

Abstract
Inherited and de novo mutations in the CARD14 gene promote the development of psoriasis, an inflammatory disease of the skin. Caspase recruitment domain-containing protein 14 (CARD14) is a member of the CARMA protein family that includes the structurally related CARD11 adaptor that mediates NF-κB activation by antigen receptors. We investigated the mechanism by which CARD14 mutation in psoriasis activates NF-κB. In contrast with wild-type CARD14, CARD14E138A and CARD14G117S psoriasis mutants interacted constitutively with BCL10 and MALT1, and triggered BCL10- and MALT1-dependent activation of NF-κB in keratinocytes. These alterations disrupted the inhibitory effect of the CARD14 linker region (LR) on NF-κB activation by facilitating BCL10 binding. Therefore, psoriasis mutations activated CARD14 by a mechanism analogous to oncogenic CARD11 mutations in non-Hodgkin B cell lymphomas. CARD14E138A also stimulated MALT1 paracaspase activity and activated both ERK1/2 and p38α MAP kinases. Inhibition of MALT1 with mepazine reduced CARD14E138A-induced expression of specific psoriasis-associated transcripts in keratinocytes. Our results establish the mechanism whereby gain-of-function CARD14 variants, which induce psoriatic disease in affected individuals, activate pro-inflammatory signalling.
Keywords: caspase recruitment domain-containing protein 14 (CARD14), keratinocytes, mucosa-associated lymphoid tissue lymphoma translocation protein 1 (MALT1), NF-κB, psoriasis
INTRODUCTION
Psoriasis is a chronic inflammatory skin disease that arises through interactions between hyper-proliferative keratinocytes and activated immune cells that infiltrate the skin [1]. The disease affects 2–4% of adults in Europe and USA, of which 20–30% also develop psoriatic arthritis (PsA). Genome-wide association studies (GWAS) have identified multiple genetic loci that individually confer low risk for the development of psoriasis [2]. Many of these genes highlight the importance of the immune system in disease development, encoding proteins involved in antigen presentation, T-cell polarization and innate immunity. However, identified genetic loci explain less than 20% of disease variance suggesting that rare variants may contribute to the remaining portion, in addition to further unknown low risk loci and genetic interactions.
We have discovered highly penetrant de novo and inherited dominant gain-of-function mutations in the CARD14 gene that promote the development of psoriasis and PsA [3,4]. Among these, the de novo CARD14E138A mutation was identified in a child with early-onset generalized pustular psoriasis and is considered severe. The CARD14G117S mutation, which leads to point mutation and altered splicing of the CARD14 transcript, was identified in a family of European descent and a Tunisian cohort with multiple cases of psoriasis and PsA [4,5]. A second multiply affected family from Taiwan harboured a mutation in the exon 3 splice donor site that affected splicing in a similar manner to the CARD14G117S mutation [4]. Although these pathogenic variants are rare, the common CARD14 polymorphism (R820W) exceeds genome-wide significance for association with psoriasis and PsA [6–8]. These findings indicate that CARD14 variants may have a more widespread role in the pathogenesis of psoriasis and PsA and are not restricted to rare families with highly penetrant mutations.
CARD14 (CARMA2) is a member of the CARD-containing membrane-associated guanylate kinase (MAGUK) protein (CARMA) family of scaffolding proteins [9]. This includes CARD11 (CARMA1) and CARD10 (CARMA3), which play critical roles in the activation of NF-κB transcription factors in response to ligation of antigen receptors and G-protein-coupled receptors (GPCRs) respectively. NF-κB transcription factors, composed of dimers of Rel polypeptides, regulate gene expression by binding to κB elements in the promoters and enhancers of multiple target genes that regulate immune and inflammatory responses [10]. CARD14 has a similar domain structure to CARD11 and CARD10 (Figure 1A), comprising an N-terminal CARD domain, followed by a coiled-coil (CC) domain and a C-terminal MAGUK domain (PDZ-SH3-GUK), and activates NF-κB when overexpressed in HEK293 cells [11]. A shorter isoform CARD14sh [12] is expressed most highly in the skin [4]. CARD14sh lacks the C-terminal MAGUK domain of CARD14, similar to the related protein CARD9 that mediates activation of NF-κB by the C-type lectin receptor dectin-1 [9].
Figure 1. Psoriasis-associated CARD14 mutants require BCL10 and MALT1 to activate NF-κB.
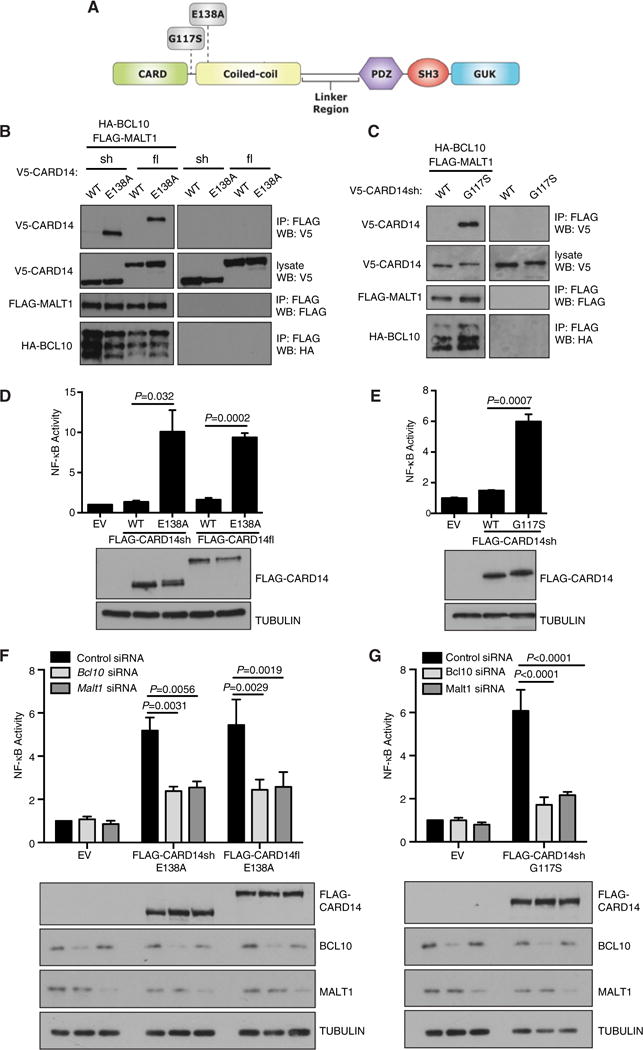
(A) Schematic of CARD14 domains and psoriasis-associated mutations. (B, C) HEK293 cells were co-transfected with vectors encoding the indicated V5-CARD14 variants (0.1–1.2 μg), FLAG-MALT1 (1 μg) and HA-BCL10 (0.6–1.2 μg). Anti-FLAG immunoprecipitates were immunoblotted with the indicated antibodies. Representative of at least two independent experiments. (D, E) HaCaT keratinocytes were co-transfected with vectors encoding the indicated FLAG-CARD14 variants (0.125–0.2 μg) or EV, pNFκB-Luc and pRL-TK. After 24 h, NF-κB activity was determined by luciferase assay (mean ± S.E.M. of three independent experiments). FLAG-CARD14 expression was determined by immunoblotting of lysates. Significance determined by t test. (F, G) HaCaT keratinocytes were transfected with Bcl10 or Malt1 siRNA pools or control non-targeting pool. After 48 h, cells were transfected with FLAG-CARD14 variants (0.05–0.1 μg), pNFκB-Luc and pRL-TK vectors and cultured for a further 24 h. NF-κB activity was determined as in (D) (mean ± S.E.M. of three independent experiments). Representative expression of FLAG-CARD14, endogenous BCL10 and endogenous MALT1 was determined by immunoblotting of corresponding lysates. Significance determined by two-way ANOVA.
The structural similarity to CARD11, CARD10 and CARD9 suggests a role for CARD14 in NF-κB activation. Consistent with this, wild type (WT) CARD14 overexpression activates an NF-κB reporter in HEK293 cells [11]. Furthermore, we have shown that transfected CARD14 stimulates NF-κB in keratinocytes and endothelial cells, and that this activity is amplified when CARD14 harbours psoriasis-associated mutations [3,4]. CARD14 overexpression additionally induces the expression of a subset of psoriasis-associated genes in keratinocytes. NF-κB is constitutively active in psoriatic skin, and GWAS have identified genetic alterations in components of the NF-κB pathway in psoriasis and PsA [13]. Thus, CARD14 probably plays an important role in psoriasis pathogenesis through its ability to trigger NF-κB-dependent-inflammation within the skin. Consistent with this model, we have found that CARD14 is highly expressed in keratinocytes of the basal and supra-basal skin epithelial layers [4]. CARD14 is additionally expressed in dermal, lymphatic and aortic endothelial cells [14], suggesting that CARD14 mutations may contribute to cardiovascular and other systemic comorbidities associated with psoriasis.
The mechanism by which the related family member CARD11 activates canonical NF-κB in T-cells has been studied in detail [9]. Following T-cell receptor (TCR) stimulation, CARD11 is activated by recruitment to the plasma membrane via its MAGUK domain, and phosphorylation of its linker region (LR) by protein kinase C (PKC) θ. This induces a conformational change that allows CARD11 to recruit the preformed complex of BCL10 (B-cell lymphoma protein 10) and MALT1 (mucosa-associated lymphoid tissue lymphoma translocation protein 1), a paracaspase [15]. TCR-induced formation of the CARD11– BCL10–MALT1 (CBM) complex is critical for TCR activation of NF-κB transcription factors. Similarly, the CBM complex is essential for B-cell receptor (BCR) activation of NF-κB. BCL10 and MALT1 are also required for CARD10 activation of NF-κB by GPCRs in fibroblasts, indicating that a CARD10–BCL10– MALT1 complex may play an analogous role to the CARD11– BCL10–MALT1 complex in antigen-stimulated lymphocytes.
In this study, we investigated the mechanism by which highly penetrant psoriasis-inducing CARD14 mutations stimulate NF-κB. We show that E138A and G117S psoriasis mutations promoted CARD14 interaction with BCL10 and MALT1, and that mutant CARD14 activation of NF-κB in keratinocytes was dependent on expression of both of these proteins. Mutations were found to activate constitutive CARD14 signalling by disrupting the inhibitory effect of the linker domain located between the coiled-coil and PDZ domains. CARD14 psoriasis variants also stimulated MALT1 paracaspase activity, and experiments with the MALT1 inhibitor mepazine demonstrated that this was required for maximal CARD14 activation of NF-κB and CARD14-induced expression of specific pro-inflammatory cytokine and chemokine gene transcripts.
EXPERIMENTAL
Expression constructs
Human CARD14fl cDNA was purchased from TrueORF (RC217455), human CARD14sh cDNA was purchased from Capital Biosciences (DHC-3073-M11). E138A (c.413A>C) and G117S (c.349G>A also causing 66 bp insertion of intronic sequence after exon 3) mutations were introduced as previously described [4]. All CARD14 inserts were cloned into pcDNA3 (Invitrogen) with either a C-terminal V5 (GKPIPNPLLGLDST) or 3xFLAG (DYKDHDGDYKDHDIDYKDDDDK) tag (Sigma). Linker region deleted (ΔLR) CARD14 constructs were generated by deletion of c.1228–1722 using overlap extension PCR. Deletion of this sequence was determined by CARD14 domain boundary prediction (JPred and Uniprot). Human MALT1 (isoform A) cDNA was purchased from GeneCopoeia (EXT0622-M11) and cloned into pcDNA3 with an N-terminal 3xFLAG tag. C464A mutation (c.1471–1473, TGT>GCT) was introduced into MALT1 by site directed mutagenesis. Human BCL10 cDNA was cloned into pcDNA3 with an N-terminal HA (YPYDVPDYA) or 3xFLAG tag.
Reagents
Antibodies used in this study were: anti-V5 (Serotec MCA1360); anti-FLAG M2 (Sigma F1804); anti-HA (Roche 11867423001); anti-BCL10 (sc-56); anti-MALT1 (sc-46677); anti-TUBULIN (TAT-1); p105 (CST 4717); phospho-p105 (CST 4806); extracellular signal-regulated kinase 1/2 (ERK1/2) (sc-93, sc-154); phospho-ERK1/2 (CST 9101); p38 (CST 9212); phospho-p38 (CST 4511); phospho-JNK (Invitrogen 44682); JNK (CST 9252). Mepazine was purchased from Calbiochem, PD0325901 and VX-745 were from Selleck Chemicals, BI605906 was obtained through the MRC PPU Reagents and Services facility (MRC PPU, College of Life Sciences, University of Dundee, Scotland, mrcppureagents.dundee.ac.uk).
Immunoprecipitations
1×106 HEK293 cells were seeded in 90 mm dishes (10 ml per dish) in DMEM, 10% FCS, 2 mM L-glutamine, 100 U penicillin, 0.1 mg/ml streptomycin. The next day cells were transfected using Lipofectamine (Invitrogen) following the manufacturer’s instructions. cDNA amounts of various constructs are indicated in corresponding figure legends. After 24 h cells were lysed in 1 ml co-immunoprecipitation (coIP) buffer (50 mM Tris pH 7.5, 150 mM NaCl, 1% Triton-X, 10% glycerol, 2 mM EDTA, 10 mM sodium fluoride, 1 mM sodium pyrophosphate, 10 mM β-glycerophosphate, 0.1 mM sodium vanadate, 10 mM iodoacetamide and protease inhibitor cocktail [Roche 11836170001]). Lysates were centrifuged at 18800 g for 10 min to clear debris. Protein levels were quantified (Pierce BCA protein assay, Thermo Scientific) and normalized. Lysates were precleared with IgG Agarose (Sigma) for 1 h rotating, 4°C. Fifty-five microlitres of precleared lysate was saved for analysis by Western blotting. Remaining precleared lysate was incubated with a 10 μl bead volume of anti-FLAG M2 affinity gel (Sigma) overnight with rotation, 4°C. Beads were subsequently washed (5 min rotating, 4°C) twice with high salt buffer (50 mM Tris pH 7.5, 500 mM NaCl, 1% Triton-X, 10% glycerol, 2 mM EDTA, 10 mM sodium fluoride, 1 mM sodium pyrophosphate, 10 mM β-glycerophosphate, 0.1 mM sodium vanadate, 10 mM iodoacetamide) and four times with coIP buffer. Beads were dried and eluted three times by incubation with 10–15 μl glycine buffer (0.2 M glycine, 0.05% NP-40, pH 2.5) for 3 min at RT with agitation. Pooled eluates were neutralized with Tris pH 8, boiled for 5 min with Laemmli buffer and resolved by SDS-PAGE for analysis.
NF-κB luciferase assays
1×105 HaCaT keratinocytes were seeded in 24-well plates (500 μl per well) in DMEM, 10% FCS, 2 mM L-glutamine, 100 U penicillin, 0.1 mg/ml streptomycin overnight. Cells were then co-transfected with 3xFLAG-CARD14 cDNA, pNF-κB-Luc reporter (0.25 μg, Clontech 631904) and pRL-TK control reporter (0.05 μg, Promega E2241) using Lipofectamine 2000 (Invitrogen) following the manufacturer’s instructions. CARD14 cDNA amounts per transfection are indicated in corresponding figure legends and total cDNA was normalized across experiments with pcDNA3 empty vector (EV). Twenty-four hours after transfection cells were lysed in 60 μl Passive Lysis Buffer (Promega) and analysed using the Dual Luciferase Reporter Assay System (Promega) according to the manufacturer’s instructions. Relative NF-κB activation was determined by normalizing pNF-κB-Luc reporter activity to pRL-TK activity, and then dividing all values to activity induced by EV alone. CARD14 expression levels in corresponding total cell lysates were determined by Western blotting. BCL10 and MALT1 knockdown efficiency was calculated by densitometric quantification of immunoblots (GS800 Calibrated Densitometer, Quantity One software). Relative BCL10 and MALT1 expression was normalized to loading control values (tubulin), and then scaled according to expression with control siRNA. Average knockdown was calculated within each experiment, and then averaged across three independent experiments.
siRNA knockdown
HaCaT keratinocytes were seeded in 24-well plates at 3.75 × 104 cells per well overnight. The following day cells were transfected with 50 μM ON-TARGET plus siRNA SMARTpools targeting Bcl10 (sequences: GCCACCAGAUCUACAGUUA; CGAACAACCUCUCCAGAUC; GGGCAUCCACUGUCAUGUA; AAUCAUAGCUGAGAGACAU), Malt1 (sequences: GGGAGUAUAUGGGUUAUUA; GCAGUGUUCUCUUAAGGUA; GCAAAUCUGUGUUGAACCA; GGUAAUCCAAGUAAUGUUA) or non-targeting control pool (sequences: UGGUUUACAUGUCGACUAA; UGGUUUACAUGUUGUGUGA; UGGUUUACAUGUUUUCUGA; UGGUUUACAUGUUUUCCUA) using Lipofectamine 2000 following the manufacturer’s instructions. Forty-eight hours later, cells were transfected for NF-κB luciferase assays as described above.
MALT1 activity assay
HEK293 cells were transfected for 24 h as described above. Cells were lysed in 600 μl MALT1 coIP buffer (25 mM HEPES pH 7.5, 150 mM NaCl, 0.2% NP-40, 10% glycerol, 1 mM DTT, 10 mM sodium fluoride, 8 mM β-glycerophosphate and 300 μM sodium vanadate) without protease inhibitors. Lysates were centrifuged at 18800 g for 10 min to remove debris. Protein levels were quantified and normalized. Lysates were precleared with streptavidin agarose resin (Thermo Scientific) for 1 h, 4°C with rotation. Forty microlitres precleared lysate was saved for analysis by Western blotting. Remaining lysate was incubated with 0.1 μM biotinylated ABP (activity based probe) 7 [16] for 50 min, RT with rotation. Twelve microlitres bead volume of streptavidin agarose resin was added for 2 h, 4°C with rotation. Beads were washed four times with MALT1 coIP buffer. Protein was eluted by boiling beads for 5 min in Laemmli buffer and resolved by SDS-PAGE for analysis.
Generation and protein analysis of tetracycline inducible HaCaT CARD14E138A keratinocytes
3xFLAG tagged CARD14 cDNA was cloned into the pcDNA5/TO vector (Invitrogen) and transfected into HaCaT-TR cells (a kind gift from Caroline Hill; [17], which stably express the tetracycline repressor (TR) (pcDNA6/TR, Invitrogen). Cells were expanded for 4–6 weeks in DMEM, 10% tetracycline free (TF) FCS, 2 mM L-glutamine, 100 U penicillin, 0.1 mg/ml streptomycin, 10 μg/ml blasticidin (for pcDNA6/TR maintenance) and 500 μg/ml hygromycin (for CARD14-pcDNA5/TO selection). For protein analysis, cells were seeded in 24-well plates at 1×105 cells per well in DMEM, 0.1% TF FCS, 2 mM L-glutamine, 100 U penicillin, 0.1 mg/ml streptomycin overnight. Cells were then incubated in DMEM for 5 h to minimize basal signalling levels prior to the addition of 1 μg/ml tetracycline (Sigma). At the indicated times cells were lysed in 60 μl radioimmunoprecipitation assay (RIPA) buffer (50 mM Tris-HCl pH 7.5, 150 mM NaCl, 2 mM EDTA, 1 mM sodium pyrophosphate, 50 mM sodium fluoride, 1 mM sodium vanadate, 0.1% SDS, 1% Triton-X, 0.5% deoxycholate and protease inhibitor cocktail). Total cell lysates were boiled in Laemmli buffer and resolved by SDS-PAGE for analysis.
Quantitative PCR
HaCaT CARD14E138A cells were seeded in 24-well plates at 1×105 cells per well in DMEM, 1% TF FCS, 2 mM L-glutamine, 100 U penicillin, 0.1 mg/ml streptomycin overnight. Cells were then incubated in DMEM in the presence of DMSO or indicated pharmacological inhibitor for 1 h prior to the addition of 1 μg/ml tetracycline. Cells were lysed 6 h later in 350 μl RLT buffer (QIAGEN), 1% 2-mercaptoethanol. RNA was purified using RNAeasy mini kit (QIAGEN) according to the manufacturer’s instructions. SuperScript VILO cDNA synthesis kit (Thermo Fisher) was used to transcribe cDNA according to the manufacturer’s instructions. Quantitative real-time PCR was carried out using the TaqMan Assay system (Applied Biosystems). Primer probes used were: IL8 (Hs00174103_m1); CCL20 (Hs00355476_m1); IL36γ (Hs00219742_m1). Expression levels were normalized to the housekeeping gene 18S (Hs99999901_s1) and calculated using the 2(−ΔΔCT) method.
Statistical analysis
Students t test (two-tailed, unpaired) and two-way ANOVA (with Bonferroni multiple testing correction) analyses were conducted using GraphPad Prism version 6.00 for Mac OS X, GraphPad Software, La Jolla, California, USA, www.graphpad.com.
RESULTS
CARD14 activation of NF-κB requires BCL10 and MALT1
Secondary structure similarity of CARD11 and CARD10 suggested that CARD14 would form a complex with BCL10 and MALT1 to activate NF-κB (Figure 1A). To investigate this, full length (fl) and short (sh) forms of V5-CARD14 were expressed together with FLAG-MALT1 and HA-BCL10 in HEK293 cells. WT CARD11 does not interact with BCL10/MALT1 to form active signalling complexes unless cells are stimulated via specific upstream receptors, such as the TCR [18]. In line with this, immunoblotting revealed little detectable coIP of WT V5-CARD14fl or V5-CARD14sh with FLAG-MALT1/HA-BCL10 (Figure 1B). However, introduction of the E138A psoriasis-associated CARD14 mutation (Figure 1A) induced clear association of FLAG-MALT1/HA-BCL10 with both V5-CARD14fl and V5-CARD14sh (Figure 1B). Similarly, the V5-CARD14shG117S psoriasis variant co-purified with FLAG-MALT1/HA-BCL10 complexes (Figure 1C). Therefore, a common effect of two psoriasis-associated CARD14 mutations was formation of a CARD14–BCL10–MALT1 complex, without the need for upstream agonist stimulation.
We next tested the effect of the CARD14 psoriasis mutations on activation of an NF-κB reporter in HaCaT keratinocytes, a cell type that plays an important role in psoriasis aetiology and expresses CARD14 [1,4]. Both FLAG-CARD14flE138A and FLAG-CARD14shE138A activated NF-κB, whereas WT FLAG-CARD14 had only a modest or no stimulatory effect (Figure 1D). FLAG-CARD14shG117S also strongly activated the NF-κB reporter (Figure 1E). SiRNA knockdown of endogenous BCL10 (77.63% knockdown ± 2.3 [S.E.M., n = 3]), significantly reduced activation of an NF-κB reporter by each of the CARD14 psoriasis variants (Figures 1F and 1G). Similarly, MALT1 knockdown (70.70% knockdown ±1.90 [S.E.M., n = 3]) significantly decreased NF- κB reporter activation by FLAG-CARD14flE138A, FLAG-CARD14shE138A and FLAG-CARD14shG117S. Together the results in this section suggested that formation of a complex with BCL10 and MALT1 was required for activation of NF-κB by CARD14 alterations seen in psoriasis.
E138A and G117S psoriasis mutations disrupt CARD14 autoinhibition
In the basal state, CARD11 signalling activity is held in check by an inhibitory domain (ID), located between the coiled-coil and PDZ domains, that interacts with the CARD and coiled-coil domains [19]. CARD11 autoinhibition is relieved following antigen receptor stimulation by the inducible phosphorylation of the ID by PKC isoforms, which triggers formation of a CARD11–BCL10–MALT1 complex [20]. Oncogenic mutations in CARD11 in several types of lymphoma also interrupt ID-mediated inhibition [21]. These mutations are located within the CARD11 coiled-coil domain and disrupt ID intramolecular interactions, bypassing the requirement for antigen-receptor induced ID phosphorylation for CARD11 activation of NF-κB.
Psoriasis-associated activating mutations in CARD14 are located predominantly within or adjacent to the coiled-coil domain [3,4]. The comparable secondary structure of CARD14 suggested a similar activating mechanism to oncogenic CARD11 mutations. Initially, the effect of removing the LR between the coiled-coil and PDZ domains of WT FLAG-CARD14 was tested. Analogous to CARD11 [19], this promoted FLAG-CARD14 stimulation of NF-κB in HaCaT keratinocytes (Figure 2A). However, LR deletion in FLAG-CARD14flE138A and FLAG-CARD14shE138A psoriasis variants did not alter their ability to activate NF-κB. Similarly, LR deletion did not alter NF-κB activation by FLAG-CARD14shG117S (Figure 2B). Consistently, the stimulatory effects of the E138A and G117S mutations on NF-κB activation were significantly reduced in the absence of the LR (Figure 2C). These results support the hypothesis that psoriasis mutations interfere with autoinhibitory function of the LR.
Figure 2. Abrogation of CARD14 inhibitory LR activity by deletion or psoriasis mutation induces BCL10 association.
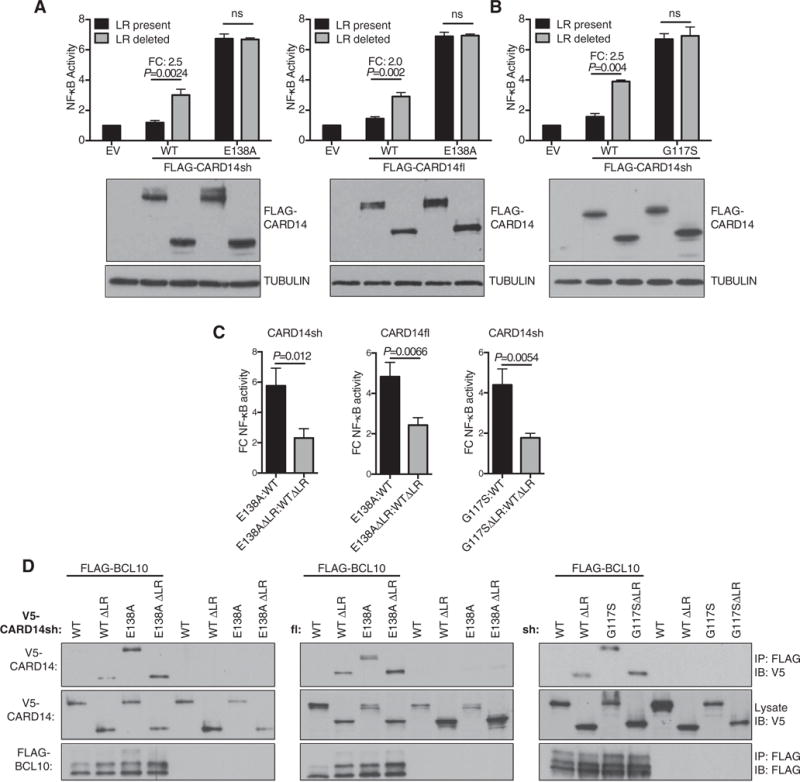
(A, B) HaCaT keratinocytes were co-transfected with FLAG-CARD14 (0.125–0.2 μg), pNFκB-Luc and pRL-TK plasmids. NF-κB activity was determined as in Figure 1(D) (mean ± S.E.M. of three independent experiments). Representative expression of FLAG-CARD14 and tubulin in corresponding lysates was determined by immunoblotting. Significance determined by−two-way ANOVA. FC; fold change. (C) FC NF-κB activation of mutant compared with WT CARD14, in the presence or absence of the LR (calculated from data in A and B). Mean ± S.E.M. of three independent experiments. Significance determined by t test. (D) HEK293 cells were co-transfected with vectors encoding the indicated V5-CARD14 variants (0.1–1 μg) and FLAG-BCL10 (0.6–0.8 μg). Anti-FLAG immunoprecipitates were immunoblotted with the indicated antibodies. Representative of three independent experiments.
Deletion of the inhibitory linker domain induces binding of CARD11 to BCL10 and co-recruitment of MALT1 to promote the formation of CARD11–BCL10–MALT1 complex [19]. To determine whether the linker domain of CARD14 functioned in a similar way, HEK293 cells were co-transfected with expression constructs encoding WT or LR-deleted V5-CARD14 and FLAG-BCL10. Immunoblotting of anti-FLAG immunoprecipitates revealed that FLAG-BCL10 co-purified with V5-CARD14sh/fl ΔLR but not V5-CARD14 (Figure 2D). V5-CARD14sh/flE138A also co-immunoprecipitated with FLAG-BCL10, but this was not further increased by LR deletion. Similarly, V5-CARD14shG117S interacted with BCL10 and this was not increased by deletion of the LR (Figure 2D). Together these results suggested that E138A and G117S psoriasis mutations abrogated LR inhibition, which facilitated BCL10 binding and NF-κB activation.
CARD14E138A activates MALT1 paracaspase activity
In addition to functioning as a scaffolding protein, MALT1 is a paracaspase that cleaves substrates after arginine residues [15]. MALT1 proteolytic activity is induced following T-cell antigen receptor stimulation. This promotes the cleavage of a limited number of proteins, including A20, CYLD and RelB that negatively regulate NF-κB activation. MALT1 is also activated in diffuse large B-cell lymphomas that rely on chronic B-cell receptor signalling [22], and MALT1 paracaspase activity promotes lymphoma survival by inactivating negative regulators of NF-κB [23]. Furthermore, expression of oncogenic CARD11 is sufficient to trigger MALT1 activation in BJAB cells [16].
In this and the following sections, we focused on the E138A psoriasis CARD14 mutation, which is the most pathogenic variant [3,4]. To determine whether CARD14 could activate MALT1 paracaspase activity, HEK293 cells were co-transfected with expression vectors encoding V5-CARD14fl or V5-CARD14sh together with HA-BCL10 and FLAG-MALT1 vectors. MALT1 activity in cell lysates was then measured with a biotin-coupled activity based probe (ABP) that covalently modifies the active centre of MALT1 [16]. Immunoblotting of ABP streptavidin pulldowns indicated that the E138A variant of both V5-CARD14fl and V5-CARD14sh activated FLAG-MALT1, although activation of FLAG-MALT1 by WT versions of these proteins was very weak or absent (Figure 3A). Catalytically inactive C464A FLAG-MALT1 mutant was virtually undetectable in ABP pulldowns from lysates of cells co-expressing V5-CARD14flE138A or V5-CARD14shE138A and HA-BCL10, confirming the specificity of the MALT1 assay.
Figure 3. CARD14E138A mutation activates MALT1 paracaspase activity.
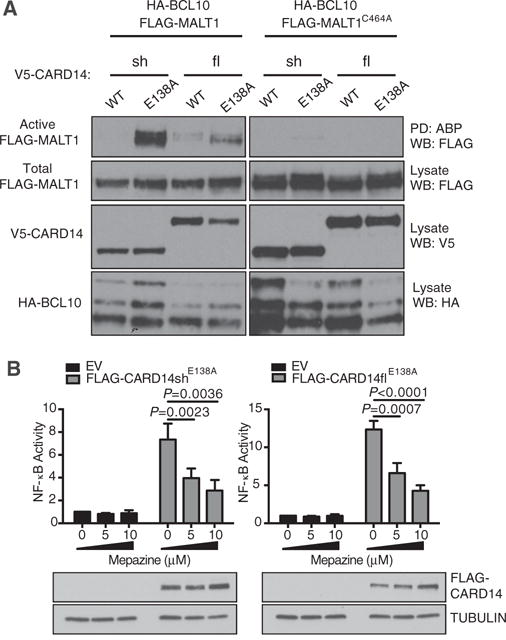
(A) HEK293 cells were co-transfected with vectors encoding V5-CARD14 variants (0.1–0.4 μg), WT or C464 FLAG-MALT1 (0.8–1.2 μg) and HA-BCL10 (0.8–1.4 μg). Active MALT1 was assayed by ABP pull down and immunoblotting. Representative of three independent experiments. (B) HaCaT keratinocytes were co-transfected with FLAG-CARD14 variant (0.125–0.2 μg) or EV, pNFκB-Luc and pRL-TK vectors. 5 h after transfection, cells were treated with mepazine or DMSO vehicle and lysed after further 24 h culture. NF-κB activity was determined as in Figure 1(D) (mean ± S.E.M. of three independent experiments). Representative expression of FLAG-CARD14 was determined by immunoblotting of corresponding lysates. Significance determined by two-way ANOVA.
MALT1 protease activity can be inhibited by the phenothiazine mepazine by a non-competitive mechanism [23]. To determine whether MALT1 paracaspase activity was required for CARD14 activation of NF-κB, HaCaT cells were transiently transfected with vectors encoding FLAG-CARD14shE138A or FLAG-CARD14flE138A and an NF-κB reporter and cultured in the presence of different concentrations of mepazine. NF-κB activation by both FLAG-CARD14shE138A and FLAG-CARD14flE138A was inhibited by mepazine, in a dose-dependent fashion (Figure 3B). Thus optimal stimulation of NF-κB by CARD14 was dependent on MALT1 paracaspase activity.
Maximal CARD14E138A-induced gene expression depends on MALT1 paracaspase activity and MAP kinase signalling
Induction of B-cell lymphoproliferation by an oncogenic CARD11 mutant is dependent on activation of both NF-κB and c-Jun N-terminal kinase (JNK) signalling cascades [24]. To determine whether CARD14E138A activated mitogen-activated protein (MAP) kinase pathways in addition to NF-κB, HaCaT cells stably expressing Tet repressor (HaCaT-TR cells) [17] were transfected with FLAG-CARD14E138A cDNA subcloned into pcDNA5/TO and selected with hygromycin for 4–6 weeks (HaCaT-CARD14E138A cells). Culture of the resulting cells with tetracycline rapidly induced the expression of FLAG-CARD14E138A (Figure 4A). With similar kinetics, tetracycline treatment of HaCaT-CARD14E138A cells stimulated inhibitor of NF-κB kinase 2 (IKK2) phosphorylation of NF-κB1 p105 [25]. FLAG-CARD14E138A expression also triggered activation loop phosphorylation of both ERK1/2 and p38α MAP kinases, although JNK activation loop phosphorylation was only weakly detectable after prolonged exposure. Culture of parental HaCaT-TRs cells with tetracycline did not induce phosphorylations of NF-κB1 p105, ERK1/2 or p38α ruling out any effects of tetracycline alone on signalling. Thus FLAG-CARD14E138A stimulated the activation of ERK1/2 and p38α MAP kinase signalling pathways in keratinocytes, in addition to IKK2/NF-κB.
Figure 4. CARD14E138A-induced gene expression is dependent on IKK2, MALT1, ERK1/2 and p38α activity.
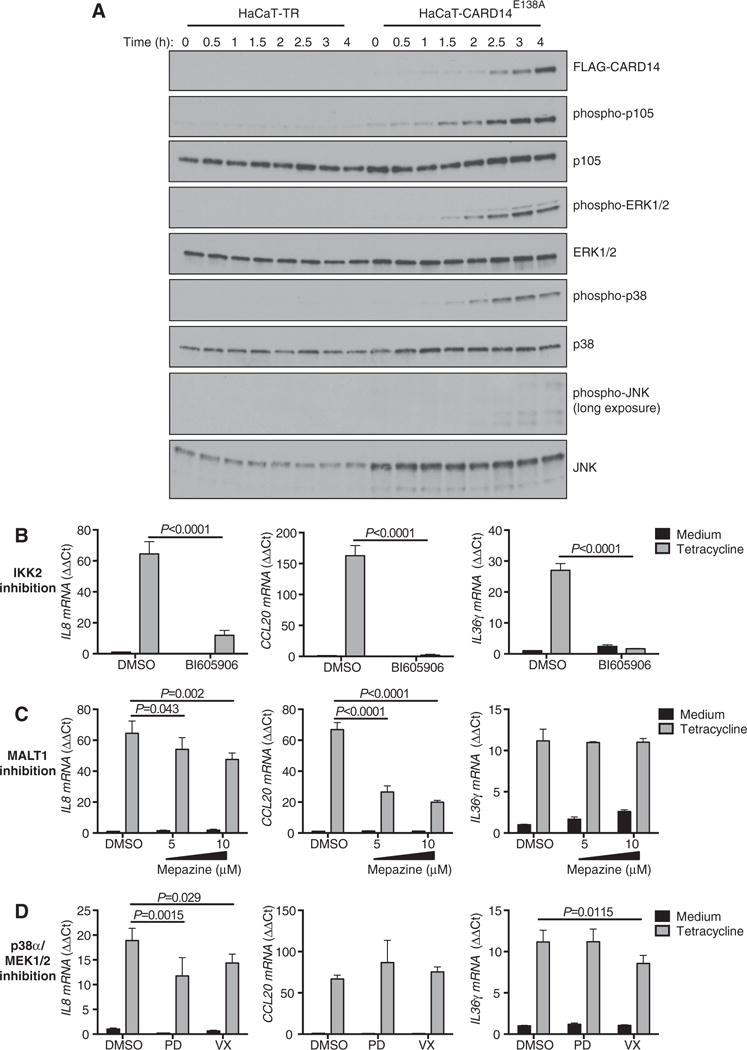
(A) HaCaT-TR or HaCaT-CARD14E138A cells were treated with tetracycline for the indicated times. CARD14E138A expression and phosphorylation of indicated proteins was determined by immunoblotting. Representative of three independent experiments using two independently generated stable cultures. (B–D) HaCaT-CARD14E138A cells were pre-treated with BI605906 (10 μM, IKK2 inhibitor), mepazine (MALT1 inhibitor), PD0325901 (0.1 μM, MEK1/2 inhibitor), VX-745 (1 μM, p38α inhibitor) or DMSO vehicle for 1 h, and cultured for a further 6 h after subsequent tetracycline addition. Gene expression was assayed by qRT-PCR (mean ± S.D., n = 3). Significance determined by two-way ANOVA. Representative of at least two independent experiments using independently generated stable cultures.
We have previously shown that CARD14E138A induces the expression in HEK001 keratinocytes of IL8, CCL20 and IL36γ mRNAs [4], which are also elevated in psoriatic skin. The contribution of the IKK complex, MALT1 paracaspase activity and MAP kinase signalling to CARD14E138A-induced expression of these genes was investigated in HaCaT-CARD14E138A cells using pharmacological inhibitors and qRT-PCR. Treatment of cells with the IKK2 inhibitor BI605906 [26] essentially blocked expression of each of the genes following CARD14E138A induction with tetracycline (Figure 4B). Inhibition of MALT1 paracaspase activity with mepazine [23] also substantially reduced FLAG-CARD14E138A-induced CCL20 mRNA expression and mildly impaired IL8 induction, while not affecting IL36γ mRNA levels. Inhibition of ERK1/2 activation with the MEK1/2 inhibitor PD0325901 or p38α catalytic activity with VX-745 fractionally reduced IL8 and IL36γ mRNA levels induced by FLAG-CARD14E138A. These results indicate that CARD14E138A principally promoted pro-inflammatory gene expression in keratinocytes via activation of IKK and NF-κB, although activation of ERK1/2 and p38α MAP kinase pathways was also required for maximal gene expression. In addition, MALT1 paracaspase activity was needed for optimal expression of specific target genes by CARD14E138A, reflecting the contribution of MALT1 activity to CARD14E138A activation of NF-κB.
DISCUSSION
CARD14 is the causative gene within psoriasis susceptibility locus 2 (PSORS2) [3,4]. Dominant inherited gain-of-function mutations in CARD14 result in enhanced activation of NF-κB in keratinocytes, leading to psoriasis. In the current study, we demonstrated that CARD14 psoriasis mutations induced the association of CARD14 with BCL10 and MALT1 to form a CARD14–BCL10–MALT1 complex that constitutively activated NF-κB. Each mutation abrogated an inhibitory effect of the LR between the coiled-coil and PDZ domains, facilitating BCL10 binding to promote NF-κB activation. CARD14E138A also activated the paracaspase activity of MALT1, which was required for optimal NF-κB activation and maximal expression of pro-inflammatory genes in keratinocytes.
The mechanism by which E138A and G117S mutations activate CARD14 signalling is analogous to that described for oncogenic CARD11 mutations in ABC DLBCL and other lymphomas [21]. These disrupt an autoinhibitory interaction involving the CARD11 LR (known as the ID), resulting in constitutive formation of a CARD11–BCL10–MALT1 complex that activates NF-κB. However, activating CARD14 mutations in psoriasis are not linked to an increased incidence of skin cancer, which probably reflects differences in the physiology of keratinocytes compared with B-cells. To mediate their function in adaptive immune responses, B-cells must generate high affinity antibodies. Many of the genetic lesions that promote lymphomagenesis result from the aberrant activity of the RAG V(D)J recombinase and activation-induced cytidine deaminase (AID) enzymes that rearrange and mutate immunoglobulin genes in normal B-cells [27]. Oncogenic CARD11 mutations cooperate with mutations in other genes to transform B-cells, including MYD88, BCL6 and PRDM1 [21]. It is possible that the lack of RAG and AID enzymes in keratinocytes make generation of secondary activating mutations to cooperate with mutant CARD14 significantly less likely.
Although there are similarities between the effects of oncogenic CARD11 mutations and psoriasis mutations in CARD14, the downstream consequences are not identical. A recent study has shown that the L225LI gain-of-function CARD11 mutant activates JNK and NF-κB to promote B-cell proliferation [24]. In contrast, CARD14E138A expression in keratinocytes activated NF-κB, ERK1/2 and p38α, but activation of JNK was barely detectable. Furthermore, although NF-κB activation was an absolute requirement for the expression of IL8, CCL20 and IL36γ downstream of CARD14E138A, activation of ERK1/2 and p38α was required for maximal induction of IL8 and IL36γ. Interestingly, increased levels of activated ERK1/2 and p38α are detected in lesional psoriatic skin compared with non-lesional skin, although JNK activation is not altered [28]. These results suggest that CARD11 and CARD14 are wired differently to downstream effectors. MALT1 inhibition by mepazine also exerted differential effects on the induction of CARD14E138A induced genes leading to reduced CCL20 and IL8 induction whereas IL36γ was unaffected. It is possible that we did not observe stronger effects of MALT1 inhibition as NF-κB activation was only partially inhibited by mepazine. This was demonstrated in our transient transfection system where mepazine-mediated inhibition of CARD14sh/flE138A-induced NF-κB reporter activity was partial. Thus, residual NF-κB activity may have been sufficient to fully induce IL36γ expression. Alternatively, as two critical posttranscriptional regulators, Regnase-1 and Roquin1/2, were recently identified as MALT1 substrates [29,30], it is possible that MALT1-dependent regulation of mRNA stability could also contribute to the differential effects on gene expression. It will be important in future studies to elucidate the components of the signalling pathways that link CARD14 to the downstream activation of NF-κB, MALT1, ERK1/2 and p38α in keratinocytes. In order to understand the normal regulation of CARD14, it will also be necessary to determine whether wild type CARD14 activation involves phosphorylation of the inhibitory linker domain by PKC isoforms like CARD11 and to identify physiological upstream activating receptors.
Psoriasis is usually considered to be a multi-factorial disease [2] and current models of psoriasis pathogenesis propose a complex interplay between activated keratinocytes and immune cells, with a key role for T-lymphocytes [1]. As CARD14 expression is largely restricted to keratinocytes and endothelial cells [4,14], it is likely that gain-of-function CARD14 mutations promote psoriasis by generating an environment of chronic pro-inflammatory signalling and cytokine/chemokine expression in the skin. This will lead to the recruitment of immune cells including T-cells producing IL-17 and IL-22 cytokines, which enhance keratinocyte activation and sustain skin inflammation [1]. However, whether gain-of-function CARD14 mutations are sufficient for the initiation (as opposed maintenance) of psoriatic disease is currently unclear. CARD14E138A and CARD14G117S activate NF-κB when expressed in keratinocytes in vitro (this study and [4]). However, the age of onset of psoriatic disease in patients harbouring these mutations is variable [3,4]. Thus additional factors may contribute to psoriasis pathogenesis in individuals harbouring CARD14 mutations, which may be environmental triggers or possibly epigenetic alterations. The fact that CARD14 pathogenic alterations are highly penetrant suggests that additional genetic factors are unlikely to be required, although they could contribute to disease severity or onset of PsA. Consistent with this hypothesis, it has recently been proposed that the onset of CARD14G117S associated psoriatic disease may be earlier in the presence of the psoriasis-risk allele HLA-C*06:02 [31].
Currently, anti-TNF biologics are one of the most effective treatments for both psoriasis and psoriatic arthritis (NICE guidelines [CG153], 2012). However, anti-TNF therapies are expensive, require injection and can impair the immune response to pathogens. In addition, a fraction of patients do not respond to anti-TNF, or their response diminishes over time [32]. Furthermore, patients have been described in which psoriatic lesions are exacerbated following anti-TNF therapy [33]. The new biologic ustekinumab (which binds the p40 subunit of IL-12 and IL-23) ameliorates psoriasis in the majority of patients [34]. However, anti-p40 is less effective for PsA, and in a subset of patients arthritic symptoms worsen with anti-p40 therapy [35]. Similarly, although anti-IL-17 is very effective in treatment of psoriasis [36], its effects on PsA are more modest [37]. Our study has demonstrated that MALT1 paracaspase activity is required for activation of NF-κB and induced expression of key pro-inflammatory genes by rare gain-of-function CARD14 variants. The common R820W variant of CARD14 predisposes individuals to psoriasis in European and Chinese populations [3,12], placing CARD14 in the pathway to common psoriasis. This raises the interesting possibility that MALT1 inhibitors might have beneficial effects for treatment of psoriasis and PsA.
Acknowledgments
We thank Caroline Hill for providing the HaCaT-TR cell line.
FUNDING
This work was supported by the National Institutes of Health (USA) [grant number AR050266 (to A.M.B.)]; the Wilhelm-Sander Stiftung (GER) [grant number 2012.075.02 (to D.K.)]; and The Francis Crick Institute (UK) (to S.C.L.).
Abbreviations
- AID
activation-induced cytidine deaminase
- CARD14
caspase recruitment domain-containing protein 14
- CARMA
CARD-containing MAGUK protein
- coIP
co-immunoprecipitation
- ERK1/2
extracellular signal-regulated kinase 1/2
- EV
empty vector
- IKK2
inhibitor of NF-κB (IκB) kinase 2
- JNK
c-Jun N-terminal kinase
- MAGUK
membrane-associated guanylate kinase
- MAP kinase
mitogen-activated protein kinase
- PKC
protein kinase C
- TCR
T-cell receptor
- TR
tetracycline (Tet) repressor
- WT
wild type
Footnotes
AUTHOR CONTRIBUTION
Ashleigh Howes co-designed the study, performed the majority of the experiments and co-wrote the manuscript. Paul O’Sullivan, Felix Breyer and Ashavari Ghose performed some experiments. Li Cao performed preliminary experiments on the project. Daniel Krappmann advised on methodology and experimental design, and critically reviewed the manuscript. Anne Bowcock co-supervised the study and critically reviewed the manuscript. Steven Ley co-designed, co-supervised and co-wrote the manuscript.
References
- 1.Lowes MA, Suarez-Farinas M, Krueger JG. Immunology of psoriasis. Ann Rev Immunol. 2014;32:227–255. doi: 10.1146/annurev-immunol-032713-120225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Harden JL, Krueger JG, Bowcock AM. The immunogenetics of psoriasis: a comprehensive review. J Autoimmun. 2015;64:66–73. doi: 10.1016/j.jaut.2015.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Jordan CT, Cao L, Roberson ED, Duan S, Helms CA, Nair RP, Duffin KC, Stuart PE, Goldgar D, Hayashi G, et al. Rare and common variants in CARD14, encoding an epidermal regulator of NF-κB, in psoriasis. Am J Hum Genet. 2012;90:796–808. doi: 10.1016/j.ajhg.2012.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Jordan CT, Cao L, Roberson ED, Pierson KC, Yang CF, Joyse CE, Ryan C, Duan S, Helms CA, Liu Y, et al. PSORS2 is due to mutations in CARD14. Am J Hum Genet. 2012;90:784–795. doi: 10.1016/j.ajhg.2012.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ammar M, Jordan CT, Cao L, Lim E, Bouchlaka Souissi C, Jrad A, Omrane I, Kouidhi S, Zaraa I, Anbunathan H, et al. CARD14 alterations in Tunisian patients with psoriasis and further characterization in European cohorts. Br J Dermatol. 2016;174:330–337. doi: 10.1111/bjd.14158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tsoi LC, Spain SL, Knight J, Ellinghaus E, Stuart PE, Capon F, Ding J, Li Y, Tejasvi T. Identification of 15 new psoriasis susceptibility loci highlights the role of innate immunity. Nat Genet. 2012;44:1341–1348. doi: 10.1038/ng.2467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cheng H, Li Y, Zuo XB, Tang HY, Tang XF, Gao JP, Sheng YJ, Yin XY, Zhou FS, Zhang C, et al. Identification of a missense variant in LNPEP that confers psoriasis risk. J Invest Dermatol. 2014;134:359–365. doi: 10.1038/jid.2013.317. [DOI] [PubMed] [Google Scholar]
- 8.Bowes J, Budu-Aggrey A, Huffmeier U, Uebe S, Steel K, Hebert HL, Wallace C, Massey J, Bruce IN, Bluett J, et al. Dense genotyping of immune-related susceptibility loci reveals new insights into the genetics of psoriatic arthritis. Nat Commun. 2015;6:6046. doi: 10.1038/ncomms7046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jiang C, Lin X. Regulation of NF-κB by the CARD proteins. Immunol Rev. 2012;246:141–153. doi: 10.1111/j.1600-065X.2012.01110.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ghosh S, May MJ, Kopp EB. NF-kB and Rel proteins: evolutionary conserved mediators of immune responses. Annu Rev Immunol. 1998;16:225–260. doi: 10.1146/annurev.immunol.16.1.225. [DOI] [PubMed] [Google Scholar]
- 11.Bertin J, Wang L, Guo Y, Jacobson MD, Poyet JL, Srinivasula SM, Merriam S, DiStefano PS, Alnemri ES. CARD11 and CARD14 are novel caspase recruitment domain (CARD) / membrane-associated guanylate kinase (MAGUK) family members that interact with BCL10 and activate NF-κB. J Biol Chem. 2001;276:11877–11882. doi: 10.1074/jbc.M010512200. [DOI] [PubMed] [Google Scholar]
- 12.Scudiero I, Zotti T, Ferravante A, Vessichelli M, Vito P, Stilo R. Alternative splicing of CARMA2/CARD14 transcripts generates protein variants with differential effect on NF-κB activation and endoplasmic reticulum stress-induced cell death. J Cell Pysiol. 2011;226:3121–3131. doi: 10.1002/jcp.22667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Goldminz AM, Au SC, Kim N, Gottlieb AB, Lizzul PF. NF-kB: an essential transcription factor in psoriasis. J Derm Sci. 2013;69:89–94. doi: 10.1016/j.jdermsci.2012.11.002. [DOI] [PubMed] [Google Scholar]
- 14.Harden JL, Lewis SM, Pierson KC, Suarez-Farinas M, Lentini T, Ortenzio FS, Zaba LC, Goldbach-Mansky R, Bowcock AM, Lowes MA. CARD14 expression in dermal endothelial cells in psoriasis. PLoS One. 2014;9:e111255. doi: 10.1371/journal.pone.0111255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Afonina IS, Elton L, Carpentier I, Beyaert R. MALT1 – a universal soldier: multiple strategies to ensure NF-kB activation and target gene expression. FEBS J. 2015;282:3286–3297. doi: 10.1111/febs.13325. [DOI] [PubMed] [Google Scholar]
- 16.Eitelhuber AC, Vosyka O, Nagel D, Bognar M, Lenze D, Lammens K, Schlauderer F, Hlahla D, Hopfner KP, Lenz G, et al. Activity-based probes for detection of active MALT1 paracaspase in immune cells and lymphomas. Chem Biol. 2015;22:139–147. doi: 10.1016/j.chembiol.2014.10.021. [DOI] [PubMed] [Google Scholar]
- 17.Levy L, Hill CS. Smad4 dependency defines two classes of transforming growth factor beta (TGFb) target genes and distinguishes TGFb-induced epithelial-mesenchymal transition from its antiproliferative and migratory responses. Mol Cell Biol. 2005;25:8108–8125. doi: 10.1128/MCB.25.18.8108-8125.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jiang C, Lin X. Regulation of NF-kappaB by the CARD proteins. Immunol Rev. 2012;246:141–153. doi: 10.1111/j.1600-065X.2012.01110.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.McCully RR, Pomerantz JL. The protein kinase C-responsive inhibitory domain of CARD11 functions in NF-kB activation to regulate the association of multiple signaling cofactors that differentially depend on Bcl10 and MALT1 for association. Mol Cell Biol. 2008;28:5668–5686. doi: 10.1128/MCB.00418-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sommer K, Guo B, Pomerantz JL, Bandaranayake AD, Moreno-Garcia ME, Ovechkina YL, Rawlings DJ. Phosphorylation of the CARMA1 linker controls NF-kB activation. Immunity. 2005;23:561–574. doi: 10.1016/j.immuni.2005.09.014. [DOI] [PubMed] [Google Scholar]
- 21.Lamason RL, McCully RR, Lew SM, Pomerantz JL. Oncogenic CARD11 mutations induce hyperactive signaling by disrupting autoinhibition by the PKC-responsive inhibitory domain. Biochem. 2010;49:8240–8250. doi: 10.1021/bi101052d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lim KH, Yang Y, Staudt LM. Pathogenetic importance and therapeutic implications of NF-kB in lymphoid malignancies. Immunol Rev. 2012;246:359–378. doi: 10.1111/j.1600-065X.2012.01105.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Nagel D, Spranger S, Vincendeau M, Grau M, Raffegerst S, Kloo B, Hlahla D, Neuenschwander M, von Kries JP, Hadian K, et al. Pharmacologic inhibition of MALT1 protease by phenothiazines as a therapeutic approach for the treatment of aggressive ABC-DLBCL. Cancer Cell. 2012;22:825–837. doi: 10.1016/j.ccr.2012.11.002. [DOI] [PubMed] [Google Scholar]
- 24.Knies N, Alankus B, Weilemann A, Tzankov A, Brunner K, Ruff T, Kremer M, Keller UB, Lenz G, Ruland J. Lymphomagenic CARD11/BCL10/MALT1 signaling drives malignant B-cell proliferation via cooperative NF-kB and JNK activation. Proc Natl Acad Sci USA. 2015;112:E7230–E7238. doi: 10.1073/pnas.1507459112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lang V, Janzen J, Fischer GZ, Soneji Y, Beinke S, Salmeron A, Allen H, Hay RT, Ben-Neriah Y, Ley SC. bTrCP-mediated proteolysis of NF-kB1 p105 requires phosphorylation of p105 serines 927 and 932. Mol Cell Biol. 2003;23:402–413. doi: 10.1128/MCB.23.1.402-413.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Clark K, Peggie M, Plater L, Sorcek RJ, Young ERR, Madwed JB, Hough J, McIver EG, Cohen P. Novel cross-talk within the IKK family controls innate immunity. Biochem J. 2011;434:93–104. doi: 10.1042/BJ20101701. [DOI] [PubMed] [Google Scholar]
- 27.Robbiani DF, Nussenzweig MC. Chromosome translocation, B cell lymphoma, and activation-induced cytidine deaminase. Ann Rev Pathol Mech Dis. 2012;8:79–103. doi: 10.1146/annurev-pathol-020712-164004. [DOI] [PubMed] [Google Scholar]
- 28.Johansen C, Kragballe K, Westergaard M, Henningsen J, Kristiansen K, Iverson L. The mitogen-activated protein kinase p38 and ERK1/2 are increased in lesional psoriatic skin. Br J Dermatol. 2005;152:37–42. doi: 10.1111/j.1365-2133.2004.06304.x. [DOI] [PubMed] [Google Scholar]
- 29.Uehata T, Iwasaki H, Vandenbron A, Matsushita K, Hernandez-Cuellar E, Kuniyoshi K, Satoh T, Mino T, Suzuki Y, Standley DM, et al. Malt1-induced cleavage of regnase-1 in CD4 + helper T cells regulates immune activation. Cell. 2013;153:1036–1049. doi: 10.1016/j.cell.2013.04.034. [DOI] [PubMed] [Google Scholar]
- 30.Jeltsch KM, Hu D, Brenner S, Zöller J, Heinz GA, Nagel D, Vogel KU, Rehage N, Warth SC, Edelmann SL, et al. Cleavage of roquin and regnase-1 by the paracaspase MALT1 releases their cooperatively repressed targets to promote Th17 differentiation. Nat Immunol. 2014;15:1079–1089. doi: 10.1038/ni.3008. [DOI] [PubMed] [Google Scholar]
- 31.Eskin-Schwartz M, Basel-Vanagaite L, David M, Lagovsky I, Ben-Amitai D, Smirin-Yosef P, Atzmony L, Hodak E. Intra-familial variation in clinical phenotype of CARD14-related psoriasis. Acta Derm-Venereol. 2016 doi: 10.2340/00015555-2405. [DOI] [PubMed] [Google Scholar]
- 32.Leman J, Burden AD. Sequential use of biologics in the treatment of moderate-to-severe plaque psoriasis. Br J Dermatol. 2012;167:12–20. doi: 10.1111/j.1365-2133.2012.11209.x. [DOI] [PubMed] [Google Scholar]
- 33.Callamer AN, Guerrero KT, Henning JS, Battafarano DF. Psoriatic skin lesions induced by tumor necrosis factor antagonist therapy: a literature review and potential mechanisms of action. Arthritis Rheum. 2008;59:996–1001. doi: 10.1002/art.23835. [DOI] [PubMed] [Google Scholar]
- 34.Olivieri I, D’Angelo S, Palazzi C, Padula A. Advances in the management of psoriatic arthritis. Nat Rev Rheumatol. 2014;10:531–542. doi: 10.1038/nrrheum.2014.106. [DOI] [PubMed] [Google Scholar]
- 35.de Souza A, Ali-Shaw T, Reddy SM, Fiorentino D, Strober BE. Inflammatory arthritis following ustekinumab treatment for psoriasis: a report of two cases. Br J Dermatol. 2013;168:210–212. doi: 10.1111/j.1365-2133.2012.11206.x. [DOI] [PubMed] [Google Scholar]
- 36.Belge K, Brück J, Ghoreschi K. Advances in treating psoriasis. F1000 Prime Rep. 2014;6:4. doi: 10.12703/P6-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.McInnes IB, Sieper J, Braun J, Emery P, van der Heijde D, Isaacs JD, Dahmen G, Wollenhaupt J, Schulze-Koops H, Kogan J, et al. Efficacy and safety of secukinumab, a fully human anti-interleukin-17A monoclonal antibody, in patients with moderate-to-severe psoriatic arthritis: a 24-week, randomised, double-blind, placebo-controlled, phase II proof-of-concept trial. Ann Rheum Dis. 2014;73:349–356. doi: 10.1136/annrheumdis-2012-202646. [DOI] [PubMed] [Google Scholar]