

Abstract
Microfluidic culture of primary adipose tissue allows for reduced sample and reagent volumes as well as constant media perfusion of the cells. By continuously flowing media over the tissue, microfluidic sampling systems can more accurately mimic vascular flow in vivo. Quantitative measurements can be performed on or off chip to provide time-resolved secretion data, furthering insight into the dynamics of the function of adipose tissue. Buoyancy resulting from the large lipid storage capacity in this tissue presents a unique challenge for culture, and it is important to account for this buoyancy during microdevice design. Herein, we describe approaches for microfluidic device fabrication that utilize 3D-printed interface templating to help counteract cell buoyancy. We apply such methods to the culture of both isolated, dispersed primary adipocytes and epididymal adipose explants. To facilitate more widespread adoption of the methodology, the devices presented here are designed for user-friendly operation. Only handheld syringes are needed to control flow, and devices are inexpensive and disposable.
Keywords: Adipose tissue, Microfluidics, 3D printing, Cell culture, Explants, Secretion, Hormones, Glycerol, Insulin signaling, Free fatty acids
1 Introduction
Microfluidic systems have been shown to offer exclusive advantages over traditional methods for chemical and biochemical analysis [1, 2]. Systems can be engineered to integrate multiple steps of standard sample preparation and analysis onto one microdevice, and some devices even furnish analytical tools that are unavailable in their macro-scale counterparts [3, 4]. Recent work in cellular co-culture and organ-on-a-chip platforms have confirmed that the scale of microfluidics is ideal to culture and interrogate tissues [5, 6], and some have even reconstituted human biological function on microdevices that outperform animal models [7]. Others have shown that microfluidics is useful for studying pancreatic islets [8–10], and our group has contributed by developing easy-to-use microdevices for islet stimulation and secretion sampling [11–13].
Despite the inherent advantages available in microfluidic cell culture, fewer studies [14] have focused on miniaturizing adipose tissue culture and sampling. Recently, we have demonstrated that by customizing the cell-to-chip interfaces to counteract buoyancy, both isolated adipocytes [15] and adipose tissue explants [13] can be adapted to miniaturized systems. These devices have permitted adiponectin secretion quantification and even time-resolved glycerol secretion measurements on small amounts of endocrine tissue. With these miniaturized methods, a single mouse can provide enough tissue for over an order of magnitude more measurements when compared to macro-scale culture methods, validating a role for microfluidics in adipose tissue biology.
In this chapter, we present detailed approaches for adipose tissue culture and sampling on microfluidic devices. Chip fabrication approaches utilize 3D-printed interface templating to help counteract cell buoyancy for both dispersed primary adipocytes and epididymal adipose explants. To enhance simplicity of use, devices are designed for user-friendly operation with handheld syringes, thus requiring no electrical interfacing or fluidic valving. These inexpensive and disposable devices should be more adaptable for routine use by non-experts, helping to extend the benefits of microscale analysis to more laboratories that study adipocyte biology.
2 Materials
2.1 Photolithography for Silicon Wafer Master Fabrication
4″ Silicon wafers, N-type, <100>, 500 +/− 25 μm, 1SP (Polishing Corporation of America).
Transparency design (FineLine Imaging).
Glass plate for design mounting.
Oven (see Note 1).
Rotary shaker.
Glass dish for cleaning 4″ wafers.
Wafer tweezers (150 SA WAFER, Vomm Germany).
1.0 M sulfuric acid.
Distilled, deionized H2O (ddH2O).
Hot plate.
N2 gas line or supply.
Spin coater (WS-400BZ-6NPP/Lite, Laurell Technologies).
Spin coater alignment tool (Thingiverse.com, EasleyLab) (see Note 2).
SU-8 negative photoresist and developer (MicroChem).
Photolithography system with ultraviolet light source for photocuring of SU-8 (see Note 3).
UV blocking safety glasses.
Isopropyl alcohol (IPA).
2.2 PDMS Microchip Devices
Sylgard 184® elastomer base and curing agent (Dow Corning).
Silicon wafers.
Tygon tubing (0.02 inch ID, 0.06 inch OD).
Blunt-ended needles (22 G).
60-mL plastic, luer lock syringes.
3-mL luer lock syringes.
Plasma cleaner (Harrick; basic cleaner, PDC-32G).
Vacuum desiccator and pump.
Aluminum foil.
Double-sided tape.
Oven or heated rocker (see Note 1).
100-mm Petri dishes.
2.3 Manually Fabricated Interface Templates
Polymer casting resin (Smooth-On, Inc.; Smooth-Cast 310).
Silicone oil.
Disposable biopsy punches with plunger (Militex®, prod # 15110).
Vacuum desiccator and pump.
Plasma cleaner (Harrick; basic cleaner, PDC-32G).
2.4 3D-Printed Interface Templates
3D sketching software (e.g., SketchUp, OpenSCAD) or predesigned files (see Note 2).
3D printer (MakerBot Replicator 2).
Printer filament (Makerbot, polylactic acid).
2.5 Fat Pad Isolation and Culture
Disposable culture tubes, 12 × 75 mm.
3 mL luer lock, sterile syringe.
Polypropylene mesh, 210 μm (Spectra Mesh, prod # 145880).
Sterile 96-well plate with lid.
18G × 1 1/2 inch needles.
2 mm disposable biopsy punch with plunger.
Distilled, deionized H2O (ddH2O).
Phosphate-HEPES Buffer: bovine serum albumin (BSA), d-glucose, HEPES, NaCl, CaCl2–2 H2O, MgSO4–7 H2O, KH2PO4, Na2HPO4, pH: 7.2–7.4.
Adipocyte Media: nystatin suspension (10,000 units/mL), penicillin-streptomycin (10,000 U/mL), 100× minimal essential media, nonessential amino acids solution (MEM NEAA), fetal bovine serum (FBS) heat inactivated, dulbecco’s modified eagle medium (DMEM, low glucose), dulbecco’s modified eagle medium (DMEM, without glucose and phenol red), fatty acid free bovine serum albumin (FAF BSA), sodium pyruvate, l-glutamine, nalgene® rapid-flow™ filter units, and bottle top filters (PES membrane, 0.2 μm pore size, sterile).
Serum Media: Add 6 mL of FBS, nystatin, penicillin-streptomycin, and MEM NEAA to 476 mL of low glucose DMEM. Filter into sterile container and store at 4°C.
Serum Free Media: Add 6 mL of nystatin, penicillin-streptomycin, and MEM NEAA; 5 mL of sodium pyruvate; 10 mL of L-glutamine; and 1 g of FAF BSA to 467 mL of glucose and phenol red free DMEM.
Collagenase solution: Weigh 10 mg of collagenase type I into a 15 mL conical tube and add 8 mL of phosphate-HEPES buffer. Mix gently by inversion and place in water bath.
3 Methods
3.1 Photolithography for Silicon Wafer Master Fabrication (See Note 4)
Carefully remove new wafer and place in glass dish. Add in enough 1.0 M sulfuric acid to just cover the wafer in the dish (Fig. 1a).
Secure the dish on a rotary shaker. Turn on shaking at ~250 rpm for 30 min.
At the end of the acid wash, remove the wafer from the dish with wafer tweezers and rinse both the wafer and the dish with ample ddH2O.
Return the wafer to the dish and cover it with ddH2O. Turn on shaking at ~250 rpm for 30 min.
Remove the wafer from the dish with wafer tweezers, carefully dry with N2 gas, and place in an oven at 200°C for 15 min (Fig. 1b).
Remove the wafer from the oven, place on a clean Kimwipe, and allow it to cool to room temperature.
Center wafer on spin coater with a wafer alignment tool (see Note 2). Pour SU-8 slowly onto the wafer’s center to avoid air bubbles (~1 mL SU-8 per inch of substrate diameter). Choose the grade of SU-8 (based on viscosity) which corresponds to desired channel height.
Spin the wafer at 500 rpm with an acceleration of 100 rpm s−1 for 30 s (Fig. 1c).
For the second spin cycle, the spin speed depends on the desired microchannel depth. Exact spin coating details can be found in the manufacturer’s instructions (Microchem). However, the first spin coating cycle (step 8) will remain constant.
For edge bead removal, a methanol-soaked Kimwipe or clean-room wipe can be used to remove any bulk SU-8 accumulated at the edges of the wafer (see Note 5).
Transfer the wafer to the hot plate (95°C) for the soft baking step of the SU-8. Soft bake times can be found in the manufacturer’s instructions (Microchem) (see Note 6).
Place the wafer in the center of the UV lithography exposure platform.
Tape the microchannel mask transparency to the backside of the square glass tile (Fig. 1d).
Gently place the slide centered on the top of the wafer so that the patterned side of the mask gently contacts the SU-8. Ensure that there are no air gaps between the mask and the SU-8 on the wafer (Fig. 1e).
Expose the wafer to UV light using the exposure settings appropriate for the photolithography system in use.
Lift the glass slide from the wafer without also lifting the wafer.
Transfer the wafer back to the 95°C hot plate for the post exposure bake. Post exposure bake times can be found in the manufacturer’s instructions (see Note 6).
Place the wafer on a clean Kimwipe or another surface with low thermal conductivity, allow it to passively cool to room temperature, then transfer it to a round glass dish.
Pour enough SU-8 developer into the dish to cover the wafer. Manually shake the dish for the manufacturer’s listed development time.
Rinse both the dish and wafer with fresh developer for ~10 s, followed by an IPA rinse to remove any residual developer. Intricate or high-density channel designs may require forceful rinsing with developer, which can be accomplished using disposable transfer pipettes (Fig. 1f).
Carefully dry the wafer with N2 gas. If any white film forms, rinse again with fresh developer for ~10 s followed by another IPA rinse.
Once dry and clean, the wafer is ready for use as a PDMS channel master (Fig. 1g).
Fig. 1.

Photolithography steps. (a) Wafer is acid cleaned on a rotary shaker. (b) Wafer is baked at 200°C to remove residual water. (c) Spin coating of SU-8 onto clean wafer. (d) Microchannel mask transparency is taped onto a dust-free glass slide. (e) Wafer and design placed into UV lithography system for exposure step. (f) Photoresist development step. (g) Finilazed SU-8 master wafer, ready for PDMS soft lithography
3.2 PDMS Microchip Devices
Mix precursors of polydimethylsiloxane (PDMS) base and curing agent (10:1 respectively) in a plastic weigh boat. For a single silicon wafer, ~45.0 g total of PDMS is sufficient. Stir PDMS precursors with a disposable wooden spatula until sufficiently mixed.
Place weigh boat into a degassing chamber and allow to degas until the polymer is bubble free. While the PDMS is degassing, rinse the silicon wafer with methanol and dry with N2 gas.
Cut a square of aluminum foil large enough to form a boat that contains the silicon wafer. Place a strip of double-sided tape on the bottom of the wafer and attach it to the center of the aluminum foil boat. Gently push the wafer flat onto the center of the foil square to ensure attachment, and carefully fold up the foil around the edges of the wafer to form a boat-like structure that can hold liquid PDMS precursors.
Slowly pour the degassed PDMS precursor liquid onto the top of the wafer. If any additional bubbles are formed in the polymer, a second degassing can be applied to remove them.
At this point, interface templates can be placed over the appropriate structures in the PDMS to define the reservoir regions above channel designs (see Figs. 2b, 3b, and 4). Degas one final time to remove any air trapped between the wafer and the template.
Place the wafer, templates, and PDMS in an oven to cure the polymer. Adjust the final placement of the template by gently pushing the structures into the PDMS, and avoid lifting them from the PDMS to prevent air bubbles. Curing times around 2 h are sufficient for ovens at or above 50°C. PDMS can be cured at 60–80°C when using manually fabricated interface templates, but the oven should be kept near 50°C when using 3D-printed interface templates (see Note 7).
Carefully peel and remove the aluminum foil from the PDMS and the wafer. A razor blade can be used to remove excess pieces of foil or PDMS from the wafer’s edges.
Gently begin to lift the PDMS from the wafer, starting at the edges and working around the circumference of the wafer. Once the edges have been released, slowly lift the bulk of the PDMS from the wafer, minimizing the pressure applied to the wafer, which can be easily broken.
Rinse the wafer with methanol and dry with N2 gas. Return the water to a 100 mm petri dish for storage until the next usage.
Cut the bulk PDMS down to the single unit device pieces with a razor, making sure to leave clean edges for optimum plasma bonding to the glass floor.
With the channel sides facing up, begin punching inlet and outlet holes in the microchip. Rinse thoroughly with methanol and dry with N2 gas. Apply and remove Scotch tape to the channeled side of the chip several times to remove any remaining dust particles or debris. If there are multiple chips to be cleaned and bonded, tape can be left on the cleaned chips to prevent any contamination.
Glass slides for plasma bonding should also be rinsed with methanol, dried with N2 gas, and taped clean before use. The glass slides serve as the channel floors and help to stabilize the device.
Remove any remaining tape and place both the glass slide and the PDMS chip into the plasma oxidation chamber. Surfaces that will be bonded should be uncovered and facing up (i.e., channeled side up).
Plasma oxidize the slide and chip for 1 min at medium power. Carefully remove the slide and chip, making sure not to touch the activated surfaces. Grasp the chip by the sides and turn so that the channels are facing the glass slide. In a rolling motion, apply the PDMS to the middle section of the glass slide. Rolling, instead of simply placing the whole piece of PDMS down at once, helps prevent trapping of air bubbles between the chip and the glass slide.
Ensure that there are no air bubbles by viewing the chip contact through the glass slide. Medium pressure by hand can be applied to ensure full contact. If pressed too firmly, channels could collapse and become bonded to the glass as well. Do not disassemble the glass and PDMS to retry the bonding, as covalent bonds between the two surfaces will have already formed, and the chip will be damaged beyond use. Regions of thin PDMS membranes where interface templates have been removed often need to be gently pushed with a blunt probe to achieve full contact for bonding to the glass substrate. Cross-sections of typical devices are shown in Fig. 2.
Once the chip is bonded, fill the main reservoir with serum free media containing BSA. Let the chips incubate with the solution for at least 1 h. The BSA will adsorb to the freshly cleaned surfaces and channels to prevent nonspecific binding during experiments. This step should be carried out the day of plating collagen-based adipocytes or the day of experiments for tissue explants. Do not allow the chips to dry out once they have been treated with media.
Before plating adipocytes and collagen on the chip, remove residual serum free media from the reservoir. Immediately begin plating the cells in collagen as per Subheading 3.5.
If adipose tissue explants are to be used on the chips, remove excess media from the reservoir and transfer the explants via forceps to the punched inlet. Before adding back fresh media, place the 3D-printed explant trap into the reservoir to prevent the explant from floating. Continue to Subheading 3.7 for microfluidic sampling procedures.
Fig. 2.
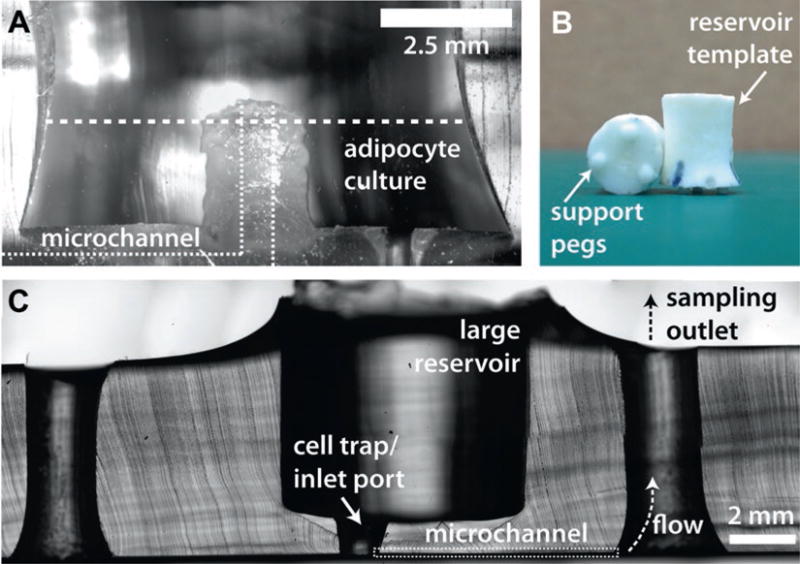
Cross-sections of typical devices. (a) A cross-section of a device made with a manually fabricated interface template. (b) Examples of manually fabricated interface templates, showing support pegs that define reservoir height. (c) A cross-section of a device made with a 3D-printed interface template
Fig. 3.
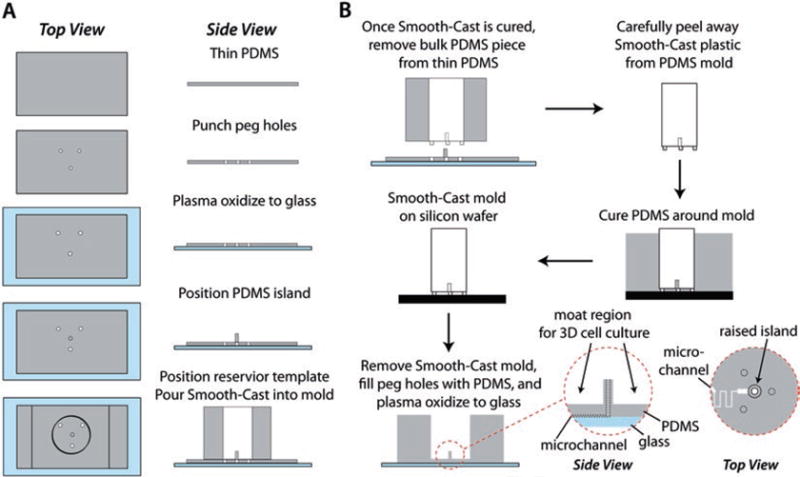
Manual fabrication method for the Smooth-Cast 310 interface template. (a) The PDMS mold for the insert serves as an initial proxy for the final device, including the raised island and moat region customized for culture of primary adipocytes in collagen. (b) The resultant template is used to define a customized macro-to-micro interface to microfluidic channels for secretion sampling (reproduced from Ref. [15] with permission from The Royal Society of Chemistry)
Fig. 4.

3D-printed interface templates. Shown here are a 3D software rendering (left) and an actual printed template (right) for explant culture
3.3 Manually Fabricated Interface Templates (Fig. 3)
Punch 1 mm diameter holes in a 1 mm thick slab of PDMS to create support legs for the template mold. The thickness of the slab will define the depth of PDMS between the large reservoir and microfluidic channels in the resulting microfluidic device, and this parameter can be altered to accommodate experimental needs (Fig. 3a).
Plasma oxidize the slab and a glass microscope slide. Place the PDMS in the center of the slide to covalently bond the two (Fig. 3a).
Punch a 2–3 mm diameter plug out of PDMS and place the plug in the center of the mold form. This plug will define the raised island in the large reservoir (Fig. 3a).
The body of the mold should be punched into a thicker piece of PDMS (~2 cm height). This piece should be centered over the support legs to reversibly seal (not plasma oxidize) the two pieces of PDMS and complete the mold (Fig. 3a, bottom).
Measure pre-polymer solutions of Smooth-Cast (part A and part B), and degas the unmixed components for 10 min. Once degassed, combine 100 μL of each part (1:1 by volume) and pipette the solution into the small support peg section of the PDMS mold; degas the prepolymer once more. Due to the small size of the support pegs, it is imperative that the Smooth-Cast is properly degassed to prevent a brittle mold foundation. After degassing, mix the remaining Smooth-Cast (~1 mL), and carefully add it to the bulk of the mold. Degas to remove any additional air trapped in the body of the mold, and allow to cure overnight.
To remove the Smooth-Cast template, lift the thick slab of PDMS from the thin slab on the glass slide. Carefully peel the template from the PDMS mold, being careful to not tear the PDMS or break the support pegs. Any extra pieces of PDMS stuck to the template can be removed with an X-Acto™ knife or a blunt-ended needle (Fig. 3b, top).
The manually fabricated interface template is now ready to template adipocyte culturing reservoirs (Fig. 3b) into PDMS devices during curing of PDMS on the SU-8 patterned wafer (see Subheading 3.2, step 5). Examples and usage of these templates were reported by Godwin et al. [15], and an image of two templates is shown in Fig. 2b.
3.4 3D-Printed Interface Templates
To use predesigned interface templates, choose the appropriate 3D part from those available. Refer to our laboratory’s profile on Thingiverse website (see Note 2).
Alternatively, templates can be customized or modified, exploiting the rapid prototyping feature of 3D printing systems. Using appropriate 3D CAD software (e.g., SketchUp, OpenSCAD), design 3D structure of the custom interface template.
Print the interface template using a 3D printer. Templates and parts shown in this chapter were printed using a MakerBot Replicator 2 with PLA filament. 3D renderings and sample prints of these templates are shown in Fig. 4.
The 3D-printed interface template is now ready to template adipocyte culturing reservoirs into PDMS devices during curing of PDMS on the SU-8 patterned wafer (see Subheading 3.2, step 5).
3.5 Adipocyte Digestion and Microfluidic Culture
Immediately before surgery, make the collagenase solution. Place the collagenase, phosphate-HEPES buffer, and serum media in 37°C water bath.
Remove epididymal fat pads from C57BL/6J male mice and transfer to 4 mL of pre-warmed phosphate-HEPES buffer.
Place the extracted fat into a 60 mm dish to record the wet weight of the tissue.
Transfer the extracted fat into a 2 mL snap cap tube. Mince the tissue in the tube for 2 min using surgical scissors.
For every 1 g of fat, add 2 mL of collagenase to the sample. Pipette the suspension up and down several times, then place the tube in a rocking water bath set at 120 rpm and 37°C.
Allow the tissue to digest for 30 min in the water bath. To ensure adequate digestion, pipette the suspension up and down every 10 min during the digestion.
Prepare a funnel with 210 μm Spectra mesh. Pipette the digested tissue over the filter and into a 5 mL glass test tube. Wash the cells with 3–4 mL of phosphate-HEPES buffer.
Centrifuge the cell suspension at 900 rpm for 6 min at 4°C.
Remove the infranatant with an 18G 1 1/2 inch needle and small syringe.
Wash cells with an additional 3–4 mL of phosphate-HEPES buffer and repeat centrifuging and infranatant removal.
After the second wash, add 3–4 mL of serum media and centrifuge at 1000 rpm for 6 min at room temperature.
Remove infranatant and add 3–4 mL of serum media.
Place the tube with freshly washed cells in the CO2 incubator for 30 min. Lightly cover the top of the tube with Parafilm.
Immediately before the incubation is complete, mix 4.5 mL of collagen, 450 μL 10× MEM, and 3–5 μL of 1.0 M NaOH. Upon addition of the 10× MEM, the solution will turn yellow. Due to the viscosity of collagen, make sure to vortex thoroughly. When adding the NaOH, a light pink end color is desired. The freshly mixed collagen will begin to harden and can only be used for ~30 minutes before it becomes too viscous for plating.
Pipette the fat cells floating on the top layer of the serum media and transfer to a 2 mL snap cap tube. Record the total volume of cells collected.
Add the collagen directly to the cell solution (10 μL of cells to 45 μL collagen). Pipette the solution to ensure homogeneity and begin plating (55 μL/microwell). Gently shake the chips to cover the surface evenly with cells. Continuously pipette to mix the stock suspension during the plating process.
After all the microdevice wells have been plated with cell suspension and collagen, place the chips into the CO2 incubator for 60 min. The collagen and cell mixture will have solidified in this time, and serum media can be slowly added to each well. An example of digested and reconstituted adipose tissue cultured on a microdevice is shown in Fig. 5a.
Allow the cells to sit in the incubator overnight, at which time the cells and devices will be ready for microfluidic secretion sampling.
Fig. 5.

Adipocytes cultured on 8-channel microfluidic sampling devices. (a) Top-down view image of digested and reconstituted adipose tissue cultured on a microdevice made with a 3D-printed interface template. Inset shows zoomed version of adipocytes in collagen. (b) 3D renderings and (c) example prints of adipose tissue explant traps. (d) Image of a trapped adipose tissue explant cultured on a microdevice and held in place with an explant trap. Inset shows zoomed version of trapped explant
3.6 Explant Culture on Microdevices
Place phosphate-HEPES and serum media in a water bath at 37°C before the surgery.
Remove epididymal fat pads from C57BL/6J male mice and transfer to 4 mL of pre-warmed phosphate-HEPES buffer.
Transfer the fat pads to a 60 mm dish containing fresh phosphate-HEPES buffer. Carefully cut away any large veins from the tissue and discard them.
Punch 2 mm diameter plugs of tissue using biopsy punches with plungers. Transfer each explant as it is punched into a glass test tube with phosphate-HEPES buffer. Continue punching until sufficient tissue has been collected or the remaining tissue is insufficient for accurate punching.
Centrifuge the explants at 1000 rpm for 3 min. Remove the infranatant using a 18-gauge needle and a 3 mL syringe. Replace with 4 mL of phosphate-HEPES buffer. Repeat this process two additional times, replacing the buffer with 4 mL of serum media.
After the final rinse, transfer each individual explant to separate wells on a 96-well plate containing 200 μL serum media. Place the 96-well plate into the CO2 incubator for 30 min. To counteract buoyancy during preparative culture, add customized 3D-printed explant traps (see Note 2) into each well and place the plate back into the incubator [13]. 3D renderings and example prints of these traps are shown in Fig. 5b.
To maintain the explants, wash with fresh serum media in the morning and the evening. Explants can be used for experiments for up to 7–10 days if maintained in this way.
Select a BSA-treated device (see Subheading 3.2, step 16), and remove approximately 90 % of the BSA buffer so that only the smaller tissue capture reservoir contains buffer. Transfer an explant from the 96-well plate using surgical tweezers into the capture reservoir of the microdevice. Add customized 3D-printed explant trap (see Note 2) to counteract buoyancy, then add the appropriate starting solution, depending on the experiment. At this point, devices will be ready for microfluidic secretion sampling of the trapped explant. An example of a trapped adipose tissue explant cultured on a microdevice is shown in Fig. 5c.
3.7 Adipocyte Secretion Sampling in Microfluidic Devices
Pre-warm all solutions and media in a water bath at 37°C.
Wash cells three times with fresh serum media, and allow them to incubate in the media for at least 1 h.
While the cells are bathing in fresh media, prepare treatment media buffer. If the final sample solution will be used in a homogeneous assay, serum free media without phenol red can be used to prevent readout issues with colored solutions.
A variety of treatments and timings can be applied to these small portions of tissue on the microdevice, depending upon the experiment of choice. As an example, we have shown in Fig. 6 that glycerol secretion is strongly dependent upon insulin and glucose concentrations at physiologically relevant levels. Importantly, up to an order of magnitude more experiments can be carried out on single mouse using this microfluidic system, compared to standard sampling techniques.
When the cultured adipocytes or explants are ready for sampling, insert Tygon tubing into the outlet holes on the chip. On the opposite end of the tubing, slide in a blunt-ended needle at least 3–4 mm.
Attach a 60 mL syringe to the luer lock end of the blunt needle, with the syringe already set to the starting volume. Carefully apply vacuum to the chip via the syringe and place a lock or stopper on the plunger of the syringe to prevent its movement.
After sampling has completed, remove the syringe from the tubing to stop the vacuum.
To collect the sample from the tubing, place a 3 mL luer lock syringe on the end of the tubing, but do not apply any vacuum. Have an opened, 1.7 mL snap cap tube ready for the transfer. Gently pull the tubing from the chip with the 3 mL syringe still in place. Hold the open end of the tubing in the snap cap tube and slowly apply pressure from the 3 mL syringe to the tubing to transfer the sample volume.
Depending on the assay format, samples can be immediately analyzed or placed in a freezer at −20°C until future analysis.
Fig. 6.
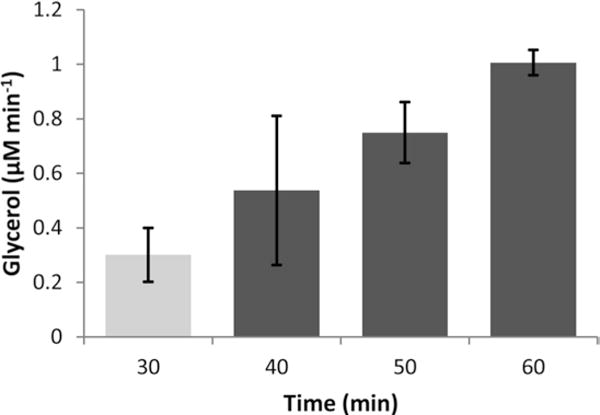
Time-resolved glycerol secretion from an adipose tissue explant using 3D-templated reservoirs and a 3D-printed tissue trap on a microfluidic device under continuous, vacuum-driven flow. After switching from high insulin and glucose (2 nM insulin, 19 mM glucose) to low insulin and glucose (50 pM insulin, 3.5 mM glucose) solutions at the 30 min time point, glycerol secretion rates increased. Average data is shown for three explants, with error bars representing standard deviations
Acknowledgments
Support for the work was provided by the National Institutes of Health (R01 DK093810) as well as by the Department of Chemistry and Biochemistry and the College of Science and Mathematics at Auburn University. The authors would like to thank Mark D. Holtan and Tesfagebriel Hagos for assistance with device fabrication photographs. We also extend thanks to Dr. Leah A. Godwin for initiating the work in this area.
4 Notes
Convection ovens will provide more consistent results with photolithography or device curing, but a standard laboratory oven should suffice for these devices.
3D-printed templates and accessory devices mentioned in this chapter are currently available for free download from our laboratory’s profile on the Thingiverse website (http://www.thingiverse.com/; EasleyLab). Parts are available in STL format and are print-ready. Specifically, we printed these parts using a Makerbot Replicator 2, a desktop 3D printer that retails for less than $2000.
Photolithography can be achieved using a standard mask aligner and UV light source, if available. Most electrical engineering departments will have access to such a system. However, these systems can cost greater than $20,000 and require continued maintenance. Following the 2014 publication by Groisman et al. [16], we built a customized UV lithography source based on 365 nm LEDs, which cost less than $1000 and is essentially maintenance free. All SU-8 masters for microdevices used in the chapter were fabricated using this inexpensive UV lithography system.
Due to the photosensitivity of photoresist, all steps involving SU-8 should be carried out in minimal light or with UV filters over light fixtures. These steps are also preferably carried out in a clean environment with particulate filtered air, although a microelectronics quality cleanroom is not required.
Once the wafer is coated with SU-8, be mindful to not contact the wafer with the tweezers or any other objects in areas where channel designs will be patterned.
To avoid thermally compromising the SU-8/silicon adhesion and reduce bubble formation, it may be necessary to start the hot plate at a lower temperature then ramp slowly to the required baking temperature (e.g., 95°C). Cooling ramps will also help maintain the adhesion, thus rapid cooling should be avoided.
PDMS precursors can be cured at higher temperatures than 50°C and are often cured at 80°C in our laboratory and others. However, when using 3D-printed interface templates, the oven temperature must be kept below the glass transition temperature of the PLA material, which is around 60–65°C. This precaution will avoid warping of the 3D-printed template structure.
References
- 1.Lee JN, Park C, Whitesides GM. Solvent compatibility of poly(dimethylsiloxane)-based microfluidic devices. Anal Chem. 2003;75:6544–6554. doi: 10.1021/ac0346712. [DOI] [PubMed] [Google Scholar]
- 2.Whitesides GM, Ostuni E, Takayama S, et al. Soft lithography in biology and biochemistry. Annu Rev Biomed Eng. 2001;3:335–373. doi: 10.1146/annurev.bioeng.3.1.335. [DOI] [PubMed] [Google Scholar]
- 3.Easley CJ, Karlinsey JM, Landers JP. On-chip pressure injection for integration of infrared-mediated DNA amplification with electrophoretic separation. Lab Chip. 2006;6:601–610. doi: 10.1039/b600039h. [DOI] [PubMed] [Google Scholar]
- 4.Kim J, Johnson M, Hill P, Gale BK. Microfluidic sample preparation: cell lysis and nucleic acid purification. Integr Biol. 2009;1:574–586. doi: 10.1039/b905844c. [DOI] [PubMed] [Google Scholar]
- 5.Gao Y, Majumdar D, Jovanovic B, et al. A versatile valve-enabled microfluidic cell coculture platform and demonstration of its applications to neurobiology and cancer biology. Biomed Microdevices. 2011;13:539–548. doi: 10.1007/s10544-011-9523-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Halldorsson S, Lucumi E, Gómez-Sjöberg R, Fleming RMT. Advantages and challenges of microfluidic cell culture in polydimethylsiloxane devices. Biosens Bioelectron. 2015;63:218–231. doi: 10.1016/j.bios.2014.07.029. [DOI] [PubMed] [Google Scholar]
- 7.Bhatia SN, Ingber DE. Microfluidic organs-on-chips. Nat Biotechnol. 2014;32:760–772. doi: 10.1038/nbt.2989. [DOI] [PubMed] [Google Scholar]
- 8.Dishinger JF, Reid KR, Kennedy RT. Quantitative monitoring of insulin secretion from single islets of Langerhans in parallel on a microfluidic chip. Anal Chem. 2009;81:3119–3127. doi: 10.1021/ac900109t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Adewola AF, Wang Y, Harvat T, et al. A multi-parametric islet perifusion system within a microfluidic perifusion device. J Vis Exp. 2010;35:e1649. doi: 10.3791/1649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yi L, Wang X, Dhumpa R, et al. Integrated perfusion and separation systems for entrainment of insulin secretion from islets of Langerhans. Lab Chip. 2015;15:823–832. doi: 10.1039/c4lc01360c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Easley CJ, Rocheleau JV, Head WS, Piston DW. Quantitative measurement of zinc secretion from pancreatic islets with high temporal resolution using droplet-based microfluidics. Anal Chem. 2009;81:9086–9095. doi: 10.1021/ac9017692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Godwin LA, Pilkerton ME, Deal KS, et al. Passively operated microfluidic device for stimulation and secretion sampling of single pancreatic islets. Anal Chem. 2011;83:7166–7172. doi: 10.1021/ac201598b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Brooks JC, Ford KI, Holder DH, et al. Macro-to-micro interfacing to microfluidic channels using 3D-printed templates: application to time-resolved secretion sampling of endocrine tissue. Analyst. 2016 doi: 10.1039/C6AN01055E. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Moraes C, Labuz JM, Leung BM, et al. On being the right size: scaling effects in designing a human-on-a-chip. Integr Biol. 2013;5:1149–1161. doi: 10.1039/c3ib40040a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Godwin LA, Brooks JC, Hoepfner LD, et al. A microfluidic interface for the culture and sampling of adiponectin from primary adipocytes. Analyst. 2015;140:1019–1025. doi: 10.1039/c4an01725k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Erickstad M, Gutierrez E, Groisman A. A low-cost low-maintenance ultraviolet lithography light source based on light-emitting diodes. Lab Chip. 2015;15:57–61. doi: 10.1039/c4lc00472h. [DOI] [PubMed] [Google Scholar]