

Abstract
Background
Thrombomodulin (TM), a transmembrane glycoprotein highly expressed in endothelial cells (ECs), is a potent anticoagulant maintaining circulation homeostasis. Under inflammatory states, TM expression is drastically reduced in ECs while vascular smooth muscle cells (VSMCs) show a robust expression of TM. The functional role of TM in VSMCs remains elusive.
Methods
We examined the role of TM in VSMCs activities in human aortic VSMCs stimulated with platelet-derived growth factor-BB (PDGF-BB). Using rat embryonic aorta-derived A7r5 VSMCs which do not express TM, the role of the chondroitin sulfate (CS) moiety of TM in VSMCs was delineated with cells expressing wild-type TM and the CS-devoid TM mutant.
Results
Expression of TM enhanced cell migration and adhesion/spreading onto type I collagen, but had no effect on cell proliferation. Knocking down TM with short hairpin RNA reduced PDGF-stimulated adhesion and migration of human aortic VSMCs. In A7r5 cells, TM-mediated cell adhesion was eradicated by pretreatment with chondroitinase ABC which degrades CS moiety. Furthermore, the TM mutant (TMS490, 492A) devoid of CS moiety failed to increase cell adhesion, spreading or migration. Wild-type TM, but not TMS490, 492A, increased focal adhesion kinase (FAK) activation during cell adhesion, and TM-enhanced cell migration was abolished by a function-blocking anti-integrin β1 antibody.
Conclusion
Chondroitin sulfate modification is required for TM-mediated activation of β1-integrin and FAK, thereby enhancing adhesion and migration activity of VSMCs.
Electronic supplementary material
The online version of this article (10.1186/s12929-018-0415-7) contains supplementary material, which is available to authorized users.
Keywords: Thrombomodulin, Chondroitin sulfate moiety, Migration, Adhesion, Vascular smooth muscle cells
Background
Migration and proliferation of vascular smooth muscle cells (VSMCs) are key events in the intimal thickening of atherosclerosis and neointimal formation induced by vascular injury [1]. During vascular remodeling, VSMCs in the media transform from a quiescent, contractile phenotype into an activated, synthetic phenotype and migrate to the intima. VSMCs in the intima may undergo replication, active production of extracellular matrix (ECM) or even secretion of pro-inflammatory factors, resulting in intimal thickening and atherosclerosis progression. Aberrant migration and proliferation of VSMCs can be triggered by excessive secretion of various cytokines and growth factors [2].
Thrombomodulin (TM), a transmembrane glycoprotein in endothelial cells (ECs), is a critical player in maintaining vascular thromboresistance. TM acts as a cofactor in protein C activation through regulating thrombin substrate affinity and plays a critical role in the anticoagulant pathway [3]. Under physiological conditions, TM is highly expressed on the endothelial surface and is considered an endothelial marker. In disease states associated with inflammation, such as atherosclerosis and vein graft thrombosis, TM expression in ECs is markedly downregulated, leading to thrombotic complications and an increase in local inflammation and matrix proteolysis [4, 5]. Interestingly, in advanced atherosclerotic plaques, TM is detected in VSMCs of both the intima and the media [6, 7]. Thus, intimal VSMCs were proposed to be a relevant source of TM and form a surface relatively resistant to thrombosis. We previously found that platelet-derived growth factor (PDGF)-BB, the most potent mitogen and chemoattractant of VSMCs in vitro, induces TM expression in VSMCs through Src kinase and PI3-kinase/Akt/mTOR-dependent signaling pathway. Furthermore, PDGF-BB and TM co-localize in the media and the neointima following carotid ligation-induced injury [8]. Similarly, TM expression is induced in distension-caused urinary bladder injuries and is involved in the PDGF-stimulated migration of urinary bladder smooth muscle cells [9]. Recent evidence further showed that the forced expression of TM in TM-deficient A2058 melanoma cells enhances cell adhesion, migration, and invasion through the activation of focal adhesion kinase (FAK) [10].
TM consists of five domains, including an N-terminal lectin-like domain, followed by six epidermal growth factor (EGF)-like repeats, a serine/threonine–rich domain, a transmembrane domain, and a cytoplasmic domain [11]. The activation of protein C requires thrombin binding to the EGF-like domain and is enhanced by a second site binding to the chondroitin sulfate (CS) moiety attached to the serine/threonine–rich domain [12]. In addition to an anticoagulant function, previous studies have revealed multiple biological roles for TM including anti-inflammation [13], cell-cell adhesion [14], cell-matrix adhesion and migration [10], cell proliferation, and angiogenesis [15], which are linked to different domains of TM. While TM peptides consisting of EGF-like repeats and serine/threonine-rich domains stimulate cell proliferation, migration, and angiogenesis [15], whether the CS moiety of TM participates in these processes remains unclear. Previous studies have suggested that proteoglycans containing CS play a significant role in regulating cell adhesion and migration in a variety of cell types [16–19]. The CS moiety on CD44, a transmembrane glycoprotein, modulates cell migration on type I collagen in melanoma cells, which is abolished by chondroitinase ABC (ChABC), an enzyme that cleaves CS from the core protein [16]. Similarly, CD44-associated CS proteoglycan mediates EC migration and adhesion on fibrinogen [17]. In addition, CS proteoglycans regulate cell growth, differentiation, and migration of neural precursor cells [20].
We hypothesized that the CS moiety of TM has functional roles in VSMCs adhesion and migration analogous to the CS of other membrane proteins. We tested our hypothesis by examining the effect of ChABC and the CS-devoid TM mutant expression on VSMCs adhesion and migration. Our results indicate that TM expression promotes cell spreading, adhesion, and migration involving the activation of β1-integrin and FAK and the CS moiety is required for these processes.
Methods
Cell culture
Human aortic VSMCs (HASMCs) purchased from Cascade Biologics (Portland, OR, USA) were amplified in medium 231 with smooth muscle growth supplement (Cascade Biologics). A7r5 cells were purchased from the American Type Culture Collection (Manassas, VA, USA). HASMCs and A7r5 cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM, Life Technologies, Inc.) containing 10% fetal bovine serum (FBS, HyClone), 25 mM HEPES, Glutamax, 100 U/ml penicillin, and 100 μg/ml streptomycin at 37 °C under 5% CO2 humidified atmosphere. HASMCs were used between 3 and 6 passages.
Lentivirus-based knockdown of TM expression
pLKO.1-puro-shTM, pCMV-ΔR8.91 and pMD.G were obtained from National RNAi core facility (Academia Sinica, Taiwan). The lentivirus stock was prepared as previously described [21]. HASMCs were infected with a viral stock in complete medium for 5 days and were processed for the subsequent experiments.
Transient transfection
A7r5 cells at 40-60% confluence were transfected with Fugene HD (Roche Applied Science) at a 2.5:1 ratio to plasmid DNA. The amount of DNA was adjusted if necessary. The medium was replaced 12 or 24 h later, and the cells were harvested 48 or 72 h later. Transfection efficiency was consistently 30–50% as determined by green fluorescent protein expression.
Immunoblotting analysis
Following treatments, VSMCs were lysed and immunoblotting was performed as previously described [22]. TM, FAK phosphorylation (Tyr397), and FAK expression were detected by incubation with a primary antibody for TM (mouse clone D-3, Santa Cruz Biotechnology), pFAK-Y397 (rabbit polyclonal antiserum, Upstate) or FAK (rabbit polyclonal antiserum, BD Transduction Lab.), followed by horseradish peroxidase (HRP)-conjugated goat anti-mouse or anti-rabbit IgG (Vector Laboratories). Protein bands were visualized by enhanced chemiluminescence using ECL-Plus (PerkinElmer, Inc.) and quantified densitometrically. Expression of α-tubulin (mouse clone B-5-1-2, Sigma) or β-actin (mouse clone AC-15, Sigma) was used as loading control.
Site-directed mutagenesis
TM DNA within the pEGFP-N1 vector was mutated at serine 490 and 492 to alanine using the QuikChange™ site-directed mutagenesis kit from Stratagene (La Jolla, CA, USA) following the manufacturer’s instructions. DNA sequencing confirmed the mutations.
Adhesion assay
The ability of VSMCs to adhere to ECM molecules was quantified as previously described [23]. Clear-bottom 96-well plates were coated with 40 μg/ml collagen type I or 5 μg/ml fibronectin in PBS for 16 h at 4 °C. Non-specific binding was blocked with 2% BSA in PBS. HASMCs were stimulated with 10 ng/ml human recombinant PDGF-BB (R&D Systems) for 6 h before adhesion assay. A7r5 cells were transfected with 1 μg of pEGFP, pEGFP-TM, or pEGFP-TMS490,492A for 12-24 h and cultured in regular medium for 36-48 h. Cells were trypsinized, washed, and treated both with and without ChABC (0.5 U/ml) for 1 h at 37 °C. The cells (104 cells/100 μl) were added to the coated 96-well plates and incubated for 30 min at 37 °C. Non-adherent cells were removed by washing with DMEM. Adherent cells were fixed with 4% paraformaldehyde, stained with 0.1% crystal violet for 25 min, and solubilized with 0.5% triton X-100. The optical density at 595 nm (OD 595) was determined using a microplate reader.
Wound healing assay
Fully confluent cell cultures were wounded with a 100-μl sterile pipette tip as previously described [24]. Wound healing assay was conducted in a CO2 incubator. Each scratch was randomly photographed at four separate sites along the length with time-lapse video microscopy over the succeeding 24 h. Migration velocity of A7r5 cells expressing GFP, TM-GFP, or TMS490, 492A-GFP across the wound edges was measured manually. In some experiments, a hamster IgM (clone Ha2/5, BD Pharmingen) or the control IgM (G235-1) was used. The migration rate of HASMCs was determined by the ratio of the wound area at 24 h to the initial wound area.
Invasion assay
To determine the invasive ability of A7r5 cells, the upper sides of the transwell polycarbonate membrane filters, with 8-μm pore size (Corning Inc.), were coated with 40 μg/ml collagen type I or 5 μg/ml fibronectin. Cells (20,000/well) in serum-free medium were seeded in the upper chamber, and the bottom chamber contained medium with 10% FBS. Cells were incubated for 16 h at 37 °C. Following incubation, the cells that had invaded and attached to the lower surface of the membrane were fixed with 4% paraformaldehyde and stained with 4′,6-diamidino-2-phenylindole (DAPI, diluted 1000× in PBS) (Sigma-Aldrich). Cell number was counted in five random fields per membrane under an inverted microscope.
TM and pFAK immunofluorescence
HASMCs were serum-starved for 36 h and treated with or without PDGF-BB (10 ng/ml) for 6 h. A7r5 cells were transfected with pEGFP, pEGFP-TM or pEGFP-TMS490,492A (1 μg) for 12 h and cultured in fresh medium for 36-60 h. Cells were detached and seeded onto 40 μg/ml type I collagen-coated cover slip for 3 h. The cells were fixed for 10 min with 4% paraformaldehyde and permeabilized for 15 min with 0.1% Triton X-100 in phosphate buffered saline (PBS)/BSA. The cells were incubated at 4 °C overnight with anti-TM (mouse clone D-3, Santa Cruz Biotechnology) or anti-pFAKY397 antibody (BD611807, 1:100 in 3% BSA/PBS), followed by Alexa 546–conjugated goat anti–mouse IgG (Molecular Probes; 1:200). The cell nuclei were stained with DAPI, and cells were observed under a confocal microscope (Leica TCS SP5 II).
Ki67 immunofluorescence staining
A7r5 cells were transfected with pEGFP, pEGFP-TM or pEGFP-TMS490,492A for 12 h and cultured for 12 h. Following a 48 h serum starvation, cells were treated with 10 ng/ml PDGF-BB for 24 h. The cells were fixed for 10 min with 4% paraformaldehyde and permeabilized for 15 min with 0.1% Triton X-100 in PBS/BSA. The cells were incubated for 1 h with an anti-Ki-67 antibody (Novocastra, NCL-Ki67-MM1, 1:100 in 3% BSA/PBS), followed by Alexa 546–conjugated goat anti–mouse IgG (Molecular Probes; 1:100). The nuclei were stained with DAPI, and cells were observed under an inverted fluorescence microscope (Leica IRE-2).
Construction of lentivirus-based GFP-tagged TM and TMS490, 492A
Human TM and TMS490, 492A were amplified and subcloned from pEGFP-N1-TM vector [14] and pEGFP-N1-TMS490, 492A vector. TM-EGFP and TMS490, 492A-EGFP fragments were cut from pEGFP-N1 using EcoRI restriction endonuclease. pLVX-TM-GFP-puro and pLVX-TMS490, 492A-GFP-puro vectors were generated by subcloning the TM-EGFP and TMS490, 492A-EGFP into pLVX-IRES-puro (Clontech) vector pre-treated with EcoRI. Sequences of both constructs were confirmed by DNA sequencing.
Establishing stable cell lines
For lentivirus production, plasmids pXPAS2, pMD2G, and pLVX-TM-GFP-puro (or pLVX-TMS490, 492A-GFP-puro) were co-transfected into 293 T cell with Fugene HD, and supernatants containing lentiviral particles were collected at 48, 72, and 96 h following transfection. A7r5 cells at 50-60% confluence were transduced with lentivirus-containing supernatants. At 48 h post-transduction, 1 μg/ml puromycin was added to select cells stably expressing TM-GFP, TMS490, 492A-GFP or vehicle.
Statistical analysis
Data are presented as mean ± SEM of n independent experiments. Statistical analysis was performed with Student’s t test for comparison between two groups. For comparisons among multiple groups, one-way ANOVA, followed by Dunnett multiple comparison was used. P values smaller than 0.05 were considered significant.
Results
VSMCs expressed TM both with and without chondroitin sulfate (CS) moiety
We previously reported that HASMCs express TM under PDGF stimulation but not at quiescence [8]. On the contrary, A7r5 cells did not express TM mRNA in the presence or absence of PDGF treatment (Additional file 1: Supplementary data, Table S1 and Figure S1). Therefore, we applied both HASMCs and A7r5 cells to examine the functional roles of TM in VSMCs. We first examined TM expression in PDGF-stimulated HASMCs and A7r5 cells transfected with TM cDNA. Compared to quiescent cells (Fig. 1a, Lane 1, serum starvation for 48 h), PDGF treatment profoundly increased TM expression in HASMCs. TM mainly existed as a ~ 100 kDa form, but a diffused, high-molecular-mass band of approximately 180-200 kDa was also present (Fig. 1a, Lane 2). TM possesses four potential sites for O-linked glycosylation, which supports the post-translational attachment of a CS moiety, a stretch of approximately 20 repeating disaccharide units with a trisaccharide terminus [25]. HASMCs treated with ChABC (0.5 U/ml) substantially reduced the high-molecular-mass form (Fig. 1a, Lane 3), indicating that TM expressed in HASMCs was modified by CS.
Fig. 1.
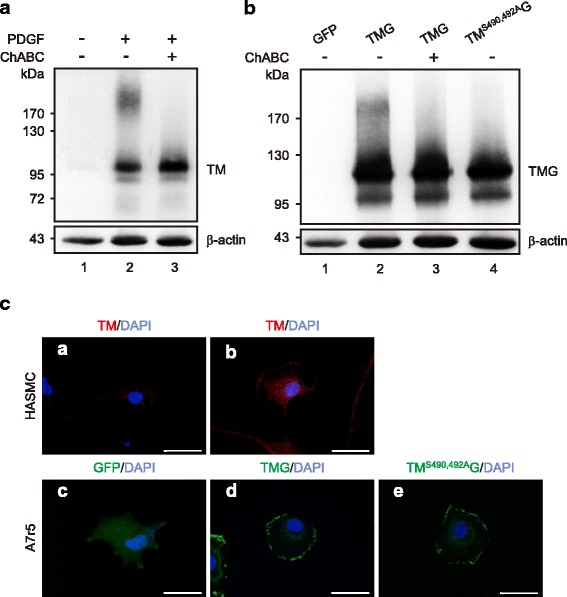
Thrombomodulin (TM) expression, glycosylation, and localization in HASMCs and A7r5 transfected with TM cDNA constructs. a HASMCs were serum-starved for 48 h (Lane 1) and then stimulated with PDGF-BB (10 ng/ml) for 6 h without (Lane 2) or with (Lane 3) ChABC (0.5 U/ml) treatment for 1 h. b A7r5 cells were transfected with pEGFP (Lane 1), pEGFP-TM (Lane 2) or pEGFP-TMS490,492A (Lane 4). The cells overexpressing TM were treated with ChABC before harvest (Lane 3). Western blotting was performed with antibodies specific to either human TM (Top) or β-actin (Bottom). c The localization of TM in VSMCs. a and b, HASMCs were serum-starved for 36 h and treated without (a) or with (b) PDGF-BB for 6 h. c, d, and e, A7r5 cells transfected with pEGFP (c), pEGFP-TM (d) or pEGFP- TMS490,492A (e) were fixed, processed for TM immunofluorescence (a and b), and examined under a confocal microscope. Scale bar: 50 μm
The CS moiety of TM is indispensable for TM to perform its anticoagulant activity [12]. However, additional roles for the CS moiety of TM have not been defined. To address this question, we generated the TM mutation of Ser490 and Ser492 to Ala, which was shown to block the CS modification of TM [26]. Transfection with pEGFP-TMS490, 492A had no effect on TM expression (Fig. 1b, Lanes 2 and 4). A7r5 cells expressing TM-GFP exhibited a pattern similar to that of PDGF-stimulated HASMCs with both bands showing higher molecular mass resulting from the GFP tag. The high-molecular-mass bands were eliminated with ChABC treatment. In addition, A7r5 cells expressing TMS490, 492A exhibited only the low-molecular-mass band. These results suggested that TM underwent CS modification in both HASMCs and A7r5 cells through serine 490 and 492. The cellular localization of TM in PDGF-stimulated HASMCs and TM-expressing A7r5 cells was examined. TM was mainly localized on the plasma membrane (Fig. 1c, b & d) and CS-deficient TMS490, 492A displayed a similar distribution to wild-type TM (Fig. 1c, e).
The CS moiety mediates TM-enhanced VSMCs adhesion and spreading
TM functions as a Ca2+-dependent cell-to-cell adhesion molecule [14] and was recently found to bind to fibronectin through the lectin-like domain [10]. To assess the effect of TM on VSMCs adhesion to the ECM, we performed cell adhesion assay on plates coated with type I collagen which is a major ECM in the neointima [27]. HASMCs were infected with a lentiviral vector encoding luciferase shRNA (shLuc) or TM shRNA (shTM). The infection with shTM, but not shLuc, for 3 to 5 days markedly decreased TM expression in HASMCs, showing the specificity of the engineered lentivirus (Fig. 2a). The adhesion of TM-depleted HASMCs was markedly suppressed (41.0% ± 9.6% of control, P < 0.05) (Fig. 2b). Cell surface CD44-related CS proteoglycan was reported to mediate melanoma cell adhesion and migration on type I collagen [16]. Therefore, we hypothesized that TM affects the adhesive and/or migratory capabilities of VSMCs through the CS component and examined it with A7r5 adhesion onto type I collagen. Expression of TM-GFP significantly increased A7r5 adhesion (Fig. 2c, 163.5% ± 15.9% of GFP, P < 0.01), which was reversed by ChABC treatment (26.3% ± 5.7% of TM-GFP, P < 0.05). Moreover, expression of TMS490, 492A-GFP had no effect on cell adhesion onto type I collagen. To examine whether TM-enhanced cell adhesion of VSMCs is specific to type I collagen, we compared A7r5 cells adhesion to type I collagen-, fibronectin-, and non-coated surface. TM expression markedly enhanced cell adhesion to both ECM molecules with stronger effect detected when fibronectin was used. In contrast, expression of TMS490, 492A-GFP inhibited cell adhesion onto fibronectin. As a comparison, expression of another TM mutant with lectin-like domain deletion (TMΔL-GFP) enhanced cell adhesion to both molecules less effectively compared to TM-GFP expression (Additional file 1: Supplementary data, Figure S2). These results indicated that TM promotes VSMCs adhesion and the CS moiety mediates the effect. Next, we examined whether TM affects cell spreading. A7r5 cells expressing GFP, TM-GFP or TMS490, 492A-GFP were re-suspended and allowed to adhere onto immobilized type I collagen for 30 min. Cells expressing TM-GFP exhibited accelerated spreading and appeared flattened with extended lamellipodia. In contrast, most GFP- and TMS490, 492A-GFP-expressing cells remained round. The spreading area of TM-expressing cells was increased (2.3 ± 0.18 fold control), whereas cells expressing CS-deficient TMS490, 492A showed no difference in spreading area (Fig. 2d).
Fig. 2.
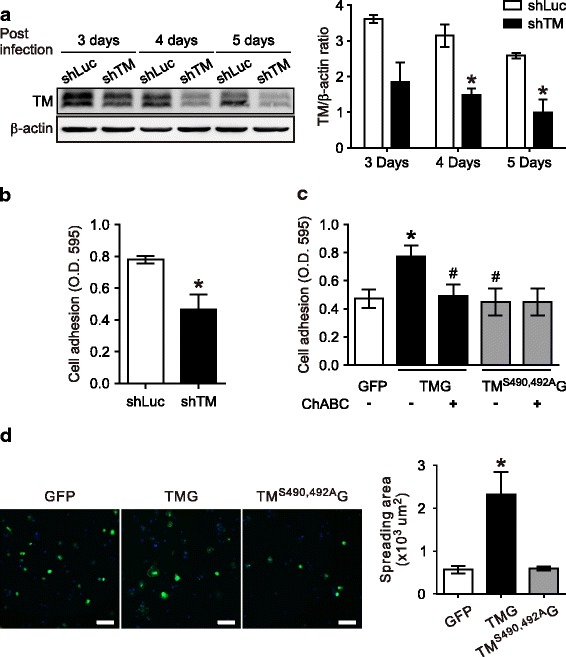
Thrombomodulin enhanced VSMCs adhesion and spreading to type I collagen in a chondroitin sulfate-dependent manner. a HASMCs were infected with lentiviral shRNA constructs targeting luciferase (shLuc) or TM (shTM) for 1-3 days, serum-starved for 2 days, and treated with PDGF for 6 h. TM expression levels were analyzed with Western blotting using β-actin as the loading control. The top panel shows a representative result and the bottom panel summarizes the results of three independent experiments expressed as mean ± SEM. b HASMCs infected with lentivirus carrying luciferase or TM shRNA were seeded on a 96-well plate coated with type I collagen, allowed to adhere for 0.5 h at 37 °C, and stained with crystal violet. Results were obtained from 4 independent experiments and expressed as mean ± SEM, *P < 0.05 compared with shLuc. c and d A7r5 cells transfected with pEGFP, pEGFP-TM, or pEGFP-TMS490,492A were cultured for 36 h. In (c), cells were treated with or without ChABC (0.5 U/ml) for 1 h before adhesion assay. Results were obtained from six independent experiments and expressed as mean ± SEM. In (d), cells were detached, seeded to type I collagen-coated dish for 30 min, and processed for fluorescence microscopy. The left panel shows representative results for cells expressing GFP, TM-GFP, and TMS490,492A-GFP (green) with DAPI nuclear staining; the right panel summarizes the spreading area for each group from at least 3 independent experiments. *P < 0.05, compared with the GFP-expressing group; #P < 0.05, compared with the TM-GFP-expressing group. Scale bar: 100 μm
The CS moiety is required for TM-enhanced VSMCs migration
VSMCs migration across the internal elastic lamella into the intima is a key process in neointima formation [28]. To investigate the role of TM in VSMCs migration, wound healing assay was performed under PDGF treatment for 24 h in HASMCs infected with lentivirus encoding luciferase or TM shRNA. Knocking down TM expression in HASMCs attenuated wound closure compared to the control group (Fig. 3a, TM knockdown: 40.4% ± 6.1%; Luc knockdown: 70.5% ± 9.7%, P < 0.05). To further elucidate the role of the TM CS moiety in VSMCs migration, A7r5 cells expressing TM-GFP, TMS490, 492A-GFP or GFP were analyzed with wound-healing assay. Cells expressing TM-GFP exhibited markedly higher migration velocity (8.6 ± 0.72 μm/h) compared with the GFP control (5.0 ± 0.3 μm/h), whereas cells expressing TMS490, 492A-GFP (6.3 ± 0.9 μm/h) showed no effect on cell mobility (Fig. 3b). We further examined the effect of expressing TM-GFP, TMS490, 492A-GFP, and TMΔL-GFP on A7r5 cells invasion through type I collagen- and fibronectin-coated transwells. Similar to that found with adhesion assay, TM-enhanced cell invasion was more pronounced with fibronectin coating. Expression of TMS490, 492A-GFP inhibited cell invasion through fibronectin-coated wells, whereas TMΔL-GFP was less effective than TM-GFP in enhancing cell invasion (Fig. 3c). These results indicated that the CS moiety mediates TM-promoted mobility of VSMCs.
Fig. 3.
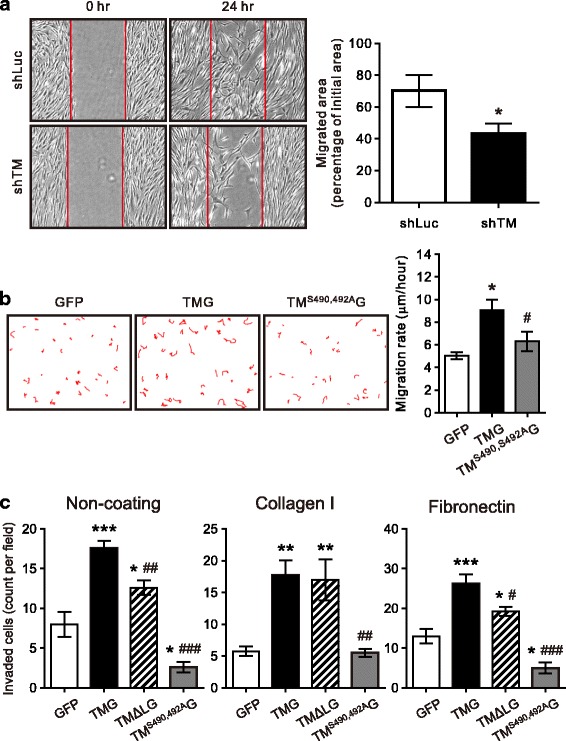
Thrombomodulin is involved in VSMCs migration. a HASMCs infected with a lentivirus encoding luciferase (shLuc) or TM (shTM) shRNA were cultured in regular medium. Cell migration was analyzed by a wound-healing assay. Each monolayer was scratched, incubated for 24 h, and followed by image acquisition. The open area (scratch) was quantified with ImageJ software from 5 different experiments and expressed as mean ± SEM. *P < 0.05 compared with shLuc. b A7r5 cells transfected with pEGFP, pEGFP-TM or pEGFP-TMS490, 492A were cultured for 36 h and serum-starved for 24 h. Wound healing assay was performed under PDGF-BB treatment for 24 h. Cell motility was quantified from 4 independent experiments and expressed as mean ± SEM. c A7r5 cells transfected with pEGFP, pEGFP-TM, pEGFP-TMΔL or pEGFP-TMS490, 492A underwent invasion assay in transwells without coating or coated with type I collagen or fibronectin. Cells seeded in the upper chamber were incubated for 16 h, those attached to the lower surface of the membrane were fixed, stained with DAPI, and GFP-expressing cells counted. *P < 0.05, **P < 0.01, ***P < 0.001, compared with the GFP-expressing group; #P < 0.05, ##P < 0.01, ###P < 0.001, compared with the TM-GFP-expressing group
TM-enhanced VSMCs migration involves β1-integrin and focal adhesion kinase
CS on CD44 is implicated in mediating EC adhesion and migration [17]. Furthermore, another CS-containing glycoprotein, PG-M/versican, binds to β1-integrin, thereby activating FAK and cell adhesion [29]. We next explored whether β1-integrin is involved in TM-increased cell mobility. A7r5 cells expressing GFP-tagged TM or TMS490, 492A were applied to wound healing assay in the presence of PDGF and control IgM or a function-blocking antibody against rat integrin β1 (Ha 2/5). Blocking integrin β1 activation eliminated the migration promoted by TM-GFP, whereas the mobility of cells expressing TMS490, 492A-GFP was not affected (Fig. 4). These results suggest that TM-promoted cell migration is mediated by β1 integrin activation and CS modification is required for this effect.
Fig. 4.
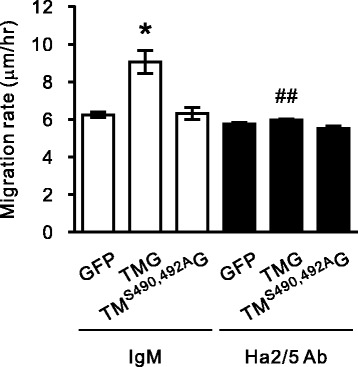
The blockade of β1-integrin function eliminated thrombomodulin-enhanced VSMCs migration. A7r5 cells transfected with pEGFP, pEGFP-TM or pEGFP-TMS490, 492A were cultured for 36 h and serum-starved for 24 h. A wound healing assay was performed under PDGF treatment for 24 h after pretreating cells with 20 mg/mL of control IgM or anti-β1 integrin antibody, Ha2/5. Cell motility was quantified from at least 3 different experiments and expressed as mean ± SEM. *P < 0.05, compared with the GFP-expressing group; ##P < 0.01, compared with the TM-GFP-expressing group
We then examined whether TM affects FAK activation. Immunofluorescence of phosphorylated FAK was more intense in TM-GFP-expressing A7r5 cells than cells expressing GFP or TMS490, 492A-GFP and can be seen at focal adhesions of pseudopods (Fig. 5a). Furthermore, FAK phosphorylation was significantly increased in cells stably expressing TM-GFP, whereas cells expressing TMS490, 492A-GFP exhibited no difference from control. In contrast to FAK activation, the expression of FAK was not affected by TM expression in VSMCs (Fig. 5b and c).
Fig. 5.
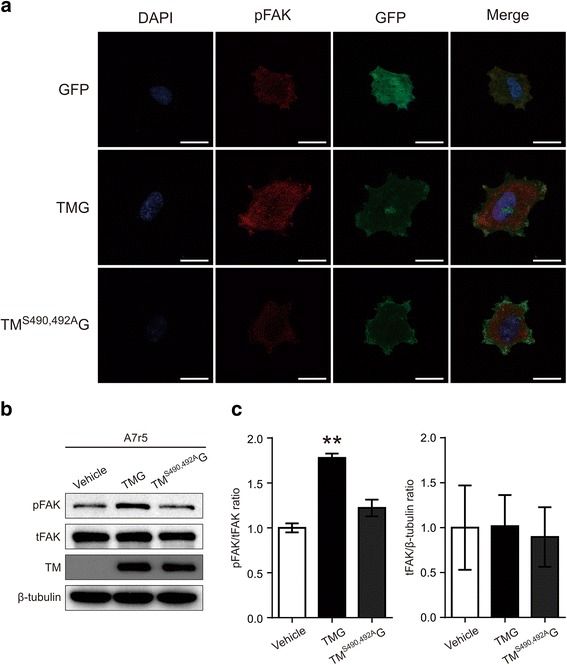
Thrombomodulin (TM) expression increased FAK phosphorylation in A7r5 cells upon adhesion. a A7r5 cells transfected with pEGFP, pEGFP-TM, or pEGFP-TMS490,492A were detached and seeded to type I collagen-coated cover slip for 3 h. Cells were processed for immunofluorescence of FAKY397 phosphorylation, stained with DAPI, and observed under a confocal microscope. Scale bar: 20 μm. b and c A7r5 cells, stably expressing TM-GFP, TMS490, 492A-GFP or GFP, were cultured to confluence. Following detachment, cells were resuspended and attached to type I collagen-coated dish for 3 h. Cell lysates were analyzed for FAK phosphorylation and expression. b shows a representative result of immunoblotting; (c) summarizes the quantitative results of FAK phosphorylation ratio and FAK expression normalized by α-tubulin levels expressed as mean ± SEM (n = 3)
TM expression has no effect on PDGF-stimulated proliferation of VSMCs
TM was shown to decrease the proliferation of tumor cell lines derived from patients with malignant melanoma [30]. On the contrary, recombinant TM containing 6 EGF-like repeats increases DNA synthesis in ECs and VSMCs [6, 15]. To determine whether TM affects VSMCs proliferation, A7r5 cells expressing GFP-tagged TM or CS-deficient TMS490, 492A were stimulated with PDGF and their entry into S phase was indicated by Ki-67 staining. The percentage of Ki-67-positive nuclei was calculated among GFP-expressing cells. In control cells, 15.8% ± 1.6% nuclei were Ki-67-positive and no difference was detected in the percentage of Ki-67-positive cells among GFP-, TM-GFP- or TMS490, 492A-GFP-expressing cells (Additional file 1: Supplementary data, Figure S3A and B). To substantiate these results, the percentage of Ki-67-positive cells was examined in A7r5 cells stably expressing vector only, TM-GFP, and TMS490, 492A-GFP. Likewise, no difference was detected in the percentage of Ki-67-positive cells (Fig. 6a and b) or cell number among three groups (Fig. 6c). These results suggested that TM expression did not affect VSMCs proliferation.
Fig. 6.
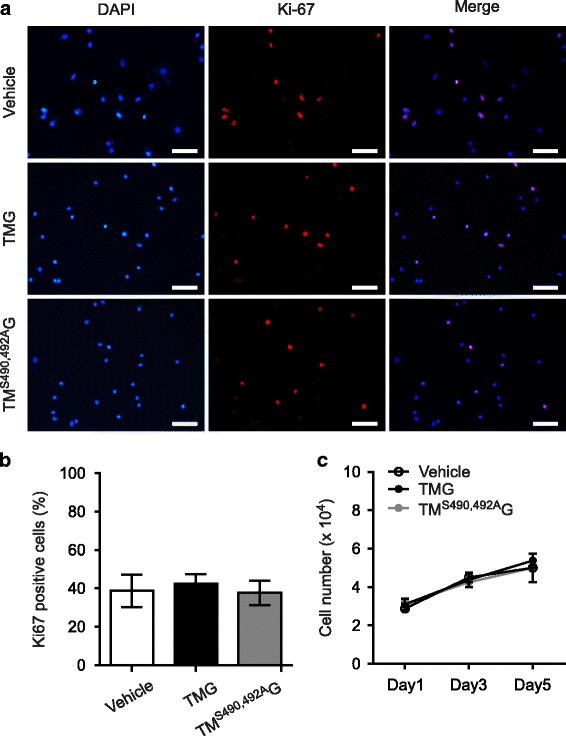
Thrombomodulin expression did not affect PDGF-stimulated proliferation of A7r5 cells. A7r5 cells were transfected with pEGFP, pEGFP-TM, or pEGFP-TMS490,492A, serum-starved for 48 h, and treated with PDGF-BB for 24 h. Cells were fixed and stained with DAPI (blue) and Ki-67(red). a shows representative results; (b) summarizes the ratio of Ki-67-positive cells; (c) summarizes total cell numbers in cells expressing vector alone, TM-GFP, and TMS490, 492A-GFP from at least 3 independent experiments. Results were expressed as mean ± SEM. Scale bar: 100 μm
Discussion
In this study we described a new function of TM in VSMCs. We showed that TM enhances cell adhesion, spreading, and migration of VSMCs. Moreover, we provide evidence showing that the CS moiety at the serine/threonine-rich domain of TM mediates these functions.
Smooth muscle cell migration is an integral part of vascular development, atherogenesis, and injury-induced vascular remodeling [1, 31]. Migration is a multistep process which involves cell protrusion, adhesion, spreading, and contraction [32]. During cell migration, cell adhesion to the substratum and interaction with ECM proteins play a central role in the mobile process. Extensive evidence indicates that soluble TM has roles in cell migration, thereby promoting angiogenesis and wound healing. A soluble recombinant TM fragment consisting of the second and third domains (rTMD23) stimulates ECs migration in vitro and angiogenesis both in vitro and in vivo [15]. Similarly, recombinant TM fragments consisting of the three N-terminal domains (rTMD123) and rTMD23 promote cutaneous wound healing [33]. Both cell spreading and wound closure are slower in TM-deficient keratinocytes compared to their wild-type counterpart, suggesting a role for TM in cell migration [33]. Indeed, TM was shown to mediate PDGF-stimulated migration activity of cultured bladder smooth muscle cells [9]. Furthermore, forced expression of TM promotes cell adhesion and migration of TM-deficient A2058 melanoma cells over a fibronectin-coated surface [10]. Results from this study agree fully with those detected in A2058 cells and suggest that TM enhances VSMCs motility, at least in part, through promoting cell adhesion. It’s noteworthy that TM was reported to inhibit the migration and invasion of different cancer cells [34–37], probably through its role in preventing epithelial-mesenchymal transition [35]. The mechanisms underlying these apparently opposite roles of TM in epithelial cancer cells and VSMCs warrant further investigation. Furthermore, the impact of TM-enhanced VSMCs migration activity in vascular remodeling remains to be determined as previous evidence showed that TM overexpression inhibits arterial overdilation-induced neointima formation [38].
The CS moiety bound to the Ser/Thr-rich domain is indispensable in the anti-coagulant function of TM, but whether the CS moiety of TM performs other functions remained elusive. Our results showing that removing the CS moiety, either through ChABC-catalyzed degradation or via expressing the CS-devoid mutant, eliminated the TM effect on VSMCs adhesion and migration establish a new role for the CS moiety in mediating TM-enhanced cell migration. These results are compatible with previous studies showing that CS proteoglycans facilitate the cell-cell and cell-ECM interaction during cell adhesion and migration [16–19, 39–41]. Given our previous results that the lectin-like domain of TM binds to fibronectin [10], we were surprised to find that CS deficiency, not the lectin-like domain deletion, eliminates VSMCs adhesion and migration on fibronectin (Additional file 1: Figure S2 and Fig. 3C). Furthermore, TM-enhanced cell adhesion and migration were more pronounced with fibronectin coating than collagen coating. These results implicate the CS moiety of TM in binding fibronectin, a notion supported by an earlier study showing that CS of melanoma cells binds to a specific domain of fibronectin and can either act independently or coordinate with an integrin to enhance melanoma cell adhesion to fibronectin [40].
CS proteoglycans coordinate with integrins to function in cell adhesion and migration [39–41]. Reduction of endogenous CS proteoglycan expression inhibits the motility, migration, and adhesion of fibrosarcoma cells, whereas treatment with exogenous CS proteoglycans dose-dependently stimulates cell motility and migration via a JNK-dependent pathway [18]. A recent study showed that chondroitin/dermatan sulfate proteoglycans play a key role in the adhesion and migration of VSMCs. The DS-epi1−/− VSMCs, which exhibit reduced iduronic acid both on the cell surface and in secreted CS proteoglycans, have fewer focal adhesion sites, decrease directional migration, and reduce the activation and expression of FAK compared to the wild-type VSMCs [19]. The activation of FAK downstream of β1 integrin signaling is a major pathway underlying cell migration activity. Our results that β1 integrin mediates TM-enhanced VSMCs migration and the fact that TM-enhanced FAK activation is dependent on the CS moiety again point to the activation of β1 integrin-FAK in mediating TM-enhanced VSMCs migration. In this context, the serine/threonine-rich domain of TM was shown to interact with β2 integrin expressed on peripheral blood mononuclear cells in vitro [42]. Further study is required to unravel how the CS moiety of TM interacts with integrin-FAK pathway as we were unable to detect the binding complex between TM and β1 integrin (CP Pai and MJ Jiang, unpublished results).
The role of TM in cell proliferation appears to vary depending on the form of TM expressed and secreted by cells. A soluble TM fragment consisting of the EGF-like repeats (rTMD2) stimulates proliferation in various cell types, including ECs, VSMCs, and Swiss 3T3 cells [6, 15, 43], and was shown to act through a fibroblast growth factor receptor in ECs [44]. On the contrary, membrane-bound TM overexpression and a soluble recombinant TM fragment consisting of the entire extracellular domains inhibit both thrombin-stimulated VSMC proliferation in culture [45, 46] and vascular injury-induced neointima formation [38, 47]. These contradictory results illustrate the complex roles of TM reflecting the interaction of various domains of TM with other proteins and possibly among different TM domains. Our results are consistent with those reported in cultured urinary bladder smooth muscle cells, both showing no effect of increased TM expression on PDGF-stimulated cell proliferation [9]. It is well established that TM soluble fragments are detected in serum and urine in various forms and concentrations depending on physiological conditions [48]. Therefore, the role of TM in VSMCs proliferation in vivo may vary under different pathophysiological conditions.
Conclusion
The CS moiety at the serine/threonine-rich domain mediates TM-enhanced VSMCs adhesion, spreading, and migration activity through the activation of β1 integrin and a FAK pathway.
Additional file
Supplementary data. (DOCX 315 kb)
Acknowledgments
We thank Ms. Hui-Chun Huang and Dr. Chia-Ching Wu for their assistance in confocal microscopy, Dr. Kuan-Chieh Wang for providing constructive suggestions to the manuscript, and Mr. John JN Jeng for editorial assistance. TM shRNA clones and lentiviral vectors were provided by the RNAi core of Academia Sinica, Taiwan.
Funding
This study was supported by the Ministry of Science and Technology (NSC 95-2752-B-006-005-PAE, NSC98-2320-B-006-025-MY3, and NSC102-2320-B-006-045) of Taiwan.
Availability of data and materials
All materials are available.
Abbreviations
- BSA
Bovine serum albumin
- ChABC
Chondroitinase ABC
- CS
Chondroitin sulfate
- ECM
Extracellular matrix
- ECs
Endothelial cells
- EGF
Epidermal growth factor
- FAK
Focal adhesion kinase
- GAPDH
Glyceraldehyde 3-phosphate dehydrogenase
- GFP
Green fluorescence protein
- HASMCs
Human aortic smooth muscle cells
- PBS
Phosphate buffered saline
- PDGF
Platelet-derived growth factor
- shRNA
Short hairpin-type RNA interference
- TM
Thrombomodulin
- VSMCs
Vascular smooth muscle cells
Authors’ contributions
VCP and ICL performed major experiments, analyzed data, and wrote the paper. YWH performed pilot experiments and data analysis. ICT and HPC performed part of experiments and data analysis. GYS and HLW provided reagents and experimental ideas. MJJ designed experiments and wrote the paper. All authors read and approved the final manuscript.
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Footnotes
Electronic supplementary material
The online version of this article (10.1186/s12929-018-0415-7) contains supplementary material, which is available to authorized users.
Contributor Information
Vincent Chunpeng Pai, Email: chunpeng.pai@gmail.com.
I-Chung Lo, Email: antique0476@gmail.com.
Yan wun Huang, Email: strawberry10071@hotmail.com.
I-Ching Tsai, Email: a3513240@yahoo.com.tw.
Hui-Pin Cheng, Email: Cerberus_sin@yahoo.com.tw.
Guey-Yueh Shi, Email: gyshi@mail.ncku.edu.tw.
Hua-Lin Wu, Email: halnwu@mail.ncku.edu.tw.
Meei Jyh Jiang, Email: mjiang@mail.ncku.edu.tw.
References
- 1.Bennett MR, Sinha S, Owens GK. Vascular smooth muscle cells in atherosclerosis. Circ Res. 2016;118(4):692–702. doi: 10.1161/CIRCRESAHA.115.306361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Doran AC, Meller N, CA MN. Role of smooth muscle cells in the initiation and early progression of atherosclerosis. Arterioscler Thromb Vasc Biol. 2008;28(5):812–819. doi: 10.1161/ATVBAHA.107.159327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Owen WG, Esmon CT. Functional properties of an endothelial cell cofactor for thrombin-catalyzed activation of protein C. J Biol Chem. 1981;256(11):5532–5535. [PubMed] [Google Scholar]
- 4.Laszik ZG, Zhou XJ, Ferrell GL, Silva FG, Esmon CT. Down-regulation of endothelial expression of endothelial cell protein C receptor and thrombomodulin in coronary atherosclerosis. Am J Pathol. 2001;159(3):797–802. doi: 10.1016/S0002-9440(10)61753-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kim AY, Walinsky PL, Kolodgie FD, Bian C, Sperry JL, Deming CB, Peck EA, Shake JG, Ang GB, Sohn RH, Esmon CT, Virmani R, Stuart RS, Rade JJ. Early loss of thrombomodulin expression impairs vein graft thromboresistance: implications for vein graft failure. Circ Res. 2002;90(2):205–212. doi: 10.1161/hh0202.105097. [DOI] [PubMed] [Google Scholar]
- 6.Tohda G, Oida K, Okada Y, Kosaka S, Okada E, Takahashi S, Ishii H, Miyamori I. Expression of thrombomodulin in atherosclerotic lesions and mitogenic activity of recombinant thrombomodulin in vascular smooth muscle cells. Arterioscler Thromb Vasc Biol. 1998;18(12):1861–1869. doi: 10.1161/01.ATV.18.12.1861. [DOI] [PubMed] [Google Scholar]
- 7.Yoshii Y, Okada Y, Sasaki S, Mori H, Oida K, Ishii H. Expression of thrombomodulin in human aortic smooth muscle cells with special reference to atherosclerotic lesion types and age differences. Med Electron Microsc. 2003;36(3):165–172. doi: 10.1007/s00795-003-0212-5. [DOI] [PubMed] [Google Scholar]
- 8.Lo IC, Lin TM, Chou LH, Liu SL, Wu LW, Shi GY, MJ WHLJ. Ets-1 mediates platelet-derived growth factor-BB-induced thrombomodulin expression in human vascular smooth muscle cells. Cardiovasc Res. 2009;81(4):771–779. doi: 10.1093/cvr/cvn351. [DOI] [PubMed] [Google Scholar]
- 9.Ramachandran A, Ranpura SA, Gong EM, Mulone M, Cannon GM, Jr, Adam RM. An Akt- and Fra-1-dependent pathway mediates platelet-derived growth factor-induced expression of thrombomodulin, a novel regulator of smooth muscle cell migration. Am J Pathol. 2010;177(1):119–131. doi: 10.2353/ajpath.2010.090772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hsu YY, Shi GY, Wang KC, Ma CY, Cheng TL, Wu HL. Thrombomodulin promotes focal adhesion kinase activation and contributes to angiogenesis by binding to fibronectin. Oncotarget. 2016;7(42):68122–68139. doi: 10.18632/oncotarget.11828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Conway EM. Thrombomodulin and its role in inflammation. Semin Immunopathol. 2012;34(1):107–125. doi: 10.1007/s00281-011-0282-8. [DOI] [PubMed] [Google Scholar]
- 12.Tsiang M, Lentz SR, Sadler JE. Functional domains of membrane-bound human thrombomodulin. EGF-like domains four to six and the serine/threonine-rich domain are required for cofactor activity. J Biol Chem. 1992;267(9):6164–6170. [PubMed] [Google Scholar]
- 13.Conway EM, Van de Wouwer M, Pollefeyt S, Jurk K, Van Aken H, De Vriese A, Weitz JI, Weiler H, Hellings PW, Schaeffer P, Herbert JM, Collen D, Theilmeier G. The lectin-like domain of thrombomodulin confers protection from neutrophil-mediated tissue damage by suppressing adhesion molecule expression via nuclear factor kappaB and mitogen-activated protein kinase pathways. J Exp Med. 2002;196(5):565–577. doi: 10.1084/jem.20020077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Huang HC, Shi GY, Jiang SJ, Shi CS, Wu CM, Yang HY, Wu HL. Thrombomodulin-mediated cell adhesion: involvement of its lectin-like domain. J Biol Chem. 2003;278(47):46750–46759. doi: 10.1074/jbc.M305216200. [DOI] [PubMed] [Google Scholar]
- 15.Shi CS, Shi GY, Chang YS, Han HS, Kuo CH, Liu C, Huang HC, Chang YJ, Chen PS, Wu HL. Evidence of human thrombomodulin domain as a novel angiogenic factor. Circulation. 2005;111(13):1627–1636. doi: 10.1161/01.CIR.0000160364.05405.B5. [DOI] [PubMed] [Google Scholar]
- 16.Faassen AE, Schrager JA, Klein DJ, Oegema TR, Couchman JR, JB MC. A cell surface chondroitin sulfate proteoglycan, immunologically related to CD44, is involved in type I collagen-mediated melanoma cell motility and invasion. J Cell Biol. 1992;116(2):521–531. doi: 10.1083/jcb.116.2.521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Henke CA, Roongta U, Mickelson DJ, Knutson JR, JB MC. CD44-related chondroitin sulfate proteoglycan, a cell surface receptor implicated with tumor cell invasion, mediates endothelial cell migration on fibrinogen and invasion into a fibrin matrix. J Clin Invest. 1996;97(11):2541–2552. doi: 10.1172/JCI118702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fthenou E, Zong F, Zafiropoulos A, Dobra K, Hjerpe A, Tzanakakis GN. Chondroitin sulfate a regulates fibrosarcoma cell adhesion, motility and migration through JNK and tyrosine kinase signaling pathways. In vivo. 2009;23(1):69–76. [PubMed] [Google Scholar]
- 19.Bartolini B, Thelin MA, Svensson L, Ghiselli G, van Kuppevelt TH, Malmstrom A, Maccarana M. Iduronic acid in chondroitin/dermatan sulfate affects directional migration of aortic smooth muscle cells. PLoS One. 2013;8(7):e66704. doi: 10.1371/journal.pone.0066704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gu WL, Fu SL, Wang YX, Li Y, Lu HZ, Xu XMLPH. Chondroitin sulfate proteoglycans regulate the growth, differentiation and migration of multipotent neural precursor cells through the integrin signaling pathway. BMC Neurosci. 2009;10:128. doi: 10.1186/1471-2202-10-128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Liu WH, Yeh SH, Lu CC, Yu SL, Chen HY, Lin CY, Chen DS, Chen PJ. MicroRNA-18a prevents estrogen receptor-alpha expression, promoting proliferation of hepatocellular carcinoma cells. Gastroenterology. 2009;136(2):683–693. doi: 10.1053/j.gastro.2008.10.029. [DOI] [PubMed] [Google Scholar]
- 22.Lo IC, Shih JM, Jiang MJ. Reactive oxygen species and ERK 1/2 mediate monocyte chemotactic protein-1-stimulated smooth muscle cell migration. J Biomed Sci. 2005;12(2):377–388. doi: 10.1007/s11373-005-1703-2. [DOI] [PubMed] [Google Scholar]
- 23.Lessan K, Aguiar DJ, Oegema T, Siebenson L, Skubitz AP. CD44 and beta1 integrin mediate ovarian carcinoma cell adhesion to peritoneal mesothelial cells. Am J Pathol. 1999;154(5):1525–1537. doi: 10.1016/S0002-9440(10)65406-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Enomoto A, Murakami H, Asai N, Morone N, Watanabe T, Kawai K, Murakumo Y, Usukura J, Kaibuchi K, Takahashi M. Akt/PKB regulates actin organization and cell motility via Girdin/APE. Dev Cell. 2005;9(3):389–402. doi: 10.1016/j.devcel.2005.08.001. [DOI] [PubMed] [Google Scholar]
- 25.Bourin MC, Lundgren-Akerlund E, Lindahl U. Isolation and characterization of the glycosaminoglycan component of rabbit thrombomodulin proteoglycan. J Biol Chem. 1990;265(26):15424–15431. [PubMed] [Google Scholar]
- 26.Lin JH, McLean K, Morser J, Young TA, Wydro RM, Andrews WH, Light DR. Modulation of glycosaminoglycan addition in naturally expressed and recombinant human thrombomodulin. J Biol Chem. 1994;269(40):25021–25030. [PubMed] [Google Scholar]
- 27.Farb A, Kolodgie FD, Hwang JY, Burke AP, Tefera K, Weber DK, Wight TN, Virmani R. Extracellular matrix changes in stented human coronary arteries. Circulation. 2004;110(8):940–947. doi: 10.1161/01.CIR.0000139337.56084.30. [DOI] [PubMed] [Google Scholar]
- 28.Ferns GA, Raines EW, Sprugel KH, Motani AS, Reidy MA, Ross R. Inhibition of neointimal smooth muscle accumulation after angioplasty by an antibody to PDGF. Science. 1991;253(5024):1129–1132. doi: 10.1126/science.1653454. [DOI] [PubMed] [Google Scholar]
- 29.Wu Y, Chen L, Zheng PS, Yang BB. Beta 1-Integrin-mediated glioma cell adhesion and free radical-induced apoptosis are regulated by binding to a C-terminal domain of PG-M/versican. J Biol Chem. 2002;277(14):12294–12301. doi: 10.1074/jbc.M110748200. [DOI] [PubMed] [Google Scholar]
- 30.Zhang Y, Weiler-Guettler H, Chen J, Wilhelm O, Deng Y, Qiu F, Nakagawa K, Klevesath M, Wilhelm S, Bohrer H, Nakagawa M, Graeff H, Martin E, Stern DM, Rosenberg RD, Ziegler R, Nawroth PP. Thrombomodulin modulates growth of tumor cells independent of its anticoagulant activity. J Clin Invest. 1998;101(7):1301–1309. doi: 10.1172/JCI925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gerthoffer WT. Mechanisms of vascular smooth muscle cell migration. Circ Res. 2007;100(5):607–621. doi: 10.1161/01.RES.0000258492.96097.47. [DOI] [PubMed] [Google Scholar]
- 32.Ridley AJ, Schwartz MA, Burridge K, Firtel RA, Ginsberg MH, Borisy G, Parsons JT, Horwitz AR. Cell migration: integrating signals from front to back. Science. 2003;302(5651):1704–1709. doi: 10.1126/science.1092053. [DOI] [PubMed] [Google Scholar]
- 33.Cheng TL, Wu YT, Lai CH, Kao YC, Kuo CH, Liu SL, Hsu YY, Chen PK, Cho CF, Wang KC, Lin WL, Chang BI, Chen CM, Weiler H, Shi GY, Wu HL. Thrombomodulin regulates keratinocyte differentiation and promotes wound healing. J Invest Dermatol. 2013;133(6):1638–1645. doi: 10.1038/jid.2013.8. [DOI] [PubMed] [Google Scholar]
- 34.Huang MT, Wei PL, Liu JJ, Liu DZ, Huey-Chun H, An J, Wu CC, Wu CH, Ho YS, Yang YY, Chang YJ. Knockdown of thrombomodulin enhances HCC cell migration through increase of ZEB1 and decrease of E-cadherin gene expression. Ann Surg Oncol. 2010;17(12):3379–3385. doi: 10.1245/s10434-010-1163-4. [DOI] [PubMed] [Google Scholar]
- 35.Kao YC, Wu LW, Shi CS, Chu CH, Huang CW, Kuo CP, Sheu HM, Shi GY, Wu HL. Downregulation of thrombomodulin, a novel target of snail, induces tumorigenesis through epithelial-mesenchymal transition. Mol Cell Biol. 2010;30(20):4767–4785. doi: 10.1128/MCB.01021-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Liu PL, Tsai JR, Chiu CC, Hwang JJ, Chou SH, Wang CK, Wu SJ, Chen YL, Chen WC, Chen YH, Chong IW. Decreased expression of thrombomodulin is correlated with tumor cell invasiveness and poor prognosis in nonsmall cell lung cancer. Mol Carcinog. 2010;49(10):874–881. doi: 10.1002/mc.20663. [DOI] [PubMed] [Google Scholar]
- 37.Zheng N, Huo Z, Zhang B, Meng M, Cao Z, Wang Z, Zhou Q. Thrombomodulin reduces tumorigenic and metastatic potential of lung cancer cells by up-regulation of E-cadherin and down-regulation of N-cadherin expression. Biochem Biophys Res Commun. 2016;476(4):252–259. doi: 10.1016/j.bbrc.2016.05.105. [DOI] [PubMed] [Google Scholar]
- 38.Waugh JM, Li-Hawkins J, Yuksel E, Kuo MD, Cifra PN, Hilfiker PR, Geske R, Chawla M, Thomas J, Shenaq SM, Dake MD, Woo SL. Thrombomodulin overexpression to limit neointima formation. Circulation. 2000;102(3):332–337. doi: 10.1161/01.CIR.102.3.332. [DOI] [PubMed] [Google Scholar]
- 39.Fukushi J, Makagiansar IT, Stallcup WB. NG2 proteoglycan promotes endothelial cell motility and angiogenesis via engagement of galectin-3 and alpha3beta1 integrin. Mol Biol Cell. 2004;15(8):3580–3590. doi: 10.1091/mbc.E04-03-0236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Iida J, Skubitz AP, Furcht LT, Wayner EA, McCarthy JB. Coordinate role for cell surface chondroitin sulfate proteoglycan and alpha 4 beta 1 integrin in mediating melanoma cell adhesion to fibronectin. J Cell Biol. 1992;118(2):431–444. doi: 10.1083/jcb.118.2.431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Li H, Leung TC, Hoffman S, Balsamo J, Lilien J. Coordinate regulation of cadherin and integrin function by the chondroitin sulfate proteoglycan neurocan. J Cell Biol. 2000;149(6):1275–1288. doi: 10.1083/jcb.149.6.1275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kawamoto E, Okamoto T, Takagi Y, Honda G, Suzuki K, Imai H, Shimaoka M. LFA-1 and Mac-1 integrins bind to the serine/threonine-rich domain of thrombomodulin. Biochem Biophys Res Commun. 2016;473(4):1005–1012. doi: 10.1016/j.bbrc.2016.04.007. [DOI] [PubMed] [Google Scholar]
- 43.Hamada H, Ishii H, Sakyo K, Horie S, Nishiki K, Kazama M. The epidermal growth factor-like domain of recombinant human thrombomodulin exhibits mitogenic activity for Swiss 3T3 cells. Blood. 1995;86(1):225–233. [PubMed] [Google Scholar]
- 44.Kuo CH, Sung MC, Chen PK, Chang BI, Lee FT, Cho CF, Hsieh TT, Huang YC, Li YH, Shi GY, Luo CY, Wu HL. FGFR1 mediates recombinant thrombomodulin domain-induced angiogenesis. Cardiovasc Res. 2015;105(1):107–117. doi: 10.1093/cvr/cvu239. [DOI] [PubMed] [Google Scholar]
- 45.Grinnell BW, Berg DT. Surface thrombomodulin modulates thrombin receptor responses on vascular smooth muscle cells. Am J Phys. 1996;270(2 Pt 2):H603–H609. doi: 10.1152/ajpheart.1996.270.2.H603. [DOI] [PubMed] [Google Scholar]
- 46.Li J, Garnette CS, Cahn M, Claytor RB, Rohrer MJ, Dobson JG, Jr, Gerlitz B, Cutler BS. Recombinant thrombomodulin inhibits arterial smooth muscle cell proliferation induced by thrombin. J Vasc Surg. 2000;32(4):804–813. doi: 10.1067/mva.2000.107992. [DOI] [PubMed] [Google Scholar]
- 47.Li JM, Singh MJ, Itani M, Vasiliu C, Hendricks G, Baker SP, Hale JE, Rohrer MJ, Cutler BS, Nelson PR. Recombinant human thrombomodulin inhibits arterial neointimal hyperplasia after balloon injury. J Vasc Surg. 2004;39(5):1074–1083. doi: 10.1016/j.jvs.2003.12.030. [DOI] [PubMed] [Google Scholar]
- 48.Takano S, Kimura S, Ohdama S, Aoki N. Plasma thrombomodulin in health and diseases. Blood. 1990;76(10):2024–2029. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary data. (DOCX 315 kb)
Data Availability Statement
All materials are available.