

ABSTRACT
Nef-specific CD8+ T lymphocytes (CD8TL) are linked to extraordinary control of primate lentiviral replication, but the mechanisms underlying their efficacy remain largely unknown. The immunodominant, Mamu-B*017:01+-restricted Nef195-203MW9 epitope in SIVmac239 partially overlaps a sorting motif important for interactions with host AP-2 proteins and, hence, downmodulation of several host proteins, including Tetherin (CD317/BST-2), CD28, CD4, SERINC3, and SERINC5. We reasoned that CD8TL-driven evolution in this epitope might compromise Nef's ability to modulate these important molecules. Here, we used deep sequencing of SIV from nine B*017:01+ macaques throughout infection with SIVmac239 to characterize the patterns of viral escape in this epitope and then assayed the impacts of these variants on Nef-mediated modulation of multiple host molecules. Acute variation in multiple Nef195-203MW9 residues significantly compromised Nef's ability to downregulate surface Tetherin, CD4, and CD28 and reduced its ability to prevent SERINC5-mediated reduction in viral infectivity but did not impact downregulation of CD3 or major histocompatibility complex class I, suggesting the selective disruption of immunomodulatory pathways involving Nef AP-2 interactions. Together, our data illuminate a pattern of viral escape dictated by a selective balance to maintain AP-2-mediated downregulation while evading epitope-specific CD8TL responses. These data could shed light on mechanisms of both CD8TL-driven viral control generally and on Mamu-B*017:01-mediated viral control specifically.
IMPORTANCE A rare subset of humans infected with HIV-1 and macaques infected with SIV can control the virus without aid of antiviral medications. A common feature of these individuals is the ability to mount unusually effective CD8 T lymphocyte responses against the virus. One of the most formidable aspects of HIV is its ability to evolve to evade immune responses, particularly CD8 T lymphocytes. We show that macaques that target a specific peptide in the SIV Nef protein are capable of better control of the virus and that, as the virus evolves to escape this response, it does so at a cost to specific functions performed by the Nef protein. Our results help show how the virus can be controlled by an immune response, which could help in designing effective vaccines.
KEYWORDS: CD4, SERINC, Tetherin, viral escape, evolution, major histocompatibility complex, simian immunodeficiency virus
INTRODUCTION
Virus-specific CD8+ T lymphocytes (CD8TL) play a critical role in establishing the set point of HIV-1 and SIV replication in vivo. There are strong correlations between expression of particular major histocompatibility complex class I (MHC-I) alleles and extraordinarily low or high viral set points (1, 2), further implicating CD8TL as causative agents in viral control. The control of HIV and SIV replication by antiviral CD8TL is complicated by the immense capacity of the viruses to evolve to escape these immune pressures (3–15). In fact, CD8TL are likely dominant selective forces driving viral sequence variation in individuals and on population scales during infection with HIV-1 or SIV (16, 17). In addition, escape from CD8TL can exact a cost to viral fitness (9, 18–24). The balance among these variables—the efficacy of antiviral CD8TL, the propensity of the virus to evolve to evade them, and the specific fitness costs associated with particular escape mutations—is likely critically important in determining the viral set point in a given individual. Hence, understanding immune-mediated control of HIV/SIV replication necessitates understanding all components of this process.
Although several lines of data indicate that CD8TL targeting epitopes in HIV-1 Gag are important for enhanced viral control, likely due to functional constraints limiting viral escape (6, 9, 18, 22, 25–31), CD8TL-mediated control of SIV in rhesus macaques (RM) is better correlated with CD8TL targeting the viral Nef and Vif proteins (12, 14, 32–36). Evidence suggests that Nef-specific CD8TL might aid in HIV-1 control as well (37–39), an underappreciated effect. The Nef protein in both SIV and HIV-1 is highly immunogenic and harbors epitopes restricted by MHC-I alleles associated with control in both RM and humans (5, 12, 14, 40–42). Hence, understanding the mechanisms of CD8TL-mediated selection in Nef is important for understanding CD8TL-mediated viral control in general.
Nef represents an intriguing and potentially important target for CD8TL. In both SIV and HIV-1, Nef is a pleiotropic protein whose myriad functions are focused on immunomodulation, including the downregulation of several cell surface proteins that are involved in host immunity, such as TCR-CD3 (in most SIVs but not HIV-1) (43), CD4 (44–46), CD8αβ (47), CD28 (48), Tetherin (BST2 or CD317; in most SIVs but not HIV-1) (49, 50), MHC-I (51), MHC-II (52), CD1d (53), CD80/CD86 (54), and likely others, as well as enhancing viral infectivity by preventing virion incorporation of host serine incorporator 3 (SERINC3) and SERINC5 proteins (55–58). Modulation of several of these molecules, including CD4, CD8αβ, CD28, Tetherin, and SERINC3 and SERINC5 is effected via interactions between Nef and adaptor protein 2 (AP-2) complexes (47, 55, 59–63).
Interestingly, one of two dominant Mamu-B*017:01-restricted epitopes in Nef, Nef195-203MW9, spans the region between two characterized sorting motifs. The first of which is a “dileucine” motif (ExxxLM in SIVmac viruses) known to be important for modulation of host molecules (62, 64), likely via direct interactions with host AP-2 proteins. The other is a diacidic motif (DD in nearly all primate lentiviruses), which represents a novel AP-2 binding motif in HIV-1 Nef (63, 65). Given the importance of Nef-mediated immunomodulation to viral replication and pathogenesis in vivo, we were intrigued by whether viral escape in this epitope compromised Nef's ability to downregulate host molecules that rely on interactions between Nef and host AP-2 proteins, such as Tetherin, CD4, CD28, SERINC3, and SERINC5.
Since Tetherin directly traps budding virus particles at the cell surface, leading to increased surface Env, we also investigated whether variations that impacted Tetherin expression levels correlated with the cell surface expression of Env. Increased surface Env may increase the susceptibility of infected cells to antibody-dependent cell-mediated cytotoxicity, suggesting that virus-mediated downregulation of Tetherin serves as a means to escape these potentially important immune responses (66–68). Here, we comprehensively examined the impacts of natural variation in a CD8TL epitope, which partially overlaps a known AP-2 binding domain, on Nef's ability to perform functions that rely on this interaction and functions that do not. Variation at every residue in the epitope impacted Nef's ability to perform functions that rely on AP-2 interactions but did not impact other functions. Our data illuminate a process whereby Nef cannot effectively evolve to escape Nef195-203MW9-specific CD8TL without some loss of viral function.
RESULTS
Acute-phase CD8TL target Mamu-B*017:01-restricted Nef195-203MW9.
We measured the magnitude of Nef195-203MW9-specific CD8TL during infection with SIVmac239 in our acutely infected cohort of six rhesus macaques (RM) using IFN-γ enzyme linked immunospot (ELISpot) assays from peripheral blood mononuclear cells (PBMC) samples collected at 4 and 8 weeks postinfection. All six RM made responses to the epitope at one or both time points (Fig. 1). The magnitude of these responses varied among RM but generally increased in dominance between 4 and 8 weeks postinfection. For instance, in RM r97088 the response was undetectable at 4 weeks but high magnitude at eight (>2,000 spot-forming cells [SFC]/million PBMC).
FIG 1.
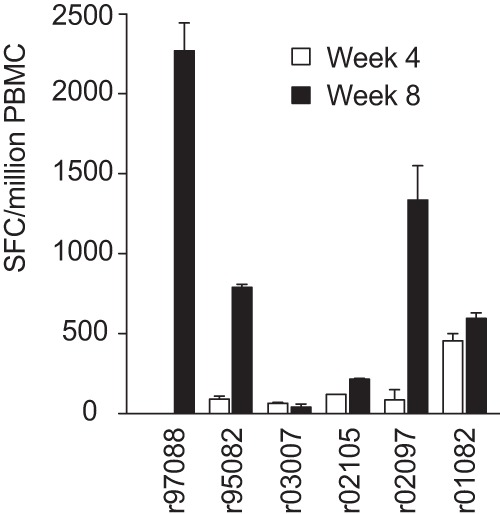
Acute-phase CD8TL responses against the Nef195-203MW9 epitope. Peptide representing the Nef195-203MW9 epitope was used at 0.01 mM to stimulate PBMC from six RM from 4 and 8 weeks postinfection. Overnight stimulations of 100,000 cells per well were tested in duplicate and followed by spot development and counting. Data shown represent the SFC per million PBMC.
Characterizing sequence variation in Nef195-203MW9 throughout infection.
To evaluate viral sequence evolution under pressure from the Nef195-203MW9-directed CD8TL response, we used deep sequencing of plasma virus at multiple time points during infection. Six RM were sampled periodically during acute and early chronic infection (ranging from 3 to 17 weeks postinfection) with one exception. RM r03007 was maintained for more than 1 year and sampled at 60 weeks postinfection. Three additional RM used in separate studies were sampled at multiple chronic phase time points (the earliest at 12 weeks postinfection and the latest at 267 weeks postinfection). Isolated viral RNA was reverse transcribed and amplified using four overlapping amplicons spanning the SIV genome as we described previously (69). Pooled reverse transcription-PCR (RT-PCR) products were sequenced on an Illumina MiSeq instrument, and this data set has recently been published. Methods of sequencing an analysis were as described previously (69) and similar to previous studies (70).
Nonsynonymous variation at ≥1% was found at every residue in the Nef195-203MW9 epitope (Fig. 2A), while nonsynonymous variation above 0.5% was never detected in nucleotides encoding highly conserved amino acids near the Nef195-203MW9 epitope, including in the diacidic motif. We identified a limited number of variants within the Nef195-203MW9 epitope (Fig. 2B). For instance, variation at M195 (p1 of the epitope) nearly always showed a change from methionine (M) to isoleucine (I) or valine (V) and frequently was a mixture of both mutants, along with residual wild-type virus. In fact, variant nucleotides were not fixed in the viral population as late as 17 weeks postinfection (Fig. 2B, RM r02105) in our acutely infected cohort and 35 weeks in RM rh1937 in our chronically infected cohort, followed by nearly complete fixation of the M-to-I mutant at 167 weeks postinfection in rh1937. Similarly, nonsynonymous variants at H196 nearly always encoded an H196Q change, with similarly limited variations at other positions in the epitope.
FIG 2.

Viral evolution in the Nef195-203MW9 epitope. (A) Heat map generated from the deep sequence data showing the frequency of nonsynonymous variation at each residue in the epitope at each sampled time point. Only nonsynonymous mutations that represented more than once and ≥1% of nucleotides at each location are shown. (B) Pie charts showing the frequency of specific amino acid substitutions at each residue in the epitope from each sample are shown. The identity of the amino acids is shown by the pie slices below each residue.
Most detected variants in Nef195-203MW9 confer escape from CD8TL.
We next sought to determine whether variants detected in Nef195-203MW9 conferred escape from CD8TL that target this epitope. We tested cryopreserved PBMC from week 8 from three animals in our study with relatively high magnitude Nef195-203MW9-specific responses for reactivity to dilutions of peptides representing the most common variants detected in our animals. In all animals, the M195V variant completely abrogated the response, as did the H196Q and W203C variants. All variants resulted in reduced reactivity in PBMC from animal r97088 (Fig. 3A) and, with the exception of S201A, all other variants resulted in reduced reactivity relative to the wild-type Nef195-203MW9 peptide in cumulative data from all three animals (Fig. 3B).
FIG 3.

Evolution in Nef195-203MW9 confers escape from CD8TL. (A) PBMC from animal r97088 8 weeks after SIV infection were tested for reactivity to dilutions of peptides representing the wild-type Nef195-203MW9 peptide (WT) and the most common variants identified by deep sequencing in our cohort. (B) Reactivity to variant peptides assessed with week 8 PBMC from three animals from our cohort, tested in duplicate IFN-γ ELISpot wells. Shown are the reactivities relative to the wild type.
Primary CD4 T cells express Tetherin.
Tetherin is induced as part of the type I interferon (IFN) antiviral pathway, and its expression in primary CD4 T cells would not be expected in the absence of exogenously added IFN-α, although these cells might upregulate it upon infection with SIV. We used transcriptome sequencing (RNA-seq) to measure global transcription in primary CD4 T cells either activated with concanavalin A (ConA) or left untreated and infected with SIVmac239. Surprisingly, the BST2/Tetherin gene was unambiguously expressed in these cells (Fig. 4A). Despite not being treated with exogenous IFNs, transcriptional profiles in these cells were consistent with IFN stimulation as several IFN-stimulated genes (ISGs) were expressed, including known HIV/SIV restriction factors such as Apobec3G, TRIM5, and SAMHD1, as well as genes selectively induced by IFN-γ (such as IRF1) and those induced selectively by type I IFNs such as OAS1 (Fig. 4B). These data were collected as part of a separate experiment that did not involve uninfected cells, so we cannot rule out the role of SIV infection in upregulating ISGs. However, the Tetherin protein was also easily detected on the surface of both infected and uninfected primary CD4 T cells using a fluorescently labeled monoclonal antibody (MAb). These data suggest that SIV infection alone was not the cause of its upregulation, but rather it was induced either in vivo prior to cell isolation or due to an unknown aspect of our isolation or culture conditions.
FIG 4.

RNA-seq analysis of ISG expression in primary CD4 T cells. Directional whole transcriptome libraries were sequenced from SIV-infected primary CD4 T cells that were either activated with ConA or left nonactivated. (A) Sashimi plot showing BST2/Tetherin coverage depth and exon splicing from a representative animal. Coverage depth is shown by peaks above the axis, and splicing events are shown as arcs below the axis. The numbers of reads that aligned across the splice junctions are depicted below the arcs. (B) Transcript abundance expressed as transcripts per million reads of HIV/SIV restriction factor genes, known ISGs, and genes of interest in this study, CD4 and CD28, for comparison.
Multiple variants in Nef195-203MW9 impact surface Tetherin and CD28, but not CD3 or MHC-I modulation in primary CD4 T cells.
To identify the functional consequences of escape in the Nef195-203MW9 epitope, we introduced the most common Nef195-203MW9 variants identified from our cohorts into both the SIVmac239-encoding plasmid, which was used to produce infectious SIV stocks bearing the introduced mutations, and an expression construct (pCGCG) that expresses both Nef and green fluorescent protein (GFP) from the same bicistronic mRNA by way of an internal ribosomal entry site. Whenever possible, we measured the Nef-mediated modulation of specific host proteins in primary CD4 T cells infected in culture with SIVmac239 and our Nef variants. We were able to assess the impacts of these mutants on Nef-mediated modulation of CD3, MHC-I, CD28, and Tetherin using these cells. An additional two Nef functions, CD4 downregulation and inhibition of SERINC5-mediated reductions in viral infectivity, were measured using cell lines transfected with our pCGCG-Nef vectors.
To determine the impact of the mutants on Nef-mediated CD3, MHC-I, CD28, and Tetherin downmodulation, we used our Nef mutant viruses to infect ConA-activated, primary CD4 T cells using the spinoculation technique (71). After 36 h, we used MAbs to label the surfaces of the cells for the expression of these proteins. After fixation and permeabilization, cells were labeled with a fluorescein isothiocyanate (FITC)-labeled MAb against the Gag p27 protein. We used differential gating of p27− and p27+ (uninfected and infected, respectively) cells (Fig. 5A) to assess surface Tetherin, CD3, CD28, and MHC-I molecules using flow cytometry. We measured the n-fold downregulation of each molecule relative to uninfected cells from a minimum of four healthy macaques. Cells from each animal were tested singly, and in Fig. 5B to E the error bars represent variations between animals.
FIG 5.

Specific variants of Nef195-203MW9 compromise Tetherin and CD28 modulation but not CD3 or MHC-I. (A) Schematic showing how the downregulation of surface molecules was detected. (B) n-fold decrease in the MFI values of surface Tetherin expression in Gag p27+ cells relative to Gag p27− (uninfected) cells in the same samples. Cells from at least four RM were infected with each virus. (C to E) Same as in panel B but surface CD28 (C), CD3 (D), and MHC-I (E) levels were measured. Above each bar graph is a representative histogram showing the strategy for assessing surface downregulation of each molecule. For each tested molecule, the n-fold downregulation was compared between each variant and wild-type SIVmac239 using a paired t test (*, P < 0.05).
While neither variant detected at M195 (M195I and M195V) had a significant impact on downregulation of any of the tested molecules, the variants tested at all other residues in the epitope (H196Q, P197T, A198D, Q199R, T200A, S201A, Q202K, and W203C) significantly reduced Nef's ability to downregulate Tetherin (Fig. 5B). Most also impacted the ability of Nef to downregulate host CD28 molecules (Fig. 5C). Importantly, the variant with the greatest impact on Nef-mediated Tetherin and CD28 downregulation was H196Q. H196 is the N-terminal anchor residue, critical for binding of the peptide to the Mamu-B*017:01 molecule, while variation of the C-terminal anchor residue (W203C) also reduced both Nef-mediated Tetherin and CD28 downregulatory capacity. We found that mutations in this epitope had no effect on expression of CD3 or MHC-I (Fig. 5D and E).
Viral evolution in Nef195-203MW9 modulates cell surface Env expression.
We next sought to determine whether selected variants in Nef195-203MW9 that impacted Nef-mediated Tetherin expression also lead to increased cell surface Env expression. To test this, we surface labeled cells with a rhesus-derived monoclonal antibody against SIV gp120 (clone 1.9C) and stained intracellularly for Gag p27 and used the same gating strategy as with other cell surface markers to determine the impact of our identified mutants on surface Env expression. We found that most variants that impacted surface Tetherin expression also led to increased Env expression with the most profound impact on Env expression coming from the H196Q and W203C variants (Fig. 6A shows representative data from cells from a single animal, while Fig. 6B shows the analysis with cells from multiple animals). H196 and W203 are the anchor residues that bind the Nef195-203MW9 peptide to the Mamu-B*017:01 molecule, and variation in these residues completely abrogated recognition of the epitope by CD8TL.
FIG 6.

Impact of mutations in the Nef195-203MW9 epitope on surface Envelope expression. (A) MFIs of surface Env expression in Gag p27+ cells infected with SIVmac239 and the indicated mutants. (B) n-fold increases in surface Envelope expression in cells infected with each mutant virus compared to cells infected with wild-type SIVmac239. *, P < 0.05 for each measure compared to cells infected with wild-type SIVmac239 tested using a two-tailed t test.
Multiple variants of Nef195-203MW9 modulate cell surface CD4 expression.
Using SIV-infected CD4 cells, we found that surface expression of CD4 was reduced in infected cells even in the absence of Nef (SIVmac239Δnef) possibly due to direct interactions between Env and CD4 (Fig. 7A), negating our ability to assess variants of Nef to modulate surface CD4 expression. Thus, to detect impacts of the variants on CD4 downregulation, we devised an alternate strategy that used introduction of the mutations into the pCGCG Nef construct, which expresses both GFP and Nef via a bicistronic mRNA. To detect the impacts of the variants on CD4 downregulation, we transfected TZM-bl (CD4-expressing HeLa cells) with the pCGCG Nef vectors. Forty-eight hours later, we labeled the surfaces of the cells with an anti-CD4 antibody. We used flow cytometry to analyze CD4 expression in Nef-expressing cells, identified by the coexpression of GFP in transfected cells (Fig. 7B). By comparing the median fluorescence intensity (MFI) of CD4 expression in GFP+ and GFP− cells in the same sample, we calculated the downregulation of CD4 by each Nef variant. The H196Q variant had the most significant impact on Nef's ability to downregulate CD4 (Fig. 7C). Generally, the pattern of variants that impacted CD4 downregulation mirrored that of variants that impacted CD28 downregulation, with the exception of P197T, which impacted CD28 but not CD4 downregulation.
FIG 7.

Impact of mutations in the Nef195-203MW9 epitope on surface CD4 downregulation. (A) Surface CD4 expression in primary CD4 T cells infected with SIVmac239 or SIVmac239Δnef. (B) Surface CD4 expression in TZM-bl cells (CD4 expressing HeLa cells) transfected with pCGCG vectors that express wild-type SIV Nef or the indicated mutants. (C) n-fold decreases in surface CD4 expression in GFP+ cells relative to GFP− cells. n-fold values were compared between each variant and wild-type SIV Nef. *, P < 0.05 for each comparison tested using a two-tailed t test. The G2A variant of Nef, which lacks a myristoylation signal and is defective for most Nef activities, was included as a negative control in these experiments.
Variation in every residue in Nef195-203MW9 impacts Nef-mediated anti-SERINC activity.
We next tested whether our identified variants in Nef195-203MW9 negatively impacted Nef's ability to enhance viral infectivity by countering the restriction factor, SERINC5 (56, 58). Proviral SIVmac239 Δnef plasmid was transfected into JTag SERINC3−/− cells that express SERINC5 and trans-complemented with pCGCG-empty vector or pCGCG-Nef constructs. At 72 h posttransfection, the viruses in the supernatant were collected and quantified by p27 enzyme-linked immunosorbent assay (ELISA), and the infectivity was assayed on TZM-bl reporter cells with p27-normalized viruses. The anti-SERINC5 activity of each Nef195-203MW9 variant was impaired with variants of residues closest to the dileucine and diacidic motifs with the strongest impacts (Fig. 8).
FIG 8.
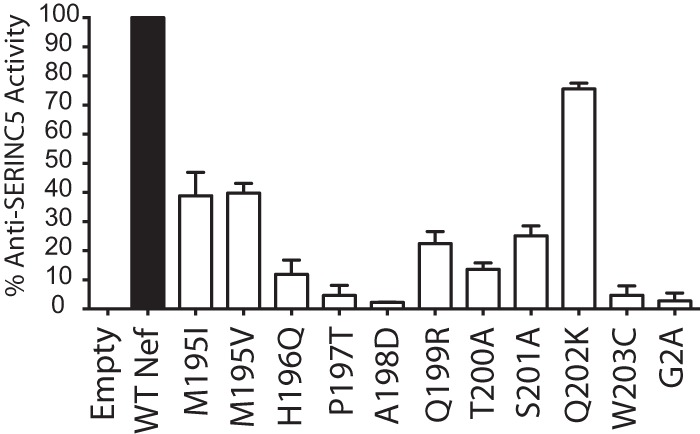
Impact of mutations in the Nef195-203MW9 epitope on SERINC5-mediated reduction in viral infectivity. The percent anti-SERINC5 activities of individual Nef mutants are reported as a function of the infectivity of SIV in the presence of the Nef mutants relative to the infectivity in the presence of WT Nef and empty vector. The G2A variant of Nef, which lacks a myristoylation signal and is defective for most Nef activities, was included as a negative control in these experiments.
Detected variants in Nef195-203MW9 do not globally impact Nef protein stability and do not impact viral replication in vitro.
To assess the impact of the tested mutations on Nef protein stability, we infected ConA-activated primary CD4 T cells with the mutant viruses and labeled cells intracellularly with both an anti-Nef monoclonal antibody and an anti-Gag p27 antibody. We then used flow cytometry to measure the median fluorescent intensity of Nef expression in p27+ cells 36 h after infection (Fig. 9A). None of the tested mutants significantly impacted Nef stability (Fig. 9B).
FIG 9.

Viral evolution in Nef195-203MW9 does not compromise Nef stability and does not impact viral replication in vitro. (A and B) Activated CD4 T cells isolated from healthy RM were infected with SIVmac239 and variants. After 36 h, the cells were labeled intracellularly with MAbs against the SIV Nef and Gag proteins. (A) Representative data showing the MFI values of Nef expression in Gag p27+ cells as measured by flow cytometry. (B) Assay as shown in panel A, with data combined from triplicate samples. (C) All variants were tested for replicative capacity in activated primary CD4 T cells using quantitative RT-PCR of supernatant viruses 2, 4, and 8 days after infection. (D) Viral competition assays between wild-type SIVmac239 and the M195I, M195V, and H196Q variants. Identical input amounts of competing viruses were intended, but the inoculum was sequenced to assess true input ratios (day 0), followed by sequencing on days 2 and 7 after infection. Shown is the fraction of clones with wild-type sequence in the Nef195-203MW9 epitope in competitions starting with equal amounts of wild-type SIVmac239 versus the M195I variant (top), versus the M195V variant (middle), and versus the H196Q variant (bottom).
To assess the impacts of our identified Nef195-203MW9 variants on viral replication kinetics, we used quantitative RT-PCR to measure viral RNA in supernatant of infected CD4 T cells over a 1-week assay. None of the variants was significantly different from the wild type. We also conducted competition assays with equal ratios of wild-type SIVmac239 and virus harboring the M195I, M195V, and H196Q variants. We infected ConA-activated primary CD4 T cells with the wild type or the variant viruses and cloned and sequenced a PCR amplicon spanning the Nef195-203MW9 epitope in supernatant virus on days zero (the inoculum), 2 and 7 days after infection. The ratios of variant viruses in the assays stayed relatively constant throughout the assays, suggesting no impact on viral replicative capacity (Fig. 9C).
DISCUSSION
We identified a pattern of variation in the Nef195-203MW9 epitope that suggest that viral evolution in this epitope is a result of an interplay of selective forces to effectively escape CD8TL immunity while maintaining optimal modulation of molecules such as Tetherin. This epitope completely encompasses the region spanning the ExxxLM motif (ExxxLL in HIV-1) and the diacidic (DD) motif, both of which are sorting signals important for interaction with host adaptor proteins, which Nef uses to facilitate clathrin-mediated endocytosis and modulation of multiple surface proteins, including Tetherin, CD4, CD8, CD28, SERINC3, and SERINC5 and possibly others (55, 56, 62, 63, 65, 72). Hence, immune targeting of the Nef195-203MW9 epitope might be important for viral control due to fitness costs associated with viral escape in this critical region of the Nef protein. Interestingly, CD8TL targeting of nearly the same epitope in Mauritian cynomolgus macaques has been associated with control of SIV, further demonstrating the importance of this region of Nef (42).
Several mutations we identified in the Nef195-203MW9 epitope impaired Nef's ability to modulate Tetherin, CD4, CD28, and SERINC5, while others only impacted modulation of a subset of these molecules, suggesting genetic separation of these Nef functions and that the loss of direct binding between Nef and AP-2 likely does not fully explain the compromised functions in this study. Thus, viral evolution in Nef195-203MW9 likely impacts direct interactions between Nef and AP-2 but may also impact other interactions as well. For instance, the P197T variant significantly impacted Nef's ability to modulate Tetherin, CD28, and SERINC5 but did not impact CD4 downregulation. Several of the residues we identified as being important for Tetherin and/or CD4 modulation were recently shown to impact the same molecules through the use of alanine substitutions (64), while our data demonstrated overlapping but not identical impacts. For instance, Serra-Moreno et al. reported that positions Q199 and T200 in Nef (p5 and p6 in the epitope) were important for Tetherin but not CD4 downregulation (64). Our results show that these residues are important for both Tetherin and CD4 downregulation. It is not entirely surprising that our approach of using mutants selected in vivo would have results that differed from those using alanine scanning and highlight the need to address Nef functions with multiple approaches, particularly given the immunogenicity of Nef and the propensity of it to evolve to escape CD8TL responses.
It is interesting that the sole residue in the Nef195-203MW9 epitope that could vary with the least impact on tested functions is the methionine in the first position. This residue forms part of what is typically characterized as a dileucine motif (ExxxLL in HIV-1, ExxxLM in SIVmac239) that binds host AP-2 proteins. However, the M195I variant at this position also did not provide complete escape from CD8TL but was the most commonly selected variant. These data underscore the balance of providing escape with a minimum of loss of function. In contrast, variation in residues near the motif, such as H196Q, which provided complete escape but dramatically impacted all functions known to rely on AP-2 binding. The AP-2 protein tolerates more variation in the recognition sequence of targeted molecules than other adaptor proteins, such as AP-1 and AP-3 (73), which might explain our data. Our data are nonetheless important in that they highlight the process by which selection must favor a compromise between loss of function with escape from immune responses. CD8TL that target specific regions of HIV-1 Gag are correlated with viral control, likely due to fitness costs associated with viral evolution in targeted epitopes (25, 74, 75). Our data suggest that similar mechanisms could be important for CD8TL that target the viral Nef protein, which is inherently more variable than Gag and, as such, often ignored as an important CD8TL target. Many CDTL escape mutations in Gag are associated with extraepitopic mutations that compensate, or partially compensate, for the functional consequences of variation within the epitope. We tested whether any of the variants we characterized were commonly associated with any specific variants outside the epitope. Since variation at every residue in the Nef195-203MW9 epitope negatively impacted Nef's ability to perform at least one of the tested AP-2 dependent functions, extraepitopic variants might compensate to restore or partially restore these functions. Although extensive sequence variation occurred in Nef outside the Nef195-203MW9 epitope in all animals, we found no evidence of compensatory variation in Nef that associated with any of the variants we tested, i.e., no specific variants outside the epitope were found to co-occur with variants within the epitope. In fact, the most common variants observed outside the epitope were found within the Nef165–173IW9 epitope, which is also restricted by Mamu-B*017:01 and which also evolves in the face of acute CD8TL immune pressure (69).
Unfortunately, the three-dimensional structure of the flexible loop of SIV Nef (which contains the Nef195-203MW9 epitope) has not been determined. However, the structure of HIV-1 Nef complexed with human AP-2 is known (63), and this structure clearly showed that the dileucine motif in Nef (LM in SIVmac239) was physically anchored to the AP-2 protein (60). However, the H166 residue (homologous to H196 in SIVmac239 Nef) protruded away from AP-2 but formed part of a small loop, presumably important for the dileucine anchor. Without similar data for SIV Nef, we cannot be certain of the role of this H residue in SIV Nef interactions with macaque AP-2, and our data do not directly address how variation in Nef195-203MW9 impacts interactions between Nef and AP-2 proteins. Regardless, these data further support an important role for this residue in AP-2 interactions despite not interacting directly with the AP-2 protein.
One of the most recently discovered functions of Nef is its ability to enhance virion infectivity by preventing host SERINC3 and SERINC5 proteins from being packaged into viral particles. Given this function relies on Nef interactions with AP-2 proteins (55, 56, 76), it stands to reason that variants that impact downregulation of Tetherin, CD4, and CD28 might also impact SERINC3 and SERINC5 modulation. Indeed, we found that this function may have been the most impacted by variation in the Nef195-203MW9 epitope, including the M195I and M195V variants, which were the most dominant residues selected for in our cohort.
Finally, we found that the N-terminal anchor residue in the epitope, H196, was critical for all tested functions. This amino acid is conserved across nearly all sequenced primate lentiviral Nef proteins (77), suggesting strong stabilizing selection. Hence, targeting this epitope could limit pathways of escape enabling enhanced viral control. It is well known that viral escape can occur in nonanchor residues, but variant viruses harboring such mutations can also be targeted by de novo, variant-specific CD8TL responses. Indeed, we showed previously that variation at p1 of this epitope, specifically the M195I variant, maintained the ability to bind the Mamu-B*017:01 molecule and was recognized by CD8TL in chronically infected macaques, suggesting that the most common escape variant in the epitope may not confer complete escape from host immunity (14).
Together, our data assess the immunovirology of the interaction between the Mamu-B*017:01 molecule and SIV. Intriguingly from a molecular immunology perspective, the mutation in Nef195-203MW9 that most dramatically impaired downregulation of Tetherin, CD4, and CD28 was in the anchor residue that binds the Nef195-203MW9 peptide to the Mamu-B*017:01 molecule. This same residue might also anchor the Nef196-203HW8 peptide to the Mafa-A1*063 molecule, which is also associated with enhanced control of SIVmac239 in Mauritian cynomolgus macaques (42). Hence, our data link the molecular immunology of the MHC-associated lentiviral control with specific functions and motifs of the SIV Nef protein. Taken together, our data suggest that Nef-specific CD8TL can control viral replication by targeting epitopes in which viral escape is associated with significant loss of critical functions. Hence, particular regions of the Nef protein might be beneficial components of vaccines designed to induce potent CD8TL responses.
MATERIALS AND METHODS
Viral RNA isolation, RT-PCR, Illumina sequencing, and analysis.
Viral RNA was isolated as previously described (69) using the QIAamp viral RNA minikit (Qiagen, Valencia, CA) or the QIAamp UltraSens virus kit for low viral load samples (specifically from week 60 from RM r03007). We reverse transcribed and PCR amplified SIV vRNA from select time points as described previously (78, 79) using Superscript III one-step RT-PCR (Life Technologies) and four overlapping PCR amplicons that, together, span the genome. RT-PCR-amplified products were isolated using agarose gel electrophoresis, followed by isolation of fragments using a Qiagen MinElute gel extraction kit. Fragments were quantified using Qubit reagents (Life Technologies, Carlsbad, CA). All amplicons from a single viral genome were pooled in equimolar amounts. Approximately 1 ng of DNA was subjected to tagmentation (simultaneous fragmentation and adaptor ligation) using the Nextera XT DNA prep kit. After cleaning of the DNA using an Agencourt AMPure system, the samples were PCR amplified to add Illumina-compatible adaptors onto each fragment, followed by further cleanup. DNA fragments were then sequenced by using an Illumina MiSeq instrument. Data analysis was performed using Geneious software version 10.0.2 (created by Biomatters). After pairing bidirectional sequence reads, sequence reads were trimmed and mapped to the SIVmac239 genome. Nonsynonymous polymorphisms were pursued in subsequent assays if they were represented multiple times and at a minimum frequency of 1% of sequences at a given nucleotide. We used this cutoff previously with this same sequence data set (69).
ELISpot assays.
IFN-γ ELISpot assays were performed as previously described (80) using 100,000 PBMC per well in 96-well plates (Mabtech, Stockholm, Sweden) assayed in duplicate. Peptide representing the Nef195-203MW9 epitope and variants thereof were synthesized by GenScript (Piscataway, NJ) and used in assays at 0.01 mM or in 10-fold dilutions.
Site-directed mutagenesis and mutant virus and plasmid production.
Mutations identified via deep sequencing were engineered into a plasmid encoding the 3′ end of the SIVmac239 genome, as well as the pCGCG-Nef plasmid, as described previously (81) using a Stratagene QuikChange site directed mutagenesis kit (Agilent, Santa Clara, CA) and according to the manufacturer's protocol with primers that harbored the mutant residue near the center of the primer. Virus was produced by transfecting plasmids into Vero cells using Lipofectamine 2000 (Life Technologies). Twenty-four hours after transfection, Vero cells were overlaid with CEMx174 cells, and virus was harvested at the peak of syncytium formation (typically 10 to 14 days posttransfection). The viruses were then sequenced to verify maintenance of the mutations through production. At 24 h after complete medium replacement, virus stocks were obtained by freezing filtered culture media from the infected CEMx174 cells at −80°C for short-term storage and in vapor-phase liquid nitrogen for longer-term storage. Introduced mutations were verified by Sanger sequencing of both the plasmid DNA and the harvested viral stocks prior to use in assays.
CD4+ T cell isolation, infection, and functional assays.
Primary PBMC were harvested from SIV-naive Indian rhesus macaques from the Tulane National Primate Center's specific-pathogen-free breeding colony. CD4+ T cells were isolated from the PBMC using nonhuman primate CD4 microbeads (Miltenyi Biotec, Bergisch Gladbach, Germany). CD4+ T cells were then activated using ConA (Sigma, St. Louis, MO) for 3 days in RPMI 1640 medium (Cellgro; Corning, Corning, NY) containing 15% FCS (Atlas Biologicals, Fort Collins, CO) and 50 U/ml IL-2 (Peprotech, Rocky Hill, NJ). Activated cells were infected using the spinoculation technique (71) with virus purified through 20% sucrose as described previously (82). At 36 h after infection, the cells were surface labeled for the following molecules: CD4 (Qdot605, NHP Reagent Reference Program, www.nhpreagents.org/), CD3 (PE-Cy7; BD Biosciences, Franklin Lakes, NJ), Tetherin (PE; BioLegend, San Diego, CA), and MHC-I (W6/32, Alexa 647; BioLegend). After fixation, the cells were washed and permeabilized, followed by intracellular staining with an FITC-labeled antibody against the Gag p27 molecule (NIH AIDS Reagent Program and conjugated with FITC at the Wisconsin National Primate Research Center).
Measurements of surface Env expression.
Surface Envelope expression was measured using flow cytometry as described previously (83, 84). Briefly, the cells were incubated at room temperature with the rhesus anti-gp120 MAb clone 1.9C for 30 min, followed by two washes and then incubated with a phycoerythrin (PE)-labeled anti-rhesus secondary antibody for 30 min, followed by fixation and acquisition. Flow cytometry was performed on a BD Fortessa or LSRII instrument, and data were analyzed using FlowJo software (vX.07).
RNA-seq on SIV-infected CD4 T cells.
CD4 T cells were isolated and infected as described above. At 36 h after infection, total RNA was isolated using the RNeasy minikit with genomic DNA eliminator columns (Qiagen). The rRNA was removed, and strand-specific cDNA libraries were constructed using the Illumina TruSeq kit. Samples were multiplexed at up to six samples per lane and sequenced on an Illumina HiSeq 2000 instrument. Sequence reads were aligned to the rhesus macaque (MMul8.0.1) genome using a STAR aligner (85), and transcript abundance estimates were generated using RSEM (86). Data, including coverage depth and splicing patterns, were visualized using Integrated Genome Viewer (Broad Institute).
TZM-bl CD4 downregulation assay.
TZM-bl cells were obtained from the NIH AIDS Reagent Program. TZM-bl cells were cultured in Dulbecco modified Eagle medium with 10% fetal bovine serum (FBS). For the CD4 downregulation assays, cells were plated in 48-well plates and transfected with 1 μg of each pCGCG vector using the GenJet transfection reagent for HeLa cells (SignaGen). At 48 h after transfection, cells were removed from the plate using trypsin and labeled with a BV605-labeled anti-CD4 antibody (clone OCT-4; BioLegend) for 30 min. The cells were then washed once using phosphate-buffered saline supplemented with 10% FBS and fixed. Data were acquired on a BD LSRII instrument and analyzed using FlowJo software (vX.07). Preliminary analysis showed that Nef and GFP were expressed in a 1:1 ratio, so gating on GFP-positive cells served as a surrogate for Nef expression. The MFI values for GFP-positive cells were compared to those for GFP-negative cells in each sample. Data were analyzed using GraphPad Prism and are displayed as the n-fold downregulation relative to untransfected (GFP-negative) cells. Downregulation was compared between each mutant pCGCG Nef vector and wild-type pCGCG Nef using a two-tailed t test, and the differences were considered significant if the P value was <0.05.
Infectivity assay to assess SERINC5 antagonism.
JTag SERINC3−/− cells were kindly provided by H. Gottlinger and were cultured in RPMI 1640 supplemented with 10% FBS, 2 mM l-glutamine, and penicillin/streptomycin. To assess the anti-SERINC5 activity of SIV Nef mutants, 100,000 JTag SERINC3−/− cells were plated and transfected with 450 ng of SIVmac239Δnef and 50 ng of pCGCG-Nef expression constructs, or pCGCG-empty constructs using GenJet Jurkat transfection reagent (SignaGen) according to the manufacturer's instructions. At 72 h posttransfection, the supernatant was collected, and p27 in the supernatant was quantified using SIV p27 antigen capture ELISA kit (ABL Inc.). The infectivity of the resultant viruses was assayed with 0.5 ng equivalent of p27 applied to 10,000 TZM-bl cells in duplicate. The luciferase activities of the infected cells were assayed 72 h postransduction. The percent anti-SERINC5 activities of the individual Nef mutants were normalized to the pCGCG-WT Nef and pCGCG-empty vector as follows: % anti-SERINC5 activity = (infectivity with Nef mutant − infectivity with empty vector)/(infectivity with WT Nef − infectivity with empty vector).
Viral replication and competition assays.
One million ConA-activated CD4+ T cells were infected with 100 pg of each indicated virus for 4 h, followed by plating in duplicate in 0.5 ml on medium with 50 U/ml IL-2. Half of the medium was replaced every other day, and virus was quantified on days 2, 4, and 8 postinfection according to previously described methods (87). Competition assays entailed the same infection protocol but using equal amounts of both wild-type SIVmac239 and the indicated variants. RT-PCR was conducted with PCR primers flanking the Nef195-203MW9 epitope (forward primer, GGATACTCGCAATCCCCAGG; reverse primer, CCTCTGACAGGCCTGACTTG) using a SuperScript III one-step RT-PCR kit (Life Technologies). PCR amplicons were cloned into Top10 cells, and 32 clones were picked for each time point and sequenced.
ACKNOWLEDGMENTS
This study was supported by NIAID grant AI112447 to N.J.M. The funders had no role in study design, data collection and interpretation, or the decision to submit the work for publication.
We thank James Robinson for kindly providing the 1.9C anti-gp120 monoclonal antibody and David Watkins for providing samples.
REFERENCES
- 1.Carrington M, O'Brien S. 2003. The influence of HLA genotype on AIDS. Annu Rev Med 54:535–551. doi: 10.1146/annurev.med.54.101601.152346. [DOI] [PubMed] [Google Scholar]
- 2.Goulder P, Watkins D. 2008. Impact of MHC class I diversity on immune control of immunodeficiency virus replication. Nat Rev Immunol 8:619–630. doi: 10.1038/nri2357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Goulder P, Watkins D. 2004. HIV and SIV CTL escape: implications for vaccine design. Nat Rev Immunol 4:630–640. doi: 10.1038/nri1417. [DOI] [PubMed] [Google Scholar]
- 4.O'Connor D, Allen T, Vogel T, Jing P, DeSouza I, Dodds E, Dunphy E, Melsaether C, Mothe B, Yamamoto H, Horton H, Wilson N, Hughes A, Watkins D. 2002. Acute-phase cytotoxic T lymphocyte escape is a hallmark of simian immunodeficiency virus infection. Nat Med 8:493–499. doi: 10.1038/nm0502-493. [DOI] [PubMed] [Google Scholar]
- 5.Goulder P, Phillips R, Colbert R, McAdam S, Ogg G, Nowak M, Giangrande P, Luzzi G, Morgan B, Edwards A, McMichael A, Rowland-Jones S. 1997. Late escape from an immunodominant cytotoxic T-lymphocyte response associated with progression to AIDS. Nat Med 3:212–217. doi: 10.1038/nm0297-212. [DOI] [PubMed] [Google Scholar]
- 6.Goepfert P, Lumm W, Farmer P, Matthews P, Prendergast A, Carlson J, Derdeyn C, Tang J, Kaslow R, Bansal A, Yusim K, Heckerman D, Mulenga J, Allen S, Goulder P, Hunter E. 2008. Transmission of HIV-1 Gag immune escape mutations is associated with reduced viral load in linked recipients. J Exp Med 205:1009–1017. doi: 10.1084/jem.20072457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Feeney M, Tang Y, Roosevelt K, Leslie A, McIntosh K, Karthas N, Walker B, Goulder P. 2004. Immune escape precedes breakthrough human immunodeficiency virus type 1 viremia and broadening of the cytotoxic T-lymphocyte response in an HLA-B27-positive long-term-nonprogressing child. J Virol 78:8927–8930. doi: 10.1128/JVI.78.16.8927-8930.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Draenert R, Le Gall S, Pfafferott K, Leslie A, Chetty P, Brander C, Holmes E, Chang S, Feeney M, Addo M, Ruiz L, Ramduth D, Jeena P, Altfeld M, Thomas S, Tang Y, Verrill C, Dixon C, Prado J, Kiepiela P, Martinez-Picado J, Walker B, Goulder P. 2004. Immune selection for altered antigen processing leads to cytotoxic T lymphocyte escape in chronic HIV-1 infection. J Exp Med 199:905–915. doi: 10.1084/jem.20031982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Crawford H, Prado J, Leslie A, Hue S, Honeyborne I, Reddy S, van der Stok M, Mncube Z, Brander C, Rousseau C, Mullins J, Kaslow R, Goepfert P, Allen S, Hunter E, Mulenga J, Kiepiela P, Walker B, Goulder P. 2007. Compensatory mutation partially restores fitness and delays reversion of escape mutation within the immunodominant HLA-B*5703-restricted Gag epitope in chronic human immunodeficiency virus type 1 infection. J Virol 81:8346–8351. doi: 10.1128/JVI.00465-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Carlson JM, Listgarten J, Pfeifer N, Tan V, Kadie C, Walker BD, Ndung'u T, Shapiro R, Frater J, Brumme ZL, Goulder PJ, Heckerman D. 2012. Widespread impact of HLA restriction on immune control and escape pathways of HIV-1. J Virol 86:5230–5243. doi: 10.1128/JVI.06728-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Evans D, O'Connor D, Jing P, Dzuris J, Sidney J, da Silva J, Allen T, Horton H, Venham J, Rudersdorf R, Vogel T, Pauza C, Bontrop R, DeMars R, Sette A, Hughes A, Watkins D. 1999. Virus-specific cytotoxic T-lymphocyte responses select for amino-acid variation in simian immunodeficiency virus Env and Nef. Nat Med 5:1270–1276. doi: 10.1038/15224. [DOI] [PubMed] [Google Scholar]
- 12.Loffredo J, Friedrich T, Leon E, Stephany J, Rodrigues D, Spencer S, Bean A, Beal D, Burwitz B, Rudersdorf R, Wallace L, Piaskowski S, May G, Sidney J, Gostick E, Wilson N, Price D, Kallas E, Piontkivska H, Hughes A, Sette A, Watkins D. 2007. CD8+ T cells from SIV elite controller macaques recognize Mamu-B*08-bound epitopes and select for widespread viral variation. PLoS One 2:e1152. doi: 10.1371/journal.pone.0001152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Maness N, Valentine L, May G, Reed J, Piaskowski S, Soma T, Furlott J, Rakasz E, Friedrich T, Price D, Gostick E, Hughes A, Sidney J, Sette A, Wilson N, Watkins D. 2007. AIDS virus-specific CD8+ T lymphocytes against an immunodominant cryptic epitope select for viral escape. J Exp Med 204:2505–2512. doi: 10.1084/jem.20071261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Maness N, Yant L, Chung C, Loffredo J, Friedrich T, Piaskowski S, Furlott J, May G, Soma T, Leon E, Wilson N, Piontkivska H, Hughes A, Sidney J, Sette A, Watkins D. 2008. Comprehensive immunological evaluation reveals surprisingly few differences between elite controller and progressor Mamu-B*17-positive simian immunodeficiency virus-infected rhesus macaques. J Virol 82:5245–5254. doi: 10.1128/JVI.00292-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mudd P, Ericsen A, Burwitz B, Wilson N, O'Connor D, Hughes A, Watkins D. 2012. Escape from CD8+ T cell responses in Mamu-B*00801+ macaques differentiates progressors from elite controllers. J Immunol 188:3364–3370. doi: 10.4049/jimmunol.1102470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.O'Connor D, McDermott A, Krebs K, Dodds E, Miller J, Gonzalez E, Jacoby T, Yant L, Piontkivska H, Pantophlet R, Burton D, Rehrauer W, Wilson N, Hughes A, Watkins D. 2004. A dominant role for CD8+-T-lymphocyte selection in simian immunodeficiency virus sequence variation. J Virol 78:14012–14022. doi: 10.1128/JVI.78.24.14012-14022.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Allen T, Altfeld M, Geer S, Kalife E, Moore C, O'sullivan K, Desouza I, Feeney M, Eldridge R, Maier E, Kaufmann D, Lahaie M, Reyor L, Tanzi G, Johnston M, Brander C, Draenert R, Rockstroh J, Jessen H, Rosenberg E, Mallal S, Walker B. 2005. Selective escape from CD8+ T-cell responses represents a major driving force of human immunodeficiency virus type 1 (HIV-1) sequence diversity and reveals constraints on HIV-1 evolution. J Virol 79:13239–13249. doi: 10.1128/JVI.79.21.13239-13249.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kelleher A, Long C, Holmes E, Allen R, Wilson J, Conlon C, Workman C, Shaunak S, Olson K, Goulder P, Brander C, Ogg G, Sullivan J, Dyer W, Jones I, McMichael A, Rowland-Jones S, Phillips R. 2001. Clustered mutations in HIV-1 gag are consistently required for escape from HLA-B27-restricted cytotoxic T lymphocyte responses. J Exp Med 193:375–386. doi: 10.1084/jem.193.3.375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Leslie A, Pfafferott K, Chetty P, Draenert R, Addo M, Feeney M, Tang Y, Holmes E, Allen T, Prado J, Altfeld M, Brander C, Dixon C, Ramduth D, Jeena P, Thomas S, St John A, Roach T, Kupfer B, Luzzi G, Edwards A, Taylor G, Lyall H, Tudor-Williams G, Novelli V, Martinez-Picado J, Kiepiela P, Walker B, Goulder P. 2004. HIV evolution: CTL escape mutation and reversion after transmission. Nat Med 10:282–289. doi: 10.1038/nm992. [DOI] [PubMed] [Google Scholar]
- 20.Martinez-Picado J, Prado J, Fry E, Pfafferott K, Leslie A, Chetty S, Thobakgale C, Honeyborne I, Crawford H, Matthews P, Pillay T, Rousseau C, Mullins J, Brander C, Walker B, Stuart D, Kiepiela P, Goulder P. 2006. Fitness cost of escape mutations in p24 Gag in association with control of human immunodeficiency virus type 1. J Virol 80:3617–3623. doi: 10.1128/JVI.80.7.3617-3623.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Matthews P, Prendergast A, Leslie A, Crawford H, Payne R, Rousseau C, Rolland M, Honeyborne I, Carlson J, Kadie C, Brander C, Bishop K, Mlotshwa N, Mullins J, Coovadia H, Ndung'u T, Walker B, Heckerman D, Goulder P. 2008. Central role of reverting mutations in HLA associations with human immunodeficiency virus set point. J Virol 82:8548–8559. doi: 10.1128/JVI.00580-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wright JK, Naidoo VL, Brumme ZL, Prince JL, Claiborne DT, Goulder PJ, Brockman MA, Hunter E, Ndung'u T. 2012. Impact of HLA-B*81-associated mutations in HIV-1 Gag on viral replication capacity. J Virol 86:3193–3199. doi: 10.1128/JVI.06682-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Friedrich TC, Dodds EJ, Yant LJ, Vojnov L, Rudersdorf R, Cullen C, Evans DT, Desrosiers RC, Mothe BR, Sidney J, Sette A, Kunstman K, Wolinsky S, Piatak M, Lifson J, Hughes AL, Wilson N, O'Connor DH, Watkins DI. 2004. Reversion of CTL escape-variant immunodeficiency viruses in vivo. Nat Med 10:275–281. doi: 10.1038/nm998. [DOI] [PubMed] [Google Scholar]
- 24.Friedrich TC, Frye CA, Yant LJ, O'Connor DH, Kriewaldt NA, Benson M, Vojnov L, Dodds EJ, Cullen C, Rudersdorf R, Hughes AL, Wilson N, Watkins DI. 2004. Extraepitopic compensatory substitutions partially restore fitness to simian immunodeficiency virus variants that escape from an immunodominant cytotoxic-T-lymphocyte response. J Virol 78:2581–2585. doi: 10.1128/JVI.78.5.2581-2585.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kiepiela P, Ngumbela K, Thobakgale C, Ramduth D, Honeyborne I, Moodley E, Reddy S, de Pierres C, Mncube Z, Mkhwanazi N, Bishop K, van der Stok M, Nair K, Khan N, Crawford H, Payne R, Leslie A, Prado J, Prendergast A, Frater J, McCarthy N, Brander C, Learn G, Nickle D, Rousseau C, Coovadia H, Mullins J, Heckerman D, Walker B, Goulder P. 2007. CD8+ T-cell responses to different HIV proteins have discordant associations with viral load. Nat Med 13:46–53. doi: 10.1038/nm1520. [DOI] [PubMed] [Google Scholar]
- 26.Brockman MA, Brumme ZL, Brumme CJ, Miura T, Sela J, Rosato PC, Kadie CM, Carlson JM, Markle TJ, Streeck H, Kelleher AD, Markowitz M, Jessen H, Rosenberg E, Altfeld M, Harrigan PR, Heckerman D, Walker BD, Allen TM. 2010. Early selection in Gag by protective HLA alleles contributes to reduced HIV-1 replication capacity that may be largely compensated for in chronic infection. J Virol 84:11937–11949. doi: 10.1128/JVI.01086-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Brumme ZL, Tao I, Szeto S, Brumme CJ, Carlson JM, Chan D, Kadie C, Frahm N, Brander C, Walker B, Heckerman D, Harrigan PR. 2008. Human leukocyte antigen-specific polymorphisms in HIV-1 Gag and their association with viral load in chronic untreated infection. AIDS 22:1277–1286. doi: 10.1097/QAD.0b013e3283021a8c. [DOI] [PubMed] [Google Scholar]
- 28.Julg B, Williams K, Reddy S, Bishop K, Qi Y, Carrington M, Goulder P, Ndung'u T, Walker B. 2010. Enhanced anti-HIV functional activity associated with Gag-specific CD8 T-cell responses. J Virol 84:5540–5549. doi: 10.1128/JVI.02031-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kloverpris H, Payne R, Sacha J, Rasaiyaah J, Chen F, Takiguchi M, Yang O, Towers G, Goulder P, Prado J. 2013. Early antigen presentation of protective HIV-1 KF11Gag and KK10Gag epitopes from incoming viral particles facilitates rapid recognition of infected cells by specific CD8+ T cells. J Virol 87:2628–2638. doi: 10.1128/JVI.02131-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Payne RP, Kloverpris H, Sacha JB, Brumme Z, Brumme C, Buus S, Sims S, Hickling S, Riddell L, Chen F, Luzzi G, Edwards A, Phillips R, Prado JG, Goulder PJ. 2010. Efficacious early antiviral activity of HIV Gag- and Pol-specific HLA-B 2705-restricted CD8+ T cells. J Virol 84:10543–10557. doi: 10.1128/JVI.00793-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Schneidewind A, Brockman M, Yang R, Adam R, Li B, Le Gall S, Rinaldo C, Craggs S, Allgaier R, Power K, Kuntzen T, Tung C, LaBute M, Mueller S, Harrer T, McMichael A, Goulder P, Aiken C, Brander C, Kelleher A, Allen T. 2007. Escape from the dominant HLA-B27-restricted cytotoxic T-lymphocyte response in Gag is associated with a dramatic reduction in human immunodeficiency virus type 1 replication. J Virol 81:12382–12393. doi: 10.1128/JVI.01543-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Loffredo J, Bean A, Beal D, Leon E, May G, Piaskowski S, Furlott J, Reed J, Musani S, Rakasz E, Friedrich T, Wilson N, Allison D, Watkins D. 2008. Patterns of CD8+ immunodominance may influence the ability of Mamu-B*08-positive macaques to naturally control simian immunodeficiency virus SIVmac239 replication. J Virol 82:1723–1738. doi: 10.1128/JVI.02084-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Loffredo J, Sidney J, Bean A, Beal D, Bardet W, Wahl A, Hawkins O, Piaskowski S, Wilson N, Hildebrand W, Watkins D, Sette A. 2009. Two MHC class I molecules associated with elite control of immunodeficiency virus replication, Mamu-B*08 and HLA-B*2705, bind peptides with sequence similarity. J Immunol 182:7763–7775. doi: 10.4049/jimmunol.0900111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mothe B, Sidney J, Dzuris J, Liebl M, Fuenger S, Watkins D, Sette A. 2002. Characterization of the peptide-binding specificity of Mamu-B*17 and identification of Mamu-B*17-restricted epitopes derived from simian immunodeficiency virus proteins. J Immunol 169:210–219. doi: 10.4049/jimmunol.169.1.210. [DOI] [PubMed] [Google Scholar]
- 35.Mudd P, Martins M, Ericsen A, Tully D, Power K, Bean A, Piaskowski S, Duan L, Seese A, Gladden A, Weisgrau K, Furlott J, Kim Y, Veloso de Santana M, Rakasz E, Iii S, Wilson N, Bonaldo M, Galler R, Allison D, Piatak M Jr, Haase A, Lifson J, Allen T, Watkins D. 2012. Vaccine-induced CD8+ T cells control AIDS virus replication. Nature 491:129–133. doi: 10.1038/nature11443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Frahm N, Adams S, Kiepiela P, Linde C, Hewitt H, Lichterfeld M, Sango K, Brown N, Pae E, Wurcel A, Altfeld M, Feeney M, Allen T, Roach T, St John M, Daar E, Rosenberg E, Korber B, Marincola F, Walker B, Goulder P, Brander C. 2005. HLA-B63 presents HLA-B57/B58-restricted cytotoxic T-lymphocyte epitopes and is associated with low human immunodeficiency virus load. J Virol 79:10218–10225. doi: 10.1128/JVI.79.16.10218-10225.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chakraborty S, Rahman T, Chakravorty R. 2014. Characterization of the protective HIV-1 CTL epitopes and the corresponding HLA class I alleles: a step towards designing a CTL-based HIV-1 vaccine. Adv Virol 2014:321974. doi: 10.1155/2014/321974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mann JK, Byakwaga H, Kuang XT, Le AQ, Brumme CJ, Mwimanzi P, Omarjee S, Martin E, Lee GQ, Baraki B, Danroth R, McCloskey R, Muzoora C, Bangsberg DR, Hunt PW, Goulder PJ, Walker BD, Harrigan PR, Martin JN, Ndung'u T, Brockman MA, Brumme ZL. 2013. Ability of HIV-1 Nef to downregulate CD4 and HLA class I differs among viral subtypes. Retrovirology 10:100. doi: 10.1186/1742-4690-10-100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Mwimanzi P, Markle TJ, Martin E, Ogata Y, Kuang XT, Tokunaga M, Mahiti M, Pereyra F, Miura T, Walker BD, Brumme ZL, Brockman MA, Ueno T. 2013. Attenuation of multiple Nef functions in HIV-1 elite controllers. Retrovirology 10:1. doi: 10.1186/1742-4690-10-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Adland E, Carlson JM, Paioni P, Kloverpris H, Shapiro R, Ogwu A, Riddell L, Luzzi G, Chen F, Balachandran T, Heckerman D, Stryhn A, Edwards A, Ndung'u T, Walker BD, Buus S, Goulder P, Matthews PC. 2013. Nef-specific CD8+ T cell responses contribute to HIV-1 immune control. PLoS One 8:e73117. doi: 10.1371/journal.pone.0073117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Salgado M, Brennan T, O'Connell K, Bailey J, Ray S, Siliciano R, Blankson J. 2010. Evolution of the HIV-1 nef gene in HLA-B*57 positive elite suppressors. Retrovirology 7:94. doi: 10.1186/1742-4690-7-94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Budde ML, Greene JM, Chin EN, Ericsen AJ, Scarlotta M, Cain BT, Pham NH, Becker EA, Harris M, Weinfurter JT, O'Connor SL, Piatak M Jr, Lifson JD, Gostick E, Price DA, Friedrich TC, O'Connor DH. 2012. Specific CD8+ T cell responses correlate with control of simian immunodeficiency virus replication in Mauritian cynomolgus macaques. J Virol 86:7596–7604. doi: 10.1128/JVI.00716-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Khalid M, Yu H, Sauter D, Usmani S, Schmokel J, Feldman J, Gruters R, van der Ende M, Geyer M, Rowland-Jones S, Osterhaus A, Kirchhoff F. 2012. Efficient Nef-mediated downmodulation of TCR-CD3 and CD28 is associated with high CD4+ T cell counts in viremic HIV-2 infection. J Virol 86:4906–4920. doi: 10.1128/JVI.06856-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ilyinskii PO, Daniel MD, Simon MA, Lackner AA, Desrosiers RC. 1994. The role of upstream U3 sequences in the pathogenesis of simian immunodeficiency virus-induced AIDS in rhesus monkeys. J Virol 68:5933–5944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Garcia JV, Miller AD. 1991. Serine phosphorylation-independent downregulation of cell surface CD4 by nef. Nature 350:508–511. doi: 10.1038/350508a0. [DOI] [PubMed] [Google Scholar]
- 46.Foster JL, Anderson SJ, Frazier AL, Garcia JV. 1994. Specific suppression of human CD4 surface expression by Nef from the pathogenic simian immunodeficiency virus SIVmac239open. Virology 201:373–379. doi: 10.1006/viro.1994.1303. [DOI] [PubMed] [Google Scholar]
- 47.Heigele A, Schindler M, Gnanadurai C, Leonard J, Collins K, Kirchhoff F. 2012. Down-modulation of CD8αβ is a fundamental activity of primate lentiviral Nef proteins. J Virol 86:36–48. doi: 10.1128/JVI.00717-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bell I, Schaefer TM, Trible RP, Amedee A, Reinhart TA. 2001. Down-modulation of the costimulatory molecule, CD28, is a conserved activity of multiple SIV Nefs and is dependent on histidine 196 of Nef. Virology 283:148–158. doi: 10.1006/viro.2001.0872. [DOI] [PubMed] [Google Scholar]
- 49.Zhang F, Wilson SJ, Landford WC, Virgen B, Gregory D, Johnson MC, Munch J, Kirchhoff F, Bieniasz PD, Hatziioannou T. 2009. Nef proteins from simian immunodeficiency viruses are tetherin antagonists. Cell Host Microbe 6:54–67. doi: 10.1016/j.chom.2009.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Jia B, Serra-Moreno R, Neidermyer W, Rahmberg A, Mackey J, Fofana I, Johnson W, Westmoreland S, Evans D. 2009. Species-specific activity of SIV Nef and HIV-1 Vpu in overcoming restriction by Tetherin/BST2. PLoS Pathog 5:e1000429. doi: 10.1371/journal.ppat.1000429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Schwartz O, Marechal V, Le Gall S, Lemonnier F, Heard J. 1996. Endocytosis of major histocompatibility complex class I molecules is induced by the HIV-1 Nef protein. Nat Med 2:338–342. doi: 10.1038/nm0396-338. [DOI] [PubMed] [Google Scholar]
- 52.Stumptner-Cuvelette P, Morchoisne S, Dugast M, Le Gall S, Raposo G, Schwartz O, Benaroch P. 2001. HIV-1 Nef impairs MHC class II antigen presentation and surface expression. Proc Natl Acad Sci U S A 98:12144–12149. doi: 10.1073/pnas.221256498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Chen N, McCarthy C, Drakesmith H, Li D, Cerundolo V, McMichael A, Screaton G, Xu X. 2006. HIV-1 downregulates the expression of CD1d via Nef. Eur J Immunol 36:278–286. doi: 10.1002/eji.200535487. [DOI] [PubMed] [Google Scholar]
- 54.Chaudhry A, Das S, Hussain A, Mayor S, George A, Bal V, Jameel S, Rath S. 2005. The Nef protein of HIV-1 induces loss of cell surface costimulatory molecules CD80 and CD86 in APCs. J Immunol 175:4566–4574. doi: 10.4049/jimmunol.175.7.4566. [DOI] [PubMed] [Google Scholar]
- 55.Trautz B, Pierini V, Wombacher R, Stolp B, Chase AJ, Pizzato M, Fackler OT. 2016. The antagonism of HIV-1 Nef to SERINC5 particle infectivity restriction involves the counteraction of virion-associated pools of the restriction factor. J Virol 90:10915–10927. doi: 10.1128/JVI.01246-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Usami Y, Wu Y, Gottlinger HG. 2015. SERINC3 and SERINC5 restrict HIV-1 infectivity and are counteracted by Nef. Nature 526:218–223. doi: 10.1038/nature15400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Rosa A, Chande A, Ziglio S, De Sanctis V, Bertorelli R, Goh SL, McCauley SM, Nowosielska A, Antonarakis SE, Luban J, Santoni FA, Pizzato M. 2015. HIV-1 Nef promotes infection by excluding SERINC5 from virion incorporation. Nature 526:212–217. doi: 10.1038/nature15399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Matheson NJ, Sumner J, Wals K, Rapiteanu R, Weekes MP, Vigan R, Weinelt J, Schindler M, Antrobus R, Costa AS, Frezza C, Clish CB, Neil SJ, Lehner PJ. 2015. Cell surface proteomic map of HIV infection reveals antagonism of amino acid metabolism by Vpu and Nef. Cell Host Microbe 18:409–423. doi: 10.1016/j.chom.2015.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Greenberg M, Bronson S, Lock M, Neumann M, Pavlakis G, Skowronski J. 1997. Colocalization of HIV-1 Nef with the AP-2 adaptor protein complex correlates with Nef-induced CD4 downregulation. EMBO J 16:6964–6976. doi: 10.1093/emboj/16.23.6964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Swigut T, Shohdy N, Skowronski J. 2001. Mechanism for down-regulation of CD28 by Nef. EMBO J 20:1593–1604. doi: 10.1093/emboj/20.7.1593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Brenner M, Munch J, Schindler M, Wildum S, Stolte N, Stahl-Hennig C, Fuchs D, Matz-Rensing K, Franz M, Heeney J, Ten Haaft P, Swigut T, Hrecka K, Skowronski J, Kirchhoff F. 2006. Importance of the N-distal AP-2 binding element in Nef for simian immunodeficiency virus replication and pathogenicity in rhesus macaques. J Virol 80:4469–4481. doi: 10.1128/JVI.80.9.4469-4481.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Zhang F, Landford W, Ng M, McNatt M, Bieniasz P, Hatziioannou T. 2011. SIV Nef proteins recruit the AP-2 complex to antagonize Tetherin and facilitate virion release. PLoS Pathog 7:e1002039. doi: 10.1371/journal.ppat.1002039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Ren X, Park SY, Bonifacino JS, Hurley JH. 2014. How HIV-1 Nef hijacks the AP-2 clathrin adaptor to downregulate CD4. Elife 3:e01754. doi: 10.7554/eLife.01754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Serra-Moreno R, Zimmermann K, Stern L, Evans D. 2013. Tetherin/BST-2 antagonism by Nef depends on a direct physical interaction between Nef and tetherin, and on clathrin-mediated endocytosis. PLoS Pathog 9:e1003487. doi: 10.1371/journal.ppat.1003487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Lindwasser OW, Smith WJ, Chaudhuri R, Yang P, Hurley JH, Bonifacino JS. 2008. A diacidic motif in human immunodeficiency virus type 1 Nef is a novel determinant of binding to AP-2. J Virol 82:1166–1174. doi: 10.1128/JVI.01874-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Arias JF, Heyer LN, von Bredow B, Weisgrau KL, Moldt B, Burton DR, Rakasz EG, Evans DT. 2014. Tetherin antagonism by Vpu protects HIV-infected cells from antibody-dependent cell-mediated cytotoxicity. Proc Natl Acad Sci U S A 111:6425–6430. doi: 10.1073/pnas.1321507111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Arias JF, Colomer-Lluch M, von Bredow B, Greene JM, MacDonald J, O'Connor DH, Serra-Moreno R, Evans DT. 2016. Tetherin antagonism by HIV-1 group M Nef proteins. J Virol doi: 10.1128/JVI.01465-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.von Bredow B, Arias JF, Heyer LN, Gardner MR, Farzan M, Rakasz EG, Evans DT. 2015. Envelope glycoprotein internalization protects human and simian immunodeficiency virus-infected cells from antibody-dependent cell-mediated cytotoxicity. J Virol 89:10648–10655. doi: 10.1128/JVI.01911-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Weiler AM, Das A, Akinyosoye O, Cui S, O'Connor SL, Scheef EA, Reed JS, Panganiban AT, Sacha JB, Rakasz EG, Friedrich TC, Maness NJ. 2016. Acute viral escape selectively impairs Nef-mediated major histocompatibility complex class I downmodulation and increases susceptibility to antiviral T cells. J Virol 90:2119–2126. doi: 10.1128/JVI.01975-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Wilker PR, Dinis JM, Starrett G, Imai M, Hatta M, Nelson CW, O'Connor DH, Hughes AL, Neumann G, Kawaoka Y, Friedrich TC. 2013. Selection on haemagglutinin imposes a bottleneck during mammalian transmission of reassortant H5N1 influenza viruses. Nat Commun 4:2636. doi: 10.1038/ncomms3636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.O'Doherty U, Swiggard W, Malim M. 2000. Human immunodeficiency virus type 1 spinoculation enhances infection through virus binding. J Virol 74:10074–10080. doi: 10.1128/JVI.74.21.10074-10080.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Chaudhuri R, Lindwasser OW, Smith WJ, Hurley JH, Bonifacino JS. 2007. Downregulation of CD4 by human immunodeficiency virus type 1 Nef is dependent on clathrin and involves direct interaction of Nef with the AP2 clathrin adaptor. J Virol 81:3877–3890. doi: 10.1128/JVI.02725-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Rodionov DG, Honing S, Silye A, Kongsvik TL, von Figura K, Bakke O. 2002. Structural requirements for interactions between leucine-sorting signals and clathrin-associated adaptor protein complex AP3. J Biol Chem 277:47436–47443. doi: 10.1074/jbc.M207149200. [DOI] [PubMed] [Google Scholar]
- 74.Prado J, Honeyborne I, Brierley I, Puertas M, Martinez-Picado J, Goulder P. 2009. Functional consequences of human immunodeficiency virus escape from an HLA-B*13-restricted CD8+ T-cell epitope in p1 Gag protein. J Virol 83:1018–1025. doi: 10.1128/JVI.01882-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Wang Y, Li B, Carlson J, Streeck H, Gladden A, Goodman R, Schneidewind A, Power K, Toth I, Frahm N, Alter G, Brander C, Carrington M, Walker B, Altfeld M, Heckerman D, Allen T. 2008. Protective HLA class I alleles restricting acute-phase CD8+ T cell responses are associated with viral escape mutations located in highly conserved regions of HIV-1. J Virol 83:1845–1855. doi: 10.1128/JVI.01061-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Heigele A, Kmiec D, Regensburger K, Langer S, Peiffer L, Sturzel CM, Sauter D, Peeters M, Pizzato M, Learn GH, Hahn BH, Kirchhoff F. 2016. The potency of Nef-mediated SERINC5 antagonism correlates with the prevalence of primate lentiviruses in the wild. Cell Host Microbe 20:381–391. doi: 10.1016/j.chom.2016.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Lauck M, Switzer WM, Sibley SD, Hyeroba D, Tumukunde A, Weny G, Shankar A, Greene JM, Ericsen AJ, Zheng H, Ting N, Chapman CA, Friedrich TC, Goldberg TL, O'Connor DH. 2014. Discovery and full genome characterization of a new SIV lineage infecting red-tailed guenons (Cercopithecus ascanius schmidti) in Kibale National Park, Uganda. Retrovirology 11:55. doi: 10.1186/1742-4690-11-55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Bimber B, Dudley D, Lauck M, Becker E, Chin E, Lank S, Grunenwald H, Caruccio N, Maffitt M, Wilson N, Reed J, Sosman J, Tarosso L, Sanabani S, Kallas E, Hughes A, O'Connor D. 2010. Whole-genome characterization of human and simian immunodeficiency virus intrahost diversity by ultradeep pyrosequencing. J Virol 84:12087–12092. doi: 10.1128/JVI.01378-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Clack NG, O'Connor DH, Huber D, Petreanu L, Hires A, Peron S, Svoboda K, Myers EW. 2012. Automated tracking of whiskers in videos of head fixed rodents. PLoS Comput Biol 8:e1002591. doi: 10.1371/journal.pcbi.1002591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Wilson N, Reed J, Napoe G, Piaskowski S, Szymanski A, Furlott J, Gonzalez E, Yant L, Maness N, May G, Soma T, Reynolds M, Rakasz E, Rudersdorf R, McDermott A, O'Connor D, Friedrich T, Allison D, Patki A, Picker L, Burton D, Lin J, Huang L, Patel D, Heindecker G, Fan J, Citron M, Horton M, Wang F, Liang X, Shiver J, Casimiro D, Watkins D. 2006. Vaccine-induced cellular immune responses reduce plasma viral concentrations after repeated low-dose challenge with pathogenic simian immunodeficiency virus SIVmac239. J Virol 80:5875–5885. doi: 10.1128/JVI.00171-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Maness N, Wilson N, Reed J, Piaskowski S, Sacha J, Walsh A, Thoryk E, Heidecker G, Citron M, Liang X, Bett A, Casimiro D, Watkins D. 2010. Robust, vaccine-induced CD8+ T lymphocyte response against an out-of-frame epitope. J Immunol 184:67–72. doi: 10.4049/jimmunol.0903118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Sacha J, Watkins D. 2010. Synchronous infection of SIV and HIV in vitro for virology, immunology and vaccine-related studies. Nat Protoc 5:239–246. doi: 10.1038/nprot.2009.227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Yuste E, Reeves JD, Doms RW, Desrosiers RC. 2004. Modulation of Env content in virions of simian immunodeficiency virus: correlation with cell surface expression and virion infectivity. J Virol 78:6775–6785. doi: 10.1128/JVI.78.13.6775-6785.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Postler TS, Bixby JG, Desrosiers RC, Yuste E. 2014. Systematic analysis of intracellular trafficking motifs located within the cytoplasmic domain of simian immunodeficiency virus glycoprotein gp41. PLoS One 9:e114753. doi: 10.1371/journal.pone.0114753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, Gingeras TR. 2013. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29:15–21. doi: 10.1093/bioinformatics/bts635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Li B, Dewey C. 2011. RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinformatics 12:323. doi: 10.1186/1471-2105-12-323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Monjure CJ, Tatum CD, Panganiban AT, Arainga M, Traina-Dorge V, Marx PA Jr, Didier ES. 2014. Optimization of PCR for quantification of simian immunodeficiency virus genomic RNA in plasma of rhesus macaques (Macaca mulatta) using armored RNA. J Med Primatol 43:31–43. doi: 10.1111/jmp.12088. [DOI] [PMC free article] [PubMed] [Google Scholar]