

Summary
Sodium ions are endogenous allosteric modulators of many G protein-coupled receptors (GPCRs). Mutation of key residues in the sodium binding motif causes a striking effect on G protein signaling. We report the crystal structures of agonist complexes for two variants in the first sodium coordination shell of the human A2A adenosine receptor (A2AAR), D522.50N and S913.39A. Both structures present an overall active-like conformation; however, the variants show key changes in the activation motif NPxxY. Changes in the hydrogen bonding network in this microswitch suggest a possible mechanism for modified G protein signaling and enhanced thermal stability. These structures, signaling data, and thermal stability analysis with a panel of pharmacological ligands provide a basis for understanding the role of the sodium-coordinating residues on stability and G protein signaling. Utilizing the D2.50N variant is a promising method for stabilizing class A GPCRs to accelerate structural efforts and drug discovery.
Keywords: GPCR, cell signaling, adenosine receptor, allosteric modulators, crystallography, structural biology, sodium binding
eTOC Blurb
White and Eddy et al. report agonist-bound structures of human A2AAR variants that disrupt allosteric sodium effects. The structures reveal changes in hydrogen bonding near a conserved activation motif that correspond to striking differences in signaling, providing a rational for increased variant receptor stability.
Introduction
Endogenous allosteric modulators of G protein-coupled receptors (GPCRs) are important effectors of receptor signaling and include lipids, ions, and intracellular proteins (Changeux and Christopoulos, 2016; van der Westhuizen et al., 2015). In particular, sodium ions have been proposed to be an endogenous allosteric modulator for many GPCRs (Katritch et al., 2014). Observations in mouse brain extracts over 40 years ago first demonstrated the allosteric effects of sodium on opioid receptors (Pert et al., 1973). Since those initial observations, sodium’s allosteric effects have been reported from in vitro and in vivo assays for over 15 GPCRs, including adrenergic receptors (Ceresa and Limbird, 1994), adenosine receptors (Gutierrez-de-Teran et al., 2013), dopamine receptors (Neve, 1991), and others as reviewed in the literature (Katritch et al., 2014).
Allosteric effects of sodium have been found to be largely mediated by the negatively charged residue D2.50 (Ceresa and Limbird, 1994; Fenalti et al., 2014; Katritch et al., 2014; Liu et al., 2012; Massink et al., 2015; Werling et al., 1986) located in transmembrane helix II (superscripts indicate the Ballesteros-Weinstein nomenclature (Ballesteros and Weinstein, 1995). D2.50 is one of the most highly conserved amino acids in class A GPCRs (Katritch et al., 2014). Replacement of D2.50 with a neutral amino acid diminished the ability of sodium to compete with agonist binding and concurrently affected G protein signaling for more than twenty-five different GPCRs (Hishinuma et al., 2017; Katritch et al., 2014). In the δ-opioid receptor (DOR), a D2.50 variant switched the efficacy of an antagonist to an arrestin-biased agonist (Fenalti et al., 2014). For the human A2A adenosine receptor (A2AAR), replacement of D2.50 with alanine completely abolished G protein-dependent signaling (Massink et al., 2015).
D2.50 forms the core of a cluster of residues that coordinate a sodium ion that was first observed in a high resolution crystal structure of A2AAR in complex with an antagonist (Liu et al., 2012) and subsequently observed in a DOR-antagonist complex (Fenalti et al., 2014). In those structures, other residues coordinating sodium are S3.39, which also directly coordinates sodium, and W6.48, N7.49, N7.45, and S7.46, which indirectly coordinate sodium through a network of water molecules (Figure 1). All of these sodium-coordinating residues are highly conserved among class A GPCRs (Katritch et al., 2014). In A2AAR, replacement of any of these sodium pocket residues with alanine altered agonist-induced G protein signaling (Massink et al., 2015).
Figure 1. Allosteric sodium binding pocket and sodium coordinating residues in the 1.8 Å A2AAR crystal structure.

(a) Side view of the overall crystal structure of A2AAR in complex with the antagonist ZM241385. A2AAR is shown as cartoon representation and ZM241385 is shown in blue stick representation (PDB ID 4EIY) (Liu et al., 2012). Water molecules are shown as red spheres and sodium is shown as a blue sphere. (b) Expansion of the allosteric sodium pocket. Sodium is shown near the center as a blue sphere and water molecules are shown as red spheres. Residues coordinating with the sodium ion or nearby water molecules are labeled. Dashed lines indicate polar contacts and helices are labeled with Roman numerals.
Sodium-coordinating residues also strongly influence thermal unfolding temperatures of GPCRs. Replacement of one or more of the sodium coordinating residues increased denaturation temperatures for several GPCRs, including the neurotensin receptor (NTSR1) (Shibata et al., 2009), κ-opioid receptor (KOR) (Schutz et al., 2016), and α1a adrenergic receptor (α1aAR) (Dodevski and Pluckthun, 2011). For the purinergic P2Y1 receptor (P2Y1R), replacement of an endogenous D7.49 with asparagine improved protein expression yield and was required for crystallization of a P2Y1R-antagonist complex (Zhang et al., 2015a). Recently, observations in A2AAR by nuclear magnetic resonance (NMR) spectroscopy showed that replacement of D2.50 with asparagine drastically altered conformational dynamics at the A2AAR intracellular surface without altering the receptor’s conformation at the extracellular surface (Reference to Eddy et al. paper under review).
Motivated by these numerous observations, we sought to provide a structural basis for how the sodium-coordinating residues regulate G protein signaling and affect denaturation temperatures of human A2AAR. A2AAR is a validated drug target for Parkinson’s Disease (Cieslak et al., 2008), chronic obstructive pulmonary disease (Bonneau et al., 2006), and a promising target for cancer co-immunotherapies (Leone et al., 2015). Thus, the role of the allosteric sodium site in A2AAR drug discovery is of relevant interest. We determined crystal structures of A2AAR variants D2.50N and S3.39A in complex with the full agonist UK432097 at 2.6 Å and 2.9 Å resolution, respectively (Figure 2). Comparison of the structures was complemented by protein thermal stability and cAMP signaling assays, which showed significant differences among the A2AAR variants. As the residues comprising the allosteric sodium pocket are highly conserved, these data likely provide fresh insights into regulation of G protein signaling by allosteric sodium coordinating residues for many class A GPCRs and provide new tools for GPCR drug discovery. Our results suggest utility for the D2.50N variant as an exciting approach for accelerated crystallization and drug screening efforts for class A GPCRs.
Figure 2. Functional and structural comparisons of A2AAR with the variants A2AAR–D52N and A2AAR–S91A.

(a) UK432097 competition binding with [3H] CGS21680 and (b) cAMP accumulation upon stimulation with UK432097. Data in (a) and (b) are shown as means ± S.D. Experiments were conducted three times, each in triplicate. A2AAR is shown in grey, A2AAR–D52N is shown in green, and A2AAR–S91A is shown in blue. (c–e) Superposition of side views of the overall structure of D52N–UK432097 (green) with (c) A2AAR–ZM241385 (yellow; PDB ID 3EML), (d) A2AAR–UK432097 (grey; PDB ID 3QAK), and (e) S91A–UK432097 (blue). The dashed line between helices V and VI indicates the position of the T4L fusion protein. Ligands are colored as follows: ZM241385 in orange, UK432097 bound to D52N–UK432097 in purple, UK432097 bound to A2AAR in cyan, and UK432097 bound to S91A–UK432097 in yellow. (f–h) Intracellular views of D52N–UK432097 superimposed on (f) A2AAR–ZM241385, (g) A2AAR–UK432097, and (h) S91A–UK432097; with the same coloring scheme used in panels (c–e). Helices are numbered with Roman numerals. In (f), arrows indicate rearragnements in the positions of helices at the intracellular surface between A2AAR–ZM241385 and D52N–UK432097. See also Figures S1 and S2 and Table 2.
Results
Functional characterization of A2AAR sodium-site variants
In the high-resolution crystal structure of antagonist-bound A2AAR, D522.50 and S913.39 were the only two residues observed to directly coordinate the sodium ion (Figure 1) (Liu et al., 2012). Thus, we focused on variants of these two residues in the current study. We measured ligand binding with A2AAR and the variants A2AAR–D52N and A2AAR–S91A with the antagonist ZM241385 and agonist NECA (Table 1). A2AAR, A2AAR–D52N, and A2AAR–S91A all showed similar affinity for the antagonist ZM241385 and agonists CGS21680 and UK432097 (Table 1 and Figure 2a). Affinity of the agonist NECA was slightly higher for the A2AAR–D52N variant (KD: 9.16 nM, logKD: −8.03 ± 0.10) relative to A2AAR (KD: 23.4 nM, logKD: −7.63 ± 0.11) and A2AAR–S91A (KD: 132 nM, logKD: −6.88 ± 0.08). To investigate the allosteric role of sodium on agonist binding, we performed the same binding assays in the presence of a physiologically-relevant sodium concentration (150 mM). The presence of sodium decreased the affinity of the agonist NECA for A2AAR, and had no effect on agonist affinity for A2AAR–D52N and A2AAR–S91A (Table 1).
Table 1.
| Receptor | KD [3H] ZM241385 (nM)/logKD |
KD [3H] NECA 0 mM NaCl (nM)/logKD |
KD [3H] NECA 150 mM NaCl (nM)/logKD |
KD [3H] CGS21680 (nM) |
Ki UK432097 (nM) |
EC50 UK432097 (nM) |
|---|---|---|---|---|---|---|
| A2AAR (WT) | 7.69 nM −8.11 ± 0.06 | 23.4 nM −7.63 ± 0.11 | 290 nM −6.5 ± 0.14 | 13.6 ± 3.1 nM | 17.3 ± 5.9 nM | 0.45 ± 0.08 nM |
| A2AAR–D52N | 9.63 nM −8.02 ± 0.05 | 9.16 nM −8.03 ± 0.10 | 10.4 nM −7.98 ± 0.11 | 7.35 ± 0.30 nM | 22.4 ± 2.5 nM | No Signaling |
| A2AAR–S91A | 7.0 ± 0.2 nM* | 132 nM −6.88 ± 0.08 | 138 nM −6.86 ± 0.07 | 17.3 ± 6.12 nM | 14.7 ± 0.7 nM | 0.75 ± 0.09 nM |
| A2AAR–D52N–T4L | ND | ND | ND | 12.0 ± 3.6 nM | 25.2 ± 3.5 nM | ND |
| A2AAR–S91A–T4L | ND | ND | ND | 10.0 ± 4.9 nM | 20.0 ± 3.7 nM | ND |
Radioligand binding and G protein signaling effects of A2AAR variants. ZM241385 and NECA KD measurements were done via homologous competition, and CGS21680 KD measurement was done using saturation binding. KD, Ki, and EC50 values were calculated in Prism 7.0 software (GraphPAD, San Diego, CA) and data is displayed as means ± S.D. and 3 experiments were conducted in triplicate. Error is shown for either nM or logKD depending on assay method as detailed in the methods section.
Denotes data previously published11.
ND – not determined.
We examined G protein signaling for the A2AAR variants by measuring cAMP accumulation via receptor activation upon stimulation with the agonist UK432097. A2AAR–D52N evidenced no cAMP accumulation upon UK432097 stimulation, similar to what was observed with A2AAR–D52A in an earlier report (Massink et al., 2015), while A2AAR–S91A had similar activity to A2AAR (Figure 2b and Table 1). While A2AAR–S91A signaling was previously found to have higher basal and lower Emax signaling upon CGS21680 agonist stimulation (Massink et al., 2015), differences reported here may be due to using different cell types, agonists, and assay conditions (for example, adenosine deaminase was included in our experiments to eliminate the influence of endogenous adenosine).
Crystal structures of A2AAR sodium-site variants in complex with an agonist present an overall active-like conformation
We determined the crystal structures of A2AAR–D52N and A2AAR–S91A in complex with the full agonist UK432097. To compare these structures with the previously published crystal structure of A2AAR in complex with the agonist UK432097 (A2AAR–UK432097; PDB ID 3QAK)(Xu et al., 2011), we employed a nearly identical construct, where the A2AAR third intracellular loop (ICL3) was replaced with T4-lysozyme to facilitate crystallization(Xu et al., 2011). The only change effected to this earlier construct was the single amino acid replacement D52N or S91A. To ensure the crystallization constructs have similar binding affinities as A2AAR and A2AAR variants without the T4L ICL3 fusion we determined the affinity of the agonists CGS21680 and UK432097 prior to crystallization. A2AAR, A2AAR–D52N–T4L, and A2AAR–S91A–T4L all had very similar affinities (KD) for CGS21680, 13.6 ± 3.1 nM, 12.0 ± 3.6 nM, and 10.0 ± 4.9 nM, respectively (Table 1). The agonist UK432097, co-crystallized in complex with each construct, also showed similar affinities (Ki) for A2AAR, A2AAR–D52N–T4L, A2AAR–S91A–T4L, 17.3 ± 5.9 nM, 25.2 ± 3.5 nM, and 20.0 ± 3.7 nM, respectively (Table 1).
Crystallization of A2AAR–D52N–T4L and A2AAR–S91A–T4L bound to the agonist UK432097 was carried out in lipid cubic phase (LCP) (Caffrey and Cherezov, 2009) (Figure S1), and the structures were refined to a final resolution of 2.6 Å and 2.9Å, respectively (Figure 2c–h). Direct structural comparisons were made only for constructs that have this identical T4L fusion partner for consistency and to remove the possibility of introducing bias by using different fusion partners. Throughout the manuscript, we refer to the co-crystal structures of UK432097 bound to A2AAR–D52N–T4L and A2AAR–S91A–T4L as D52N–UK432097 and S91A–UK432097, respectively.
Comparing the structure of the agonist-bound D52N–UK432097 complex with the previously published structures of A2AAR–T4L in complex with the antagonist ZM241385 (A2AAR–ZM241385; PDB ID 3EML) (Jaakola et al., 2008) and A22AAR–T4L in complex with the agonist UK432097 (A2AAR–UK432097; PDB ID 3QAK)20 showed that the overall structure of the variant protein presented hallmarks of an active-like state (Figure 2c–d). Specifically, the positions of helices at the intracellular surface of the D52N–UK432097 complex were displaced from the antagonist bound structure in the same directions expected for an active-like conformation. The alignment of D52N–UK432097 and A2AAR–ZM241385 was performed after removal of fusion partner atoms and the root mean square deviation (RMSD) was 1.33 Å (264 aligned Cα atoms). Furthermore, the superposition of D52N–UK432097 and A2AAR–UK432097 revealed an overall strong agreement between the two structures with an RMSD of 0.32 Å (263 aligned Cα atoms). Residues in the ligand binding pocket and the binding pose of UK432097 appeared to be nearly identical between A2AAR–UK432097 and D52N–UK432097 agonist complexes (Figure 2d and Figure S2a–b), indicating the D52N replacement did not interfere with the conformation of the agonist in the binding pocket.
We also solved a crystal structure of the variant A2AAR–S91A in complex with the agonist UK432097 using the same T4L fusion construct design, referred to as S91A–UK432097. Crystallization of S91A–UK432097 was also performed in LCP (Caffrey and Cherezov, 2009) and the structure was refined to 2.9 Å. The S91A–UK432097 also showed active-like features that were very similar to both D52N–UK432097 (RMSD 0.44 Å, 263 aligned Cα atoms) (Figure 2e) and A2AAR–UK432097 (RMSD 0.46 Å, 261 aligned Cα atoms). The density maps of D52N–UK432097 and S91A–UK432097 are shown in Figure S2c–d.
Residues in the NPxxY motif of D52N–UK432097 are neither in an entirely inactive or active-like conformation
The relative side chain orientations in GPCR microswitches are sensitive to differences of the pharmacological efficacies of bound ligands (Katritch et al., 2012). Rearrangements in side chains of the microswitches are thought to precede larger structural rearrangements of helical backbones required for the receptor to recognize intracellular partner proteins. We compared the structures of antagonist bound A2AAR–ZM241385 (PDB ID 3EML), agonist bound A2AAR–UK432097 (PDB ID 3QAK), and agonist bound D52N-UK432097 in four conserved structural motifs: the PIF motif formed between helices III, V, and VI; the CWxP motif in helix VI; the DRY motif in helix III, and the NPxxY motif in helix VII (Figure 3). Strong electron densities in the microswitch regions of D52N–UK432097 (Figure S2c) allowed us to confidently compare backbone and side chain orientations for residues in these regions. We observed strong overlap between A2AAR–UK432097 and D52N–UK432097 for three microswitch regions: PIF, CWxP, and DRY. Because there was strong agreement in these regions between the A2AAR–UK432097 and D52N-UK432097 (Figure 3a–c), we concluded that side chain orientations for D52N–UK432097 were in an active-like conformation.
Figure 3. Residues in the NPxxY motif of D52N–UK432097 are neither in an entirely inactive or active-like conformation.

(a–d) Four conserved structural motifs (i.e. microswitches) labeled according to the amino acid types found in each motif. Within each panel, a superposition of A2AAR–ZM241385 (yellow; PDB ID 3EML) with D52N–UK432097 (green) is on the left, and a superposition of A2AAR–UK432097 (grey; PDB ID 3QAK) with D52N–UK432097 (green) is on the right. Amino acids comprising each structural motif are shown in stick representation and labeled by the Ballesteros-Weinstein nomenclature. Positions of the microswitches in the global structures are indicated by boxes in the middle superposition of all three structures. The location of residue D52N is indicated in the global view of the structure and labeled. Additionally, the binding sites of the orthosteric ligand (UK432097, cyan sticks), sodium (blue sphere), and G protein are shown in the center composite panel, where a surface representation of the mini Gαs and its contacts with A2AAR from the previously published A2AAR–NECA–mini Gαs complex structure (PDB ID 5G35) (Carpenter et al., 2016) are shown. See also Figure S2.
Comparing the RMSD in the positions of Cα atoms between the structures of A2AAR–UK432097 and D52N–UK432097 revealed that the largest differences occurred for residues in the NPxxY motif in helix VII and for residues immediately preceding this motif (Figure 4a). The structural rearrangement begins at S2817.46, which has an altered Cα position in D52N–UK432097 relative to A2AAR–UK432097 of 2.4 Å. Furthermore, the Cα positions for several subsequent residues are also altered: V2827.47 by 2.9 Å, V2837.48 by 2.2 Å, N2847.49 by 1.5 Å, and P2857.50 by 1.1 Å. D52N–UK432097 residues S2817.46, V2827.47, and V2837.48 have side chain conformations that resemble those for the antagonist-bound receptor A2AAR–ZM241385 (Figure 4b–c), and residues N2847.49 and P2857.50 are neither in inactive or active-like conformations. At the end of the NPxxY motif, F2867.51 through Y2887.53, we again observe a strong agreement between the agonist-bound structures, A2AAR–UK432097 and D52N–UK432097, that extends through the conformations of the remaining residues from the intracellular surface to the C-terminus (Figure 4a).
Figure 4. Comparison of the positions of Cα atoms between A2AAR–UK432097, D52N–UK432097, and A2AAR–ZM241385.

All three structures were aligned globally and the root-mean-square deviation (RMSD) was calculated between Cα atoms of the same residue number between (a) A2AAR–UK432097 and D52N–UK432097 and (b) A2AAR–UK432097 and A2AAR–ZM241385, and (c) D52N–UK432097 and A2AAR–ZM241385. Residue number is plotted on the horizontal axis and RMSD is plotted on the vertical axis. The observed RMSDs are colored according to the legends in the figure panels. Roman numerals indicate helix numbers, blue shading indicates positions of intracellular loops, and red shading indicates positions of extracellular loops.
The structural rearrangement near the NPxxY motif of D52N–UK432097 was observed in the difference electron density maps (|Fo|-|Fc|) between D52N–UK432097 and A2AAR–UK432097 (Figure S2e). The difference electron density maps show clear rearrangements near S2817.46, P2857.50, and the site of mutation N522.50 (Figure S2e). Similar differences were observed in the |Fo|-|Fc| maps using the molecular replacement (MR) solution with A2AAR–UK432097 as a search model. We observe negative electron densities at S2817.46, and positive electron densities near V2827.47, V2837.48, N2847.49, and P2857.50 consistent with our final D52N–UK432097 structure (Figure S2f). Additionally, we solved the structure of D52N–UK432097 using A2AAR–UK432097 or A2AAR–ZM241385 as search models, and obtained the same results, confirming that the structural rearrangements near the NPxxY motif are not due to phase bias.
The two most significant differences between the D52N–UK432097 and S91A–UK432097 structures is the rotamer position of S2817.46 as well as differences in Cα position of residues in the NPxxY motif (Figure S2a–d). Because the electron density maps were more ambiguous in the NPxxY motif for the S91A–UK432097 structure (Figure S2c–d), we limited detailed comparisons of side chain conformations and rotameric states to the A2AAR– ZM241385, A2AAR–UK432097, and D52N–UK432097 complexes.
Changes in the hydrogen bonding network observed among antagonist-bound A2AAR–ZM241385, agonist-bound A2AAR–UK432097, and agonist-bound D52N–UK432097 structures
To gain insight into the effects of the D52N amino acid replacement on helix-helix interactions, we investigated the hydrogen bond network between helix VII and neighboring helices I, II, III, and VI (Figure 5 and Figure S3), near the site of structural rearrangement in the D52N–UK432097 structure discussed in the above text. In the antagonist-bound structure of A2AAR–ZM241385 (PDB ID 3EML) (Jaakola et al., 2008), S2817.46 formed hydrogen bonds between N241.50 and D522.50. In the agonist-bound conformation, A2AAR–UK432097 (PDB ID 3QAK), a change in the sidechain rotamer of S2817.46 caused a loss of these hydrogen bonds, while additional hydrogen bonds are formed between N2807.45 and W2466.48, and between D522.50 and S913.39 (Figure 5 and Figure S3). By comparison, the overall hydrogen bond network in this region of the agonist-bound variant D52N–UK432097 is more similar to the antagonist-bound A2AAR–ZM241385 inactive state structure than the agonist-bound A2AAR–UK432097. In D52N–UK432097, S2817.46 formed a hydrogen bond with N241.50 and formed two hydrogen bonds between N522.50. N522.50 formed a hydrogen bond with S913.39; however, the hydrogen bond between N2807.45 and W2466.48 observed in the agonist-bound A2AAR-UK432097 is absent in the agonist-bound variant D52N–UK432097. These results detail consequences of the N522.50 replacement on structural rearrangement in helix VII.
Figure 5. Comparison of hydrogen bonding networks in A2AAR–ZM241385, A2AAR–UK432097, and D52N–UK432097.

A schematic is presented of cross sectional views of helices I–III, VI, and VII, indicated by circles with Roman numerals. Amino acids involved in hydrogen bonding between helices are indicated by colored dots on each circle and labeled. Dashed lines between helices indicate the presence of a hydrogen bond, and the thickness of the dashed line indicates the number of hydrogen bonds (i.e., two hydrogen bonds between N52 and S281 are observed in D52-UK432097 and only one is observed in A2AAR-ZM241385). See also Figures S3 and S4.
Analysis of A2AAR thermal denaturation temperatures corroborates prominent effects of D52 and influence of sodium on agonist complexes
We measured thermal denaturation temperatures (Tm) for A2AAR and A2AAR variants in complex with various ligands using a thiol-specific fluorochrome (CPM). This dye produces a signal when it conjugates to cysteine residues buried in the receptor transmembrane helices that become exposed to an aqueous environment upon heating the protein (Alexandrov et al., 2008). To investigate the effect of the sodium allosteric site on receptor melting temperatures, we included five A2AAR variants in the thermal denaturation experiments. Purified A2AAR–BRIL reconstituted into DDM/CHS mixed micelles was added to a panel of ligands at saturating concentrations (25 µM for UK432097 and 100 µM for all other ligands) or no ligand added (apo) all in the presence of 150 mM NaCl. In these conditions, A2AAR–D52N showed the highest denaturation temperature for all but one ligand complex (A2AAR–N284A in complex with UK432097 showed the overall highest Tm of nearly 75 °C) (Table S1). The most striking effect was for apo A2AAR–D52N, which showed a nearly 8 °C increase in the Tm compared to apo A2AAR (Figure 6a). Notably, for the variant A2AAR–D52A, we observed a melting temperature that was slightly lower than A2AAR and markedly lower than A2AAR–D52N for all ligands studied. This striking difference can be rationalized by the loss of hydrogen bonds in A2AAR–D52A, which are observed in A2AAR–D52N between N522.50 and residues in helices II, III, and VII (Figure 5 and Figure S3). The measured Tm values for Figure 6a are given in Table S1.
Figure 6. Thermal stability and function of A2AAR and allosteric site variants.

(a) Fluorescence thermal shift assays for A2AAR-BRIL and variants in complex with 5 different ligands and for no ligand added (apo). The mean changes in melting temperature (ΔTm) relative to A2AAR are displayed with error bars representing S.E.M. (n = 3) and performed at 150 mM NaCl. Tm values are listed in Table S1 and statistical analysis shown in Figure S5. (b) Tm values calculated from fluorescence thermal shift assays for A2AAR (without a fusion partner protein) for complexes with 10 ligands and apo. Ligands are identified below each column and the assay was performed at 75 mM NaCl. (c) Tm values for A2AAR-BRIL complexes with ligands in the absence (solid bars) and presence (checkered bars) of sodium (150 mM or 500 mM), as indicated. See also Figure S6.
There was a statistically significant differences in Tm between receptor variants for each ligand condition tested as determined by one-way ANOVA: Apo [F(5,17) = 14.18, P < 0.0001], ZM241385 [F(5,16) = 5.724, P = 0.0033], UK432097 [F(5,13) = 139.4, P < 0.0001], NECA [F(5,14) = 26.12, P < 0.0001], Theophylline [F(5,14) = 4.485, P = 0.0119]. Dunnett’s multiple comparison tests revealed that the D522.50N variant is significantly different from A2AAR for Apo (P < 0.001), ZM241385 (P < 0.05), UK432097 (P < 0.01), and NECA (P < 0.05) (Figure S5).
Additionally, we recorded thermal denaturation experiments for A2AAR in complex with a larger selection of ligands at a lower sodium concentration of 75 mM (Figure 6b), and in the absence and presence of sodium at variable concentrations (Figure 6c) to investigate the effect of different sodium concentrations on receptor stability. At 75 mM NaCl, the highest melting temperatures were observed for complexes with either full or partial agonists, which were six to ten degrees higher than melting temperatures reported for A2AAR in complex with the same or similar agonists and partial agonists in the presence of 800 mM NaCl (Xu et al., 2011). Melting temperatures for A2AAR in complex with antagonists were similar to previously reported melting temperatures for A2AAR antagonist complexes in the presence of 800 mM NaCl (Jaakola et al., 2008; Xu et al., 2011). Experiments using variable sodium concentrations confirmed the sensitivity of agonist complexes to sodium and indicated a greater effect of sodium on agonist complexes than antagonist complexes. The melting temperatures of A2AAR in complex with the full agonist UK432097 were 77.7 °C, 73.2 °C, and 69.7 °C at 0, 150, and 500 mM NaCl, respectively (Figure 6c). This effect was not observed when using 150 or 500 mM choline chloride (Figure S6). This trend of sodium on A2AAR melting temperatures may reflect changes in rates of ligand association or dissociation specific to agonist complexes (see Discussion).
Discussion
The combination of structural, biophysical, and pharmacological data presented here provide a basis to delineate the role of the D2.50N mutation on A2AAR thermal stability, the structure of the NPxxY activation motif, and inhibition of G protein signaling. Additionally, the D2.50N mutation desensitizes the response of A2AAR agonist binding to increasing sodium concentration, which has been documented in other receptors including DOR (Fenalti et al., 2014). We propose that this sodium desensitization and loss of G protein signaling are manifested by D2.50N, and that loss of G protein signaling is due to subtle changes in interhelical packing facilitated by specific interactions with N2.50.
Local conformational changes observed in the NPxxY motif for D52N–UK432097 suggest coupling between the NPxxY motif and position D2.50 that may contribute to changes in signaling for the variant protein. It has been proposed that maintaining the structural integrity of the NPxxY motif in native class A receptors is critical for signal propagation from the orthosteric ligand binding site to the intracellular surface (Fritze et al., 2003; Hulme, 2013; Trzaskowski et al., 2012). In earlier studies, replacement of residues in the NPxxY motif result in apparent attenuation or complete loss of G protein signaling (Borroto-Escuela et al., 2011; Bouley et al., 2003; Galés et al., 2000; Govaerts et al., 2001; Lu et al., 2001; Raitio et al., 2005). Replacement of N7.49 with alanine did not significantly perturb ligand binding affinities but caused to a striking loss of G protein signaling for a number of receptors. In the CB2 cannabinoid receptor, replacement of S7.46 with alanine causes of loss of agonist-induced signaling for some ligands (Raitio et al., 2005). Thus, differences in signaling for the D52N–UK432097 complex and local structural rearrangements in the NPxxY motif and residues immediately preceding appear consistent with mutational studies that disrupted the same region. It has been proposed that the NPxxY region facilitates signaling through changes in the network of hydrogen-bonded water molecules (Pardo et al., 2007), which may rationalize the required structural integrity of this region.
Comparison of the agonist-bound variant D52N–UK432097 crystal structure with the crystal structures of A2AAR–UK432097 (PDB ID 3QAK) and A2AAR–ZM241385 (PDB ID 3EML) provides fresh insight into the mechanism of how agonist binding at the orthosteric ligand cavity leads to conformational changes and interactions of helices that lead to receptor activation. Starting at the orthosteric binding pocket, the ribose moiety of agonist UK432097 makes unique contacts with S2777.42 and H2787.43 in helix VII. As this ribose moiety is characteristic of all A2AAR full agonists, structures of A2AAR in complex with other agonists also revealed the same critical contacts between the agonists and helix VII (Lebon et al., 2011b). While important agonist-receptor interactions occur at helix VII, a recent structure of an A2AAR ternary complex with an agonist, NECA, and engineered G protein revealed a larger outward rearrangement of helix VI at the intracellular surface (Carpenter et al., 2016). From these observations, we propose that a key step in signal propagation from the orthosteric site involves interactions between helices VI and VII near the orthosteric pocket and the NPxxY activation motif (Figure 7a–b). Though additional interactions are likely also involved in activation, these observations are intriguing in the context of analyzing patterns of interaction networks among class A GPCRs (Venkatakrishnan et al., 2016; Venkatakrishnan et al., 2013). For the D52N–UK432097 agonist complex, loss of interactions between helices VI and VII and concurrent gain of interactions between helices VII and II are correlated with a loss of G protein signaling and increased thermal stability (Figure 5 and Figure 7d). Additionally, the protonation state of D2.50 may be involved in A2AAR signaling, as was suggested by recent analysis of crystal structures of class A GPCR-ligand complexes and computational modeling (Vickery O, Carvalheda C, Zaidi SA, Pisliakov A, Katritch V, Zachariae U. Intracellular Passage of Na+ In An Active State G Protein Coupled Receptor. Structure. 2017 in press). This study suggests for inactive conformations, D2.50 is negatively charged and forms coordinative bonds with sodium, while agonist-binding facilitates D2.50 protonation and intracellular transfer of sodium.
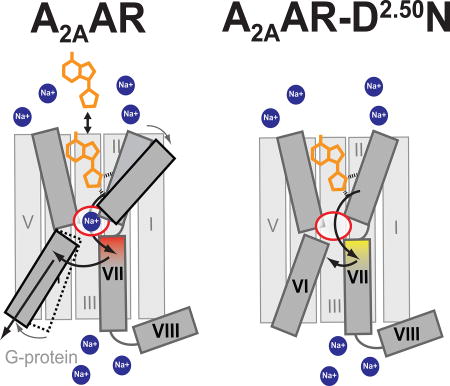
Figure 7. Schematic of effects of a range of sodium concentrations on A2AAR agonist recognition and binding and proposed mechanism of D52N mutation on G protein signaling.

A2AAR is shown in grey in complex with an agonist (orange) in the presence of (a) low sodium concentration (< 150 mM), (b) physiologically-relevant sodium concentration (150 mM), and (c) high sodium concentration (> 150 mM). The allosteric sodium pocket is shown as a red oval in the center of the protein. Sodium ions are shown as blue circles. (a) Highlights a hypothetical pathway of signal transduction from the orthosteric ligand binding pocket through the NPxxY motif in helix VII, shaded in red, to the intracellular surface of helix VI. (a–c) Highlights the allosteric effect of sodium on agonist binding. (d) A2AAR–D52N in complex with an agonist and is not affected by sodium concentration. The NPxxY motif is shaded in yellow, reflecting the structural changes observed in the crystal structure of D52N–UK432097, and arrows indicate incomplete transfer of signal from the orthosteric ligand binding pocket.
The schematic in Figure 7 summarizes experimental conditions from the literature (Xu et al., 2011) and current study, and highlights the effects of a spectrum of sodium concentrations on A2AAR agonist binding. The extreme boundaries of sodium concentrations provide a mechanistic understanding of A2AAR function that will inform future experimental design (Figure 7a–c). At a lower sodium concentration of 75 mM NaCl, all A2AAR complexes with either partial or full agonists had a higher melting temperature than almost all antagonist complexes, even for the antagonist ZM241385 that has been co-crystallized with A2AAR (Figure 6b). Furthermore, A2AAR in complex with UK432097 showed a trend of decreasing melting temperatures with increasing concentration of sodium (150 mM and higher) (Figure 6c). Typically, antagonists are thought to have a more stabilizing effect on receptors than agonists (Chun et al., 2012; Zhang et al., 2015b), but our data suggests that may only be the case at sodium concentrations higher than physiologically relevant levels (≥150 mM NaCl).
From these collective observations, we propose that the collapsed sodium pocket observed in A2AAR agonist-bound states facilitates stronger helix-helix interactions at the intersection between helices II, III, VI, and VII. Coordination of sodium at this intersection maintains these interactions but decreases their strength. Additionally, the coordination of sodium at this site possibly modulates the conformation of the orthosteric pocket in subtle ways that could result in suboptimal interactions between agonists and receptor (Figure 7c). The notion of suboptimal agonist-receptor interactions is supported by the observation of decreased ligand binding affinities for agonists, but not antagonists, in the presence of physiological concentrations of sodium (150 mM) (Katritch et al., 2014; Pert et al., 1973). This was also observed for the agonist NECA binding to A2AAR, but not for variants A2AAR–D52N and A2AAR–S91A (Table 1).
The data reported here suggest an important utility of the D2.50N mutation in GPCR construct design for drug discovery and biophysical assays that has not yet been explored fully. The D2.50N single point mutation increases the Tm of all A2AAR ligand complexes, and there is an especially large increase in the Tm for the apo receptor (Figure 6a). While A2AAR–D52N exhibits no G protein signaling (Figure 2b), the conformations of residues in the ligand binding pocket and binding pose of the bound agonist are identical to A2AAR–UK432097. Also, ligand binding affinities of antagonist ZM241385 and agonist UK432097 were nearly identical between A2AAR and A2AAR–D52N (Table 1). Because D2.50 is one of the most highly conserved amino acids among all class A GPCRs (Katritch et al., 2014), introducing a single point mutation at this location may provide a rapid and straightforward means for improving the overall stability and expression of many GPCRs and may accelerate the design of GPCR constructs for crystallization trials. Further, this method may also improve efforts for fragment-based screening of small molecules that target GPCRs, for example by NMR (Chen et al., 2012) or surface plasmon resonance (SPR) (Shepherd et al., 2014), because these methods rely on immobilized GPCRs that have relatively higher denaturation temperatures.
Replacement of D2.50 has been shown to be beneficial to the stabilization of both the κ-opioid receptor (KOR) and neurotensin receptor 1 (NTSR1) in a recent study utilizing the directed evolution approach of GPCR stabilization (Schutz et al., 2016). It is notable that the directed evolution approach utilized no prior knowledge about which mutations could potentially be beneficial; however evolved constructs for both KOR and NTSR1 included substitutions at D2.50. Other methods of stabilizing GPCRs, including systematic alanine scanning (Lebon et al., 2011a) or the directed evolution approach (Schutz et al., 2016), have been essential for structure determination of unique GPCRs. However, such methods can be time and resource consuming and may stabilize receptor interactions with ligands of specific chemical or pharmacological properties, which can limit experimental probing with multiple ligands. Here we propose an alternative approach of a single mutant, D2.50N, which significantly boosts receptor stability, increases the melting temperature of apo GPCRs and GPCR-ligand complexes, and maintains ligand binding affinities similar to the native receptor. Thus, utilization of the D2.50N mutation across the spectrum of class A GPCRs may improve the robustness of screening platforms and accelerate drug discovery.
STAR METHODS
CONTACT FOR REAGENTS AND RESOURCE SHARING
Further information and requests for reagents may be directed to, and will be fulfilled by the Lead Author, Dr. Raymond C. Stevens (stevens@usc.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Microbes
E. coli cells were cultured in LB medium, and P. pastoris cells were cultured in BMGY and BMMY media.
Cell Lines
Sf9 cells were cultured in ESF 921 media. HEK cells were cultured in DMEM media. Insect and mammalian cells are periodically tested for mycoplasma using MycoAlert™ Mycoplasma Detection kit (Lonza). The cell lines used were authenticated by suppliers and were chosen to remain consistent with previous studies and they are well validated for these assays.
METHODS DETAILS
Construct Design and Cloning
All constructs produced in both Sf9 and Pichia pastoris used in this study were truncated after residue 316 and contained an N-terminal FLAG tag and C-terminal 10 X His tag. For A2AAR produced in Pichia pastoris, the glycosylation site N154 was replaced with Q. A2AARBRIL was identical to a previously published construct (Chun et al., 2012; Liu et al., 2012) where residues 1–106 of the thermostabilized apocytochrome b562 from E. coli (M7W, H102I, K106L) were inserted between K209 to G218 in the third intracellular loop of A2AAR, and variants of this construct were generated by single amino acid replacement. A2AAR-T4L was identical to a previously published construct used to determine the crystal structure of A2AAR in complex with UK432097 (Xu et al., 2011), where A2AAR residues K209-A221 were replaced by a cysteine-free variant of bacteriophage T4 lysozyme (C54T, C97A). PCR-based site-directed mutagenesis (QuickChange II, Stratagene, CA) was used to generate all variants, and primers used for site-directed mutagenesis were obtained from Integrated DNA Technology (San Diego, CA).
Protein Production and Purification
High-titer recombinant baculovirus were generated with the Bac-to-Bac system (Invitrogen) and used to infect Sf9 cells (ATCC). Insect cell membranes were disrupted in hypotonic buffer (10 mM HEPES pH 7.0, 10 mM MgCl2, 20 mM KCl, and protease inhibitors) and then washed in high osmotic buffer (10 mM HEPES pH 7.0, 10 mM MgCl2, 20 mM KCl, 1 M NaCl, protease inhibitors).
Isolated Sf9 membranes were resuspended in buffer with 2.0 mg/ml iodoacetamide (Sigma), 4 mM theophylline (Sigma), and a 1:1000 v/v protease inhibitor solution (500 µM 4-benzenesulfonyl fluoride hydrochloride AEBSF, 1 µM E-64, 1 µM Leupeptin, 150 nM Aprotinin). Protein was extracted from membranes in buffer containing 0.5% w/v n-Dodecyl β-D-maltoside (DDM) (Anatrace) and 0.1% cholesteryl hemisuccinate (CHS) (Sigma) for 3 hours at 4°C, and unsolubilized material was separated by centrifugation at 150,000×g for 30 minutes. The supernatant was incubated overnight at 4°C with Co2+-charged immobilized metal resin (TALON; Clontech). After washing with 20 column volumes of buffer 1 (50 mM HEPES at pH 7.0, 500 mM NaCl, 10 mM MgCl2, 1 mM DDM, 0.2 mM CHS, 30 mM imidazole, 8 mM ATP), the protein-bound IMAC resin was washed with buffer 2 (50 mM HEPES at pH 7.0, 250 mM NaCl, 0.5 mM DDM, 0.1 mM CHS, 30 mM imidazole, 27 mM glycerol) and protein was eluted with buffer 2 containing 250 mM imidazole. For crystallization, the agonist UK432097 was added to buffers 1, 2 and elution buffer at a final concentration of 25 µM. Protein purity and approximate protein concentration were determined by analytical size-exclusion chromatography.
For Pichia pastoris-produced A2AAR (1–316), the gene was cloned into a pPIC9K vector (Invitrogen) at BamHI and NotI restriction sites. The construct was transformed via electroporation into the Bg12 strain (BioGrammatics, Carlsbad, CA). Protein purification of A2AAR produced from Pichia pastoris was performed identically to Sf9-produced protein with the exception of a slightly longer solubilization period of approximately 4 hours. Protein purified from Pichia pastoris was used for the thermal stability assays (Figure 6b).
For probing the effect of monovalent cations on receptor thermal stability, sodium chloride was replaced in all buffers with the same concentration of choline chloride or potassium chloride, as indicated in the text.
Theophylline (Sigma) was dissolved in deionized water at a final concentration of 100 mM. Stock solutions of other ligands were prepared for UK432097 (25 mM; Axon Medchem, VA), ZM241385 (100 mM; Tocris), and NECA (50 mM; Tocris).
Fluorescent Thermal Shift Assays - CPM Assays
Thermal shift assays were carried out as described previously (Alexandrov et al., 2008). N-[4-(7-diethylamino-4-methyl-3-coumarinyl)phenyl]maleimide (CPM; Invitrogen) was dissolved at 4 mg/ml in dimethylformamide (Sigma) and further diluted at a ratio of 1:1000 v/v in sample buffer that consisted of 50 mM HEPES pH 7.0, 150 mM NaCl, 0.05% DDM, and 0.01% CHS (Figure 6a), or in the presence of 75 mM NaCl (Figure 6b). For studying the effects of sodium on receptor melting temperatures (Figure 6C and S6), the receptor was purified with KCl instead of NaCl followed by a desalting step to replace KCl with 75 mM choline chloride. These samples were then diluted in assay buffer containing salt conditions specified in the figures (Figure 6C and S6). Approximately 5 µg of protein with no ligand added (apo) was incubated on ice with CPM buffer containing ligand in a total volume of 200 µL. Assays were performed with a Cary Eclipse spectrofluorometer using quartz cuvettes (Starna Cells, Inc., Atascadero, CA) over a linear temperature range from 20°C to 90°C heated at a constant rate of 2°C/min. The excitation wavelength was 384 nm, and the emission wavelength was 470 nm.
Signaling Assay - cAMP assay
CHO cells (ATCC) were grown in DMEM and F12 (1:1) supplemented with 10% fetal bovine serum, 100 Units/ml penicillin, 100 µg/ml streptomycin, and 2 µmol/ml glutamine. Transfection of the wild-type or mutant A2AAR was performed in 6-well plate (0.5 µg plasmids/well) using Lipofectamine 2000. After 48 hours, cells were treated with assay buffer containing rolipram (10 µM) and adenosine deaminase (3 units/ml) for 30 min followed by the addition of agonists and incubated for 20 min. The reaction was terminated upon removal of the supernatant, and addition of 100 µl Tween-20 (0.3%). Intracellular cAMP levels were measured with an ALPHAScreen cAMP assay kit as instructed by the manufacturer (PerkinElmer).
Radioligand Binding Assay
HEK293 cells (ATCC) were cultured in DMEM supplemented with 10% fetal bovine serum, 100 Units/ml penicillin, 100 µg/ml streptomycin, and 2 µmol/ml glutamine. Lipofectamine 2000 was used for the transfection of the wild-type or mutant A2AAR. Transfected cells were detached from plates by scraping into cold PBS and centrifuged at 250 g for 5 min. The pellets were resuspended in ice-cold Tris HCl buffer (50 mM, pH 7.4) and then homogenized. After homogenization and suspension, cells were centrifuged at 1000 g for 10 min, and the pellet was discarded. The suspension was then re-centrifuged at 20,000 g for 60 min at 4°C. The pellets were resuspended in buffer containing 3 Units/ml adenosine deaminase and incubated at 37°C for 30 min. The aliquots of membrane preparations were stored at −80°C until the binding experiments. The protein concentration was measured using the Bradford assay. For saturation binding, [3H] CGS21680 (ranging from 2 to 100 nM) was incubated with membrane preparations (5–10 µg proteins/tube) for 60 min at 25°C in a total assay volume of 200 µL of 50 mM Tris HCl (pH 7.4) containing 10 mM MgCl2. Nonspecific binding was determined using 10 µM NECA. For displacement binding assays, membrane preparations (10–20 µg proteins/tube) were incubated at 25°C for 60 min with a final concentration of 5 nM [3H] CGS21680 in a mixture containing 50 µL of increasing concentrations of a test ligand in a total assay volume of 200 µL of 50 mM Tris HCl, pH 7.4, containing 10 mM MgCl2. Nonspecific binding was determined using 10 µM of NECA. The reaction was terminated by filtration with GF/B filters. Filters were placed in scintillation vials containing 5 ml of Hydrofluor scintillation buffer and counted using a PerkinElmer Tricarb 2810TR Liquid Scintillation Analyzer.
Homologous Radioligand Binding Assays
Radioligand binding assays were performed as previously described (Massink et al., 2015). Briefly, ligand binding was measured using washed membranes from HEK 293 cells (FreeStyle 293-F, ThermoFisher, R79007) transiently expressing wild-type A2AAR or pointmutant A2AAR constructs. Cells were dounce homogenized (50 mM Tris HCl, pH 7.4) and centrifuged for 15 min at 30,000 g. The pellet was resuspended in assay buffer in the presence of adenosine deaminase (0.8 IU/ml, Roche) to break down endogenous adenosine. Membranes were stored in 250 µl aliquots at −80 °C. Binding assays were carried out in a total volume of 0.125 ml in 96-well plates with a binding buffer with or without 150 mM NaCl as indicated (50 mM Tris HCl, 1 U/ml adenosine deaminase, pH 7.4) containing 0.5 nM [3H] ZM241385 (American Radiolabelled Chemicals, Inc., St. Louis, MO) or 1.0 nM [3H] NECA (Perkin Elmer) for 60 min at room temperature. Membranes were harvested over 0.3% polyethyleneimine-treated, 96-well filter mats using a 96-well Filtermate harvester (Perkin Elmer) and washed three times with cold buffer (50 mM Tris HCl, pH 7.4). Filter mats were dried, wax scintillant was melted onto each filter, and radioactivity was counted in a MicroBeta2 TriLux plate scintillation counter (Perkin Elmer). ZM241385 and NECA binding affinities (KD) for wild-type and D522.50N were determined using homologous competition binding. All cell lines used arrived with a certificate of authenticity from the supplier and morphology and growth rates are constantly monitored. After thirty passages the cells are discarded. Insect and mammalian cells are periodically tested for mycoplasma using MycoAlert™ Mycoplasma Detection kit (Lonza). The cell lines used here were chosen to remain consistent with previous studies and they are well validated for these assays.
Crystallization of A2AAR variants
Solutions containing purified A2AAR variants in complex with UK432097 were concentrated and reconstituted into lipidic cubic phase (LCP) by mixing in a Hamilton gas tight syringe as described previously (Caffrey and Cherezov, 2009). The protein-LCP mixture contained 40% (w/w) protein solution, 54% (w/w) monoolein (Sigma) and 6% (w/w) cholesterol (Avanti Polar Lipids). All crystallization trials were performed in 96-well glass sandwich plates by an NT8-LCP in meso crystallization robot (Formulatrix) using 40 nl protein/LCP drops each overlaid with 0.8 µl precipitant solution. Crystallization trials were conducted at room temperature (~23 °C), and plates stored and imaged at 20 °C in an incubator imager (RockImager 1000, Formulatrix). Diffraction quality crystals were obtained in precipitant conditions containing 100 mM sodium citrate pH 5, 24–27% (v/v) polyethylene glycol (PEG) 400, 30–80 mM MgCl2, 5% (v/v) Jeffamine M-600 pH 7 (Hampton). Crystals appeared about 1 hour after crystallization setup and continued to grow to full size (~200 µm in length) within one week. Crystals were harvested directly from LCP using MicroMounts (MiTeGen) and flash frozen in liquid nitrogen.
Data collection and structure determination
X-ray diffraction data were collected on the 23ID-D and 23ID-B beamlines (GM/CA-CAT) at the Advanced Photon Source (Argonne, IL) using a 20 µm minibeam with a Pilatus3-6M and Eiger-16m, respectively. Crystals embedded in LCP were not visible in the mesophase after flash-freezing in liquid nitrogen, so a previously reported common rastering and datacollection strategy was used as described (Cherezov et al., 2009). We collected 30–50 frames with 0.2–0.5° oscillation per crystal to minimize radiation damage with a 1s exposure using an unattenuated beam. Data were integrated, scaled, and merged using HKL2000 (Otwinowski and Minor, 1997). Data from 24 crystals and 25 crystals were merged for D52N-UK432097 and S91A-UK432097, respectively. Diffraction of D52N–UK432097 yielded a 92.3% complete dataset at 2.6 Å and S91A–UK432097 crystals yielded a 91.9 % complete dataset at 2.9 Å. In order to minimize phase bias from the model, the initial structures were obtained by molecular replacement (MR) using PHASER (McCoy et al., 2007) in the CPP4 suite using A2AAR and T4L as separate search models from either A2AAR–ZM241385 or A2AAR–UK432097 structures (3EML (Jaakola et al., 2008) or 3QAK (Xu et al., 2011)). The results between the two search models were very similar, and our final structures were obtained using A2AAR-ZM241385 (3EML) as the search model. The resulting models of A2AAR–T4L variants were refined with phenix.refine (PHENIX) followed by manual examination and rebuilding of the refined coordinates in COOT (Emsley et al., 2010) using both sigma-A weighted 2|Fo|-|Fc| and |Fo|-|Fc| maps. The data collection and refinement statistics are shown in Table 2.
Table 2.
Data collection and refinement statistics
| D52N-UK432097 PDB 5WF5 |
S91A-UK432097 PDB 5WF6 |
|
|---|---|---|
| Data collection | ||
| Number of crystals | 24 | 25 |
| Space group | P21 | P21 |
| Cell dimensions | ||
| a, b, c (Å) | 47.69, 77.77, 86.49 | 47.78, 78.22, 86.48 |
| β (°) | 100.05 | 101.20 |
| No. Total Reflections | 78572 | 42494 |
| No. Unique Reflections | 17786 | 13000 |
| Resolution (Å) | 46.80–2.60 (2.69–2.60)* | 44.88–2.90 (3.00–2.90)* |
| Rsym or Rmerge | 0.180 (0.880) | 0.218 (0.710) |
| I / σI | 8.1 (1.1) | 4.6 (1.3) |
| Completeness (%) | 92.3 (69.4) | 91.9 (74.5) |
| Redundancy | 4.4 (3.0) | 3.3 (2.3) |
| Highest Shell CC1/2 | 0.50 | 0.52 |
| Refinement | ||
| Resolution (Å) | 29.9–2.60 | 29.49–2.90 |
| No. reflections / test set | 17709 / 907 | 12850 / 660 |
| Rwork / Rfree | 0.214 / 0.251 | 0.254 / 0.289 |
| No. atoms | ||
| Protein | 3479 | 3405 |
| UK432097 | 57 | 57 |
| Lipids and others | 29 | 11 |
| WILSON B-factors (Å2) | 75.6 | 92.2 |
| Average B-factors (Å2) | ||
| A2a | 82.9 | 98.7 |
| T4L | 80.0 | 92.5 |
| UK432097 | 77.3 | 89.2 |
| Lipids and others | 124.8 | 111.3 |
| R.m.s. deviations | ||
| Bond lengths (Å) | 0.003 | 0.003 |
| Bond angles (°) | 0.46 | 0.48 |
There were no Ramachandran outliers for any structures.
Values in parentheses are for highest-resolution shell.
QUANTIFICATION AND ANALYSIS
Fluorescent Thermal Shift Assays - CPM Assays
All thermal shift data were analyzed with the program Prism (version 7.0 for Windows, GraphPad Software, La Jolla, CA). To determine melting temperatures (Tm), the data were normalized and then fit to a Boltzmann sigmoidal function and the means ± S.D. are shown in Table S1. All CPM experiments were carried out in triplicate or more for statistical analysis.
The data were presented in a bar graph as means ± S.E.M. and assessed using one-way analysis of variance (ANOVA) to determine the effect of receptor variants on melting temperature for a range of drug treatments (Figure S5). For each drug treatment a Dunnett’s multiple comparison test was performed to determine if variant receptors had a significant effect on melting temperatures relative to A2AAR.
Radioligand Binding and Functional Assays
Binding and functional parameters were calculated using Prism 7.0 software (GraphPAD, San Diego, CA). IC50 values obtained from competition curves were converted to Ki values using the Cheng-Prusoff equation. Data were expressed as mean ± S.D. and done three times in triplicate.
Homologous Radioligand Binding Assays
The data were analysed by Prism 7.0 (GraphPad Software) to give KD values and reported as the mean ± S.D. and done three times or more in triplicate.
DATA AVAILABILITY
Coordinates and structure factors have been deposited in the Protein Data Bank for the A2AAR variant structures D52N–UK432097 (PDB ID 5WF5) and S91A–UK432097 (PDB ID 5WF6). The PDB accession codes 4EIY, 3QAK, 3EML, and 5G35 were referenced in this study. The UniProt accession codes P29274 for human A2AAR was used in this study. All other data are available from the corresponding authors on reasonable request.
Supplementary Material
Highlights.
X-ray structures of A2AAR variants D2.50N and S3.39A agonist complexes
A2AAR-D2.50N shows striking loss of G protein signaling
Structural changes near activation motif correspond with loss of signaling
D2.50N improves GPCR stability for accelerating drug discovery
Acknowledgments
KLW was funded by the Weike Zhang Postdoctoral Fellowship at USC. MTE acknowledges funding from an American Cancer Society postdoctoral fellowship. The authors also acknowledge additional support from the USC Bridge Undergraduate Science (BUGS) research program (to AD and TL). ZGG and KAJ thank NIDDK Intramural Res. (ZIADK031117) for support. The authors would like to thank Vadim Cherezov for feedback on the manuscript, Jeffrey Velasquez for assistance with construct cloning, Meihua Chu, Kelly Villers, and Chris Hanson for baculovirus expression, and Angela Walker for assistance with manuscript preparation.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Author Contributions
KLW, MTE, and RCS designed the project. KLW and MTE optimized, purified, and crystallized the receptors in LCP. KLW collected and processed synchrotron data for D52N–UK43209 and S91A–UK43209. KLW and GWH solved and refined structures. ZGG conducted radioligand binding and G protein signaling assays. KLW performed homologous radioligand binding assays with ZM241385 and NECA. KLW, MTE, TL, and AD generated and purified A2AAR and A2AAR variant constructs. KLW, MTE, TL, and AD performed thermal denaturation assays. KLW, MTE, and NP analyzed the structures. KLW and MTE wrote the manuscript with contributions from RCS, KAJ, VK, and all other coauthors.
Declaration of Interests
Authors have no conflicts of interests.
References
- Alexandrov AI, Mileni M, Chien EYT, Hanson MA, Stevens RC. Microscale Fluorescent Thermal Stability Assay for Membrane Proteins. Structure. 2008;16:351–359. doi: 10.1016/j.str.2008.02.004. [DOI] [PubMed] [Google Scholar]
- Ballesteros JA, Weinstein H. Integrated methods for the construction of three-dimensional models and computational probing of structure-function relations in G proteincoupled receptors. Methods Neurosci. 1995;25:366–428. [Google Scholar]
- Bonneau O, Wyss D, Ferretti S, Blaydon C, Stevenson CS, Trifilieff A. Effect of adenosine A2A receptor activation in murine models of respiratory disorders. Am J Physiol Lung Cell Mol Physiol. 2006;290:L1036–1043. doi: 10.1152/ajplung.00422.2005. [DOI] [PubMed] [Google Scholar]
- Borroto-Escuela DO, Romero-Fernandez W, García-Negredo G, Correia PA, Garriga P, Fuxe K, Ciruela F. Dissecting the conserved NPxxY motif of the M3 muscarinic acetylcholine receptor: critical role of Asp-7.49 for receptor signaling and multiprotein complex formation. Cell Physiol Biochem. 2011;28:1009–1022. doi: 10.1159/000335788. [DOI] [PubMed] [Google Scholar]
- Bouley R, Sun TX, Chenard M, McLaughlin M, McKee M, Lin HY, Brown D, Ausiello DA. Functional role of the NPxxY motif in internalization of the type 2 vasopressin receptor in LLC-PK1 cells. Am J Physiol Cell Physiol. 2003;285:C750–C762. doi: 10.1152/ajpcell.00477.2002. [DOI] [PubMed] [Google Scholar]
- Caffrey M, Cherezov V. Crystallizing membrane proteins using lipidic mesophases. Nat Protoc. 2009;4:706–731. doi: 10.1038/nprot.2009.31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carpenter B, Nehmé R, Warne T, Leslie AGW, Tate CG. Structure of the adenosine A(2A) receptor bound to an engineered G protein. Nature. 2016;536:104–107. doi: 10.1038/nature18966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ceresa BP, Limbird LE. Mutation of an aspartate residue highly conserved among G-protein-coupled receptors results in nonreciprocal disruption of alpha 2-adrenergic receptor-G-protein interactions. A negative charge at amino acid residue 79 forecasts alpha 2A-adrenergic receptor sensitivity to allosteric modulation by monovalent cations and fully effective receptor/G-protein coupling. J Biol Chem. 1994;269:29557–29564. [PubMed] [Google Scholar]
- Changeux J-P, Christopoulos A. Allosteric Modulation as a Unifying Mechanism for Receptor Function and Regulation. Cell. 2016;166:1084–1102. doi: 10.1016/j.cell.2016.08.015. [DOI] [PubMed] [Google Scholar]
- Chen D, Errey JC, Heitman LH, Marshall FH, IJzerman AP, Siegal G. Fragment Screening of GPCRs Using Biophysical Methods: Identification of Ligands of the Adenosine A 2AReceptor with Novel Biological Activity. ACS Chem. Biol. 2012;7:2064–2073. doi: 10.1021/cb300436c. [DOI] [PubMed] [Google Scholar]
- Cherezov V, Hanson MA, Griffith MT, Hilgart MC, Sanishvili R, Nagarajan V, Stepanov S, Fischetti RF, Kuhn P, Stevens RC. Rastering strategy for screening and centring of microcrystal samples of human membrane proteins with a sub-10 microm size X-ray synchrotron beam. J R Soc Interface. 2009;6(Suppl 5):S587–597. doi: 10.1098/rsif.2009.0142.focus. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chun E, Thompson AA, Liu W, Roth CB, Griffith MT, Katritch V, Kunken J, Xu F, Cherezov V, Hanson MA, et al. Fusion partner toolchest for the stabilization and crystallization of G protein-coupled receptors. Structure. 2012;20:967–976. doi: 10.1016/j.str.2012.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cieslak M, Komoszynski M, Wojtczak A. Adenosine A(2A) receptors in Parkinson's disease treatment. Purinergic Signal. 2008;4:305–312. doi: 10.1007/s11302-008-9100-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dodevski I, Pluckthun A. Evolution of three human GPCRs for higher expression and stability. J Mol Biol. 2011;408:599–615. doi: 10.1016/j.jmb.2011.02.051. [DOI] [PubMed] [Google Scholar]
- Emsley P, Lohkamp B, Scott WG, Cowtan K. Features and development of Coot. Acta Crystallogr D Biol Crystallogr. 2010;66:486–501. doi: 10.1107/S0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fenalti G, Giguere PM, Katritch V, Huang X-P, Thompson AA, Cherezov V, Roth BL, Stevens RC. Molecular control of δ-opioid receptor signalling. Nature. 2014;506:191–196. doi: 10.1038/nature12944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fritze O, Filipek S, Kuksa V, Palczewski K, Hofmann KP, Ernst OP. Role of the conserved NPxxY(x)5,6F motif in the rhodopsin ground state and during activation. Proc Natl Acad Sci USA. 2003;100:2290–2295. doi: 10.1073/pnas.0435715100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galés C, Kowalski-Chauvel A, Dufour MN, Seva C, Moroder L, Pradayrol L, Vaysse N, Fourmy D, Silvente-Poirot S. Mutation of Asn-391 within the Conserved NP XXY Motif of the Cholecystokinin B Receptor Abolishes G qProtein Activation without Affecting Its Association with the Receptor. J. Biol. Chem. 2000;275:17321–17327. doi: 10.1074/jbc.M909801199. [DOI] [PubMed] [Google Scholar]
- Govaerts C, Lefort A, Costagliola S, Wodak SJ, Ballesteros JA, Van Sande J, Pardo L, Vassart G. A Conserved Asn in Transmembrane Helix 7 Is an On/Off Switch in the Activation of the Thyrotropin Receptor. J. Biol. Chem. 2001;276:22991–22999. doi: 10.1074/jbc.M102244200. [DOI] [PubMed] [Google Scholar]
- Gutierrez-de-Teran H, Massink A, Rodriguez D, Liu W, Han GW, Joseph JS, Katritch I, Heitman LH, Xia L, Ijzerman AP, et al. The role of a sodium ion binding site in the allosteric modulation of the A(2A) adenosine G protein-coupled receptor. Structure. 2013;21:2175–2185. doi: 10.1016/j.str.2013.09.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hishinuma S, Kosaka K, Akatsu C, Uesawa Y, Fukui H, Shoji M. Asp73-dependent and -independent regulation of the affinity of ligands for human histamine H1 receptors by Na. Biochem Pharmacol. 2017;128:46–54. doi: 10.1016/j.bcp.2016.12.021. [DOI] [PubMed] [Google Scholar]
- Hulme EC. GPCR activation: a mutagenic spotlight on crystal structures. Trends Pharmacol. Sci. 2013;34:67–84. doi: 10.1016/j.tips.2012.11.002. [DOI] [PubMed] [Google Scholar]
- Jaakola V-P, Griffith MT, Hanson MA, Cherezov V, Chien EYT, Lane JR, IJzerman AP, Stevens RC. The 2.6 angstrom crystal structure of a human A2A adenosine receptor bound to an antagonist. Science. 2008;322:1211–1217. doi: 10.1126/science.1164772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katritch V, Cherezov V, Stevens RC. Diversity and modularity of G protein-coupled receptor structures. Trends Pharmacol. Sci. 2012;33:17–27. doi: 10.1016/j.tips.2011.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katritch V, Fenalti G, Abola EE, Roth BL, Cherezov V, Stevens RC. Allosteric sodium in class A GPCR signaling. Trends Biochem. Sci. 2014;39:233–244. doi: 10.1016/j.tibs.2014.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lebon G, Bennett K, Jazayeri A, Tate CG. Thermostabilisation of an agonist-bound conformation of the human adenosine A(2A) receptor. J Mol Biol. 2011a;409:298–310. doi: 10.1016/j.jmb.2011.03.075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lebon G, Warne T, Edwards PC, Bennett K, Langmead CJ, Leslie AGW, Tate CG. Agonist-bound adenosine A2A receptor structures reveal common features of GPCR activation. Nature. 2011b;474:521–525. doi: 10.1038/nature10136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leone RD, Lo Y-C, Powell JD. A2aR antagonists: Next generation checkpoint blockade for cancer immunotherapy. CSBJ. 2015;13:265–272. doi: 10.1016/j.csbj.2015.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu W, Chun E, Thompson AA, Chubukov P, Xu F, Katritch V, Han GW, Roth CB, Heitman LH, AP IJ, et al. Structural basis for allosteric regulation of GPCRs by sodium ions. Science. 2012;337:232–236. doi: 10.1126/science.1219218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu ZL, Saldanha JW, Hulme EC. Transmembrane domains 4 and 7 of the M(1) muscarinic acetylcholine receptor are critical for ligand binding and the receptor activation switch. J. Biol. Chem. 2001;276:34098–34104. doi: 10.1074/jbc.M104217200. [DOI] [PubMed] [Google Scholar]
- Massink A, Gutierrez-de-Teran H, Lenselink EB, Ortiz Zacarias NV, Xia L, Heitman LH, Katritch V, Stevens RC, AP IJ. Sodium ion binding pocket mutations and adenosine A2A receptor function. Mol Pharmacol. 2015;87:305–313. doi: 10.1124/mol.114.095737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC, Read RJ. Phaser crystallographic software. J Appl Crystallogr. 2007;40:658–674. doi: 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neve KA. Regulation of dopamine D2 receptors by sodium and pH. Mol. Pharmacol. 1991;39:570–578. [PubMed] [Google Scholar]
- Otwinowski Z, Minor W. Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- Pardo L, Deupi X, Dölker N, López-Rodríguez ML, Campillo M. The Role of Internal Water Molecules in the Structure and Function of the Rhodopsin Family of G Protein-Coupled Receptors. Chembiochem. 2007;8:19–24. doi: 10.1002/cbic.200600429. [DOI] [PubMed] [Google Scholar]
- Pert CB, Pasternak G, Snyder SH. Opiate agonists and antagonists discriminated by receptor binding in brain. Science. 1973;182:1359–1361. doi: 10.1126/science.182.4119.1359. [DOI] [PubMed] [Google Scholar]
- Raitio KH, Salo OMH, Nevalainen T, Poso A, Järvinen T. Targeting the cannabinoid CB2 receptor: mutations, modeling and development of CB2 selective ligands. Curr. Med. Chem. 2005;12:1217–1237. doi: 10.2174/0929867053764617. [DOI] [PubMed] [Google Scholar]
- Schutz M, Schoppe J, Sedlak E, Hillenbrand M, Nagy-Davidescu G, Ehrenmann J, Klenk C, Egloff P, Kummer L, Pluckthun A. Directed evolution of G protein-coupled receptors in yeast for higher functional production in eukaryotic expression hosts. Sci Rep. 2016;6:21508. doi: 10.1038/srep21508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shepherd CA, Hopkins AL, Navratilova I. Fragment screening by SPR and advanced application to GPCRs. Prog. Biophys. Mol. Biol. 2014;116:113–123. doi: 10.1016/j.pbiomolbio.2014.09.008. [DOI] [PubMed] [Google Scholar]
- Shibata Y, White JF, Serrano-Vega MJ, Magnani F, Aloia AL, Grisshammer R, Tate CG. Thermostabilization of the neurotensin receptor NTS1. J Mol Biol. 2009;390:262–277. doi: 10.1016/j.jmb.2009.04.068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trzaskowski B, Latek D, Yuan S, Ghoshdastider U, Debinski A, Filipek S. Action of molecular switches in GPCRs--theoretical and experimental studies. Curr. Med. Chem. 2012;19:1090–1109. doi: 10.2174/092986712799320556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Westhuizen ET, Valant C, Sexton PM, Christopoulos A. Endogenous Allosteric Modulators of G Protein-Coupled Receptors. J. Pharmacol. Exp. Ther. 2015;353:246–260. doi: 10.1124/jpet.114.221606. [DOI] [PubMed] [Google Scholar]
- Venkatakrishnan AJ, Deupi X, Lebon G, Heydenreich FM, Flock T, Miljus T, Balaji S, Bouvier M, Veprintsev DB, Tate CG, et al. Diverse activation pathways in class A GPCRs converge near the G-protein-coupling region. Nature. 2016;536:484–487. doi: 10.1038/nature19107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venkatakrishnan AJ, Deupi X, Lebon G, Tate CG, Schertler GF, Babu MM. Molecular signatures of G-protein-coupled receptors. Nature. 2013;494:185–194. doi: 10.1038/nature11896. [DOI] [PubMed] [Google Scholar]
- Werling LL, Brown SR, Puttfarcken P, Cox BM. Sodium regulation of agonist binding at opioid receptors. II. Effects of sodium replacement on opioid binding in guinea pig cortical membranes. Mol. Pharmacol. 1986;30:90–95. [PubMed] [Google Scholar]
- Xu F, Wu H, Katritch V, Han GW, Jacobson KA, Gao Z-G, Cherezov V, Stevens RC. Structure of an agonist-bound human A2A adenosine receptor. Science. 2011;332:322–327. doi: 10.1126/science.1202793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang D, Gao ZG, Zhang K, Kiselev E, Crane S, Wang J, Paoletta S, Yi C, Ma L, Zhang W, et al. Two disparate ligand-binding sites in the human P2Y1 receptor. Nature. 2015a;520:317–321. doi: 10.1038/nature14287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Stevens RC, Xu F. The importance of ligands for G protein-coupled receptor stability. Trends Biochem Sci. 2015b;40:79–87. doi: 10.1016/j.tibs.2014.12.005. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Coordinates and structure factors have been deposited in the Protein Data Bank for the A2AAR variant structures D52N–UK432097 (PDB ID 5WF5) and S91A–UK432097 (PDB ID 5WF6). The PDB accession codes 4EIY, 3QAK, 3EML, and 5G35 were referenced in this study. The UniProt accession codes P29274 for human A2AAR was used in this study. All other data are available from the corresponding authors on reasonable request.