

Abstract

Covalent organic frameworks (COFs) are a new class of crystalline organic polymers that have garnered significant recent attention as highly promising H2 evolution photocatalysts. This Perspective discusses the advances in this field of energy research while highlighting the underlying peremptory factors for the rational design of readily tunable COF photoabsorber–cocatalyst systems for optimal photocatalytic performance.
Fossil fuels have been the driving force for economic growth in our world since the dawn of the industrial revolution. At present, more than 80% of the world energy requirement is derived from fossil fuels. However, overexploitation and hence the ever increasing depletion of these natural resources, in addition to the anthropogenic climate change caused by the release of greenhouse gases by combustion of fossil fuels, is a matter of profound concern. Of the renewable alternative energy resources available, solar power is arguably the most promising one. However, solar energy is diffused and thus requires large collection areas for harvesting meaningful amounts. Also, solar energy is intermittent in nature. Thus, as a probable primary energy source, it would need to be coupled to energy storage mechanisms in an exceptional scale. In nature, photosynthesis converts solar energy into stored chemical energy in the form of carbohydrate fuels and oxygen. While too complex to duplicate in all its detail, it is an excellent inspiration to keep pace with the increasing energy demands on our planet, as it offers a blueprint for the design of artificial photosynthetic systems where the goal is to use (and hence convert) solar energy to make solar fuels like H2 by driving thermodynamically uphill chemical reactions like splitting of water (Scheme 1), as shown in eqs 1–3.1−9
| 1 |
| 2 |
| 3 |
Scheme 1. Artificial Photosynthetic Water Splitting.
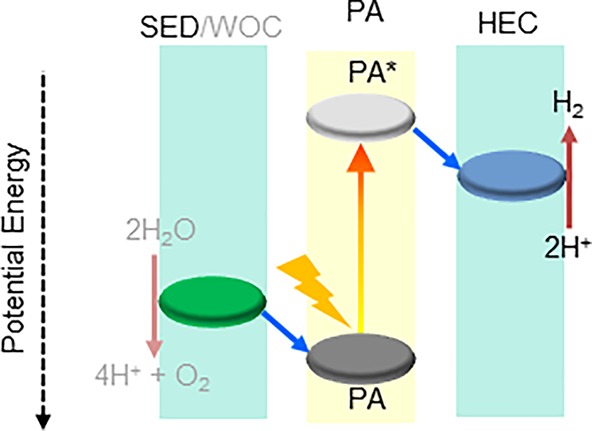
SED, sacrificial electron donor; WOC, water oxidation catalyst; PA, photoabsorber; HEC, hydrogen evolution catalyst.
Under standard conditions, the free-energy change of 237.2 kJ/mol for the conversion of one molecule of water to H2 and 1/2O2 (eq 3) corresponds to ΔE = 1.23 V per electron transferred. Thus, for a photosystem to drive this reaction upon photoexcitation, it must absorb light with photon energies >1.23 eV, corresponding to wavelengths ∼ ≤1000 nm. This process should produce two and four electron–hole pairs per molecule of H2 and O2, respectively. An ideal photosystem, with its band gap larger than that required to split water, and with appropriately positioned conduction band and valence band energies with respect to E(H+/H2) and E(O2/H2O), respectively, should be able to drive the hydrogen evolution (eq 1) and the oxygen evolution (eq 2) reactions using e–/h+ generated upon illumination. Honda and Fujishima were the first to report water splitting by band gap excitation of titanium dioxide in 1972.10 Substantial progress has been made in subsequent years, but the intense complications associated with the complete water-splitting reaction has led to only a handful of successful systems.4 On the other hand, studying the oxidative and the reductive half reactions separately enables detailed investigations and optimizations and thus greatly facilitates the ultimate endeavor.
A typical photocatalytic hydrogen production scheme (Scheme 1) starts with absorption of light by the photosensitizer to generate electron–hole pairs. Charge separation occurs subsequently; a cocatalyst is usually added for carrying out the proton reduction reaction, while a sacrificial electron donor is added as a source of electrons, replacing water as a thermodynamically and kinetically challenging reducing agent. The sacrificial donor then regenerates the photosensitizer by undergoing irreversible decomposition and thus prevents back electron transfer. Direct photocatalytic hydrogen production following this mechanism has been explored under homogeneous conditions and using particulate photoabsorbers alike, each with their pros and cons. Molecular photocatalytic systems based on redox active metal complexes are highly tunable, but they are poorly stable and have comparatively low efficiencies.1,11−14 Heterogeneous systems, on the other hand, have limited light-harvesting abilities and tunability.9,15−18 However, they are robust and long-lived and show decent photocatalytic efficiencies.
The past few years have witnessed increasing interest in organic polymers for photocatalytic H2 evolution, the study of which had been dominated by inorganic materials so far.17−20 Graphitic carbon nitride, represented by Liebig’s “Melon” is the most prominent example in this category.20,21 While it features good H2 evolution activity, the scope for fine-tuning the structure and photophysical properties, and hence H2 evolution activity, is rather limited and mechanistic insights are accordingly scarce.20−25 This is because carbon nitrides, made by polycondensation of the precursors at high temperatures, are mostly amorphous or semicrystalline 1D or 2D polymers with a large dispersity index. In addition, the molecular backbone of carbon nitrides is composed of either heptazine or triazine units, thus limiting their molecular tunability. The need to overcome these inherent limitations with carbon nitrides, while still retaining the well-defined molecular backbone in a heterogeneous system, marks the advent of covalent organic framework (COF) photocatalysis (Scheme 2).
Scheme 2. Photocatalytic H2 Evolution with Metallic Pt (left) and Molecular Cocatalysts (right).
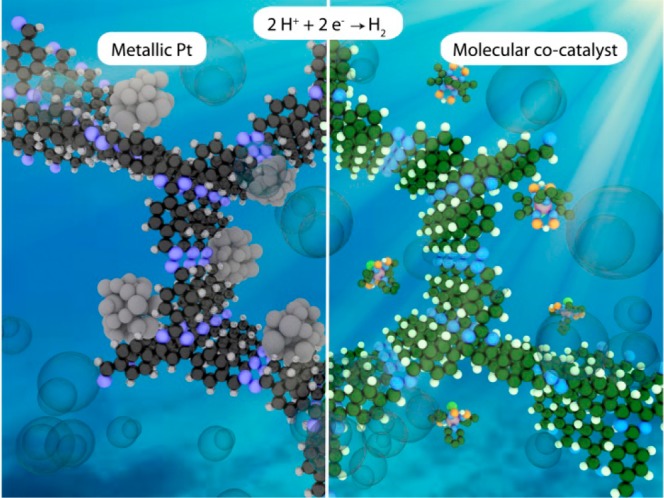
The sacrificial electron donor molecules have been omitted for clarity.
In 2005, Yaghi and co-workers showed the utility of topological design principles in reticulation of molecular building blocks via covalent bonds, to form crystalline COFs.26 COFs were thus a new class of highly porous organic polymers with 2D or 3D network topologies, similar to metal–organic frameworks, but composed solely of light elements and potentially more robust in nature. The suitably chosen functionalized molecular building units are linked to each other in a reversible fashion by thermodynamically controlled dynamic covalent chemistry.27−31 The reversibility in bond formation under the network-forming reaction conditions imparts self-healing ability for the repair of structural defects and facilitates reorganization of the framework structure to produce long-range order and crystallinity, not seen in typical organic polymers which are formed by kinetically driven, irreversible bond formation reactions such as C–C cross coupling.
COFs are one of the most significant discoveries pertaining to heterogeneous photocatalysis because (i) they are composed of molecular building blocks and hence possess almost unlimited chemical tunability of the different functions fundamental to the photocatalytic process, namely, light harvesting, charge separation, charge transport and electrocatalysis. (ii) They possess permanent, nanometer-sized structural pores which can be precisely tuned by choice of appropriate molecular building blocks and their reticulation. The high structural porosity entails high surface areas, enabling both rapid diffusion of charges to the surface and a very high interaction surface for enhanced accessibility of sensitizers, electrolytes, sacrificial components, and cocatalysts throughout the sample. (iii) Unlike molecular systems, the photoactive building blocks can be locked in a rigid architecture, and this can enhance the lifetimes of the excited states by preventing deactivation through collisions. Possible conjugation, both in-plane and in the stacking direction, can also contribute to increased charge carrier mobility. (iv) The crystallinity, in other words, the local and the long-range order in these systems, facilitates charge transport, can prevent recombination of charge carriers, and minimizes charge trapping at defect sites. (v) COFs are composed of covalent bonds and thus are very stable and robust. They are largely impermeable to solvents and, with appropriately chosen linking schemes, can be stable to hydrolysis, extremes of pH, and oxidative and reductive environments, and (vi) being composed of lightweight elements, COFs have an extremely low density and can offer high gravimetric performance. The exceptional blend of solid-state character together with modularity, porosity, and crystallinity means that COFs actually have the potential to be avant-garde in photocatalysis research.18,32−34
In 2013, Jiang and co-workers synthesized a squaraine linked porphyrin COF featuring extended π-conjugation and charge-carrier mobility. With a 1,3-diphenylisobenzofuran label, this COF showed steady generation of singlet oxygen from molecular oxygen.35 Molecular oxygen being in the triplet state, this showed that a triplet excited state of the COF photocatalyst, which did not contain any noble metals, can be populated upon visible light excitation and can actually be harvested in a subsequent reaction.
Indeed, this ability of COFs to harvest light energy laid the foundation for their development as platforms for photocatalytic hydrogen evolution. In 2014, we reported the first COF (Figure 1a) observed to produce H2 in the presence of metallic platinum as the proton reduction catalyst when irradiated with visible light.36 The hydrazone-linked TFPT-COF, based on 1,3,5-tris(4-formyl-phenyl)triazine (TFPT) and 2,5-diethoxy-terephthalohydrazide building blocks, shows a much smaller dihedral angle of 7.7° between the central triazine and the peripheral phenyl rings as compared to its benzene analogue with a dihedral angle of 38.3°. This planarity of the molecular building block translates into a largely planar structure, potentially enabling enhanced stacking interactions and thus charge transport in the axial direction as evident from the interlayer distance of 3.37 Å corresponding to typical van der Waals interactions between aromatic rings. The relatively small optical band gap of 2.8 eV enables significant light absorption in the visible region, thus rendering platinized TFPT-COF an effective H2-evolving photocatalytic system. With ascorbic acid as the sacrificial donor in water, H2 evolved at a steady rate of 230 μmol h–1 g–1, and in 52 h (Figure 1c), the amount of H2 produced was more than the amount of H2 present in the COF itself, thus showing that H2 production is photocatalytic and does not originate from the decomposition of the COF. The COF was also seen to be recyclable, at least three times, with no appreciable decrease in H2 evolution activity. H2 evolution at specific wavelengths of irradiation was found to follow the absorption spectrum of TFPT-COF, thus suggesting band gap excitation to be the source of charge carrier separation. H2PtCl6 was used as the platinum precursor, and TEM images of the COF post photocatalysis showed photodeposition of Pt nanoparticles of roughly 5 nm size (Figure 1d). While photoactivity was retained, the COF lost its crystallinity as seen in a 92 h postphotocatalysis sample, likely because of exfoliation in water. Interestingly though, the amorphous product filtered out of the photocatalysis reaction mixture could be reconverted to the crystalline and porous TFPT-COF by subjecting it to the original synthesis conditions without addition of new building blocks, thus suggesting that the connectivity of the COF remained intact throughout the catalytic conversion. The H2 evolution activity could be improved by replacing the sacrificial electron donor ascorbic acid with triethanolamine (TEOA), however at an expense of a quicker deactivation of the COF. With 10 vol % TEOA, the H2 evolution rate was 1970 μmol h–1 g–1 (Figure 1c) corresponding to a quantum efficiency of 2.2%. This rate was almost 3 times higher than those with benchmark photocatalytic systems such as Pt-modified amorphous melon,37 other carbon nitrides,38 and crystalline poly(triazine imide).37
Figure 1.
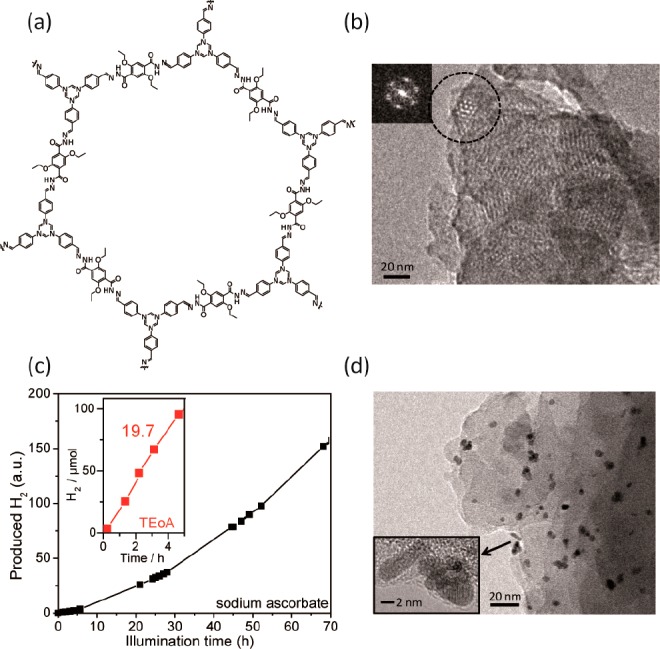
Molecular structure (a) of the TFPT-COF hexagonal pore as seen by TEM at 300 kV (b). Visible light-mediated H2 evolution (c) with TFPT-COF using sodium ascorbate donor and Pt cocatalyst. The inset shows H2 evolution using TEOA as an electron donor. Photodeposited Pt nanoparticles (d) on TFPT-COF after photocatalysis for 84 h. Adapted with permission from ref (36). Copyright 2014 Royal Society of Chemistry.
As discussed before, the most remarkable feature of COFs pertinent to photocatalysis research is their tunability down to the atomic level in an otherwise heterogeneous backbone.28,29,31,39 This was exemplified in the engineering of azine-linked Nx-COFs with triphenylaryl nodes for photocatalytic water reduction.40 Four COFs, with the number of nitrogen atoms in the central aryl ring increasing from 0 to 3, were synthesized by reaction of the corresponding aldehydes with hydrazine under reversible conditions (Figure 2a). Substitution of the C–H units with N atoms gradually decreased the dihedral angle between the central aryl ring and the peripheral phenyl rings in the COF nodes, thus increasing planarity. As a direct result, the peaks in the powder X-ray diffraction (PXRD) pattern become sharper and the stacking reflection at 2θ = 26° becomes more and more prominent along the series from N0- to N3-COF, thus indicating a gradual increase in crystallinity with increasing nitrogen content (Figure 2b). This finding is significant in that it shows that a bulk property such as crystallinity can be controlled precisely by a modulation at the molecular, i.e., the building block level. The porosity of the COFs as measured by the BET surface area increased as well along this series, again reflecting the increasing degree of order with increasing nitrogen content. The increase in planarity and hence crystallinity can effect more facile exciton migration not only along the COF plane but also along the axial direction. The observed increase in planarity also leads to an obvious increase in electronic conjugation; however, with the simultaneous increase in the electron-deficient character of the central aryl ring acting against this trend, all four COFs were found to have essentially identical optical band gaps of around 2.6–2.7 eV. The light-harvesting ability of the COFs was therefore ruled out as a variable for the photocatalytic water reduction activity which was studied under visible light irradiation using TEOA donor and photodeposited platinum nanoparticles as electrocatalyst. Interestingly, the H2 evolution rate showed a 4-fold increase with each isolobal substitution of C–H with N atoms in the central aryl ring. Thus, the H2 evolution rate increased from a mere 23 μmol h–1 g–1 for N0-COF to 90, 438, and finally to 1703 μmol h–1 g–1 for N1-, N2-, and N3-COFs, respectively (Figure 2c, Table 1). Unlike TFPT-COF, the postphotocatalysis samples did not show any significant structural change in the material; framework connectivity, structure, and crystallinity were nearly fully retained with only a slight loss in the long-range order. Also, upon replacing the sacrificial donor with ascorbic acid, long-term experiments with N3-COF for over 120 h showed sustained H2 evolution and thus evidence the remarkable stability of the COF under photocatalytic conditions.
Figure 2.

Molecular structure (a) of the hexagonal pore of Nx- and PTP-COF. For the Nx-COFs, the crystallinity increases gradually from N0- to N3-COF as seen in the PXRD pattern (b). The H2 evolution rate with Pt cocatalyst and TEOA donor (c) analogously increases by 4 times for every additional N atom in the central aryl ring. The stability of the radical anion consonantly increases (d) as one goes from N0- to N3-COF. Four different conformations are possible around torsion angle A in PTP-CHO (e) as opposed to only two around torsion angle C in N3-CHO. Additional D–A type interactions (f) and H-bonding interactions (g) can be seen in single-crystal structure solutions of PTP-H. All of these possibly contribute to the lower crystallinity of PTP-COF. Panels a–d are adapted with permission from ref (40). Copyright 2015 by Nature Publishing Group. Panels e–g are adapted with permission from ref (41). Copyright 2017 Royal Society of Chemistry.
Table 1. Summary of H2 Evolution Activity of COF-Based Photocatalytic Systems.
| COF | HEC | SED | other conditions | solvent | illumin-ation | activity (μmol h–1 g–1) | AQE | TONa | ref |
|---|---|---|---|---|---|---|---|---|---|
| TFPT-COF | Pt | 1 wt % sodium ascorbate | water | >420 nm | 230 | (36) | |||
| TFPT-COF | Pt | 10 vol % TEOA | water | >420 nm | 1970 | 2.2–3.9% at 500 nm | (36) | ||
| N0-COF | Pt | 1 vol % TEOA | PBS buffer at pH 7 | >420 nm | 23 | 0.001% at 450 nm | (40) | ||
| N1-COF | Pt | 1 vol % TEOA | PBS buffer at pH 7 | >420 nm | 90 | 0.077% at 450 nm | (40) | ||
| N2-COF | Pt | 1 vol % TEOA | PBS buffer at pH 7 | >420 nm | 438 | 0.19% at 450 nm | (40) | ||
| N3-COF | Pt | 1 vol % TEOA | PBS buffer at pH 7 | >420 nm | 1703 | 0.44% at 450 nm | (40) | ||
| PTP-COF | Pt | 1 vol % TEOA | PBS buffer at pH 7 | AM 1.5 | 83.83 | (41) | |||
| N2-COF | Co-1b | 1 vol % TEOA | pH 8, 60 equiv dmgH2 | 4:1 ACN/H2O | AM 1.5 | 782 | 0.16% at 400 nm | 54.4 | (60) |
| N2-COF | Co-2c | 1 vol % TEOA | pH 10 | 4:1 ACN/H2O | AM 1.5 | 414 | 9.79 | (60) | |
| N1-COF | Co-1 | 1 vol % TEOA | pH 8 | 4:1 ACN/H2O | AM 1.5 | 100 | 2.03 | (60) | |
| N3-COF | Co-1 | 1 vol % TEOA | pH 8 | 4:1 ACN/H2O | AM 1.5 | 163 | 5.65 | (60) | |
| COF-42 | Co-1 | 1 vol % TEOA | pH 8 | 4:1 ACN/H2O | AM 1.5 | 233 | 5.79 | (60) |
TON is based on the cobaloxime cocatalyst.
Co-1: [Co(dmgH)2pyCl].
Co-2: [Co(dmgBF2)2(OH2)2]
Computational methods are a powerful tool in predicting and analyzing electronic properties of COF photocatalysts pertinent to their photocatalytic activity. Using representative semiextended model systems for the Nx-COF series, it was found that, in line with the observed H2 evolution activities, the highest occupied molecular orbital (HOMO) was stabilized gradually from N0- to N3-COF, suggesting a progressive increase in the thermodynamic driving force for hole extraction by TEOA. The simultaneous decrease in the lowest unoccupied molecular orbital (LUMO) energy, however, indicated a gradual decrease in the driving force for electron transfer to Pt up the series, contrary to the increasing H2 evolution rate. Molecular orbital calculations in unit cells of Nx-COFs after optimization with periodic single-point conditions at the DFTB+/mio-1-0 level of theory reveal that the HOMO is localized only on the azine-linker moiety, thus suggesting it to be a possible hole-quenching site through hydrogen bonding interactions with TEOA. The LUMO was seen to be delocalized across the conjugated π system of the framework. For hydrazone-terminated model hexagons on the PBE0-D3/def2-SVP level of theory, electron affinities were of the order of −2 eV, rendering anionic quenching of the photoexcited COF as the likely reaction pathway. The ionization potential values were estimated to be very high, around +10 eV in vacuum. Thus, oxidative quenching of the photoexcited COF, i.e., the intermediate formation of a radical cationic COF by electron transfer to the cocatalyst, seems unlikely. Assuming the formation of the radical anion to be the rate-determining step in the overall photocatalytic process, the increasingly electron-poor character of the central aryl ring translated into a progressive increase in the stability of the radical anion, going from the N0- to the N3-COF model systems (Figure 2d), and this was found to be in line with the observed trend in the H2 production activity of the COFs. Increased stability of the COF radical anion also suggests more effective charge separation. This highlights the importance of the interface between the COF and the electron donor, i.e., the necessity of efficient hole-transfer reactivity by optimizing the COF–sacrificial donor interactions, for the design of a potent photocatalytic system.
The complex interplay of structural, morphological, and electronic factors for photocatalytic H2 evolution in COF-based systems is further demonstrated with PTP-COF, having the same total number of N atoms as N3-COF, but distributed instead on the peripheral rather than the central ring (Figure 2a).41 Under similar conditions as for the Nx-COFs, PTP-COF produces H2 at a rate of only 83.83 μmol h–1 g–1 after an initial activation period corresponding to the photodeposition of Pt nanoparticles. The lower symmetry of PTP-COF, compared to that of N3-COF, most likely leads to disorder in the former system due to a higher number of possible molecular conformations involving the torsion angle between the central and the peripheral aryl rings in the PTP nodes (Figure 2e). Such molecular arrangements could have a detrimental influence on the stacking interactions, thus causing stacking disorder. Further disorder can be induced into the PTP-system by a possible donor (phenyl)–acceptor (pyridyl) stacking interactions, as opposed to face-to-face interactions in the N3- system (Figure 2f). Also, the higher basicity of pyridyl Ns in the PTP nodes, compared to that of the triazine Ns in the N3-nodes, could lead to oligomers or molecules occluding the pores of PTP-COF (Figure 2g). As a consequence of a possible combination of these factors, the overall crystallinity of PTP-COF was seen to be very low as compared to that of N3-COF, resulting in a BET surface area of only 84.21 m2 g–1 for the former, the theoretical surface area being 2147 m2 g–1. The low crystallinity and porosity further induce morphological changes: as compared to small, well-dispersible aggregates for N3-COF, PTP-COF forms large spheres and macroscopic intergrown monoliths that are very hard to disperse in water during photocatalysis. Because dispersibility affects the efficiency of light absorption and scattering, the amount of COF photosensitizer needed to absorb all light now becomes a variable and puts comparison of the H2 evolution rate with N3-COF in perspective. Photophysical measurements and quantum chemical calculations call attention to additional factors responsible for the poor performance of PTP-COF: As compared to N3-COF, measured fluorescence quantum yield and lifetime imply possibly a less efficient nonradiative deactivation of the photoexcited state of PTP-COF via charge-transfer pathways involved in H2 evolution. That the nonradiative excited-state decay rates might actually correspond to these charge-transfer steps was confirmed in an analogous experiment where the emission quantum yield of PTP-COF was observed to be significantly lower and its luminescence decay significantly faster under photocatalytic conditions (i.e., with added TEOA and photodeposited Pt), compared to that in water. Interestingly, while all Nx-COFs have a band gap of around 2.6–2.7 eV, PTP-COF has a band gap of only 2.1 eV and thus a more extended absorption in the visible region. However, the undermining factors discussed above, which apparently challenge charge transport and the efficiency of light absorption in PTP-COF, seemingly emasculate this effect. In addition, from the calculations of frontier molecular orbitals for the PTP-CHO building block at the PBE0-D3/def2-TZVP level of theory, the HOMO and the LUMO were seen to have a similar spatial extent, thus rendering facile charge recombination another possible channel reducing the efficiency of the photocatalytic system. In addition, the calculated vertical electron affinities of the Nx-CHO and the PTP-CHO units show that the anion radical of PTP-CHO is significantly destabilized compared to that of N3–CHO. This is because the pyridine moieties in the PTP motifs cannot stabilize the negative charges as effectively as the central triazine rings in the N3 system.
It is thus evident that there are a myriad of variables that need to be modulated and orchestrated to have the “perfect” H2 evolving COF photocatalyst. This includes structural factors such as crystallinity and porosity on the one hand and optoelectronic factors like charge separation, charge migration, charge recombination, and stability of the radical cationic or anionic intermediates on the other. Our research highlights the structure–property–activity relationship in such systems and accentuates the importance of the best possible optimization of the said factors for best performance. While these can be potential hurdles, they can be actually engineered to the desired extent at a molecular level in COFs, as mentioned at the beginning.
The development of robust COFs is the most basic step toward the development of photocatalytically active systems. In this context, it is important to remember that crystallinity in COFs is generated through reversible bond formation.28,29 Under conditions of dynamic covalent chemistry, bond breaking is thus as facile as the bond formation process, and stability and crystallinity act in opposite ways. The choice of the linkage in a COF, as well as the COF synthesis conditions (including choice of catalyst, solvent and temperature), is thus of significant importance, and linkages which are more prone to hydrolysis, such as boronic acid ones, might render the photocatalytic system unstable.29,30,42 In that regard, supramolecular interactions to strengthen the intra- and interlayer interactions31,43−46 and irreversible lock-in strategies such as post synthetic stabilization of crystalline COFs47 could be promising tactics to rigidify the framework with a desired complexity. The competition between stability and crystallinity quite reasonably generates COFs with structural disorder and defects,48−50 the roles of which in the charge-transfer processes during photocatalysis need to be explored in detail and precisely controlled, as this could be the key to establish a precise structure–property correlation.
COF as the photosensitizing scaffold has to be able to harvest light energy efficiently and transfer charges to the electrocatalyst. For optimal performance, this mandates extended absorption in the visible and near-infrared region while still maintaining the driving force necessary for proton reduction as well as the overpotential for electron transfer. Conjugation, in other words the delocalization of the π-electron system, both in the axial direction and in-plane, should lower the band gap and also render charge transport more efficient by quick dissipation of the excitation energy,51,52 thus emphasizing the importance of planar and conjugated chromophores as the building blocks. An appropriate choice of the linker is also necessary for a fully conjugated COF layer. Achieving efficient charge separation is another challenge in such low dielectric constant polymers53 that typically show facile recombination of charges created upon photoexcitation. In that regard, our frontier molecular orbital calculations of model oligomeric systems indicate that electron-rich terminal groups could actually assist in charge separation.40 Another way to circumvent this issue would be to work with molecules having long-lived excited states to possibly increase the excited-state lifetimes of the COF. However, while systematic and thorough studies are yet to be done, our studies have generated examples where COFs with longer-lived excited states are less efficient as H2 evolution photocatalysts.41 It must however be mentioned that it is very difficult to ascribe the H2 evolution activity to a single variable, as discussed above.
The unique advantage of COFs over molecular systems is their ability to transport photogenerated charges efficiently, thus reducing the likelihood of recombination.54,55 An interesting research exemplifying the superior charge transport properties of COFs was reported by Banerjee and co-workers where the authors used the COF synthesized from 1,3,5-triformylphloroglucinol and 2,5-dimethyl-p-phenylenediamine as a support for CdS nanoparticles.56 For a CdS:COF ratio of 90:10, the authors observed a H2 evolution rate of 3678 μmol h–1 g–1, which was ascribed by emission quenching experiments and Mott–Schottky measurements to an efficient transport of the photogenerated electrons from the CdS photosensitizer via the COF layers, which further prevented charge recombination losses seen in bare CdS. A H2 evolution rate of only 128 μmol h–1 g–1 was observed for CdS alone under the same conditions. In more general terms, the charge transport and the carrier mobilities can be maximized in a COF by improving the overall crystallinity; by refining stacking interactions for optimal overlap of π-orbitals in the axial direction; and by increasing in-plane conjugation preferably with precursors having high native charge carrier mobilities, such as thiophene, perylene, etc.
Optimization of the interaction of the COF with the sacrificial electron donor is necessary as well for optimal H2 evolution photocatalysis (Scheme 3). The appropriate choice of the donor could be very specific for a particular COF photosensitizer and has to be optimized for high cage escape yields (for reductive quenching) and faster degradation than charge recombination, in addition to the solvent, pH, concentrations, etc.57 While quantum chemical calculations point to a reductive quenching mechanism in the azine-based COF photocatalytic systems developed by us,40 a thorough photophysical investigation of the mechanism and identification of the reaction intermediates is necessary. This could be all the more important because of the possible role of these Lewis basic electron donors in some other steps in the intricate photocatalytic cycle or its possible role in poisoning the nanoparticulate electrocatalysts.
Scheme 3. Representative Variables That Need To Be Optimized for Maximizing Photocatalytic H2 Evolution Efficiency of COF-Based Systems.

Optimization of the COF–electrocatalyst interface would be equally important for efficient H2 evolution photocatalysis (Scheme 3). This is because without an added electrocatalyst, COFs have not yet been observed to produce H2. While charge recombination is an aspect, the major factor seems to be the kinetic overpotential associated with the charge-transfer and bond formation processes for H2 evolution. Thus, in the absence of dedicated catalytic sites right at the COF backbone, suitable cocatalysts for hydrogen evolution need to be identified. Metallic Pt, with a large work function and a low Fermi level, is traditionally employed as the electron sink to trap electrons from the COF.58 It further provides effective proton reduction sites and makes H2 formation facile. In this regard, coordination sites for platinum on the COF backbone might lead to more specific interaction of platinum with the COF and can result in improved charge transfer. This was observed in studies with phenyl-triazine oligomers (PTOs) where the smaller oligomers were observed to be more efficient H2 evolution photocatalysts because of the increased number of terminal nitrile moieties which possibly act as coordination sites for platinum while also assisting in the dispersion of the photocatalyst by H-bonding.59 Unfortunately, platinum is an extremely rare element and hence very expensive. In the long run, it thus needs to be replaced with cocatalysts based on earth-abundant nonprecious elements.11,12 In our recent work we demonstrated the feasibility of this approach using Nx-COFs and the hydrazone-based COF-42 as photosensitizers and a series of molecular cobaloxime cocatalysts as biomimetic hydrogenase mimics (Figure 3a,b, Table 1).60 Composed entirely of molecular building blocks, this system represents the first single-site heterogeneous COF photocatalyst with a unique level of molecular tunability. H2 evolution activity was found to be dependent on the solvent used, and acetonitrile was observed to be important for better performance. Further dependencies on reaction pH, choice of sacrificial donor, and the crystallinity and porosity of the COF were noted. Using N2-COF as the photosensitizer and chloro(pyridine)cobaloxime cocatalyst, H2 evolution rates as high as 782 μmol h–1 g–1 were obtained, corresponding to an apparant quantum efficiency (AQE) of 0.16% at 400 nm. The H2 evolution rates were comparable to the previously discussed benchmark photocatalytic systems like Pt-modified amorphous melon,37 other graphitic carbon nitrides,38 and crystalline poly(triazine imide) (PTI).37 The turnover number (TON) was 54.4 at 20 h. Interestingly, for the same mol % of metallic Pt as the cobaloxime catalyst Co-1, a three times lower H2 evolution rate was observed with the former when measured under the same conditions in 4:1 acetonitrile/water. However, the H2 evolution rate of N2-COF with Pt cocatalyst was more than 8 times higher in water than in 4:1 acetonitrile/water, and a poorer distribution of Pt nanoparticles on the COF surface and/or a poorer photodeposition from the hexachloroplatinic acid precursor in the latter solvent was observed to be the probable reason. Photodeposition of Pt nanoparticles,61 their distribution on the COF surface, as well as the sizes of the nanoparticle clusters, only the surface atoms of which are catalytically active, thus are important factors that affect the H2 evolution rate in such cases and will need to be optimized. The decisive role of the kinetics and thermodynamics of the charge-transfer processes between the COF photosensitizer and the cocatalyst was illustrated in the lower H2 evolution rate with N3-COF than N2-COF, using Co-1 cocatalyst (Figure 3b). With molecular cocatalysts, an important advantage will be the possibility of studying the photocatalytic processes in detail and resolving the reaction intermediates experimentally to further fine-tune the photocatalytic activity of the hybrid system. Using COF-42 photosensitizer and Co-1 cocatalyst, successive reduction of CoIII to CoII and then to CoI and/or the final formation of the possible H2 evolving CoIII–H species could be verified (Figure 3c,d). The CoIII–H and/or CoII–H species were further observed to produce H2 in a heterolytic pathway (Figure 3e). Characterization of the H2 evolving species and optimization of its integrity during photocatalysis with molecular cocatalysts will be important. This is because many molecular cocatalysts are known to be photoreduced to the corresponding metallic species during photocatalysis which instead act as the heterogeneous H2-evolving species.62 Furthermore, the deactivation of the catalyst, for example by formation of cobalt oxide from cobaloximes, could be a limiting factor in the long run.63 With cocatalysts like cobaloximes featuring labile ligands and the molecular heterogeneous structure of COFs having potential coordinating framework atoms, it is important to probe whether the complex actually binds to the COF, because if it binds it can mediate an inner sphere electron transfer to the catalyst. As studied for the N2-COF system with Co-1 cocatalyst, we could confirm that neither metallic Co nor cobalt oxide nanoparticles were formed during photocatalysis, nor does the catalyst bind chemically to the COF at any stage during photocatalysis. Improving upon this weak and nonspecific interaction between the cocatalyst and the COF by covalently binding the two could be the next step forward. A more directional binding is expected to optimize the kinetics of electron transfer to the cocatalyst and surpass the diffusion-controlled limits. Our work also shows that the simpler path of optimizing the COF and the cocatalyst as independent modules has potential as well. With molecular cocatalysts the biggest challenge is however the search for a system that is stable and has limited photodecomposition over time.
Figure 3.
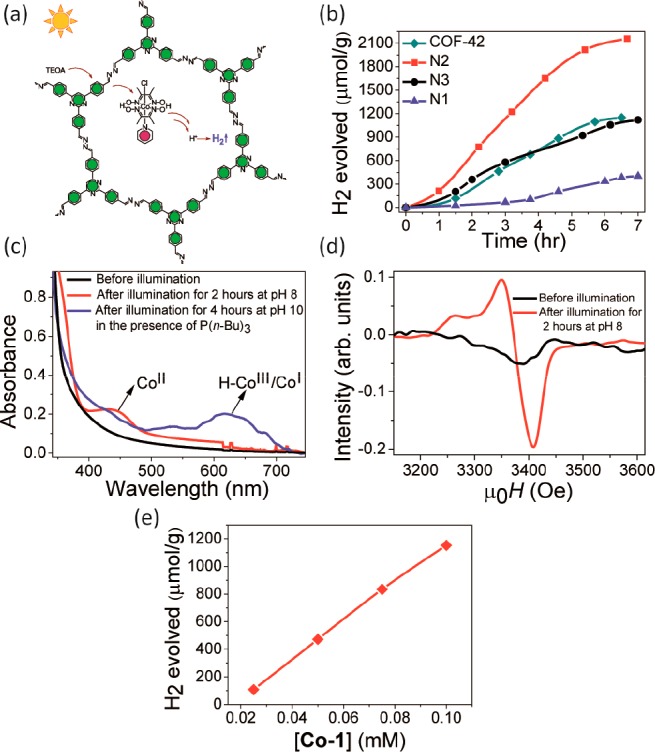
(a) Photocatalytic H2 evolution with N2-COF and Co-1, (b) H2 evolution rates with the Nx-COFs and with COF-42 photosensitizers using Co-1 cocatalyst and TEOA donor, and (c) spectrophotometrically monitoring the reduced CoII state and subsequent formation of the CoI and/or H–CoIII state using COF-42 and Co-1 cocatalyst. (d) The paramagnetic CoII state formed during photocatalysis can be observed in the X-band EPR spectrum. (e) The CoIII–H and/or the CoII–H species are shown to produce H2 by a heterolytic pathway involving a single cobalt center in the reaction involving N2-COF and Co-1 cocatalyst. Reprinted from ref (60). Copyright 2017 American Chemical Society.
In conclusion, the molecular and hence the modular nature of the heterogeneous COF backbone creates enormous prospects for H2 evolution photocatalysis as demonstrated by the first promising steps outlined above. However, these results mark just the beginning of a prospering area of research, and every aspect of these complex architectures needs to be scrutinized to push the limits of COF photocatalysis further. Optimization of the solid-state factors such as robustness, crystallinity, porosity, and defect engineering of COFs will be important and are expected to ameliorate the desired bottom-up design for enhancing the light-harvesting and charge transport properties of such materials (Scheme 3). Thus, the development of this field will be driven by the overall progress in COF research; its success will be contingent on our ability to engineer ordered complexity within a stable, photoactive COF framework.
Acknowledgments
Financial support was provided by an ERC Starting Grant (Project COF Leaf, Grant Number 639233), the Max Planck Society, the cluster of excellence Nanosystems Initiative Munich, and the Center for Nanoscience (CeNS). C.O. acknowledges financial support by the excellence cluster EXC114 (DFG).
Biographies
Tanmay Banerjee has been a scientist in the Lotsch Group at MPI for Solid State Research, Stuttgart since 2016. He received his Ph.D. from National Chemical Laboratory, Pune, India in 2014. His research interests include electron- and energy-transfer processes pertinent to photocatalysis and DSSC applications.
Kerstin Gottschling received her B.Sc. and M.Sc. from the Ludwig-Maximilians-Universität München (LMU) and has worked as a Ph.D. candidate in the group of Bettina V. Lotsch at MPI for Solid State Research since 2015. Her research interests include synthesis and modification of porous 2D frameworks for applications in (photo)catalysis.
Gökcen Savasci received his B.Sc. and M.Sc. degrees from the Ludwig-Maximilians-Universität München (LMU) and works as a PhD candidate in the group of Christian Ochsenfeld at LMU since 2014. His research interests include electronic structure and dynamics of covalent organic frameworks for photocatalysis.
Christian Ochsenfeld received his Ph.D. from University of Karlsruhe in 1994. After postdoctoral work at UC Berkeley, he was a Liebig- and Emmy-Noether-fellow at University of Mainz and became professor for theoretical chemistry at University of Tübingen in 2001. Since 2010 he has held the chair of theoretical chemistry at LMU Munich. Since 2017 he has also been a Max-Planck-Fellow at MPI-FKF, Stuttgart and an elected member of the “International Academy of Quantum Molecular Sciences” (IAQMS) and the board of the “World Association of Theoretical and Computational Chemists” (WATOC). His research focuses on developing and applying linear-scaling quantum-chemical methods for studying molecular processes for large, complex systems such as COFs and DNA/RNA.
Bettina V. Lotsch received her Ph.D. from LMU Munich in 2006. After a postdoctoral stay at the University of Toronto she was appointed associate professor at LMU Munich. Since 2017 she has been Director at the Max Planck Institute for Solid State Research in Stuttgart. Her research interests are at the interface between solid-state chemistry, nanochemistry, and molecular chemistry and include porous frameworks and 2D materials for sensing and energy conversion. Bettina was named Fellow of the Royal Society of Chemistry in 2014 and is recipient of an ERC Starting grant (2014) and the EU-40 Materials Prize 2017 from the European Materials Research Society (EMRS).
The authors declare no competing financial interest.
References
- Berardi S.; Drouet S.; Francàs L.; Gimbert-Suriňach C.; Guttentag M.; Richmond C.; Stoll T.; Llobet A. Molecular Artificial Photosynthesis. Chem. Soc. Rev. 2014, 43, 7501–7519. 10.1039/C3CS60405E. [DOI] [PubMed] [Google Scholar]
- Tachibana A. Y.; Vayssieres L.; Durrant J. R. Artificial Photosynthesis for Solar Water-Splitting.. Nat. Photonics 2012, 6, 511–518. and references therein 10.1038/nphoton.2012.175. [DOI] [Google Scholar]
- Jafari T.; Moharreri E.; Amin A. S.; Miao R.; Song W.; Suib S. L. Photocatalytic Water Splitting—The Untamed Dream: A Review of Recent Advances. Molecules 2016, 21, 900. 10.3390/molecules21070900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen S.; Takata T.; Domen K. Particulate Photocatalysts for Overall Water Splitting. Nat. Rev. Mater. 2017, 2, 17050. 10.1038/natrevmats.2017.50. [DOI] [Google Scholar]
- Jiang C.; Moniz S. J. A.; Wang A.; Zhang T.; Tang J. Photoelectrochemical Devices for Solar Water Splitting – Materials and Challenges. Chem. Soc. Rev. 2017, 46, 4645–4660. 10.1039/C6CS00306K. [DOI] [PubMed] [Google Scholar]
- Roger I.; Shipman M. A.; Symes M. D. Earth-Abundant Catalysts for Electrochemical and Photoelectrochemical Water Splitting. Nat. Rev. Chem. 2017, 1, 0003. 10.1038/s41570-016-0003. [DOI] [Google Scholar]
- Li X.; Hao X.; Abudula A.; Guan G. Nanostructured Catalysts for Electrochemical Water Splitting: Current State and Prospects. J. Mater. Chem. A 2016, 4, 11973–12000. 10.1039/C6TA02334G. [DOI] [Google Scholar]
- Brennaman M. K.; Dillon R. J.; Alibabaei L.; Gish M. K.; Dares C. J.; Ashford D. L.; House R. L.; Meyer G. J.; Papanikolas J. M.; Meyer T. J. Finding the Way to Solar Fuels with Dye-Sensitized Photoelectrosynthesis Cells. J. Am. Chem. Soc. 2016, 138, 13085–13102. 10.1021/jacs.6b06466. [DOI] [PubMed] [Google Scholar]
- Tran P. D.; Wong L. H.; Barber J.; Loo J. S. C. Recent Advances in Hybrid Photocatalysts for Solar Fuel Production. Energy Environ. Sci. 2012, 5, 5902–5918. 10.1039/c2ee02849b. [DOI] [Google Scholar]
- Fujishima A.; Honda K. Electrochemical Photolysis of Water at a Semiconductor Electrode. Nature 1972, 238, 37–38. 10.1038/238037a0. [DOI] [PubMed] [Google Scholar]
- Du A. P.; Eisenberg R. Catalysts Made of Earth-Abundant Elements (Co, Ni, Fe) for Water Splitting: Recent Progress and Future Challenges. Energy Environ. Sci. 2012, 5, 6012–6021. 10.1039/c2ee03250c. [DOI] [Google Scholar]
- Eckenhoff W. T.; McNamara W. R.; Du P.; Eisenberg R. Cobalt Complexes as Artificial Hydrogenases for the Reductive Side of Water Splitting. Biochim. Biophys. Acta, Bioenerg. 2013, 1827, 958–973. 10.1016/j.bbabio.2013.05.003. [DOI] [PubMed] [Google Scholar]
- Wang F.; Wang W.-G.; Wang H.-Y.; Si G.; Tung C.-H.; Wu L.-Z. Artificial Photosynthetic Systems Based on [FeFe]-Hydrogenase Mimics: the Road to High Efficiency for Light-Driven Hydrogen Evolution. ACS Catal. 2012, 2, 407–416. 10.1021/cs200458b. [DOI] [Google Scholar]
- Deponti E.; Natali M. Photocatalytic Hydrogen Evolution with Ruthenium Polypyridine Sensitizers: Unveiling the Key Factors to Improve Efficiencies. Dalton Trans. 2016, 45, 9136–9147. 10.1039/C6DT01221C. [DOI] [PubMed] [Google Scholar]
- Tentu R. D.; Basu S. Photocatalytic Water Splitting for Hydrogen Production. Curr. Opin. Electrochem 2017, 5, 56–62. 10.1016/j.coelec.2017.10.019. [DOI] [Google Scholar]
- Ahmad H.; Kamarudin S. K.; Minggu L. J.; Kassim M. Hydrogen from Photo-Catalytic Water Splitting Process: A review. Renew. Renewable Sustainable Energy Rev. 2015, 43, 599–610. 10.1016/j.rser.2014.10.101. [DOI] [Google Scholar]
- Zhang G.; Lan Z.-A.; Wang X. Conjugated Polymers: Catalysts for Photocatalytic Hydrogen Evolution. Angew. Chem., Int. Ed. 2016, 55, 15712–15727. 10.1002/anie.201607375. [DOI] [PubMed] [Google Scholar]
- Vyas V. S.; Lau V. W.; Lotsch B. V. Soft Photocatalysis: Organic Polymers for Solar Fuel Production. Chem. Mater. 2016, 28, 5191–5204. 10.1021/acs.chemmater.6b01894. [DOI] [Google Scholar]
- Meier C. B.; Sprick R. S.; Monti A.; Guiglion P.; Lee J.-S. M.; Zwijnenburg M. A.; Cooper A. I. Structure-Property Relationships for Covalent Triazine-Based Frameworks: The Effect of Spacer Length on Photocatalytic Hydrogen Evolution From Water. Polymer 2017, 126, 283–290. 10.1016/j.polymer.2017.04.017. [DOI] [Google Scholar]
- Ong W.-J.; Tan L.-L.; Ng Y. H.; Yong S.-T.; Chai S.-P. Graphitic Carbon Nitride (g-C3N4)-Based Photocatalysts for Artificial Photosynthesis and Environmental Remediation: Are We a Step Closer To Achieving Sustainability?. Chem. Rev. 2016, 116, 7159–7329. 10.1021/acs.chemrev.6b00075. [DOI] [PubMed] [Google Scholar]
- Cao S.; Low J.; Yu J.; Jaroniec M. Polymeric Photocatalysts Based on Graphitic Carbon Nitride. Adv. Mater. 2015, 27, 2150–2176. 10.1002/adma.201500033. [DOI] [PubMed] [Google Scholar]
- Corp K. L.; Schlenker C. W. Ultrafast Spectroscopy Reveals Electron-Transfer Cascade That Improves Hydrogen Evolution with Carbon Nitride Photocatalysts. J. Am. Chem. Soc. 2017, 139, 7904–7912. and references therein 10.1021/jacs.7b02869. [DOI] [PubMed] [Google Scholar]
- Ehrmaier J.; Karsili T. N. V.; Sobolewski A. L.; Domcke W. Mechanism of Photocatalytic Water Splitting with Graphitic Carbon Nitride: Photochemistry of the Heptazine–Water Complex. J. Phys. Chem. A 2017, 121, 4754–4764. 10.1021/acs.jpca.7b04594. [DOI] [PubMed] [Google Scholar]
- Lau V. W.; Yu V. W.; Ehrat F.; Botari T.; Moudrakovski I.; Simon T.; Duppel V.; Medina E.; Stolarczyk J. K.; Feldmann J.; et al. Urea-Modified Carbon Nitrides: Enhancing Photocatalytic Hydrogen Evolution by Rational Defect Engineering. Adv. Energy Mater. 2017, 7, 1602251. 10.1002/aenm.201602251. [DOI] [Google Scholar]
- Lau V. W.; Moudrakovski I.; Botari T.; Weinberger S.; Mesch M. B.; Duppel V.; Senker J.; Blum V.; Lotsch B. V. Rational Design of Carbon Nitride Photocatalysts by Identification of Cyanamide Defects as Catalytically Relevant Sites. Nat. Commun. 2016, 7, 12165. 10.1038/ncomms12165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cote A. P.; Benin A. I.; Ockwig N. W.; O’Keefe M.; Matzger A. J.; Yaghi O. M. Porous, Crystalline, Covalent Organic Frameworks. Science 2005, 310, 1166–1170. 10.1126/science.1120411. [DOI] [PubMed] [Google Scholar]
- Diercks C. S.; Yaghi O. M. The Atom, the Molecule, and the Covalent Organic Framework. Science 2017, 355, eaal1585. 10.1126/science.aal1585. [DOI] [PubMed] [Google Scholar]
- Jiang J.; Zhao Y.; Yaghi O. M. Covalent Chemistry Beyond Molecules. J. Am. Chem. Soc. 2016, 138, 3255–3265. 10.1021/jacs.5b10666. [DOI] [PubMed] [Google Scholar]
- Feng X.; Ding X.; Jiang D. Covalent Organic Frameworks. Chem. Soc. Rev. 2012, 41, 6010–6022. 10.1039/c2cs35157a. [DOI] [PubMed] [Google Scholar]
- Ding S.-Y.; Wang W. Covalent Organic Frameworks (COFs): From Design to Applications. Chem. Soc. Rev. 2013, 42, 548–568. 10.1039/C2CS35072F. [DOI] [PubMed] [Google Scholar]
- Huang N.; Wang P.; Jiang D. Covalent Organic Frameworks: a Materials Platform for Structural and Functional Designs. Nat. Rev. Mater. 2016, 1, 16068. 10.1038/natrevmats.2016.68. [DOI] [Google Scholar]
- Banerjee T.; Bennett T.; Butler K.; Easun T. L.; Eddaoudi M.; Forgan R.; Gagliardi L.; Hendon C.; Jorge M.; Lamberti C.; et al. Electronic, Magnetic and Photophysical Properties of MOFs and COFs: General Discussion. Faraday Discuss. 2017, 201, 87–99. 10.1039/C7FD90043K. [DOI] [PubMed] [Google Scholar]
- Nandi S.; Singh S. K.; Mullangi D.; Illathvalappil R.; George L.; Vinod C. P.; Kurungot S.; Vaidhyanathan R. Low Band Gap Benzimidazole COF Supported Ni3N as Highly Active OER Catalyst. Adv. Energy Mater. 2016, 6, 1601189. 10.1002/aenm.201601189. [DOI] [Google Scholar]
- Mullangi D.; Dhavale V.; Shalini S.; Nandi S.; Collins S.; Woo T.; Kurungot S.; Vaidhyanathan R. Low-Overpotential Electrocatalytic Water Splitting with Noble-Metal-Free Nanoparticles Supported in a sp3 N-Rich Flexible COF. Adv. Energy Mater. 2016, 6, 1600110. 10.1002/aenm.201600110. [DOI] [Google Scholar]
- Nagai A.; Chen X.; Feng X.; Ding X.; Guo Z.; Jiang D. A Squaraine-Linked Mesoporous Covalent Organic Framework. Angew. Chem., Int. Ed. 2013, 52, 3770–3774. 10.1002/anie.201300256. [DOI] [PubMed] [Google Scholar]
- Stegbauer L.; Schwinghammer K.; Lotsch B. V. A Hydrazone-Based Covalent Organic Framework for Photocatalytic Hydrogen Production. Chem. Sci. 2014, 5, 2789–2793. 10.1039/C4SC00016A. [DOI] [Google Scholar]
- Schwinghammer K.; Tuffy B.; Mesch M. B.; Wirnhier E.; Martineau C.; Taulelle F.; Schnick W.; Senker J.; Lotsch B. V. Triazine-Based Carbon Nitrides for Visible-Light-Driven Hydrogen Evolution. Angew. Chem., Int. Ed. 2013, 52, 2435–2439. 10.1002/anie.201206817. [DOI] [PubMed] [Google Scholar]
- Zhang J.; Chen X.; Takanabe K.; Maeda K.; Domen K.; Epping J. D.; Fu X.; Antonietti M.; Wang X. Synthesis of a Carbon Nitride Structure for Visible-Light Catalysis by Copolymerization. Angew. Chem., Int. Ed. 2010, 49, 441–444. 10.1002/anie.200903886. [DOI] [PubMed] [Google Scholar]
- Bisbey R. P.; Dichtel W. R. Covalent Organic Frameworks as a Platform for Multidimensional Polymerization. ACS Cent. Sci. 2017, 3, 533–543. 10.1021/acscentsci.7b00127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vyas V. S.; Haase F.; Stegbauer L.; Savasci G.; Podjaski F.; Ochsenfeld C.; Lotsch B. V. A Tunable Azine Covalent Organic Framework Platform for Visible Light-Induced Hydrogen Generation. Nat. Commun. 2015, 6, 8508. 10.1038/ncomms9508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haase F.; Banerjee T.; Savasci G.; Ochsenfeld C.; Lotsch B. V. Structure–Property–Activity Relationships in a Pyridine Containing Azine-Linked Covalent Organic Framework for Photocatalytic Hydrogen Evolution. Faraday Discuss. 2017, 201, 247–264. 10.1039/C7FD00051K. [DOI] [PubMed] [Google Scholar]
- DeBlase C. R.; Dichtel W. R. Moving Beyond Boron: The Emergence of New Linkage Chemistries in Covalent Organic Frameworks. Macromolecules 2016, 49, 5297–5305. 10.1021/acs.macromol.6b00891. [DOI] [Google Scholar]
- Chen X.; Addicoat M.; Irle S.; Nagai A.; Jiang D. Control of Crystallinity and Porosity of Covalent Organic Frameworks by Managing Interlayer Interactions Based on Self-Complementary π-Electronic Force. J. Am. Chem. Soc. 2013, 135, 546–549. 10.1021/ja3100319. [DOI] [PubMed] [Google Scholar]
- Kandambeth S.; Shinde D. B.; Panda M. K.; Lukose B.; Heine T.; Banerjee R. Enhancement of Chemical Stability and Crystallinity in Porphyrin-Containing Covalent Organic Frameworks by Intramolecular Hydrogen Bonds. Angew. Chem., Int. Ed. 2013, 52, 13052–13056. 10.1002/anie.201306775. [DOI] [PubMed] [Google Scholar]
- Chen X.; Addicoat M.; Jin E.; Zhai L.; Xu H.; Huang N.; Guo Z.; Liu L.; Irle S.; Jiang D. Locking Covalent Organic Frameworks with Hydrogen Bonds: General and Remarkable Effects on Crystalline Structure, Physical Properties, and Photochemical Activity. J. Am. Chem. Soc. 2015, 137, 3241–3247. 10.1021/ja509602c. [DOI] [PubMed] [Google Scholar]
- Kandambeth S.; Mallick A.; Lukose B.; Mane M. V.; Heine T.; Banerjee R. Construction of Crystalline 2D Covalent Organic Frameworks with Remarkable Chemical (Acid/Base) Stability via a Combined Reversible and Irreversible Route. J. Am. Chem. Soc. 2012, 134, 19524–19527. 10.1021/ja308278w. [DOI] [PubMed] [Google Scholar]
- Waller P. J.; Lyle S. J.; Osborn Popp T. M.; Diercks C. S.; Reimer J. A.; Yaghi O. M. Chemical Conversion of Linkages in Covalent Organic Frameworks. J. Am. Chem. Soc. 2016, 138, 15519–15522. 10.1021/jacs.6b08377. [DOI] [PubMed] [Google Scholar]
- Lukose B.; Kuc A.; Heine T. The Structure of Layered Covalent-Organic Frameworks. Chem. - Eur. J. 2011, 17, 2388–2392. 10.1002/chem.201001290. [DOI] [PubMed] [Google Scholar]
- Zwaneveld N. A. A.; Pawlak R.; Abel M.; Catalin D.; Gigmes D.; Bertin D.; Porte L. Organized Formation of 2D Extended Covalent Organic Frameworks at Surfaces. J. Am. Chem. Soc. 2008, 130, 6678–6679. 10.1021/ja800906f. [DOI] [PubMed] [Google Scholar]
- Gutzler R.; Walch H.; Eder G.; Kloft S.; Heckl W. M.; Lackinger M. Surface Mediated Synthesis of 2D Covalent Organic Frameworks: 1,3,5-tris(4-bromophenyl)benzene on Graphite(001), Cu(111), and Ag(110). Chem. Commun. 2009, 4456–4458. 10.1039/b906836h. [DOI] [PubMed] [Google Scholar]
- Butchosa C.; McDonald T. O.; Cooper A. I.; Adams D. J.; Zwijnenburg M. A. Shining a Light on s-Triazine-Based Polymers. J. Phys. Chem. C 2014, 118, 4314–4324. 10.1021/jp411854f. [DOI] [Google Scholar]
- Guo J.; Xu Y.; Jin S.; Chen L.; Kaji T.; Honsho Y.; Addicoat M. A.; Kim J.; Saeki A.; Ihee H.; et al. Conjugated Organic Framework with Three-Dimensionally Ordered Stable Structure and Delocalized π Clouds. Nat. Commun. 2013, 4, 2736. 10.1038/ncomms3736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medina D. D.; Petrus M. L.; Jumabekov A. N.; Margraf J. T.; Weinberger S.; Rotter J. M.; Clark T.; Bein T. Directional Charge-Carrier Transport in Oriented Benzodithiophene Covalent Organic Framework Thin Films. ACS Nano 2017, 11, 2706–2713. 10.1021/acsnano.6b07692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding X.; Guo J.; Feng X.; Honsho Y.; Guo J.; Seki S.; Maitarad P.; Saeki A.; Nagase S.; Jiang D. Synthesis of Metallophthalocyanine Covalent Organic Frameworks That Exhibit High Carrier Mobility and Photoconductivity. Angew. Chem., Int. Ed. 2011, 50, 1289–1293. 10.1002/anie.201005919. [DOI] [PubMed] [Google Scholar]
- Wan S.; Gándara F.; Asano A.; Furukawa H.; Saeki A.; Dey S. K.; Liao L.; Ambrogio M. W.; Botros Y. Y.; Duan X.; et al. Covalent Organic Frameworks with High Charge Carrier Mobility. Chem. Mater. 2011, 23, 4094–4097. 10.1021/cm201140r. [DOI] [Google Scholar]
- Thote J.; Aiyappa H. B.; Deshpande A.; Diaz D. D.; Kurungot S.; Banerjee R. A Covalent Organic Framework–Cadmium Sulfide Hybrid as a Prototype Photocatalyst for Visible-Light-Driven Hydrogen Production. Chem. - Eur. J. 2014, 20, 15961–15965. 10.1002/chem.201403800. [DOI] [PubMed] [Google Scholar]
- Pellegrin Y.; Odobel F. Sacrificial Electron Donor Reagents for Solar Fuel Production. C. R. Chim. 2017, 20, 283–295. 10.1016/j.crci.2015.11.026. [DOI] [Google Scholar]
- Yang J.; Wang D.; Han H.; Li C. Roles of Cocatalysts in Photocatalysis and Photoelectrocatalysis. Acc. Chem. Res. 2013, 46, 1900–1909. 10.1021/ar300227e. [DOI] [PubMed] [Google Scholar]
- Schwinghammer K.; Hug S.; Mesch M. B.; Senker J.; Lotsch B. V. Phenyl-triazine Oligomers for Light-Driven Hydrogen Evolution. Energy Environ. Sci. 2015, 8, 3345–3353. 10.1039/C5EE02574E. [DOI] [Google Scholar]
- Banerjee T.; Haase F.; Savasci G.; Gottschling K.; Ochsenfeld C.; Lotsch B. V. Single-Site Photocatalytic H2 Evolution from Covalent Organic Frameworks with Molecular Cobaloxime Co-Catalysts. J. Am. Chem. Soc. 2017, 139, 16228–16234. 10.1021/jacs.7b07489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang Z.; Zhang Z.; Shangguan W.; Isaacs M. A.; Durndell L. J.; Parlett C. M. A.; Lee A. F. Photodeposition as a Facile Route to Tunable Pt Photocatalysts for Hydrogen Production: on the Role of Methanol. Catal. Sci. Technol. 2016, 6, 81–88. 10.1039/C5CY01364J. [DOI] [Google Scholar]
- Lei P.; Hedlund M.; Lomoth R.; Rensmo H.; Johansson O.; Hammarström L. The Role of Colloid Formation in the Photoinduced H2 Production with a RuII-PdII Supramolecular Complex: A Study by GC, XPS, and TEM. J. Am. Chem. Soc. 2008, 130, 26–27. 10.1021/ja0776780. [DOI] [PubMed] [Google Scholar]
- Cao S.-W.; Liu X.-F.; Yuan Y.-P.; Zhang Z.-Y.; Fang J.; Loo S. C. J.; Barber J.; Sum T. C.; Xue C. Artificial Photosynthetic Hydrogen Evolution Over g-C3N4 Nanosheets Coupled with Cobaloxime. Phys. Chem. Chem. Phys. 2013, 15, 18363–18366. 10.1039/c3cp53350f. [DOI] [PubMed] [Google Scholar]