

Abstract
Background
Plague is a flea-borne zoonotic and invasive disease caused by a gram negative coccobacillus bacterium called Yersinia pestis. Plague has caused three devastating pandemics globally namely: the Justinian, Black Death and Oriental plague. The disease in the Eastern Province of Zambia has been reported in Nyimba and Sinda Districts in the past 15 years. The aim of this study was to investigate the molecular epidemiology of plague in the two affected districts. Polymerase Chain Reaction (PCR), targeting Plasminogen activator gene (pla gene) of Y. pestis, was performed on suspected human bubo aspirates (n = 7), rodents (n = 216), shrews (n = 27) and fleas (n = 1494). Of these, one positive sample from each source or host was subjected to sequencing followed by phylogenetic analysis.
Results
The plasminogen activator gene (pla gene) of Y. pestis was detected in 42.8% bubo aspirates, 6.9% rodents, 3.7% shrew and 0.8% fleas. The fleas were from pigs (n = 4), goats (n = 5) and rodents (n = 3). The sequencing and phylogenetic analysis suggested that the pla gene of Y. pestis in Nyimba and Sinda was similar and the isolates demonstrated a high degree of evolutionary relationship with Antiqua strains from the Republic of Congo and Kenya.
Conclusion
It can be concluded that pla gene of Y. pestis was present in various hosts in the two districts and the strains circulating in each district were similar and resembles those in the Republic of Congo and Kenya.
Electronic supplementary material
The online version of this article (10.1186/s12866-017-1146-8) contains supplementary material, which is available to authorized users.
Keywords: Yersinia pestis, Plague, Phylogenetic analysis, Zambia
Background
Plague is a flea-borne zoonotic disease caused by Yersinia pestis, a gram-negative coccobacillus, non-motile and non-spore-forming bacterium. The genus Yersinia is a member of the family Enterobacteriaceae, which consists of 11 species, of which Y. pestis, Y. pseudotuberculosis and Y. enterocolitica are human pathogens. Yersinia pestis is mainly transmitted by flea vectors, particularly Xenopsylla spp., but can also be transmitted mechanically by other flea species and hematophagous arthropods such as ticks and lice during epizootic periods [1, 2]. The bacteria infect mammals and rodents, which are considered to be the natural reservoirs once they survive the initial infection. The bacterium is believed to have originated from Central Asia, Transbajkalian and Mongolian steppes in the former Soviet Union (FSU) and spread out globally [3, 4]. Yersinia pestis has caused one of the most devastating historical pandemics of the world. It has caused three plague pandemics that affected many people, who succumbed to the infection [5]. Though these pandemics originated from the same place, Justinian plague strain was constrained to be the direct ancestor of those associated with the second and third pandemics, also known as the Black Death and the Oriental plague, respectively. The Black Death pandemic was the result of a separate emergence of Y. pestis from rodents into the human population and it gave birth to subsequent Y. pestis infections, including those that were responsible for the Oriental plague. The Black death spread in Europe and Africa, and probably back to China and the causal agent re-emerged as a new strain causing the outbreaks of plague that followed [6]. The third plague pandemic, probably started in the Yunnan province of China [7]. This episode spread to Hong Kong and later established fresh rodent foci in Asia, Africa, and North America, giving rise to an extant strain of the bacterium [8].
In Central and Southern Africa, there were possibly two routes in which the spread of the disease could have transpired. The first route of strain 1.ANT group in the second pandemic came through Egypt down to East and Central Africa, including the Uganda and Republic of Congo (Congo- Brazzaville). The second route of 1.ORI group could be in the third pandemic and spread through Madagascar and Southern Africa during the nineteenth century. It has been documented by Ziwa et al. [9], that plague was introduced to Eastern, Central and Southern Africa from the Middle East or the Far East by medieval traders, including slave and ivory caravans or via pilgrims to and from Egypt and Saudi Arabia.
Zambia, being a landlocked country, could probably be the intersection of the two routes of transmission, and it is most likely that the two different strains of the bacteria, 1.ANT and 1.ORI groups are present in this country (Fig. 1). Zambia experienced plague outbreak in three zones, namely: Eastern, Southern and North western [10–12]. In the Eastern part of the country, four districts have reported Plague outbreaks since 1917. These are Chama, Lundazi, Sinda and Nyimba [11]. However, in the past 15 years, the disease has occurred in the latter two districts, with the latest outbreak occurring in the Nyimba district in March 2015 [13]. No efforts have been made so far to identify the biotype or biovars of the bacterium in the area. The objective of this study was, therefore, to investigate the molecular epidemiology of plague among different potential reservoir hosts and their flea vectors in Sinda and Nyimba districts.
Fig. 1.
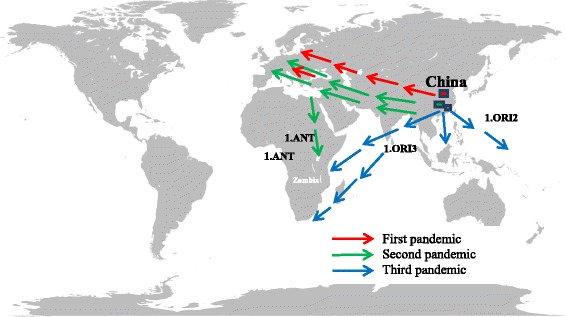
Geographical spread of Yersinia pestis from the suspected sources to Africa
There are three biotypes (biovars) of Y. pestis, which can also be recognised by their distinct biochemical characteristics in their ability to reduce nitrates (NO3−) to nitrites (NO2−) and ferment glycerol. Biotype Antiqua is positive for both of the biochemical characteristics, Orientalis biovar forms nitrites from nitrates but does not ferment glycerol because of a 93 bp deletion in the glycerol 3-phosphate dehydrogenase (glpD) gene, while Medievalis biovar ferments glycerol, but does not form nitrites from nitrates due to a G to T nucleotide mutation that results in a stop codon in the napA gene [14, 15].
Based on the whole genome sequences to discover Single Nucleotide Polymorphisms (SNP), a global analysis demonstrated that the three Y. pestis strains could be separated into several populations with distinctive geographic patterns, including 0.ANT (Antiqua; Asia), 0.PE (Pestoides in group of Antiqua; Angola and Asia), 1.ORI (Orientalis; North and South America, Madagascar, Southeast Asia), 2.MED (Mediaevalis; Asia), 1.ANT (Antiqua; East and Central Africa), and 2.ANT (Antiqua; Asia), 3.ANT (Antiqua; China and Mongolia) and 4.ANT (Antiqua; Mongolia) [4, 6].
Strains of the three biotypes demonstrate no difference in their virulence or pathogenesis in animals and humans [16]. Regardless of the biotypes or biovars, the bacterium possesses three plasmids which contribute to its virulence namely; Calcium Dependant (pCD1), Murine Toxin/Fraction1 (MT1/pFra1) and Pesticin, Coagulase and Plasminogen activator (pPCP1). Plasmid PCP1 has 9.5 kb and is made up of five different genes namely: i) Pesticin (1074 bp pst), ii) Pesticin immunity- a transcriptional regulator gene (426 bp pim), iii) the replication regulation proteins (195 bp rop), iv) IS100 (made up of 1,02 kb transposase and 782 bp ATP-binding protein) and v) Plasminogen activator gene (939 bp pla gene) [17, 18]. The pla gene, only found in Y. pestis, is 1.4 kb and has 936 bp open reading frame (ORF). It is responsible for cleaving fibrin deposits that trap the microorganisms, consequently, helps to facilitate dissemination of the bacteria in the host. This gene is targeted for molecular diagnosis and epidemiology of the disease in this study as described elsewhere [19–21].
Methods
Study areas
This study was carried out in the Eastern part of Zambia in the two districts of Sinda and Nyimba, both of which have a recent history of the plague outbreaks. These areas experience high rainfall between the months of January and March [11]. The study was conducted between March 2015 and August 2016.
Sample collection
Domestic animals
The villages were selected randomly and a maximum of 20 animals from each species, including pigs, goats and sheep, were sampled in each village upon obtaining the verbal consent from the livestock keepers (as per the approved Plague Research provided in Assurance No. FWA0000338). Depending on the species, each animal was assigned a unique number. The first number was picked randomly followed by a systematic technique (Systematic Random Sampling). Each restrained domestic animal was laid on a white plastic sheet and inspected for flea infestation. The animal was brushed with cotton wool soaked in 90% diethyl ether, to anaesthetise the ectoparasites, and then scrubbed with most an appropriate animal brush to remove fleas and other ectoparasites from its fur. Fleas fell onto the white plastic sheet and those which remained attached to the animal skin/fur were gently removed with a pair of fine forceps and put in small vials containing 70% ethanol. Animals below the age of six months and those which came from other villages, in the past six months, were excluded from the study.
Rodent and shrew trapping
Each selected village was divided into six arbitrary zones from which three zones were selected at random. Sherman’s live traps (50 × 65 × 157 mm) baited with peanut butter mixed with soya flour were set at a distance of 10 m apart in the nearby bushes and left overnight. Wire cage traps (145 × 100 × 230 mm) (Hoga-lab, Kyoto, Japan), baited with fish, Stolothrissa tanganicae (Kapenta), and tomatoes, were set in selected houses in the zones. Traps were inspected the following morning and captured animals were taken to a nearby mobile laboratory for organ and flea collection. Trapping continued for three consecutive days in the same area as previously described elsewhere [13].
Collection of fleas from rodents and shrews
Fleas and other ectoparasites were collected from these animals by introducing the latter into the plastic bag with a cotton wool soaked in 90% diethyl ether. Once the animal and ectoparasites became anaethetise, they were transferred to a silver basin, where the animal was brushed with the toothbrush to disengage the fleas and other ectoparasites. Fleas which plummeted into the basin were gently picked using either a fine camel brush or a pair of forceps into serum vials with 70% ethanol, as a preservative. Other ectoparasites were also collected into separate vials containing 70% ethanol.
Collection of organs and bubo aspirates
Each rodent or shrew was aseptically dissected and organs (spleen, liver, lung, kidney and heart) were collected, divided into two parts in two separate vials and stored at −20 °C until required for use. After a verbal consent from the patients and/or their guardian, clinically suspected human plague cases were examined and about 0.5 ml of bubo aspirate was collected and inoculated into 5 ml of Brain Heart Infusion (BHI) medium (Oxoid, Hampshire, England).
Flea identification
Fleas were pooled (1 to 5) according to their species and location, and from each pool, one to two fleas were removed, processed, and identified using main key features such as pronotal combs, genal combs, and the shape of head and reproductive organs (spermathecae in females and penis plates in males) as described by Kilonzo, [22].
DNA extraction, PCR and sequencing
DNA extraction and PCR processing, from the rodent organs, human bubo aspirates and fleas, were performed as previously described [23, 24] and five positive samples were selected for the purpose of sequencing to determine their biovars or biotypes (Table 1).
Table 1.
Flea vector species and their PCR results
| District | Host/source of fleas | Host sampled (n) | Fleas collected | Species of fleas collected | SFI | No. of PCR positive |
|---|---|---|---|---|---|---|
| Nyimba | Rodents | 120 | 15 | Xenopsylla cheopis | 0.12 | 3 |
| Shrews | 17 | 0 | – | – | – | |
| Pigs | 2 | 3 | Ctenocephalides canis | 1.5 | 0 | |
| Pigs | 9 | 7 | Echidnophaga gallinacea | 0.8 | 4 | |
| Goats | 83 | 16 | Ctenocephalides canis | 0.19 | 5 | |
| Sinda | Pigs | 121 | 382 | Echidnophaga gallinacea | 3.2 | 0 |
| Pigs | 124 | 1064 | Echidnophaga larina | 8.6 | 0 | |
| Goats | 232 | 0 | – | – | – | |
| Sheep | 31 | 0 | – | – | – | |
| Rodents | 96 | 7 | Xenopsylla cheopis | 0.07 | 0 | |
| Shrews | 10 | 0 | – | – | – | |
| Total | 845 | 1494 | 12 |
DNA amplification was done using forward primers Yp2 pla1 (5’ ATC TTA CTT TCC GTG AGA AG3’) and reverse primer Yp2 pla2 (5’ CTT GGA TGT TGA GCT TCC TA 3′), which amplifies 479 bp region corresponding to nucleotides 971 to 990 and 1431 to 1450 of the pla locus sequence, respectively [18, 25]. Using a Wizard_SV Gel and Clean-up system (Promega), PCR products were purifed following the manufacturer’s instructions. These products were sequenced with a BigDye Terminator Cycle Sequencing Ready Reaction Kit V3.1 and were further purified using ethanol/EDTA/sodium acetate precipitation, and separated on a 3130 Genetic Analyzer (Applied Biosystems). Assembling and editing of the obtained nucleotide sequences was accomplished by using GENETYX ATGC software, version 4.0.10 (GENETYX Co., Tokyo, Japan), and were equated with other pla genes of Y. pestis elsewhere using basic local alignment search tool (BLAST) in MEGA 6 software as described previously [26].
Phylogenetic data analysis
Molecular evolutionary analyses were conducted using Molecular Evolutionary Genetics Analysis version (MEGA) version 6.0 [27]. Phylogenetic analysis of the derived sequences was performed with the Y. pestis pla gene reference sequences after an alignment was made via ClustalW. In the constructed phylogenetic tree, the sequences obtained from this study were compared with other reference sequences of Y. pestis pla gene acquired from the GenBank. This was accomplished by a homology search of pla gene of Y. pestis using BLAST technique [28]. The topological reliability of the trees was inferred by the bootstrap method with 1000 replicates.
Results
Molecular detection of pla gene of Y. pestis
A total of 216 rodents, 27 shrews, 245 pigs, 232 goats and 31 sheep were sampled for fleas, where a total of 22 fleas from rodents, 1456 fleas from pigs and 16 from goats, were collected. There were no fleas collected from shrews and sheep. Organs were collected from the rodents (n = 216) and the shrews (n = 27). Seven human bubo aspirates were also collected. Altogether, Echidnophaga larina (Jordan & Rothschild) (n = 1064), Xenopsylla cheopis (Rothschild) (n = 22), Echidnophaga gallinacea (Westwood) (n = 389) and Ctenocephaides canis (Curtis) (n = 19) were collected from the two districts.
The pla gene was detected in 6.9% rodents, 3.7% shrews, 42.9% human bubo aspirates and 0.94% fleas (Tables 1 and 2). The results also show that Mastomys natalensis had the highest number of positive Y. pestis pla gene (Table 3).
Table 2.
PCR results from rodents, shrews and humans
| District | Animal spp | No. of animals sampled | PCR positive | Positive (%) |
|---|---|---|---|---|
| Nyimba | Rodents | 120 | 6 | 5.0 |
| Shrews | 17 | 1 | 5.9 | |
| Human | 7 | 3 | 42.8 | |
| Sinda | Rodents | 96 | 9 | 9.4 |
| Shrews | 10 | 0 | 0 | |
| Total | 250 | 19 |
Table 3.
PCR results of tissues from rodents (per specie) and shrews
| Rodent spp | Sinda district | Nyimba district | ||
|---|---|---|---|---|
| No. sampled | No. positive (PCR) | No. sampled | No. positive (PCR) | |
| Mastomys natalensis | 52 | 5 | 68 | 5 |
| Gerbillurus spp | 22 | 3 | 0 | 0 |
| Rattus rattus | 19 | 0 | 34 | 1 |
| Crocidura spp | 10 | 0 | 19 | 1 |
| Saccostomus spp. (Pouched mouse) | 3 | 1 | 31 | 0 |
| Steatomys parvus (Fat mouse) | 0 | 0 | 5 | 0 |
| Total | 106 | 9 (9.4%) | 137 | 7 (5.1%) |
Sequencing and phylogenetic analysis
From the 31 positive amplicons for pla gene of Y. pestis, five amplicons one from each species were selected for sequencing; Homo sapiens (Human), E. gallinacea, X. cheopis, Mastomys natalensis and Gerbillinus spp. (Table 4). A phylogenetic tree was constructed with the known strains of Y. pestis pla gene, which included: Antiqua CP009903.1 (1.ANT), Nairobi CP010294.1 (1.ANT), Nepal 516 CP000307.1 (2.ANT), CO92 L109969.1 (1.ORI), KIM F053945.1 (2.MED), Angola CP009936.1 (0.PE3), Eldorado CP009782.1 (1.ORI2), Microtus 9001 AE017046.1 (2.MED) and Yersinia pestis D182038 CP001592.1 (2.ANT) strains. (Nucleotide sequences of the pla gene of Y. pestis from different hosts from this study are found in Additional file 1:Table S1). Strains from Nyimba demonstrated a high degree of similarity with Y. pestis of Antiqua (1.ANT) (Accession No. CP009903.1), isolated from a human in the Republic of Congo. In contrast, the Sinda strain showed that it was (98%-99%) related to the Nairobi strain (Accession No. CP010294.1), isolated from wild rodent in Nairobi, Kenya (Fig. 2).
Table 4.
Isolation frequency and host distribution of Y. pestis isolates in this study
| Host | Species | No. of Y. pestis DNA extracts | District | |
|---|---|---|---|---|
| Host | Rodent | Mastomys natalensis | 5 | Nyimba |
| Mastomys natalensis | 5 | Sinda | ||
| Gerbillurus spp | 3 | Sinda | ||
| Rattus rattus | 1 | Nyimba | ||
| Crocidura spp | 1 | Nyimba | ||
| Saccostomus spp | 1 | Sinda | ||
| Human | Homo sapiens | 3 | Nyimba | |
| Vector | Fleas from Goat | Ctenocephalides canis | 5 | Nyimba |
| Fleas from Pigs | Echidnophaga gallinacea | 4 | Nyimba | |
| Fleas from Rodents | Xenopsylla cheopis | 3 | Nyimba | |
| Total | 31 | |||
Fig. 2.
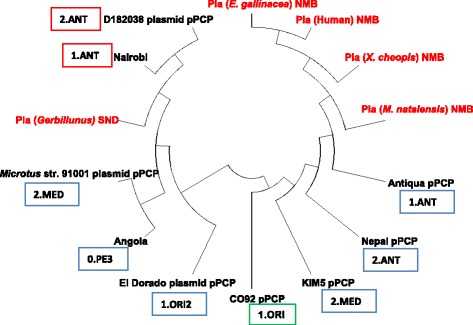
Dendrogram showing the evolutionary relationship of Yersinia pestis strains from Sinda and Nyimba districts using the Maximum Likelihood clustering method. ANT = Antiqua, MED = Medievalis, ORI=Orientalis, PE = Pestoide, NMB=Nyimba, SND = Sinda
Discussion
The presence of pla gene of Y. pestis in rodents, fleas, a shrew and human samples was an indication that the bacterium was present and was the cause of the disease outbreak in Nyimba, where sampling was done just after an outbreak of plague. The results showed that during the outbreak different species of flea vectors have the potential to acquire and mechanically transmit the bacterium to other animals as previously described [29]. In contrast, in Sinda district, the flea vectors were negative for pla gene, suggesting that during the quiescent period, fleas did not harbour the bacterium, although the latter was still isolated from the rodents, which are the natural reservoirs of Y. pestis. It is, therefore, established that the bacterium was in circulation among the reservoir hosts and the disease could break out anytime if environmental conditions for both the hosts and flea vectors, become favourable.
From our findings, it is demonstrated the isolates from Nyimba districts were closely related to those of Antiqua (1.ANT) from the Republic of Congo, isolated from human samples. The Nyimba isolates may have originated from China and migrated during the trade voyages as the steamships sailed from the infected regions to non-infected areas of the world [30]. This is consistent with the findings by Morelli et al. [4], that the isolates in the East and Central Africa involve the 1.ANT group, which they estimated was between 628 and 6914 years and predated the trade voyages in China [8, 31]. Intercontinental trade carried large consignments of cereals which provided a suitable environment for rodents harbouring flea-vector, Rattus rattus and Xenopsylla cheopis respectively, which are primary natural reservoirs and vectors of the Y. pestis [30].
The Sinda isolate was closely related to the Nairobi strain [32] (Fig. 2). This entails that the strain may have originally spread from China to East Africa through the Nile route of Arab traders down south to Central Africa during the second pandemic [4, 26]. Both strains may have migrated forth south probably along the Great Rift Valley during the seasonal migration of rodents along Rukwa valley down to Luangwa valley [33]. The plague outbreak in Nyimba occurred in the Luangwa valley, whereas Sinda is a few kilometres from the Valley but closer to Mozambique. As previously described by Davis, [34], there was a similarity between the Luangwa Valley rodent species and those in Southern Tanganyika (current Tanzania) and Nyasaland (current Malawi) near the border with Northern Rhodesia (current Zambia). Our findings are consistent with those described by Davis et al. [33], that the strain isolated from Mukomba in Lundazi district in eastern Zambia, were biochemically glycerol positive and reduced nitrates to nitrites indicating that it was Antiqua strain of Y. pestis isolated [15]. This bacterium may have been transported along the valley from Northern to Southern Luangwa by either rodents or by human migration [30, 35].
Conclusions
It is concluded that Y. pestis was present in the study area and the isolates from Nyimba district were similar despite coming from different hosts or sources. The Nyimba isolates demonstrated a high degree of evolutionary relationship with Antiqua (1.ANT) strain from the Republic of Congo, while the Sinda strain was evolutionary similar to Nairobi (1.ANT) strain in Kenya.
Additional file
Nucleotide sequences of pla gene of Y. pestis from this study. (DOCX 34 kb)
Acknowledgements
The authors would like to thank the Director, Department of Veterinary Services in the Ministry of Fisheries and Livestock for supporting this work, members of staff of the Nyanje RCZ Mission Hospital, Chinambi Health Center, District Veterinary Offices and Community health personnel in Nyimba and Sinda districts for organizing the villagers and the sampling team. We would like to thank the villagers for their co-operation during the study.
Funding
This work was supported by a PhD grant from the Intra-ACP Academic mobility scheme Africa (Mwalimu Nyerere) and the “Caribbean and Pacific under the commission of the European Union” and the project “Enhancing community of practice in one Health for Infectious diseases through post-graduate training” Grant No. 2012-3166/001, of the Sokoine University of Agriculture (SUA), Morogoro, Tanzania.
However, the funding body had no role in study design, sample collection, analysis and interpretation of data, and in the decision to publish the manuscript.
Availability of data and materials
All data generated or analysed during this study are included in this published article and available at the Additional file 1: Table S1.
Abbreviations
- BLAST
Basic Local Alignment Search Tool
- MEGA
Molecular Evolutionary Genetic Analysis
- PCR
Polymerase Chain Reaction
Authors’ contributions
NSS, HMB and MSR designed and conducted the research and wrote the manuscript; ME, NSS and ML were involved in sample collection and analysis; HMB, MG, KSB, NSS, MSR and SE conducted some of the laboratory experiments and data analyses; MG, KSB, MSR and SE read and edited the manuscript. All authors read and approved the final version of the manuscript.
Ethics approval and consent to participate
Ethical approval to conduct this study was sought from the Institutional Ad hoc committee of the Sokoine University of Agriculture, Tanzania and the Biomedical Research Ethics Committee (BREC), Zambia (Assurance No. is FWA00000338). Verbal consent to participate in the study was granted by the patients and/or their guardian, and persons whose animals and houses were sampled. The reason for not obtaining the written consent to participate from patients, was that the samples used in this study were collected for routine diagnosis, which was approved by the Ethics Committee.
Consent for publication
Not Applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Footnotes
Electronic supplementary material
The online version of this article (10.1186/s12866-017-1146-8) contains supplementary material, which is available to authorized users.
Contributor Information
Stanley S. Nyirenda, Phone: +260 977 801 323, Email: stanleynyirenda@yahoo.co.uk
Bernard M. Hang′ombe, Email: mudenda68@yahoo.com
Edgar Simulundu, Email: esikabala@yahoo.com.
Evans Mulenga, Email: ntongo2004@yahoo.co.uk.
Ladslav Moonga, Email: ladslavm@yahoo.com.
Robert S. Machang′u, Email: rrshandrack2014@gmail.com, Email: rmachangu2013@yahoo.co.uk
Gerald Misinzo, Email: gmisinzo@gmail.com.
Bukheti S. Kilonzo, Email: bskilonzo@yahoo.com
References
- 1.Kiefer D, Dalantai G, Damdindorj T, Riehm JM, Tomaso H, Zöller L, Dashdavaa O, Pfister K, Scholz HC. Phenotypical characterization of Mongolian Yersinia pestis strains. Vector Borne Zoonotic Dis. 2012;12(3):183–188. doi: 10.1089/vbz.2011.0748. [DOI] [PubMed] [Google Scholar]
- 2.Ratovonjato S, Rajerison J, Rahelinirina M, Boyer R. Yersinia pestis in Pulex irritans fleas during plague outbreak, Madagascar. Emerg Infect Dis. 2014;20:1414–1415. doi: 10.3201/eid2008.130629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Li Y, Cui Y, Hauck Y, Platonov ME, Dai E, Song Y, Guo Z. Genotyping and Phylogenetic analysis of Yersinia pestis by MLVA: insights into the worldwide expansion of Central Asia plague foci. PLoS One. 2009;4(6):e6000. doi: 10.1371/journal.pone.0006000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Morelli PG, Song Y, Mazzoni CJ, Eppinger M, Roumagnac R, Wagner DM, Feldkamp M, Kusecek B, Vogler AJ, Li Y, Cui Y, Thomson NR, Jombart T, Leblois EC, Lichtner P, Rahalison L, Petersen JM, Balloux F, Keim P, Wirth T, Ravel J, Yang R, Achtman M. Phylogenetic diversity and historical patterns of pandemic spread of Yersinia pestis. Nat Genet. 2010;42(12):1140–1143. doi: 10.1038/ng.705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Harbeck M, Seifert L, Hänsch S, Wagner DM, Birdsell D, Parise KL, Wiechmann I, Grupe G, Thomas A, Keim P, Zöller L, Bramanti B, Riehm JM, Scholz HC. Yersinia pestis DNA from skeletal remains from the 6th century AD reveals insights into Justinianic plague. PLoS Pathog. 2013;9(5):e1003349. doi: 10.1371/journal.ppat.1003349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cui Y, Yu C, Yan Y, Li D, Li Y, Jombart T, Weinert LA. Historical variations in mutation rate in an epidemic pathogen, Yersinia pestis. Proc Natl Acad Sci. 2013;110(2):577–582. doi: 10.1073/pnas.1205750110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pollitzer R. Plague studies-a summary of history and a survey of the present distribution of the disease. Bull World Health Organ. 1951;4(4):475–533. [PMC free article] [PubMed] [Google Scholar]
- 8.Wagner DM, Klunk J, Harbeck M, Devault A, Waglechner N, Sahl JW, Enk J, Birdsell DN, Kuch M, Lumibao C, Poinar D, Pearson T, Fourment M, Golding B, Riehm JM, Earn DJD, Dewitte S, Rouillard J, Grupe G, Wiechmann I, Bliska JB, Keim PS, Scholz HC, Holmes EC, Poinar H. Yersinia pestis and the plague of Justinian 541 – 543 AD: a genomic analysis. Lancet Infect Dis. 2014;13:1–9. doi: 10.1016/S1473-3099(13)70323-2. [DOI] [PubMed] [Google Scholar]
- 9.Ziwa MH, Matee MI, Hang′ombe BM, Lyamuya EF, Kilonzo BS. Plague in Tanzania: An overview. Tanzan J Health Res. 2013;15(4):1–9. [PubMed] [Google Scholar]
- 10.Neerinckx S, Bertherat E, Leirs H. Human plague occurrences in Africa: an overview from 1877 to 2008. Trans R Soc Trop Med Hyg. 2010;104:97–103. doi: 10.1016/j.trstmh.2009.07.028. [DOI] [PubMed] [Google Scholar]
- 11.Nyirenda SS, Hang′ombe BM, Kilonzo B. Factors that precipitated human plague in Zambia from 1914 to 2014-an overview for a century (100 years) J Zoonotic Dis. 2016;1(1):1–14. [Google Scholar]
- 12.McClean KL. An outbreak of plague in northwestern province, Zambia. Clin Infect Dis. 1995;21(3):650–652. doi: 10.1093/clinids/21.3.650. [DOI] [PubMed] [Google Scholar]
- 13.Nyirenda SS, Hang′ombe BM, Kilonzo BS, Kabeta MN, Mundia C, Sinkala Y. Molecular, serological and epidemiological observations after a suspected outbreak of plague in Nyimba, eastern Zambia. Trop Dr. 2016;1(1):1–6. doi: 10.1177/0049475516662804. [DOI] [PubMed] [Google Scholar]
- 14.Haensch S, Bianucci R, Signoli M, Rajerison M, Schultz M, Kacki S, Vermunt M, Weston DA, Hurst D, Achtman M. Distinct clones of Yersinia pestis caused the black death. PLoS Pathog. 2010;6(10):1–8. doi: 10.1371/journal.ppat.1001134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Perry RD, Fetherston JD. Yersinia pestis--etiologic agent of plague. Clin Microbiol Rev. 1997;10(1):35–66. doi: 10.1128/cmr.10.1.35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chain PSG, Hu P, Malfatti S, Radnedge L, Larimer F, Vergez LM, Worsham P, Chu MC, Andersen GL. Complete genome sequence of Yersinia pestis strains Antiqua and Nepal516: evidence of gene reduction in an emerging pathogen. J Bacteriol. 2006;188(12):4453–4463. doi: 10.1128/JB.00124-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rajanna C, Revazishvili T, Rashid MH, Chubinidze S, Bakanidze L, Tsanava S, Imnadze P, Bishop-Lilly KA, Sozhamannan S, Gibbons SH, Morris GJJ, Sulakvelidze A. Characterization of pPCP1 plasmids in Yersinia Pestis strains isolated from the former soviet union. Int J Microbiol. 2010;2010:1–9. [DOI] [PMC free article] [PubMed]
- 18.Sodeinde O, Goguen J. Nucleotide sequence of the plasminogen activator gene of Yersinia pestis: relationship to ompT of Escherichia Coli and gene E of Salmonella typhimurium. Infect Immun. 1989;57(5):1517–1523. doi: 10.1128/iai.57.5.1517-1523.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Giles TA, Greenwood AD, Tsangaras K, Giles TC, Barrow PA, Hannant D, Yon L. Detection of a Yersinia pestis gene homologue in rodent samples. Peer J. 2016;4:e2216. doi: 10.7717/peerj.2216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Malek MA, Hammani A, Beneldjouzi A, Bitam I. Enzootic plague foci, Algeria. New Microbes New Infect. 2015;4:13–16. doi: 10.1016/j.nmni.2014.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bos KI, Herbig A, Sahl J, Waglechner N, Fourment M, Forrest SA, Klunk J, Schuenemann JV, Poinar D, Kuch M, Golding GB, Dutour O, Keim P, Wagner DM, Holmes EC, Krause J, Poina HN, Broca P, Etudes H. Eighteenth century Yersinia pestis genomes reveal the long-term persistence of an historical plague focus. eLife. 2016:1–11. [DOI] [PMC free article] [PubMed]
- 22.Kilonzo BS. Basic procedures for collecting, processing and identification of common fleas. Morogoro, Tanzania; 1999.
- 23.Hang′ombe BM, Nakamura I, Samui KL, Kaile D, Mweene AS, Kilonzo BS, Sawa H, Sugimoto C, Wren BW. Evidence of Yersinia pestis DNA from fleas in an endemic plague area of Zambia. BMC Res Notes. 2012;5(1):72. doi: 10.1186/1756-0500-5-72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Engelthaler DM, Gage KL, Montenieri J, Chu M, Carter LG. PCR detection of Yersinia pestis in fleas: comparison with mouse inoculation. J Clin Microbiol. 1999;37(6):1980–1984. doi: 10.1128/jcm.37.6.1980-1984.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tsukano H, Itoh KI, Suzuki S, Watanabe H. Detection and identification of Yersinia pestis by polymerase chain reaction (PCR) using multiplex primers. Microbiol Immunol. 1996;40(10):773–775. doi: 10.1111/j.1348-0421.1996.tb01140.x. [DOI] [PubMed] [Google Scholar]
- 26.Ndashe K, Simulundu E, Hang′ombe BM, Ogawa H, Takada A, Mweene AS. Molecular characterization of infectious bursal disease viruses detected in vaccinated commercial broiler flocks in Lusaka, Zambia. Arch Virol. 2016;161(3):513–519. doi: 10.1007/s00705-015-2690-x. [DOI] [PubMed] [Google Scholar]
- 27.Tamura K, Stecher G, Peterson D, Filipski A, Kumar S. MEGA 6: molecular evolutionary genetics analysis version 6.0. Mol Biol Evol. 2013;30(12):2725–2729. doi: 10.1093/molbev/mst197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bethesda (MD): National Library of Medicine (US) NC for BI. National Center for Biotechnology Information NCBI). USA; 1988. Available from: https://blast.ncbi.nlm.nih.gov/Blast.cgi/.
- 29.Andrianaivoarimanana V, Katharina K, Elissa N, Duplantie JM, Carniel E, Rajerison M, Jambou R. Understanding the persistence of plague foci in Madagascar. PLoS Negl Trop Dis. 2013;7(11):2382. doi: 10.1371/journal.pntd.0002382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Dennis E, Gage DT, Gratz KL, Poland N, Tikhomirov JD. Plague manual: epidemiology, distribution, surveillance and control. Geneva; World Health Organisation; 1999.
- 31.Liang Y, Hou X, Wang Y, Cui Z, Zhang Z, Zhu X, Xia L, Shen X, Cai H, Wang J, Xu D. Genome rearrangements of completely sequenced strains of Yersinia pestis. J Clin Microbiol. 2010;48(5):1619–1623. doi: 10.1128/JCM.01473-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ayyadurai S, Flaudrops C, Raoult D, Drancourt M. Rapid identification and typing of Yersinia pestis and other Yersinia species by matrix-assisted laser mass spectrometry. BMC Microbiol. 2010;10(1):285. doi: 10.1186/1471-2180-10-285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Davis DH, Fisher BW, Goldring F. The Luangwa Valley plague outbreaks and their significance in relation to Savannah plague in Central Africa. Bull World Health Organ. 1960;23:405–408. [PMC free article] [PubMed] [Google Scholar]
- 34.Davis DH. Plague in Africa from 1935 to 1949; a survey of wild rodents in African territories. Bull World Health Organ. 1953;9:665–700. [PMC free article] [PubMed] [Google Scholar]
- 35.Wang H, Cui Y, Wang Z, Wang X, Guo Z, Yan Y, Li C, Cui B, Xiao X, Yang Y, Qi Z, Wang G, Wei BYS, He D, Chen H, Chen G, Song Y, Yang R. A dog-associated primary pneumonic plague in Qinghai Province, China. Clin Infect Dis. 2011;52(2):185–190. doi: 10.1093/cid/ciq107. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Nucleotide sequences of pla gene of Y. pestis from this study. (DOCX 34 kb)
Data Availability Statement
All data generated or analysed during this study are included in this published article and available at the Additional file 1: Table S1.