

Abstract
Background
Chlamydia trachomatis (Ct) plasmid has been shown to encode genes essential for infection. We evaluated the population structure of Ct using whole-genome sequence data (WGS). In particular, the relationship between the Ct genome, plasmid and disease was investigated.
Results
WGS data from 157 Ct isolates deposited in the Chlamydiales pubMLST database (http://pubMLST.org/chlamydiales/) were annotated with 902 genes including the core and accessory genome. Plasmid associated genes were annotated and a plasmid MLST scheme was defined allowing plasmid sequence types to be determined. Plasmid allelic variation was investigated. Phylogenetic relationships were examined using the Genome Comparator tool available in pubMLST. Phylogenetic analyses identified four distinct Ct core genome clusters and six plasmid clusters, with a strong association between the chromosomal genotype and plasmid. This in turn was linked to ompA genovars and disease phenotype. Horizontal genetic transfer of plasmids was observed for three urogenital-associated isolates, which possessed plasmids more commonly found in isolates resulting from ocular infections. The pgp3 gene was identified as the most polymorphic plasmid gene and pgp4 was the most conserved.
Conclusion
A strong association between chromosomal genome, plasmid type and disease was observed, consistent with previous studies. This suggests co-evolution of the Ct chromosome and their plasmids, but we confirmed that plasmid transfer can occur between isolates. These data provide a better understanding of the genetic diversity occurring across the Ct genome in association with the plasmid content.
Electronic supplementary material
The online version of this article (10.1186/s12864-018-4522-3) contains supplementary material, which is available to authorized users.
Keywords: Genome sequencing, Chlamydia trachomatis, Genomics, Plasmid
Background
Chlamydia trachomatis (Ct) is responsible for the majority of bacterial sexually transmitted infections worldwide [1]. In addition, ocular Ct infections (trachoma) are the world’s leading cause of preventable blindness [2, 3]. Although there are few documented reports of antibiotic resistance in Ct and infections can be easily treated, the persistent rates of Ct globally, makes this infection an important public health priority.
Ct isolates can be grouped into 15 main genovars based on sequence data of ompA, the gene encoding the major outer membrane protein [4–6]. Specific genovars have been strongly associated with distinct disease pathologies: genovars A-C are associated with conjunctival epithelia; genovars D-K with urogenital, pharyngeal and anorectal epithelia; and genovars L1-L3 with submucosal connective tissue layers resulting in dissemination to locoregional lymph nodes and lymphogranuloma venereum (LGV) [7]. Ct clonal groups identified through different multilocus sequence typing (MLST) schemes based on 7 housekeeping genes as well as phylogenetic analyses of whole-genome sequence (WGS) data [8–11], have also shown an association between Ct strains and tissue tropism.
Ct isolates possess multiple copies of highly conserved small 7.5-kb plasmids containing both non-coding RNA of which the function is unknown, and 8 open reading frames (ORFs), designated pgp1 to pgp8 [12, 13]. The plasmid of Ct has been shown to encode genes essential for infection and transmission, consistent with the rare occurrence of plasmid-deficient clinical isolates [14–16]. The essential role of the plasmid in virulence and inflammatory responses was further demonstrated using animal models, where plasmid-deficient Ct strains or those with mutated plasmids were found to exhibit reduced pathology and decreased inflammatory responses [17–23]. All plasmid-borne genes are transcribed and at least one protein (pgp3) is known to be expressed. Putative functions have been assigned to some of the plasmid genes, based on homology to known proteins [13, 24–26], with pgp1 exhibiting homology to a DnaB like helicase, pgp5 to a partitioning protein which may regulate expression of a set of chromosomal genes, and pgp7 and − 8 identified as integrase/recombinase homologues. In contrast, pgp2, and pgp6 genes are unique to the chlamydia genus. The product of pgp3 is secreted into the host cell cytosol and is the most studied plasmid gene, both as a serological marker for past infection and as virulence factor, as it was demonstrated to play an important role in the induction of inflammatory responses [21, 27, 28]. Finally, pgp4 is a transcriptional regulator of pgp3 and of some chromosomal genes that are likely to be important for chlamydial virulence [25]. The pgp7 gene is not essential for plasmid maintenance as it was found to be interrupted in naturally occurring Ct strains resulting in the emergence and rapid spread of a new Ct variant [29]. This new variant originated in Sweden in 2006 and had a 377 bp deletion in the pgp7 gene that prevented detection of infections using plasmid based PCR diagnostics targeting this gene [13, 29]. The potential of spread and emergence of new strains due to genetic variation indicates the need for more comprehensive studies to better understand the Ct genetic population structure.
Although it is clear that the plasmid plays an important role in the pathogenesis of Ct infection, limited data is available on its genetic diversity and whether distinct plasmid types are associated with different tissue tropisms and pathologies. This study set out to characterise the population structure of Ct in association with the plasmid using WGS data from 157 isolates available in the Chlamydiales pubMLST database (http://pubMLST.org/chlamydiales/). A better understanding of genetic diversity across the Ct genome in association with plasmid content may elucidate Ct epidemiology, ultimately reducing the burden of infection.
Methods
Ct isolate collections and WGS methods
Whole-genome sequence (WGS) data from 157 Ct isolates were analysed (Additional file 1: Table S1). WGS data were obtained from published isolate collections for which plasmid sequence data were also available [9, 26, 30–32]. Short reads were obtained from the European Nucleotide Archive (ENA) and assembled de novo using VELVET in combination with VELVETOPTIMISER as previously described [33]. The resulting contigs were uploaded to the Bacterial Isolate Genome Sequence (BIGSdb) genomics platform hosted on www.pubMLST.org/chlamydiales.
The Chlamydiales pubMLST platform consists of two types of database: i) a sequence definition database that contains sequences of known alleles for loci as well as allelic profiles for specific schemes such as MLST; and, ii) an isolate database that contains isolate provenance and other metadata along with nucleotide sequences associated with that isolate [34]. Sequence definitions have been established for 902 protein-encoding genes, annotated with the CHLAM prefix, and the majority of these have been organised into schemes dependent on function (Additional file 2: Table S2). Chromosomal genes were defined using the annotated genome from Ct strain D/UW-3/CX (accession number NC_000117, [35]).
The BIGSdb software includes ‘autotagger’ and “autodefiner” tools which scan deposited WGS against defined loci identifying alleles ≥98% sequence identity. This process runs in the background and automatically updates isolate records with specific allele numbers, marking regions on assembled contigs for any of the defined loci. Loci with sequence identity < 98% are manually checked and curated.
OmpA, which encodes the major outer membrane protein, was annotated in WGS data as CHLAM0681. Nucleotide sequence data from CHLAM0681 was extracted from all WGS. Bionumerics software (version 7.5, Applied Maths, Sint-Martens-Latem, Belgium) was used to import all extracted ompA sequences to a local offline reference database of ompA sequences that had been described in previous Ct studies [10]. Based on sequence similarity of the ompA variable domains 1 and 2, ompA genovars were assigned to all isolates. Genovars A-C were considered to be ocular isolates, genovars D-K, urogenital isolates and genovar L, LGV isolates. Genovar B is known to cause both urogenital and ocular infections.
Phylogenetic analyses
Relationships among isolates were established using the Genome Comparator tool implemented within the Chlamydiales pubMLST database [34]. Genome Comparator compares groups of shared genes among isolates with any number of loci predefined in the Chlamydiales database or a reference genome. For each locus, allele sequences, designated by integers, are compared and used to generate a distance matrix that is based on the number of variable loci across the genome generating a wgMLST profile. Genome Comparator output provides lists of loci that are: i) identical, ii) variable, iii) missing, or incomplete between data sets, rapidly resolving bacterial population structures and relationships, and identifying loci that belong to the core of a particular data set [33].
Using the Genome Comparator tool, all chromosomal genes identified in the automated annotation process were compared. The set of 888 genes shared between 95% of all Ct isolates was referred to as the ‘core genome’. Varying the stringency of the core genome threshold did not have a significant impact on the results in this study, as a threshold of 90% resulted in a core genome of 889 genes while a threshold of 97.5% resulted in a core genome of 886 genes. Genome Comparator was used to compare the core genome among all isolates and to generate a distance matrix based on the number of variable loci. In addition, this was used to compare previously identified plasmid genes among all isolates. The generated distance matrix for the core genome and plasmid genes were further analysed using the NeighborNet algorithm in SplitsTree version 4.14, to investigate the phylogenetic clustering of Ct isolates according to both their core genome and plasmid loci [36]. Maximum Likelihood phylogenetic trees were also generated from concatenated aligned nucleotide sequence data derived from both core genome and plasmid loci using PhyML [37] and, the HKY85 model with 100 bootstraps. In addition, to each isolate a unique ID, the corresponding ompA genovar and the plasmid sequence type (pST) were linked to each isolate. ClonalFrameML [38] was also used with default parameters to take into account recombination events.
Ct plasmid
Sequences from the plasmid belonging to Ct strain D/SotonD6 were retrieved from plasmid pSotonD6 (HE603231) and designated as CHLAM0895 through to CHLAM0902 encoding the genes pgp1 to pgp8. Using Blast, all WGS sequence data deposited in pubMLST were annotated for these loci as described previously [34, 39, 40].
The eight Ct plasmid genes were grouped into a plasmid MLST typing scheme and plasmid sequence types (pSTs) were assigned based on identified allele variants for isolates with sequence data on all eight plasmid genes.
The number of polymorphic sites per plasmid gene, was assessed using the locus explorer tool in the database (http://pubMLST.org/chlamydiales/). Molecular Evolutionary Genetics Analysis software, version 6 (MEGA 6; http://www.megasoftware.net) was used to align all sequences based on codons in order to calculate average pairwise diversity between isolates [41]. For each gene, p-distance values estimated, both on the nucleotide and amino acid level, with pairwise deletion option selected and standard error (SE) determined with 1000 bootstrap replications. Using MEGA 6, average numbers of synonymous substitutions per synonymous site (dS) and non-synonymous substitutions per non-synonymous site (dN) were calculated by using the overall mean Kumar model [42, 43]. For dN/dS > 1 the Z-test of positive selection was applied and values of P < 0.05 were considered significant. To set a context for the Ct biological clock, dN/dS ratios were also determined for seven housekeeping genes included in the Chlamydiales MLST scheme [11] and compared these to the dN/dS ratios observed for the plasmid genes.
Results
Ct core genome analyses
Ct WGS data available in the Chlamydiales pubMLST database, were filtered to identify those which included complete plasmid sequence data, resulting in 157 isolates. Isolates dated from 1959 to 2011 and were from diverse geographical locations (Additional file 1: Table S1).
A total of 31 allelic ompA variants corresponded to 13 genovars: A, 9.6% (n = 15 isolates); B, 3.2% (n = 5); C, 1.9% (n = 3); D, 8.9% (n = 14); E, 26.8% (n = 42); F, 8.9% (n = 14); G, 8.3% (n = 13); H, 2.5% (n = 4); I, 4.5% (n = 7); J, 1.9% (n = 3); K, 9.6% (n = 15); L1, 3.8% (n = 6), and L2b, 10.2% (n = 16).
A total of 888 out of 902 loci (98.4%) were found to be shared among 95% of the 157 isolates and represented the Ct core genome (cgMLST). Based on the diversity, a distance matrix was calculated from which a NeighborNet tree was generated (Fig. 1, Additional file 3: Table S3). Four phylogenetically distinct clusters were observed, consistent with previous studies [9]. These included: Cluster I, comprising the ocular genovars A, B and C; Cluster II, the clinically more prevalent urogenital genovars D, E and F; Cluster III, the LGV genovars L1 and L2b and, Cluster IV, the rarer urogenital genovars B, D, G, H, I, J and K. WGS from two trachoma isolates (708 and 840, both genovar C), however, clustered with urogenital Ct strains in Cluster II (Fig. 1) consistent with horizontal gene transfer. Identical clusters were identified from the maximum likelihood phylogenetic analysis (Additional file 4: Figure S1).
Fig. 1.
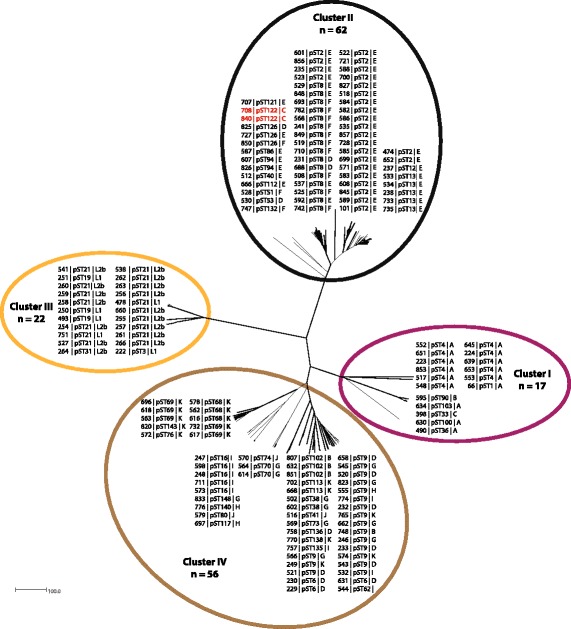
NeighborNet tree showing the core genome clustering of 157 Chlamydia trachomatis isolates. Coloured halos indicate the distinct clusters, isolates are indicated by the Chlamydiales pubMLST ID numbers, plasmid sequence type (pST) and ompA genovar. Isolates shown in red (in cluster II) indicate potential recombinants
The number of alleles present for each core gene varied from 1 to 44 (Additional file 5: Table S4). CHAM0061 (fliA) encoding a Sigma-28/WhiG family protein had the lowest number of allelic variants (n = 1), while the highest number of alleles (n = 44) was observed for CHLAM0147 encoding a hypothetical protein associated with the type III secretion system (T3SS) [44]. Overall, the most diverse genes were ompA (CHLAM0681), the polymorphic outer membrane proteins (CHLAM0412, CHLAM0413, CHLAM0414, CHLAM0812, CHLAM0869, CHLAM0870, CHLAM0871, CHLAM0872, and CHLAM0874), 11 genes associated with the T3SS and 4 genes located in the plasticity zone (CHLAM0153, CHLAM0154, CHLAM0157 and CHLAM0166) in accordance to what was described previously [44, 45]. The plasticity zone (CHLAM0152-CHLAM0177) is a region in the Ct genome which has undergone genetic reorganization to a greater extent than the rest of the chromosome and encodes enzymes required for the biosynthesis of tryptophan [46]. The more conserved genes were found to be hypothetical proteins, RNA associated genes, genes involved in DNA replication and nucleotide excision repair.
The remaining 14 genes not included in the core genome were considered to be accessory genes. Of these, 8 comprised the plasmid genes pgp1 to pgp8 that were considered accessory to assess the association between the core genome and plasmid types. Although, in this dataset, all 8 plasmid genes were present, plasmids are commonly considered to be accessory since 6.5% of Ct isolates have been described to lack plasmids [47]. The remaining 6: CHLAM0165, CHLAM0166, CHLAM0167, CHLAM0173, and CHLAM0174 encoded hypothetical proteins, while CHLAM0456 encoded the translocated actin recruiting phosphoprotein (tarp), a T3SS effector in Chlamydia [45]. The tarp gene has previously been reported as highly variable correlating with ocular, urogenital and LGV disease phenotypes [44, 45, 48]. Among isolates included in this study, tarp alleles were present in 93/157 isolates (59.2%) with a total of 36 unique allelic variants (Additional file 6: Table S6). Analysis of tarp allelic variation revealed that 28/36 (77.8%) of variants were associated with disease phenotype (ocular, urogenital or LGV). For the remaining 8, alleles 25, 33 and 36 were found in WGS data from ocular, urogenital and LGV isolates, alleles 20, 22 and 44 were shared between both ocular and urogenital isolates, allele 1 was found in urogenital and LGV isolates while allele 13 was associated with an ocular and LGV isolate. Overall, the majority of tarp alleles were specific to isolates belonging to a particular disease phenotype, although some tarp alleles were also exchanged between isolates belonging to different disease phenotypes.
Analysis of p-distance values across the core genome revealed that the following were the most divers core genome loci: DNA-binding protein CHLAM0046 (hctB, p-distance = 0.451), ribulose-phosphate 3-epimerase CHLAM0121 (araD, p-distance = 0.500), rRNA methylase CHLAM0133 (p-distance = 0.063), the plasticity zone genes: CHLAM0155 (p-distance = 0.011), CHLAM0157 (p-distance = 0.099), CHLAM0171 (p-distance = 0.064) and hypothetical protein CHLAM0326 (p-distance = 0.039) (Table 1).
Table 1.
Genomic diversity for 888 core loci of 157 Chlamydia trachomatis isolates
| Locia | p-distance |
|---|---|
| CHLAM0046 | 0.451 |
| CHLAM0121 | 0.500 |
| CHLAM0133 | 0.063 |
| CHALM0155 | 0.011 |
| CHALM0157 | 0.099 |
| CHLAM0171 | 0.064 |
| CHLAM0326 | 0.039 |
aThe remaining 881 loci had a p-distance< 0.01
Ct plasmid analyses
The gene pgp4 (CHLAM0900) was the most conserved plasmid gene with 3 alleles containing only 2 polymorphic sites (Table 2). In contrast, pgp6 (CHLAM902) was the most diverse, with 17 allelic variants, followed by: pgp2 with 14, pgp3 and pgp1 with 12, pgp5 and pgp7 with 13 and pgp8 with 11 unique allelic variants. In addition, all plasmid genes had between 15 and 23 polymorphic sites except for pgp4. Further analysis revealed that pgp3 (CHLAM0899), which has also been described as a virulence-associated gene [21, 27, 28], was the most polymorphic (p-distance = 0.008; SE ± 0.002 and amino acid level 0.016; SE ± 0.004). Although the majority of the ORFs in the plasmid are known to be non-coding, in order to identify any putative associations with lineages or understand where such associations stemmed from, we sought to investigate whether selection pressure was evident in any of these ORFs. Therefore, the ratio between the rate of non-synonymous (dN) and synonymous (dS) substitutions per (non-) synonymous nucleotide site was determined. This ranged from 0.083 (pgp8) to 2.00 (pgp5) indicating that plasmid genes were not subject to strong positive selection. Gene pgp5 which is suggested to regulate the expression of some chromosomal genes, showed a dN/dS = 2. We subsequently tested the null hypothesis of no selection (H0: dN = dS) versus the positive selection hypothesis (H1: dN > dS) using the Z-test: Z = (dN − dS)/√(Var(dS) + Var(dN)), but this value was not statistically significant and therefore did not indicate positive selection. The dN/dS ratios from the plasmid genes (0.083 to 2.00) were similar to those observed for theseven housekeeping genes included in the Chlamydiales MLST scheme (gatA, oppA, hflX, gidA, enoA, hemN, and fumC) highlighting the sequence conservation seen in the plasmid genes. Ratios from these housekeeping genes ranged from 0.167 (fumC) to 1.500 (oppA_3). Gene oppA_3 showed a dN/dS = 1.500, but this value was not statistically significant on the Z-test and therefore did not indicate positive selection (Table 3).
Table 2.
Polymorphism analysis and allelic variance of the plasmid genes derived from 157 Chlamydia trachomatis isolates
| Overall Mean Distance (pairwise deletion; bootstrap = 1000 replicates) | ||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Nucleotide | Amino Acid | P-distance; Non- and Synonymous substitutions | ||||||||||||||
| Gene (ORF)a | Length (bp)b | No. alleles | No. polymorphic sites | No. of differences | [SE] | P-distance | [SE] | No. of differences | [SE] | P-distance | [SE] | Mean dS | [SE] | Mean dN | [SE] | dN/dSc |
| pgp1 (ORF3) | 1356 | 12 | 18 | 4.965 | 1.402 | 0.004 | 0.001 | 1.602 | 0.682 | 0.004 | 0.002 | 0.007 | 0.002 | 0.002 | 0.001 | 0.286 |
| pgp2 (ORF4) | 1065 | 14 | 20 | 4.420 | 1.125 | 0.004 | 0.001 | 1.043 | 0.553 | 0.003 | 0.002 | 0.009 | 0.003 | 0.001 | 0.001 | 0.111 |
| pgp3 (ORF5) | 795 | 12 | 23 | 6.133 | 1.488 | 0.008 | 0.002 | 4.172 | 1.122 | 0.016 | 0.004 | 0.007 | 0.003 | 0.007 | 0.002 | 1.000 |
| pgp4 (ORF6) | 309 | 3 | 2 | 0.363 | 0.262 | 0.001 | 0.001 | 0.243 | 0.239 | 0.002 | 0.002 | 0.001 | 0.001 | 0.001 | 0.001 | 1.000 |
| pgp5 (ORF7) | 795 | 13 | 15 | 2.720 | 0.934 | 0.003 | 0.001 | 2.111 | 0.872 | 0.008 | 0.003 | 0.002 | 0.001 | 0.004 | 0.002 | 2.000 |
| pgp6 (ORF8) | 744 | 17 | 20 | 3.003 | 1.020 | 0.004 | 0.001 | 1.415 | 0.648 | 0.006 | 0.003 | 0.006 | 0.003 | 0.003 | 0.001 | 0.500 |
| pgp7 (ORF1) | 918 | 13 | 23 | 4.523 | 1.227 | 0.005 | 0.001 | 1.715 | 0.739 | 0.006 | 0.002 | 0.010 | 0.004 | 0.002 | 0.001 | 0.200 |
| pgp8 (ORF2) | 993 | 11 | 18 | 4.346 | 1.223 | 0.004 | 0.001 | 0.476 | 0.428 | 0.001 | 0.001 | 0.012 | 0.004 | 0.001 | 0.001 | 0.083 |
aORFs designation according to Thomas et al., 1997. Microbiology. 143, 1847–1854
bLength in base pairs relative to the sequence of the plasmid of the E/Bour strain (GenBank No. HE603212)
cThe value of dN/dS > 1 obtained for pgp5 was not statistically significant by the Z-test of positive selection
Table 3.
Polymorphism analysis and allelic variance among 157 Chlamydia trachomatis isolates of seven housekeeping genes present in the Chlamydiales MLST scheme
| Overall Mean Distance (pairwise deletion; bootstrap = 1000 replicates) | ||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Nucleotide | Amino Acid | P-distance; Non- and Synonymous substitutions | ||||||||||||||
| Genea | Length (bp)b | No. alleles | No. polymorphic sites | No. of differences | [SE] | P-distance | [SE] | No. of differences | [SE] | P-distance | [SE] | Mean dS | [SE] | Mean dN | [SE] | dN/dSc |
| enoA (CHLAM0587) | 1275 | 5 | 7 | 1.847 | 0.915 | 0.001 | 0.001 | 0.867 | 0.554 | 0.002 | 0.001 | 0.002 | 0.002 | 0.001 | 0.001 | 0.500 |
| fumC (CHLAM0855) | 1392 | 17 | 20 | 3.040 | 1.136 | 0.002 | 0.001 | 0.358 | 0.111 | 0.001 | 0.000 | 0.006 | 0.002 | 0.001 | 0.000 | 0.167 |
| gatA (CHLAM0003) | 1476 | 11 | 10 | 1.464 | 0.686 | 0.001 | 0.000 | 0.503 | 0.450 | 0.001 | 0.001 | 0.002 | 0.001 | 0.001 | 0.000 | 0.500 |
| gidA (CHLAM0498) | 1833 | 15 | 22 | 5.933 | 1.576 | 0.003 | 0.001 | 2.652 | 1.014 | 0.004 | 0.002 | 0.005 | 0.002 | 0.002 | 0.001 | 0.400 |
| hemN (CHLAM0746) | 1374 | 8 | 7 | 1.282 | 0.633 | 0.001 | 0.000 | 0.880 | 0.560 | 0.002 | 0.001 | 0.001 | 0.001 | 0.001 | 0.001 | 1.000 |
| hflX (CHLAM0379) | 1344 | 16 | 20 | 3.330 | 1.059 | 0.002 | 0.001 | 2.158 | 0.836 | 0.005 | 0.002 | 0.002 | 0.001 | 0.002 | 0.001 | 1.000 |
| oppA_3 (CHLAM0193) | 1557 | 12 | 20 | 4.469 | 1.353 | 0.003 | 0.001 | 3.314 | 1.182 | 0.006 | 0.002 | 0.002 | 0.001 | 0.003 | 0.001 | 1.500 |
aCHLAM refers to the gene annotated in the pubMLST database
bLength in base pairs relative to the sequence of the plasmid of the E/Bour strain (GenBank No. HE603212)
cThe value of dN/dS > 1 obtained for oppA_3 was not statistically significant by the Z-test of positive selection
A total of 47 unique pSTs were randomly assigned (Additional file 3: Table S3, Fig. 1 and Additional file 4: Figure S1). Sequence comparison of plasmid genes identified six phylogenetic clusters numbered 1 to 6 (Fig. 2 and Additional file 7: Figure S2). These clusters were associated with core genome clusters; all isolates of plasmid cluster 2, 4, and 6 fell into core genome clusters II, III and IV, respectively (Fig. 3 and Additional file 8: Figure S3). All isolates except three (564, 570, and 614), of plasmid cluster 1 grouped with those from core genome cluster I. The three exceptions grouped with isolates of core genome cluster IV, suggesting horizontal plasmid transfer. In addition, plasmid clusters 3 and 6 formed sub-branches in core genome cluster IV. Overall, each core genome cluster had one dominating pST (Figs. 1 and 4, Additional file 3: Table S3). Potential recombination events were further assessed using ClonalFrameML, which results indicating that this was restricted to isolates within clusters with recombination apparent to four particular regions to the genome (Additional file 9: Figure S4).
Fig. 2.
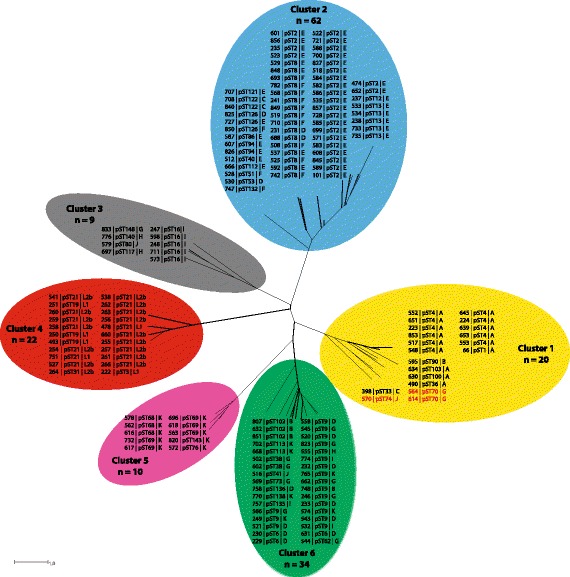
NeighborNet tree showing clustering of the plasmid sequence types (pST) generated from plasmid genes pgp1 to pgp8 of 157 Chlamydia trachomatis isolates. Coloured halos indicate the distinct clusters, isolates are indicated by the Chlamydiales pubMLST ID numbers, pST and ompA genovar. Isolates shown in red (cluster 1) indicate potential plasmid exchange compared to the core genome clusters
Fig. 3.
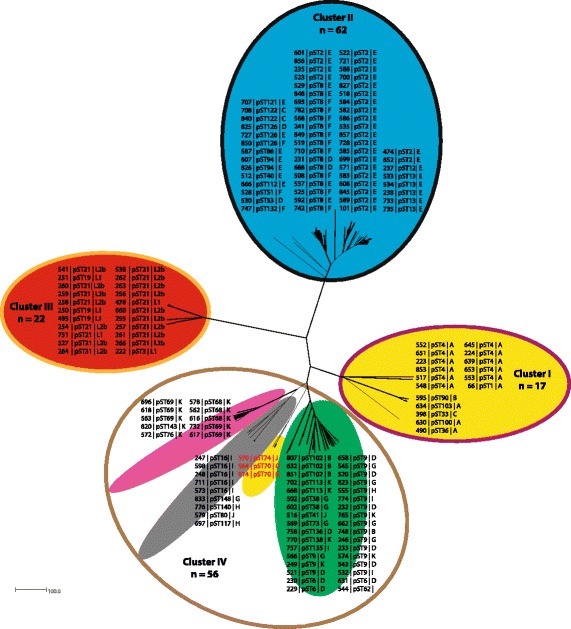
NeighborNet tree showing the comparison between core genome and plasmid clustering of 157 Chlamydia trachomatis isolates. Coloured halos indicate the distinct clusters from Fig. 1, isolates are indicated by the Chlamydiales pubMLST ID numbers, plasmid sequence type (pST) and ompA genovar. Isolates shown in red indicate potential plasmid exchanges compared to the plasmid clusters in Fig. 2
Fig. 4.
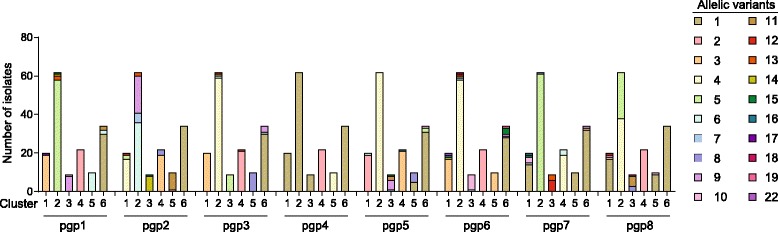
Graph showing the allelic variants for each single plasmid gene (pgp1–8) with respect to the observed plasmid clusters (cluster 1 to 6) among 157 Chlamydia trachomatis isolates. Each allelic variant received a unique colour as shown in the colour legend. Allelic numbers were randomly assigned to each unique allelic variant of a single plasmid gene (pgp1–8). Specific allelic numbers can be assigned to multiple plasmid genes
Analysis of the allelic variation in each single plasmid gene (pgp1 to pgp8) with respect to the observed plasmid clusters revealed that exchange of alleles between plasmid clusters was limited. Plasmid clusters possessed specific pgp1, 2, 3, and 5 alleles that were unique to each cluster (Fig. 4, Additional file 10: Table S5). For example, plasmids in cluster 1 contained pgp1 alleles 3 and 17 which were not found in any of the other plasmid clusters, while cluster 3 had pgp1 alleles 9 and 10. The allelic variation of pgp6, 7 and 8 was specific for the clinically most prevalent urogenital cluster (cluster 2), one cluster containing some of the less frequently occurring genital genovars (cluster 3) and the LGV cluster (cluster 4), but exchange of allelic variants was observed among clusters 1, 5 and 6. Finally, the allelic variation of the pgp4 gene was only specific for the LGV cluster (cluster 4) and a urogenital cluster containing the less frequently occurring genovar K isolates (cluster 5), but exchange of pgp4 allelic variants was observed among clusters 1, 2, 3 and 6.
Discussion
Advances in sequencing technologies and increasing availability of WGS data provide unique opportunities for improving our understanding of Ct infection and epidemiology. Pivotal to this is the ability to rapidly extract strain information from WGS data including ompA genovar, plasmid type and MLST sequence type such that global surveillance of Ct infections can be achieved. In this study, a catalogue of genes both core and accessory to the Ct genome was generated on the web-accessible http://pubMLST.org/chlamydiales/ website providing tools for Ct surveillance in an open database. Data presented here reveal that Ct core genomes were strongly associated with distinct Ct plasmid types (Figs. 1, 2, 4 and Additional files 4 and 7: Figures S1 and S2). Four core genome clusters were apparent following cgMLST analyses consistent with a previous study [9], however, two recombinant isolates were also apparent. These were two trachoma isolates with urogenital backbones that had been identified previously by Andersson et al. [30].
Ct is known to have a closed conserved pan-genome due to persistence of these bacteria in isolated niches with limited access to the global microbial gene pool [49]. In this dataset, a total of 888 genes were found to be core, with only 14 accessory genes consistent with the effects of genome reduction known to have occurred in Ct [50]. All of the accessory genes encoded hypothetical proteins with the exception of the plasmid genes and CHLAM0456, which encodes tarp. None of the hypothetical genes correlated with disease phenotype. Subsequent analysis of tarp allelic variants revealed that the majority of alleles (77.8%) were associated with distinct disease phenotypes (ocular, urogenital, or LGV), although some alleles were also shared between isolates from different disease phenotypes. The gene tarp has been suggested to contribute to tissue tropism [44, 45, 48]. However, these results indicate that it is not the sole gene driving these phenotypes. The most variable gene was CHLAM0147 encoding a T3SS effector involved in endosomal trafficking by recruiting nutrient-rich endocytic vesicles via a non-fusogenic pathway to the chlamydial inclusion [44]. Ct T3SS are activated when Ct attaches to a host cell, after which T3SS are used to deliver an arsenal of bacterial gene-encoded effector proteins into the cytosol of the host cell [44, 48, 51]. The exact molecular mechanisms of T3SS remains to be elucidated, but it is probable that these genetically diverse T3SS genes function together to favour specific tissue tropisms [44, 45].
The most conserved gene was fliA encoding sigma-28, for which the exact function remains to be elucidated. It has been suggested that expression of sigma-28 occurs in response to cellular stress, such as nutrient deprivation within the chlamydial inclusion [52, 53]. Overall, the most variable core genes were ompA, the polymorphic outer membrane proteins, genes associated with T3SS and the plasticity zone, which all have been suggested to contribute to tissue tropism and disease severity due to their high polymorphic variation [44, 45]. The more conserved genes encoded hypothetical proteins for which no known functions have been described, but have been suggested to play an important role in the complex Ct-host interactions [44, 45]. The highly-conserved nature of these genes suggests that mutations may have deleterious effects on biological fitness. Polymorphic genes included hctB (CHLAM0046), araD (CHLAM121), CHLAM133, CHLAM0155, CHLAM0157, CHLAM172 and CHLAM326. CHLAM0046 (hctB) encodes a DNA-binding protein thought to mediate the chromatin compaction [54]. This gene is described to vary among Ct genovars due to internal deletions from a region of the hctB gene encoding lysine- and alanine-rich pentameric repeats [55]. The genes CHLAM0155, CHLAM0157 and CHLAM0172 are all part of a 20.3-kb highly polymorphic genomic region encoding toxin-like genes known as the plasticity zone [45]. The chlamydia plasticity zone has also been known to vary among genovars in accordance with known phylogenetic tissue tropism (urogenital, ocular and LGV) [44, 45]. The CHLAM0326 gene encodes a hypothetical protein without a known function, but this gene has been associated with rectal tropism of Ct genovar G isolates [56]. Finally, the genes araD (CHLAM0121) and CHLAM0133 are known to encode a ribulose-phosphate 3-epimerase and rRNA methylase, but variation has not been linked to specific Ct strains [57].
A total of 47 unique pSTs were identified (Fig. 2, Additional file 3: Table S3) which aggregated into six distinct plasmid clusters, five of which were comparable to those of Harris et al. [9]. The additional cluster consisted solely of genovar K isolates and was likely the result of Ct strains that had previously not been sampled. In comparison to the clusters observed following cgMLST analyses, plasmid analyses showed that the rarer ompA genovars (genovars B, D, G, H, I, J, and K) formed three distinct plasmid clusters. Moreover, three urogenital isolates with genovars G and J were identified that contained plasmids clustering with ocular isolates (genovars A-C), suggesting that exchange of plasmids may have occurred between urogenital and trachoma isolates. Overall, our findings are in agreement with previous studies that suggested co-evolution of Ct plasmids and their chromosome and demonstrated that, although rare, exchange of a plasmid can occur [9, 12]. In addition, allelic variation in some of the plasmid genes was specific and distinct for each cluster, while variation in other genes was mainly specific for the LGV cluster since the allelic variants of the remaining clusters were shared. Potential recombination events were further assessed using ClonalFrameML, which revealed evidence of recombination within the clusters that was restricted to four particular regions in the genome. Further examination should reveal the exact genomic location of these recombination hotspots and their subsequent contribution to C. trachomatis pathogenesis and evolution (Additional file 9: Figure S4).
The observed polymorphic variation was in agreement with previous analyses [9, 13, 58] and comparison to the polymorphic variation found in seven housekeeping genes included in the Chlamydiales MLST scheme highlighted the sequence conservation of the Ct plasmid genes. Although the plasmid genes were highly conserved, much of the diversity appeared to be restricted to one plasmid gene known to be associated with chlamydial virulence. This gene, pgp3, which is associated with increased Ct inflammatory responses [21, 27, 28], was the most polymorphic gene (p-distance = 0.008) and is known to be secreted in the host cell cytosol and its diversity is possibly a result of immune selection [12, 21, 27, 28]. In contrast, pgp4, which functions as a transcriptional regulator of pgp3 and some chromosomally encoded genes, was identified as the most conserved gene. The high sequence conservation of this gene suggests that pgp4 is essential for virulence and infection, although previous studies have demonstrated that pgp4 was dispensable for growth in vitro [25, 59]. Finally, we observed that exchange of alleles between plasmid clusters was very limited. Some alleles were specific to each plasmid cluster, with, in particular, distinct plasmid alleles among LGV isolates. As Ct harbours multiple plasmid copies, these results may be useful to design plasmid assays which can distinguish clinically relevant Ct strain types, for instance to detect the Swedish truncated plasmid variant or to differentiate between LGV/non-LGV isolates. The latter is of clinical importance since LGV infections are more invasive and require extended treatment [9, 12, 60].
A limitation of our study was that it used previously sequenced and stored isolates, for which limited epidemiological or geographical data were available. Since these isolates were originally selected for different objectives, the data available could not be extrapolated to all included isolates. Previous molecular epidemiological studies on Ct using MLST showed no association between Ct strain types and symptoms, anatomical locations, gender or geographical location. Most strain types were also globally distributed with similar ompA genovar distributions [10, 61–63]. These studies therefore suggest that the population structure of Ct is comparable among different human populations and that the effect of selection bias would be minimal. However, MLST is limited by the fact that only a small (but polymorphic) fragment of the genome is used for typing, so samples, that are indistinguishable by MLST type, may still contain (considerable) genomic diversity in the rest of the genome. Future studies should therefore include a well-defined population with known epidemiological and clinical data to gain a better understanding of Ct epidemiology in relation to the population structure while also analysing the whol genome in association with MLST.
In conclusion, a strong association between Ct core genome and plasmid types was observed, consistent with co-evolution of Ct plasmids and their chromosome. Moreover, we suggest that, although rare, plasmid exchange may occur between isolates. Finally, we showed that exchange of alleles between plasmid clusters was limited. Future research should apply the gene-by-gene approach to a well-defined population with known epidemiological and clinical data, as this will enhance our understanding of chlamydial transmission and disease.
Additional files
List of samples used in this study and associated metadata. (XLSX 20 kb)
List of annotated loci present in the Chlamydiales pubMLST database. (XLSX 60 kb)
Plasmid data of the 157 C. trachomatis isolates. Coding is according to the Chlamydiales pubMLST database (http://pubMLST.org/chlamydiales/). The samples are sorted by cluster and plasmid sequence type. (DOCX 30 kb)
Maximum Likelihood phylogenetic tree derived from core genes of the 157 Chlamydia trachomatis isolates in this study (plasmid omitted) (DOCX 380 kb)
List of genome genes and functions with respect to the allelic variance. (XLSX 58 kb)
The number of allelic variants for the CHLAM0456 (tarp) gene in respect to disease phenotype (ocular, urogenital, LGV). The samples are sorted by allelic variant. (DOCX 15 kb)
Maximum Likelihood phylogenetic tree derived from concatenated, aligned nucleotide sequence data from plasmid loci. (DOCX 325 kb)
Maximum Likelihood phylogenetic tree derived from concatenated, aligned nucleotide sequence data from both core and plasmid loci. (DOCX 393 kb)
ClonalFrameML Recombination-corrected. Maximum Likelihood phylogenetic tree derived from concatenated, aligned nucleotide sequence. Dark blue horizontal bars indicate recombination events, light blue indicates lack of substitution, and colours ranging from white to red indicate substitutions with increasing levels of homoplasy. (DOCX 857 kb)
The number of allelic variants for each plasmid gene (pgp1 to 8) in respect to the observed plasmid clusters among 157 C. trachomatis isolates. The samples are sorted by plasmid gene and allelic variant. (DOCX 21 kb)
Acknowledgements
Not applicable.
Funding
OBH and MCJM were funded by a Wellcome Institutional Strategic Support Fund (WTISSF) and the Oxford Martin School, University of Oxford (H2RXJo00); KAJ was funded by Wellcome Trust Biomedical Resource Grant (104992). These funding bodies had no role in study design, analysis, decision to publish, or preparation of the manuscript.
Availability of data and materials
All data generated or analysed during this study are included in this published article and its supplementary information files. Sequence data and links to the public records holding the raw sequences are linked to ID numbers published on the pubMLST website (https://pubmlst.org/chlamydiales/).
Abbreviations
- BIGSdb
Bacterial Isolate Genome Sequence database
- cgMLST
Core Genome MLST
- Ct
C. trachomatis
- ENA
European Nucleotide Archive
- LGV
Lymphogranyloma venereum
- MLST
Multilocus sequence typing
- ORF
Open reading frame
- pST
Plasmid Sequence Type
- T3SS
Type III secretion system
- tarp
translocated actin recruiting phosphoprotein
- WGS
Whole Genome Sequences
Authors’ contributions
BV, SMB, YP, KAJ, MCJM, AvdE and OBH were responsible for the study design; BV and OBH were responsible for the genome analysis; BV, SMB, YP, KAJ, MCJM, AvdE and OBH contributed to the interpretation of the data. BV, SMB, YP, KAJ, MCJM and OBH were responsible for the first draft of the manuscript. All authors were responsible for critical review and approval of the manuscript
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Footnotes
Electronic supplementary material
The online version of this article (10.1186/s12864-018-4522-3) contains supplementary material, which is available to authorized users.
Contributor Information
Bart Versteeg, Phone: +44 (0)20 7636 8636, Email: aalbertus.versteeg@lshtm.ac.uk.
Sylvia M. Bruisten, Email: sbruisten@ggd.amsterdam.nl
Yvonne Pannekoek, Email: y.pannekoek@amc.uva.nl.
Keith A. Jolley, Email: keith.jolley@zoo.ox.ac.uk
Martin C. J. Maiden, Email: martin.maiden@zoo.ox.ac.uk
Arie van der Ende, Email: a.vanderende@amc.uva.nl.
Odile B. Harrison, Email: odile.harrison@zoo.ox.ac.uk
References
- 1.Newman L, Rowley J, Vander HS, Wijesooriya NS, Unemo M, Low N, Stevens G, Gottlieb S, Kiarie J, Temmerman M. Global estimates of the prevalence and incidence of four curable sexually transmitted infections in 2012 based on systematic review and global reporting. PLoS One. 2015;10(12):e0143304. doi: 10.1371/journal.pone.0143304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Mariotti SP, Pascolini D, Rose-Nussbaumer J. Trachoma: global magnitude of a preventable cause of blindness. Br J Ophthalmol. 2009;93(5):563–568. doi: 10.1136/bjo.2008.148494. [DOI] [PubMed] [Google Scholar]
- 3.Taylor HR, Burton MJ, Haddad D, West S, Wright H. Trachoma. Lancet. 2014;384(9960):2142–2152. doi: 10.1016/S0140-6736(13)62182-0. [DOI] [PubMed] [Google Scholar]
- 4.Barnes RC, Wang SP, Kuo CC, Stamm WE. Rapid immunotyping of Chlamydia trachomatis with monoclonal antibodies in a solid-phase enzyme immunoassay. J Clin Microbiol. 1985;22(4):609–613. doi: 10.1128/jcm.22.4.609-613.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Batteiger BE, Newhall WJ, Terho P, Wilde CE, III, Jones RB: Antigenic analysis of the major outer membrane protein of Chlamydia trachomatis with murine monoclonal antibodies. Infect Immun 1986, 53(3):530-533. [DOI] [PMC free article] [PubMed]
- 6.Caldwell HD, Schachter J. Antigenic analysis of the major outer membrane protein of Chlamydia spp. Infect Immun. 1982;35(3):1024–1031. doi: 10.1128/iai.35.3.1024-1031.1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.de Vries HJ, Smelov V, Middelburg JG, Pleijster J, Speksnijder AG, Morre SA. Delayed microbial cure of lymphogranuloma venereum proctitis with doxycycline treatment. Clin Infect Dis. 2009;48(5):e53–e56. doi: 10.1086/597011. [DOI] [PubMed] [Google Scholar]
- 8.Dean D, Bruno WJ, Wan R, Gomes JP, Devignot S, Mehari T, de Vries HJ, Morre SA, Myers G, Read TD, et al. Predicting phenotype and emerging strains among Chlamydia trachomatis infections. Emerg Infect Dis. 2009;15(9):1385–1394. doi: 10.3201/eid1509.090272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Harris SR, Clarke IN, Seth-Smith HM, Solomon AW, Cutcliffe LT, Marsh P, Skilton RJ, Holland MJ, Mabey D, Peeling RW, et al. Whole-genome analysis of diverse Chlamydia trachomatis strains identifies phylogenetic relationships masked by current clinical typing. Nat Genet. 2012;44(4):413–419. doi: 10.1038/ng.2214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Herrmann B, Isaksson J, Ryberg M, Tangrot J, Saleh I, Versteeg B, Gravningen K, Bruisten S. Global multilocus sequence type analysis of Chlamydia trachomatis strains from 16 countries. J Clin Microbiol. 2015;53(7):2172–2179. doi: 10.1128/JCM.00249-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pannekoek Y, Morelli G, Kusecek B, Morre SA, Ossewaarde JM, Langerak AA, van der Ende A. Multi locus sequence typing of Chlamydiales: clonal groupings within the obligate intracellular bacteria Chlamydia trachomatis. BMC Microbiol. 2008;8:42. doi: 10.1186/1471-2180-8-42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Seth-Smith HM, Harris SR, Persson K, Marsh P, Barron A, Bignell A, Bjartling C, Clark L, Cutcliffe LT, Lambden PR, et al. Co-evolution of genomes and plasmids within Chlamydia trachomatis and the emergence in Sweden of a new variant strain. BMC Genomics. 2009;10:239. doi: 10.1186/1471-2164-10-239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Thomas NS, Lusher M, Storey CC, Clarke IN. Plasmid diversity in Chlamydia. Microbiology. 1997;143(Pt 6):1847–1854. doi: 10.1099/00221287-143-6-1847. [DOI] [PubMed] [Google Scholar]
- 14.Stothard DR, Williams JA, Van Der Pol B, Jones RB. Identification of a Chlamydia trachomatis serovar E urogenital isolate which lacks the cryptic plasmid. Infect Immun. 1998;66(12):6010–6013. doi: 10.1128/iai.66.12.6010-6013.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Farencena A, Comanducci M, Donati M, Ratti G, Cevenini R. Characterization of a new isolate of Chlamydia trachomatis which lacks the common plasmid and has properties of biovar trachoma. Infect Immun. 1997;65(7):2965–2969. doi: 10.1128/iai.65.7.2965-2969.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Peterson EM, Markoff BA, Schachter J, de la Maza LM. The 7.5-kb plasmid present in Chlamydia trachomatis is not essential for the growth of this microorganism. Plasmid. 1990;23(2):144–148. doi: 10.1016/0147-619X(90)90033-9. [DOI] [PubMed] [Google Scholar]
- 17.Carlson JH, Whitmire WM, Crane DD, Wicke L, Virtaneva K, Sturdevant DE, Kupko JJ, III, Porcella SF, Martinez-Orengo N, Heinzen RA et al: The Chlamydia trachomatis plasmid is a transcriptional regulator of chromosomal genes and a virulence factor. Infect Immun 2008, 76(6):2273-2283. [DOI] [PMC free article] [PubMed]
- 18.Kari L, Whitmire WM, Olivares-Zavaleta N, Goheen MM, Taylor LD, Carlson JH, Sturdevant GL, Lu C, Bakios LE, Randall LB, et al. A live-attenuated chlamydial vaccine protects against trachoma in nonhuman primates. J Exp Med. 2011;208(11):2217–2223. doi: 10.1084/jem.20111266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.O'Connell CM, Ingalls RR, Andrews CW, Jr, Scurlock AM, Darville T. Plasmid-deficient Chlamydia muridarum fail to induce immune pathology and protect against oviduct disease. J Immunol. 2007;179(6):4027–4034. doi: 10.4049/jimmunol.179.6.4027. [DOI] [PubMed] [Google Scholar]
- 20.Olivares-Zavaleta N, Whitmire W, Gardner D, Caldwell HD. Immunization with the attenuated plasmidless Chlamydia trachomatis L2(25667R) strain provides partial protection in a murine model of female genitourinary tract infection. Vaccine. 2010;28(6):1454–1462. doi: 10.1016/j.vaccine.2009.11.073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ramsey KH, Schripsema JH, Smith BJ, Wang Y, Jham BC, O'Hagan KP, Thomson NR, Murthy AK, Skilton RJ, Chu P, et al. Plasmid CDS5 influences infectivity and virulence in a mouse model of Chlamydia trachomatis urogenital infection. Infect Immun. 2014;82(8):3341–3349. doi: 10.1128/IAI.01795-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Russell M, Darville T, Chandra-Kuntal K, Smith B, Andrews CW, Jr, O'Connell CM. Infectivity acts as in vivo selection for maintenance of the chlamydial cryptic plasmid. Infect Immun. 2011;79(1):98–107. doi: 10.1128/IAI.01105-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sigar IM, Schripsema JH, Wang Y, Clarke IN, Cutcliffe LT, Seth-Smith HM, Thomson NR, Bjartling C, Unemo M, Persson K, et al. Plasmid deficiency in urogenital isolates of Chlamydia trachomatis reduces infectivity and virulence in a mouse model. Pathog Dis. 2014;70(1):61–69. doi: 10.1111/2049-632X.12086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hatt C, Ward ME, Clarke IN. Analysis of the entire nucleotide sequence of the cryptic plasmid of Chlamydia trachomatis serovar L1. Evidence for involvement in DNA replication. Nucleic Acids Res. 1988;16(9):4053–4067. doi: 10.1093/nar/16.9.4053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Song L, Carlson JH, Whitmire WM, Kari L, Virtaneva K, Sturdevant DE, Watkins H, Zhou B, Sturdevant GL, Porcella SF, et al. Chlamydia trachomatis plasmid-encoded Pgp4 is a transcriptional regulator of virulence-associated genes. Infect Immun. 2013;81(3):636–644. doi: 10.1128/IAI.01305-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Thomson NR, Holden MT, Carder C, Lennard N, Lockey SJ, Marsh P, Skipp P, O'Connor CD, Goodhead I, Norbertzcak H, et al. Chlamydia trachomatis: genome sequence analysis of lymphogranuloma venereum isolates. Genome Res. 2008;18(1):161–171. doi: 10.1101/gr.7020108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chen D, Lei L, Lu C, Galaleldeen A, Hart PJ, Zhong G. Characterization of Pgp3, a Chlamydia trachomatis plasmid-encoded immunodominant antigen. J Bacteriol. 2010;192(22):6017–6024. doi: 10.1128/JB.00847-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Li Z, Chen D, Zhong Y, Wang S, Zhong G. The chlamydial plasmid-encoded protein pgp3 is secreted into the cytosol of Chlamydia-infected cells. Infect Immun. 2008;76(8):3415–3428. doi: 10.1128/IAI.01377-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Unemo M, Seth-Smith HMB, Cutcliffe LT, Skilton RJ, Barlow D, Goulding D, Persson K, Harris SR, Kelly A, Bjartling C, et al. The Swedish new variant of Chlamydia trachomatis: genome sequence, morphology, cell tropism and phenotypic characterization. Microbiology. 2010;156(Pt 5):1394–1404. doi: 10.1099/mic.0.036830-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Andersson P, Harris SR, Smith HMBS, Hadfield J, O'Neill C, Cutcliffe LT, Douglas FP, Asche LV, Mathews JD, Hutton SI, et al. Chlamydia trachomatis from Australian aboriginal people with trachoma are polyphyletic composed of multiple distinctive lineages. Nat Commun. 2016;7 [DOI] [PMC free article] [PubMed]
- 31.Hadfield J, Harris SR, Seth-Smith HMB, Parmar S, Andersson P, Giffard PM, Schachter J, Moncada J, Ellison L, Vaulet MLG, et al. Comprehensive global genome dynamics of Chlamydia trachomatis show ancient diversification followed by contemporary mixing and recent lineage expansion. Genome Res. 2017; [DOI] [PMC free article] [PubMed]
- 32.Seth-Smith HM, Harris SR, Skilton RJ, Radebe FM, Golparian D, Shipitsyna E, Duy PT, Scott P, Cutcliffe LT, O'Neill C, et al. Whole-genome sequences of Chlamydia trachomatis directly from clinical samples without culture. Genome Res. 2013;23(5):855–866. doi: 10.1101/gr.150037.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bratcher HB, Corton C, Jolley KA, Parkhill J, Maiden MC. A gene-by-gene population genomics platform: de novo assembly, annotation and genealogical analysis of 108 representative Neisseria meningitidis genomes. BMC Genomics. 2014;15:1138. doi: 10.1186/1471-2164-15-1138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Maiden MC, Harrison OB. Population and functional genomics of Neisseria revealed with gene-by-gene approaches. J Clin Microbiol. 2016;54(8):1949–1955. doi: 10.1128/JCM.00301-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Stephens RS, Kalman S, Lammel C, Fan J, Marathe R, Aravind L, Mitchell W, Olinger L, Tatusov RL, Zhao Q, et al. Genome sequence of an obligate intracellular pathogen of humans: Chlamydia trachomatis. Science. 1998;282(5389):754–759. doi: 10.1126/science.282.5389.754. [DOI] [PubMed] [Google Scholar]
- 36.Huson DH, Bryant D. Application of phylogenetic networks in evolutionary studies. Mol Biol Evol. 2006;23(2):254–267. doi: 10.1093/molbev/msj030. [DOI] [PubMed] [Google Scholar]
- 37.Guindon S, Gascuel O. A simple, fast, and accurate algorithm to estimate large phylogenies by maximum likelihood. Syst Biol. 2003;52(5):696–704. doi: 10.1080/10635150390235520. [DOI] [PubMed] [Google Scholar]
- 38.Didelot X, Wilson DJ. ClonalFrameML: efficient inference of recombination in whole bacterial genomes. PLoS Comput Biol. 2015;11(2):e1004041. doi: 10.1371/journal.pcbi.1004041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Jolley KA, Maiden MC. BIGSdb: scalable analysis of bacterial genome variation at the population level. BMC Bioinformatics. 2010;11:595. doi: 10.1186/1471-2105-11-595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Jolley KA, Hill DM, Bratcher HB, Harrison OB, Feavers IM, Parkhill J, Maiden MC. Resolution of a meningococcal disease outbreak from whole-genome sequence data with rapid web-based analysis methods. J Clin Microbiol. 2012;50(9):3046–3053. doi: 10.1128/JCM.01312-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tamura K, Peterson D, Peterson N, Stecher G, Nei M, Kumar S. MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol Biol Evol. 2011;28(10):2731–2739. doi: 10.1093/molbev/msr121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Nei MKS. Molecular evolution and phylogenetics. Oxford: Oxford University Press; 2000. [Google Scholar]
- 43.Ferreira R, Borges V, Nunes A, Borrego MJ, Gomes JP. Assessment of the load and transcriptional dynamics of Chlamydia trachomatis plasmid according to strains' tissue tropism. Microbiol Res. 2013;168(6):333–339. doi: 10.1016/j.micres.2013.02.001. [DOI] [PubMed] [Google Scholar]
- 44.Nunes A, Borrego MJ, Gomes JP. Genomic features beyond Chlamydia trachomatis phenotypes: what do we think we know? Infect Genet Evol. 2013;16:392–400. doi: 10.1016/j.meegid.2013.03.018. [DOI] [PubMed] [Google Scholar]
- 45.Abdelsamed H, Peters J, Byrne GI. Genetic variation in Chlamydia trachomatis and their hosts: impact on disease severity and tissue tropism. Future Microbiol. 2013;8(9):1129–1146. doi: 10.2217/fmb.13.80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Taylor LD, Nelson DE, Dorward DW, Whitmire WM, Caldwell HD. Biological characterization of Chlamydia trachomatis plasticity zone MACPF domain family protein CT153. Infect Immun. 2010;78(6):2691–2699. doi: 10.1128/IAI.01455-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Yeow TC, Wong WF, Sabet NS, Sulaiman S, Shahhosseini F, Tan GM, Movahed E, Looi CY, Shankar EM, Gupta R, et al. Prevalence of plasmid-bearing and plasmid-free Chlamydia trachomatis infection among women who visited obstetrics and gynecology clinics in Malaysia. BMC Microbiol. 2016;16:45. doi: 10.1186/s12866-016-0671-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lutter EI, Bonner C, Holland MJ, Suchland RJ, Stamm WE, Jewett TJ, McClarty G, Hackstadt T. Phylogenetic analysis of Chlamydia trachomatis tarp and correlation with clinical phenotype. Infect Immun. 2010;78(9):3678–3688. doi: 10.1128/IAI.00515-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Rouli L, Merhej V, Fournier PE, Raoult D. The bacterial pangenome as a new tool for analysing pathogenic bacteria. New Microbes New Infect. 2015;7:72–85. doi: 10.1016/j.nmni.2015.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Nunes A, Gomes JP. Evolution, phylogeny, and molecular epidemiology of Chlamydia. Infect Genet Evol. 2014;23:49–64. doi: 10.1016/j.meegid.2014.01.029. [DOI] [PubMed] [Google Scholar]
- 51.Abdelrahman YM, Belland RJ. The chlamydial developmental cycle. FEMS Microbiol Rev. 2005;29(5):949–959. doi: 10.1016/j.femsre.2005.03.002. [DOI] [PubMed] [Google Scholar]
- 52.Yu HH, Tan M. Sigma28 RNA polymerase regulates hctB, a late developmental gene in Chlamydia. Mol Microbiol. 2003;50(2):577–584. doi: 10.1046/j.1365-2958.2003.03708.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Yu HH, Kibler D, Tan M. In silico prediction and functional validation of sigma28-regulated genes in Chlamydia and Escherichia Coli. J Bacteriol. 2006;188(23):8206–8212. doi: 10.1128/JB.01082-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Klint M, Thollesson M, Bongcam-Rudloff E, Birkelund S, Nilsson A, Herrmann B. Mosaic structure of intragenic repetitive elements in histone H1-like protein Hc2 varies within serovars of Chlamydia trachomatis. BMC Microbiol. 2010;10:81. doi: 10.1186/1471-2180-10-81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Hackstadt T, Brickman TJ, Barry CE, III, Sager J: Diversity in the Chlamydia trachomatis histone homologue Hc2. Gene 1993, 132(1):137-141. [DOI] [PubMed]
- 56.Jeffrey BM, Suchland RJ, Quinn KL, Davidson JR, Stamm WE, Rockey DD. Genome sequencing of recent clinical Chlamydia trachomatis strains identifies loci associated with tissue tropism and regions of apparent recombination. Infect Immun. 2010;78(6):2544–2553. doi: 10.1128/IAI.01324-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Joseph SJ, Didelot X, Rothschild J, de Vries HJ, Morre SA, Read TD, Dean D. Population genomics of Chlamydia trachomatis: insights on drift, selection, recombination, and population structure. Mol Biol Evol. 2012;29(12):3933–3946. doi: 10.1093/molbev/mss198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Comanducci M, Ricci S, Cevenini R, Ratti G. Diversity of the Chlamydia trachomatis common plasmid in biovars with different pathogenicity. Plasmid. 1990;23(2):149–154. doi: 10.1016/0147-619X(90)90034-A. [DOI] [PubMed] [Google Scholar]
- 59.Gong S, Yang Z, Lei L, Shen L, Zhong G. Characterization of Chlamydia trachomatis plasmid-encoded open reading frames. J Bacteriol. 2013;195(17):3819–3826. doi: 10.1128/JB.00511-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.de Vries HJ, Zingoni A, Kreuter A, Moi H, White JA. 2013 European guideline on the management of lymphogranuloma venereum. J Eur Acad Dermatol Venereol. 2015;29(1):1–6. doi: 10.1111/jdv.12461. [DOI] [PubMed] [Google Scholar]
- 61.Bom RJ, van den Hoek A, Wang Q, Long F, de Vries HJ, Bruisten SM. High-resolution typing reveals distinct Chlamydia trachomatis strains in an at-risk population in Nanjing, China. Sex Transm Dis. 2013;40(8):647–649. doi: 10.1097/01.OLQ.0000431355.73217.df. [DOI] [PubMed] [Google Scholar]
- 62.Versteeg B, van Rooijen MS, SvdL MF, de Vries HJ, Bruisten SM. No indication for tissue tropism in urogenital and anorectal Chlamydia trachomatis infections using high-resolution multilocus sequence typing. BMC Infect Dis. 2014;14(1):464. doi: 10.1186/1471-2334-14-464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Versteeg B, Himschoot M, van den Broek IV, Bom RJ, Speksnijder AG, SvdL MF, Bruisten SM. Urogenital Chlamydia trachomatis strain types, defined by high-resolution multilocus sequence typing, in relation to ethnicity and urogenital symptoms among a young screening population in Amsterdam, The Netherlands. Sex Transm Infect. 2015;91(6):415–422. doi: 10.1136/sextrans-2014-051790. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
List of samples used in this study and associated metadata. (XLSX 20 kb)
List of annotated loci present in the Chlamydiales pubMLST database. (XLSX 60 kb)
Plasmid data of the 157 C. trachomatis isolates. Coding is according to the Chlamydiales pubMLST database (http://pubMLST.org/chlamydiales/). The samples are sorted by cluster and plasmid sequence type. (DOCX 30 kb)
Maximum Likelihood phylogenetic tree derived from core genes of the 157 Chlamydia trachomatis isolates in this study (plasmid omitted) (DOCX 380 kb)
List of genome genes and functions with respect to the allelic variance. (XLSX 58 kb)
The number of allelic variants for the CHLAM0456 (tarp) gene in respect to disease phenotype (ocular, urogenital, LGV). The samples are sorted by allelic variant. (DOCX 15 kb)
Maximum Likelihood phylogenetic tree derived from concatenated, aligned nucleotide sequence data from plasmid loci. (DOCX 325 kb)
Maximum Likelihood phylogenetic tree derived from concatenated, aligned nucleotide sequence data from both core and plasmid loci. (DOCX 393 kb)
ClonalFrameML Recombination-corrected. Maximum Likelihood phylogenetic tree derived from concatenated, aligned nucleotide sequence. Dark blue horizontal bars indicate recombination events, light blue indicates lack of substitution, and colours ranging from white to red indicate substitutions with increasing levels of homoplasy. (DOCX 857 kb)
The number of allelic variants for each plasmid gene (pgp1 to 8) in respect to the observed plasmid clusters among 157 C. trachomatis isolates. The samples are sorted by plasmid gene and allelic variant. (DOCX 21 kb)
Data Availability Statement
All data generated or analysed during this study are included in this published article and its supplementary information files. Sequence data and links to the public records holding the raw sequences are linked to ID numbers published on the pubMLST website (https://pubmlst.org/chlamydiales/).