

Abstract
Alzheimer’s disease (AD) is a neurodegenerative disorder that is characterized by amyloid plaques in patients’ brain tissue. The plaques are mainly made of β-amyloid peptides and trace elements including Zn2+, Cu2+, and Fe2+. Some studies have shown that AD can be considered a type of metal dyshomeostasis. Among metal ions involved in plaques, numerous studies have focused on copper ions, which seem to be one of the main cationic elements in plaque formation. The involvement of copper in AD is controversial, as some studies show a copper deficiency in AD, and consequently a need to enhance copper levels, while other data point to copper overload and therefore a need to reduce copper levels. In this paper, the role of copper ions in AD and some contradictory reports are reviewed and discussed.
Keywords: Alzheimer’s disease, copper, zinc, neurodegenerative disorder, amyloid plaques, cholesterol, calcium
Introduction
Alzheimer’s disease (AD) is a progressive neurodegenerative disorder described in 1907 by Alois Alzheimer. He observed amyloid plaques and neurofibrillary tangles (NFTs) in the brain of patients showing signs of dementia (Sarell, 2010). Today, AD is the most prevalent neurodegenerative disease affecting 10% of people aged 65+ and 50% of people aged 80+ (Zhang et al., 2011). In 2016, it was estimated that there were about 47 million AD patients in the world and this number is expected to increase to more than 130 million by 2050. The World Alzheimer Report evaluated the annual social and economic cost of dementia to be US$ 818 billion worldwide in 2015 and this amount is expected to increase to one trillion by 2018 (Unzeta et al., 2016).
Alzheimer’s disease is ultimately lethal, characterized by the developing damage of neuronal tissues in the brain. Signs include memory loss, paranoia, loss of reasoning powers and confusion (Sarell, 2010). Unfortunately, AD is recognized only after the manifestation of cognitive signs, which may be too late for effective treatment (Leskovjan et al., 2011). Moreover, approved drugs have inconsiderable effects on patients’ well-being, which may be because many factors are responsible for AD (Conte-Daban et al., 2016; Pickart et al., 2017). Indeed, there are several theories to demonstrate the cause of AD supported by empirical data, which have been reviewed by Armstrong (2013).
In brain regions affected in AD, such as the cortex and hippocampus, extracellular senile plaques, and intracellular NFTs accumulate (Zhang et al., 2011). Senile plaques or amyloid plaques, as the name implies, are mainly composed of small peptides called β-amyloid (Masters et al., 1985). The latter is produced from β-amyloid precursor protein (APP) through successive cleavages first by β-secretase at residue 671 and then by γ-secretase at residues 711 or 713 (residue numbering according to the APP770 isoform) in amyloidogenic pathway. Alternatively, APP molecules can be cleaved by α-secretase within the β-amyloid domain at residue 687 and prevent β-amyloid production in non-amyloidogenic pathway (Selkoe, 2001; Zhang et al., 2011). There are three main isoforms of APP including APP695, APP751, and APP770 with APP695 being expressed at high levels in brain compared to other isoforms (Nalivaeva and Turner, 2013). Furthermore, NFTs, mainly composed of tau proteins, is the other important factor that accumulate in AD brain (Buée et al., 2000). Tau proteins mostly expressed in neurons have the ability to induce microtubule assembly in vitro (Weingarten et al., 1975; Cleveland et al., 1977). In fact, microtubules, one of the main components of the cytoskeletal system, are involved in the maintenance of neuronal morphology and the formation of axonal and dendritic processes (Gendron and Petrucelli, 2009). One of the first and most severely injured brain areas in AD is the hippocampus, which is associated with neurogenesis and long-term memory storage. It is also thought to be more susceptible to metal disturbance than other brain areas. Another brain region that suffers from damage in AD due to plaque pathology is the cortex, associated with functions such as argumentation, feeling, and language (Leskovjan et al., 2011).
β-amyloid aggregations into senile plaques are one of the main characteristics of AD (Wan et al., 2011). A considerable co-localization of adenosine receptors and β-amyloid has been reported in senile plaques (Angulo et al., 2003). Adenosine, a purine ribonucleoside that has neuromodulatory and neuroprotective properties (Rahman, 2009), affects various important brain functions such as sleep, cognition, memory, and neurodegeneration (De Mendonça and Ribeiro, 1996; Porkka-Heiskanen, 1999; Ribeiro et al., 2002; Rahman, 2009). Adenosine is involved in numerous neurological disorders including AD (Cortés et al., 2015; Maiuolo et al., 2016). It exerts its various effects via its receptors, and thus managing its receptor agonists and antagonists significantly influences learning and memory (Ohno and Watanabe, 1996; Kopf et al., 1999; Corodimas and Tomita, 2001; Hauber and Bareiß, 2001; Pereira et al., 2002). On one hand, deamination of adenosine to inosine by adenosine deaminase (ADA) is one of the metabolic pathways for the catabolism of adenosine in the brain (Boison, 2006). On the other hand, ADA acts as an allosteric modulator of adenosine receptors (Cortés et al., 2015). Because of ADA’s involvement in different health disorders, the development of ADA inhibitors as feasible therapeutic agents has been considered in many studies (Cristalli et al., 2001; Saboury et al., 2003, 2004, 2005; Ataie et al., 2004, 2007; Terasaka et al., 2004a,b; Da Settimo et al., 2005; Ajloo et al., 2007; Ujjinamatada et al., 2008; La Motta et al., 2009; Bazl et al., 2012). Recently, ADA inhibitors have been proposed in perinatal hypoxia–ischemia brain injury treatment (Pimentel et al., 2013).
Polyvalent metal cations such as copper, zinc, and iron are found in high concentrations in senile plaques in AD patients’ brain (Smith et al., 1997; Lovell et al., 1998; Sayre et al., 2000; Suh et al., 2000; Dong et al., 2003; Miller et al., 2006). Furthermore, some studies in mouse models of AD revealed that in spite of accumulation of copper in senile plaques in the mouse models with neurodegeneration including 5 × FAD and CVN (Bourassa et al., 2013), no copper accumulation is observed in PSAPP mouse model with slight neurodegeneration (Bourassa et al., 2013; James et al., 2017). Considerable data point to dyshomeostasis of zinc and copper ions as the main factor of AD pathogenesis (Deibel et al., 1996; Lovell et al., 1998; Cherny et al., 1999, 2001; González et al., 1999; Huang et al., 1999, 2004; Sayre et al., 2000; Bayer et al., 2003; Phinney et al., 2003; Ritchie et al., 2003; Pajonk et al., 2005; Kessler et al., 2006; Ma et al., 2006; Maynard et al., 2006, 2002; Miller et al., 2006; Cater et al., 2008; Donnelly et al., 2008; Hung et al., 2009; Leskovjan et al., 2009; Hozumi et al., 2011; Mao et al., 2012; Arnal et al., 2013a,b; Pal et al., 2013; Singh et al., 2013) and indicate that copper metabolism proteins are associated with AD (Phinney et al., 2003; Southon et al., 2013; Pal et al., 2014).
Different authors have put forward various models of the toxicity of copper involvement in AD. The most accredited one proposes the gain-of-function of β-amyloid (Bush et al., 2003; Bush and Tanzi, 2008) after binding Cu2+ (Multhaup et al., 1996). Alternative and more recent hypotheses (Lee et al., 2005; Cavaleri, 2015; Kepp, 2016) propose a protective role of β-amyloid against an excess of toxic metals within the brain, designating β-amyloid loss-of-function as a pathogenic process in the disease (Hua et al., 2011; Kepp, 2016). APP is thought to possess a normal function in metal export from neurons, and a putative loss of the soluble, functional β-amyloid monomer could cause copper build-up in the cell (Kepp, 2016).
Scientific evidence has shown that metal ion binding to β-amyloid accelerates amyloid aggregation, which could finally damage the neurons in AD (Pithadia and Lim, 2012). The involvement of copper in AD is controversial, as some studies show copper deficiency in AD, and consequently a need to enhance copper levels (Borchardt et al., 1999; Kessler et al., 2005, 2008a,b; Exley, 2006; Jiao and Yang, 2007; Vural et al., 2010; Kaden et al., 2011; Exley et al., 2012), while other data point to copper overload and therefore a need to reduce copper levels (Cherny et al., 2001; Sparks et al., 2006; Hua et al., 2011; Luo et al., 2011; Ceccom et al., 2012; Eskici and Axelsen, 2012; Brewer, 2014; Squitti et al., 2014b; Yu et al., 2015). An aberrant copper homeostasis with an increase in the labile pool of copper and a decrease in the copper bound to protein is the main up-dated interpretation (Kepp, 2016; Squitti et al., 2016). In this paper, the role of metal ions, particularly copper, in AD is reviewed and discussed.
Copper Ion Toxicity in AD
The key event in AD is the formation of fibrils and plaques in AD patients’ brain. Plaques are mainly made of β-amyloid peptide, the natural peptide that is produced in the brain and exists at nanomolar concentration levels in cerebrospinal fluid (CSF) and serum (Masters et al., 1985; Vigo-Pelfrey et al., 1993). On the other hand, a high concentration of trace metals, including copper, is observed in amyloid plaques (Miller et al., 2006). Interestingly, some data show that copper distribution in the brain does not correspond to β-amyloid plaques distribution in TASTPM mice model (Torres et al., 2016) with plaque pathology but not appreciable neuronal loss (Howlett et al., 2004). Copper is a necessary trace metal in nervous system development since disruption of its homeostasis leads to neurodegenerative disorders like Menkes and Wilson’s diseases (Waggoner et al., 1999). Cu2+ ions bind to β-amyloid peptides with high affinity (Atwood et al., 2000; Sarell et al., 2009; Barritt and Viles, 2015; Mital et al., 2015; Drew, 2017) and increase the proportions of β-sheet and α-helix structures in amyloid peptides, which can be responsible for β-amyloid aggregation (Dai et al., 2006). Various concentrations of Cu2+ ions enhance fibril formation while binding of copper ions to β-amyloid noticeably increases its toxicity for cells (Dai et al., 2006; Sarell et al., 2010). In addition, substoichiometric concentrations of Cu2+ are more toxic to cells (Sarell et al., 2010).
Fibril formation is highly pH-dependent and Cu2+ ions cause it to occur at physiological pH. However, the formation of amorphous aggregates dominates in acidic conditions (Jun et al., 2009; Sarell, 2010; Lv et al., 2013). In a proton-rich environment, β-amyloid (Aβ40) possesses two copper binding sites, and its second bound Cu2+ion causes the formation of amorphous aggregates by preventing the conformational transition of β-amyloid into amyloid fibrils (Jun et al., 2009).
The production of Reactive Oxygen Species (ROS) is a key factor in β-amyloid toxicity toward neurons, which is dependent on metal ion redox properties. Copper ions in complex with β-amyloid fibrils produce hydrogen peroxide, in the presence of biological reducing agents (Parthasarathy et al., 2014). When the ratio of copper to peptide increases, hydrogen peroxide levels and the production of hydroxyl radicals increase, and the morphology of aggregates changes from fibrillar to amorphous (Mayes et al., 2014). Although previous studies have presented ROS as fatal molecules provoking neurodegeneration, the accumulated evidence shows that some ROS act as essential molecules in processes underlying cognition and memory formation (Klann, 1998; Yermolaieva et al., 2000; Knapp and Klann, 2002a,b; Kamsler and Segal, 2003, 2004; Hu et al., 2006; Kishida and Klann, 2007). On the other hand, some results imply that the copper-amyloid complex produces fewer ROS than free copper ions (Nakamura et al., 2007). According to in vitro data, oligomeric and fibrillar forms of β-amyloid inhibit H2O2 generation at higher concentrations of Cu2+. In addition, the fibrillar form generates less H2O2 than the oligomeric form (Fang et al., 2010).
Copper toxicity in AD brains is attributed to the oxidized form of copper ions, i.e., Cu2+, (Brewer, 2015; Greenough et al., 2016). In contrast, other data show that copper ions are only transported in their reduced form, i.e., Cu1+, (Macreadie, 2008). Some studies suggest that Cu2+ bypasses the liver (Brewer, 2015). Otherwise, some data show that the removal of Cu1+ from β-amyloid, hinders the formation of oligomers and prevents ROS production (Atrián-Blasco et al., 2015). A study of the affinity of the soluble copper-binding domain of the β-amyloid peptide for Cu1+shows that it binds to β-amyloid stronger than Cu2+ suggesting Cu1+ is the relevant in vivo oxidation state (Feaga et al., 2011). Both Cu1+ and Cu2+ inhibit β-amyloid degradation by insulin-degrading enzyme, but Cu1+ cations act as irreversible inhibitors (Grasso et al., 2011). Copper ion by reduction from Cu2+ to Cu1+ protects proteins against free radicals (Deloncle and Guillard, 2014).
A meta-analysis (Schrag et al., 2011) and subsequent studies (James et al., 2012; Szabo et al., 2016) demonstrated that the concentration of total copper is decreased in the brain of AD patients, while the concentration of labile copper is increased in the most affected regions of the AD brain (James et al., 2012). In addition, AD cortical tissues (James et al., 2012) and the cortex of mice with Traumatic Brain Injury show an elevated binding capacity for Cu2+ (Peng et al., 2015). Another study shows that in APPsw/0 mouse model of AD, which shows parenchymal plaques but no neuronal loss (Elder et al., 2010; Sasaguri et al., 2017), unlike in the control mouse in which the metal accumulates in the capillaries, copper ions accumulate in brain parenchyma. These ions could bind to β-amyloid and stimulate β-sheet conformation, aggregation, and toxicity (Singh et al., 2013).
In the “amyloid cascade hypothesis,” plaque formation is a main event in AD pathology but it is sometimes preceded by neurodegeneration, and plaque clearance by immunization of AD patients does not prevent disease progression (Chui et al., 1999; Oddo et al., 2003; Holmes et al., 2008; Bayer and Wirths, 2010; Bittner et al., 2012; Wright et al., 2013; Xie et al., 2013; Jung et al., 2015). Moreover, some studies show that senile plaques exist in cognitively normal people (Jack et al., 2010; Sperling et al., 2011; Swerdlow, 2011; Esparza et al., 2013) and, despite an equivalent plaque presence, the concentration of brain amyloid oligomers is higher in AD patients than in normal cases. The “Toxic oligomers hypothesis” explains these events by suggesting that small, diffusible oligomers are responsible for toxicity, and not the amyloid plaques (Naylor et al., 2008; Sarell, 2010). The oligomers derived from cell culture have unusually high chemical stability and resist degradation into monomers by various degrading agents, supporting the existence of covalent cross-links between the oligomers (Podlisny et al., 1995; Walsh et al., 2002; Lesné et al., 2006; Naylor et al., 2008). Based on in vitro experiments, Cu2+ binding to β-amyloid can lead to the formation of dityrosine-linked dimers of β-amyloids found in AD (Atwood et al., 2004; Haeffner et al., 2005; Bush and Tanzi, 2008; Streltsov et al., 2008; Al-Hilaly et al., 2013). In the presence of Cu2+, the dimer conformation changes from parallel to anti-parallel and is stabilized by the occupied copper binding sites (Hane et al., 2013). However, the same authors showed later that Cu2+ at nanomolar concentrations has no effect on peptide–peptide affinity in the amyloid dimer (Hane et al., 2016). Other authors have demonstrated that binding of Cu2+ ions induces structural changes in the amyloid dimer resulting in N-termini interactions within it (Lv et al., 2013). The mutant dimer that is unable to produce cross-links provides supporting evidence for the toxicity of Cu2+cross-linked dimers because the mutant dimer’s properties are the same as those of the wild type dimer except that it has no neurotoxicity (Barnham, 2004; Figure 1). In addition, other studies suggest that the toxicity of the cross-linked dimer is due to enhanced membrane binding (Ciccotosto et al., 2004).
FIGURE 1.
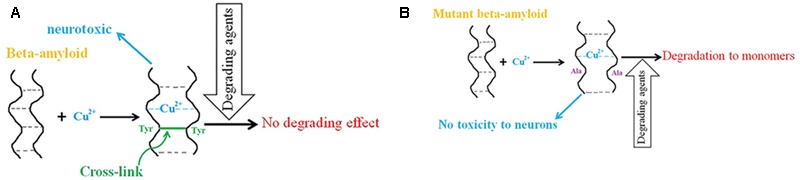
The role of copper in β-amyloid neurotoxicity in AD. (A) Copper binding to β-amyloid peptides leads to the formation of dityrosine-linked β-amyloid dimers, which resist degradation into monomers and have neurotoxic properties. (B) Mutant β-amyloids (Tyr to Ala) in the presence of copper ions have no toxicity effect on neurons and degrade to monomers by degrading agents.
Elimination of Cu2+ from β-amyloid prevents amyloid aggregation in vitro (Wu et al., 2008; Behbehani et al., 2012), and promotes β-amyloid degradation, and prevents H2O2 formation. Hence, it also decreases cell mortality (Wu et al., 2008). Because of these positive effects of copper elimination, some studies have targeted copper chelators as suitable drugs (Moret et al., 2006; Geng et al., 2012; Nguyen et al., 2014; Savelieff et al., 2014; Hauser-Davis et al., 2015; Hung et al., 2015; Yang et al., 2016). However, the most recently published review on copper chelation therapy states that the results in human clinical trials are discouraging (Drew, 2017), even though some authors have refuted this interpretation (Squitti et al., 2017a). Furthermore, studies in Tg2576 mouse model of AD, which shows parenchymal plaques but no neuronal loss (Elder et al., 2010; Sasaguri et al., 2017), show that although the use of chelator helps to prevent AD, it is inefficient in AD treatment suggesting that systemic copper removal is useful only in the early stages of the disease (Quinn et al., 2010). Interestingly, there is some evidence that making changes in brain copper uptake in the primary stages can have a considerable effect on amyloid pathology (Lang et al., 2013).
Until 2012, a number of ambiguous results published previously fueled a debate about copper levels in AD patients. Overall, six meta-analyses have been carried out in the last 6 years to evaluate copper concentrations in AD in different biological matrices (serum, plasma, and cerebrospinal fluid). These meta-analyses, combining data collected from studies published between 1984 and 2017 (Bucossi et al., 2011; Ventriglia et al., 2012; Schrag et al., 2013; Squitti et al., 2014a; Wang et al., 2015; Li et al., 2017), provide unequivocal results: total copper (Bucossi et al., 2011; Ventriglia et al., 2012; Schrag et al., 2013; Squitti et al., 2014a; Wang et al., 2015; Li et al., 2017) and “free” copper (Squitti et al., 2014a) are higher in the serum–plasma of AD patients in comparison with healthy controls. More specifically, the large stand most recent meta-analysis (total pool of subjects analyzed: 2128 AD vs. 2889 healthy controls) includes a total of 35 studies: 18 report an increase, 14 no difference, and one a decrease in values of copper in the serum–plasma in AD compared to healthy controls (Li et al., 2017). Three additional studies appeared after the publication of this consensus result (Guan et al., 2017; Pu et al., 2017; Talwar et al., 2017), reporting increased concentrations of Cu2+ in AD patients vs. controls.
Recent studies have contributed to unraveling further the initial controversy, demonstrating that the increased concentration of serum copper in AD can be explained by the increased concentrations of the plasma fraction of the “free” copper pool in the blood, which is detected in only 50–60% of AD patients (Squitti et al., 2016; Szabo et al., 2016; Tecchio et al., 2016; Talwar et al., 2017). An older study also indicated that serum copper concentration rises in a special type of AD (González et al., 1999). Some studies have proposed a genetic basis for this AD subtype as an explanation of this observation (González et al., 1999; Liu et al., 2013; Squitti et al., 2013, 2017b; Mercer et al., 2017).
Brain Copper Deficiency in AD
A significant reduction in copper ion levels is observed in the hippocampus and amygdale areas of AD patients compared to age-matched control subjects (Deibel et al., 1996). In addition, a reduction in net copper is found in the brain of TgCRND8 AD mice model (Phinney et al., 2003), and this model exhibits parenchymal amyloid deposition but no neuronal loss (Sasaguri et al., 2017). As mentioned above, a meta-analysis indicates that copper is significantly decreased in the brain of AD patients (Schrag et al., 2011). An analysis of the human brains of deceased patients with dementia concludes that defective regions have a very low copper content (Pickart et al., 2017). The copper content of aged human brains has a significant negative correlation with the degree of severity of amyloid plaques (Exley et al., 2012). Based on the results showing a significant reduction in copper ion in AD patients compared to controls (Giacoppo et al., 2014), it has been hypothesized that AD is a result of copper deficiency (Klevay, 2008). An alternative, and more comprehensive, interpretation, which can explain the copper quantification results of meta-analyses in serum–plasma and the meta-analysis in the brain, is that the copper decrease in the brain is a sign of an aberrant copper homeostasis, which resembles Wilson’s disease (Fujiwara et al., 2006). Interestingly, some data indicate that copper deficiency in AD patients is independent of their diet (Giacoppo et al., 2014).
In the presence and absence of copper, APP molecules are cleaved in non-amyloidogenic and amyloidogenic pathways, respectively. The latter pathway results in amyloid production. Some data show that copper ions inhibit amyloid production by interacting with a γ-secretase complex (Gerber et al., 2017) or by affecting APP dimerization (Kong et al., 2008). Otherwise, copper ions enhance APP exposure on the cell surface by both increasing its exocytosis and decreasing its endocytosis (Acevedo et al., 2011). Cu deficiency, as observed in AD patients, enhances β-amyloid production and accumulation by inducing the amyloidogenic processing of APP (Bayer et al., 2003). However, its molecular mechanism is still unclear (Wild et al., 2017).
The mutations in genes encoding proteins required for copper ion uptake in mammalian systems lead to early-onset familial AD (Southon et al., 2013). On the other hand, enhancement of intracellular copper levels through addition of dietary copper in APP/PS1 transgenic AD mice (Crouch et al., 2009) and APP23 transgenic mice (Bayer et al., 2003) with parenchymal plaques but no neuronal loss (Elder et al., 2010; Sasaguri et al., 2017), causes a reduction in AD pathology. Based on the report of low brain copper in several neurodegenerative disorders, impairment in copper protection against free radicals has been proposed as the main cause of these disorders (Deloncle and Guillard, 2014).
Copper and Zinc Ions in AD
A significant elevation in Zn2+ is found in the AD hippocampus and amygdale area (Deibel et al., 1996), and in the AD neuropil compared to controls (Lovell et al., 1998), although some data indicate an elevation in Zn2+ content in the AD brain cortex (Religa et al., 2006). Accordingly, a meta-analysis revealed no significant changes in zinc content in the AD neocortex (Schrag et al., 2011). Nevertheless, three meta-analyses (Ventriglia et al., 2015; Wang et al., 2015; Li et al., 2017) were recently carried out on plasma and serum zinc levels in AD patients and healthy controls. One study (Ventriglia et al., 2015) (the pooled sample size included 777 AD vs. 1728 healthy controls) analyzed a total of 16 studies and concludes that AD patients show a decrease in serum zinc levels compared to healthy controls. Ventriglia et al.’ (2015) study (the pooled sample size included 287 AD vs. 166 healthy controls) analyzed a total of five studies on plasma zinc: two indicate a decrease and three no significant differences in plasma zinc in AD patients compared to controls. The other study (Wang et al., 2015) (the pool of subjects included 862 AD patients vs. 1705 controls) analyzed a total of 17 studies: 10 indicate a decrease, and seven no significant differences in serum zinc in AD patients compared to controls. The most recent meta-analysis by Li et al. (2017) analyzed 22 studies with a total pool of 1027 patients with AD and 1949 healthy controls: three studies indicate an increase, 18 studies a decrease, and one study no differences in serum zinc between AD and healthy controls. Interestingly, an older study found that a rise in serum Zn2+ occurs in a special type of AD (González et al., 1999). As the absence of fibrinogen in serum is the main difference between serum and plasma compositions and fibrinogen-β-amyloid interactions are involved in AD progression (Cortes-Canteli et al., 2012; Derakhshankhah et al., 2016), it is likely that the reduction in serum Zn2+ in AD patients is due to its interactions with fibrinogen. The rise in zinc levels is accompanied by a rise in tissue amyloid levels (Religa et al., 2006). AD brain tissue contains hot spots of metal ions especially enriched by copper and zinc ions. In 2006, it was reported for the first time that β-amyloid plaques and hot spots of accumulated metal ions co-localize (Miller et al., 2006).
It has been suggested that zinc ions induce β-amyloid aggregation in vitro whereas Cu2+ ions inhibit it through competing with zinc for histidine residues. The strongest inhibitory effect occurs at a copper: β-amyloid molar ratio of about four. Above this value, copper ions themselves induce aggregation (Suzuki et al., 2001). Interestingly, previous studies have shown that β-amyloid binds to three or four Cu2+ ions at pH 7.0 (Atwood et al., 1998). Studies on synthetic Aβ(1–40) and Aβ(1–42) peptides show that at physiological pH, β-amyloid binds the same ratio of copper and zinc ions, whereas in acidic conditions copper ions replace zinc ions (Figure 2; Atwood et al., 2000; Roberts et al., 2012). Cu2+ and Zn2+ ions inhibit β-amyloid fibrillization, promoting instead the formation of non-fibrillar aggregates in vitro. In addition, zinc ions have a threefold stronger inhibitory effect than copper ions (Tõugu et al., 2009). Both Cu2+ and Zn2 ions prevent the formation of soluble fibrils if incubated with Aβ(1–42) in vitro (Bolognin et al., 2011). Some authors have also reported that small, soluble oligomers play the main role in β-amyloid neurotoxicity (Tabner et al., 2011). Cu2+ ions form stable and soluble 1:1 complexes with β-amyloid (Aβ40) while Zn2+ ions cause their partial aggregation (Tõugu et al., 2008). Cu2+-induced aggregates are toxic to neurons only in the presence of ascorbate, while monomers and zinc-induced aggregates are not toxic (Tõugu et al., 2009).
FIGURE 2.
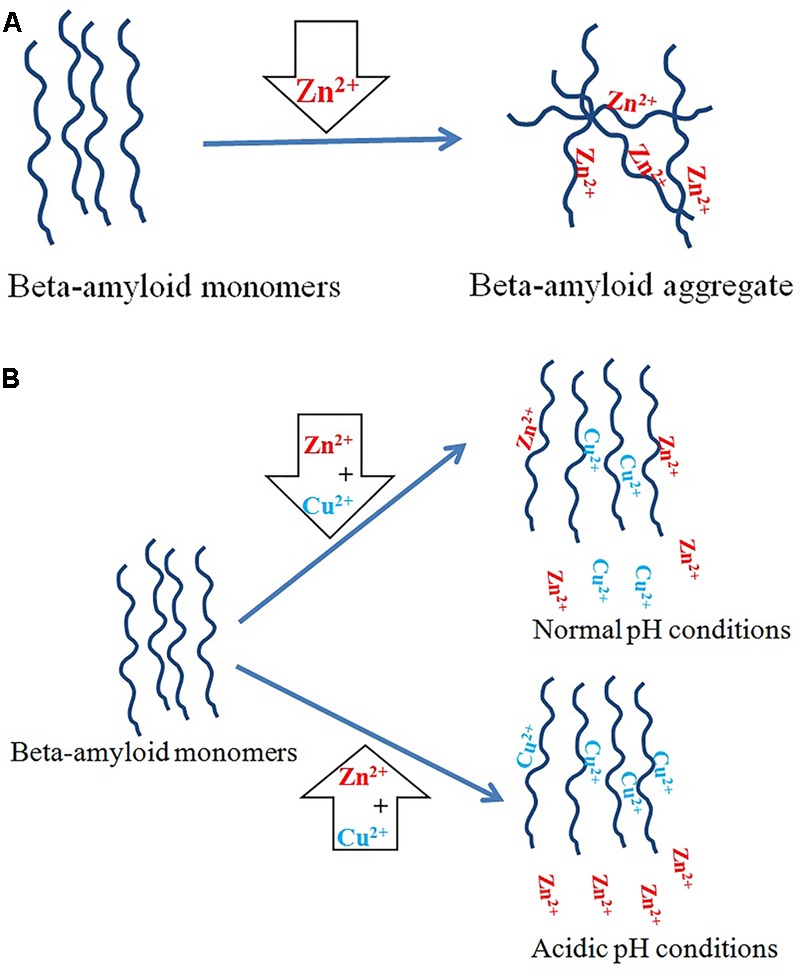
Copper and zinc ions in AD. (A) Zinc ions promote β-amyloid aggregation. (B) Copper ions have an inhibitory effect on aggregation induced by zinc ions. Under physiological conditions, β-amyloid binds the same ratio of copper and zinc ions, but in acidic pH copper ions replace zinc ions overall.
The role of zinc supplementation in AD remains controversial according to a systematic review in 2012 reporting that Zn2+ in the diet has no effect on cognitive decline (Loef et al., 2012) even though the Zenith and the Zincage studies suggested some effects (Simpson et al., 2005; Marcellini et al., 2006; Maylor et al., 2006). However, 3xTg-AD mice model with background neuropathology similar to AD patients (Sasaguri et al., 2017) displayed a delay in memory impairment (Corona et al., 2010). Furthermore, Tg2576, TgCRND8 and CRND8/E4 models exhibited a potentiationin memory impairment (Linkous et al., 2009; Railey et al., 2011; Flinn et al., 2014). Moreover, Tg2576 mice and Sprague Dawley rats showed a lower brain copper and amyloid burden (Harris et al., 2014), and a higher cognitive performance, respectively (Sandusky-Beltran et al., 2017).
Selective release of Cu2+ and Zn2+ ions inside the cells by complexes can decrease the level of β-amyloids (Donnelly et al., 2008). B-amyloid peptides in the presence of physiological concentrations of copper and zinc ions are degraded but at higher concentrations of these cations they aggregate (Strozyk et al., 2009). In agreement with this finding, the use of a copper–zinc chelator rapidly causes a considerable decrease in the deposition of brain amyloid in APP2576 transgenic mice (Cherny et al., 2001). Other authors suggest that the addition of chelators to β-amyloid aggregates induces their rapid fibrillization in vitro. In addition, a long incubation of non-fibrillar aggregates transforms them into fibrillar forms (Tõugu et al., 2009). These findings confirm that the fibrillar form of β-amyloid is its most stable state (Tõugu et al., 2009; Breydo et al., 2016). Zn2+ and Cu2+ ions induce refolding of β-sheets into α-helix structures leading to oligomerization and membrane penetration of amyloid peptides (Curtain et al., 2001). Copper and zinc ions both induce a β-amyloid conformational change but zinc has a greater effect (Yao et al., 2012).
Iron Ions in AD
Iron is a redox active metal and its brain level varies according to specific regions of the brain. Commonly, the brain areas responsible for motor functions are found to have higher iron concentrations than non-motor related areas (Kozlowski et al., 2012). Iron also has a high concentration at the periphery of amyloid plaques (Greenough et al., 2013). Nevertheless, there is controversy about the level of iron outside plaques in the AD brain (Schrag et al., 2011). Contrary to copper and zinc, very few structural studies have been reported on iron coordination to β-amyloid (Hureau, 2012). While copper and zinc co-purify with β-amyloid extracted from plaques, iron does not co-localize within β-amyloid deposits (Grundke-Iqbal et al., 1990; Quintana et al., 2006; Kozlowski et al., 2012) and in vitro studies have shown that β-amyloid binds to iron with low affinity (Viles, 2012; Greenough et al., 2013). After Louis Goodman’s case studies performed in the 1950s, a relationship between regions of AD pathology and iron accumulation was proposed (Everett et al., 2014). Some data show that β-amyloid reduces Fe3+–Fe2+ in solution in vitro (Khan et al., 2006; Everett et al., 2014). While some authors argue that the direct coordination of Fe3+ to β-amyloid is impossible at natural pH and that Fe2+ is air-sensitive and oxidizes to Fe3+ during measurements (Hureau, 2012), others report a potential pro-aggregating function for Fe2+ and Fe3+ (Masters and Selkoe, 2012) and some data show a higher affinity for β-amyloid to Fe2+ relative to transferrin (Jiang et al., 2009). Three meta-analyses have been carried out in the last 4 years to assess iron concentrations in AD in serum, cerebrospinal fluid, and brain (Tao et al., 2014; Wang et al., 2015; Li et al., 2017). Tao et al.’s (2014) meta-analysis (total pool of subjects analyzed: 1813 AD vs. 2401 healthy controls) includes a total of 43 studies; 21 consider serum iron; seven consider CSF iron and 19 investigate iron in various brain areas in AD. Their results shows that serum iron significantly decreases in AD compared to controls. CSF iron shows no difference while some specific brain areas show an increase in iron concentration. Wang et al.’s (2015) meta-analysis (total pool of subjects analyzed: 1084 AD vs. 1319 healthy controls) includes a total of 18 studies showing no difference in serum iron but, after exclusion of the study producing high heterogeneity, their results conclude that serum iron levels are significantly lower in AD subjects. Li et al.’s (2017) meta-analysis (total pool of subjects analyzed: 1379 AD vs. 1664 healthy controls) includes 25 studies that show overall no significant difference in serum iron between AD and controls but, after excluding two studies with high heterogeneity, serum iron is significantly lower in AD cases.
Copper and Lipid Rafts in AD
There is considerable evidence that the amyloidogenic pathway takes place in lipid rafts, which are specific membrane domains enriched in cholesterol. The enzymes that cleave APP to β-amyloid peptides are found in lipid rafts (Riddell et al., 2001; Hung et al., 2009). Cellular copper deficiency results in an accumulation of copper ions in cholesterol-rich lipid rafts. In fact, the copper level in lipid rafts is inversely related to cellular copper, which results in enhanced copper-amyloid complex formation under copper deficiency conditions in AD (Cater et al., 2008; Hung et al., 2009).
Clearly, diet is a critical factor in the progression of AD. Several studies indicate the key role of fat in AD (Grant, 1997; Sparks and Schreurs, 2003; Morris et al., 2006; Brewer, 2012). The effective role of Cu2+ and cholesterol overload in neurodegeneration has been reported (Arnal et al., 2013b; Wong et al., 2014). In rats treated with copper and cholesterol, a significant change in visuo-spatial memory is detected (Arnal et al., 2013b) while the administration of dietary cholesterol plus copper-supplemented drinking water (in the form of copper sulfate) induces an accumulation of β-amyloid in rabbit brain (Larry, 2004). The binding of Cu2+ ions to β-amyloid causes oxidation of cholesterol, and the generation of H2O2 (Hung et al., 2009) and other lipid peroxidation products accumulating in AD patients’ brain (Murray et al., 2007) and in Tg2576 transgenic mouse model (Puglielli et al., 2005). In fact, previous unsuccessful therapeutic attempts and recent findings regarding β-amyloid accumulation at lipid rafts have led to a new hypothesis that neurotoxicity in AD is the result of the association of small soluble amyloid oligomers with the plasma membrane (Drolle et al., 2014; Kotler et al., 2014; Arbor et al., 2016). B-amyloid membrane binding is mediated by its interactions with phosphatidylserine (Ciccotosto et al., 2011).
A highly toxic isoform of β-amyloid that accumulates in AD patients’ brain has a great capacity to induce lipid peroxidation. It also alters the calcium influx by binding to cell membranes. This isoform does not lead to an increase in ROS but it causes instead a reduction in plasma membrane integrity and an increase in dityrosine-β-amyloid oligomers (Gunn et al., 2016). Other studies have suggested that the toxicity of the cross-linked dimer is due to enhanced membrane binding (Ciccotosto et al., 2004).
Simulation experiments show that increased cell membrane cholesterol results in some changes in the membrane; namely, an enhancement of surface hydrophobicity and a reduction in bilayer mobility. These membrane changes induce amyloid binding to the cell membrane and cause β-amyloid to refold into a helical or unstructured form. B-amyloid is stabilized on the membrane surface or inserted into the bilayer with the help of calcium ions (Yu and Zheng, 2012). In fact, β-amyloid peptides compose the oligomeric pores in the membrane via the cholesterol-binding domain (Lashuel et al., 2002; Di Scala et al., 2014). Channel formation is cholesterol-dependent and occurs in the presence of at least 30% cholesterol in lipid bilayer membranes. These pores are ion channels that disrupt calcium homeostasis in neural cells and have led to the return of the “calcium hypothesis” of AD (Di Scala et al., 2014, 2016; Figure 3). In the absence of copper, β-amyloids are cleared to the blood even if there are increased cholesterol levels, while in the presence of copper β-amyloid accumulate in the brain (Sparks, 2007). Otherwise, copper ions in the absence of β-amyloid are not toxic to cells (Sarell et al., 2010).
FIGURE 3.
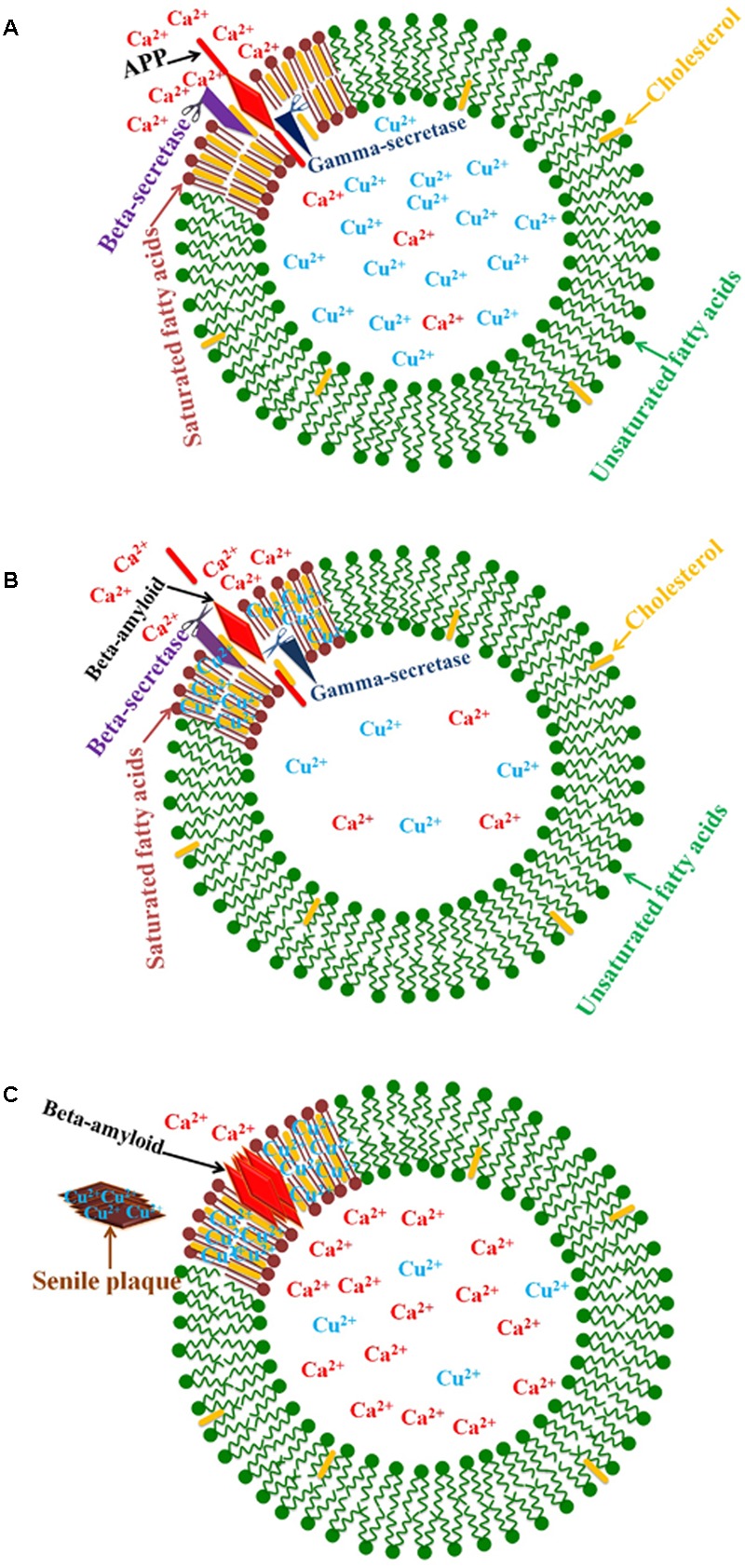
Schematic diagram of the relationship between copper and lipid rafts in AD. (A) The enzymes that cleave APP to β-amyloid peptides are found in lipid rafts. (B) Cellular copper deficiency results in copper ions accumulating in cholesterol-rich lipid rafts, and β-amyloid production increases because of greater enzyme activity due to a rising copper concentration. (C) β-amyloid peptides compose the oligomeric pores in membranes via the cholesterol-binding domain. These pores are calcium channels that disrupt calcium homeostasis in neural cells.
Conclusion
Alzheimer’s disease is a progressive degenerative disease characterized by the presence of senile plaques in the AD patient’s brain. It is a multifactorial disease with a number of genetic and environmental factors likely involved. Previous studies have identified these plaques as toxic elements in AD. However, other studies have demonstrated that 20–40% of healthy people possess senile plaques in their brains (Jack et al., 2010). Moreover, cell death sometimes precedes plaque formation. A number of studies have focused on ROS generation by a copper-amyloid complex and propose ROS as toxic elements in AD. Still other authors have shown that ROS are the basis of memory formation. In addition, fibrils and oligomers at higher copper ion concentrations inhibit the production of ROS. As an alternative, the toxic oligomers hypothesis seems strong, showing that soluble oligomers interact with cell membranes, inducing in them the formation of calcium channels. Collectively, it seems that a disruption of copper control mechanisms occurs in AD, affecting the compartmentalization of the metal in different tissues and organs. Some authors have observed that the increased levels of the labile pool of copper in the brain (James et al., 2012) and in the periphery (Squitti et al., 2014a) bring about a deficiency of copper in the brain and an increase in the labile pool of copper in the brain and in “free copper” in the blood. This is the picture implied by the conditions of Wilson’s disease (Fujiwara et al., 2006). A possible interpretation is that a copper deficiency in brain cells is harmful because of amyloid production, and then copper deficiency leads to a rise in copper levels on lipid rafts. In conditions of copper deficiency, the amyloid-copper complex increases in lipid rafts because of higher levels of both copper and amyloid as well as the high affinity of amyloid peptides for copper. Copper ion binding and proximity to the cell membrane induce the refolding of amyloid peptides, and finally their oligomerization and interactions with cell membranes. The root of copper deficiency in the brain cells seems to be an important factor in AD. Since it is accompanied by copper enrichment in lipid rafts, one can argue that an elevation in lipid raft domains could lead to copper deficiency in the brain, thus targeting lipid rafts could be an effective therapeutic approach. Indeed, some data show that disrupting lipid rafts (by omega-3 fatty acids) delays the incidence of the disease (Cooper, 2003; Dannenberger et al., 2013; Arbor et al., 2016).
Author Contributions
The review was written by SB with assistance and feedback from AS, RS, TH, and MS. All authors approved the final version of the manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
- Acevedo K. M., Hung Y. H., Dalziel A. H., Li Q. X., Laughton K., Wikhe K., et al. (2011). Copper promotes the trafficking of the amyloid precursor protein. J. Biol. Chem. 286 8252–8262. 10.1074/jbc.M110.128512 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ajloo D., Saboury A. A., Haghi-Asli N., Ataei-Jafarai G., Moosavi-Movahedi A. A., Ahmadi M., et al. (2007). Kinetic, thermodynamic and statistical studies on the inhibition of adenosine deaminase by aspirin and diclofenac. J. Enzyme Inhib. Med. Chem. 22 395–406. 10.1080/14756360701229085 [DOI] [PubMed] [Google Scholar]
- Al-Hilaly Y. K., Williams T. L., Stewart-Parker M., Ford L., Skaria E., Cole M., et al. (2013). A central role for dityrosine crosslinking of Amyloid-β in Alzheimer’s disease. Acta Neuropathol. Commun. 1:83. 10.1186/2051-5960-1-83 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Angulo E., Casadó V., Mallol J., Canela E. I., Viñals F., Ferrer I., et al. (2003). A1 adenosine receptors accumulate in neurodegenerative structures in Alzheimer disease and mediate both amyloid precursor protein processing and tau phosphorylation and translocation. Brain Pathol. 13 440–451. 10.1111/j.1750-3639.2003.tb00475.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arbor S. C., Lafontaine M., Cumbay M. (2016). Amyloid-beta Alzheimer targets — protein processing, lipid rafts, and amyloid-beta pores. Yale J. Biol. Med. 89 5–21. [PMC free article] [PubMed] [Google Scholar]
- Armstrong R. A. (2013). What causes Alzheimer’s disease? Folia Neuropathol. 51 169–188. 10.5114/fn.2013.37702 [DOI] [PubMed] [Google Scholar]
- Arnal N., Castillo O., De Alaniz M. J. T., Marra C. A. (2013a). Effects of copper and/or cholesterol overload on mitochondrial function in a rat model of incipient neurodegeneration. Int. J. Alzheimers Dis. 2013:645379. 10.1155/2013/645379 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnal N., Morel G. R., De Alaniz M. J. T., Castillo O., Marra C. A. (2013b). Role of copper and cholesterol association in the neurodegenerative process. Int. J. Alzheimers Dis. 2013:414817. 10.1155/2013/414817 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ataie G., Bagheri S., Divsalar A., Saboury A. A., Safarian S., Namaki S., et al. (2007). A kinetic comparison on the inhibition of adenosine deaminase by purine drugs. Iran. J. Pharm. Res. 6 43–50. [Google Scholar]
- Ataie G., Safarian S., Divsalar A., Saboury A. A., Moosavi-Movahedi A. A., Ranjbar B., et al. (2004). Kinetic and structural analysis of the inhibition of adenosine deaminase by acetaminophen. J. Enzyme Inhib. Med. Chem. 19 71–78. 10.1080/14756360310001632741 [DOI] [PubMed] [Google Scholar]
- Atrián-Blasco E., Cerrada E., Conte-Daban A., Testemale D., Faller P., Laguna M., et al. (2015). Copper(I) targeting in the Alzheimer’s disease context: a first example using the biocompatible PTA ligand. Metallomics 7 1229–1232. 10.1039/C5MT00077G [DOI] [PubMed] [Google Scholar]
- Atwood C. S., Moir R. D., Huang X., Scarpa R. C., Bacarra N. M. E., Romano D. M., et al. (1998). Dramatic aggregation of Alzheimer by Cu(II) is induced by conditions representing physiological acidosis. J. Biol. Chem. 273 12817–12826. 10.1074/jbc.273.21.12817 [DOI] [PubMed] [Google Scholar]
- Atwood C. S., Perry G., Zeng H., Kato Y., Jones W. D., Ling K. Q., et al. (2004). Copper mediates dityrosine cross-linking of Alzheimer’s Amyloid-β. Biochemistry 43 560–568. 10.1021/bi0358824 [DOI] [PubMed] [Google Scholar]
- Atwood C. S., Scarpa R. C., Huang X., Moir R. D., Jones W. D., Fairlie D. P., et al. (2000). Characterization of copper interactions with Alzheimer Amyloid β peptides. J. Neurochem. 75 1219–1233. 10.1046/j.1471-4159.2000.0751219.x [DOI] [PubMed] [Google Scholar]
- Barnham K. J. (2004). Tyrosine gated electron transfer is key to the toxic mechanism of Alzheimer’s disease -amyloid. FASEB J. 18 1427–1429. 10.1096/fj.04-1890fje [DOI] [PubMed] [Google Scholar]
- Barritt J. D., Viles J. H. (2015). Truncated amyloid-β(11-40/42) from Alzheimer disease binds Cu2+ with a femtomolar affinity and influences fiber assembly. J. Biol. Chem. 290 27791–27802. 10.1074/jbc.M115.684084 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bayer T. A., Schafer S., Simons A., Kemmling A., Kamer T., Tepests R., et al. (2003). Dietary Cu stabilizes brain superoxide dismutase 1 activity and reduces amyloid A production in APP23 transgenic mice. Proc. Natl. Acad. Sci. U.S.A. 100 14187–14192. 10.1073/pnas.2332818100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bayer T. A., Wirths O. (2010). Intracellular accumulation of amyloid-beta – a predictor for synaptic dysfunction and neuron loss in Alzheimer’s disease. Front. Aging Neurosci. 2:8 10.3389/fnagi.2010.00008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bazl R., Ganjali M. R., Saboury A., Foroumadi A., Nourozi P., Amanlou M. (2012). A new strategy based on pharmacophore-based virtual screening in adenosine deaminase inhibitors detection and in-vitro study. DARU J. Pharm. Sci. 20:64. 10.1186/2008-2231-20-64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Behbehani G. R., Barzegar L., Mohebbian M., Saboury A. A. (2012). A comparative interaction between copper ions with Alzheimer’s amyloid peptide and human serum albumin. Bioinorg. Chem. Appl. 2012:208641. 10.1155/2012/208641 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bittner T., Burgold S., Dorostkar M. M., Fuhrmann M., Wegenast-Braun B. M., Schmidt B., et al. (2012). Amyloid plaque formation precedes dendritic spine loss. Acta Neuropathol. 124 797–807. 10.1007/s00401-012-1047-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boison D. (2006). Adenosine kinase, epilepsy and stroke: mechanisms and therapies. Trends Pharmacol. Sci. 27 652–658. 10.1016/j.tips.2006.10.008 [DOI] [PubMed] [Google Scholar]
- Bolognin S., Messori L., Drago D., Gabbiani C., Cendron L., Zatta P. (2011). Aluminum, copper, iron and zinc differentially alter amyloid-Aβ 1-42 aggregation and toxicity. Int. J. Biochem. Cell Biol. 43 877–885. 10.1016/j.biocel.2011.02.009 [DOI] [PubMed] [Google Scholar]
- Borchardt T., Camakaris J., Cappai R., Masters C. L., Beyreuther K., Multhaup G. (1999). Copper inhibits beta-amyloid production and stimulates the non-amyloidogenic pathway of amyloid-precursor-protein secretion. Biochem. J. 344(Pt 2) 461–467. [PMC free article] [PubMed] [Google Scholar]
- Bourassa M. W., Leskovjan A. C., Tappero R. V., Farquhar E. R., Colton C. A., Van Nostrand W. E., et al. (2013). Elevated copper in the amyloid plaques and iron in the cortex are observed in mouse models of Alzheimer’s disease that exhibit neurodegeneration. Biomed. Spectrosc. Imaging 2 129–139. 10.3233/BSI-130041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brewer G. J. (2012). Copper excess, zinc deficiency, and cognition loss in Alzheimer’s disease. Biofactors 38 107–113. 10.1002/biof.1005 [DOI] [PubMed] [Google Scholar]
- Brewer G. J. (2014). Alzheimer’s disease causation by copper toxicity and treatment with zinc. Front. Aging Neurosci. 6:92 10.3389/fnagi.2014.00092 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brewer G. J. (2015). Divalent copper as a major triggering agent in Alzheimer’s disease. J. Alzheimers Dis. 46 593–604. 10.3233/JAD-143123 [DOI] [PubMed] [Google Scholar]
- Breydo L., Kurouski D., Rasool S., Milton S., Wu J. W., Uversky V. N., et al. (2016). Structural differences between amyloid beta oligomers. Biochem. Biophys. Res. Commun. 477 700–705. 10.1016/j.bbrc.2016.06.122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bucossi S., Ventriglia M., Panetta V., Salustri C., Pasqualetti P., Mariani S., et al. (2011). Copper in Alzheimer’s disease: a meta-analysis of serum, plasma, and cerebrospinal fluid studies. J. Alzheimers Dis. 24 175–185. 10.3233/JAD-2010-101473 [DOI] [PubMed] [Google Scholar]
- Buée L., Bussière T., Buée-Scherrer V., Delacourte A., Hof P. R. (2000). Tau protein isoforms, phosphorylation and role in neurodegenerative disorders. Brain Res. Rev. 33 95–130. 10.1016/S0165-0173(00)00019-9 [DOI] [PubMed] [Google Scholar]
- Bush A. I., Masters C. L., Tanzi R. E. (2003). Copper, -amyloid, and Alzheimer’s disease: tapping a sensitive connection. Proc. Natl. Acad. Sci. U.S.A. 100 11193–11194. 10.1073/pnas.2135061100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bush A. I., Tanzi R. E. (2008). Therapeutics for Alzheimer’s disease based on the metal hypothesis. Neurotherapeutics 5 421–432. 10.1016/j.nurt.2008.05.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cater M. A., McInnes K. T., Li Q.-X., Volitakis I., Fontaine S. L. A., Mercer J. F. B., et al. (2008). Intracellular copper deficiency increases amyloid-β secretion by diverse mechanisms. Biochem. J. 412 141–152. 10.1042/BJ20080103 [DOI] [PubMed] [Google Scholar]
- Cavaleri F. (2015). Paradigm shift redefining molecular, metabolic and structural events in Alzheimer’s disease involves a proposed contribution by transition metals. Defined lengthy preclinical stage provides new hope to circumvent advancement of disease- and age-related neurodegeneration. Med. Hypotheses 84 460–469. 10.1016/j.mehy.2015.01.044 [DOI] [PubMed] [Google Scholar]
- Ceccom J., Coslédan F., Halley H., Francès B., Lassalle J. M., Meunier B. (2012). Copper chelator induced efficient episodic memory recovery in a non-transgenic Alzheimer’s mouse model. PLOS ONE 7:e43105. 10.1371/journal.pone.0043105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cherny R. A., Atwood C. S., Xilinas M. E., Gray D. N., Jones W. D., McLean C. A., et al. (2001). Treatment with a copper-zinc chelator markedly and rapidly inhibits β-amyloid accumulation in Alzheimer’s disease transgenic mice. Neuron 30 665–676. 10.1016/S0896-6273(01)00317-8 [DOI] [PubMed] [Google Scholar]
- Cherny R. A., Legg J. T., McLean C. A., Fairlie D. P., Huang X., Atwood C. S., et al. (1999). Aqueous dissolution of Alzheimer’s disease Abeta amyloid deposits by biometal depletion. J. Biol. Chem. 274 23223–23228. 10.1074/jbc.274.33.23223 [DOI] [PubMed] [Google Scholar]
- Chui D. H., Tanahashi H., Ozawa K., Ikeda S., Checler F., Ueda O., et al. (1999). Transgenic mice with Alzheimer presenilin 1 mutations show accelerated neurodegeneration without amyloid plaque formation. Nat. Med. 5 560–564. 10.1038/8438 [DOI] [PubMed] [Google Scholar]
- Ciccotosto G. D., Tew D., Curtain C. C., Smith D., Carrington D., Masters C. L., et al. (2004). Enhanced toxicity and cellular binding of a modified amyloid β peptide with a methionine to valine substitution. J. Biol. Chem. 279 42528–42534. 10.1074/jbc.M406465200 [DOI] [PubMed] [Google Scholar]
- Ciccotosto G. D., Tew D. J., Drew S. C., Smith D. G., Johanssen T., Lal V., et al. (2011). Stereospecific interactions are necessary for Alzheimer disease amyloid-β toxicity. Neurobiol. Aging 32 235–248. 10.1016/j.neurobiolaging.2009.02.018 [DOI] [PubMed] [Google Scholar]
- Cleveland D. W., Hwo S. Y., Kirschner M. W. (1977). Purification of tau, a microtubule-associated protein that induces assembly of microtubules from purified tubulin. J. Mol. Biol. 116 207–225. 10.1016/0022-2836(77)90213-3 [DOI] [PubMed] [Google Scholar]
- Conte-Daban A., Day A., Faller P., Hureau C. (2016). How Zn can impede Cu detoxification by chelating agents in Alzheimer’s disease: a proof-of-concept study. Dalton Trans. 45 15671–15678. 10.1039/C6DT02308H [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper J. L. (2003). Dietary lipids in the aetiology of Alzheimer’s disease. Drugs Aging 20 399–418. 10.2165/00002512-200320060-00001 [DOI] [PubMed] [Google Scholar]
- Corodimas K. P., Tomita H. (2001). Adenosine A1 receptor activation selectively impairs the acquisition of contextual fear conditioning in rats. Behav. Neurosci. 115 1283–1290. 10.1037/0735-7044.115.6.1283 [DOI] [PubMed] [Google Scholar]
- Corona C., Masciopinto F., Silvestri E., Del Viscovo A., Lattanzio R., La Sorda R., et al. (2010). Dietary zinc supplementation of 3xTg-AD mice increases BDNF levels and prevents cognitive deficits as well as mitochondrial dysfunction. Cell Death Dis. 1:e91. 10.1038/cddis.2010.73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cortés A., Gracia E., Moreno E., Mallol J., Lluís C., Canela E. I., et al. (2015). Moonlighting adenosine deaminase: a target protein for drug development. Med. Res. Rev. 35 85–125. 10.1002/med.21324 [DOI] [PubMed] [Google Scholar]
- Cortes-Canteli M., Zamolodchikov D., Ahn H. J., Strickland S., Norris E. H. (2012). Fibrinogen and altered hemostasis in Alzheimer’s disease. J. Alzheimers Dis. 32 599–608. 10.3233/JAD-2012-120820 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cristalli G., Costanzi S., Lambertucci C., Lupidi G., Vittori S., Volpini R., et al. (2001). Adenosine deaminase: functional implications and different classes of inhibitors. Med. Res. Rev. 21 105–128. [DOI] [PubMed] [Google Scholar]
- Crouch P. J., Wai L., Adlard P. A., Cortes M., Lal V., Filiz G. (2009). Increasing Cu bioavailability inhibits Aβ oligomers and tau phosphorylation. Proc. Natl. Acad. Sci. U.S.A. 106 381–386. 10.1073/pnas.0809057106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curtain C. C., Ali F., Volitakis I., Cherny R. A., Norton R. S., Beyreuther K., et al. (2001). Alzheimer’s disease amyloid-β binds copper and zinc to generate an allosterically ordered membrane-penetrating structure containing superoxide dismutase-like subunits. J. Biol. Chem. 276 20466–20473. 10.1074/jbc.M100175200 [DOI] [PubMed] [Google Scholar]
- Da Settimo F., Primofiore G., La Motta C., Taliani S., Simorini F., Marini A. M., et al. (2005). Novel, highly potent adenosine deaminase inhibitors containing the pyrazolo[3,4-d]pyrimidine ring system. Synthesis, structure-activity relationships, and molecular modeling studies. J. Med. Chem. 48 5162–5174. 10.1021/jm050136d [DOI] [PubMed] [Google Scholar]
- Dai X. L., Sun Y. X., Jiang Z. F. (2006). Cu(II) potentiation of Alzheimer Aβ1-40 cytotoxicity and transition on its secondary structure. Acta Biochim. Biophys. Sin. 38 765–772. 10.1111/j.1745-7270.2006.00228.x [DOI] [PubMed] [Google Scholar]
- Dannenberger D., Nuernberg G., Renne U., Nuernberg K., Langhammer M., Huber K., et al. (2013). High-fat diets rich in u-3 or u-6 polyunsaturated fatty acids have distinct effects on lipid profiles and lipid peroxidation in mice selected for either high body weight or leanness. Nutrition 29 765–771. 10.1016/j.nut.2012.10.010 [DOI] [PubMed] [Google Scholar]
- De Mendonça A., Ribeiro J. A. (1996). Adenosine and neuronal plasticity. Life Sci. 60 245–251. 10.1016/S0024-3205(96)00544-9 [DOI] [PubMed] [Google Scholar]
- Deibel M. A., Ehmann W. D., Markesbery W. R. (1996). Copper, iron, and zinc imbalances in severely degenerated brain regions in Alzheimer’s disease: possible relation to oxidative stress. J. Neurol. Sci. 143 137–142. 10.1016/S0022-510X(96)00203-1 [DOI] [PubMed] [Google Scholar]
- Deloncle R., Guillard O. (2014). Is brain copper deficiency in Alzheimer’s, Lewy body, and Creutzfeldt Jakob diseases the common key for a free radical mechanism and oxidative stress-induced damage? J. Alzheimers Dis. 43 1149–1156. 10.3233/JAD-140765 [DOI] [PubMed] [Google Scholar]
- Derakhshankhah H., Hajipour M. J., Barzegari E., Lotfabadi A., Ferdousi M., Saboury A. A., et al. (2016). Zeolite nanoparticles inhibit Aβ-fibrinogen interaction and formation of a consequent abnormal structural clot. ACS Appl. Mater. Interfaces 8 30768–30779. 10.1021/acsami.6b10941 [DOI] [PubMed] [Google Scholar]
- Di Scala C., Troadec J. D., Lelièvre C., Garmy N., Fantini J., Chahinian H. (2014). Mechanism of cholesterol-assisted oligomeric channel formation by a short Alzheimer β-amyloid peptide. J. Neurochem. 128 186–195. 10.1111/jnc.12390 [DOI] [PubMed] [Google Scholar]
- Di Scala C., Yahi N., Boutemeur S., Flores A., Rodriguez L., Chahinian H., et al. (2016). Common molecular mechanism of amyloid pore formation by Alzheimer’s β-amyloid peptide and α-synuclein. Sci. Rep. 6:28781. 10.1038/srep28781 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong J., Atwood C. S., Anderson V. E., Siedlak S. L., Smith M. A., Perry G., et al. (2003). Metal binding and oxidation of amyloid-β within isolated senile plaque cores: Raman microscopic evidence. Biochemistry 42 2768–2773. 10.1021/bi0272151 [DOI] [PubMed] [Google Scholar]
- Donnelly P. S., Caragounis A., Du T., Laughton K. M., Volitakis I., Cherny R. A., et al. (2008). Selective intracellular release of copper and zinc ions from bis(thiosemicarbazonato) complexes reduces levels of Alzheimer disease amyloid-beta peptide. J. Biol. Chem. 283 4568–4577. 10.1074/Jbc.M705957200 [DOI] [PubMed] [Google Scholar]
- Drew S. C. (2017). The case for abandoning therapeutic chelation of copper ions in Alzheimer’s disease. Front. Neurosci. 11:317. 10.3389/fnins.2017.00317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drolle E., Hane F., Lee B., Leonenko Z. (2014). Atomic force microscopy to study molecular mechanisms of amyloid fibril formation and toxicity in Alzheimer’s disease. Drug Metab. Rev. 46 207–223. 10.3109/03602532.2014.882354 [DOI] [PubMed] [Google Scholar]
- Elder G. A., Gama Sosa M. A., De Gasperi R. (2010). Transgenic mouse models of Alzheimer’s disease. Mt. Sinai J. Med. 77 69–81. 10.1002/msj.20159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eskici G., Axelsen P. H. (2012). Copper and oxidative stress in the pathogenesis of Alzheimer’s disease. Biochemistry 51 6289–6311. 10.1021/bi3006169 [DOI] [PubMed] [Google Scholar]
- Esparza T. J., Zhao H., Cirrito J. R., Cairns N. J., Bateman R. J., Holtzman D. M., et al. (2013). Amyloid-beta oligomerization in Alzheimer dementia versus high-pathology controls. Ann. Neurol. 73 104–119. 10.1002/ana.23748 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Everett J., Cespedes E., Shelford L. R., Exley C., Collingwood J. F., Dobson J., et al. (2014). Ferrous iron formation following the co-aggregation of ferric iron and the Alzheimer’s disease peptide beta-amyloid (1-42). J. R. Soc. Interface 11:20140165. 10.1098/rsif.2014.0165 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Exley C. (2006). Aluminium and iron, but neither copper nor zinc, are key to the precipitation of beta-sheets of Aβ42 in senile plaque cores in Alzheimer’s disease. J. Alzheimers Dis. 10 173–177. 10.3233/JAD-2006-102-305 [DOI] [PubMed] [Google Scholar]
- Exley C., House E., Polwart A., Esiri M. M. (2012). Brain burdens of aluminum, iron, and copper and their relationships with amyloid-β pathology in 60 human brains. J. Alzheimers Dis. 31 725–730. 10.3233/JAD-2012-120766 [DOI] [PubMed] [Google Scholar]
- Fang C. L., Wu W. H., Liu Q., Sun X., Ma Y., Zhao Y. F., et al. (2010). Dual functions of β-amyloid oligomer and fibril in Cu(II)-induced H2O2 production. Regul. Pept. 163 1–6. 10.1016/j.regpep.2010.05.001 [DOI] [PubMed] [Google Scholar]
- Feaga H. A., Maduka R. C., Foster M. N., Szalai V. A. (2011). Affinity of Cu+ for the copper-binding domain of the amyloid-β peptide of Alzheimer’s disease. Inorg. Chem. 50 1614–1618. 10.1021/ic100967s [DOI] [PubMed] [Google Scholar]
- Flinn J. M., Lorenzo Bozzelli P., Adlard P. A., Railey A. M. (2014). Spatial memory deficits in a mouse model of late-onset Alzheimer’s disease are caused by zinc supplementation and correlate with amyloid-beta levels. Front. Aging Neurosci. 6:174 10.3389/fnagi.2014.00174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujiwara N., Iso H., Kitanaka N., Kitanaka J., Eguchi H., Ookawara T., et al. (2006). Effects of copper metabolism on neurological functions in Wistar and Wilson’s disease model rats. Biochem. Biophys. Res. Commun. 349 1079–1086. 10.1016/j.bbrc.2006.08.139 [DOI] [PubMed] [Google Scholar]
- Gendron T. F., Petrucelli L. (2009). The role of tau in neurodegeneration. Mol. Neurodegener. 4:13. 10.1186/1750-1326-4-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geng J., Li M., Wu L., Ren J., Qu X. (2012). Liberation of copper from amyloid plaques: making a risk factor useful for Alzheimers disease treatment. J. Med. Chem. 55 9146–9155. 10.1021/jm3003813 [DOI] [PubMed] [Google Scholar]
- Gerber H., Wu F., Dimitrov M., Osuna G. M. G., Fraering P. C. (2017). Zinc and copper differentially modulate amyloid precursor protein processing by γ-secretase and amyloid-β peptide production. J. Biol. Chem. 292 3751–3767. 10.1074/jbc.M116.754101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giacoppo S., Galuppo M., Calabrò R. S., D’Aleo G., Marra A., Sessa E., et al. (2014). Heavy metals and neurodegenerative diseases: an observational study. Biol. Trace Elem. Res. 161 151–160. 10.1007/s12011-014-0094-5 [DOI] [PubMed] [Google Scholar]
- González C., Martín T., Cacho J., Breñas M. T., Arroyo T., García-Berrocal B., et al. (1999). Serum zinc, copper, insulin and lipids in Alzheimer’s disease epsilon 4 apolipoprotein E allele carriers. Eur. J. Clin. Invest. 29 637–642. 10.1046/j.1365-2362.1999.00471.x [DOI] [PubMed] [Google Scholar]
- Grant W. B. (1997). Dietary links to Alzheimer’s disease. Alzheimers Dis. Rev. 2 42–55. [DOI] [PubMed] [Google Scholar]
- Grasso G., Pietropaolo A., Spoto G., Pappalardo G., Tundo G. R., Ciaccio C., et al. (2011). Copper(I) and Copper(II) inhibit Aβ peptides proteolysis by insulin-degrading enzyme differently: implications for metallostasis alteration in Alzheimer’s disease. Chemistry 17 2752–2762. 10.1002/chem.201002809 [DOI] [PubMed] [Google Scholar]
- Greenough M. A., Camakaris J., Bush A. I. (2013). Metal dyshomeostasis and oxidative stress in Alzheimer’s disease. Neurochem. Int. 62 540–555. 10.1016/j.neuint.2012.08.014 [DOI] [PubMed] [Google Scholar]
- Greenough M. A., Ramírez Munoz A., Bush A. I., Opazo C. M. (2016). Metallo-pathways to Alzheimer’s disease: lessons from genetic disorders of copper trafficking. Metallomics 8 831–839. 10.1039/c6mt00095a [DOI] [PubMed] [Google Scholar]
- Grundke-Iqbal I., Fleming J., Tung Y. C., Lassmann H., Iqbal K., Joshi J. G. (1990). Ferritin is a component of the neuritic (senile) plaque in Alzheimer dementia. Acta Neuropathol. 81 105–110. 10.1007/BF00334497 [DOI] [PubMed] [Google Scholar]
- Guan C., Dang R., Cui Y., Liu L., Chen X., Wang X., et al. (2017). Characterization of plasma metal profiles in Alzheimer’s disease using multivariate statistical analysis. PLOS ONE 12:e0178271 10.1371/journal.pone.0178271s [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gunn A. P., Wong B. X., Johanssen T., Griffith J. C., Masters C. L., Bush A. I., et al. (2016). Amyloid-Beta peptide Aβ3pE-42 induces lipid peroxidation, membrane permeabilization and calcium-influx in neurons. J. Biol. Chem. 42 6134–6145. 10.1074/jbc.M115.655183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haeffner F., Smith D. G., Barnham K. J., Bush A. I. (2005). Model studies of cholesterol and ascorbate oxidation by copper complexes: relevance to Alzheimer’s disease β-amyloid metallochemistry. J. Inorg. Biochem. 99 2403–2422. 10.1016/j.jinorgbio.2005.09.011 [DOI] [PubMed] [Google Scholar]
- Hane F., Tran G., Attwood S. J., Leonenko Z. (2013). Cu2+ affects amyloid-β (1-42) aggregation by increasing peptide-peptide binding forces. PLOS ONE 8:e59005. 10.1371/journal.pone.0059005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hane F. T., Hayes R., Lee B. Y., Leonenko Z. (2016). Effect of copper and zinc on the single molecule self-affinity of Alzheimer’s amyloid-beta peptides. PLOS ONE 11:e0147488. 10.1371/journal.pone.0147488 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris C. J., Voss K., Murchison C., Ralle M., Frahler K., Carter R., et al. (2014). Oral zinc reduces amyloid burden in Tg2576 mice. J. Alzheimers Dis. 41 179–192. 10.3233/JAD-131703 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hauber W., Bareiß A. (2001). Facilitative effects of an adenosine A1/A2 receptor blockade on spatial memory performance of rats: selective enhancement of reference memory retention during the light period. Behav. Brain Res. 118 43–52. 10.1016/S0166-4328(00)00307-7 [DOI] [PubMed] [Google Scholar]
- Hauser-Davis R. A., de Freitas L. V., Cukierman D. S., Cruz W. S., Miotto M. C., Landeira-Fernandez J., et al. (2015). Disruption of zinc and copper interactions with Aβ(1–40) by a non-toxic, isoniazid-derived, hydrazone: a novel biometal homeostasis restoring agent in Alzheimer’s disease therapy? Metallomics 7 743–747. 10.1039/C5MT00003C [DOI] [PubMed] [Google Scholar]
- Holmes C., Boche D., Wilkinson D., Yadegarfar G., Hopkins V., Bayer A., et al. (2008). Long-term effects of Aβ42 immunisation in Alzheimer’s disease: follow-up of a randomised, placebo-controlled phase i trial. Lancet 372 216–223. 10.1016/S0140-6736(08)61075-2 [DOI] [PubMed] [Google Scholar]
- Howlett D. R., Richardson J. C., Austin A., Parsons A. A., Bate S. T., Davies D. C., et al. (2004). Cognitive correlates of Abeta deposition in male and female mice bearing amyloid precursor protein and presenilin-1 mutant transgenes. Brain Res. 1017 130–136. 10.1016/j.brainres.2004.05.029 [DOI] [PubMed] [Google Scholar]
- Hozumi I., Hasegawa T., Honda A., Ozawa K., Hayashi Y., Hashimoto K., et al. (2011). Patterns of levels of biological metals in CSF differ among neurodegenerative diseases. J. Neurol. Sci. 303 95–99. 10.1016/j.jns.2011.01.003 [DOI] [PubMed] [Google Scholar]
- Hu D., Serrano F., Oury T. D., Klann E. (2006). Aging-dependent alterations in synaptic plasticity and memory in mice that overexpress extracellular superoxide dismutase. J. Neurosci. 26 3933–3941. 10.1523/JNEUROSCI.5566-05.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hua H., Münter L., Harmeier A., Georgiev O., Multhaup G., Schaffner W. (2011). Toxicity of Alzheimer’s disease-associated Aβ peptide is ameliorated in a Drosophila model by tight control of zinc and copper availability. Biol. Chem. 392 919–926. 10.1515/BC.2011.084 [DOI] [PubMed] [Google Scholar]
- Huang X., Atwood C. S., Moir R. D., Hartshorn M. A., Tanzi R. E., Bush A. I. (2004). Trace metal contamination initiates the apparent auto-aggregation, amyloidosis, and oligomerization of Alzheimer’s Aβ peptides. J. Biol. Inorg. Chem. 9 954–960. 10.1007/s00775-004-0602-8 [DOI] [PubMed] [Google Scholar]
- Huang X., Cuajungco M. P., Atwood C. S., Hartshorn M. A., Tyndall J. D., Hanson G. R., et al. (1999). Cu(II) potentiation of Alzheimer Aß neurotoxicity. J. Biol. Chem. 274 37111–37116. 10.1074/jbc.274.52.37111 [DOI] [PubMed] [Google Scholar]
- Hung V. W. S., Bressan L. P., Seo K., Kerman K. (2015). Electroanalysis of natural compounds as copper chelating agents for Alzheimer’s disease therapy. Electroanalysis 27 2670–2678. 10.1002/elan.201500138 [DOI] [Google Scholar]
- Hung Y. H., Robb E. L., Voltakis I., Ho M., Evin G., Li Q. X., et al. (2009). Paradoxical condensation of copper with elevated β-amyloid in lipid rafts under cellular copper deficiency conditions. Implications for Alzheimer disease. J. Biol. Chem. 284 21899–21907. 10.1074/jbc.M109.019521 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hureau C. (2012). Coordination of redox active metal ions to the amyloid precursor protein and to amyloid beta peptides involved in Alzheimer disease. Part 1: an overview. Coord. Chem. Rev. 256 2164–2174. 10.1016/j.ccr.2012.03.037 [DOI] [Google Scholar]
- Jack C. R., Knopman D. S., Jagust W. J., Shaw L. M., Aisen P. S., Weiner M. W., et al. (2010). Hypothetical model of dynamic biomarkers of the Alzheimer’s pathological cascade. Lancet Neurol. 9 119–128. 10.1016/S1474-4422(09)70299-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- James S. A., Churches Q. I., De Jonge M. D., Birchall I. E., Streltsov V., McColl G., et al. (2017). Iron, copper, and zinc concentration in Aβ plaques in the APP/PS1 mouse model of Alzheimer’s disease correlates with metal levels in the surrounding neuropil. ACS Chem. Neurosci. 8 629–637. 10.1021/acschemneuro.6b00362 [DOI] [PubMed] [Google Scholar]
- James S. A., Volitakis I., Adlard P. A., Duce J. A., Masters C. L., Cherny R. A., et al. (2012). Elevated labile Cu is associated with oxidative pathology in Alzheimer disease. Free Radic. Biol. Med. 52 298–302. 10.1016/j.freeradbiomed.2011.10.446 [DOI] [PubMed] [Google Scholar]
- Jiang D., Li X., Williams R., Patel S., Men L., Wang Y., et al. (2009). Ternary complexes of iron, amyloid-β, and nitrilotriacetic acid: binding affinities, redox properties, and relevance to iron-induced oxidative stress in Alzheimer’s disease. Biochemistry 48 7939–7947. 10.1021/bi900907a [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiao Y., Yang P. (2007). Mechanism of copper(II) inhibiting Alzheimer’s amyloid beta-peptide from aggregation: a molecular dynamics investigation. J. Phys. Chem. B 111 7646–7655. 10.1021/jp0673359 [DOI] [PubMed] [Google Scholar]
- Jun S., Gillespie J. R., Shin B. K., Saxena S. (2009). The second Cu(II)-binding site in a proton-rich environment interferes with the aggregation of amyloid-β(1-40) into amyloid fibrils. Biochemistry 48 10724–10732. 10.1021/bi9012935 [DOI] [PubMed] [Google Scholar]
- Jung C. K. E., Keppler K., Steinbach S., Blazquez-Llorca L., Herms J. (2015). Fibrillar amyloid plaque formation precedes microglial activation. PLOS ONE 10:e0119768. 10.1371/journal.pone.0119768 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaden D., Bush A. I., Danzeisen R., Bayer T. A., Multhaup G. (2011). Disturbed copper bioavailability in Alzheimer’s disease. Int. J. Alzheimers Dis. 2011:345614. 10.4061/2011/345614 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamsler A., Segal M. (2003). Hydrogen peroxide modulation of synaptic plasticity. J. Neurosci. 23 269–276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamsler A., Segal M. (2004). Hydrogen peroxide as a diffusible signal molecule in synaptic plasticity. Mol. Neurobiol. 29 167–178. 10.1385/MN:29:2:167 [DOI] [PubMed] [Google Scholar]
- Kepp K. P. (2016). Alzheimer’s disease due to loss of function: a new synthesis of the available data. Prog. Neurobiol. 143 36–60. 10.1016/j.pneurobio.2016.06.004 [DOI] [PubMed] [Google Scholar]
- Kessler H., Bayer T. A., Bach D., Schneider-Axmann T., Supprian T., Herrmann W., et al. (2008a). Intake of copper has no effect on cognition in patients with mild Alzheimer’s disease: a pilot phase 2 clinical trial. J. Neural Transm. 115 1181–1187. 10.1007/s00702-008-0080-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kessler H., Pajonk F.-G., Bach D., Schneider-Axmann T., Falkai P., Herrmann W., et al. (2008b). Effect of copper intake on CSF parameters in patients with mild Alzheimer’s disease: a pilot phase 2 clinical trial. J. Neural Transm. 115 1181–1187. 10.1007/s00702-008-0080-1 [DOI] [PubMed] [Google Scholar]
- Kessler H., Pajonk F. G., Meisser P., Schneider-Axmann T., Hoffmann K. H., Supprian T., et al. (2006). Cerebrospinal fluid diagnostic markers correlate with lower plasma copper and ceruloplasmin in patients with Alzheimer’s disease. J. Neural Transm. 113 1763–1769. 10.1007/s00702-006-0485-7 [DOI] [PubMed] [Google Scholar]
- Kessler H., Pajonk F. G., Supprian T., Falkai P., Multhaup G., Bayer T. A. (2005). The role of copper in the pathophysiology of Alzheimer’s disease. Nervenarzt 76 581–585. 10.1007/s00115-004-1849-6 [DOI] [PubMed] [Google Scholar]
- Khan A., Dobson J. P., Exley C. (2006). Redox cycling of iron by Abeta42. Free Radic. Biol. Med. 40 557–569. 10.1016/j.freeradbiomed.2005.09.013 [DOI] [PubMed] [Google Scholar]
- Kishida K. T., Klann E. (2007). Sources and targets of reactive oxygen species in synaptic plasticity and memory. Antioxid. Redox Signal. 9 233–244. 10.1089/ars.2007.9.ft-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klann E. (1998). Cell-permeable scavengers of superoxide prevent long-term potentiation in hippocampal area CA1. J. Neurophysiol. 80 452–457. 10.1152/jn.1998.80.1.452 [DOI] [PubMed] [Google Scholar]
- Klevay L. M. (2008). Alzheimer’s disease as copper deficiency. Med. Hypotheses 70 802–807. 10.1016/j.mehy.2007.04.051 [DOI] [PubMed] [Google Scholar]
- Knapp L. T., Klann E. (2002a). Role of reactive oxygen species in hippocampal long-term potentiation: contributory or inhibitory? J. Neurosci. Res. 70 1–7. [DOI] [PubMed] [Google Scholar]
- Knapp L. T., Klann E. (2002b). Potentiation of hippocampal synaptic transmission by superoxide requires the oxidative activation of protein kinase C. J. Neurosci. 22 674–683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kong G. K.-W., Miles L. A., Crespi G. A. N., Morton C. J., Ng H. L., Barnham K. J., et al. (2008). Copper binding to the Alzheimer’s disease amyloid precursor protein. Eur. Biophys. J. 37 269–279. 10.1007/s00249-007-0234-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kopf S. R., Melani A., Pedata F., Pepeu G. (1999). Adenosine and memory storage: effect of A(1) and A(2) receptor antagonists. Psychopharmacology 146 214–219. 10.1007/s002130051109 [DOI] [PubMed] [Google Scholar]
- Kotler S. A., Walsh P., Brender J. R., Ramamoorthy A. (2014). Differences between amyloid-β aggregation in solution and on the membrane: insights into elucidation of the mechanistic details of Alzheimer’s disease. Chem. Soc. Rev. 43 6692–6700. 10.1039/C3CS60431D [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kozlowski H., Luczkowski M., Remelli M., Valensin D. (2012). Copper, zinc and iron in neurodegenerative diseases (Alzheimer’s, Parkinson’s and prion diseases). Coord. Chem. Rev. 256 2129–2141. 10.1016/j.ccr.2012.03.013 [DOI] [Google Scholar]
- La Motta C., Sartini S., Mugnaini L., Salerno S., Simorini F., Taliani S., et al. (2009). Exploiting the pyrazolo[3,4-d]pyrimidin-4-one ring system as a useful template to obtain potent adenosine deaminase inhibitors. J. Med. Chem. 52 1681–1692. 10.1021/jm801427r [DOI] [PubMed] [Google Scholar]
- Lang M., Fan Q., Wang L., Zheng Y., Xiao G., Wang X., et al. (2013). Inhibition of human high-affinity copper importer Ctr1 orthologous in the nervous system of Drosophila ameliorates Aβ 42-induced Alzheimer’s disease-like symptoms. Neurobiol. Aging 34 2604–2612. 10.1016/j.neurobiolaging.2013.05.029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larry S. D. (2004). Cholesterol, copper, and accumulation of thioflavine S-reactive Alzheimer’s-like amyloid beta in rabbit brain. J. Mol. Neurosci. 24 97–104. 10.1385/JMN:24:1:097 [DOI] [PubMed] [Google Scholar]
- Lashuel H. A., Hartley D., Petre B. M., Walz T., Lansbury P. T. (2002). Neurodegenerative disease: amyloid pores from pathogenic mutations. Nature 418 291–291. 10.1038/418291a [DOI] [PubMed] [Google Scholar]
- Lee H. G., Castellani R. J., Zhu X., Perry G., Smith M. A. (2005). Amyloid-β in Alzheimer’s disease: the horse or the cart? Pathogenic or protective? Int. J. Exp. Pathol. 86 133–138. 10.1111/j.0959-9673.2005.00429.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leskovjan A. C., Kretlow A., Lanzirotti A., Barrea R., Vogt S., Miller L. M. (2011). Increased brain iron coincides with early plaque formation in a mouse model of Alzheimer’s disease. Neuroimage 55 32–38. 10.1016/j.neuroimage.2010.11.073 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leskovjan A. C., Lanzirotti A., Miller L. M. (2009). Amyloid plaques in PSAPP mice bind less metal than plaques in human Alzheimer’s disease. Neuroimage 47 1215–1220. 10.1016/j.neuroimage.2009.05.063 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lesné S., Koh M. T., Kotilinek L., Kayed R., Glabe C. G., Yang A., et al. (2006). A specific amyloid-β protein assembly in the brain impairs memory. Nature 440 352–357. 10.1038/nature04533 [DOI] [PubMed] [Google Scholar]
- Li D.-D., Zhang W., Wang Z.-Y., Zhao P. (2017). Serum copper, zinc, and iron levels in patients with Alzheimer’s disease: a meta-analysis of case-control studies. Front. Aging Neurosci. 9:300 10.3389/fnagi.2017.00300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linkous D. H., Adlard P. A., Wanschura P. B., Conko K. M., Flinn J. M. (2009). The effects of enhanced zinc on spatial memory and plaque formation in transgenic mice. J. Alzheimers Dis. 18 565–579. 10.3233/JAD-2009-1162 [DOI] [PubMed] [Google Scholar]
- Liu H. P., Lin W. Y., Wang W. F., Tsai C. H., Wu W. C., Chiou M. T., et al. (2013). Genetic variability in copper-transporting P-type adenosine triphosphatase (ATP7B) is associated with Alzheimer’s disease in a Chinese population. J. Biol. Regul. Homeost. Agents 27 319–327. [PubMed] [Google Scholar]
- Loef M., von Stillfried N., Walach H. (2012). Zinc diet and Alzheimer’s disease: a systematic review. Nutr. Neurosci. 15 2–12. 10.1179/1476830512Y.0000000010 [DOI] [PubMed] [Google Scholar]
- Lovell M. A., Robertson J. D., Teesdale W. J., Campbell J. L., Markesbery W. R. (1998). Copper, iron and zinc in Alzheimer’s disease senile plaques. J. Neurol. Sci. 158 47–52. 10.1016/S0022-510X(98)00092-6 [DOI] [PubMed] [Google Scholar]
- Luo Y. F., Zhang J., Liu N. Q., Luo Y., Zhao B. L. (2011). Copper ions influence the toxicity of β-amyloid(1-42) in a concentration-dependent manner in a Caenorhabditis elegans model of Alzheimer’s disease. Sci. China Life Sci. 54 527–534. 10.1007/s11427-011-4180-z [DOI] [PubMed] [Google Scholar]
- Lv Z., Condron M. M., Teplow D. B., Lyubchenko Y. L. (2013). Nanoprobing of the effect of Cu2+ cations on misfolding, interaction and aggregation of amyloid β peptide. J. Neuroimmune Pharmacol. 8 262–273. 10.1007/s11481-012-9416-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma Q., Li Y., Du J., Liu H., Kanazawa K., Nemoto T., et al. (2006). Copper binding properties of a tau peptide associated with Alzheimer’s disease studied by CD, NMR, and MALDI-TOF MS. Peptides 27 841–849. 10.1016/j.peptides.2005.09.002 [DOI] [PubMed] [Google Scholar]
- Macreadie I. (2008). Copper transport and Alzheimer’s disease. Eur. Biophys. J. 37 295–300. 10.1007/s00249-007-0235-2 [DOI] [PubMed] [Google Scholar]
- Maiuolo J., Oppedisano F., Gratteri S., Muscoli C., Mollace V. (2016). Regulation of uric acid metabolism and excretion. Int. J. Cardiol. 213 8–14. 10.1016/j.ijcard.2015.08.109 [DOI] [PubMed] [Google Scholar]
- Mao X., Ye J., Zhou S., Pi R., Dou J., Zang L., et al. (2012). The effects of chronic copper exposure on the amyloid protein metabolisim associated genes’ expression in chronic cerebral hypoperfused rats. Neurosci. Lett. 518 14–18. 10.1016/j.neulet.2012.04.030 [DOI] [PubMed] [Google Scholar]
- Marcellini F., Giuli C., Papa R., Gagliardi C., Dedoussis G., Herbein G., et al. (2006). Zinc status, psychological and nutritional assessment in old people recruited in five European countries: zincage study. Biogerontology 7 339–345. 10.1007/s10522-006-9048-4 [DOI] [PubMed] [Google Scholar]
- Masters C. L., Selkoe D. J. (2012). Biochemistry of amyloid β-protein and amyloid deposits in Alzheimer disease. Cold Spring Harb. Perspect. Med. 2:a006262. 10.1101/cshperspect.a006262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masters C. L., Simms G., Weinman N. A., Multhaup G., McDonald B. L., Beyreuther K. (1985). Amyloid plaque core protein in Alzheimer disease and Down syndrome. Proc. Natl. Acad. Sci. U.S.A. 82 4245–4249. 10.1073/pnas.82.12.4245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayes J., Tinker-Mill C., Kolosov O., Zhang H., Tabner B. J., Allsop D. (2014). β-Amyloid fibrils in Alzheimer disease are not inert when bound to copper ions but can degrade hydrogen peroxide and generate reactive oxygen species. J. Biol. Chem. 289 12052–12062. 10.1074/jbc.M113.525212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maylor E. A., Simpson E. E. A., Secker D. L., Meunier N., Andriollo-Sanchez M., Polito A., et al. (2006). Effects of zinc supplementation on cognitive function in healthy middle-aged and older adults: the ZENITH study. Br. J. Nutr. 96 752–760. 10.1079/bjn20061911 [DOI] [PubMed] [Google Scholar]
- Maynard C. J., Cappai R., Volitakis I., Cherny R. A., Masters C. L., Li Q. X., et al. (2006). Gender and genetic background effects on brain metal levels in APP transgenic and normal mice: implications for Alzheimer beta-amyloid pathology. J. Inorg. Biochem. 100 952–962. 10.1016/j.jinorgbio.2006.02.010 [DOI] [PubMed] [Google Scholar]
- Maynard C. J., Cappai R., Volitakis I., Cherny R. A., White A. R., Beyreuther K., et al. (2002). Overexpression of Alzheimer’s disease amyloid-β opposes the age-dependent elevations of brain copper and iron. J. Biol. Chem. 277 44670–44676. 10.1074/jbc.M204379200 [DOI] [PubMed] [Google Scholar]
- Mercer S. W., Wang J., Burke R. (2017). In vivo modeling of the pathogenic effect of copper transporter mutations that cause Menkes and Wilson diseases, motor neuropathy, and susceptibility to Alzheimer’s disease. J. Biol. Chem. 292 4113–4122. 10.1074/jbc.M116.756163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller L. M., Wang Q., Telivala T. P., Smith R. J., Lanzirotti A., Miklossy J. (2006). Synchrotron-based infrared and X-ray imaging shows focalized accumulation of Cu and Zn co-localized with β-amyloid deposits in Alzheimer’s disease. J. Struct. Biol. 155 30–37. 10.1016/j.jsb.2005.09.004 [DOI] [PubMed] [Google Scholar]
- Mital M., Wezynfeld N. E., Frączyk T., Wiloch M. Z., Wawrzyniak U. E., Bonna A., et al. (2015). A functional role for Aβ in metal homeostasis? N-truncation and high-affinity copper binding. Angew. Chem. Int. Ed. Engl. 54 10460–10464. 10.1002/anie.201502644 [DOI] [PubMed] [Google Scholar]
- Moret V., Laras Y., Pietrancosta N., Garino C., Quéléver G., Rolland A., et al. (2006). 1,1′-Xylyl bis-1,4,8,11-tetraaza cyclotetradecane: a new potential copper chelator agent for neuroprotection in Alzheimer’s disease. Its comparative effects with clioquinol on rat brain copper distribution. Bioorg. Med. Chem. Lett. 16 3298–3301. 10.1016/j.bmcl.2006.03.026 [DOI] [PubMed] [Google Scholar]
- Morris M. C., Evans D. A., Tangney C. C., Bienias J. L., Schneider J. A., Wilson R. S., et al. (2006). Dietary copper and high saturated and trans fat intakes associated with cognitive decline. Arch. Neurol. 63 1085–1088. 10.1001/archneur.63.8.1085 [DOI] [PubMed] [Google Scholar]
- Multhaup G., Schlicksupp A., Hesse L., Beher D., Ruppert T., Masters C. L., et al. (1996). The amyloid precursor protein of Alzheimer’s disease in the reduction of copper(II) to copper(I). Science 271 1406–1409. 10.1126/science.271.5254.1406 [DOI] [PubMed] [Google Scholar]
- Murray I. V. J., Liu L., Komatsu H., Uryu K., Xiao G., Lawson J. A., et al. (2007). Membrane-mediated amyloidogenesis and the promotion of oxidative lipid damage by amyloidβ proteins. J. Biol. Chem. 282 9335–9345. 10.1074/jbc.M608589200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura M., Shishido N., Nunomura A., Smith M. A., Perry G., Hayashi Y., et al. (2007). Three histidine residues of amyloid-beta peptide control the redox activity of copper and iron. Biochemistry 46 12737–12743. 10.1021/bi701079z [DOI] [PubMed] [Google Scholar]
- Nalivaeva N. N., Turner A. J. (2013). The amyloid precursor protein: a biochemical enigma in brain development, function and disease. FEBS Lett. 587 2046–2054. 10.1016/j.febslet.2013.05.010 [DOI] [PubMed] [Google Scholar]
- Naylor R., Hill A. F., Barnham K. J. (2008). Is covalently crosslinked Abeta responsible for synaptotoxicity in Alzheimer’s disease? Curr. Alzheimer Res. 5 533–539. 10.2174/156720508786898433 [DOI] [PubMed] [Google Scholar]
- Nguyen M., Robert A., Sournia-Saquet A., Vendier L., Meunier B. (2014). Characterization of new specific copper chelators as potential drugs for the treatment of Alzheimer’s disease. Chemistry 20 6771–6785. 10.1002/chem.201402143 [DOI] [PubMed] [Google Scholar]
- Oddo S., Caccamo A., Shepherd J. D., Murphy M. P., Golde T. E., Kayed R., et al. (2003). Triple-transgenic model of Alzheimer’s disease with plaques and tangles: intracellular Aβ and synaptic dysfunction. Neuron 39 409–421. 10.1016/S0896-6273(03)00434-3 [DOI] [PubMed] [Google Scholar]
- Ohno M., Watanabe S. (1996). Working memory failure by stimulation of hippocampal adenosine A1 receptors in rats. Neuroreport 7 3013–3016. 10.1097/00001756-199611250-00043 [DOI] [PubMed] [Google Scholar]
- Pajonk F.-G., Kessler H., Supprian T., Hamzei P., Bach D., Schweickhardt J., et al. (2005). Cognitive decline correlates with low plasma concentrations of copper in patients with mild to moderate Alzheimer’s disease. J. Alzheimers Dis. 8 23–27. 10.3233/JAD-2005-8103 [DOI] [PubMed] [Google Scholar]
- Pal A., Badyal R. K., Vasishta R. K., Attri S. V., Thapa B. R., Prasad R. (2013). Biochemical, histological, and memory impairment effects of chronic copper toxicity: a model for non-Wilsonian brain copper toxicosis in Wistar rat. Biol. Trace Elem. Res. 153 257–268. 10.1007/s12011-013-9665-0 [DOI] [PubMed] [Google Scholar]
- Pal A., Kumar A., Prasad R. (2014). Predictive association of copper metabolism proteins with Alzheimer’s disease and Parkinson’s disease: a preliminary perspective. Biometals 27 25–31. 10.1007/s10534-013-9702-7 [DOI] [PubMed] [Google Scholar]
- Parthasarathy S., Yoo B., McElheny D., Tay W., Ishii Y. (2014). Capturing a reactive state of amyloid aggregates: Nmr-based characterization of copper-bound Alzheimer disease amyloid β-fibrils in a redox cycle. J. Biol. Chem. 289 9998–10010. 10.1074/jbc.M113.511345 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng F., Muzik O., Gatson J., Kernie S. G., Diaz-Arrastia R. (2015). Assessment of traumatic brain injury by increased 64Cu uptake on 64CuCl2 PET/CT. J. Nucl. Med. 56 1252–1257. 10.2967/jnumed.115.154575 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pereira G. S., Mello e Souza T., Vinadé E. R. C., Choi H., Rodrigues C., Battastini A. M. O., et al. (2002). Blockade of adenosine A1 receptors in the posterior cingulate cortex facilitates memory in rats. Eur. J. Pharmacol. 437 151–154. 10.1016/S0014-2999(02)01307-9 [DOI] [PubMed] [Google Scholar]
- Phinney A. L., Drisaldi B., Schmidt S. D., Lugowski S., Coronado V., Liang Y., et al. (2003). In vivo reduction of amyloid-beta by a mutant copper transporter. Proc. Natl. Acad. Sci. U.S.A. 100 14193–14198. 10.1073/pnas.2332851100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pickart L., Vasquez-Soltero J. M., Margolina A. (2017). The effect of the human peptide GHK on gene expression relevant to nervous system function and cognitive decline. Brain Sci. 7:E20. 10.3390/brainsci7020020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pimentel V. C., Gomes J. L., Zanini D., Abdalla F. H., Da Costa P., Gonçalves J. F., et al. (2013). Evaluation of acetylcholinesterase and adenosine deaminase activities in brain and erythrocytes and proinflammatory cytokine levels in rats submitted to neonatal hypoxia-ischemia model. Mol. Cell. Biochem. 378 247–255. 10.1007/s11010-013-1615-9 [DOI] [PubMed] [Google Scholar]
- Pithadia A. S., Lim M. H. (2012). Metal-associated amyloid-β species in Alzheimer’s disease. Curr. Opin. Chem. Biol. 16 67–73. 10.1016/j.cbpa.2012.01.016 [DOI] [PubMed] [Google Scholar]
- Podlisny M. B., Ostaszewski B. L., Squazzo S. L., Koo E. H., Rydell R. E., Teplow D. B., et al. (1995). Aggregation of secreted amyloid beta-protein into sodium dodecyl sulfate-stable oligomers in cell culture. J. Biol. Chem. 270 9564–9570. 10.1074/jbc.270.16.9564 [DOI] [PubMed] [Google Scholar]
- Porkka-Heiskanen T. (1999). Adenosine in sleep and wakefulness. Ann. Med. 31 125–129. 10.3109/07853899908998788 [DOI] [PubMed] [Google Scholar]
- Pu Z., Xu W., Lin Y., He J., Huang M. (2017). Oxidative stress markers and metal ions are correlated with cognitive function in Alzheimer’s disease. Am. J. Alzheimers Dis. Other Demen. 32 353–359. 10.1177/1533317517709549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puglielli L., Friedlich A. L., Setchell K. D. R., Nagano S., Opazo C., Cherny R. A., et al. (2005). Alzheimer disease beta-amyloid activity mimics cholesterol oxidase. J. Clin. Invest. 115 2556–2563. 10.1172/JCI23610.2556 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinn J. F., Harris C. J., Cobb K. E., Domes C., Ralle M., Brewer G., et al. (2010). A copper-lowering strategy attenuates amyloid pathology in a transgenic mouse model of Alzheimer’s disease. J. Alzheimers Dis. 21 903–914. 10.3233/JAD-2010-100408 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quintana C., Bellefqih S., Laval J. Y., Guerquin-Kern J. L., Wu T. D., Avila J., et al. (2006). Study of the localization of iron, ferritin, and hemosiderin in Alzheimer’s disease hippocampus by analytical microscopy at the subcellular level. J. Struct. Biol. 153 42–54. 10.1016/j.jsb.2005.11.001 [DOI] [PubMed] [Google Scholar]
- Rahman A. (2009). The role of adenosine in Alzheimer’s disease. Curr. Neuropharmacol. 7 207–216. 10.2174/157015909789152119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Railey A. M., Groeber C. M., Flinn J. M. (2011). The effect of metals on spatial memory in a transgenic mouse model of Alzheimer’s disease. J. Alzheimers Dis. 24 375–381. 10.3233/JAD-2011-101452 [DOI] [PubMed] [Google Scholar]
- Religa D., Strozyk D., Cherny R. A., Volitakis I., Haroutunian V., Winblad B., et al. (2006). Elevated cortical zinc in Alzheimer disease. Neurology 67 69–75. 10.1212/01.wnl.0000223644.08653.b5 [DOI] [PubMed] [Google Scholar]
- Ribeiro J. A., Sebastião A. M., De Mendonça A. (2002). Adenosine receptors in the nervous system: pathophysiological implications. Prog. Neurobiol. 68 377–392. 10.1016/S0301-0082(02)00155-7 [DOI] [PubMed] [Google Scholar]
- Riddell D. R., Christie G., Hussain I., Dingwall C. (2001). Compartmentalization of β-secretase (Asp2) into low-buoyant density, noncaveolar lipid rafts. Curr. Biol. 11 1288–1293. 10.1016/S0960-9822(01)00394-3 [DOI] [PubMed] [Google Scholar]
- Ritchie C. W., Bush A. I., Mackinnon A., Macfarlane S., Mastwyk M., MacGregor L., et al. (2003). Metal-protein attenuation with iodochlorhydroxyquin (clioquinol) targeting Abeta amyloid deposition and toxicity in Alzheimer disease: a pilot phase 2 clinical trial. Arch. Neurol. 60 1685–1691. 10.1001/archneur.60.12.1685 [DOI] [PubMed] [Google Scholar]
- Roberts B. R., Ryan T. M., Bush A. I., Masters C. L., Duce J. A. (2012). The role of metallobiology and amyloid-β peptides in Alzheimer’s disease. J. Neurochem. 120(Suppl. 1) 149–166. 10.1111/j.1471-4159.2011.07500.x [DOI] [PubMed] [Google Scholar]
- Saboury A. A., Bagheri S., Ataie G., Amanlou M., Moosavi-Movahedi A. A., Hakimelahi G. H., et al. (2004). Binding properties of adenosine deaminase interacted with theophylline. Chem. Pharm. Bull. 52 1179–1182. 10.1248/cpb.52.1179 [DOI] [PubMed] [Google Scholar]
- Saboury A. A., Bagheri S., Ataie G., Moosavi-Movahedi A. A., Hakimelahi G. H., Cristalli G., et al. (2005). Kinetic and thermodynamic study on the inhibition of adenosine deaminase by theobromine. Asian J. Chem. 17 233–239. [Google Scholar]
- Saboury A. A., Divsalar A., Ataie G., Amanlou M., Moosavi-Movahedi A. A., Hakimelahi G. H. (2003). Inhibition study of adenosine deaminase by caffeine using spectroscopy and isothermal titration calorimetry. Acta Biochim. Pol. 50 849–855. [PubMed] [Google Scholar]
- Sandusky-Beltran L. A., Manchester B. L., McNay E. C. (2017). Supplementation with zinc in rats enhances memory and reverses an age-dependent increase in plasma copper. Behav. Brain Res. 333 179–183. 10.1016/j.bbr.2017.07.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarell C. J. (2010). The Copper-Amyloid-beta-Peptide Complex of Alzheimer’s Disease: Affinity, Structure, Fibril Formation and Toxicity. London: University of London. [Google Scholar]
- Sarell C. J., Syme C. D., Rigby S. E. J., Viles J. H. (2009). Copper(II) binding to amyloid-β fibrils of Alzheimer’s disease reveals a picomolar affinity: stoichiometry and coordination geometry are independent of Aβ oligomeric form. Biochemistry 48 4388–4402. 10.1021/bi900254n [DOI] [PubMed] [Google Scholar]
- Sarell C. J., Wilkinson S. R., Viles J. H. (2010). Substoichiometric levels of Cu2+ ions accelerate the kinetics of fiber formation and promote cell toxicity of amyloid-β from Alzheimer disease. J. Biol. Chem. 285 41533–41540. 10.1074/jbc.M110.171355 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasaguri H., Nilsson P., Hashimoto S., Nagata K., Saito T., De Strooper B., et al. (2017). APP mouse models for Alzheimer’s disease preclinical studies. EMBO J. 36 2473–2487. 10.15252/embj.201797397 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Savelieff M. G., Liu Y., Senthamarai R. R. P., Korshavn K. J., Lee H. J., Ramamoorthy A., et al. (2014). A small molecule that displays marked reactivity toward copper-versus zinc-amyloid-β implicated in Alzheimer’s disease. Chem. Commun. 50 5301–5303. 10.1039/c3cc48473d [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sayre L. M., Perry G., Harris P. L. R., Liu Y., Schubert K. A., Smith M. A. (2000). In situ oxidative catalysis by neurofibrillary tangles and senile plaques in Alzheimer’s disease. J. Neurochem. 74 270–279. 10.1046/j.1471-4159.2000.0740270.x [DOI] [PubMed] [Google Scholar]
- Schrag M., Mueller C., Oyoyo U., Smith M. A., Kirsch W. M. (2011). Iron, zinc and copper in the Alzheimer’s disease brain: a quantitative meta-analysis. Some insight on the influence of citation bias on scientific opinion. Prog. Neurobiol. 94 296–306. 10.1016/j.pneurobio.2011.05.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schrag M., Mueller C., Zabel M., Crofton A., Kirsch W. M., Ghribi O., et al. (2013). Oxidative stress in blood in Alzheimer‘s disease and mild cognitive impairment: a meta-analysis. Neurobiol. Dis. 59 100–110. 10.1016/j.nbd.2013.07.005 [DOI] [PubMed] [Google Scholar]
- Selkoe D. J. (2001). Alzheimer’s disease: genes, proteins, and therapy. Physiol. Rev. 81 741–766. 10.1152/physrev.2001.81.2.741 [DOI] [PubMed] [Google Scholar]
- Simpson E. E. A., O’Connor J. M., Livingstone M. B. E., Rae G., Stewart-Knox B. J., Andriollo-Sanchez M., et al. (2005). Health and lifestyle characteristics of older European adults: the ZENITH study. Eur. J. Clin. Nutr. 59 S13–S21. 10.1038/sj.ejcn.1602292 [DOI] [PubMed] [Google Scholar]
- Singh I., Sagare A. P., Coma M., Perlmutter D., Gelein R., Bell R. D., et al. (2013). Low levels of copper disrupt brain amyloid-β homeostasis by altering its production and clearance. Proc. Natl. Acad. Sci. U.S.A. 110 14771–14776. 10.1073/pnas.1302212110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith M. A., Harris P. L., Sayre L. M., Perry G. (1997). Iron accumulation in Alzheimer disease is a source of redox-generated free radicals. Proc. Natl. Acad. Sci. U.S.A. 94 9866–9868. 10.1073/pnas.94.18.9866 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Southon A., Greenough M. A., Ganio G., Bush A. I., Burke R., Camakaris J. (2013). Presenilin promotes dietary copper uptake. PLOS ONE 8:e62811. 10.1371/journal.pone.0062811 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sparks D. L. (2007). Cholesterol metabolism and brain amyloidosis: evidence for a role of copper in the clearance of Abeta through the liver. Curr. Alzheimer Res. 4 165–169. 10.2174/156720507780362119 [DOI] [PubMed] [Google Scholar]
- Sparks D. L., Friedland R., Petanceska S., Schreurs B. G., Shi J., Perry G., et al. (2006). Trace copper levels in the drinking water, but not zinc or aluminum influence CNS Alzheimer-like pathology. J. Nutr. Health Aging 10 247–254. [PMC free article] [PubMed] [Google Scholar]
- Sparks D. L., Schreurs B. G. (2003). Trace amounts of copper in water induce beta-amyloid plaques and learning deficits in a rabbit model of Alzheimer’s disease. Proc. Natl. Acad. Sci. U.S.A. 100 11065–11069. 10.1073/pnas.1832769100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sperling R. A., Aisen P. S., Beckett L. A., Bennett D. A., Craft S., Fagan A. M., et al. (2011). Toward defining the preclinical stages of Alzheimer’s disease: recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement. 7 280–292. 10.1016/j.jalz.2011.03.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Squitti R., Polimanti R., Siotto M., Bucossi S., Ventriglia M., Mariani S., et al. (2013). ATP7B variants as modulators of copper dyshomeostasis in Alzheimer’s disease. Neuromolecular Med. 15 515–522. 10.1007/s12017-013-8237-y [DOI] [PubMed] [Google Scholar]
- Squitti R., Salustri C., Rongioletti M., Siotto M. (2017a). Commentary: the case for abandoning therapeutic chelation of copper ions in Alzheimer’s disease. Front. Neurol. 8:503 10.3389/fneur.2017.00503 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Squitti R., Simonelli I., Ventriglia M., Siotto M., Pasqualetti P., Rembach A., et al. (2014a). Meta-analysis of serum non-ceruloplasmin copper in Alzheimer’s disease. J. Alzheimers Dis. 38 809–822. 10.3233/JAD-131247 [DOI] [PubMed] [Google Scholar]
- Squitti R., Siotto M., Polimanti R. (2014b). Low-copper diet as a preventive strategy for Alzheimer’s disease. Neurobiol. Aging 35(Suppl. 2) S40–S50. 10.1016/j.neurobiolaging.2014.02.031 [DOI] [PubMed] [Google Scholar]
- Squitti R., Siotto M., Arciello M., Rossi L. (2016). Non-ceruloplasmin bound copper and ATP7B gene variants in Alzheimer’s disease. Metallomics 8 863–873. 10.1039/C6MT00101G [DOI] [PubMed] [Google Scholar]
- Squitti R., Ventriglia M., Gennarelli M., Colabufo N. A., El Idrissi I. G., Bucossi S., et al. (2017b). Non-ceruloplasmin copper distincts subtypes in Alzheimer’s disease: a genetic study of ATP7B frequency. Mol. Neurobiol. 54 671–681. 10.1007/s12035-015-9664-6 [DOI] [PubMed] [Google Scholar]
- Streltsov V. A., Titmuss S. J., Epa V. C., Barnham K. J., Masters C. L., Varghese J. N. (2008). The structure of the amyloid-beta peptide high-affinity copper II binding site in Alzheimer disease. Biophys. J. 95 3447–3456. 10.1529/biophysj.108.134429 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strozyk D., Launer L., Adlard P. (2009). Zinc and copper modulate Alzheimer Aβ levels in human cerebrospinal fluid. Neurobiol. Aging 30 1069–1077. 10.1016/j.neurobiolaging.2007.10.012.Zinc [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suh S. W., Jensen K. B., Jensen M. S., Silva D. S., Kesslak P. J., Danscher G., et al. (2000). Histochemically-reactive zinc in amyloid plaques, angiopathy, and degenerating neurons of Alzheimer’s diseased brains. Brain Res. 852 274–278. 10.1016/S0006-8993(99)02096-X [DOI] [PubMed] [Google Scholar]
- Suzuki K., Miura T., Takeuchi H. (2001). Inhibitory effect of copper(II) on zinc(II)-induced aggregation of amyloid β-peptide. Biochem. Biophys. Res. Commun. 285 991–996. 10.1006/bbrc.2001.5263 [DOI] [PubMed] [Google Scholar]
- Swerdlow R. H. (2011). Brain aging, Alzheimer’s disease, and mitochondria. Biochim. Biophys. Acta 1812 1630–1639. 10.1016/j.bbadis.2011.08.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szabo S. T., Jean Harry G., Hayden K. M., Szabo D. T., Birnbaum L. (2016). Comparison of metal levels between postmortem brain and ventricular fluid in Alzheimer’s disease and nondemented elderly controls. Toxicol. Sci. 150 292–300. 10.1093/toxsci/kfv325 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tabner B. J., Mayes J., Allsop D. (2011). Hypothesis: soluble A β oligomers in association with redox-active metal ions are the optimal generators of reactive oxygen species in Alzheimer’s disease. Int. J. Alzheimers Dis. 2011:546380. 10.4061/2011/546380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Talwar P., Grover S., Sinha J., Chandna P., Agarwal R., Kushwaha S., et al. (2017). Multifactorial analysis of a biomarker pool for Alzheimer disease risk in a North Indian population. Dement. Geriatr. Cogn. Disord. 44 25–34. 10.1159/000477206 [DOI] [PubMed] [Google Scholar]
- Tao Y., Wang Y., Rogers J. T., Wang F. (2014). Perturbed iron distribution in Alzheimer’s disease serum, cerebrospinal fluid, and selected brain regions: a systematic review and meta-analysis. J. Alzheimers Dis. 42 679–690. 10.3233/JAD-140396 [DOI] [PubMed] [Google Scholar]
- Tecchio F., Vecchio F., Ventriglia M., Porcaro C., Miraglia F., Siotto M., et al. (2016). Non-ceruloplasmin copper appears a distinct subtype of Alzheimer’s disease: a study of EEG-derived brain activity. Curr. Alzheimer Res. 13 1374–1384. 10.2174/1567205013666160603001131 [DOI] [PubMed] [Google Scholar]
- Terasaka T., Kinoshita T., Kuno M., Nakanishi I. (2004a). A highly potent non-nucleoside adenosine deaminase inhibitor: efficient drug discovery by intentional lead hybridization. J. Am. Chem. Soc. 126 34–35. 10.1021/ja038606l [DOI] [PubMed] [Google Scholar]
- Terasaka T., Kinoshita T., Kuno M., Seki N., Tanaka K., Nakanishi I. (2004b). Structure-based design, synthesis, and structure-activity relationship studies of novel non-nucleoside adenosine deaminase inhibitors. J. Med. Chem. 47 3730–3743. [DOI] [PubMed] [Google Scholar]
- Torres J. B., Andreozzi E. M., Dunn J. T., Siddique M., Szanda I., Howlett D. R., et al. (2016). PET imaging of copper trafficking in a mouse model of Alzheimer disease. J. Nucl. Med. 57 109–114. 10.2967/jnumed.115.162370 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tõugu V., Karafin A., Palumaa P. (2008). Binding of zinc(II) and copper(II) to the full-length Alzheimer’s amyloid-β peptide. J. Neurochem. 104 1249–1259. 10.1111/j.1471-4159.2007.05061.x [DOI] [PubMed] [Google Scholar]
- Tõugu V., Karafin A., Zovo K., Chung R. S., Howells C., West A. K., et al. (2009). Zn(II)- and Cu(II)-induced non-fibrillar aggregates of amyloid-β (1-42) peptide are transformed to amyloid fibrils, both spontaneously and under the influence of metal chelators. J. Neurochem. 110 1784–1795. 10.1111/j.1471-4159.2009.06269.x [DOI] [PubMed] [Google Scholar]
- Ujjinamatada R. K., Phatak P., Burger A. M., Hosmane R. S. (2008). Inhibition of adenosine deaminase by analogues of adenosine and inosine, incorporating a common heterocyclic base, 4(7)-amino-6(5)H-imidazo[4,5-d] pyridazin-7(4)one. J. Med. Chem. 51 694–698. 10.1021/jm700931t [DOI] [PubMed] [Google Scholar]
- Unzeta M., Esteban G., Bolea I., Fogel W. A., Ramsay R. R., Youdim M. B. H., et al. (2016). Multi-target directed donepezil-like ligands for Alzheimer’s disease. Front. Neurosci. 10:205 10.3389/fnins.2016.00205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ventriglia M., Brewer G. J., Simonelli I., Mariani S., Siotto M., Bucossi S., et al. (2015). Zinc in Alzheimer’s disease: a meta-analysis of serum, plasma, and cerebrospinal fluid studies. J. Alzheimers Dis. 46 75–87. 10.3233/JAD-141296 [DOI] [PubMed] [Google Scholar]
- Ventriglia M., Bucossi S., Panetta V., Squitti R. (2012). Copper in Alzheimer’s disease: a meta-analysis of serum, plasma, and cerebrospinal fluid studies. J. Alzheimers Dis. 30 981–984. 10.3233/JAD-2012-120244 [DOI] [PubMed] [Google Scholar]
- Vigo-Pelfrey C., Lee D., Keim P., Lieberburg I., Schenk D. B. (1993). Characterization of beta-amyloid peptide from human cerebrospinal fluid. J. Neurochem. 61 1965–1968. 10.1111/j.1471-4159.1993.tb09841.x [DOI] [PubMed] [Google Scholar]
- Viles J. H. (2012). Metal ions and amyloid fiber formation in neurodegenerative diseases. Copper, zinc and iron in Alzheimer’s, Parkinson’s and prion diseases. Coord. Chem. Rev. 256 2271–2284. 10.1016/j.ccr.2012.05.003 [DOI] [Google Scholar]
- Vural H., Demirin H., Kara Y., Eren I., Delibas N. (2010). Alterations of plasma magnesium, copper, zinc, iron and selenium concentrations and some related erythrocyte antioxidant enzyme activities in patients with Alzheimer’s disease. J. Trace Elem. Med. Biol. 24 169–173. 10.1016/j.jtemb.2010.02.002 [DOI] [PubMed] [Google Scholar]
- Waggoner D. J., Bartnikas T. B., Gitlin J. D. (1999). The role of copper in neurodegenerative disease. Neurobiol. Dis. 6 221–230. 10.1006/nbdi.1999.0250 [DOI] [PubMed] [Google Scholar]
- Walsh D. M., Klyubin I., Fadeeva J. V., Cullen W. K., Anwyl R., Wolfe M. S., et al. (2002). Naturally secreted oligomers of amyloid β protein potently inhibit hippocampal long-term potentiation in vivo. Nature 416 535–539. 10.1038/416535a [DOI] [PubMed] [Google Scholar]
- Wan L., Nie G., Zhang J., Luo Y., Zhang P., Zhang Z., et al. (2011). β-Amyloid peptide increases levels of iron content and oxidative stress in human cell and Caenorhabditis elegans models of Alzheimer disease. Free Radic. Biol. Med. 50 122–129. 10.1016/j.freeradbiomed.2010.10.707 [DOI] [PubMed] [Google Scholar]
- Wang Z.-X., Tan L., Wang H.-F., Ma J., Liu J., Tan M.-S., et al. (2015). Serum iron, zinc, and copper levels in patients with Alzheimer’s disease: a replication study and meta-analyses. J. Alzheimers Dis. 47 565–581. 10.3233/JAD-143108 [DOI] [PubMed] [Google Scholar]
- Weingarten M. D., Lockwood A. H., Hwo S. Y., Kirschner M. W. (1975). A protein factor essential for microtubule assembly. Proc. Natl. Acad. Sci. U.S.A. 72 1858–1862. 10.1073/pnas.72.5.1858 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wild K., August A., Pietrzik C. U., Kins S. (2017). Structure and synaptic function of metal binding to the amyloid precursor protein and its proteolytic fragments. Front. Mol. Neurosci. 10:21. 10.3389/fnmol.2017.00021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong B. X., Hung Y. H., Bush A. I., Duce J. A. (2014). Metals and cholesterol: two sides of the same coin in Alzheimer’s disease pathology. Front. Aging Neurosci. 6:91 10.3389/fnagi.2014.00091 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright A. L., Zinn R., Hohensinn B., Konen L. M., Beynon S. B., Tan R. P., et al. (2013). Neuroinflammation and neuronal loss precede Aβ plaque deposition in the hAPP-J20 mouse model of Alzheimer’s disease. PLOS ONE 8:e59586. 10.1371/journal.pone.0059586 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu W. H., Lei P., Liu Q., Hu J., Gunn A. P., Chen M. S., et al. (2008). Sequestration of copper from β-amyloid promotes selective lysis by cyclen-hybrid cleavage agents. J. Biol. Chem. 283 31657–31664. 10.1074/jbc.M804722200 [DOI] [PubMed] [Google Scholar]
- Xie H., Hou S., Jiang J., Sekutowicz M., Kelly J., Bacskai B. J. (2013). Rapid cell death is preceded by amyloid plaque-mediated oxidative stress. Proc. Natl. Acad. Sci. U.S.A. 110 7904–7909. 10.1073/pnas.1217938110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang T., Yang L., Zhang C., Wang Y., Ma X., Wang K., et al. (2016). A copper–amyloid-β targeted fluorescent chelator as a potential theranostic agent for Alzheimer’s disease. Inorg. Chem. Front. 3 1572–1581. 10.1039/C6QI00268D [DOI] [Google Scholar]
- Yao F., Zhang R., Tian H., Li X. (2012). Studies on the interactions of copper and zinc ions with β-amyloid peptides by a surface plasmon resonance biosensor. Int. J. Mol. Sci. 13 11832–11843. 10.3390/ijms130911832 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yermolaieva O., Brot N., Weissbach H., Heinemann S. H., Hoshi T. (2000). Reactive oxygen species and nitric oxide mediate plasticity of neuronal calcium signaling. Proc. Natl. Acad. Sci. U.S.A. 97 448–453. 10.1073/pnas.97.1.448 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu J., Luo X., Xu H., Ma Q., Yuan J., Li X., et al. (2015). Identification of the key molecules involved in chronic copper exposure-aggravated memory impairment in transgenic mice of Alzheimer’s disease using proteomic analysis. J. Alzheimers Dis. 44 455–469. 10.3233/JAD-141776 [DOI] [PubMed] [Google Scholar]
- Yu X., Zheng J. (2012). Cholesterol promotes the interaction of Alzheimer β-amyloid monomer with lipid bilayer. J. Mol. Biol. 421 561–571. 10.1016/j.jmb.2011.11.006 [DOI] [PubMed] [Google Scholar]
- Zhang Y., Thompson R., Zhang H., Xu H. (2011). APP processing in Alzheimer’s disease. Mol. Brain 4:3. 10.1186/1756-6606-4-3 [DOI] [PMC free article] [PubMed] [Google Scholar]