

Abstract
The nephrotoxicity of cisplatin limits its clinical application. Schizandrin B (SchB) has been demonstrated to have a variety of potential cytoprotective activities. The present study explored the molecular mechanisms by which SchB inhibits the dichlorodiammine platinum (DDP)-induced apoptosis of HK-2 proximal tubule epithelial cells. In vitro assays demonstrated that SchB increased the viability of HK-2 cells, alleviated the cis-DDP-induced activation of caspase-3, reduced apoptosis and improved the nuclear morphology of HK-2 cells. Additionally, the mechanism underlying the cis-DDP-induced apoptosis was indicated to involve the activation of p53, c-Jun-N-terminal kinase (JNK) and p38 signaling. Furthermore, SchB was demonstrated to activate extracellular signal-regulated kinase (ERK) and nuclear factor κB (NF-κB) signaling, and induce the expression of survivin. The inhibition of ERK and NF-κB signaling using U0126 and pyrollidine dithiocarbamate, respectively, inhibited the expression of survivin, whereas blocking the expression of survivin using small interfering RNA inhibited the alleviating effect of SchB on cis-DDP-induced apoptosis as indicated by a reduction in cleaved caspase-3 expression. In conclusion, SchB regulates ERK/NF-κB signaling to induce the expression of survivin, thereby alleviating cis-DDP-induced renal injury.
Keywords: schizandrin B, cisplatin nephrotoxicity, survivin, HK-2 cells
Introduction
Cisplatin (cis-dichlorodiammine platinum, cis-DDP) is widely used in the treatment of various solid tumors, for example, testicular cancer, in which remission rates of up to 90% have been obtained (1). However, the development of drug resistance and undesirable side-effects have limited the clinical application of DDP. These side-effects include nephrotoxicity, neurotoxicity and ototoxicity (2). DDP-induced nephrotoxicity primarily occurs in the renal proximal tubule, with manifestations of epithelial cell necrosis, glomerular sclerosis, renal dysfunction and renal failure (3–6). Therefore, the development of therapeutic strategies and drugs that relieve the side-effects of DDP may enable expansion of its clinical applications.
Previous studies have confirmed that DDP-induced kidney injury is primarily caused by apoptosis (7) due to oxidative stress injury and the inflammatory infiltration of epithelial cells in the renal proximal tubule (8). In a previous study conducted by the present research team, it was shown that DDP induces apoptosis by causing oxidative stress in HK-2 proximal tubule epithelial cells (9). Mitogen-activated protein kinase (MAPK) signaling pathways have been demonstrated to serve a key role in this pathological process. MAPKs are composed of three key kinases, namely extracellular signal-regulated kinase (ERK), c-Jun-N-terminal kinase (JNK) and p38 (10,11). Following induction by DDP, renal tissues produce large amounts of reactive oxygen species (ROS) and inflammatory factors, which activate and initiate apoptosis through MAPK signaling (12,13). The nuclear factor-κB (NF-κB) signaling pathway inhibits apoptosis. The activation of NF-κB and its translocation to the nucleus regulates the expression of Fas-associated death domain-like interleukin-1β-converting enzyme-inhibitory protein (FLIP) (14) and X-linked inhibitor-of-apoptosis protein (XIAP) (15,16). The expression of FLIP then inhibits the activation of caspase-8 (17), whereas the expression of XIAP inhibits the function of second mitochondria-derived activator of caspase (also known as Diablo) (18). Therefore, abrogation of DPP-induced renal cell apoptosis may be achieved by blocking MAPK signaling or activating NF-κB signaling.
Schizandrin B (SchB) is extracted from the fruit of Magnoliaceae Schisandra chinensis (Turcz.) Baill (19). SchB has been shown to alleviate damage in a number of different types of tissues and cells, including hepatocytes (20,21), nerve cells (22), renal tissues (23) and cardiomyocytes (24,25). Additionally, SchB has been demonstrated to have a potent anticancer effect (26). However, the cytoprotective mechanism of SchB has not been fully elucidated. Therefore, the effect of SchB on DDP-exposed proximal tubular epithelial HK-2 cells was evaluated in the present study with the aim of elucidating the protective mechanism of SchB.
Materials and methods
Antibodies and reagents
Cis-DDP and SchB were purchased from Sigma-Aldrich (Merck KGaA, Darmstadt, Germany). The primary antibodies targeting cleaved-caspase-3 (cat. no. 9661), ERK (cat. no. 4695), phospho (p)-ERK (cat. no. 4370), IκB kinase (IKK)β (cat. no. 8943), IKKα (cat. no. 2682), p-IKKα/β (cat. no 2697), inhibitor of NF-κBα (IκBα; cat. no. 4812), p-IκBα (cat. no. 2859), NF-κB p65 (cat. no. 8242), p-NF-κB p65 (cat. no. 3033), survivin (cat. no. 2808) and GAPDH (cat. no: 5174), all used at 1:1,000 dilution, were purchased from Cell Signaling Technology, Inc. (Danvers, MA, USA). IRDye 800CW goat anti-mouse secondary antibodies (P/N 926-80010; 1:5,000 dilution) and Alexa IRDye 700RD goat anti-rabbit secondary antibodies (P/N 925-68070; 1:5,000 dilution) were purchased from LI-COR Biosciences (Lincoln, NE, USA), and 4′,6-diamidino-2-phenylindole (DAPI), pyrollidine dithiocarbamate (PDTC) and U0126 were purchased from Beyotime Institute of Biotechnology (Shanghai, China).
Cell culture
HK-2 cells were purchased from American Type Culture Collection (Manassas, VA, USA). The cells were cultured in keratinocyte serum-free medium containing 0.05 mg/ml bovine pituitary extract and 5 ng/ml human recombinant epidermal growth factor (all HyClone; GE Healthcare Life Sciences, Logan, UT, USA). The culture was incubated under an atmosphere of 5% CO2 at 37°C and passaged the next day.
Cell viability assay
HK-2 cells (1×106 cells/well) were seeded in 96-well plates. Following stimulation with cis-DDP and/or SchB as indicated, the medium was exchanged, and 10 µl Cell Counting kit (CCK)-8 test solution (Dojindo Molecular Technologies, Inc., Kumamoto, Japan) was added to each well for 1 h at 37°C The absorbance of the culture solution was measured at 450 nm using a microplate reader.
Apoptosis rate assay
HK-2 cells (1×107 cells/well) were seeded into 6-well plates, and SchB or cis-DDP was added 24 h later. Following the appropriate incubation period, the cells were stained with Annexin V-fluorescein isothiocyanate and propidium iodide (Beyotime Institute of Biotechnology) for 30 min. A flow cytometer (BD FACSVerse; BD Biosciences, Franklin Lakes, NJ, USA) was used to determine the apoptotic rate using CellQuest Pro 3.3 software (BD Biosciences).
DAPI staining
HK-2 cells (1×107 cells/well) were seeded into 6-well plates, and SchB or cis-DDP was added 24 h later. Following the appropriate incubation period, the cells were fixed in 4% paraformaldehyde for 30 min at room temperature and then stained with DAPI (Beyotime Institute of Biotechnology) for 1 h at room temperature. An Olympus IX71 inverted microscope (Olympus Corporation, Tokyo, Japan) was used to evaluate the nuclear morphology.
Double-wavelength in-cell western blotting assay
HK-2 cells (1×106 cells/well) were seeded in a 96-well plate. Following treatment with cis-DPP and/or SchB as indicated, the cells were fixed with 4% paraformaldehyde for 20 min at room temperature and then washed three times with 0.1% Triton-100%/phosphate-buffered saline (PBS). The cells were then incubated with rabbit and mouse antibodies against cleaved-caspase-3 and GAPDH, respectively, overnight at 4°C, followed by incubation with goat anti-rabbit antibody labelled with Alexa Fluor® 700 or goat anti-mouse antibody labelled with IRDye 800 for 2 h at room temperature. The results were observed using an Odyssey imaging system (LI-COR Biosciences).
Western blotting
Following the aforementioned treatments, the HK-2 cells were washed with pre-chilled PBS and then lysed with cell lysis buffer (Beyotime Institute of Biotechnology) on ice for 40 min. Next, the lysate was collected and mixed with 2X loading buffer for sodium dodecyl sulfate-polyacrylamide (12% separation gel and 4% concentrated gel) gel electrophoresis. The proteins were then transferred to a nitrocellulose membrane, and the membrane was incubated in blocking solution (5% skimmed milk) at room temperature for 2 h. The membrane was incubated overnight at 4°C with the primary antibody, and then incubated with a fluorescent secondary antibody for 1 h at room temperature and imaged using the Odyssey imaging system (LI-COR Biosciences).
Cell transfection and interference
Small interfering RNAs (siRNAs) against survivin (Survivin RNAi-1: Sense, ACA GAC AGA CAG UCU GUG GU and antisense, UUU GUC UGU CUG UCA GAC ACC; Survivin RNAi-2: Sense, CAG ACA GAC AGU CUG UGG UUU and antisense, UUGUCUGUCUGUCAGACACCA) and a pGL3-human survivin promoter luciferase (pG3L-hSurvivin-Luc) plasmid were synthesized by Shanghai GenePharma Co., Ltd. (Shanghai, China). The survivin siRNAs or pGL3-hSur-vivin-Luc and Lipofectamine® 2000 (Gibco; Thermo Fisher Scientific, Inc., MA, USA) were mixed in opti-MEM (Gibco; Thermo Fisher Scientific, Inc.) and incubated with HK-2 cells for 72 h. Survivin expression was detected by western blotting. The luciferase activity of pG3L-hSurvivin-Luc was measured using a GloMax 20/20 Luminometer (Promega Corporation, Madison, WI, USA).
Signaling pathway screening
HK-2 cells were pre-incubated with SchB for 2 h and/or stimulated with cis-DDP for various time periods at 37°C, as indicated. The cell lysates were collected and analyzed using PathScan Antibody Array kits (Cell Signaling Technology, Inc.) according to the instructions provided by the manufacturer. Fluorescence emission points on the chip were observed at 680 nm using an Odyssey imaging system. The grey-scale value was calculated and statistically analyzed.
Statistical analysis
All data are presented as the mean ± standard deviation. One-way analysis of variance followed by Dunnett’s test were used for inter-group comparisons. Statistical analysis of the data was performed using SPSS 13.0 software (SPSS, Inc., Chicago, IL, USA). P<0.05 was considered to indicate a statistically significant difference.
Results
Effects of SchB on the cis-DDP-induced loss of viability of HK-2 cells
In a previous study, in vitro experiments indicated that SchB is able to alleviate the toxicity of DDP to HK-2 cells (27). In the present study, the effect of SchB on the cis-DDP-induced apoptosis of HK-2 cells was further evaluated. To test the effect of SchB on the viability of HK-2 cells, the cells were incubated with SchB at various concentrations (0, 2.5, 5, 10, 20, 40 and 80 µM) for 24 h. Then, cell viability was determined using a CCK-8 assay. The experimental results are shown in Fig. 1A. When the concentration of SchB was 20 or 40 µM, the cell viability was significantly increased compared with that of the untreated cells. However, a SchB concentration of 80 µM was toxic to the cells, and significantly reduced their viability. To evaluate the time-dependent response to SchB, HK-2 cells were incubated with 40 µM SchB for various time periods (0, 6, 12, 18, 24, 30, 36 and 42 h). As shown in Fig. 1B, SchB significantly increased the viability of the HK-2 cells compared with that of the untreated cells when the incubation time was ≥18 h. To evaluate the effect of cis-DPP on cell viability, HK-2 cells were incubated with cis-DDP at different concentrations (0, 2.5, 5, 10, 20 and 30 µM) for 24 h, and cell viability was determined. As shown in Fig. 1C, concentrations of cis-DDP ≥5 µM were significantly cytotoxic to the HK-2 cells, and cis-DDP concentrations ≥10 µM strongly inhibited the viability of the HK-2 cells. Therefore, 10 µM cis-DDP was considered the toxic dose for subsequent experiments. To evaluate the effect of SchB on that of cis-DPP, HK-2 cells were pre-incubated with different concentrations of SchB (0, 2.5, 5, 10, 20, 40 and 80 µM) for 2 h and then stimulated with 10 µM cis-DDP for 24 h. The viability of the cells was subsequently determined. The results indicated that SchB concentrations ≥10 µM significantly alleviated the reduction in HK-2 cell viability induced by cis-DDP. A SchB concentration of 80 µM exhibited no additional impact compared with that of a 40 µM concentration on the decreased viability of the HK-2 cells induced by cis-DDP. Therefore, 10, 20 and 40 µM SchB were used as the low, medium and high dose groups, respectively, in the subsequent experiments.
Figure 1.

Effects of SchB on the cis-DDP-induced loss of viability of HK-2 cells. (A) HK-2 cells were incubated with SchB at different concentrations (0, 2.5, 5, 10, 20, 40 and 80 µM) for 24 h. Cell viability was then determined using a Cell Counting kit-8 assay. (B) HK-2 cells were incubated with 40 µM SchB for 0, 6, 12, 18, 24, 30, 36 and 42 h, and the cell viability was then determined. (C) HK-2 cells were incubated with cis-DDP at different concentrations (0, 2.5, 5, 10, 20 and 30 µM) for 24 h, and cell viability was then determined. (D) Cells were pre-incubated with different concentrations of SchB (0, 2.5, 5, 10, 20, 40 and 80 µM) for 2 h and then stimulated with 10 µM cis-DDP for 24 h. The viability of the cells was determined. Data are presented as the mean ± standard deviation (n=3). (A-C) *P<0.05 and **P<0.01 vs. the control group (0 µM SchB); (D) *P<0.05 and **P<0.01 vs. the group treated with cis-DDP alone. SchB, schizandrin B; cis-DDP, cis-dichlorodiammine platinum.
Effects of SchB on the cis-DDP-induced apoptosis of HK-2 cells
The effects of SchB on the cis-DDP-induced apoptosis of HK-2 cells were evaluated. Following the pre-incubation of HK-2 cells with 10, 20 and 40 µM SchB for 2 h, the cells were stimulated with 10 µM cis-DDP for 24 h and stained with DAPI to observe the nuclear morphology. The results (Fig. 2A) demonstrated that cis-DDP induced nuclear DNA shrinkage and fragmentation in the HK-2 cells, whereas SchB significantly attenuated the cis-DDP-induced nuclear condensation and fragmentation. Following co-staining of the HK-2 cells with Annexin V-fluorescein isothiocyanate and propidium iodide, the apoptotic and survival rates of the HK-2 cells were determined by flow cytometry. The results (Fig. 2B) demonstrate that SchB significantly reduced the cis-DDP-induced apoptosis of HK-2 cells. Caspase-3 is an executive apoptotic protein (28). Following the pre-incubation of HK-2 cells with 10, 20 and 40 µM SchB for 2 h, the cells were stimulated with 10 µM cis-DDP for 24 h. Then, the activation of caspase-3 was determined by in-cell western blot analysis. The results (Fig. 2C) indicate that the activation of cleaved caspase-3 was increased during cis-DDP stimulation. However, when the cells were pre-incubated with SchB, the activation of caspase-3 by cis-DDP was significantly attenuated compared with that in the cells that did not undergo SchB pretreatment. These results indicate that SchB effectively blocked the cis-DDP-induced apoptosis of HK-2 cells.
Figure 2.

Effects of SchB on the cis-DDP-induced apoptosis of HK-2 cells. (A) Following the pre-incubation of HK-2 cells with 10, 20 and 40 µM SchB for 2 h, the cells were stimulated with 10 µM cis-DDP for 24 h and then stained with DAPI to observe nuclear morphology (magnification, ×400). (B) Following co-staining of the HK-2 cells with Annexin V-FITC and PI, the apoptotic rates of the HK-2 cells were determined by flow cytometry. (C) Following the pre-incubation of HK-2 cells with 10, 20 and 40 µM SchB for 2 h, the cells were stimulated with cis-DDP for 24 h. The activation of caspase-3 was then determined by in-cell western blot analysis. Data are presented as the mean ± standard deviation (n=3). *P<0.05 and **P<0.01 vs. the blank control group or cis-DDP-control group. SchB, schizandrin B; cis-DDP, cis-dichlorodiammine platinum.
Effects of SchB and cis-DDP on the apoptosis/survival signaling pathway of HK-2 cells
The signaling targets in HK-2 cells on which SchB may exert cytoprotective effects were screened. HK-2 cells were used to create a SchB (40 µM) incubation group, a cis-DDP (10 µM) incubation group and a SchB (40 µM) + cis-DDP (10 µM) co-incubation group, which were incubated for 1, 2, 4, 8 and 16 h. The activation of apoptosis/survival signal transduction pathways was determined using protein chips. As shown in Fig. 3A and B, at 1 h after cis-DDP stimulation, p53, p38 and JNK were phosphorylated to different extents. After 16 h, cis-DDP induced the activation of cleaved caspases-3 and -7, leading to poly(ADP-ribose) polymerase (PARP) degradation. These results suggest that cis-DDP may have induced the apoptosis of HK-2 cells via the activation of p53, p38 and JNK signaling. Incubation with SchB alone did not activate p53, p38 and JNK and exhibited no effect on caspases-3 and -7 or PARP. However, with SchB incubation, ERK was strongly phosphorylated in the HK-2 cells, with decreased expression of total IκBα and increased expression of phosphorylated (p)-lκBα. Notably, following incubation with SchB for 16 h, survivin protein was strongly expressed. Survivin is an inhibitor of apoptosis (29) and may be the key protein involved in the protective effect of SchB against DDP nephrotoxicity. When the HK-2 cells were co-incubated with SchB (40 µM) and cis-DDP (10 µM), and when combined with the effects of p-p38, p-JNK, p-lκBα, p-ERK and survivin, the cleaved-caspases-3 and -7 and cleaved-PARP apoptosis proteins were significantly inhibited. On the basis of these results (Fig. 3B), it appears that ERK and NF-κB signaling, and survivin serve important roles in the alleviation of cis-DDP-induced apoptosis by SchB in HK-2 cells. The predicted signaling pathways in the cis-DDP-induced apoptosis and SchB-induced survival of HK-2 cells are shown in Fig. 3C.
Figure 3.

Effects of SchB and cis-DDP on the apoptosis/survival signaling pathway of HK-2 cells. HK-2 cells were used to establish a SchB (40 µM) incubation group, a cis-DDP (10 µM) incubation group and a SchB (40 µM) + cis-DDP (10 µM) co-incubation group and the cells were incubated for 1, 2, 4, 8 and 16 h. (A) The activation of associated signal transduction pathways was determined using protein chips. (B) Heat-maps of the protein chip results. (C) The predicted signaling pathways in the cis-DDP-induced apoptosis and SchB-induced survival of HK-2 cells. SchB, schizandrin B; cis-DDP, cis-dichlorodiammine platinum; ERK, extracellular signal-regulated kinase; JNK, c-Jun-N-terminal kinase; NF-κB, nuclear factor κB; IκBα, inhibitor of NF-κBα; PARP, poly(ADP-ribose) polymerase; p, phosphorylated.
Effects of the activation of ERK/NF-κB signaling by SchB on the cis-DDP-induced apoptosis of HK-2 cells
To confirm the activation of ERK and NF-κB by SchB, HK-2 cells were incubated with 40 µM SchB for 0, 0.5, 1, 2, 4 and 8 h, and the cell lysates were collected. Western blotting was performed to determine the phosphorylation of ERK, IKKα/β, IκBα and p65. As shown in Fig. 4A, SchB activated ERK and NF-κB signaling at the five time points tested, with maximum activation for ERK, IKKα/β, IκBα and p65 at 0.5, 2, 2 and 8 h, respectively. The HK-2 cells were incubated with 10, 20 and 40 µM SchB for 0.5, 2, 2 and 8 h, and the phosphorylation levels of ERK, IKKα/β, IκBα and p65, respectively, were determined. As shown in Fig. 4B, SchB activated ERK and NF-κB signaling at the three concentrations tested. To confirm the roles of activated ERK and NF-κB signaling in the DDP-induced apoptosis of HK-2 cells, the HK-2 cells were incubated with ERK inhibitor U0126 (10 µM) or NF-κB inhibitor PDTC (20 µM) for 2 h, followed by incubation with SchB for 4 h and stimulation with cis-DDP for 24 h. Changes in the expression of cleaved caspase-3 protein were determined (Fig. 5). As shown in Fig. 5A, SchB effectively attenuated the cis-DDP-induced expression of cleaved caspase-3, indicating that it reduced apoptosis, whereas U0126 and PDTC inhibited the anti-apoptotic effect of SchB to different extents. These results suggest that the activation of ERK and NF-κB signaling serves a critical role in the inhibitory effect of SchB against cis-DDP-induced apoptosis in HK-2 cells.
Figure 4.

Signaling via ERK/NF-κB activation in the cis-DDP-induced apoptosis of HK-2 cells. (A) HK-2 cells were incubated with 40 µM SchB for 0, 0.5, 1, 2, 4 and 8 h, and the cell lysates were collected. Western blotting was performed to determine the phosphorylation of ERK, IKKα/β, IκBα and p65. (B) HK-2 cells were incubated with 10, 20 and 40 µM SchB for 0.5, 2, 2 and 8 h, and the phosphorylation of ERK, IKKα/β, IκBα and p65 was determined, respectively. Data are presented as the mean ± standard deviation (n=3). *P<0.05 and **P<0.01 vs. the control group [(A), 0 h; (B), 0 µM]. SchB, schizandrin B; cis-DDP, cis-dichlorodiammine platinum; ERK, extracellular signal-regulated kinase; NF-κB, nuclear factor κB; IκBα, inhibitor of NF-κBα; IKK, IκB kinase; p, phosphorylated.
Figure 5.

Effects of survivin on the cis-DDP-induced apoptosis of HK-2 cells. (A) The hSurvivin RNAi sequence was transfected into HK-2 cells. Transfected and untransfected cells were then incubated with 40 µM SchB alone or with PDTC or U0126 for 4 h and stimulated with cis-DDP for 24 h. The activation of cleaved-caspase-3 protein was determined. (B) Survivin RNAi sequences were synthesized and transfected into HK-2 cells, which were incubated with 40 µM SchB for 16 h. The expression of survivin was then determined. Data are presented as the mean ± standard deviation (n=3). *P<0.05 and **P<0.01 vs. the control group. SchB, schizandrin B; cis-DDP, cis-dichlorodiammine platinum; PDTC, pyrollidine dithiocarbamate; RNAi, RNA interference; hSurvivin, human survivin.
Effect of the regulation of survivin expression by NF-κB on the cis-DDP-induced apoptosis of HK-2 cells
Survivin is an anti-apoptotic protein with a molecular weight of 16 kDa. Survivin binds to and inhibits caspase-3 activity, thereby inhibiting apoptosis and promoting cell division (29,30). The aforementioned antibody chip results indicate that SchB might act as a survivin inducer in an anti-apoptotic role. Thus, the effect of survivin activation on cis-DDP nephrotoxicity was evaluated. HK-2 cells were incubated with 0, 10, 20 and 40 µM SchB for 16 h, and the expression of survivin protein was determined. As shown in Fig. 6A, SchB significantly activated survivin expression at the three concentrations tested. The expression of survivin was also determined following the incubation of HK-2 cells with 40 µM SchB for 0, 1, 4, 8 and 16 h. As shown in Fig. 6B, SchB significantly increased survivin expression at the three time points tested. HK-2 cells were incubated with ERK inhibitor U0126 or NF-κB inhibitor PDTC for 2 h, followed by incubation with SchB for 16 h, and then the expression of survivin was determined. As shown in Fig. 6C, PDTC and U0126 inhibited the SchB-induced expression of survivin.
Figure 6.

ERK/NF-κB mediates SchB-induced survivin expression in HK-2 cells. (A) HK-2 cells were incubated with 0, 10, 20 and 40 µM SchB for 16 h, and the expression of survivin protein was determined. (B) HK-2 cells were incubated with 40 µM SchB for 0, 1, 4, 8 and 16 h, and the expression of survivin protein was determined. (C) HK-2 cells were incubated with ERK inhibitor U0126 and NF-κB inhibitor PDTC for 2 h, followed by incubation with SchB for 16 h, and then the expression of survivin was determined. (D) The luciferase reporter gene pGL3-hSurvivin promoter luciferase was transfected into HK-2 cells. These cells were then incubated with 10, 20 and 40 µM SchB for 16 h or pre-incubated with U0126 and PDTC and subsequently incubated with 40 µM SchB. The fluorescence intensity of the HK-2 cells was then detected using a fluorescence microplate reader. Data are presented as the mean ± standard deviation (n=3). *P<0.05 and **P<0.01. vs. the control group [(A), 0 µM; (B), 0 h] or as indicated. SchB, schizandrin B; cis-DDP, cis-dichlorodiammine platinum; PDTC, pyrollidine dithiocarbamate; ERK, extracellular signal-regulated kinase; NF-κB, nuclear factor κB.
Subsequently, a survivin promoter luciferase reporter construct (pGL3-hSurvivin-Luc) was constructed and transfected into HK-2 cells. The transfected cells were incubated with 10, 20 or 40 µM SchB for 16 h, or pre-incubated with 10 µM U0126 or 20 µM PDTC and subsequently incubated with 40 µM SchB for 16 h. As shown in Fig. 6D, SchB induced pGL3-hSurvivin-Luc fluorescence in a dose-dependent manner. Pre-incubation with PDTC or U0126 significantly inhibited the SchB-induced fluorescence intensity of the luciferase reporter gene compared with that in the unpretreated cells. These results indicate that the activation of NF-κB and ERK regulated the expression of survivin. To confirm the effect of survivin activation on the cis-DDP-induced apoptosis of HK-2 cells, survivin siRNA was synthesized for the RNA interference (RNAi) of survivin. Survivin siRNA was transfected into HK-2 cells, which were incubated with 40 µM SchB for 16 h. The expression of survivin was then determined. As shown in Fig. 5B, hSurvivin RNAi successfully inhibited the expression of survivin. The interference efficiencies, presented as the fractional reductions in survivin expression, for Survivin RNAi-1 were 0.82 and 0.85 at 1 and 2 µg/ml. The interference efficiencies of Survivin RNAi-2 were 0.69 and 0.58 at 1 and 2 µg/ml. Therefore, 2 µg/ml Survivin RNAi-2 was selected for the subsequent experiments. The survivin siRNA was transfected into HK-2 cells, which were then incubated with 40 µM SchB for 4 h and stimulated with cis-DDP for 24 h. The activation of cleaved caspase-3 protein was then determined. As shown in Fig. 5A, SchB effectively inhibited the activation of cleaved caspase 3, indicating that it abrogated the cis-DDP-induced apoptosis of HK-2 cells. Thus, blocking the expression of survivin is indicated to inhibit the anti-apoptotic effect of SchB, which indicates that survivin serves an anti-apoptotic role as an activation target of SchB. In summary, it appears that SchB alleviated the cis-DDP-induced apoptosis of HK-2 cells by activating NF-κB/ERK and thereby regulating the expression of survivin.
Discussion
Since the FDA approved platinum compounds for the treatment of various solid tumors in the 1970s, DDP has been an important front-line anticancer drug, but its severe side have greatly limited its clinical application. The development of a suitable treatment strategy to alleviate the side-effects of DDP should greatly increase its clinical applicability. SchB is a naturally existing small molecule that has been shown to have a variety of cytoprotective activities (31). To the best of our knowledge, the present study is the first to provide evidence that SchB is able to alleviate DDP-induced renal injury by activating the ERK/NF-κB signaling pathway in vitro.
DDP-induced apoptosis is a well-known process for killing proliferative cancer cells. However, the excessive re-absorption of DDP by the renal proximal tubule may reduce the proliferation rate of the epithelial cells in the tubule (32,33). In addition, an excessive concentration of DDP may induce renal tubular and peripheral tissue cells to produce inflammatory factors, such as tumor necrosis factor-α, or large amounts of ROS and reactive nitrogen clusters; to induce mitochondrial apoptotic pathways and death receptor apoptotic pathways; to activate caspases-3, -6, -9 and -12; and to eventually induce epithelial cell apoptosis in the renal proximal tubule (34,35). In the present study, 10 µM DDP significantly decreased cell viability, activated caspase-3, induced nuclear condensation and fragmentation, and increased the apoptosis rate of HK-2 cells. However, when the HK-2 cells were pre-incubated with SchB, the DDP-induced apoptosis was significantly inhibited. These experimental results confirm the potential therapeutic effect of SchB on cis-DDP-induced renal injury.
The nephrotoxicity of cis-DDP is associated with multiple apoptosis-related signaling pathways, the most important of which are MAPKs (36); for example, p53 serves a key role in DDP-induced apoptosis (37). The experiments conducted in the present study verified the possible involvement of SchB in the protection of HK-2 cells. Protein chip screening was used to determine the possible target pathways via which SchB acts as an anti-apoptotic agent. Under cis-DDP stimulation, p53, p38 and JNK were phosphorylated to varying extents, with subsequent enhanced activation of caspases-3 and -7 and PARP degradation. Thus, p53, JNK and p38 may mediate the DDP-induced apoptosis of HK-2 cells. When pre-incubated with SchB, the cis-DDP-induced activation of caspases-3 and -7 and PARP degradation in the HK-2 cells was inhibited, but the cis-DDP-induced activation of p53, JNK and p38 was not inhibited. For the HK-2 cells incubated with SchB alone, ERK was phosphorylated, the expression of total IκBα decreased while that of p-IκBα increased, and survivin was highly activated. The CCK-8 assay demonstrated that incubation with SchB alone promoted an increase in HK-2 cell viability. Therefore, it may be speculated that with cis-DDP incubation, the activation of JNK, p38 and p53 leads to apoptosis. Furthermore, with SchB incubation, the activation of NF-κB and ERK leads to cell proliferation, and co-incubation with cis-DDP and SchB promotes HK-2 cell survival compared with that for cells incubated with cis-DDP alone. Therefore, it appears that MAPKs serve a dual role in the nephrotoxicity of cisplatin. The activation of JNK and p38 induces the apoptosis of HK-2 cells, whereas the activation of ERK protects HK-2 cells.
Survivin is a small molecular anti-apoptotic protein that binds to caspase-3 and inhibits its activation (30,38). In the present study, it was demonstrated that the incubation of HK-2 cells with SchB alone induced the expression of survivin and activated survivin promoter luciferase. When siRNA was used to interfere with the expression of survivin, the cis-DDP-induced induction of cleaved caspase-3 in the HK-2 cells was markedly inhibited, indicating that the activation of survivin by SchB may be the mechanism underlying its anti-apoptotic effects. Western blot assays confirmed that SchB activated ERK and that the ERK inhibitor U0126 downregulated the expression of survivin and blocked the anti-apoptotic effect of SchB. The anti-apoptotic effect of NF-κB has been demonstrated in a number of studies, as summarized in a previous review (39). In the present study, SchB activated NF-κB, and the NF-κB inhibitor PDTC downregulated survivin expression and the activity of the survivin promoter, which is indicative of the ability of SchB to inhibit cis-DDP-induced apoptosis. PDTC inhibited the expression of survivin more strongly than did U0126. Therefore, it appears that NF-κB signaling has a dominant role in the regulation of survivin expression. These results indicate that SchB regulates the expression of survivin via ERK/NF-κB to alleviate the cis-DDP-induced apoptosis of HK-2 cells.
In conclusion, the present study demonstrated for the first time, to the best of our knowledge, that SchB is able to alleviate the cis-DDP-induced apoptosis of HK-2 cells in vitro. SchB likely activates the ERK/NF-κB signaling pathway, which in turn, activates survivin (Fig. 7). These results suggested that SchB may serve as a survivin inducer in the treatment of DDP nephrotoxicity.
Figure 7.
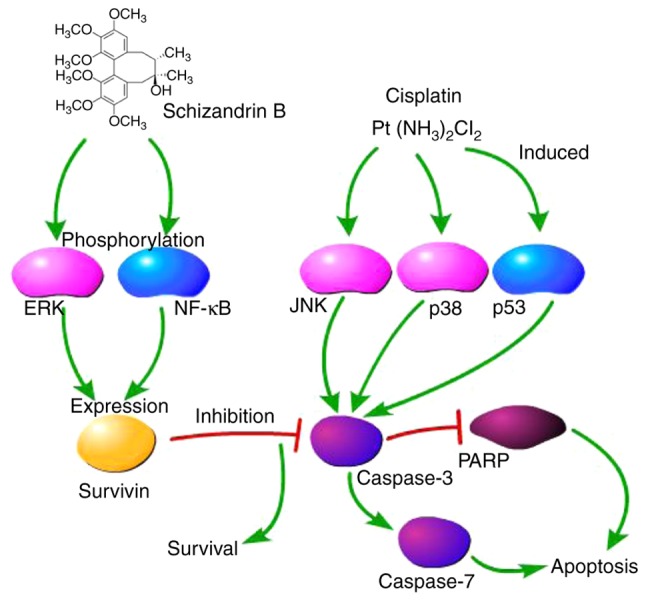
Mechanism of the cisplatin-induced nephrotoxicity attenuated by schizandrin B. ERK, extracellular signal-regulated kinase; NF-κB, nuclear factor κB; JNK, c-Jun-N-terminal kinase; PARP, poly(ADP ribose) polymerase.
Footnotes
Competing interests
The authors declare that they have no competing interests.
References
- 1.Einhorn LH. Curing metastatic testicular cancer. Proc Natl Acad Sci USA. 2002;99:4592–4595. doi: 10.1073/pnas.072067999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Karasawa T, Steyger PS. An integrated view of cisplatin-induced nephrotoxicity and ototoxicity. Toxicol Lett. 2015;237:219–227. doi: 10.1016/j.toxlet.2015.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Brock PR, Knight KR, Freyer DR, Campbell KC, Steyger PS, Blakley BW, Rassekh SR, Chang KW, Fligor BJ, Rajput K, et al. Platinum-induced ototoxicity in children: A consensus review on mechanisms, predisposition, and protection, including a new International Society of Pediatric Oncology Boston ototoxicity scale. J Clin Oncol. 2012;30:2408–2417. doi: 10.1200/JCO.2011.39.1110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.McWhinney SR, Goldberg RM, McLeod HL. Platinum neurotoxicity pharmacogenetics. Mol Cancer Ther. 2009;8:10–16. doi: 10.1158/1535-7163.MCT-08-0840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Shen DW, Pouliot LM, Hall MD, Gottesman MM. Cisplatin resistance: A cellular self-defense mechanism resulting from multiple epigenetic and genetic changes. Pharmacol Rev. 2012;64:706–721. doi: 10.1124/pr.111.005637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yao X, Panichpisal K, Kurtzman N, Nugent K. Cisplatin nephrotoxicity: A review. Am J Med Sci. 2007;334:115–124. doi: 10.1097/MAJ.0b013e31812dfe1e. [DOI] [PubMed] [Google Scholar]
- 7.Nozaki Y, Kinoshita K, Hino S, Yano T, Niki K, Hirooka Y, Kishimoto K, Funauchi M, Matsumura I. Signaling Rho-kinase mediates inflammation and apoptosis in T cells and renal tubules in cisplatin nephrotoxicity. Am J Physiol Renal Physiol. 2015;308:F899–F909. doi: 10.1152/ajprenal.00362.2014. [DOI] [PubMed] [Google Scholar]
- 8.Hagar H, Medany AE, Salam R, Medany GE, Nayal OA. Betaine supplementation mitigates cisplatin-induced nephrotoxicity by abrogation of oxidative/nitrosative stress and suppression of inflammation and apoptosis in rats. Exp Toxicol Pathol. 2015;67:133–141. doi: 10.1016/j.etp.2014.11.001. [DOI] [PubMed] [Google Scholar]
- 9.Chen Q, Peng H, Dong L, Chen L, Ma X, Peng Y, Dai S, Liu Q. Activation of the NRF2-ARE signalling pathway by the Lentinula edodes polysaccharose LNT alleviates ROS-mediated cisplatin nephrotoxicity. Int Immunopharmacol. 2016;36:1–8. doi: 10.1016/j.intimp.2016.04.007. [DOI] [PubMed] [Google Scholar]
- 10.Ma X, Dang C, Kang H, Dai Z, Lin S, Guan H, Liu X, Wang X, Hui W. Saikosaponin-D reduces cisplatin-induced nephrotoxicity by repressing ROS-mediated activation of MAPK and NF-κB signalling pathways. Int Immunopharmacol. 2015;28:399–408. doi: 10.1016/j.intimp.2015.06.020. [DOI] [PubMed] [Google Scholar]
- 11.Francescato HD, Costa RS, Silva CG, Coimbra TM. Treatment with a p38 MAPK inhibitor attenuates cisplatin nephrotoxicity starting after the beginning of renal damage. Life Sci. 2009;84:590–597. doi: 10.1016/j.lfs.2009.02.004. [DOI] [PubMed] [Google Scholar]
- 12.Mishima K, Baba A, Matsuo M, Itoh Y, Oishi R. Protective effect of cyclic AMP against cisplatin-induced nephrotoxicity. Free Radic Biol Med. 2006;40:1564–1577. doi: 10.1016/j.freeradbiomed.2005.12.025. [DOI] [PubMed] [Google Scholar]
- 13.Ramesh G, Reeves WB. p38 MAP kinase inhibition ameliorates cisplatin nephrotoxicity in mice. Am J Physiol Renal Physiol. 2005;289:F166–F174. doi: 10.1152/ajprenal.00401.2004. [DOI] [PubMed] [Google Scholar]
- 14.Shao Y, Le K, Cheng H, Aplin AE. NF-κB regulation of c-FLIP promotes TNFα-mediated RAF inhibitor resistance in melanoma. J Invest Dermatol. 2015;135:1839–1848. doi: 10.1038/jid.2015.91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tsubaki M, Ogawa N, Takeda T, Sakamoto K, Shimaoka H, Fujita A, Itoh T, Imano M, Satou T, Nishida S. Dimethyl fumarate induces apoptosis of hematopoietic tumor cells via inhibition of NF-κB nuclear translocation and down-regulation of Bcl-xL and XIAP. Biomed Pharmacother. 2014;68:999–1005. doi: 10.1016/j.biopha.2014.09.009. [DOI] [PubMed] [Google Scholar]
- 16.Yang T, Lan J, Huang Q, Chen X, Sun X, Liu X, Yang P, Jin T, Wang S, Mou X. Embelin sensitizes acute myeloid leukemia cells to TRAIL through XIAP inhibition and NF-κB inactivation. Cell Biochem Biophys. 2015;71:291–297. doi: 10.1007/s12013-014-0197-9. [DOI] [PubMed] [Google Scholar]
- 17.Hughes MA, Powley IR, Jukes-Jones R, Horn S, Feoktistova M, Fairall L, Schwabe JW, Leverkus M, Cain K, MacFarlane M. Co-operative and hierarchical binding of c-flip and caspase-8: A unified model defines how c-flip isoforms differentially control cell fate. Mol Cell. 2016;61:834–849. doi: 10.1016/j.molcel.2016.02.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Elsawy MA, Martin L, Tikhonova IG, Walker B. Solid phase synthesis of Smac/DIABLO-derived peptides using a ‘Safety-Catch’ resin: Identification of potent XIAP BIR3 antagonists. Bioorg Med Chem. 2013;21:5004–5011. doi: 10.1016/j.bmc.2013.06.055. [DOI] [PubMed] [Google Scholar]
- 19.Kwan HY, Niu X, Dai W, Tong T, Chao X, Su T, Chan CL, Lee KC, Fu X, Yi H, et al. Lipidomic-based investigation into the regulatory effect of schisandrin B on palmitic acid level in non-alcoholic steatotic livers. Sci Rep. 2015;5:9114. doi: 10.1038/srep09114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pao TT, Hsu KF, Liu KT, Chang LG, Chuang CH, Sung CY. Protective action of schizandrin B on hepatic injury in mice. Chin Med J. 1977;3:173–179. [PubMed] [Google Scholar]
- 21.Yang T, Liu S, Zheng TH, Tao YY, Liu CH. Comparative pharmacokinetics and tissue distribution profiles of lignan components in normal and hepatic fibrosis rats after oral administration of Fuzheng Huayu recipe. J Ethnopharmacol. 2015;166:305–312. doi: 10.1016/j.jep.2015.03.024. [DOI] [PubMed] [Google Scholar]
- 22.Jiang EP, Li H, Yu CR, Yu CY, Jing S, Sun HX, Wang CM, Fan XT, Chen JG, Wang S. Schisandrin B protects PC12 cells against oxidative stress of neurodegenerative diseases. Neuroreport. 2015;26:360–366. doi: 10.1097/WNR.0000000000000354. [DOI] [PubMed] [Google Scholar]
- 23.Stacchiotti A, Li Volti G, Lavazza A, Schena I, Aleo MF, Rodella LF, Rezzani R. Different role of schisandrin B on mercury-induced renal damage in vivo and in vitro. Toxicology. 2011;286:48–57. doi: 10.1016/j.tox.2011.05.005. [DOI] [PubMed] [Google Scholar]
- 24.Thandavarayan RA, Giridharan VV, Arumugam S, Suzuki K, Ko KM, Krishnamurthy P, Watanabe K, Konishi T. Schisandrin B prevents doxorubicin induced cardiac dysfunction by modulation of DNA damage, oxidative stress and inflammation through inhibition of MAPK/p53 signaling. PLoS One. 2015;10:e0119214. doi: 10.1371/journal.pone.0119214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chiu PY, Leung HY, Poon MK, Mak DH, Ko KM. (−) Schisandrin B is more potent than its enantiomer in enhancing cellular glutathione and heat shock protein production as well as protecting against oxidant injury in H9c2 cardiomyocytes. Mol Cell Biochem. 2006;289:185–191. doi: 10.1007/s11010-006-9163-1. [DOI] [PubMed] [Google Scholar]
- 26.Lv XJ, Zhao LJ, Hao YQ, Su ZZ, Li JY, Du YW, Zhang J. Schisandrin B inhibits the proliferation of human lung adenocarcinoma A549 cells by inducing cycle arrest and apoptosis. Int J Clini Exp Med. 2015;8:6926–6936. [PMC free article] [PubMed] [Google Scholar]
- 27.Bunel V, Antoine MH, Nortier J, Duez P, Stévigny C. Protective effects of schizandrin and schizandrin B towards cisplatin nephrotoxicity in vitro. J Appl Toxicol. 2014;34:1311–1319. doi: 10.1002/jat.2951. [DOI] [PubMed] [Google Scholar]
- 28.Fernandes-Alnemri T, Litwack G, Alnemri ES. CPP32, a novel human apoptotic protein with homology to Caenorhabditis elegans cell death protein Ced-3 and mammalian interleukin-1 beta-converting enzyme. J Biol Chem. 1994;269:30761–30764. [PubMed] [Google Scholar]
- 29.Reed JC, Reed SI. Survivin’ cell-separation anxiety: Nat Cell Biol. 1999;1:E199–E200. doi: 10.1038/70227. [DOI] [PubMed] [Google Scholar]
- 30.Li F, Ackermann EJ, Bennett CF, Rothermel AL, Plescia J, Tognin S, Villa A, Marchisio PC, Altieri DC. Pleiotropic cell-division defects and apoptosis induced by interference with survivin function. Nat Cell Biol. 1999;1:461–466. doi: 10.1038/70242. [DOI] [PubMed] [Google Scholar]
- 31.Dong Q, Hou H, Wu J, Chen Y. The Nrf2-ARE pathway is associated with Schisandrin B attenuating benzo(a) pyrene-Induced HTR cells damages in vitro. Environ Toxicol. 2016;31:1439–1449. doi: 10.1002/tox.22149. [DOI] [PubMed] [Google Scholar]
- 32.Filipski KK, Mathijssen RH, Mikkelsen TS, Schinkel AH, Sparreboom A. Contribution of organic cation transporter 2 (OCT2) to cisplatin-induced nephrotoxicity. Clin Pharmacol Ther. 2009;86:396–402. doi: 10.1038/clpt.2009.139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Pabla N, Murphy RF, Liu K, Dong Z. The copper transporter Ctr1 contributes to cisplatin uptake by renal tubular cells during cisplatin nephrotoxicity. Am J Physiol Renal Physiol. 2009;296:F505–F511. doi: 10.1152/ajprenal.90545.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Juo P, Kuo CJ, Yuan J, Blenis J. Essential requirement for caspase-8/FLICE in the initiation of the Fas-induced apoptotic cascade. Curr Biol. 1998;8:1001–1008. doi: 10.1016/S0960-9822(07)00420-4. [DOI] [PubMed] [Google Scholar]
- 35.Liu H, Baliga R. Endoplasmic reticulum stress-associated caspase 12 mediates cisplatin-induced LLC-PK1 cell apoptosis. J Am Soc Nephrol. 2005;16:1985–1992. doi: 10.1681/ASN.2004090768. [DOI] [PubMed] [Google Scholar]
- 36.Omar HA, Mohamed WR, Arab HH, Arafa el-SA. Tangeretin alleviates cisplatin-induced acute hepatic injury in rats: Targeting MAPKs and apoptosis. PLoS One. 2016;11:e0151649. doi: 10.1371/journal.pone.0151649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Matsumoto M, Nakajima W, Seike M, Gemma A, Tanaka N. Cisplatin-induced apoptosis in non-small-cell lung cancer cells is dependent on Bax- and Bak-induction pathway and synergistically activated by BH3-mimetic ABT-263 in p53 wild-type and mutant cells. Biochem Biophys Res Commun. 2016;473:490–496. doi: 10.1016/j.bbrc.2016.03.053. [DOI] [PubMed] [Google Scholar]
- 38.O’Connor DS, Grossman D, Plescia J, Li F, Zhang H, Villa A, Tognin S, Marchisio PC, Altieri DC. Regulation of apoptosis at cell division by p34cdc2 phosphorylation of survivin. Proc Natl Acad Sci USA. 2000;97:13103–13107. doi: 10.1073/pnas.240390697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Perkins ND. Post-translational modifications regulating the activity and function of the nuclear factor kappa B pathway. Oncogene. 2006;25:6717–6730. doi: 10.1038/sj.onc.1209937. [DOI] [PubMed] [Google Scholar]