

ABSTRACT
Replication of negative-strand RNA viruses occurs in association with discrete cytoplasmic foci called inclusion bodies. Whereas inclusion bodies represent a prominent subcellular structure induced by viral infection, our knowledge of the cellular protein components involved in inclusion body formation and function is limited. Using measles virus-infected HeLa cells, we found that the WD repeat-containing protein 5 (WDR5), a subunit of histone H3 lysine 4 methyltransferases, was selectively recruited to virus-induced inclusion bodies. Furthermore, WDR5 was found in complexes containing viral proteins associated with RNA replication. WDR5 was not detected with mitochondria, stress granules, or other known secretory or endocytic compartments of infected cells. WDR5 deficiency decreased both viral protein production and infectious virus yields. Interferon production was modestly increased in WDR5-deficient cells. Thus, our study identifies WDR5 as a novel viral inclusion body-associated cellular protein and suggests a role for WDR5 in promoting viral replication.
IMPORTANCE Measles virus is a human pathogen that remains a global concern, with more than 100,000 measles-related deaths annually despite the availability of an effective vaccine. As measles continues to cause significant morbidity and mortality, understanding the virus-host interactions at the molecular level that affect virus replication efficiency is important for development and optimization of treatment procedures. Measles virus is an RNA virus that encodes six genes and replicates in the cytoplasm of infected cells in discrete cytoplasmic replication bodies, though little is known of the biochemical nature of these structures. Here, we show that the cellular protein WDR5 is enriched in the cytoplasmic viral replication factories and enhances virus growth. WDR5-containing protein complex includes viral proteins responsible for viral RNA replication. Thus, we have identified WDR5 as a host factor that enhances the replication of measles virus.
KEYWORDS: measles virus, innate immunity, inclusion body, WD repeat-containing protein, WDR5
INTRODUCTION
Measles is typically a childhood infection and can have high morbidity and mortality. The causative agent, measles virus (MeV), is an enveloped virus belonging to the family Paramyxoviridae that includes several additional viral pathogens of humans (1–3). MeV virions contain a monopartite single-stranded negative-sense RNA genome of ∼15.9 kb that is encapsidated by the nucleocapsid (N) protein and is associated with a viral RNA-dependent RNA polymerase consisting of two subunits, the L (large) and P (phospho) proteins. Together, these viral proteins and RNA form a ribonucleoprotein (RNP) complex. The enveloped surface of the virions includes two viral membrane glycoproteins, the hemagglutinin (H) and fusion (F) proteins, that are required for viral entry into cells (4). The matrix (M) protein lies underneath the inner lipid leaflet of the envelope and is essential for particle formation by bridging the RNP complex to the glycoprotein-containing viral membrane. In addition to the above-described structural proteins, the MeV P gene also encodes two nonstructural accessory proteins, V and C, that are important for modulating the host response to infection (5, 6).
An early electron microscopic (EM) study of MeV-infected HeLa cells revealed the formation of cytoplasmic inclusion bodies similar in composition to the nucleocapsid of measles virions (7). Similar structures subsequently were seen for additional members of the family Paramyxoviridae (8–12). In the case of respiratory syncytial virus (RSV), for example, the inclusion bodies were found to contain N, P, and L proteins (13–15) as well as genomic RNA (16, 17). These findings suggest that the cytoplasmic inclusion bodies are sites of viral RNA synthesis and RNP assembly for members of the Paramyxoviridae. Inclusion bodies and their role in viral replication have also been described for other families of negative-sense RNA viruses, including Rhabdoviridae (18) and Filoviridae (19). In spite of the implied broad importance of cytoplasmic inclusion bodies in viral replication, the identification of host factors and the delineation of their functional roles in the structures remain poorly understood. Heat shock protein 70 (HSP70) was found to interact with MeV nucleocapsids (20), and overexpression of HSP70 increased viral protein production (21). It also was reported that RSV inclusion bodies impaired the cellular antiviral response by sequestration of phosphorylated p38 MAPK (mitogen-activated protein kinase) and OGT (O-linked N-acetylglucosamine transferase) (22), which in turn led to reduced MAPK-activated protein kinase 2 and suppression of stress granule formation, respectively. Whether additional host proteins reside and function within inclusion bodies is unknown.
Our laboratory has been interested in the unconventional cytoplasmic function of histone H3 lysine 4 methyltransferase (H3K4MT) subunits of the MLL family (23–25). These methyltransferases occur as a complex comprised of one catalytic subunit (SET1A, SET1B, MLL1, MLL2, MLL3, or MLL4) associated with a regulatory subcomplex consisting of WDR5, RbBP5, ASH2L, and DPY-30 (26–28). WDR5 plays a central scaffolding role in the assembly of the H3K4MT complex by bridging the catalytic and regulatory subunits. WDR5 contains 7 WD repeats that fold into a seven-bladed β-propeller structure. The top surface of the propeller forms a cavity that recognizes the catalytic subunit (29–31), and the bottom surface of the propeller mediates interaction with RbBP5-ASH2L-DPY-30 (32). Whereas the literature almost exclusively focuses on the relationship between H3K4MT complexes and cellular transcription, recent studies have begun to unveil roles of these enzymes or their subunits in other nuclear events (33). However, whether these proteins have any cytoplasmic function is little understood. One study reported that in response to infection by Sendai virus (SeV), also a cytoplasmic replicating paramyxovirus, WDR5-enhanced green fluorescent protein (EGFP) translocated to the mitochondria, where it promoted the assembly of a virus-induced signaling adapter (VISA)-associated signaling complex and led to transcriptional activation of beta interferon (IFN-β) expression (34). Because of our interest in the cytoplasmic function of H3K4MT components (23, 24), together with the host responses to MeV infection (35–37), we examined whether WDR5 played a functional role during MeV infection. We found that WDR5 localized to MeV inclusion bodies and enhanced MeV replication.
RESULTS
WDR5-EGFP fusion protein is recruited to cytoplasmic puncta in MeV-infected HeLa cells.
In HeLa cells infected with MeV, we found that the stably expressed WDR5 fusion with EGFP (WDR5-EGFP) became concentrated at cytoplasmic puncta (Fig. 1A). Uninfected cells did not show the robust puncta structures, and neither did infected cells expressing EGFP alone (Fig. 1A). To rule out the possibility that the punctate staining was an artifact caused by fixation, we also performed live-cell imaging of WDR5-EGFP and EGFP in MeV-infected cells. As shown in Fig. 1B, EGFP alone was not enriched at cytoplasmic puncta, whereas WDR5-EGFP was still concentrated at these structures in all cells detectably infected. Since the WDR5 fusion protein level remained comparable during the same period (Fig. 1C, left), the enrichment of WDR5-EGFP at the cytoplasmic puncta is most likely a result of subcellular relocalization. Likewise, the level of total endogenous WDR5 was comparable in uninfected cells and cells infected for 8, 16, or 24 h (Fig. 1C, right). Furthermore, the distribution of endogenous WDR5 between cytoplasmic and nuclear fractions was also comparable, ranging from no difference to a ∼10% increase in cytoplasm between uninfected and MeV-infected cells (data not shown) dependent upon the fractionation method employed.
FIG 1.
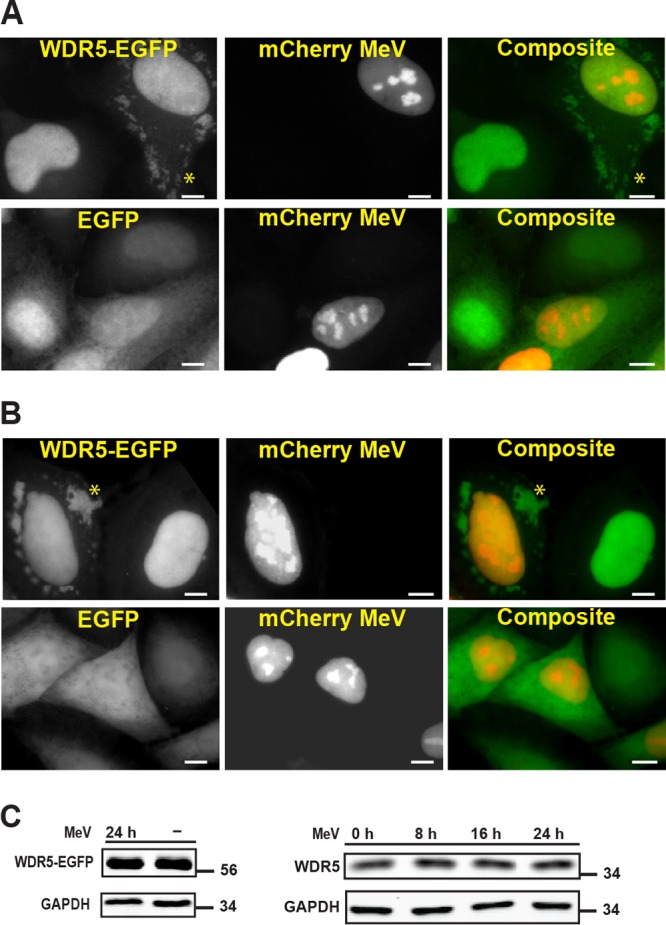
Cellular WDR5 protein is enriched in cytoplasmic puncta induced by measles virus infection. Fluorescent imaging study shows that WDR5-EGFP fusion protein compared to EGFP alone is enriched at cytoplasmic puncta (examples of puncta are shown by asterisks) to a much higher extent in HeLa cells infected with a vaccine strain (Moraten) of MeV (Mv vac2 mCherry NLS). The viral genome was engineered to contain a tag encoding a nuclear-localized mCherry protein to label infected cells. HeLa cells stably expressing WDR5-EGFP were first infected by mCherry MeV (MOI of 1) and then analyzed 24 h postinfection. (A) Cells fixed with formaldehyde prior to imaging. (B) Cells observed by live-cell imaging. Scale bars, 10 μm. (C) Western blot analysis of WDR5-EGFP fusion (left) and endogenous WDR5 (right) expression.
Other regulatory subunits of H3K4MT are not enriched within the cytoplasmic punctate structures seen in MeV-infected HeLa cells.
The above-described observation raises the question of whether the WDR5 subunit functions in the context of an H3K4MT complex or subcomplex at the punctate structures. We addressed this question by first asking whether other H3K4MT regulatory subunits are present at the sites. For this purpose, we generated HeLa cells stably expressing the EGFP fusion of either RbBP5 (the subunit most proximal to WDR5) or DPY-30 (the subunit most distal to WDR5) and found that unlike WDR5-EGFP, neither RbBP5-EGFP nor DPY-30-EGFP was detected at these structures in infected cells (Fig. 2A).
FIG 2.

Unlike WDR5-EGFP, other H3K4MT regulatory subunits are not enriched at cytoplasmic puncta in mCherry MeV-infected cells. (A) HeLa cells stably expressing RbBP5-EGFP or DPY-30-EGFP were infected and analyzed as described in the legend to Fig. 1A. (B) WDR5-EGFP HeLa cells were infected, fixed, and stained with an antibody against either RbBP5 or DPY-30 as indicated. Scale bars, 10 μm.
We next examined whether RbBP5 or DPY-30 could be detected at the puncta in cells stably expressing WDR5-EGFP. As shown in Fig. 2B, we still were unable to detect either RbBP5 or DPY-30 at the puncta structures following infection. Based on these observations, we conclude that WDR5 likely functions independently of other regulatory subunits at these cytoplasmic sites.
WDR5-EGFP fusion protein colocalizes with MeV-induced cytoplasmic inclusion bodies.
To further characterize the WDR5-EGFP puncta, we next examined whether they represent mitochondria as previously described (34). We found that the virus-induced puncta did not colocalize with either the mitochondrial marker HSP60 in fixed cells (Fig. 3A) or with MitoTracker in live cells (Fig. 3B). Moreover, these puncta also were not associated with stress granules (Fig. 3C), a cytoplasmic punctate structure often induced by viral infection, including with MeV (37, 38). Since multiple vesicular compartments exist in the cytoplasm of cells as punctate forms, we then performed a series of colocalization studies with markers of vesicular compartments. WDR5-EGFP puncta showed no overlap with markers of secretory (Fig. 3D, ER and Golgi) or endocytic (early endosomes, recycling endosomes, late endosomes, and lysosomes) compartments (Fig. 3E). We next tested whether WDR5-EGFP puncta represented cytoplasmic inclusion bodies that contain viral nucleocapsid-like structures (7, 39). Indeed, WDR5-EGFP puncta colocalized with the inclusion bodies marked by the MeV proteins N, P, C, and V (Fig. 4). Consistent with this observation, WDR5-EGFP did not colocalize with virus H protein (Fig. 4), a transmembrane envelope protein that only resides in the vesicular compartments.
FIG 3.


WDR5-EGFP-postive cytoplasmic puncta are distinct from known subcellular structures. HeLa cells stably expressing WDR5-EGFP were infected and analyzed as described in the legend to Fig. 1A (for fixed cells) or Fig. 1B (for live cells) using subcellular compartment-specific antibodies or dyes. (A) HSP60 (a mitochondrial marker); (B) MitoTracker (a mitochondrial dye for live-cell imaging); (C) G3BP1 (a stress granule marker); (D) calreticulin (an ER marker), p115 (a CGN, or cis-Golgi network, marker), GalT (a trans-Golgi marker), and Golgin97 and p230 (two TGN, or trans-Golgi network, markers); (E) EEA1 (an early endosomal marker), TfR (an early and recycling endosomal marker), CIMPR, Rab7, and Rab9 (three late endosomal markers), and Lamp1 and CD63 (two lysosomal markers). Scale bars, 10 μm.
FIG 4.

Virus-induced WDR5-EGFP-postive cytoplasmic puncta colocalize with MeV proteins N, P, C, and V but not H. HeLa cells stably expressing WDR5-EGFP were infected with mCherry-expressing MeV and analyzed as described in the legend to Fig. 1A using antibodies against specific MeV N, P, C, V, and H proteins as indicated. Scale bars, 10 μm.
Infection-induced association of WDR5 with MeV protein complexes containing L, P, N, and C proteins but not H or V protein.
To test whether WDR5-GFP possibly is associated with viral proteins, we performed a coimmunoprecipitation assay and found that the MeV L, N, and P proteins efficiently coimmunoprecipitated with WDR5-EGFP (Fig. 5). WDR5-EGFP associated with C protein as well (Fig. 5). On the other hand, very little or no H or V protein coimmunoprecipitated with WDR5-EGFP (Fig. 5). Since V protein was readily detected in the inclusion bodies by immunofluorescence but not in association with WDR5-EGFP by coimmunoprecipitation, these results suggest that WDR5-EGFP selectively associates either directly or indirectly with a complex containing L, P, and N proteins within the MeV-induced inclusion body structures.
FIG 5.
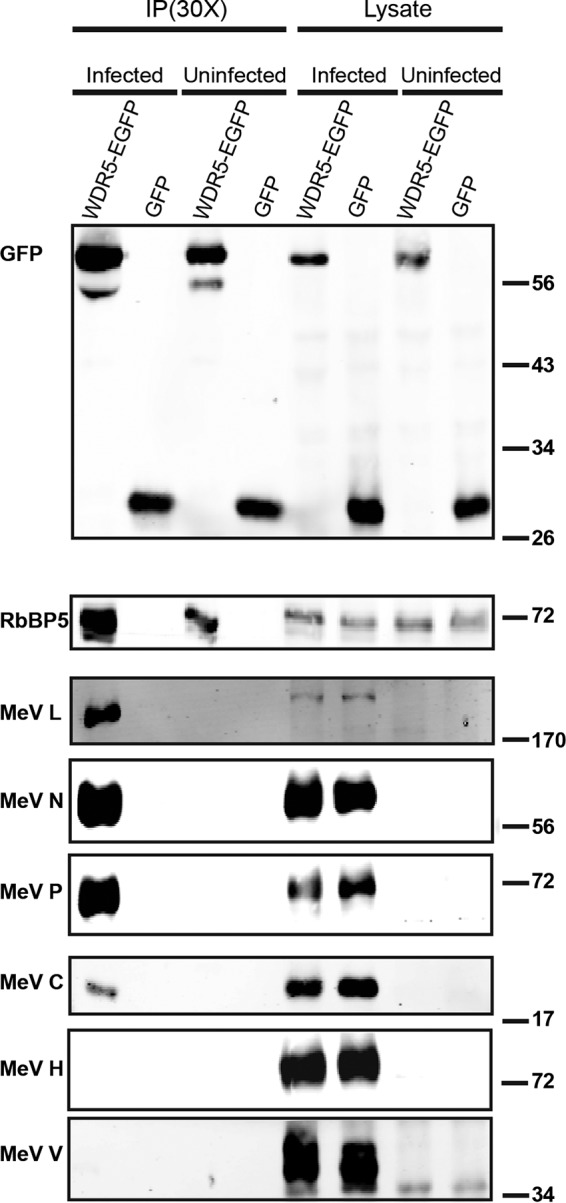
WDR5-EGFP associates in a complex with MeV proteins L, N, P, and C but not H or V. HeLa cells stably expressing either WDR5-EGFP or EGFP alone were infected using mCherry MeV (MOI of 1) or left uninfected as a control. Twenty-four hours after infection, cell extracts were prepared and WDR5-EGFP and its associated proteins were immunoprecipitated using magnetic beads conjugated to an anti-GFP antibody. The sample was then fractionated by SDS-PAGE and the blot probed with specific antibodies as indicated. Each IP lane contained the immunoprecipitate isolated from the 30-fold amount of lysate that was analyzed.
We next asked whether endogenous WDR5 likewise associates with a complex containing MeV L, P, and N proteins. Unfortunately, all commercial WDR5 antibodies that we tested displayed poor pulldown efficiency (less than 1% of total WDR5 was immunoprecipitated). As an alternative approach, we infected HeLa cells with MeV expressing a modified L protein engineered to express an N-terminal hemagglutinin (HA) tag and then probed the interaction between MeV proteins and endogenous WDR5 using magnetic beads conjugated to an HA antibody. As shown in Fig. 6, compared to the uninfected control, a much higher level of endogenous WDR5 was detected in the HA-L-containing complex. As expected, the ribonucleocapsid protein P, but not the envelope protein H, coimmunoprecipitated with the HA-tagged L protein (Fig. 6).
FIG 6.
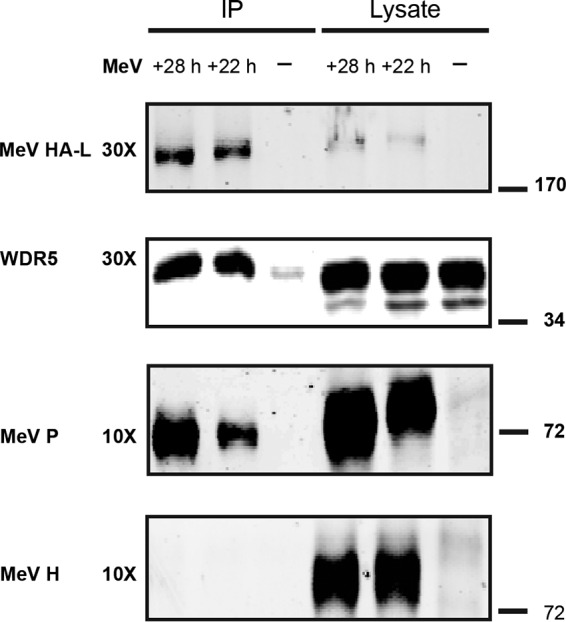
MeV protein L associates with endogenous WDR5. HeLa cells were infected using an engineered MeV (MOI of 1) that encodes an HA-tagged L protein. Cells were lysed 22 or 28 h postinfection (or left uninfected, −), and HA-L and its associated proteins were immunoprecipitated using magnetic beads conjugated to an anti-HA antibody. The purified sample then was fractionated by SDS-PAGE and probed with specific antibodies as indicated. Each IP lane contained the immunoprecipitate isolated from the 30- or 10-fold amount of lysate, as indicated, that was analyzed.
Knockdown of endogenous WDR5 results in reduced MeV replication.
To investigate the effect of WDR5 on virus growth, we assessed the effects of RNA interference (RNAi)-mediated knockdown of WDR5 on MeV protein production and the yield of infectious progeny MeV. Compared to a nontargeting control short interfering RNA (siRNA), two specific WDR5 siRNAs (siRNA1 and siRNA2) reduced viral protein levels at both time points examined, 24 h (Fig. 7A) and 48 h (Fig. 7B) postinfection. To further exclude the potential off-target effects, we also utilized another two different WDR5 siRNAs (siRNA11 and siRNA12) and obtained similar effects (Fig. 7C and D). Since these four siRNAs are produced by two different vendors and target different regions of the wdr5 gene, our data strongly suggest that WDR5 knockdown impairs viral replication as measured by viral protein accumulation. As shown in Fig. 7E and F, compared to control siRNAs, there was an associated 3- to 15-fold reduction in infectious virus yields when WDR5 was knocked down with siRNA1 or siRNA2 (Fig. 7E) or with siRNA11 or siRNA12 (Fig. 7F). The virus yield reduction was seen with parental vac2 virus at both 0.1 (data not shown) and 1.0 (Fig. 7) multiplicities of infection (MOI).
FIG 7.

Measles virus protein production and infectious virus yields are reduced in WDR5-deficient cells. HeLa cells were treated with a nontargeting control siRNA or WDR5-specific siRNA on days 1 and 3 and then infected with MeV (MOI of 1) on day 6. Infected cells were lysed 24 or 48 h after infection, and the lysates were fractionated by SDS-PAGE and analyzed using specific antibodies as indicated. Quantification of Western blots was conducted using a LI-COR Odyssey imaging system. (A to D) MeV protein abundance and WDR5 knockdown efficiency measured at 24 and 48 h postinfection from cells treated with WDR5 siRNA1 or siRNA2 (A and B) or siRNA11 or siRNA12 (C and D) compared to control siRNA. Data included in each bar chart are from at least three independent experiments, and the means and standard errors of the means are shown. Western blots are representative images to illustrate WDR5 knockdown efficiency. For Western blot data analysis, a 1-tailed t test with unequal variance was used. P values were calculated using Microsoft Excel Student's t test: *, P < 0.05; **, P < 0.001; N.S., not significant. (E and F) A parallel set of samples was collected at 48 h postinfection to determine infectious virus yields from cells treated with WDR5 siRNA1 or siRNA2 (E) or siRNA11 or siRNA12 (F) compared to control siRNA. Scatter plot infectious yield data are derived from a minimum of 5 independent experiments. For the virus titers, a 1-tailed t test with equal variance was used. *, P < 0.05; **, P < 0.001.
Knockdown of WDR5 has a marginal effect on interferon induction by MeV.
The effect of WDR5 deficiency on the induction of interferon following infection with either wild-type (WT) parental or isogenic Cko mutant MeV was measured by quantitative PCR (qPCR). Cko mutant was utilized because significant amounts of double-stranded RNA (dsRNA) accumulate during infection compared to that of the parental virus, making Cko a more robust inducer of IFN-β (40, 41). The level of IFN-β transcript was assessed at 48 h after infection of control and WDR5 knockdown cells. As shown in Fig. 8, Cko virus (Fig. 8B) displayed ∼25-fold enhanced IFN-β transcript induction compared to parental wild-type virus (Fig. 8A) in WDR5-sufficient cells as previously reported (40, 41). With both the WT and Cko viruses, a further increase in IFN-β transcript level was observed in cells following WDR5 knockdown with siRNA1 or siRNA2 compared to control WDR5-sufficient cells following infection, with a maximal increase of ∼6.5-fold with WT infection and ∼2.9-fold with Cko infection. Likewise, the induction of IFN-λ transcript was not adversely affected by the knockdown of WDR5 (data not shown).
FIG 8.
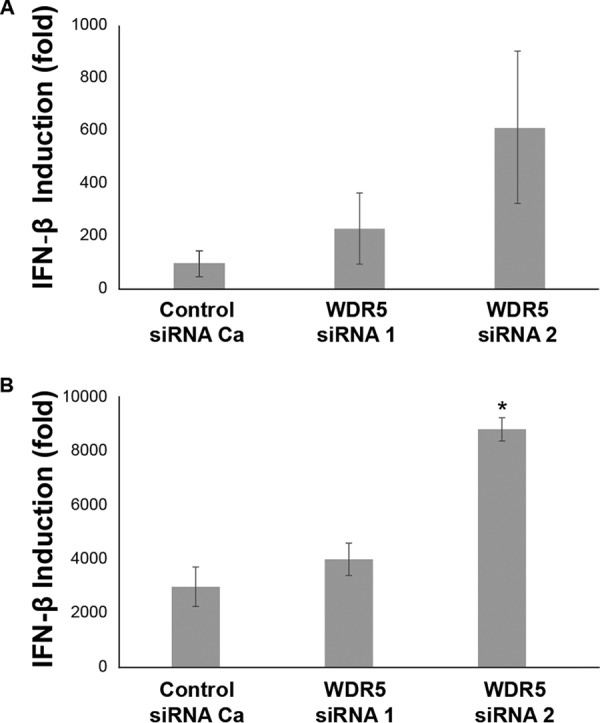
Induction of interferon is modestly increased in WDR5-deficient cells following measles virus infection. HeLa cells were transfected with a nontargeting control or WDR5-specific siRNA and subsequently infected with either a wild-type (WT) MeV (Mv vac2 mCherry NLS) (A) or an MeV mutant (Cko) in which the C gene has been knocked out (Mv vac2 mCherry Cko) (B) at an MOI of 1. Total RNA was isolated 48 h postinfection, and the mRNA encoding IFN-β was estimated by reverse transcription-qPCR. The data are from 4 (WT) or 3 (Cko) independent experiments. Significance was calculated by the Student t test with equal variance. *, P < 0.05.
MeV infection does not lead to a global change of H3K4 trimethylation.
The finding that WDR5 protein translocated to cytoplasmic inclusion bodies following infection prompted us to examine whether MeV infection also causes a change in the level of nuclear H3K4 methylation, which might in turn affect either viral replication or host responses through modulation of host gene expression. To address this question, histone H3 was acid extracted from both uninfected and infected cells, and the levels of H3K4 trimethylation were determined using a methyl-specific antibody. The data in Fig. 9A show that the global level of H3K4 trimethylation was not significantly altered following infection. Quantification from 3 independent experiments showed that the relative level of H3K4 trimethylation at 24 h postinfection in infected cells was 0.96 ± 0.05 compared to that of uninfected cells. A similar conclusion was obtained when H3K4 trimethylation was assessed using the subcellular fractionation method (Fig. 9B).
FIG 9.
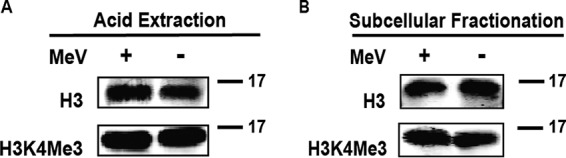
Measles virus infection does not significantly affect global H3K4 trimethylation. Cells were infected with MeV and harvested 24 h postinfection. Extracts were prepared and analyzed by Western blotting using antibodies against histone H3 (H3) or H3 containing trimethylated lysine 4 (H3K4Me3) as indicated. Samples were prepared by acid extraction (A) or subcellular fractionation (B).
DISCUSSION
Our understanding of the identity of cellular protein constituents and the functional roles played by them in replication factories of single-stranded negative-sense RNA viruses, including MeV, is limited (42). The findings reported here establish WDR5 as a component of MeV-induced cytoplasmic inclusion bodies and implicate WDR5 as a functionally important host factor in the replication of MeV. Although inclusion bodies are thought to be the major site of replication for negative-sense RNA viruses, little is known regarding how the virus-host interaction regulates the formation and function of cytoplasmic inclusion bodies. We found that WDR5, a subunit of MLL family H3K4MT, was recruited to the cytoplasmic inclusion bodies whose formation was induced by MeV infection. WDR5 also promoted virus replication. These conclusions are based on the following observations. First, WDR5-EGFP colocalized with MeV L, P, and N proteins at the cytoplasmic inclusion body structures formed following infection (Fig. 1 and 4). Second, both ectopically expressed WDR5-EGFP (Fig. 5) and endogenous WDR5 (Fig. 6) coimmunoprecipitated with viral L, P, and N proteins. Third, knockdown of WDR5 with each of the four different siRNAs reduced both the levels of viral protein production and the yields of infectious progeny virus (Fig. 7). Our findings provide added insight into the understanding of host-pathogen interactions involving MeV-induced inclusion bodies and contribute to the understanding of how host factors modulate the replication of negative-sense RNA viruses.
Although a reduced viral titer in WDR5 knockdown cells suggests a proviral effect of WDR5, exactly how the presence of WDR5 facilitates viral replication is unknown. Since no other MLL regulatory subunits in addition to WDR5 localize to the inclusion bodies (Fig. 2), it seems unlikely that the inclusion body-associated WDR5 functions in the context of a methyltransferase complex. There are precedents for WDR5 function independent of other subunits of the methyltransferase complex (24, 34). Given that WDR5 forms a complex that includes MeV L, P, and N proteins, one attractive hypothesis is that MeV hijacks WDR5 and redirects the protein to inclusion bodies where WDR5 functions as a scaffold to promote viral replication. We do not know, however, whether WDR5 is a direct or indirect binding partner of L, N, or P or whether the interaction involves other cellular proteins. An additional candidate cellular protein present at sites of negative-strand virus replication is O-linked N-acetylglucosamine transferase (OGT), which is reported to accumulate at the inclusion bodies of RSV-infected A549 cells (22) and is a putative WDR5-interacting partner (43). This is intriguing, because O-GlcNAcylation by OGT plays a role in modulating cell signaling, in part due to its ability to affect other posttranslational modifications, including protein phosphorylation (44). While we found that a commercially available OGT antibody (sc-32921) stained the inclusion bodies that we observed in MeV-infected HeLa cells, two other OGT antibodies (generously provided by Gerald Hart) did not produce signals corresponding to the MeV-induced inclusion bodies. Furthermore, the OGT inclusion body signal observed with the sc-32921 antibody in MeV-infected cells persisted even following siRNA-mediated knockdown of the OGT protein, thereby raising uncertainty about the commercial antibody specificity given the lack of validation.
Recombinant MeV expressing V protein with a tag allowed for proteomic identification of V-interacting proteins from infected cells (45, 46). WDR5 was not among them, though STAT1, STAT2, and IFIH1 were found as expected in one study (45) and cofilin in another (46). Consistent with these reports (45, 46), we found that MeV V protein was very poorly pulled down in the protein complex with WDR5-EGFP compared to the MeV L, N, and P proteins and also the C protein (Fig. 5). Our finding that the C protein, but not the V protein, was efficiently coimmunoprecipitated with the WDR5 complex that included the viral L, N, and P proteins is consistent with the notion that C functions during RNA replication to enhance the processivity of the viral L polymerase at the intergenic junctions (47). A yeast two-hybrid screen of a HeLa cDNA library identified Shc Src homology 2 domain binding protein 1 (SHCBP1) and WDR26, but not WDR5, as host proteins interacting with MeV C protein. Similar to our findings described here that knockdown of WDR5 reduced MeV growth and viral protein synthesis, knockdown of SHCBP1 likewise reduced MeV growth (48). SHCBP1 bound both C and P proteins, with C binding leading to a suppression of viral RNA polymerase activity (48).
We considered whether the nuclear function of WDR5 as a mediator of H3K4 methylation-dependent changes in host transcriptional responses was required for optimal viral replication and whether the rerouting of WDR5 to the cytoplasmic inclusion bodies represented a host defense to sequester WDR5 and thereby limit virus growth. This seems unlikely for two reasons. First, MeV growth was impaired, not enhanced, in WDR5-deficient cells (Fig. 7), suggesting a proviral rather than antiviral role for WDR5. Second, our data did not reveal significant changes in global H3K4 trimethylation in MeV-infected cells compared to uninfected cells (Fig. 9), although we cannot exclude the possibility of local methylation changes for a specific subset of genes.
While the source of the endogenous WDR5 that constitutes the puncta induced by MeV infection remains an open question, our imaging and fractionation results are consistent with the redistribution of preexisting diffuse cytoplasmic WDR5 to the cytoplasmic puncta structures rather than a nuclear-to-cytoplasmic relocalization of WDR5. Virus infection did not detectably affect the level of total endogenous WDR5 present in cells (Fig. 1C), and as discussed already and shown in Fig. 9, the nuclear methylation function of WDR5 is not detectably decreased following MeV infection. Although virus infection decreased nuclear and increased cytoplasmic endogenous WDR5, albeit modestly (no change to ∼10% change dependent upon the fractionation method), the potential significance of the subtle redistribution must be viewed in the context of the caveat of nuclear pore complex size threshold limits. Macromolecules smaller than ∼40 to 60 kDa in size may diffuse freely during fractionation between nuclear and cytoplasmic compartments (49, 50). The size of WDR5 protein, also known as BIG3, is ∼34 kDa (51).
Sendai virus infection was reported to cause a fraction of WDR5 to translocate to punctate structures, identified as mitochondria, where WDR5 was essential for assembly of the VISA-associated complex necessary for beta interferon induction in HEK293 cells (34). Knockdown of WDR5 was claimed to inhibit Sendai virus-induced IFN-β transcription (34). We were unable to confirm this finding with MeV. We did not detect the mitochondrial localization of WDR5-EGFP in MeV-infected HeLa cells, either for fixed cells or during live-cell imaging (Fig. 3A and B). Although we cannot exclude the possibility that an amount of WDR5-EGFP below the threshold of detection is translocated to the mitochondria following MeV infection, we do not believe this is the case. We readily detected the localization of WDR5 to punctate cytoplasmic structures that corresponded with viral inclusion bodies and replication factories but did not colocalize with mitochondria (Fig. 1, 3, 4, and 5). The reduced production of viral proteins and yield of infectious progeny seen in WDR5-deficient cells may not be mediated primarily via the interferon pathway. First, knockdown of WDR5 only exerted a modest effect on interferon induction by MeV, and it was an enhancing, not impairing, effect (Fig. 8). Second, WDR5 knockdown similarly reduced the replication of both wild-type parental (Fig. 7) and Cko mutant (unpublished observations) MeV in spite of the greatly increased interferon-inducing capacity of the C mutant virus (Fig. 8) (40, 41). It is unclear whether the difference in WDR5 dependency for IFN-β induction between measles and Sendai viruses, WDR5 independent seen with MeV (here) and WDR5 dependent reported with Sendai virus (34), is due to the different viruses and/or different host cells examined. Extensive work has been done on the mitochondrial antiviral signaling adaptor protein VISA, also known as IPS-1, MAVS, and CARDIF, and WDR5 did not emerge as a component of the signaling response for IFN-β induction from these studies (52, 53). Interestingly, WDR82, which, like WDR5, also is required for normal function of SETD1A/B complexes (54), is reported to enhance Sendai virus replication in HEK293 cells and macrophages (55), similar to the enhancing effect of WDR5 that we find for MeV replication (Fig. 7).
While four different siRNAs targeting WDR5 all reduced viral protein production and infectious virus yields compared to control siRNAs, we found that the reduction seen in viral proteins was not strictly proportional to the WDR5 knockdown efficiency and reduction in infectious progeny. For example, although WDR5 siRNA2 had a slightly higher knockdown efficiency and exerted a more potent effect on viral replication at 48 h postinfection, WDR5 siRNA1 decreased viral protein levels more at 24 h postinfection. Likewise, both siRNA11 and siRNA12 knocked down WDR5 with a similar high efficiency, whereas viral replication was impaired somewhat more by siRNA11 than by siRNA12. A potential explanation for these differences is the occurrence of multiple isoforms of WDR5 (NCBI). That is, it is conceivable that an siRNA that is more effective differentially knocks down one isoform over another.
Polarized epithelial cells are infected with MeV using nectin-4 as a receptor (56). In the case of these cells, virus release occurs from the apical membrane where the MeV ribonucleoprotein complexes are trafficked via Rab11-containing endosomes (2, 57). The punctate WDR5-containing structures we observed in infected cells did not show overlapping localization with endocytic compartments, including those positive for EEA1 (an early endosomal marker); TfR (an early and recycling endosomal marker); CIMPR, Rab7, and Rab9 (three late endosomal markers); or G3BP1 (a stress granule marker) (Fig. 3). Thus, the WDR5-positive structures induced by MeV infection that colocalized with MeV proteins, and that coimmunoprecipitated with the viral nucleocapsid proteins L, P, and N but not H, appear to represent a cellular site involved in the cytoplasmic replication of virus other than endocytic compartments or RNA stress granules induced by infection. There is increasing evidence that the replication machinery of negative-strand RNA viruses utilizes a number of host cell factors (42, 45, 46, 48, 57). These factors might function to affect multiple processes, including formation of the viral RNA replication complex, nucleocapsid trafficking, and shielding from host innate immune response sensors.
MATERIALS AND METHODS
Cells and virus.
CD8-CIMPR-expressing HeLa cells (23) were cultured in advanced Dulbecco's modified Eagle's medium (DMEM-RS; HyClone) supplemented with 4%, vol/vol, fetal bovine serum (Atlanta Biologicals), 2 mM glutamine (Life Technologies), and penicillin (100 μg/ml)-streptomycin (100 U/ml) (Life Technologies). HeLa cells stably expressing WDR5-EGFP (24) were grown in the same medium with additional supplement of 3 μg/ml puromycin. Cells were dissociated for passage using trypsin (0.25%)-EDTA (0.1%) (HyClone). Virus infections were carried out at a multiplicity of infection of 0.1 or 1 50% tissue culture infective dose (TCID50) per cell as indicated. Cells were harvested by scraping them into the growth medium, and the material then was subjected to three freeze-thaw cycles. Infectious virus yields then were determined by titration on Vero cells according to the Spearman-Kärber method (58, 59).
Recombinant vac2 MeV isogenic to the Moraten vaccine strain and expressing enhanced green fluorescent protein (EGFP) downstream of the viral H gene (60, 61) (EGFP MeV), the isogenic C-deficient EGFP mutant virus (Cko) (62), and the recombinant mCherry-expressing vac2 virus with a nuclear localization signal (mCherryNLS) isogenic to the Moraten vaccine strain (36) all have been described. For generation of virus expressing N-terminally HA-tagged L protein, the SpeI/NotI fragment of pB(+)MVvac2-CKO(CFL)H was transferred into pCR3 to yield pCR3-SN. This was further digested with SpeI and EcoRI and the fragment transferred into pCR3 to yield pCR3-SE. The HA tag sequence was introduced into pCR3-SE by site-directed mutagenesis using oligonucleotides (MV L-HAn forward, GTGGTTCCCC GTTATGTACC CATACGATGT TCCAGATTAC GCTGGGGACT CGCTATCTGT CAAC; MV L-HAn reverse, GTTGACAGAT AGCGAGTCCC CAGCGTAATC TGGAACATCG TATGGGTACA TAACGGGGAA CCAC), yielding pCR3-S-HA-E. The SpeI/EcoRI fragment of this vector was transferred back into pCR3-SN, yielding pCR3-S-HA-N, and the SpeI/NotI fragment of this vector was transferred back into pB(+)MVvac2-CKO(CFL)H, yielding the final vector, pB(+)MVvac2-CKO(CFL)HHAL. Virus rescue, stock generation, and titration were performed as previously described (63, 64).
Antibodies.
Primary antibodies used for Western blot analysis (WB), immunofluorescence (IF), and immunoprecipitation (IP) are the following: rabbit anti-calreticulin (C4606; IF dilution, 1:1,000; Sigma), mouse anti-p115 (612260; IF dilution, 1:750; BD Transduction Laboratory), mouse anti-β4 GalT1 (IF dilution, 1:2,000; a gift from Ulla Mandel), mouse anti-Golgin-97 (A21270; IF dilution, 1:500; Molecular Probes), mouse anti-p230 (611280; IF dilution, 1:1,000; BD Transduction Laboratory), rabbit anti-EEA1 (E4156; IF dilution, 1:200; Sigma), mouse anti-CD71 (TfR) (555534; IF dilution, 1:250; BD Pharmingen), mouse anti-CD63 (556019; IF dilution, 1:250; BD Pharmingen), mouse anti-CD222 (CIMPR) (315901; IF dilution, 1:250; BioLegend), mouse anti-Lamp1 (H4A3; IF dilution, 1:8,000; Hybridoma Bank), rabbit anti-Rab7 (9367; IF dilution, 1:250; Cell Signaling Technology), rabbit anti-Rab9 (5118; IF dilution, 1:300; Cell Signaling Technology), mouse anti-G3BP1 (ab56574; IF dilution, 1:250; Abcam), mouse anti-HSP60 (sc-376240; IF dilution, 1:150; Santa Cruz Biotechnology), rabbit anti-MeV N505 (IF dilution, 1:2,500; WB dilution, 1:5,000 [60]), rabbit anti-MeV P254 (IF dilution, 1:1,500; WB dilution, 1:3,000 [60]), rabbit anti-(trimethyl)-histone H3 (lys4) (9751; WB dilution, 1:1,000; Cell Signaling Technology), rabbit anti-WDR5 (A302-430A; WB dilution, 1:3,000; Bethyl Laboratory), mouse anti-WDR5 (sc-393080; WB dilution, 1:600; Santa Cruz Biotechnology), rabbit anti-MeV VCT (IF dilution, 1:1,500; WB dilution, 1:3,000 [65]), rabbit anti-MeV C2 (IF dilution, 1:1,500; WB dilution, 1:3,000 [65]), rabbit anti-MeV M81 (WB dilution, 1:2,000 [60]), rabbit anti-MeV HCYT (IF dilution, 1:15,00; WB dilution, 1:3,000 [66]), rabbit anti-MeV L2170 [WB dilution, 1:2,000; generated against the L protein sequence (C)EWYKLVGYSALIKD corresponding to positions 2170 to 2183, preceded by a cysteine used for coupling purposes], mouse anti-glyceraldehyde-3-phosphate dehydrogenase (GAPDH) (Y3322GAPDH; WB dilution, 1:6,000; Biochain Institute), mouse anti-mCherry (125096; WB dilution, 1:2,000; Abcam), and mouse anti-GFP (11814460001; WB dilution, 1:1,600; Roche). For labeling mitochondria in live cells, MitoTracker deep red FM (8778; Cell Signaling Technology) was used (500 nM) for 30 min at 37°C.
RNAi.
The siRNAs purchased from Qiagen were the following: AllStars negative-control siRNA (SI03650318; control siRNA Ca), WDR5 siRNA1 (SI00118916; target sequence, 5′-CAC GCT GGA CAA CAC TCT GAA-3′), and WDR5 siRNA2 (SI00118923; target sequence, 5′-TAG CCT GGT CGT CAG ATT CTA-3′). The siRNAs purchased from Life Technologies were Silencer Select negative-control no. 2 siRNA (4390843; control siRNA Cb), WDR5 siRNA11 (s21862; target sequence, 5′-AGA CGA AAG AGA TTG TAC A-3′), and WDR5 siRNA12 (s21864; target sequence, 5′-GAT GGG AAA TTT GAG AAA A-3′). The siRNAs were transfected into cells using Lipofectamine RNAiMAX (Life Technologies) at a final concentration of 10 nM (for Qiagen siRNAs) or 5 nM (for Life Technologies siRNAs). To achieve more efficient knockdown, cells were transfected twice, first on day 1 and then on day 3, before MeV infection on day 6.
Immunoprecipitation.
HeLa cells, either infected or uninfected, stably expressing EGFP (as a negative control) or WDR5-EGFP, were lysed in ice-cold NP-40 lysis buffer (50 mM Tris-HCl, pH 8.0, 150 mM NaCl, and 1%, vol/vol, NP-40) containing 1× protease inhibitor cocktail (Biotool), 1× phosphatase inhibitor cocktail (Biotool), and 5 mM EDTA. Lysates were obtained by rocking cells with lysis buffer at 4°C for 15 min and cleared by centrifugation (14,000 rpm) at 4°C, and protein concentration was determined using the CB-X protein assay kit (G-Biosciences). An equivalent amount of lysate total protein was bound to a mouse monoclonal anti-GFP antibody covalently conjugated to magnetic microbeads by rocking at 4°C for 3 h (μMACS GFP-tagged protein isolation kit; Miltenyi Biotec). The beads were washed on μMACS columns with NP-40 lysis buffer and eluted according to the manufacturer's instructions. Immunoprecipitation of HA-tagged L protein was carried out in a similar manner but using anti-HA magnetic beads (Thermo Scientific).
SDS-PAGE and Western blotting.
Unless otherwise specified, cell lysates were made using ice-cold radioimmunoprecipitation assay (RIPA) buffer (50 mM Tris-HCl, pH 8.0, 150 mM NaCl, 1% NP-40, 0.5% sodium deoxycholate, and 0.1% SDS) supplemented with 1× protease inhibitor cocktail (Biotool), 1× phosphatase inhibitor cocktail (Biotool), and 5 mM EDTA. Cytoplasmic and nuclear protein fractions were prepared either by using the Thermo Scientific subcellular fractionation kit for cultured cells (78840) according to the manufacturer's recommendations or as previously described (67, 68). Protein concentration was determined using the CB-X protein assay. An equivalent amount of total protein (for the analysis of host proteins) or an equal volume of cell lysate (for the analysis of viral proteins) was analyzed by SDS-PAGE (6 to 15% gels, depending on the proteins to be resolved). Proteins were transferred to Immobilon-FL 0.45-μm polyvinylidene difluoride (PVDF) membranes (Millipore) using an Owl HEP-1 semidry electroblotting system (Thermo Scientific). Membranes were incubated overnight at 4°C with the appropriate primary antibody diluted in a 1:1 mixture of Odyssey blocking buffer (LI-COR Biosciences) and PBST (1× phosphate-buffered saline [PBS] supplemented with 0.1% Tween 20). Membranes were washed with PBST (three times, 10 min each), incubated in the dark for 45 min with the appropriate secondary antibody [goat anti-mouse or anti-rabbit IgG (H+L) DyLight 680 conjugated; Thermo Scientific], washed with PBST (three times, 5 min each) followed by PBS for 5 min, and dried in the dark. Fluorescence imaging and quantification were performed on an Odyssey infrared imaging system (LI-COR Biosciences).
Immunofluorescence.
Unless otherwise specified, cells grown on coverslips were fixed in 3% UltraPure EM-grade formaldehyde (Polysciences, Inc.) for 10 min at 24 h after MeV infection, quenched with PBS containing 100 mM glycine (2×, 5 min each), and permeabilized with PBS containing either 0.1% saponin or 0.1% Triton X-100 for 15 min. Fixed cells were blocked in buffer (BLOK casein in PBS [G-Biosciences] containing 0.1% saponin [omitted if 0.1% Triton X-100 was used during permeabilization] and 5% goat serum) for 30 min, incubated with primary antibody diluted in blocking buffer for 2 h, washed in PBS (3×, 5 min each), incubated in fluorophore-conjugated secondary antibody diluted in blocking buffer for 1 h, washed in PBS (3×, 5 min each), incubated in PBS containing 1 μg/ml 4′,6-diamidino-2-phenylindole (DAPI) for 5 min, and washed in PBS (3×, 2 min each). Coverslips were then allowed to air dry in the dark and mounted in PPD antifade (90% glycerol, 10% PBS, 1 mg/ml p-phenylenediamine, pH 8.0). The following secondary antibodies were used at a dilution of 1:200: goat anti-mouse IgG (Alexa Fluor 488 [115-545-062], rhodamine red-X [115-295-062], and Alexa Fluor 647 [115-605-146]; Jackson ImmunoResearch) and goat anti-rabbit IgG (Alexa Fluor 488 [111-545-144], rhodamine red-X [111-295-144], and Alexa Fluor 647 [111-605-144]; Jackson ImmunoResearch).
Fluorescence microscopy.
Slides were imaged using an Olympus IX-81 fluorescence microscope with a Plan-Apochromat 60× oil total internal reflection fluorescence objective (numeric aperture, 1.45; Olympus), Semrock excitation and emission filters, and an X-Cite Exacte illumination source (Lumen Dynamics). Images were captured using a monochrome digital camera (Evolution QEi; Media Cybernetics) controlled with In Vivo acquisition software, v.3.2.0 (Media Cybernetics). Image analysis was performed using ImageJ (NIH), Photoshop, and Illustrator (Adobe).
Live-cell fluorescence microscopy.
Cells were grown on Matrigel-coated (1:500) glass-bottomed Delta T dishes (Bioptechs) and imaged using an Olympus IX-81 microscope as described above. All live-cell imaging experiments were performed at 37°C using a Delta T open dish system equipped with a heated lid and 5% CO2 perfusion (Bioptechs). Unless otherwise specified, live-cell imaging was conducted at 24 h postinfection.
Histone extraction.
Core histones were extracted through either acid extraction or the subcellular fractionation kit for cultured cells (Thermo Scientific) as noted. For samples collected via acid extraction, cells were harvested, washed with ice-cold PBS, suspended in Triton extraction buffer (TEB; PBS with 0.5% [vol/vol] Triton X-100), and rocked and cleared at 4°C, and the pellets were suspended in 0.2N HCl and rocked overnight. Total volumes of 0.2N HCl added were proportional to the protein concentrations of the TEB supernatants as determined using the CB-X protein assay. Histone extract was collected after centrifugation and denatured in a final concentration of 1× SDS loading buffer by heating at 95°C.
Interferon analysis.
IFN-β transcripts were measured by quantitative real-time PCR (qPCR) (40). Total RNA was isolated using triazole, random primed cDNA was prepared using SuperScript II (Life Technologies), and the primer pairs and cycle programs for IFN-β and glyceraldehyde-3-phosphate dehydrogenase were as previously described (41). Primer pairs for IFN-λ were as reported previously (forward, 5′-CGC CTT GGA AGA GTC ACT CA-3′; reverse, 5′-GAA GCC TCA GGT CCC AAT TC-3′) (69). The qPCRs were performed using IQ SYBR green supermix (Bio-Rad) and a Bio-Rad MyIQ real-time qPCR instrument.
Statistical analysis.
Statistical tests were performed by Student's t test using Microsoft Excel. Unless otherwise specified, data are presented as means with bars showing standard errors of the means (SEM).
ACKNOWLEDGMENTS
This work was supported in part by research grants AI-20611 to C.E.S. and AI-128037 to R.C. from the National Institute of Allergy and Infectious Diseases, NIH, and a grant from the University of California Cancer Research Coordinating Committee to D.M.
We have no conflicts of interest to declare.
D.M., C.X.G., C.K.P., R.C., and C.E.S. designed the experiments and wrote the manuscript, and C.X.G., D.M., and J.L.N. performed the experiments.
REFERENCES
- 1.King AMQ, Adams MJ, Carstens EB, Lefkowitz EJ (ed). 2012. Virus taxonomy. Classification and nomenclature of viruses. Ninth report of the International Committee on Taxonomy of Viruses. Elsevier Academic Press, San Diego, CA. [Google Scholar]
- 2.Lamb R, Parks G. 2013. Paramyxoviridae, p 957–995. In Knipe DM, Howley PM, Cohen JI, Griffin DE, Lamb RA, Martin MA, Racaniello VR, Roizman B (ed), Fields virology, 6th ed, vol 1 Lippincott Williams & Wilkins, Philadelphia, PA. [Google Scholar]
- 3.Griffin D. 2013. Measles, p 1042–1069. In Knipe DM, Howley PM, Cohen JI, Griffin DE, Lamb RA, Martin MA, Racaniello VR, Roizman B (ed), Fields virology, 6th ed, vol 1 Lippincott Williams & Wilkins, Philadelphia, PA. [Google Scholar]
- 4.Navaratnarajah CK, Leonard VH, Cattaneo R. 2009. Measles virus glycoprotein complex assembly, receptor attachment, and cell entry. Curr Top Microbiol Immunol 329:59–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cox RM, Plemper RK. 2017. Structure and organization of paramyxovirus particles. Curr Opin Virol 24:105–114. doi: 10.1016/j.coviro.2017.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Randall RE, Goodbourn S. 2008. Interferons and viruses: an interplay between induction, signalling, antiviral responses and virus countermeasures. J Gen Virol 89:1–47. doi: 10.1099/vir.0.83391-0. [DOI] [PubMed] [Google Scholar]
- 7.Nakai M, Imagawa DT. 1969. Electron microscopy of measles virus replication. J Virol 3:187–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Fearns R, Young DF, Randall RE. 1994. Evidence that the paramyxovirus simian virus 5 can establish quiescent infections by remaining inactive in cytoplasmic inclusion bodies. J Gen Virol 75:3525–3539. doi: 10.1099/0022-1317-75-12-3525. [DOI] [PubMed] [Google Scholar]
- 9.Palosaari H, Parisien J-P, Rodriguez JJ, Ulane CM, Horvath CM. 2003. STAT protein interference and suppression of cytokine signal transduction by measles virus V protein. J Virol 77:7635–7644. doi: 10.1128/JVI.77.13.7635-7644.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Nishio M, Tsurudome M, Kawano M, Watanabe N, Ohgimoto S, Ito M, Komada H, Ito Y. 1996. Interaction between nucleocapsid protein (NP) and phosphoprotein (P) of human parainfluenza virus type 2: one of the two NP binding sites on P is essential for granule formation. J Gen Virol 77:2457–2463. doi: 10.1099/0022-1317-77-10-2457. [DOI] [PubMed] [Google Scholar]
- 11.Portner A, Gupta KC, Seyer JM, Beachey EH, Kingsbury DW. 1986. Localization and characterization of Sendai virus nonstructural C and C′ proteins by antibodies against synthetic peptides. Virus Res 6:109–121. doi: 10.1016/0168-1702(86)90043-2. [DOI] [PubMed] [Google Scholar]
- 12.Ulane CM, Rodriguez JJ, Parisien J-P, Horvath CM. 2003. STAT3 ubiquitylation and degradation by mumps virus suppress cytokine and oncogene signaling. J Virol 77:6385–6393. doi: 10.1128/JVI.77.11.6385-6393.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Carromeu C, Simabuco FM, Tamura RE, Arcieri LE, Ventura AM. 2007. Intracellular localization of human respiratory syncytial virus L protein. Arch Virol 152:2259–2263. doi: 10.1007/s00705-007-1048-4. [DOI] [PubMed] [Google Scholar]
- 14.Garcıa J, Garcıa-Barreno B, Vivo A, Melero JA. 1993. Cytoplasmic inclusions of respiratory syncytial virus-infected cells: formation of inclusion bodies in transfected cells that coexpress the nucleoprotein, the phosphoprotein, and the 22K protein. Virology 195:243–247. doi: 10.1006/viro.1993.1366. [DOI] [PubMed] [Google Scholar]
- 15.Li D, Jans DA, Bardin PG, Meanger J, Mills J, Ghildyal R. 2008. Association of respiratory syncytial virus M protein with viral nucleocapsids is mediated by the M2-1 protein. J Virol 82:8863–8870. doi: 10.1128/JVI.00343-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Santangelo P, Nitin N, LaConte L, Woolums A, Bao G. 2006. Live-cell characterization and analysis of a clinical isolate of bovine respiratory syncytial virus, using molecular beacons. J Virol 80:682–688. doi: 10.1128/JVI.80.2.682-688.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lindquist ME, Lifland AW, Utley TJ, Santangelo PJ, Crowe JE. 2010. Respiratory syncytial virus induces host RNA stress granules to facilitate viral replication. J Virol 84:12274–12284. doi: 10.1128/JVI.00260-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Heinrich BS, Cureton DK, Rahmeh AA, Whelan SPJ. 2010. Protein expression redirects vesicular stomatitis virus RNA synthesis to cytoplasmic inclusions. PLoS Pathog 6:e1000958. doi: 10.1371/journal.ppat.1000958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hoenen T, Shabman RS, Groseth A, Herwig A, Weber M, Schudt G, Dolnik O, Basler CF, Becker S, Feldmann H. 2012. Inclusion bodies are a site of ebolavirus replication. J Virol 86:11779–11788. doi: 10.1128/JVI.01525-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhang X, Bourhis J-M, Longhi S, Carsillo T, Buccellato M, Morin B, Canard B, Oglesbee M. 2005. Hsp72 recognizes a P binding motif in the measles virus N protein C-terminus. Virology 337:162–174. doi: 10.1016/j.virol.2005.03.035. [DOI] [PubMed] [Google Scholar]
- 21.Carsillo T, Traylor Z, Choi C, Niewiesk S, Oglesbee M. 2006. hsp72, a host determinant of measles virus neurovirulence. J Virol 80:11031–11039. doi: 10.1128/JVI.01438-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Fricke J, Koo LY, Brown CR, Collins PL. 2013. p38 and OGT sequestration into viral inclusion bodies in cells infected with human respiratory syncytial virus suppresses MK2 activities and stress granule assembly. J Virol 87:1333–1347. doi: 10.1128/JVI.02263-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Xu Z, Gong Q, Xia B, Groves B, Zimmermann M, Mugler C, Mu D, Matsumoto B, Seaman M, Ma D. 2009. A role of histone H3 lysine 4 methyltransferase components in endosomal trafficking. J Cell Biol 186:343–353. doi: 10.1083/jcb.200902146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bailey JK, Fields AT, Cheng K, Lee A, Wagenaar E, Lagrois R, Schmidt B, Xia B, Ma D. 2015. WD repeat-containing protein 5 (WDR5) localizes to the midbody and regulates abscission. J Biol Chem 290:8987–9001. doi: 10.1074/jbc.M114.623611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bailey J, Ma D. 2016. Cellular functions of MLL/SET-family histone H3 lysine 4 methyltransferase components. Front Biol 11:10–18. doi: 10.1007/s11515-016-1390-6. [DOI] [Google Scholar]
- 26.Martin C, Zhang Y. 2005. The diverse functions of histone lysine methylation. Nat Rev Mol Cell Biol 6:838–849. doi: 10.1038/nrm1761. [DOI] [PubMed] [Google Scholar]
- 27.Shilatifard A. 2008. Molecular implementation and physiological roles for histone H3 lysine 4 (H3K4) methylation. Curr Opin Cell Biol 20:341–348. doi: 10.1016/j.ceb.2008.03.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Shilatifard A. 2012. The COMPASS family of histone H3K4 methylases: mechanisms of regulation in development and disease pathogenesis. Annu Rev Biochem 81:65–95. doi: 10.1146/annurev-biochem-051710-134100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ruthenburg AJ, Wang W, Graybosch DM, Li H, Allis CD, Patel DJ, Verdine GL. 2006. Histone H3 recognition and presentation by the WDR5 module of the MLL1 complex. Nat Struct Mol Biol 13:704–712. doi: 10.1038/nsmb1119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhang P, Lee H, Brunzelle JS, Couture J-F. 2012. The plasticity of WDR5 peptide-binding cleft enables the binding of the SET1 family of histone methyltransferases. Nucleic Acids Res 40:4237–4246. doi: 10.1093/nar/gkr1235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Patel A, Vought VE, Dharmarajan V, Cosgrove MS. 2008. A conserved arginine-containing motif crucial for the assembly and enzymatic activity of the mixed lineage leukemia protein-1 core complex. J Biol Chem 283:32162–32175. doi: 10.1074/jbc.M806317200. [DOI] [PubMed] [Google Scholar]
- 32.Odho Z, Southall SM, Wilson JR. 2010. Characterization of a novel WDR5-binding site that recruits RbBP5 through a conserved motif to enhance methylation of histone H3 lysine 4 by mixed lineage leukemia protein-1. J Biol Chem 285:32967–32976. doi: 10.1074/jbc.M110.159921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhang X, Wen H, Shi X. 2012. Lysine methylation: beyond histones. Acta Biochim Biophys Sin (Shanghai) 44:14–27. doi: 10.1093/abbs/gmr100. [DOI] [PubMed] [Google Scholar]
- 34.Wang Y-Y, Liu L-J, Zhong B, Liu T-T, Li Y, Yang Y, Ran Y, Li S, Tien P, Shu H-B. 2010. WDR5 is essential for assembly of the VISA-associated signaling complex and virus-triggered IRF3 and NF-κB activation. Proc Nat Acad Sci U S A 107:815–820. doi: 10.1073/pnas.0908967107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Pfaller CK, Li Z, George CX, Samuel CE. 2011. Protein kinase PKR and RNA adenosine deaminase ADAR1: new roles for old players as modulators of the interferon response. Curr Opin Immunol 23:573–582. doi: 10.1016/j.coi.2011.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Pfaller CK, Mastorakos GM, Matchett WE, Ma X, Samuel CE, Cattaneo R. 2015. Measles virus defective interfering RNAs are generated frequently and early in the absence of C protein and can be destabilized by adenosine deaminase acting on RNA-1-like hypermutations. J Virol 89:7735–7747. doi: 10.1128/JVI.01017-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Okonski KM, Samuel CE. 2013. Stress granule formation induced by measles virus is protein kinase PKR dependent and impaired by RNA adenosine deaminase ADAR1. J Virol 87:756–766. doi: 10.1128/JVI.02270-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ruggieri A, Dazert E, Metz P, Hofmann S, Bergeest J-P, Mazur J, Bankhead P, Hiet M-S, Kallis S, Alvisi G, Samuel CE, Lohmann V, Kaderali L, Rohr K, Frese M, Stoecklin G, Bartenschlager R. 2012. Dynamic oscillation of translation and stress granule formation mark the cellular response to virus infection. Cell Host Microbe 12:71–85. doi: 10.1016/j.chom.2012.05.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Spehner D, Kirn A, Drillien R. 1991. Assembly of nucleocapsidlike structures in animal cells infected with a vaccinia virus recombinant encoding the measles virus nucleoprotein. J Virol 65:6296–6300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.McAllister CS, Toth AM, Zhang P, Devaux P, Cattaneo R, Samuel CE. 2010. Mechanisms of protein kinase PKR-mediated amplification of beta interferon induction by C protein-deficient measles virus. J Virol 84:380–386. doi: 10.1128/JVI.02630-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Li Z, Okonski KM, Samuel CE. 2012. Adenosine deaminase acting on RNA 1 (ADAR1) suppresses the induction of interferon by measles virus. J Virol 86:3787–3794. doi: 10.1128/JVI.06307-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Novoa RR, Calderita G, Arranz R, Fontana J, Granzow H, Risco C. 2005. Virus factories: associations of cell organelles for viral replication and morphogenesis. Biol Cell 97:147–172. doi: 10.1042/BC20040058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.van Nuland R, Smits AH, Pallaki P, Jansen PW, Vermeulen M, Timmers HT. 2013. Quantitative dissection and stoichiometry determination of the human SET1/MLL histone methyltransferase complexes. Mol Cell Biol 33:2067–2077. doi: 10.1128/MCB.01742-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Dias WB, Cheung WD, Hart GW. 2012. O-GlcNAcylation of kinases. Biochem Biophys Res Commun 422:224–228. doi: 10.1016/j.bbrc.2012.04.124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Komarova AV, Combredet C, Meyniel-Schicklin L, Chapelle M, Caignard G, Camadro J-M, Lotteau V, Vidalain P-O, Tangy F. 2011. Proteomic analysis of virus-host interactions in an infectious context using recombinant viruses. Mol Cell Proteomics 10:M110.007443. doi: 10.1074/mcp.M110.007443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Koga R, Sugita Y, Noda T, Yanagi Y, Ohno S. 2015. Actin-modulating protein cofilin is involved in the formation of measles virus ribonucleoprotein complex at the perinuclear region. J Virol 89:10524–10531. doi: 10.1128/JVI.01819-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Pfaller CK, Radeke MJ, Cattaneo R, Samuel CE. 2014. Measles virus C protein impairs production of defective copyback double-stranded viral RNA and activation of protein kinase R. J Virol 88:456–468. doi: 10.1128/JVI.02572-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ito M, Iwasaki M, Takeda M, Nakamura T, Yanagi Y, Ohno S. 2013. Measles virus nonstructural C protein modulates viral RNA polymerase activity by interacting with host protein SHCBP1. J Virol 87:9633–9642. doi: 10.1128/JVI.00714-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Weis K. 2003. Regulating access to the genome: nucleocytoplasmic transport throughout the cell cycle. Cell 112:441–451. doi: 10.1016/S0092-8674(03)00082-5. [DOI] [PubMed] [Google Scholar]
- 50.Knockenhauer KE, Schwartz TU. 2016. The nuclear pore complex as a flexible and dynamic gate. Cell 164:1162–1171. doi: 10.1016/j.cell.2016.01.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Gori F, Divieti P, Demay MB. 2001. Cloning and characterization of a novel WD-40 repeat protein that dramatically accelerates osteoblastic differentiation. J Biol Chem 276:46515–46522. doi: 10.1074/jbc.M105757200. [DOI] [PubMed] [Google Scholar]
- 52.West AP, Shadel GS, Ghosh S. 2011. Mitochondria in innate immune responses. Nat Rev Immunol 11:389–402. doi: 10.1038/nri2975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Mills EL, Kelly B, O'Neill LA. 2017. Mitochondria are the powerhouses of immunity. Nat Immunol 18:488–498. doi: 10.1038/ni.3704. [DOI] [PubMed] [Google Scholar]
- 54.Lee J-H, Skalnik DG. 2008. Wdr82 is a C-terminal domain-binding protein that recruits the Setd1A histone H3-Lys4 methyltransferase complex to transcription start sites of transcribed human genes. Mol Cell Biol 28:609–618. doi: 10.1128/MCB.01356-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Zhu K, Wang X, Ju L-G, Zhu Y, Yao J, Wang Y, Wu M, Li L-Y. 2015. WDR82 negatively regulates cellular antiviral response by mediating TRAF3 polyubiquitination in multiple cell lines. J Immunol 195:5358–5366. doi: 10.4049/jimmunol.1500339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Mühlebach MD, Mateo M, Sinn PL, Prüfer S, Uhlig KM, Leonard VH, Navaratnarajah CK, Frenzke M, Wong XX, Sawatsky B, Ramachandran S, McCray PB Jr, Cichutek K, von Messling V, Lopez M, Cattaneo R. 2011. Adherens junction protein nectin-4 (PVRL4) is the epithelial receptor for measles virus. Nature 480:530–533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Nakatsu Y, Ma X, Seki F, Suzuki T, Iwasaki M, Yanagi Y, Komase K, Takeda M. 2013. Intracellular transport of the measles virus ribonucleoprotein complex is mediated by Rab11A-positive recycling endosomes and drives virus release from the apical membrane of polarized epithelial cells. J Virol 87:4683–4693. doi: 10.1128/JVI.02189-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Devaux P, Hodge G, McChesney MB, Cattaneo R. 2008. Attenuation of V-or C-defective measles viruses: infection control by the inflammatory and interferon responses of rhesus monkeys. J Virol 82:5359–5367. doi: 10.1128/JVI.00169-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kärber G. 1931. Beitrag zur kollektiven behandlung pharmakologischer reihenversuche. Naunyn-Schmiedeberg's Arch Pharmacol 162:480–483. doi: 10.1007/BF01863914. [DOI] [Google Scholar]
- 60.Toth AM, Devaux P, Cattaneo R, Samuel CE. 2009. Protein kinase PKR mediates the apoptosis induction and growth restriction phenotypes of C protein-deficient measles virus. J Virol 83:961–968. doi: 10.1128/JVI.01669-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Del Valle JR, Devaux P, Hodge G, Wegner NJ, McChesney MB, Cattaneo R. 2007. A vectored measles virus induces hepatitis B surface antigen antibodies while protecting macaques against measles virus challenge. J Virol 81:10597–10605. doi: 10.1128/JVI.00923-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Devaux P, von Messling V, Songsungthong W, Springfeld C, Cattaneo R. 2007. Tyrosine 110 in the measles virus phosphoprotein is required to block STAT1 phosphorylation. Virology 360:72–83. doi: 10.1016/j.virol.2006.09.049. [DOI] [PubMed] [Google Scholar]
- 63.Pfaller CK, Cattaneo R, Schnell MJ. 2015. Reverse genetics of Mononegavirales: how they work, new vaccines, and new cancer therapeutics. Virology 479-480:331–344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Radecke F, Spielhofer P, Schneider H, Kaelin K, Huber M, Dötsch C, Christiansen G, Billeter M. 1995. Rescue of measles viruses from cloned DNA. EMBO J 14:5773–5784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Devaux P, Cattaneo R. 2004. Measles virus phosphoprotein gene products: conformational flexibility of the P/V protein amino-terminal domain and C protein infectivity factor function. J Virol 78:11632–11640. doi: 10.1128/JVI.78.21.11632-11640.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Cathomen T, Naim HY, Cattaneo R. 1998. Measles viruses with altered envelope protein cytoplasmic tails gain cell fusion competence. J Virol 72:1224–1234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Patterson JB, Samuel CE. 1995. Expression and regulation by interferon of a double-stranded-RNA-specific adenosine deaminase from human cells: evidence for two forms of the deaminase. Mol Cell Biol 15:5376–5388. doi: 10.1128/MCB.15.10.5376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Li Z, Wolff KC, Samuel CE. 2010. RNA adenosine deaminase ADAR1 deficiency leads to increased activation of protein kinase PKR and reduced vesicular stomatitis virus growth following interferon treatment. Virology 396:316–322. doi: 10.1016/j.virol.2009.10.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Ank N, West H, Bartholdy C, Eriksson K, Thomsen AR, Paludan SR. 2006. Lambda interferon (IFN-λ), a type III IFN, is induced by viruses and IFNs and displays potent antiviral activity against select virus infections in vivo. J Virol 80:4501–4509. doi: 10.1128/JVI.80.9.4501-4509.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]