Scholar]
- François M, Copland IB, Yuan S, Romieu-Mourez R, Waller EK, Galipeau J. Cryopreserved mesenchymal stromal cells display impaired immunosuppressive properties as a result of heat-shock response and impaired interferon-γ licensing. Cytotherapy. 2012;14:147–152. doi: 10.3109/14653249.2011.623691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galipeau J. The mesenchymal stromal cells dilemma—Does a negative phase III trial of random donor mesenchymal stromal cells in steroid-resistant graft-versus-host disease represent a death knell or a bump in the road? Cytotherapy. 2013;15:2–8. doi: 10.1016/j.jcyt.2012.10.002. [DOI] [PubMed] [Google Scholar]
- Galipeau J, Krampera M, Barrett J, Dazzi F, Deans RJ, DeBruijn J, Dominici M, Fibbe WE, Gee AP, Gimble JM, Hematti P, Koh MBC, LeBlanc K, Martin I, McNiece IK, Mendicino M, Oh S, Ortiz L, Phinney DG, Planat V, Shi Y, Stroncek DF, Viswanathan S, Weiss DJ, Sensebe L. International Society for Cellular Therapy perspective on immune functional assays for mesenchymal stromal cells as potency release criterion for advanced phase clinical trials. Cytotherapy. 2015;18:151–159. doi: 10.1016/j.jcyt.2015.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guadix JA, Zugaza JL, Gálvez-martín P. Characteristics, applications and prospects of mesenchymal stem cells in cell therapy. Med Clínica (English Ed.) 2017;148:408–414. doi: 10.1016/j.medcle.2017.04.018. [DOI] [PubMed] [Google Scholar]
- Keating A. Mesenchymal stromal cells: new directions. Cell Stem Cell. 2012;10:709–716. doi: 10.1016/j.stem.2012.05.015. [DOI] [PubMed] [Google Scholar]
- Ketterl N, Brachtl G, Schuh C, Bieback K, Schallmoser K, Reinisch A, Strunk D. A robust potency assay highlights significant donor variation of human mesenchymal stem/progenitor cell immune modulatory capacity and extended radio-resistance. Stem Cell Res Ther. 2015;6:236. doi: 10.1186/s13287-015-0233-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Blanc K, Tammik C, Rosendahl K, Zetterberg E, Ringdén O. HLA expression and immunologic properties of differentiated and undifferentiated mesenchymal stem cells. Exp Hematol. 2003;31:890–896. doi: 10.1016/S0301-472X(03)00110-3. [DOI] [PubMed] [Google Scholar]
- Lyons AB, Parish CR. Determination of lymphocyte division by flow cytometry. J Immunol Methods. 1994;171:131–137. doi: 10.1016/0022-1759(94)90236-4. [DOI] [PubMed] [Google Scholar]
- Moll G, Alm JJ, Davies LC, Von Bahr L, Heldring N, Stenbeck-Funke L, Hamad OA, Hinsch R, Ignatowicz L, Locke M, Lönnies H, Lambris JD, Teramura Y, Nilsson-Ekdahl K, Nilsson B, Le Blanc K. Do cryopreserved mesenchymal stromal cells display impaired immunomodulatory and therapeutic properties? Stem Cells. 2014;32:2430–2442. doi: 10.1002/stem.1729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nombela-Arrieta C, Ritz J, Silberstein LE. The elusive nature and function of mesenchymal stem cells. Nat Rev Mol Cell Biol. 2011;12:126–131. doi: 10.1038/nrm3049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prockop DJ. The exciting prospects of new therapies with mesenchymal stromal cells. Cytotherapy. 2017;19:1–8. doi: 10.1016/j.jcyt.2016.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quah BJC, Parish CR. New and improved methods for measuring lymphocyte proliferation in vitro and in vivo using CFSE-like fluorescent dyes. J Immunol Methods. 2012;379:1–14. doi: 10.1016/j.jim.2012.02.012. [DOI] [PubMed] [Google Scholar]
- Reis M, Ogonek J, Qesari M, Borges NM, Nicholson L, Preußner L, Dickinson AM, Wang X-N, Weissinger EM, Richter A. Recent Developments in Cellular Immunotherapy for HSCT-Associated Complications. Front Immunol. 2016;7:500. doi: 10.3389/fimmu.2016.00500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schimke MM, Marozin S, Lepperdinger G. Patient-specific age: the other side of the coin in advanced mesenchymal stem cell therapy. Front Physiol. 2015;6:362. doi: 10.3389/fphys.2015.00362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider CK, et al. Challenges with advanced therapy medicinal products and how to meet them. Nat Rev Drug Discov. 2010;9:195–201. doi: 10.1038/nrd3139. [DOI] [PubMed] [Google Scholar]
- Schnitzler A, Verma A, Kehoe D, Jing D, Murrell J, Der K, Aysola M, Rapiejko P, Punreddy S, Rook MS. Bioprocessing of human mesenchymal stem/stromal cells for therapeutic use: current technologies and challenges. Biochem Eng J. 2016;108:3–13. doi: 10.1016/j.bej.2015.08.014. [DOI] [Google Scholar]
- Sensebé L, Bourin P, Tarte K. Good manufacturing practices production of mesenchymal stem/stromal cells. Hum Gene Ther. 2011;22:19–26. doi: 10.1089/hum.2010.197. [DOI] [PubMed] [Google Scholar]
- Vives J, Oliver-Vila I, Pla A. Quality compliance in the shift from cell transplantation to cell therapy in non-pharma environments. Cytotherapy. 2015;17:1009–1014. doi: 10.1016/j.jcyt.2015.02.002. [DOI] [PubMed] [Google Scholar]
- Wagner W, Wein F, Seckinger A, Frankhauser M, Wirkner U, Krause U, Blake J, Schwager C, Eckstein V, Ansorge W, Ho AD. Comparative characteristics of mesenchymal stem cells from human bone marrow, adipose tissue, and umbilical cord blood. Exp Hematol. 2005;33:1402–1416. doi: 10.1016/j.exphem.2005.07.003. [DOI] [PubMed] [Google Scholar]
- Wang Y, Chen X, Cao W, Shi Y. Plasticity of mesenchymal stem cells in immunomodulation: pathological and therapeutic implications. Nat Immunol. 2014;15:1009–1016. doi: 10.1038/ni.3002. [DOI] [PubMed] [Google Scholar]
- Woods EJ, Thirumala S, Badhe-Buchanan S, Clarke D, Mathew A. Off the shelf cellular therapeutics: factors to consider during cryopreservation and storage of human cells for clinical use. Cytotherapy. 2016;18:697–711. doi: 10.1016/j.jcyt.2016.03.295. [DOI] [PubMed] [Google Scholar]